
Single atoms supported on metal oxides for energy catalysis
Runze
Li
,
Lei
Luo
,
Xinlong
Ma
,
Wenlong
Wu
,
Menglin
Wang
and
Jie
Zeng
 *
*
Hefei National Laboratory for Physical Sciences at the Microscale, Key Laboratory of Strongly-Coupled Quantum Matter Physics of Chinese Academy of Sciences, Hefei Science Center, National Synchrotron Radiation Laboratory, Department of Chemical Physics, University of Science and Technology of China, Hefei, Anhui 230026, P. R. China. E-mail: zengj@ustc.edu.cn
First published on 3rd November 2021
Abstract
Single-atom catalysts (SACs) have attracted wide interest from researchers, as they promisingly bridge the gap between homogeneous and heterogeneous catalysts. There are various types of supports reported to anchor single metal atoms. Among them, metal oxides have received much attention because of their specific and variable properties. In this review, the unique role of typical oxide supports in anchoring isolated metal atoms and participating in catalysis is emphasized. The interactions between metal atoms and oxide supports are clarified, so as to show how to stabilize the atomic metal sites and adjust the geometric and electronic structure of single atoms reasonably. Accordingly, we highlight and summarize the progress made in the synthesis of SACs in recent years, placing special emphasis on preventing isolated metal atoms from migrating or agglomerating. In addition, the modern characterization technologies of SACs are summarized and discussed, which can provide strong evidence on the geometrical structures and electronic states of SACs. Then the applications of SACs supported on metal oxides in energy catalytic reactions are reviewed, such as CO oxidation, the water–gas shift reaction, CH4 conversion, CO2 hydrogenation, oxygen reduction reaction, hydrogen evolution reaction, and CO2 reduction reaction. Finally, the existing problems and development prospects in this field are discussed.
 Runze Li | Runze Li received his B.S. degree from University of Science and Technology of China in 2021. He is currently a Ph.D. candidate at Tsinghua University under the supervision of Prof. Dingsheng Wang. His research interests are focused on the design and synthesis of novel functional single-atom catalysts and their applications in energy conversion and storage. |
 Lei Luo | Lei Luo received his B.S. degree (2011) and master's degree (2013) from Harbin Institute of Technology, and obtained a PhD (2019) at Shanghai Jiao Tong University. He worked with Prof. Jie Zeng as a postdoctoral fellow at University of Science and Technology of China from 2019 to 2021 and was promoted to Research Associate in 2021. His research interests are focused on the design of catalytic reaction device and selective and efficient conversion of carbon-based small molecules. |
 Wenlong Wu | Wenlong Wu received his Ph.D. degree from the University of Science and Technology of China (USTC) under the supervision of Prof. Jie Zeng in 2019. Then he joined USTC and was promoted to an associate research fellow in 2021. His research interests are focused on the design and synthesis of well-defined catalysts and their applications in CO2 conversion. |
 Menglin Wang | Menglin Wang received her BS degree in materials science and engineering at the USTC in 2014. She received her PhD degree with Prof. Jie Zeng in the Department of Chemical Physics at the USTC. Then she joined USTC and was promoted to an associate research fellow in 2021. Her work now focuses on the precise fabrication of surface/interface structures of catalysts. |
 Jie Zeng | Dr Jie Zeng is a Professor of Chemistry at the University of Science and Technology of China (USTC) and PI of the Hefei National Laboratory for Physical Sciences at the Microscale (HFNL). He studied applied chemistry at the USTC (BS, 2002) and received a PhD in condensed matter physics under the tutelage of Prof. J. G. Hou and Prof. Xiaoping Wang (2007). He worked with Prof. Younan Xia as a postdoctoral fellow at Washington University in St. Louis from 2008 to 2011 and was promoted to Research Assistant Professor in 2011. In 2012, he relocated to USTC to take the position of Professor for Chemistry in Hefei National Laboratory for Physical Sciences at the Microscale. His research interests include the selective and efficient conversion of carbon-based small molecules (such as CO, CO2 and CH4) to liquid fuels and high value-added chemicals from both materials and mechanistic aspects. |
1. Introduction
Catalysis has been greatly developed in scientific research.1,2 A high specific surface area is very important for improving the performance of catalysts, especially their activity.3–6 The reactants usually interact only with the active sites on the surface, and on some occasions the inner atoms play a negligible role in the reaction.7,8 Based on this, the proportion of active surface atoms needs to be maximized. This is especially important for high-performing noble metals because they are scarce and expensive. Zhang and co-workers studied the preparation and performance of single-atom catalysts (SACs), and on this basis, put forward the concept of “single-atom catalysis” for the first time.9 Since then, single-atom catalysis has become a cutting-edge domain of heterogeneous catalysis.10 SACs are a type of catalyst that consist of isolated metal atoms and support materials.11 The catalytically active centers in SACs are isolated metal atoms, and sometimes adjacent atoms or surface functional groups, on the surface of the support. The catalytic performance of supported single atoms is strongly dependent on the interaction with the surface of the support.12 SACs are conceptually different from single site heterogeneous catalysts (SSHCs). According to its definition, a SSHC can be composed of more than one atom.13 It can be composed of atomic groups, as long as all of the groups behave identically in catalysis. SACs are equivalent to SSHCs only when all of the isolated metal atoms interact with the surface or the surface functional groups of the support in the same way. Nowadays rare-earth metals can also be used to prepare SACs.14 SACs exhibit maximized atomic utilization efficiency and can be rationally designed for target reactions on the atomic scale. However, due to the complex and changeable nature of heterogeneous metal catalysts, it is difficult for us to understand their catalytic mechanism at the molecular level, so as to develop industrial catalysts with perfect catalytic selectivity for desired products.15 Furthermore, the advantages of homogeneous and heterogeneous catalysis are combined in SACs and the selectivity can be increased.16 However, characterized by their high surface energy, isolated metal atoms can easily migrate or agglomerate into clusters, and are thus considered unstable.17–19 To improve the stability of isolated atoms, various traditional and novel synthesis methods have been developed.20–25 Advanced characterization techniques are important to develop SACs, as they can provide useful information for determining the geometries and electronic states of SACs.26 Moreover, SACs have been widely used not only in the fields of thermocatalysis,27–30 electrocatalysis,31–37 and photocatalysis,38 but also in the fields of batteries39,40 and organic chemistry.41As far as supported metal catalysts are concerned, the morphology and the valence state of the metal structure will change according to the support materials. The range of support materials of SACs has been extended from metal oxides42,43 to metal nanomaterials,44 carbon-based materials,34,36,45–55 hexagonal boron nitride (h-BN),56 zeolites,19 metal–organic frameworks (MOFs),57 covalent–organic frameworks (COFs),58 two-dimensional materials (such as MoS2 and MXene),59–63 and MoC.64,65 Among the several types of supported SACs, metal oxide supported SACs have received much attention because of the specific and variable properties observed in metal oxides, including surface pH and redox properties. The reducible oxide supports exhibit various defects, including oxygen vacancies (Ov), metal vacancies (Mv), edges, steps, and terraces, which are suggested to be suitable anchor sites.66–72 However, as all defect sites can capture metal atoms, determining the exact anchor location is quite a challenge. On the other hand, irreducible oxides (such as Al2O3) have fewer surface defects, and thus their interaction with metal species is weak. Nevertheless, the surfaces of these supports usually have abundant coordinatively unsaturated sites. For instance, there are a large number of unsaturated pentahedral Al3+ sites on Al2O3, which can stabilize noble metal Pt atoms.73 In contrast, on oxide support surfaces, transition metal atoms can coordinate with excess oxygen ions through metal–oxo/hydroxyl group (M–O(OH)) interactions.74,75 By inhibiting possible aggregation, spatially confining isolated metal species to the porous nanostructures of oxide supports is another method to maintain atomic dispersion.76,77 The support not only disperses and stabilizes the metal atoms, but also interacts with the metal atoms, which often leads to charge transfer on surfaces, a change in the metal structure and the modulation of molecular adsorption, thus affecting the performance of catalysts.23,78,79 Metal–support interactions (MSI) require very prominent charge transfer between metal species and the surfaces of the supports, leading to reversible structure change at the nanoscale. The reversible suppression of CO adsorption has been observed on Pt/TiO2 SACs.80 Although MSI between single Pt atoms and the TiO2(B) support does not occur after low temperature reduction, the adsorption of CO on single Pt atoms completely disappears when the reduction temperature reaches 600 °C. Subsequent re-oxidation at 300 °C restores the CO adsorption on nanoparticles (NPs) and single atoms. This proves that the stopping of the CO adsorption on single Pt atoms is reversible.
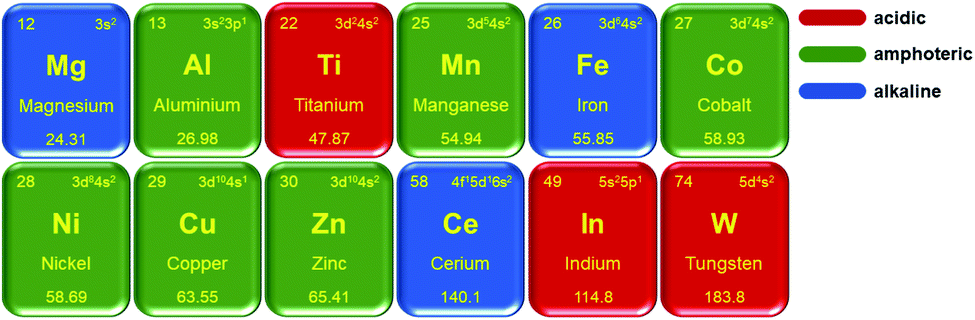
We briefly introduce several typical metal oxides as supports here. Fig. 1 shows the acidity and alkalinity of commonly used metal oxide supports. The acidity and alkalinity of metal oxides reflect their affinity to electrons and protons, respectively, thereby affecting the types of adsorbed metal groups. As electrophilic supports, acidic oxides tend to adsorb electron-rich groups such as anionic clusters, while alkaline oxides tend to adsorb electron-deficient groups.25 Meanwhile, the valence states of adsorbed metal species might be affected by the difference in the acidity and alkalinity of oxides. Iron can coordinate with oxygen easily and is a non-noble metal and one of the most abundant elements. On account of the redox properties of iron, different pretreatment methods and reaction environments can lead to different crystalline phases of iron oxides.81 Among these oxides, FeOx has been the most widely used and investigated as a support for SACs.82–88 The reconstruction of (√2 × √2)-R45° on the Fe3O4(001) surface has been determined in previous studies of SACs dispersed on Fe3O4, in which the subsurface Fe vacancies induce it, featuring pockets that can contain isolated atoms.89–91 Fe vacancies are also found in iron oxides of other crystalline phases, including Fe2O3.92 In addition to vacancies in defective iron oxide, strong covalent metal–support interactions (CMSI) between the metal atoms and the non-defective iron oxide can also stabilize isolated metal atoms due to the redox properties of iron oxide.
 | ||
| Fig. 1 Acidity and alkalinity of metal oxides. | ||
Ceria (CeO2) has also been investigated and applied as a support for SACs, due to its unique properties.93 CeO2 has a high density of vacancies.94 In addition, CeO2 stores oxygen via a reversible process. The local coordination environment and electron states of metal atom sites in SACs can be significantly modulated after thermal activation. Metal nanocrystalline structures on CeO2 supports can form covalent M–O bonds and promote the dispersion of metal atoms on the atomic scale after oxidative calcination.22,95 These properties make CeO2 a support applicable to many catalytic reactions of small molecule conversions, such as CO oxidation, semi-hydrogenation of acetylene, and CH4 reforming.67,68,96,97 For instance, single Au atoms tend to occupy surface Ce vacancies, thus forming a higher loading of metal species than in an FeOx support.72
Titanium dioxide (TiO2) is one of the most representative semiconductor materials and a suitable photochemical support.98,99 In the natural environment, rutile, anatase, and brookite are three typical crystalline phases of TiO2, of which rutile is the most stable. In thermocatalysis, electrocatalysis and chemical sensors, charge transfer can be triggered from metal atoms to the supports. This facilitates the formation of surface oxygen vacancies and Ti3+ sites of TiO2, and ultimately helps construct the active interface that results from the interaction between metal atoms and TiO2.66,76,80,100
In general, the interaction between metal atoms and oxides can be weakened by the reduction of the isolated metal cations to their metallic state, which eventually results in the formation of metal clusters or NPs. However, zinc oxide (ZnO) and copper oxides (CuO and Cu2O) are able to maintain the metallic or even negatively charged states of isolated metal atoms, which is one of their most attractive properties as supports for SACs.101–103
Al2O3 has a number of particular crystalline phases.104 In comparison to reducible oxides, Al2O3 has much weaker metal–support interactions to stabilize the isolated active metal atoms, so further modification of the support is usually required. For example, mesoporous alumina exhibits more Al3+ defects and thus improved performance when compared with traditional bulk Al2O3.73,105,106 In addition, the thermal stability of SACs can be enhanced using a MgAl2O4 spinel support during severe oxidative aging.107,108 In addition, mixing with alkaline-earth or rare-earth metal elements can also alter the bond strength between single atoms and support oxygens.109–111 The following sections will introduce these oxides in detail.
This review will focus on SACs supported by metal oxides. The performance of SACs is closely related to their geometrical and electronic structures. Therefore, we have chosen some typical and recent examples to explore the relationship between structure and performance and understand the interactions between the single metal atoms and metal oxide support. Firstly, the preparation methods of SACs supported by metal oxides will be introduced, including several mature synthesis strategies. Then, based on cutting-edge characterization technologies, the importance of characterization in understanding the structure of catalysts will be emphasized, and the commonly used characterization technologies for SACs will be introduced. Finally, we will discuss the application of SACs in some important catalytic reactions, as well as the related theoretical research and reaction mechanisms. The last section gives a brief summary and prospects in the development of SACs. Some potential problems will be put forward, and the direction for the following investigations will be pointed out.
2. Preparation of SACs supported by metal oxides
The preparation of highly-dispersed single atoms on an appropriate support is a precondition for the application of SACs. Many metal oxides, including FeOx, CeO2, TiO2, ZnO and Al2O3, are promising supports for SACs. They can not only stabilize isolated metal atoms, but also participate in reactions.11 The surface cations of a metal oxide support can be replaced with single metal atoms and because of the interactions with neighboring oxygen anions, the metal atoms can be strongly anchored. In 2011, a co-precipitation method was employed by Zhang and co-workers to prepare Pt1/FeOx, which exhibits excellent activity and stability in CO oxidation.9 Even so, preparing SACs is challenging because individual metal atoms tend to migrate and aggregate during synthesis and subsequent processing because isolated atoms usually have high surface energy, which limits the loading of isolated atoms.112 Therefore, the improvement of metal loading at an order-of-magnitude level will bring hope to the practical applications of SACs.113,114The synthetic methods generally include two categories according to the different starting metal precursors, “bottom-up” and “top-down” routes.42,115 In the “bottom-up” route, including wet-chemistry methods, atomic layer deposition (ALD), and mass-selected soft-landing techniques, mononuclear metal precursors are deposited on the support directly or mixed with support precursor materials, while in the “top-down” strategy, metal clusters, NPs or even metal bulk are precursors. The migration of free metal precursors is limited by coordination groups (mainly hydroxyl groups) or the vacancies of oxide supports in the “bottom-up” synthesis strategy.113 Such enhanced metal–oxide interactions are the key to stabilizing SACs. However, these SACs often suffer from low yield, low metal loading, inhomogeneous and ambiguously defined coordination environments for single metal atoms, and occasional requirements for complex or expensive equipment. On the other hand, the “top-down” strategy involves two steps, namely the breaking of metal–metal bonds in the NP precursors and the forming of a new strong bond with the surface of the support, with the potential to overcome the defects associated with the “bottom-up” strategy.42 We will discuss some of the methods that have been widely used to synthesize SACs in this section, with emphasis on SACs supported by metal oxides.
2.1. Wet-chemistry methods
To avoid extra expense and production scale limitations, most attention has been focused on wet-chemistry methods, such as co-precipitation,9 impregnation,116 ion-exchange,117 deposition–precipitation,118 strong electrostatic adsorption (SEA),119etc., due to their ready accessibility in most laboratories and relative ease to achieve the large-scale preparation of SACs in commercial use. The wet chemistry approach requires no special equipment and can be routinely carried out. In such a method, single metal atom species are already included in the precursor materials, and metal complexes are aimed to be anchored onto the supports without aggregation and migration during the preparation or post-treatment processes via a chemical reaction. As a result, strong metal–support interactions play an important role in preventing the aggregation of supported isolated atoms.The first studied single-atom catalyst was prepared via a co-precipitation method.9 Single atom Pt1/FeOx catalysts were synthesized in an aqueous solution of chloroplatinic acid (H2PtCl6·6H2O) mixed with ferric nitrate (Fe(NO3)3·9H2O) and sodium carbonate (Na2CO3) at 50 °C, and the resulting solution was controlled at a pH value of around 8. The obtained solid was dried for 5 h at 60 °C, calcined for 5 h at 400 °C and then reduced in 10% H2/He for 0.5 h at 200 °C. A high-surface-area FeOx nanocrystallite support with Pt metal atoms was obtained and the Pt loading was controlled to 0.17 wt%. The SACs were shown to have exceedingly high atom efficiency, distinguished stability and high activity when tested for preferential CO oxidation in a H2-rich environment. This research shows the bright prospects for the preparation of SACs using a co-precipitation method.
A key problem in the preparation of catalysts is how to increase the metal loading of SACs. Recently, Zhang and co-workers developed their preparation method for Pt1/FeOx, where the Pt loading was up to 1.8 wt%, as determined by inductively-couple plasma mass spectrometry.93 A co-precipitation method was applied in the preparation of Pt1/FeOx in an aqueous solution mixture of H2PtCl6·6H2O and Fe(NO3)3·9H2O with Na2CO3 solution under 3 h of stirring at 50 °C, and a further 2 h of static aging. After the filtered solids were washed with deionized water, the obtained samples were dried at 60 °C overnight. Therefore, carefully modulation of the preparation details of the co-precipitation method is vital for the preparation of SACs. The Pt1/FeOx catalyst exhibits excellent catalytic performance for methane combustion. In addition, many other noble metal SACs have been successfully dispersed onto FeOx, such as Ir1/FeOx, the stability of which results from the hydroxyl groups and the structural defects derived from the poorly crystallized FeOx support with a large surface area.87,88
However, the interfacial regions of the support agglomerate or the bulk of the support crystallites can bury some metal atoms, which are major problems with this method of preparing SACs.11 Thus, these buried single atoms cannot easily make contact with reactant molecules or may be wasted, significantly compromising the utilization of the reagents and the efficiency of the SACs.
Recently, Assaf and co-workers stabilized atomically-dispersed rhodium sites supported by ceria using an impregnation method.116 A CeO2-based support was synthesized prior to the method using cerium(III) nitrate hexahydrate solution mixed with zirconium(IV) oxynitrate hexahydrate and ammonium hydroxide. Rhodium(III) acetylacetonate was dissolved in ethanol and the CeO2-based support was immersed in the mixture to deposit Rh on it before the solution was dried under stirring and the obtained solid was calcined. The obtained Rh loading of Rh/CeO2 was 0.4 wt%. Rh atoms were efficiently trapped by the doped-support, preventing their agglomeration. During CO oxidation reaction, the stability of the SAC increased within a wide temperature range (30–200 °C) because O2 efficiently fills the vacancies in the material at low temperatures. CeO2, as a support, can also be used to fabricate SACs based on a Pt/CeO2 catalyst via an impregnation method for CO oxidation.127 The synthesis method of this catalyst is shown in Fig. 2A. An isolated palladium SAC supported by ceria in the reaction of the selective hydrogenation of nitroarenes without solvent using a simple impregnation strategy has also been reported.126 These successful examples indicate that in the fields of catalysis, CeO2 has drawn much attention, resulting from its redox and distinct acid–base bifunctional properties. Therefore, the adsorption properties on the surface of the catalysts and the properties of the metal species are very important.
 | ||
| Fig. 2 (A) Schematic illustration of the L-ascorbic acid (AA)-assisted reduction synthesis of the atomically dispersed Pt/CeO2 catalyst (CeO2-AA-Pt-cal). Reproduced with permission.127 Copyright 2018 American Chemical Society. (B) Support modification and Pt deposition. (i) Template grafting from toluene, (ii) x cycles of tetraethoxy orthosilicate deposition, (iii) calcination at 650 °C, (iv) SEA of PtCl62−, and (v) reduction at 250 °C. Reproduced with permission.131 Copyright 2019 AIP Publishing. (C) Synthesis approaches for producing site-isolated Pt on TiO2. Reproduced with permission.132 Copyright 2017 American Chemical Society. | ||
Single Ir atoms supported by titanium dioxide with a defective metastable phase (denoted as Ir1/def-TiO2(B)) was synthesized as a catalyst using an impregnation method.121 Def-TiO2(B) with anion vacancies was obtained via the 2 h reduction of titanium dioxide with a perfect metastable phase (per-TiO2(B)) under an atmosphere of 5% H2/N2 at 250 °C. H2IrCl6 solution was added dropwise under ultrasonic vibration to the dispersed def-TiO2(B) in water. After the solution was continuously stirred and the suspension liquid was centrifuged, the obtained solid was then washed and dried under vacuum. The as-obtained powder was reduced under a 5% H2/N2 atmosphere and the Ir1/def-TiO2(B) catalyst were obtained. In the hydrogenation of furfural, this Ir SAC exhibits excellent catalytic performance of 99% conversion and 99% selectivity to furfuryl alcohol, as well as great stability, superior to that of Ir SACs supported by mesoporous graphitic carbon nitride, as well as Ir nanoparticles. First principles simulations reveal that the regulation of the TiO2 support provides appropriate interaction strength between the reactant molecules and the active metal sites, thus leading to the excellent catalytic performance of Ir1/def-TiO2(B). Another study utilizing the cation vacancies of supports to synthesize SACs, such as a Pt SAC with up to 2.3 wt% metal loading, was reported.128 However, the dispersion of metal species on the support surface is often random.42 Therefore, producing uniformly distributed SACs may be difficult.
Nanopolyhedron ceria supported atomically dispersed gold catalysts were prepared via a deposition–precipitation method using HAuCl4 as the Au precursor.118 A surfactant-assisted co-precipitation method was used to pre-prepare the CeO2 NP support. The CeO2 support was dissolved in ultrapure water and then stirred with ammonium carbonate. Under continuous stirring, HAuCl4 solution was added dropwise to the above and fully mixed, and then the resulting solution was aged for 1 h. The filtered and washed solids from the suspension liquid were then subjected to 3 h of drying at 70 °C. Under the reaction conditions, the 1 wt% Au/CeO2 catalysts with weakly anchored gold species exhibited high reactivity for CO oxidation, resulting from the Auδ+ clusters transformed from partial atomically-dispersed gold.
A deposition–precipitation method was used to synthesize iron–ruthenium oxide samples supported by ceria.130 A hydrothermal method was first used to synthesize ceria nanorods. Under vigorous stirring, the calcined CeO2 nanorods as well as (NH4)2CO3 were dispersed in 100 mL of water. A mixed aqueous solution of Fe(NO3)3·9H2O and RuCl3·H2O was added dropwise to the above solution to form a resulting solution with a pH value of ∼9. The obtained greenish slurries were further aged at room temperature. Then, the washed powders were dried and calcined in air. When the secondary metal (ruthenium) was added, Fe dispersion on ceria nanorods was facilitated, and the selectivity of methane for the iron oxide catalyst supported by ceria in the Fischer–Tropsch synthesis (FTS) was improved. The change in the subnanometer iron oxide species to single atoms of ionic Feδ+ was confirmed after the catalytic FTS.
Although the deposition–precipitation method is very useful when preparing uniformly distributed SACs, this method may not be useful for the high loading of metal atoms. Small metal (hydroxide) clusters or NPs may form before these metal ions are adsorbed by the supports in solution, restricting the increase in loading. This encourages us to engineer anchoring sites on surfaces, including increasing the support surface area, fabricating more anchoring sites, and controlling the interactions between the metal and the support surfaces in solution.129
As shown in Fig. 2B, Notestein and co-workers prepared small Pt clusters, including a great portion of single Pt atoms, by combining H2PtCl6 SEA and oxide supports via engineering.131 Controlled amounts of SiO2, grafted with bulky organic templates in advance, were deposited onto Al2O3 to synthesize the supports (SiO2@Al2O3). Like SiO2, oxide supports with largely negative charge have small patches of Al2O3 with positive charge after the templates are removed, which is derived from the previously template covered regions. As the quantity of SiO2 increases, Pt is deposited with a loading from 1 wt% to 0.05 wt% on templated SiO2@Al2O3 using SEA with a pH of 4. Pt loading drops to near zero without a template. This appears to be a feasible method to generate patches of charge on oxide supports to create supported SACs with extremely high dispersion.
Isolated Pt atoms supported on TiO2 was prepared as a catalyst using SEA with low loadings (0.025–0.15 wt%).132 The TiO2 support was dissolved in deionized water mixed with NH4OH, resulting in a solution pH of 12.2 (Fig. 2C). Pt(NH3)4(NO3)2 solution was injected very slowly into NH4OH with constant stirring to form a precursor solution at pH = 12.2. This solution was then injected into the support solution. After post-treatment, TiO2 supported isolated Pt atoms were obtained. Compared to 1 nm clusters of metal Pt, isolated Pt shows a higher turnover frequency (TOF) with double the CO oxidation performance and an identical reaction mechanism at 200 °C. It was interpreted that the active site of CO oxidation on Pt supported by TiO2 is interfacial cationic Pt atoms, demonstrating that isolated Pt species exhibit optimal reactivity when supported on TiO2, as every atom is exposed and an interfacial site with the support is created.
It is important to point out that functional group ions usually distributes on supports heterogeneously, and the adsorption behavior of metal complexes is significantly influenced by the presence of surface defects of various types.11 Furthermore, changing the adsorption time usually leads to shifts of solution pH and consequently the quality of SACs is affected. Surfaces of the support functionalized with strong anchoring species play an important role in successfully developing stable SACs.
2.2. Physical deposition methods
The mechanism of ALD is shown in Fig. 3, which involves: (1) exposure to the first precursor, (2) purging of the reaction chamber, (3) exposure to a second reactant precursor, and (4) further purging of the reaction chamber.136 During the first half of the reaction (step A), the first precursors react with all available active sites (defects or functional groups), and the precursors are partly removed from the supports. The reaction by-product and the residues of the first precursor are then cleaned away using inert gas. During the second half of the reaction (step B), the reactant precursors react with the precursors adsorbed in step A to obtain the target materials via removal the remaining ligand from the first precursors. During this reaction, the regeneration of active sites of the surfaces for sedimentary materials deposited in the subsequent cycle is very important. The by-products and residues of the second precursor are cleaned away with inert gas to complete a reaction cycle. This cycle is repeated to achieve the desired thickness or size. During ALD, the self-limiting reaction is the key characteristic, which allows us to accurately control the size and thickness of the deposited material on the atomic scale. At present, this method is widely utilized to prepare single noble metal atom, double noble metal atom and non-noble metal atom materials.43,137,138 The first synthesis of SACs using the ALD method was achieved by Sun and co-workers.139 They reported a synthesis method using the ALD technique for single Pt atoms supported by graphene nanosheets using MeCpPtMe3 and oxygen precursors. In general, layer-by-layer growth patterns is preferred. However, deviations from ideal growth are commonly observed, including delayed nucleation in the initial film growth phase of metal oxidation or island growth.11
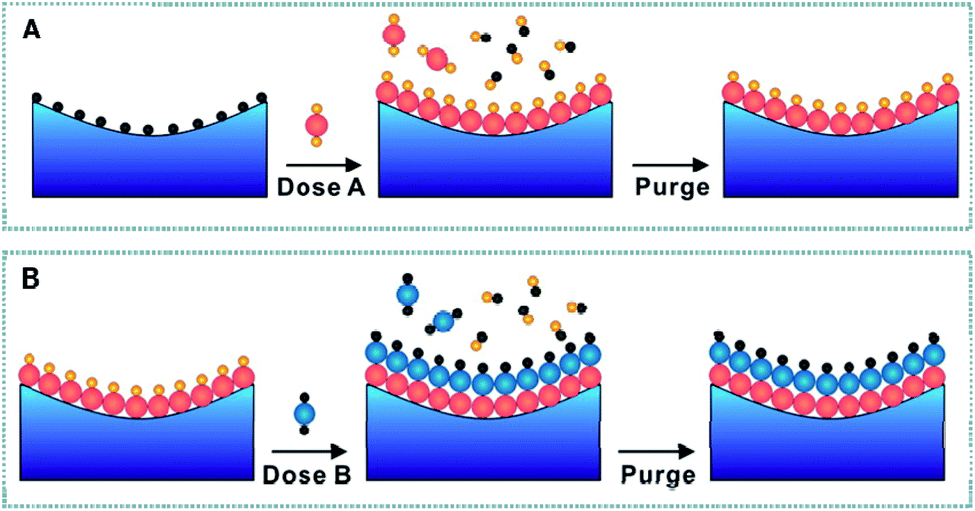 | ||
| Fig. 3 (A) General ALD binary reaction sequence. (B) Reaction scheme for Fe ALD half-reactions. Reproduced with permission.136 Copyright 2013 American Chemical Society. | ||
The deposition of single metal atoms on metal oxides by ALD technology has also been reported recently.140 Liang and co-workers used ALD in a fluidized bed reactor to prepare single Fe atom catalysts supported by TiO2 NPs using ferrocene and hydrogen as precursors. During the ALD process, they fed the chamber with Fe(Cp)2 and H2 separately via a distributor plate. During the ALD coating process, the reactor was also vibrated using a vibrator to improve particle fluidization quality. The following steps were involved in a typical ALD coating cycle: ferrocene dosing, N2 purge, evacuation; H2 dosing, N2 purge, and evacuation. The Fe content reached 1.78 wt% and the sample was denoted as 15c-Fe/TiO2 after 15 cycles of Fe ALD, and as the number of ALD cycles increased, the Fe loading of Fe/TiO2 particles increased almost linearly, which indicated that Fe species deposited uniformly during every ALD cycle. In the ALD process, the dosing time of Fe(Cp)2 is important in the formation of single Fe atoms. If the Fe dosing time of Fe(Cp)2 was short, single atoms formed, while Fe NPs formed with an extended dosing time. In the photocatalytic degradation reaction of methylene blue (MB), 2c-Fe/TiO2, the most active catalyst among the samples, exhibited a more than 6-fold activity enhancement compared with pure TiO2. Also, in terms of the non-noble metal Fe, Weissenrieder and co-workers also used an ALD method to synthesize isolated Fe1O3 sites on the surface of a Cu2O(100) single crystal with high density using the steric hindrance of the ligands.141 Combined with O2 molecules, the gas phase precursor, ferrocene (Fe(Cp)2) was used to achieve isolated Fe1O3, which occupied a single site per surface unit cell, coordinating to two oxygen atoms from the Cu2O lattice and another through abstraction from O2. Iron densities on the surface reached 0.21 per Cu2O(100) surface unit cell.
Cu single atoms supported by alumina was synthesized as a catalyst using ALD by Lu and co-workers.20 Copper(II) hexafluoroacetylacetonate hydrate (Cu(hfac)2) mixed with formalin (37% HCHO and 15% CH3OH in aqueous solution) was used as a precursor and spherical alumina powder was used as the support. On the alumina powder support, one Cu ALD cycle was performed at 250 °C to obtain the Cu1 SAC. Cu ALD was performed at 300 °C for 1, 3, and 5 cycles to obtain Cu NPs with different sizes of about 3.4, 7.3, and 9.3 nm, respectively. In the case of one cycle, the difference in temperature resulted in a difference between the Cu SAC and Cu NPs of 3.4 nm, possibly because formaldehyde is a strong reducing reagent at 300 °C when removing the Cu(hfac)2 precursor ligands. During complete conversion, the Cu SAC exhibited a high selectivity towards ethylene, reaching 91%, with excellent stability of over at least 40 h.
As a controlled catalyst preparation method for basic research, ALD is expected to be a powerful tool to study the relationships between the structure and performance of SACs supported by metal oxides. However, these methods may not, at least in the current situation, be used on a large scale, because the equipment is expensive and the yield of the catalysts is low.11 In the future, reducing the cost of ALD techniques will facilitate their wider application in SAC preparation.
During the mass-selected soft-landing procedure, the metal is vaporized by a laser evaporation source with high frequency, and a mass spectrometer is used to precisely control and load metal particles of different sizes onto the surface of the support. Taking the example of dispersed single Pt clusters on MgO(100) thin films,142 the mechanism of the mass-selected soft-landing technique was introduced. In a typical procedure, Pt clusters are produced by a laser evaporation source with high frequency, which generates high cluster currents with single positive charge. The produced metal plasma is heated by a helium pulse, and when the helium vapor experiences supersonic expansion the clusters are grown through nucleation. Subsequently, the size of the platinum clusters is selected using a quadrupole mass filter before they are deposited onto a MgO thin film.
A study of model electrodes of Ptn+ (n = 1 to 14) supported by indium tin oxide (ITO) prepared using mass-selected soft-landing technology under ultrahigh vacuum was conducted by Anderson and co-workers.21 They used a cluster beam deposition/surface analysis instrument comprising a laser vaporization cluster source, a guided ion mass-selecting beamline, and an ultra-high vacuum (UHV) system.144 The main UHV chamber includes the deposition end of the cluster beam lines, spectroscopy stations for X-ray and ultraviolet photoelectron spectroscopy and low-energy ion scattering, and several mass spectrometers for the study of gas surface reactivity. This catalyst can be used for ethanol electro-oxidation. Pt coverage was identical on each electrode, and only the size of the deposited Pt clusters was different. They proved that the oxidation rates of ethanol vary with the size of the clusters due to the change in the electronic structure of the reaction centers. However, the mass-selected soft-landing technique is expensive and low yielding, which limits its wide application and thus it is clearly not suitable, at least at present, for large-scale applications in industrial catalysis.19,42,139,145
2.3. Re-dispersion methods
 | ||
| Fig. 4 (A) Illustration of Pt nanoparticle sintering, showing how ceria can trap the mobile Pt to suppress sintering. Reproduced with permission.22 Copyright 2016 American Association for the Advancement of Science. (B) HAADF-STEM images and high-resolution HAADF-STEM images (insets) of (a) Pd-nanoparticles@ZIF-8, (b) intermediate I, (c) intermediate II and (d) Pd single atoms.146 Copyright 2018 Springer Nature. (C) Illustration of the molten-salt-mediated preparation of atomic Ni on TiO2, showing the TiO2 molecular structure. Reproduced with permission.24 Copyright 2020 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
Using in situ environmental transmission electron microscopy (ETEM), Li and co-workers for the first time observed atom trapping processes, as shown in Fig. 4B.146 ZIF-8 nanocrystals were grown on Pd NPs by mixing Pd NPs and Zn(NO3)2 solution with 2-methylimidazole solution. ZIF-8 crystals grew around Pd NPs to form Pd NPs@ZIF-8 composites. The Pd NPs@ZIF-8 nanocomposites were then heated at 900 °C for 3 h under an inert atmosphere, after which the Pd NPs were exhausted. After the first 0.5 h of heating, the crystal Pd NPs became larger and their size distribution became uneven. After 1.5 h, the number of Pd nanoparticles decreased significantly, and the Pd nanoparticles changed from a crystallized to amorphous state. With a prolongation of the pyrolysis time (3 h), the remaining NPs were completely decomposed, and ZIF-8 was decomposed and converted into nitrogen-doped carbon. This conversion also applies to Pt and Au. Density functional theory calculations showed that the high-temperature NP-to-SA conversion was driven by the more thermodynamically stable Pd–N4 structure when the moving Pd atoms are captured on the nitrogen-doped carbon defects.
2.4. Molten salt method
The molten salt method (MSM) is an ancient chemical preparation method, which has recently been adjusted to design and fabricate SACs.24,149,150 Wang and co-workers exploited a novel MSM to obtain a TiO2-supported Ni atom co-catalyst for the hydrogen evolution reaction (HER).24 In the preparation method, LiCl and KCl were mixed to dissolve these precursors into a molten salt. A liquid phase formed when the temperature reached the melting point of this salts, and uniformly dispersed Ni ions were generated on the TiO2 surface. Fig. 4C illustrates the MSM synthesis procedure for the co-catalyst on TiO2 with atomic Ni decoration. Furthermore, a metastable state of TiO2 can be generated by the molten salt with strong polarizing force, and thus Ni–O bonds between oxygen ions on the TiO2 surface and free-moving Ni2+ ions are formed.2.5. Electrochemical deposition method
Recently, Zeng and co-workers used electrochemical deposition as a universal route for fabricating SACs (Fig. 5).25 Such an electrochemical deposition process is similar to the molecular mechanisms of nucleation in solution-phase synthesis, where the support can be regarded as the solvent. On this occasion, the upper mass loading limit of the SACs corresponds to the minimum supersaturation level on the support. Isolated metal atoms nucleate into clusters once the metal mass loading on the support exceeds the minimum supersaturation. Thus, controlling mass loading below the minimum supersaturation level is crucial for the formation of SACs. In addition, the minimum supersaturation level may be related to the number of deposition sites such as vacancies, edges or steps, since isolated single atoms are usually fixed to the support via defects or strong metal–support interactions.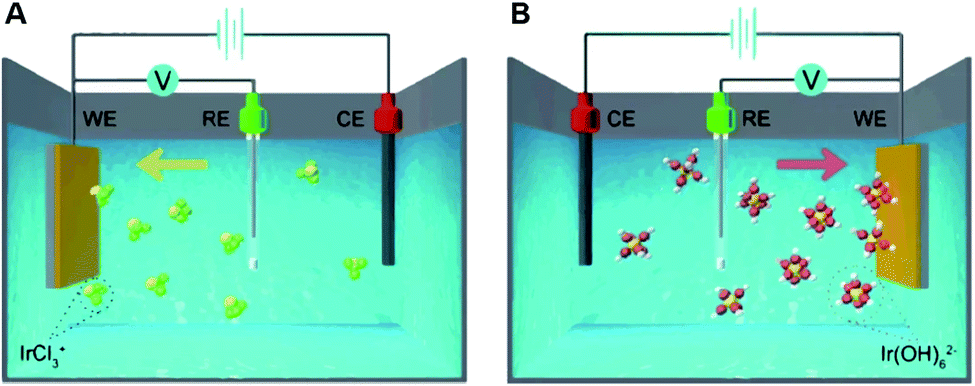 | ||
| Fig. 5 Schematic of the (A) cathodic and (B) anodic deposition of Ir species, where the yellow, green, red, and white spheres represent Ir, Cl, O, and H atoms, respectively. A standard three-electrode system was used, with Co(OH)2 nanosheets loaded on a glassy carbon electrode as the working electrode, a carbon rod as the counter electrode, and an Ag/AgCl electrode as the reference electrode. Reproduced with permission.25 Copyright 2020 Springer Nature. | ||
2.6. Other methods
In addition, there are many other useful methods that can be adopted for preparing SACs. Metal leaching via a cyanide salt method has been used for synthesizing atomically-dispersed noble metals supported on oxides, such as Au/TiO2, Au/CeO2 and Au/Fe2O3, which can be applied to the water–gas shift (WGS) reaction.151,152 A facile cation-exchange method can be used to prepare many noble metal SACs, and can even be used for large-scale production.153,154 Iced photochemical reduction can restrict the random diffusion of mononuclear metal precursors and reduce the tendency for migration and agglomeration of metal species, leading to the generation of SACs and preventing the formation of metal NPs.99,155To sum up, each method of preparing SACs has its own advantages and different applications. Wet-chemical methods are commonly used to prepare SACs and can be performed in general laboratories without special equipment. Among them, the first reported SAC was prepared via a co-precipitation method, but the metal atoms may be buried in the support during the synthesis process, resulting in a reduction in the utilization of the metal atoms. This problem also exists in sol–gel methods. Impregnation methods can be used to avoid this problem, since the metal atoms can be directly adsorbed on the oxide surface, but there are still issues using this method in controlling uniform dispersion. Deposition–precipitation methods can be effectively used to achieve uniform dispersion at very low loading, but it is difficult to increase the loading of metal atoms. In addition, strong electrostatic adsorption methods also face issues in terms of uncontrollable synthesis conditions and uneven surface sites. In contrast, physical deposition methods, including ALD and mass-selective soft-landing, can achieve controlled synthesis of SACs with uniform sites, which helps us to understand their structure–performance relationship. However, these methods are often expensive and require complex equipment, and are thus not suitable for the large-scale preparation of catalysts at this stage. Moreover, many new methods for the preparation of SACs have been developed. Re-dispersion from metal nanoparticles to single atoms is an attractive method for understanding the interactions between metal atoms and supports. The preparation of SACs via molten salt methods is a modification of an ancient synthesis method. Electrochemical deposition is a novel general synthesis method, which has valuable application in the synthesis of precious metal SACs.
3. Characterization of SACs supported by metal oxides
Advanced characterization techniques play an important role in the development of SACs, which provide useful information on the research of the structure of SACs. The source of detection can be electrons, ions, photons, neutrons, electric field, and magnetic field (Fig. 6). In general, the structures of catalysts include both geometry and electronic structures.55 The classical geometric structure of SACs refers to the existence of only isolated active metal atoms, which certifies the formation of single atoms directly according to definition. The electronic structure of SACs is mostly related to the electronic orbital states and the valence of metal atoms, which cannot offer direct evidence for the generation of isolated atoms, but offer deep insights into comprehending the structure–function relationship. | ||
| Fig. 6 Basic phenomena of characterization techniques. | ||
Directly imaging single metal atoms supported by high-surface-area materials results in the most compelling and intuitive evidence of SACs, which suggests that microscopy technologies have great applications in this field. X-ray spectroscopy can offer information about the dispersion of single atoms, the properties of the adjacent atom species, and the oxidation states of metal atoms. Infrared (IR) spectroscopy uses appropriate probe molecules to evaluate the existence and the percentage of single metal atoms on a metal oxide support to a certain degree. In addition, electron paramagnetic resonance (EPR) spectroscopy can be used to detect unsaturated coordination species on metal oxides as well as to probe the strong interactions between single atoms and the supports. Nuclear magnetic resonance (NMR) spectroscopy offers useful information about the structure of catalysts and sorbate–sorbent interactions. In situ or operando spectroscopy can provide additional information about the properties of catalysts in their working state. Each characterization technique has its unique emphasis and advantages. Therefore, it is useful to combine multiple characterization techniques when it comes to recognizing atomically dispersed single atoms, investigating the fine structure of catalysts, and exploring the distinct performance of catalysts in various working environments.
3.1. Microscopy technologies
When it comes to reliably evaluating the quality of supported metal catalysts, especially supported noble metal catalysts, integrated electron microscopy approaches have proven to be valuable. As mentioned above, microscopy technologies can provide direct information on SACs. These methods are extremely powerful for confirming whether only single atoms exist without clusters and nanoparticles, acknowledging the single metal atom locations in relation to the support surface structure, and determining the spatial distribution of isolated atoms.3 These techniques are indispensable for confirming that metal SACs have been successfully prepared in the process of developing and optimizing synthesis procedures. However, the inevitable contradictions between the enlargement of the detection area and the improvement in resolution lead to the failure of microscopic techniques to reflect the overall information of the sample convincingly.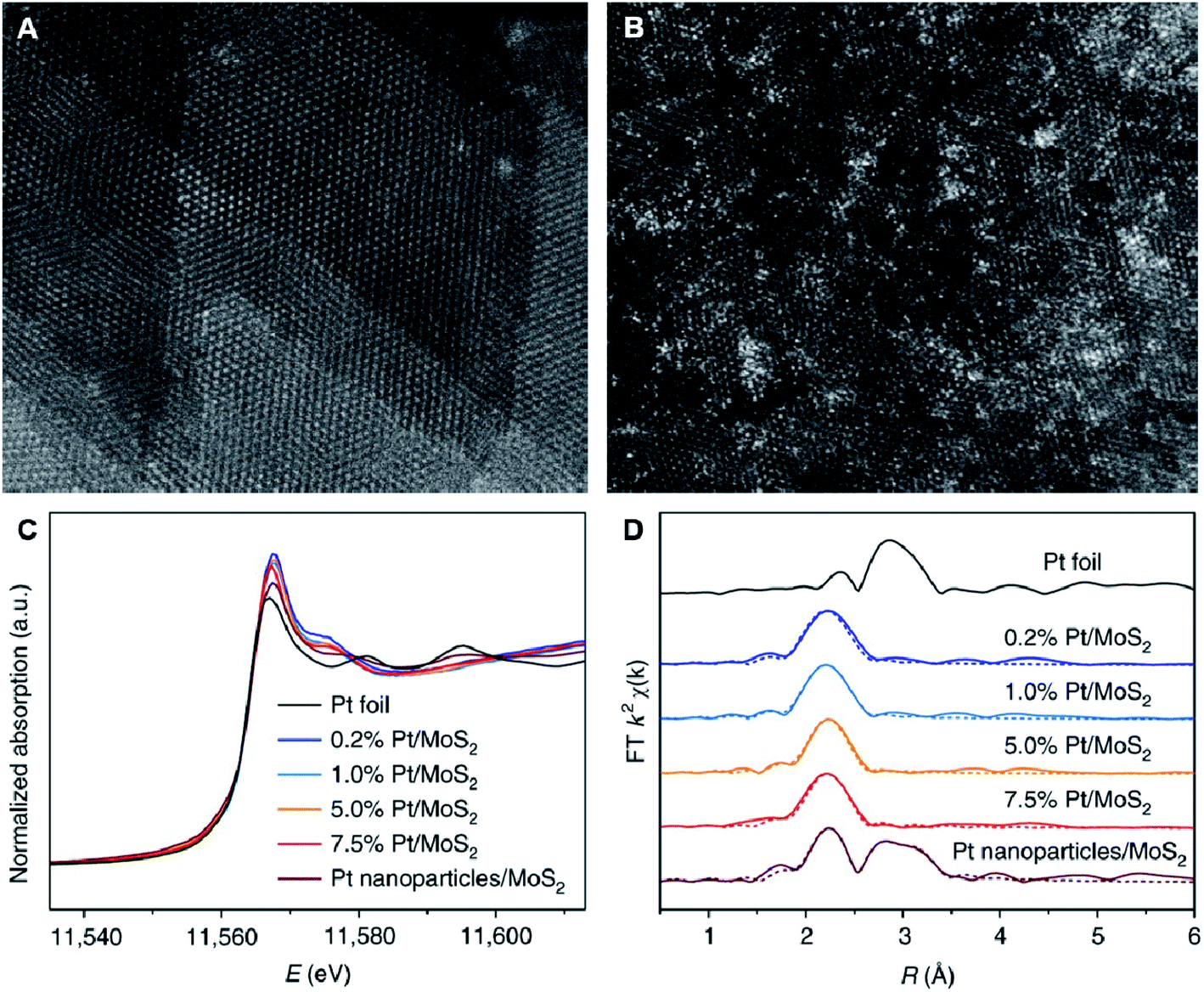 | ||
| Fig. 7 HAADF-STEM images of (A) 0.2% Pt/MoS2 and (B) 7.5% Pt/MoS2. (C) Pt K-edge XANES spectra and (D) Pt K-edge EXAFS in R space for atomically-dispersed Pt/MoS2 with different Pt loadings and Pt nanoparticles/MoS2. Pt foil was used as a reference. The dashed lines in (D) are the corresponding fitted curves. Reproduced with permission.162 Copyright 2018 Springer Nature. | ||
For example, isolated uniformly-dispersed Pt atoms were clearly observed from aberration-corrected HAADF-STEM images of Pt1–Co3O4 catalysts.163 STEM can also contribute towards investigating the dispersion and the spatial distribution of supported metal atoms. Zhang and co-workers observed isolated Pt atoms dispersed on Pt1/FeOx by aberration-corrected HAADF-STEM and a portion of the Pt atoms were found to be perfectly aligned with the Fe atomic columns.93 Moreover, in another study, they deduced that the Pt atoms were not distributed inside the individual FeOx nanocrystals by varying the beam focus settings.9 The density of 0.07 Pt atoms per nm2 in Pt1/FeOx catalyst was determined by examining a series of HAADF images, which were used to approximate the actual loading of Pt (about 0.09 Pt atoms per nm2). As a result, all of the observed single Pt atoms were confirmed to be located on the surfaces or in the adjacent subsurface of FeOx nanocrystallites.
After Au was deposited on TiO2(110) with a 0.08 monolayer (ML), an STM image of the catalyst was recorded.166 The observed smallest Au spots were assigned to Au1 species, while the larger ones were assigned to Au3 species. Some Ob-vacs were filled by isolated Au atoms on the r-TiO2 after deposition.
3.2. X-ray technology
It is worth noting that among the characterization techniques, XAS and XPS provide important information on the relationship between the structure and performance of catalytic reactions, although they cannot be used to determine the formation of SACs directly.167 However, when these techniques are applied to SACs, the results obtained by XPS are not accurate enough, but the adsorbate state splitting can reflect the strength of interactions.55As for noble metal Rh single atoms supported by CuO nanowire arrays on a copper foam (Rh SAC–CuO NAs/CF), structural and electronic information about the Rh species was acquired from XANES and EXAFS spectra at the atomic level.154 It was indicated that the valence of the CuO NA-supported Rh atoms is similar to that of Rh3+ in Rh2O3. As shown in the EXAFS spectra in R space, only a significant peak for Rh–O at around 1.5 Å in the Fourier-transformed (FT) k3-weighted EXAFS spectrum can be observed, while there is no Rh–Rh peak or Rh–O–Rh peak in the spectrum of the Rh SAC–CuO NAs, indicating the presence of atomically-dispersed Rh atoms in the CuO NAs. Similarly, as revealed by the XANES at the Ir L3-edge of Ir–NiO, the main peak is located near the same location as IrO2, indicating an Ir oxidation state of 4+ in Ir–NiO.175 After Ir doping, the oxidation state of the Ni atoms is slightly higher than 2+. By checking the EXAFS, it was found that the local coordination structure of Ir atoms has some similarities with that of IrO2, but that the second and higher shell structures are different, which may result from the formation of Ir–Ni bonds instead of Ir–Ir bonds. The EXAFS fitting of the Ni K-edge also demonstrates the same Ir–Ni scattering path, identifying the single Ir atoms on NiO. Moreover, XAS has been used to analyze other single transition metal atoms supported by different metal oxides.9,176–179
Huang and co-workers studied the XPS of Ir single atoms on a NiO matrix.154 Two sets of doublets are shown in a typical IrO2 XPS, which may largely be attributed to Ir4+, located at 62.1 and 65.0 eV and 63.1 and 66.0 eV. For Ir–NiO, the Ir 4f spectral deconvolution is a bimodal of two sets of centers that can be attributed to Ir4+ and Ir3+, at 62.3 and 65.1 eV and 61.9 and 63.8 eV, respectively. The peaks at 66.7 and 68.3 eV can be attributed to Ni 3p. Therefore, most of the Ir atoms supported by NiO are in the oxidation state of 4+, indicating the existence of highly-oxidized single Ir atoms. As revealed by the Ni 2p2/3 of NiO and Ir–NiO, after single Ir atoms are introduced, the valence state of the Ni atoms is increased. These results are consistent with XAS findings. Besides this, Shimizu and co-workers studied the role of La in a three-way catalysis process.109 They obtained the XPS spectrum for the N 1s of Pd/La(15)/Al2O3 (Pd loading is 20 wt% and La loading is 15 wt%). Four peaks at 404, 401, 398, and 393 eV were observed in the spectrum of Pd/La(15)/Al2O3 after NO adsorption, which can be attributed to the surface-adsorbed nitrite, molecularly adsorbed NO species, atomic nitrogen, and NO species bound on the Si substrate, respectively. The presence of atomic nitrogen indicates that even at 50 °C, NO can be dissociated over Pd/La(15)/Al2O3. The metallic Pd species supported by Al2O3 containing La are more deficient in electrons when compared with pristine Al2O3, which leads to a higher NO reactivity and lower CO poisoning.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) sinθ = nλ). It can be used for phase identification, crystal lattice parameter determination, and quantitative analysis of mixed crystal phases. X-ray sources in regular laboratories (mainly Cu Kα radiation), are good enough to determine the structure of large nanoparticles (generally bigger than 3 nm) or bulk materials by providing high diffraction signals. X-rays produced by synchrotron radiation have a high signal-to-noise ratio with very high energy and according to Bragg's law, they can produce lower angle diffraction peaks. Therefore, XRD is not suitable for the characterization of SACs without long-range order structure. However, single atomic alloy (SAA) catalysts with delicate structure of NPs or bulk materials are expected to be detected by synchrotron XRD, in spite of their particle size being less than 3 nm.183–187
sinθ = nλ). It can be used for phase identification, crystal lattice parameter determination, and quantitative analysis of mixed crystal phases. X-ray sources in regular laboratories (mainly Cu Kα radiation), are good enough to determine the structure of large nanoparticles (generally bigger than 3 nm) or bulk materials by providing high diffraction signals. X-rays produced by synchrotron radiation have a high signal-to-noise ratio with very high energy and according to Bragg's law, they can produce lower angle diffraction peaks. Therefore, XRD is not suitable for the characterization of SACs without long-range order structure. However, single atomic alloy (SAA) catalysts with delicate structure of NPs or bulk materials are expected to be detected by synchrotron XRD, in spite of their particle size being less than 3 nm.183–187
Cu–Pd SAA catalysts for highly-efficient NO reduction were reported by Furukawa and co-workers.188 Deposition–precipitation and/or impregnation methods were used to prepare single metal Cu and Pd and bimetallic Cu–Pd catalysts supported by γ-Al2O3. X-ray diffraction (XRD) analysis showed that the composition of the Cu–Pd solid-solution alloy phase in the catalysts was close to the feed metal ratio.
3.3. Infrared spectroscopy
When a beam of infrared light with a continuous wavelength (between 400 and 4000 cm−1) passes through a material, some of the light can be absorbed by the material. When the vibration frequency of a chemical bond in a material molecule is the same as that of infrared light, resonance occurs, and the molecule absorbs energy and transitions from the original ground state vibrational level to a vibrational level with higher energy. The absorption of infrared light by molecules is recorded, resulting in an infrared (absorption) spectrum.189,190 As a relatively straightforward technique, IR spectroscopy with extraordinary molecular and surface sensitivity is ubiquitous nowadays. The most remarkable characteristic of infrared spectroscopy is that it can be used to indirectly monitor the properties and changes in adsorption sites on the surface of a catalyst by tracking the changes in the vibration frequency and intensity of the probe molecules.191 CO is the most commonly used IR probe for characterizing the surfaces of heterogeneous catalysts.9,116,182,192–194 IR spectroscopy with CO molecules as a probe is atomically sensitive. The metal type, adsorption type (top, bridge, three- and four-fold hollow), the oxidation states, the local geometry, and the electronic environments all affect the displacement of C–O vibration frequency, suggesting the surface sensitivity of this method. Compared with the vibrational frequency of CO adsorbed on metal single atoms, that of CO adsorbed on metal clusters undergoes red-shifting due to the interactions between the CO molecules at a close distance. Time- and temperature-resolved Fourier-transform infrared (FTIR) spectroscopy can also be used to record the intermediate species of a reaction chain.116,195–197For most Pt SACs, only the absence of CO adsorption on a bridge is not very convincing to confirm the existence of Pt single atoms without clusters, as the adsorption of CO on the bridge of Pt clusters is intrinsically weak. Zhang and co-workers used FTIR spectroscopy to gain information about CO adsorption behavior.9 For the adsorption of CO on Pt/FeOx, the weak band in the FTIR spectrum at 2080 cm−1 can be attributed to linearly adsorbed CO on Ptδ+, while the strong band at 2030 cm−1 can be attributed to linearly adsorbed CO on Pt0, as shown in Fig. 8A and B. The bands at 1950 cm−1 and 1860 cm−1 can be ascribed to the adsorption of CO on the interface of Pt clusters and the support and the adsorption of CO on the bridge of two Pt atoms, respectively, suggesting the existence of dimers or Pt clusters, which are not observed in Pt1/FeOx. With an increase in CO pressure, the band at 2080 cm−1 rather than that at 2030 cm−1 is almost unchanged, which indicates the interactions among adsorbed CO molecules on Pt are lacking due to the presence of isolated Pt atoms.
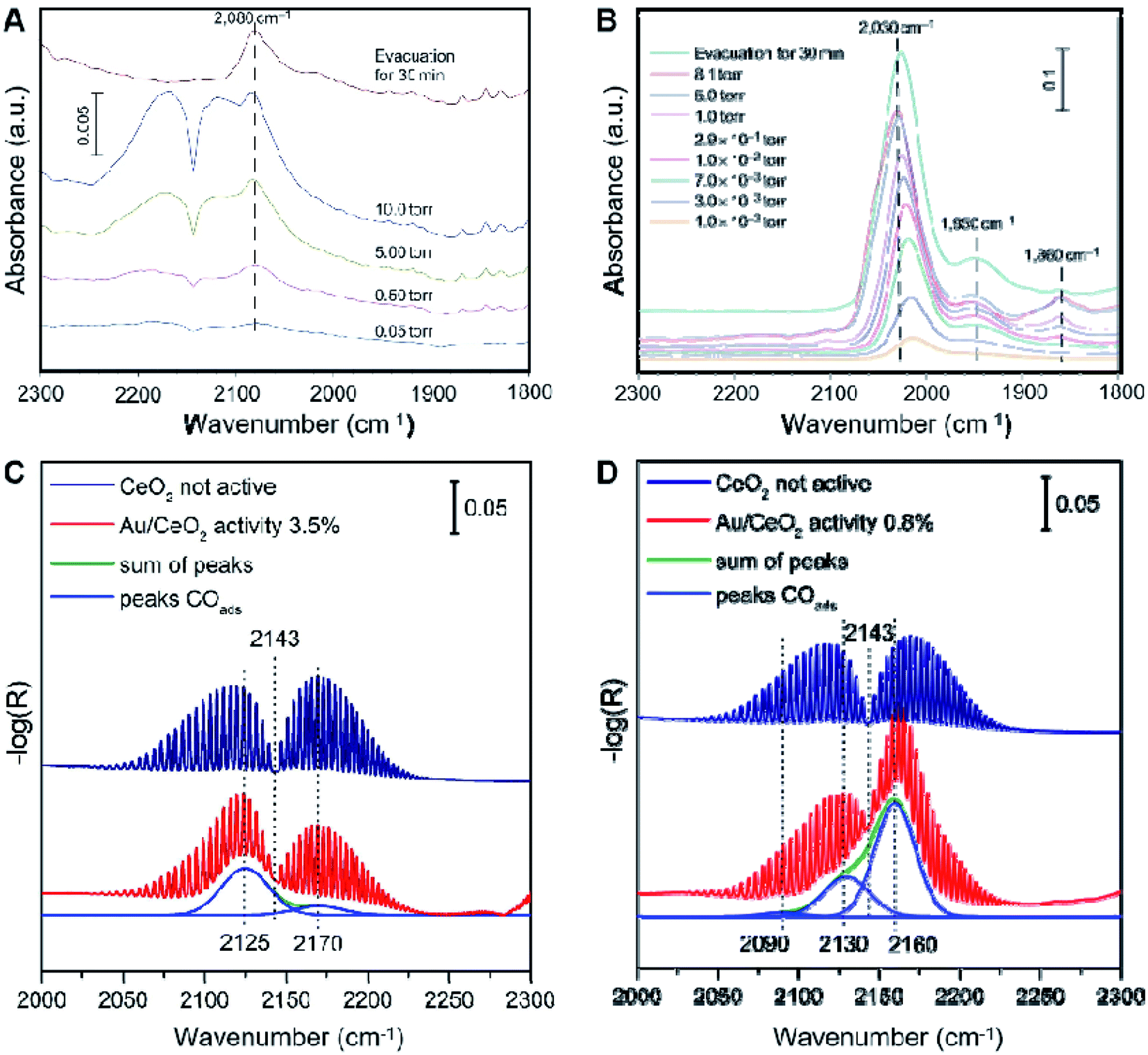 | ||
| Fig. 8 In situ FTIR spectra of CO adsorption on a Pt/FeOx catalyst with Pt loadings of (A) 0.17 wt% and (B) 2.5 wt%. Reproduced with permission.9 Copyright 2011 Springer Nature. CO stretching frequency region of operando infrared spectra under reaction conditions (red) after (C) equilibration in 25% O2/Ar flow and (D) outgassing in 25% O2/Ar at 200 °C for 1 h. Reproduced with permission.197 Copyright 2020 Elsevier. | ||
As for Au SACs, we take ceria-supported gold catalysts as an example. Operando infrared spectroscopy during the course of reactivity experiments was performed to determine the intermediate species.197 As shown in Fig. 8C and D, on the bare ceria support (blue spectra), no adsorbed CO species were observed under reaction conditions, which are detected in the region of 2154–2177 cm−1. In conjunction with density functional theory (DFT) calculations, the experimental bands observed at 2160–2170 cm−1, 2125–2130 cm−1 and 2090 cm−1 can be attributed to CO adsorbed on the ceria support [CO/CeO2(111)], CO adsorbed onto gold species in direct contact with the CeO2 support, i.e., a Olattice–Au+–CO species, and CO adsorbed on top of the gold atoms of 3D Au clusters that are not in direct contact with the support, respectively. Cationic gold was confirmed to be the active site for CO oxidation reactions in the Au/CeO2 catalysts. The proposed mechanism for CO oxidation at room temperature over Au/CeO2 catalysts can thus be revealed by combining IR spectroscopy and DFT calculations: the oxygen vacancy created by the oxidation of CO is refilled by gas phase oxygen, while the second oxygen undergoes reaction with CO.
3.4. Nuclear magnetic resonance spectroscopy
Apart from the above characterization techniques used for characterization, nuclear magnetic resonance (NMR) spectroscopy is useful for the analysis of SACs. NMR is based on the interaction between the nuclear magnetic moment and an external electromagnetic field.198,199 When the spin level of the nuclear magnetic moment is consistent with the frequency of the external electromagnetic field, the nucleus absorbs the energy and the energy level transitions to produce the corresponding resonance absorption, namely nuclear magnetic resonance. Atoms with nuclear magnetic moment have different chemical environments in molecules, and will undergo resonance absorption at different frequency positions. Therefore, NMR can be used to provide information on the properties of the components and the coordination environment between single atoms and supports.73,182 Using 27Al magic-angle spinning (MAS) NMR spectroscopy, it was verified that different preparation methods lead to different interactions between Pd atoms and Al2O3 supports, thus resulting in a unique electronic state of Pd species.182 As illustrated in Fig. 9A, a considerable drop in the intensity of the peak at 35 (δ) was shown in the spectra of Pd1/Al2O3, compared to pure Al2O3, which was attributed to Alpenta3+ during the calcination and reduction, indicating the strong interactions between Pd atoms and Alpenta3+. There was little change in the number of Alpenta3+ under the same treatment in comparison with Pd/Al2O3, revealing weak MSI. This may result from the physical barrier of Pd precursors coated by inverse nano micelles formed in the nucleation and calcination treatment by surfactant P123.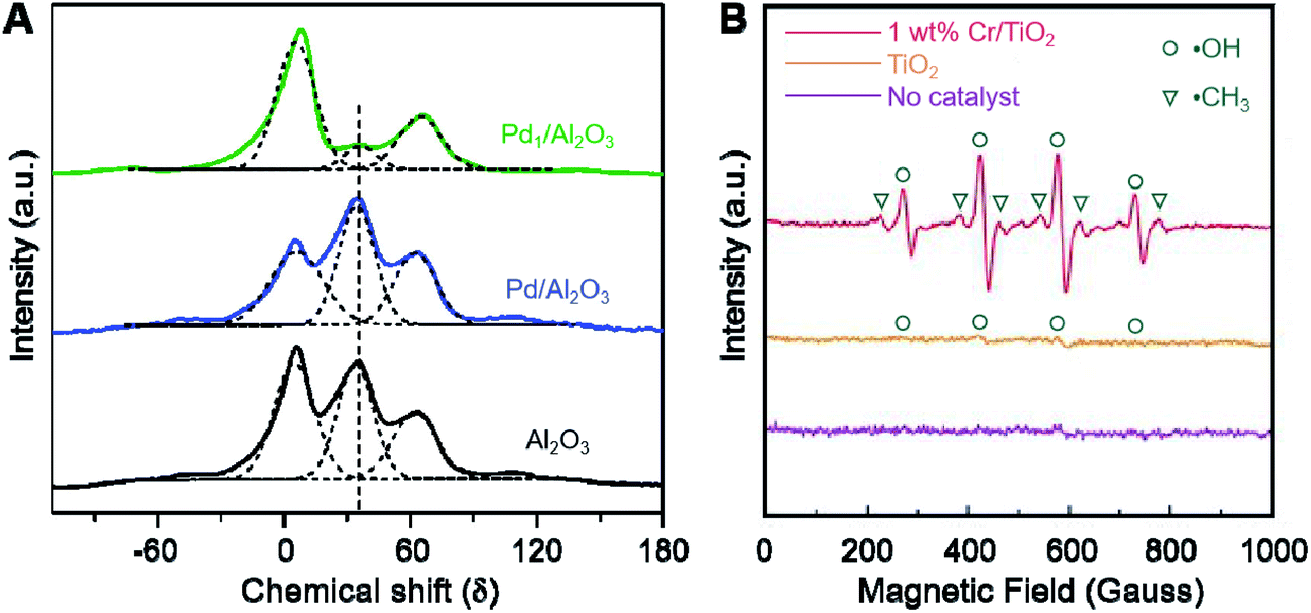 | ||
| Fig. 9 (A) 27Al MAS NMR spectra (solid lines) and their deconvolution results (dashed lines) of Pd1/Al2O3, Pd/Al2O3, and Al2O3. Reproduced with permission.182 Copyright 2020 Elsevier. (B) EPR experiments on oxygenated products in liquid phase with different catalysts. Reproduced with permission.202 Copyright 2020 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
3.5. Electron paramagnetic resonance
Since the 1960s, electron paramagnetic resonance (EPR) spectroscopy has been widely used in heterogeneous catalysis and surface chemistry for detecting paramagnetic species with unpaired electrons.200,201 It is a powerful technique that provides information about the properties, symmetry, electronic structure, and valence state changes of paramagnetic centers and their interactions with each other and reactants.163,177,202,203 Song and co-workers reported TiO2 NP-supported single Cr atoms with 1 wt% Cr.202 As shown in Fig. 9B, the radicals ˙CH3 and ˙OH were observed during the reaction. Titanium dioxide is known to produce surface-stable species of TiO2–O2− or Ti–OOH by activating H2O2, and CH3OOH may be formed by the interaction of the resulting ˙CH3 with Ti–OOH, which is also the main product of the original stage, as confirmed.In addition to the characterization techniques mentioned above, there are several other techniques that can be used to confirm the geometry and electronic structure of SACs, such as Raman and UV-Raman spectroscopies, photoluminescence spectroscopy, Auger electron spectroscopy and low energy ion scattering spectroscopy, among others.26 Furthermore, the rapidly developing in situ or operando characterization technologies can provide information on structural changes that occur under pretreatment or reaction conditions, which have become increasingly necessary and widely used to better understand the structure–property relationships of SACs. However, there is still a long way to go to realize the measurement and detection of catalysts with higher sensitivity. For example, new data analysis protocols and more powerful detectability of XAS are desired due to the limitation of supported SACs with low metal loading.204 Further improvements on the detection limit and sensitivity of IR spectroscopy are needed for the detection of weak adsorption capability on single metal atoms and SACs with extremely low loading of metal atoms.11
4. Applications of SACs supported by metal oxides
SACs have been widely used in several domains of catalysis, including thermocatalysis, electrocatalysis and photocatalysis, showing special properties different from nanocatalysts due to their unique structures. In this section, we focus on the reactions catalyzed by SACs, including CO oxidation, the water–gas shift reaction, CH4 conversion reaction, hydrogenation of CO2, oxygen reduction reaction (ORR), hydrogen evolution reaction (HER) and CO2 reduction reaction (CO2RR). SACs also exhibit distinctive performances for the electrocatalytic formic acid oxidation reaction and electrocatalytic water splitting.125,205 The mechanism of the reaction and the structure–property relationships have been investigated in detail from both experimental and theoretical aspects.4.1. CO oxidation
As a prototype reaction, catalytic CO oxidation can be used in some industrial processes, such as the removal of CO from vehicle exhausts, the removal of CO in hydrogen and proton exchange membrane fuel cells (PEMFCs), and other examples. In particular, even a small amount of residual CO may lead to a poisoned PEMFC. Therefore, the catalyst used in the preferential oxidation (PROX) of CO must be highly selective towards CO oxidation as opposed to H2 oxidation as far as possible.27 The main challenges for CO oxidation are low activity at low temperature and poor stability at high temperature, as well as high cost due to the use of noble metals. Based on the above considerations, research on the improvement in activity, selectivity, and stability of PROX catalysts, widening of the temperature window, and better water and CO2 resistance has been carried out for decades.Li and co-workers carried out comprehensive DFT studies to calculate the activity and catalytic mechanisms for CO oxidation by single-atom catalysts (M1/FeOx) with the 3d, 4d, and 5d group VIII to IB metals.83 Noticeably, a new catalyst, Pd1/FeOx, was found to exhibit high activity towards CO oxidation, predicted to be even better at CO oxidation compared with Pt1/FeOx and Ni1/FeOx previously reported. In contrast, low activities of CO oxidation were shown in other M1/FeOx SACs (M = Fe, Co, Cu; Ru, Rh, Ag; Os, Ir, Au). Moreover, it was found that the adsorption strength of CO molecules on the active sites of single atoms is the key to determining the catalytic activity of CO oxidation over SACs, since it dominates the recoverability of the surface oxygen vacancy to form a second CO2 during CO oxidation. At present, there are 3 types of mechanisms for CO oxidation: Langmuir–Hinshelhood (L–H), Eley–Rideal (E–R), and Mars–van Krevelen (M–vK) mechanisms.66,197,206,207 In realistic catalytic systems, more than one mechanism coexists. As shown in Fig. 10A, calculated results indicated that the first CO2 is formed via the L–H mechanism with high catalytic activity in M1/FeOx (M = Fe, Co, Ru, Rh, Os, Ir) via an oxygen dissociation mechanism and M1/FeOx (M = Ni, Pd, Pt) via an oxygen adsorption mechanism. On the other hand, the first CO2 was formed with relatively low catalytic activity in M1/FeOx (M = Cu, Ag, Au) via oxygen adsorption mechanism. Moreover, the second CO2 was formed via the L–H mechanism in M1/FeOx (M = Fe, Co, Rh, Pd, Au) through pathway I; M1/FeOx (M = Pt and Ir) through pathway II; and M1/FeOx (M = Ni, Cu, Ru, Ag, Os) through pathway III (Fig. 10B).
 | ||
| Fig. 10 Proposed reaction pathways of (A) the formation of the first CO2 associated with (I) oxygen dissociation on M1/FeOx (M = Fe, Co; Ru, Rh; Os, Ir) catalysts and (II) oxygen absorption on M1/FeOx (M = Ni, Cu; Pd, Ag; Pt, Au) catalysts, and (B) the formation of the second CO2 on (I) M1/FeOx (M = Fe, Co; Rh, Pd; Au) catalysts, (II) M1/FeOx (M = Ir, Pt) catalysts and (III) M1/FeOx (M = Ni, Cu; Ru, Ag; Os) catalysts, starting from the surface with the Ovac. Here, OA1 and OA2 represent the surface lattice O atoms of the support, the OB and OC atoms are derived from the dissociation of an oxygen gas molecule, and the OD atom comes from CO gas. Reproduced with permission.83 Copyright 2018 Tsinghua University Press and Springer-Verlag GmbH Germany. (C) Probing the reactivity of 0.5 ML Rh/Fe3O4(001) by TPD after sequential dosing of 13CO and 18O2. The red traces correspond to 13CO, the blue areas to 13C16O2 produced via a M–vK mechanism and the orange areas correspond to 13C16O18O produced via a L–H mechanism. Reproduced with permission.209 Copyright 2020 Royal Society of Chemistry. | ||
SACs supported by FeOx are widely used for CO oxidation in experiments.9,87,93,157,208–210 For example, CO oxidation over Au1/FeOx was tested in a fixed-bed reactor using mixed gases consisting of CO (1 vol%) and O2 (1 vol%) in He.208 The 0.03Au1/FeOx catalyst yielded a CO conversion of about 26% at room temperature (24 °C), about 70% at 50 °C, and 100% at 160 °C, and a 0.09Au/FeOx catalyst completely oxidized CO at 50 °C, indicating that Au1 supported by FeOx nanocrystallites was more active for CO oxidation. A specific rate of 8.3 molCO gAu−1 h−1 and a TOF of 0.45 s−1 were recorded for the 0.03Au1/FeOx catalysts, which were nearly 40 and 10 times, respectively, higher than those of 4.4 wt% Au/Fe2O3 catalysts. Comprehensive analyses of the used catalyst by aberration-corrected HAADF imaging indicated that after the 100 h test, no Au nanoparticles or clusters were formed, clearly confirming the rigid structure, energetic stability, and integrity of the Au1/FeOx catalysts. Parkinson and co-workers studied Rh/Fe3O4(001) model SACs for CO oxidation.209 Superoxo species were initially bound to absorbed Rh1 atoms resulting from the adsorption of O2, and Rh1 readily agglomerated to small RhxOy clusters at room temperature. However, a square-planar coordination environment preferred by absorbed Rh1 atoms was completed by the adsorption of CO, hindering O2 adsorption and stabilizing the Rh1 in place. CO oxidation takes place on both oxidized RhxOy clusters and carbonyl Rh1CO, but via very different pathways. According to TPD measurements (Fig. 10C), when catalyzing the reaction, a Langmuir–Hinshelwood mechanism is adopted by oxidized clusters at a temperature of as low as 200 K, while a Mars–van Krevelen mechanism is adopted to form CO2 at about 480 K by Rh carbonyls.
To overcome the low activity of CO oxidation at low temperatures, Li and co-workers developed single atom Pd/CoxOy on a tunable support interface induced by a crystal facet. This SAC shows very competitive activity, with 100% conversion of CO oxidation at a low temperature of 90 °C and significant 118 h stability at 60 °C.206 Isolated Pt atoms supported on TiO2 nanowires can even catalyze CO oxidation at room temperature.66 The stability at high temperatures can also be improved by depositing single Au atoms on Co3O4, which maintains the activity after a 10-cycle temperature-programmed reaction from −100 to 300 °C or one year of storage in air.207 Nowadays, to avoid high costs of noble metal catalysts, non-noble metal SACs have gradually come into use for CO oxidation.140
4.2. Water–gas shift reaction
Water–gas shift reaction (WGS) is one of the most general reactions for energy conversion, which involves the reaction CO + H2O → CO2 + H2. The produced H2-rich fuel gas can be widely used in fuel cells and other applications.65,211,212 Traditional catalysts of Cu–Zn type have been widely used for WGS, but their stability is poor at high temperature and their activity is poor at low temperature.42 Meanwhile, supported Pt, Au and other noble metals with atomic dispersion have been intensively investigated, and are considered as promising candidates for optimal metal–support interaction and can be further modified to improve performance.82,213–216 However, the high cost of noble metals restricts their applications industry. Moreover, the utilization efficiency of traditional supported catalysts made of noble metals is far from satisfactory.Ir1/FeOx SACs with dual metal active sites that drive the redox mechanism for WGS were reported by Zhang and co-workers.82 The mechanism of these SACs is different from that of the formation of intermediate carboxyl or formate via a conventional associative mechanism.214,217,218 Upon the dissociation of water, OH* is formed on a single Ir1 atom, while H* is formed on the first-neighboring O atom bonded to an Fe site. CO2 is produced by the reaction between the CO adsorbed on Ir1 sites and another O atom adjacent to it, generating an oxygen vacancy (Ovac). Then, H from adsorbed OH* migrates to Ir1 and reacts with another H* subsequently to form H2. A new WGS pathway was confirmed via the interaction between Ir1 and the sub-neighboring Fe specie, which is characterized by electron transfer from Fe3+–O⋯Ir2+–Ovac to Fe2+–Ovac⋯Ir3+–O at the active site with the participation of Ovac. The activity of Ir1/FeOx is higher than the counterparts of its cluster and nanoparticle by 1 order of magnitude and is even higher than those of Au- or Pt-based catalysts with usual high activity.88 At 300 °C, the Ir1/FeOx catalyst shows a remarkable reaction rate of 43.4 molco gIr−1 h−1, as well as a TOF of 2.31 s−1.
Single noble metal (Ru, Rh, Pd, Ag, Os, Ir, Pt, and Au) atoms supported by 5 different clusters of metal oxide ions (Al3O5+, Ce2O4+, V2O6+, FeO3−, and Ti3O7−) were studied to explore their WGS activity using DFT calculations.219 The results indicate that the d orbitals of the noble metals and the 2p orbitals of the ortho oxygen strongly hybridize, leading to an apparent charge shift of the noble metal atoms, and thus positive charge centers are formed, which are favorable for catalytic reaction. The d-band center values of Ru, Rh, Os, and Ir are generally close to the Fermi levels, so high activity can be anticipated on these metals. Among the metal oxide ions studied, RhAl3O5+ has the step with the lowest-energy barrier, which enables it to become a most promising WGS catalyst.
4.3. CH4 conversion reaction
The energy and environmental implications of converting methane, a greenhouse gas, into a usable chemical are self-evident. However, a typical unsolved problem in catalysis is that corrosive or expensive reaction media or oxidants are involved in this reaction, making it unsuitable for commercialization.220 Even though molecular oxygen can be used to directly convert methane to methanol in the gas phase under mild conditions, the conversion is usually hard to use in terms of stoichiometry or low reactivity and yield. Reactions yielding methanol, formic acid, ethylene, acetylene, or acetic acid by the reactant methane are continuously being researched in SACs.68,202,220–225 Considering the high bond energy of C–H and the highly symmetric tetrahedral structure of methane, the activation of C–H bonds is the critical step in methane upgrading reactions,225 even though the states of the active intermediate CH3 and the products are far from each other in different reaction systems.68,202,222,223,225,226As a well-studied reaction, the dry reforming of methane (DRM) has important scientific and industrial significance.226–228 Methane mixed with CO2 can be converted into value-added chemicals via intermediate syngas consisting of H2 and CO in equal proportions. The syngas can be used to synthesize long chain hydrocarbons and oxygenated chemicals including oxo-alcohols, dimethyl ether, and acetic acid. Furthermore, by conducting a WGS step in advance, a portion of CO can be reacted with H2O to give H2, and thus the H2/CO ratio can be adjusted to 2.0 before methanol synthesis and FTS.229–231 Hu and co-workers reported a synergistic role of isolated Ni1 and Ru1 sites on a CeO2 nanorod surface, namely Ce0.95Ni0.025Ru0.025O2.226 In the reforming of CH4 using CO2, two sets of isolated atom sites on the surface of this catalyst exhibit a high activity at 560 °C, with 98.5% selectivity to H2 and a production of 73.6 H2 molecules on each site per second (Fig. 11A and B). The apparent activation barrier is lower and the turnover rate to produce H2 and CO is higher compared with catalysts with only Ni1 or Ru1 sites, which evidences a synergy effect. CH4 and CO2 are activated for the formation of CO on Ni1 sites and Ru1 sites, respectively. The H atoms are first generated by activated CH4 on Ni1 sites and then H2 molecules are formed on Ru1 sites by H atom coupling.
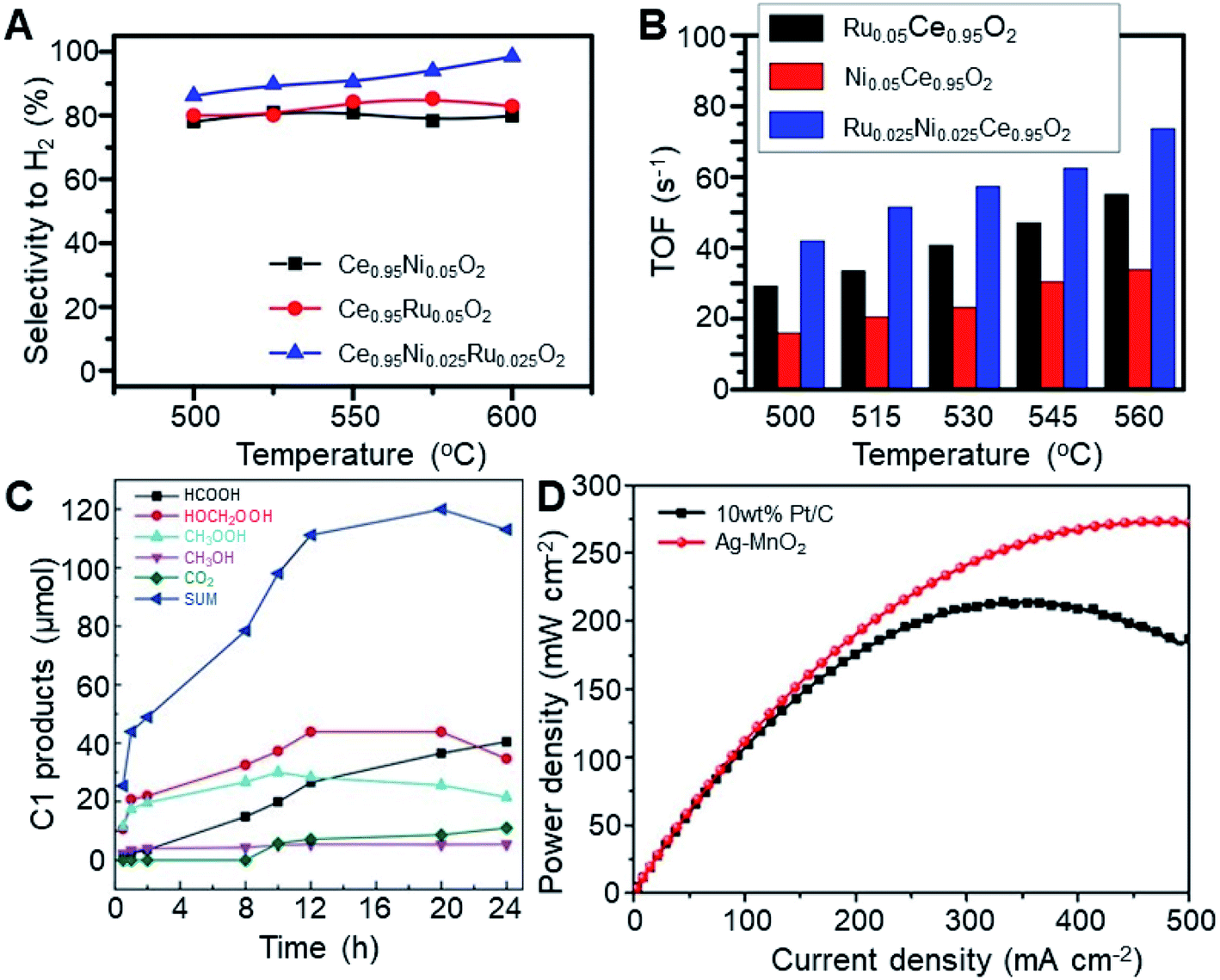 | ||
| Fig. 11 (A) Selectivity for producing H2 and (B) TOF of Ce0.95Ni0.05O2, Ce0.95Ru0.05O2, and Ce0.95Ni0.025Ru0.025O2 for the reforming of CH4 using CO2. Reproduced with permission.226 Copyright 2019 American Chemical Society. (C) Catalytic performance of the 1 wt% Cr/TiO2 catalysts for CH4 oxidation for different reaction times. Reproduced with permission.202 Copyright 2020 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. (D) Power density vs. current density for batteries employing Ag–MnO2 and Pt/C as the ORR catalysts. Reproduced with permission.241 Copyright 2019 Elsevier. | ||
The indirect route of CH4 conversion involves multiple steps, characterized by the addition of O atoms for the formation of syngas and the additional removal of O from CO for the formation of hydrocarbons. Therefore, direct methane conversion (DMC) to hydrocarbons is more economical and environmentally friendly without the intermediate step of going via syngas.232,233 Under mild conditions, single Cr atoms supported by TiO2 nanoparticles can be used as effective heterogeneous catalysts with H2O2 as an oxidant to directly oxidize methane to C1 oxygenated products (Fig. 11C).202 At 50 °C over 20 h, the highest yield of C1 oxygenation product was 57.9 mol molCr−1 with a high selectivity of 93%, which was remarkably higher than most relative catalysts. The mechanism of methane conversion can be interpreted as a methyl radical pathway, which first produces CH3OH and CH3OOH, and then HOCH2OOH and HCOOH are generated by the further oxidation of CH3OH. The Cr atoms supported by TiO2 activate the C–H bond of CH4 for the formation of a ˙CH3 radical as well as H2O2 to form ˙OH radical. C2 hydrocarbon product species can be also obtained via single atom Pt/CeO2 catalysts for direct methane coupling without oxygen, as reported by Likozar and co-workers.221
4.4. Hydrogenation of CO2
In today's world, with the consumption of non-renewable resources and the severity of environmental problems, it is crucial to reduce the dependence on fossil fuel energy and reduce carbon dioxide emissions for the sustainable development of human beings.234The hydrogenation of CO2 to turn it into liquid fuel is an effective solution.235 Currently, many studies are devoted to catalysts for the hydrogenation of CO2, including SACs supported by metal oxides,193,235–240 in which an important reaction is Fischer–Tropsch synthesis. The key to the hydrogenation of CO2 is the degree to which the carbon is coupled to the target product.
Huang and co-workers designed Ir1–In2O3 SACs with bifunctional active centers of the single atoms and the supports for highly selective hydrogenation of CO2 to ethanol.235 The catalysts are efficient for the reaction in liquid, with a high selectivity of >99% for ethanol as well as an initial TOF of 481 h−1. From the characterization results, it can be speculated that Lewis acid–base pairs are formed by the coupling of the isolated Ir atoms and adjacent oxygen vacancies, and thus the CO2 is activated and intermediate carbonyl species (CO*) are formed and adsorbed on the Ir atoms. Therefore, C–C bonds can be formed when CO* species are coupled with methoxides adsorbed on In2O3. Pd single atoms supported by a Fe3O4 surface (Pd/Fe3O4) are also efficient catalysts for the selective hydrogenation of CO2 to ethanol.240 These catalysts can even operate under atmospheric pressure. The special metal–oxide interface of single-atom Pd/Fe3O4 with a particular architecture has a favorable influence on catalytic behavior for C–C coupling.
In addition to ethanol, small molecules, such as CO and methane, can be produced by the hydrogenation of CO2.193,236,237 Serp and co-workers prepared Ni and Ru single atoms supported by TiO2 with oxygen vacancies.236 These catalysts exhibited a more active and stable performance than those atoms deposited on carbon nanotubes. By contrast, the Ni catalyst is more selective towards CO, while the Ru catalyst is more selective towards methane. For the Ru catalyst, electron-rich species orientate the selectivity towards methane, while electron-deficient species selectively produce CO, indicating a direct correlation between the hydrogenation selectivity and the electronic density of the metal.
4.5. Electrocatalysis and photocatalysis
In addition to the several applications mentioned above, SACs supported by metal oxides can also be used for electrocatalysis, such as the oxygen reduction reaction (ORR), but such reports are extremely rare. Sun and co-workers fabricated Ag atoms supported by MnO2 nanowires with atomic dispersion (Ag–MnO2), which can be used as an efficient cathode catalyst in rechargeable Zn–air batteries.241 As shown in Fig. 11D, an outstanding performance was observed in a Ag–MnO2-based Zn–air battery, exhibiting 273.2 mW cm−2 discharge peak power and 915.4 W h kg−1 energy density along with superior cycling stability, which can be attributed to the doped single Ag atoms. Atom utilization of Ag atoms in Ag–MnO2 can be maximized and abundant oxygen vacancies are induced by Ag doping, which is beneficial to carrying out the ORR with fast kinetics. As a result of the interaction of Ag and MnO2, the electronic structure of the active sites is rearranged after the Ag doping, thus improving the ORR activity of the Zn–air battery. In addition, single Pd atoms supported by a composite of MnO2 nanowires and carbon nanotubes (Pd/MnO2–CNTs) were prepared, which were applied as cathode catalysts with high efficiency in a Zn–air battery because of the synergetic effect among the Pd, MnO2, and CNTs.242 Superior charge–discharge ability was observed for the stable Zn–air liquid battery, with 190.7 mA cm−2 current density and 297.7 mW cm−2 peak power.The electrocatalytic reduction of CO2 has the potential to reduce environmental pollution and provide fuel energy by producing value-added chemicals, including alcohols and hydrocarbons, and the precise modulation of electrocatalysts on the atomic scale has achieved some improvement.68 Multiple oxygen vacancy-bound CeO2 with substitution by single atomic Cu was designed at the atomic level by Zheng and co-workers to electrocatalytically reduce CO2 into CH4, exhibiting an improved faradaic efficiency of 58%.69 Up to three oxygen vacancies can be stably enriched around each Cu site by the single Cu atoms supported on the surface of CeO2(110), producing a catalytic center with a high efficiency for CO2 adsorption and activation.
Lee and co-workers prepared Pt SACs supported by antimony-doped stannic oxide (Pt1/ATO) with a Pt loading of up to 8 wt% via a conventional incipient wetness impregnation technique.125 DFT calculations showed that it is energetically advantageous to replace Sb sites with Pt atoms in the bulk phase or at the surface of SbSn or ATO. Compared with industrial Pt/C catalysts, Pt1/ATO showed superior activity and durability in the formic acid oxidation reaction (FAOR). In CV measurements for 1800 cycles in the range of 0.05–1.4 V, Pt retained its single-atom structure and Pt1/ATO maintained high FAOR activity, even after rigorous durability tests. A full-cell direct formic acid fuel cell was prepared using Pt1/ATO as the anode catalyst, and the power of the cell was found to be one order of magnitude higher than that of Pt/C.
In the field of photocatalysis, SACs can provide an opportunity to precisely control the valence state of active sites and the coordination environment on the atomic scale, showing improved performance in many model reactions. Photocatalytic reduction of CO2 over SACs has also been reported recently.178,243 In addition to the CO2 reduction reaction, photocatalysis based on SACs has also made some achievements in other reactions.24,98,140,244 For example, Wang and co-workers designed a novel molten salt method (MSM) to prepare a TiO2-supported single Ni co catalyst for the hydrogen evolution reaction (HER).24 In the MSM process, it is found that Ni atoms on TiO2 are beneficial to the formation of oxygen vacancies, which facilitate the transfer of charge and the reaction of hydrogen evolution. Compared with a Ni co-catalyst on TiO2 synthesized via an impregnation method, the synergistic effect between the atomic Ni co-catalyst and oxygen vacancies four-fold increases the H2 evolution rate. Wang and co-workers used TiO2 nanorod arrays for anchoring atomic-scale Bi.98 Bi atoms with gradient distribution were assembled on TiO2 nanorods via a one-pot hydrothermal approach. It was pointed out that the Bi ratio relatively increased as Ti species were gradually consumed in the precursor solution, resulting in a bottom-up gradient-distributed structure. An efficient charge separation and transportation channel can be realized by such a structure, and thus the HER performance can be enhanced.
5. Conclusions and outlook
Proper synthesis methods are a prerequisite for the development of catalytic science and actual practice of SACs. In this paper, the progress of synthesis methods in recent years is reviewed and classified. Methods for preparing oxide-supported SACs are commonly classified into two categories: “bottom-up” and “top-down”. ALD and mass-selective soft-landing are two physical methods, which have high equipment costs. Wet-chemical synthesis includes several preparation methods, such as co-precipitation, impregnation, deposition–precipitation, strong electrostatic adsorption, and sol–gel methods, among others. In order to successfully prepare SACs, appropriate precursors of mononuclear metal complexes need to be carefully selected on the basis of the characteristics of the support, thus realizing the atomic dispersion and separation of the precursors, and inhibiting the atoms on the support from migrating and aggregating. Single metal atoms on the support are stabilized by enhancing metal–support interactions.The development of advanced characterization techniques can bring about better comprehension of structure–property relationships. HAADF-STEM and EXAFS play an important role in the characterization of the geometry of oxide-supported SACs, while XANES, XPS and EPR can be used to characterize the electronic structures of SACs. XRD is used to characterize SAA catalysts. IR spectra using CO as a probe is a powerful method to detect the local geometry, as well as the electronic environment. As stated in this article, many other characterization technologies can also be used to detect the structure of SACs.
Through the discussion of specific examples, we emphasized the potential applications of SACs for various reactions. At present, many important catalytic reactions depend on noble metal catalysts. Improving atom utilization is an effective approach to reduce the consumption of noble metals, which is of great importance to reduce catalyst costs and promote sustainability. As a widely used catalyst design strategy, oxide-supported SACs have been used for a variety of reactions for heterogeneous catalysis, including CO oxidation, the water–gas shift reaction, semi-hydrogenation reaction, CH4 conversion reaction, oxygen reduction reaction, hydrogen evolution reaction and CO2 reduction reaction. Fully exposed active sites, enhanced metal–support interactions, and unique electronic structures give SACs enhanced catalytic performance. In addition, due to the homogeneity and simplicity of SAC structures, this is conducive to the accurate structural recognition of the active site, which provides great potential for studying mechanisms at the atomic level via the combination of experiment and theory, and has important guiding significance for the rational design of catalysts.
Nowadays, the research on SACs is still in rapid development. In order to meet the needs of basic research and practical application of catalysts, increasing the loading of metal atoms in SACs and realizing the simple synthesis of SACs on a large scale are two crucial challenges in synthesizing SACs. In addition, how to effectively control the coordination environment of atoms supported by oxides has great significance, which provides broad prospects to optimize and improve the catalytic performance of SACs. The surfaces of oxides are generally not uniform, with the presence of multiple defects such as vacancies, edges, terraces, or grain/twin boundaries. Even on the same surface, fcc hollow sites of oxygen, hcp hollow sites of oxygen, and oxygen vacancy sites, among others, can be observed. These single atoms anchored on different surface sites lead to distinct electronic structures and coordination environments, which have a great influence on their catalytic properties. However, research into the correlations between the anchoring sites of single atoms on oxides and their catalytic performance are still lacking. It remains a grand challenge to precisely anchor single atoms on the specific sites of oxides. Moreover, the structure–property relationships between structure and performance and the catalytic mechanisms on the atomic scale still need to be further explored. The combination of theoretical calculations and in situ characterization techniques is expected to provide valuable insights towards enhancing catalytic performance and improving the structural stability of new SACs through rational selection and design. Furthermore, the stability of SACs plays an important role in evaluating the performance of catalysts. Even though existing preparation methods can ensure atomic distribution of metal species, SACs still face the risk of loss, aggregation, or reconstruction of active sites during catalysis, especially when the high temperature and reductive atmosphere in industrial catalysis challenge their stability. Therefore, more ingenious synthesis strategies are needed to construct SACs with long-term thermodynamic stability. Moreover, we should also balance the stability and activity of the catalyst, since saturating the metal atom coordination can indeed improve the stability of the catalyst, but inevitably result in a loss of intrinsic activity. What is more, more than one atom may be involved in some reactions or elementary steps, wherein the behavior of SACs remains elusive. Double atom catalysts (DACs) with a small active unit of bridge sites, triple atom catalysts (TACs) with a small unit of hollow sites and other atomic metal catalysts are expected to solve this problem.
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by the National Key Research and Development Program of China (2019YFA0405600), the National Science Fund for Distinguished Young Scholars (21925204), NSFC (U19A2015), the Fundamental Research Funds for the Central Universities, the Provincial Key Research and Development Program of Anhui (202004a05020074), the DNL Cooperation Fund, CAS (DNL202003), and the USTC Research Funds of the Double First-Class Initiative (YD2340002002).References
- Y. Zhao, W. Gao, S. Li, G. R. Williams, A. H. Mahadi and D. Ma, Joule, 2019, 3, 920–937 CrossRef CAS.
- H. Abe, J. Liu and K. Ariga, Mater. Today, 2016, 19, 12–18 CrossRef CAS.
- J. Liu, ChemCatChem, 2011, 3, 934–948 CrossRef CAS.
- R. Schlogl and S. B. Abd Hamid, Angew. Chem., Int. Ed., 2004, 43, 1628–1637 CrossRef.
- Y. Xia, Y. Xiong, B. Lim and S. E. Skrabalak, Angew. Chem., Int. Ed., 2009, 48, 60–103 CrossRef CAS PubMed.
- A. T. Bell, Science, 2003, 299, 1688–1691 CrossRef CAS.
- H. S. Taylor, Proc. R. Soc., Ser. A, 1925, 108, 105–111 CAS.
- H. Li, L. Li and Y. Li, Nanotechnol. Rev., 2013, 2, 515–528 CAS.
- B. T. Qiao, A. Q. Wang, X. F. Yang, L. F. Allard, Z. Jiang, Y. T. Cui, J. Y. Liu, J. Li and T. Zhang, Nat. Chem., 2011, 3, 634–641 CrossRef CAS PubMed.
- H. Ou, D. Wang and Y. Li, Nano Sel., 2021, 2, 492–511 CrossRef.
- J. Liu, ACS Catal., 2017, 7, 34–59 CrossRef CAS.
- J. Yang, W. Li, D. Wang and Y. Li, Small Struct., 2020, 2, 2000051 CrossRef.
- J. M. Thomas, R. Raja and D. W. Lewis, Angew. Chem., Int. Ed., 2005, 44, 6456–6482 CrossRef CAS PubMed.
- S. Ji, Y. Qu, T. Wang, Y. Chen, G. Wang, X. Li, J. Dong, Q. Chen, W. Zhang, Z. Zhang, S. Liang, R. Yu, Y. Wang, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2020, 59, 10651–10657 CrossRef CAS.
- K. Liu, R. Qin and N. Zheng, J. Am. Chem. Soc., 2021, 143, 4483–4499 CrossRef CAS.
- J. Li, W. Wang, W. Chen, Q. Gong, J. Luo, R. Lin, H. Xin, H. Zhang, D. Wang, Q. Peng, W. Zhu, C. Chen and Y. Li, Nano Res., 2018, 11, 4774–4785 CrossRef CAS.
- Y. Zhou, X. Tao, G. Chen, R. Lu, D. Wang, M.-X. Chen, E. Jin, J. Yang, H.-W. Liang, Y. Zhao, X. Feng, A. Narita and K. Mullen, Nat. Commun., 2020, 11, 5892 CrossRef CAS PubMed.
- H. Jeong, S. Shin and H. Lee, ACS Nano, 2020, 14, 14355–14374 CrossRef PubMed.
- S. Ji, Y. Chen, X. Wang, Z. Zhang, D. Wang and Y. Li, Chem. Rev., 2020, 120, 11900–11955 CrossRef CAS PubMed.
- X. Shi, Y. Lin, L. Huang, Z. Sun, Y. Yang, X. Zhou, E. Vovk, X. Liu, X. Huang, M. Sun, S. Wei and J. Lu, ACS Catal., 2020, 10, 3495–3504 CrossRef CAS.
- A. von Weber, E. T. Baxter, S. Proch, M. D. Kane, M. Rosenfelder, H. S. White and S. L. Anderson, Phys. Chem. Chem. Phys., 2015, 17, 17601–17610 RSC.
- J. Jones, H. F. Xiong, A. T. Delariva, E. J. Peterson, H. Pham, S. R. Challa, G. S. Qi, S. Oh, M. H. Wiebenga, X. I. P. Hernandez, Y. Wang and A. K. Datye, Science, 2016, 353, 150–154 CrossRef CAS.
- K. Liu, X. Zhao, G. Ren, T. Yang, Y. Ren, A. F. Lee, Y. Su, X. Pan, J. Zhang, Z. Chen, J. Yang, X. Liu, T. Zhou, W. Xi, J. Luo, C. Zeng, H. Matsumoto, W. Liu, Q. Jiang, K. Wilson, A. Wang, B. Qiao, W. Li and T. Zhang, Nat. Commun., 2020, 11, 1263 CrossRef CAS.
- M. Xiao, L. Zhang, B. Luo, M. Lyu, Z. Wang, H. Huang, S. Wang, A. Du and L. Wang, Angew. Chem., Int. Ed., 2020, 59, 7230–7234 CrossRef CAS.
- Z. Zhang, C. Feng, C. Liu, M. Zuo, L. Qin, X. Yan, Y. Xing, H. Li, R. Si, S. Zhou and J. Zeng, Nat. Commun., 2020, 11, 1215 CrossRef CAS PubMed.
- M. Che and J. C. Védrine, Characterization of Solid Materials and Heterogeneous Catalysts, 2012 Search PubMed.
- H. Zhang, S. Fang and Y. H. Hu, Catal. Rev.: Sci. Eng., 2020, 1–40 CrossRef.
- F. Doherty, H. Wang, M. Yang and B. R. Goldsmith, Catal. Sci. Technol., 2020, 10, 5772–5791 RSC.
- S. Kim, S. Jee, K. M. Choi and D.-S. Shin, Nano Res., 2020, 14, 486–492 CrossRef.
- H. Li, M. Wang, L. Luo and J. Zeng, Adv. Sci., 2019, 6, 1801471 CrossRef CAS PubMed.
- J. Yang, W. Li, D. Wang and Y. Li, Adv. Mater., 2020, 32, 2003300 CrossRef CAS.
- T. N. Nguyen, M. Salehi, L. Quyet Van, A. Seifitokaldani and D. Cao Thang, ACS Catal., 2020, 10, 10068–10095 CrossRef.
- S. Li, J. Liu, Z. Yin, P. Ren, L. Lin, Y. Gong, C. Yang, X. Zheng, R. Cao, S. Yao, Y. Deng, X. Liu, L. Gu, W. Zhou, J. Zhu, X. Wen, B. Xu and D. Ma, ACS Catal., 2020, 10, 907–913 CrossRef CAS.
- M. Huang, X. Kong, C. Wang, Z. Geng, J. Zeng and J. Bao, ChemNanoMat, 2021, 7, 2–6 CrossRef CAS.
- Y. Wang, H. Su, Y. He, L. Li, S. Zhu, H. Shen, P. Xie, X. Fu, G. Zhou, C. Feng, D. Zhao, F. Xiao, X. Zhu, Y. Zeng, M. Shao, S. Chen, G. Wu, J. Zeng and C. Wang, Chem. Rev., 2020, 120, 12217–12314 CrossRef CAS.
- H. Chen, X. Guo, X. Kong, Y. Xing, Y. Liu, B. Yu, Q.-X. Li, Z. Geng, R. Si and J. Zeng, Green Chem., 2020, 22, 7529–7536 RSC.
- L. Wang, H. Li, W. Zhang, X. Zhao, J. Qiu, A. Li, X. Zheng, Z. Hu, R. Si and J. Zeng, Angew. Chem., Int. Ed., 2017, 56, 4712–4718 CrossRef CAS PubMed.
- L. Zeng and C. Xue, Nano Res., 2020, 14, 934–944 CrossRef.
- L. Peng, L. Shang, T. Zhang and G. I. N. Waterhouse, Adv. Energy Mater., 2020, 10, 2003018 CrossRef CAS.
- Z. Zhuang, Q. Kang, D. Wang and Y. Li, Nano Res., 2020, 13, 1856–1866 CrossRef CAS.
- H. Yan, C. Su, J. He and W. Chen, J. Mater. Chem. A, 2018, 6, 8793–8814 RSC.
- R. Lang, X. Du, Y. Huang, X. Jiang, Q. Zhang, Y. Guo, K. Liu, B. Qiao, A. Wang and T. Zhang, Chem. Rev., 2020, 120, 11986–12043 CrossRef CAS PubMed.
- Y. Xu, W. Zheng, X. Liu, L. Zhang, L. Zheng, C. Yang, N. Pinna and J. Zhang, Mater. Horiz., 2020, 7, 1519–1527 RSC.
- Y. Peng, Z. Geng, S. Zhao, L. Wang, H. Li, X. Wang, X. Zheng, J. Zhu, Z. Li, R. Si and J. Zeng, Nano Lett., 2018, 18, 3785–3791 CrossRef CAS.
- E. Luo, C. Wang, Y. Li, X. Wang, L. Gong, T. Zhao, Z. Jin, J. Ge, C. Liu and W. Xing, Nano Res., 2020, 13, 2420–2426 CrossRef CAS.
- X. Yang, Y. Chen, L. Qin, X. Wu, Y. Wu, T. Yan, Z. Geng and J. Zeng, ChemSusChem, 2020, 13, 6307–6311 CAS.
- Y. Zhong, X. Kong, Z. Geng, J. Zeng, X. Luo and L. Zhang, ChemPhysChem, 2020, 21, 2051–2055 CrossRef CAS.
- P.-H. Li, M. Yang, Y.-X. Li, Z.-Y. Song, J.-H. Liu, C.-H. Lin, J. Zeng and X.-J. Huang, Anal. Chem., 2020, 92, 6128–6135 CrossRef CAS.
- S. Chen, N. Zhang, C. W. N. Villarrubia, X. Huang, L. Xie, X. Wang, X. Kong, H. Xu, G. Wu, J. Zeng and H.-L. Wang, Nano Energy, 2019, 66, 104164 CrossRef CAS.
- T. Zheng, K. Jiang, N. Ta, Y. Hu, J. Zeng, J. Liu and H. Wang, Joule, 2019, 3, 265–278 CrossRef CAS.
- Z. Geng, Y. Liu, X. Kong, P. Li, K. Li, Z. Liu, J. Du, M. Shu, R. Si and J. Zeng, Adv. Mater., 2018, 30, 1803498 CrossRef PubMed.
- J. Liu, M. Jiao, L. Lu, H. M. Barkholtz, Y. Li, Y. Wang, L. Jiang, Z. Wu, D.-J. Liu, L. Zhuang, C. Ma, J. Zeng, B. Zhang, D. Su, P. Song, W. Xing, W. Xu, Y. Wang, Z. Jiang and G. Sun, Nat. Commun., 2017, 8, 15938 CrossRef CAS.
- X. He, Q. He, Y. Deng, M. Peng, H. Chen, Y. Zhang, S. Yao, M. Zhang, D. Xiao, D. Ma, B. Ge and H. Ji, Nat. Commun., 2019, 10, 3663 CrossRef.
- M. S. Kim, J. Lee, H. S. Kim, A. Cho, K. H. Shim, T. N. Le, S. S. A. An, J. W. Han, M. I. Kim and J. Lee, Adv. Funct. Mater., 2020, 30, 1905410 CrossRef CAS.
- H. Shang, X. Zhou, J. Dong, A. Li, X. Zhao, Q. Liu, Y. Lin, J. Pei, Z. Li, Z. Jiang, D. Zhou, L. Zheng, Y. Wang, J. Zhou, Z. Yang, R. Cao, R. Sarangi, T. Sun, X. Yang, X. Zheng, W. Yan, Z. Zhuang, J. Li, W. Chen, D. Wang, J. Zhang and Y. Li, Nat. Commun., 2020, 11, 3049 CrossRef CAS.
- C. Ma, B. Liu and S. Yan, Mol. Catal., 2020, 495, 111165 CrossRef CAS.
- Y. Chen, H. Li, W. Zhao, W. Zhang, J. Li, W. Li, X. Zheng, W. Yan, W. Zhang, J. Zhu, R. Si and J. Zeng, Nat. Commun., 2019, 10, 1885 CrossRef PubMed.
- S. Wei, Y. Wang, W. Chen, Z. Li, W. C. Cheong, Q. Zhang, Y. Gong, L. Gu, C. Chen, D. Wang, Q. Peng and Y. Li, Chem. Sci., 2020, 11, 786–790 RSC.
- Y. Wang, J. Mao, X. Meng, L. Yu, D. Deng and X. Bao, Chem. Rev., 2019, 119, 1806–1854 CrossRef CAS.
- A. Alarawi, V. Ramalingam and J.-H. He, Mater. Today Energy, 2019, 11, 1–23 CrossRef CAS.
- J. Zhang, Y. Zhao, X. Guo, C. Chen, C.-L. Dong, R.-S. Liu, C.-P. Han, Y. Li, Y. Gogotsi and G. Wang, Nat. Catal., 2018, 1, 985–992 CrossRef CAS.
- V. Ramalingam, P. Varadhan, H. C. Fu, H. Kim, D. Zhang, S. Chen, L. Song, D. Ma, Y. Wang, H. N. Alshareef and J. H. He, Adv. Mater., 2019, 31, 1903841 CrossRef CAS PubMed.
- H. Su, L. Chen, Y. Chen, R. Si, Y. Wu, X. Wu, Z. Geng, W. Zhang and J. Zeng, Angew. Chem., Int. Ed., 2020, 59, 20411–20416 CrossRef CAS.
- L. Lin, Q. Yu, M. Peng, A. Li, S. Yao, S. Tian, X. Liu, A. Li, Z. Jiang, R. Gao, X. Han, Y.-W. Li, X.-D. Wen, W. Zhou and D. Ma, J. Am. Chem. Soc., 2021, 143, 309–317 CrossRef CAS PubMed.
- X. Zhang, M. Zhang, Y. Deng, M. Xu, L. Artiglia, W. Wen, R. Gao, B. Chen, S. Yao, X. Zhang, M. Peng, J. Yan, A. Li, Z. Jiang, X. Gao, S. Cao, C. Yang, A. J. Kropf, J. Shi, J. Xie, M. Bi, J. A. van Bokhoven, Y.-W. Li, X. Wen, M. Flytzani-Stephanopoulos, C. Shi, W. Zhou and D. Ma, Nature, 2021, 589, 396–401 CrossRef CAS.
- S. Hoang, Y. Guo, A. J. Binder, W. Tang, S. Wang, J. Liu, T. D. Huan, X. Lu, Y. Wang, Y. Ding, E. A. Kyriakidou, J. Yang, T. J. Toops, T. J. Pauly, R. Ramprasad and P.-X. Gao, Nat. Commun., 2020, 11, 1062 CrossRef CAS.
- D. Kunwar, S. Zhou, A. DeLaRiva, E. J. Peterson, H. Xiong, X. I. Pereira-Hernandez, S. C. Purdy, R. ter Veen, H. H. Brongersma, J. T. Miller, H. Hashiguchi, L. Kovarik, S. Lin, H. Guo, Y. Wang and A. K. Datye, ACS Catal., 2019, 9, 3978–3990 CrossRef CAS.
- P. Xie, T. Pu, A. Nie, S. Hwang, S. C. Purdy, W. Yu, D. Su, J. T. Miller and C. Wang, ACS Catal., 2018, 8, 4044–4048 CrossRef CAS.
- Y. Wang, Z. Chen, P. Han, Y. Du, Z. Gu, X. Xu and G. Zheng, ACS Catal., 2018, 8, 7113–7119 CrossRef CAS.
- A. S. Hoffman, L. M. Debefve, S. Zhang, J. E. Perez-Aguilar, E. T. Conley, K. R. Justl, I. Arslan, D. A. Dixon and B. C. Gatesr, ACS Catal., 2018, 8, 3489–3498 CrossRef CAS.
- F. Dvorak, M. F. Camellone, A. Tovt, T. Nguyen-Dung, F. R. Negreiros, M. Vorokhta, T. Skala, I. Matolinova, J. Myslivecek, V. Matolin and S. Fabris, Nat. Commun., 2016, 7, 10801 CrossRef CAS.
- B. Qiao, J. Liu, Y.-G. Wang, Q. Lin, X. Liu, A. Wang, J. Li, T. Zhang and J. Liu, ACS Catal., 2015, 5, 6249–6254 CrossRef CAS.
- Z. L. Zhang, Y. H. Zhu, H. Asakura, B. Zhang, J. G. Zhang, M. X. Zhou, Y. Han, T. Tanaka, A. Q. Wang, T. Zhang and N. Yan, Nat. Commun., 2017, 8, 16100 CrossRef CAS.
- F. Wang, J. Ma, S. Xin, Q. Wang, J. Xu, C. Zhang, H. He and X. C. Zeng, Nat. Commun., 2020, 11, 529 CrossRef CAS PubMed.
- R. Qin, L. Zhou, P. Liu, Y. Gong, K. Liu, C. Xu, Y. Zhao, L. Gu, G. Fu and N. Zheng, Nat. Catal., 2020, 3, 703–709 CrossRef CAS.
- L. Kuai, Z. Chen, S. Liu, E. Kan, N. Yu, Y. Ren, C. Fang, X. Li, Y. Li and B. Geng, Nat. Commun., 2020, 11, 48 CrossRef.
- K. Yang, Y. Liu, J. Deng, X. Zhao, J. Yang, Z. Han, Z. Hou and H. Dai, Appl. Catal., B, 2019, 244, 650–659 CrossRef CAS.
- A. Parastaev, V. Muravev, E. H. Osta, A. J. F. van Hoof, T. F. Kimpel, N. Kosinov and E. J. M. Hensen, Nat. Catal., 2020, 3, 526–533 CrossRef CAS.
- T. W. van Deelen, C. H. Mejia and K. P. de Jong, Nat. Catal., 2019, 2, 955–970 CrossRef CAS.
- B. Han, Y. Guo, Y. Huang, W. Xi, J. Xu, J. Luo, H. Qi, Y. Ren, X. Liu, B. Qiao and T. Zhang, Angew. Chem., Int. Ed., 2020, 59, 11824–11829 CrossRef CAS.
- G. S. Parkinson, Surf. Sci. Rep., 2016, 71, 272–365 CrossRef CAS.
- J.-X. Liang, J. Lin, J. Liu, X. Wang, T. Zhang and J. Li, Angew. Chem., Int. Ed., 2020, 59, 12868–12875 CrossRef CAS.
- J. Liang, Q. Yu, X. Yang, T. Zhang and J. Li, Nano Res., 2018, 11, 1599–1611 CrossRef CAS.
- J. Lin, Y. Chen, Y. Zhou, L. Li, B. Qiao, A. Wang, J. Liu, X. Wang and T. Zhang, AIChE J., 2017, 63, 4003–4012 CrossRef CAS.
- J.-X. Liang, X.-F. Yang, A. Wang, T. Zhang and J. Li, Catal. Sci. Technol., 2016, 6, 6886–6892 RSC.
- Q. Li, Z. Li, Q. Zhang, L. Zheng, W. Yan, X. Liang, L. Gu, C. Chen, D. Wang, Q. Peng and Y. Li, Nano Res., 2020, 14, 1435–1442 CrossRef.
- J.-X. Liang, J. Lin, X.-F. Yang, A.-Q. Wang, B.-T. Qiao, J. Liu, T. Zhang and J. Li, J. Phys. Chem. C, 2014, 118, 21945–21951 CrossRef CAS.
- J. Lin, A. Wang, B. Qiao, X. Liu, X. Yang, X. Wang, J. Liang, J. Li, J. Liu and T. Zhang, J. Am. Chem. Soc., 2013, 135, 15314–15317 CrossRef CAS.
- M. D. Marcinkowski, S. F. Yuk, N. Doudin, R. S. Smith, N. Manh-Thuong, B. D. Kay, V.-A. Glezakou, R. Rousseau and Z. Dohnalek, ACS Catal., 2019, 9, 10977–10982 CrossRef CAS.
- R. Bliem, J. Pavelec, O. Gamba, E. McDermott, Z. Wang, S. Gerhold, M. Wagner, J. Osiecki, K. Schulte, M. Schmid, P. Blaha, U. Diebold and G. S. Parkinson, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 92, 075440 CrossRef.
- Z. Novotny, G. Argentero, Z. Wang, M. Schmid, U. Diebold and G. S. Parkinson, Phys. Rev. Lett., 2012, 108, 216103 CrossRef PubMed.
- W. Qu, X. Liu, J. Chen, Y. Dong, X. Tang and Y. Chen, Nat. Commun., 2020, 11, 1532 CrossRef CAS.
- R. Lang, W. Xi, J.-C. Liu, Y.-T. Cui, T. Li, A. F. Lee, F. Chen, Y. Chen, L. Li, L. Li, J. Lin, S. Miao, X. Liu, A.-Q. Wang, X. Wang, J. Luo, B. Qiao, J. Li and T. Zhang, Nat. Commun., 2019, 10, 234 CrossRef.
- J. Paier, C. Penschke and J. Sauer, Chem. Rev., 2013, 113, 3949–3985 CrossRef CAS.
- L. Nie, D. Mei, H. Xiong, B. Peng, Z. Ren, X. I. P. Hernandez, A. DeLariva, M. Wang, M. H. Engelhard, L. Kovarik, A. K. Datye and Y. Wang, Science, 2017, 358, 1419–1423 CrossRef CAS.
- A. Jan, J. Shin, J. Ahn, S. Yang, K. J. Yoon, J.-W. Son, H. Kim, J.-H. Lee and H.-I. Ji, RSC Adv., 2019, 9, 27002–27012 RSC.
- C. Riley, S. Zhou, D. Kunwar, A. De La Riva, E. Peterson, R. Payne, L. Gao, S. Lin, H. Guo and A. Datye, J. Am. Chem. Soc., 2018, 140, 12964–12973 CrossRef CAS.
- Y. Pang, W. Zang, Z. Kou, L. Zhang, G. Xu, J. Lv, X. Gao, Z. Pan, J. Wang and Y. Wu, Nanoscale, 2020, 12, 4302–4308 RSC.
- H. Wei, K. Huang, D. Wang, R. Zhang, B. Ge, J. Ma, B. Wen, S. Zhang, Q. Li, M. Lei, C. Zhang, J. Irawan, L.-M. Liu and H. Wu, Nat. Commun., 2017, 8, 1490 CrossRef PubMed.
- K. P. Kuhn, I. F. Chaberny, K. Massholder, M. Stickler, V. W. Benz, H. G. Sonntag and L. Erdinger, Chemosphere, 2003, 53, 71–77 CrossRef CAS.
- A. J. Therrien, A. J. R. Hensley, M. D. Marcinkowski, R. Zhang, F. R. Lucci, B. Coughlin, A. C. Schilling, J.-S. McEwen and E. C. H. Sykes, Nat. Catal., 2018, 1, 192–198 CrossRef CAS.
- H. Zhou, X. Yang, A. Wang, S. Miao, X. Liu, X. Pan, Y. Su, L. Li, Y. Tan and T. Zhang, Chin. J. Catal., 2016, 37, 692–699 CrossRef CAS.
- X. Zhou, Q. Shen, K. Yuan, W. Yang, Q. Chen, Z. Geng, J. Zhang, X. Shao, W. Chen, G. Xu, X. Yang and K. Wu, J. Am. Chem. Soc., 2018, 140, 554–557 CrossRef CAS.
- G. Busca, Catal. Today, 2014, 226, 2–13 CrossRef CAS.
- J. M. Clary, S. A. Van Norman, H. H. Funke, D. Su, C. B. Musgrave and A. W. Weimer, Nanotechnology, 2020, 31, 175703 CrossRef CAS PubMed.
- H. Shang, W. Chen, Z. Jiang, D. Zhou and J. Zhang, Chem. Commun., 2020, 56, 3127–3130 RSC.
- Y. Lu, J. Wang, L. Yu, L. Kovarik, X. Zhang, A. S. Hoffman, A. Gallo, S. R. Bare, D. Sokaras, T. Kroll, V. Dagle, H. Xin and A. M. Karim, Nat. Catal., 2019, 2, 149–156 CrossRef CAS.
- F. Yan, C. Zhao, L. Yi, J. Zhang, B. Ge, T. Zhang and W. Li, Chin. J. Catal., 2017, 38, 1613–1620 CrossRef CAS.
- Y. Jing, Z. Cai, C. Liu, T. Toyao, Z. Maeno, H. Asakura, S. Hiwasa, S. Nagaoka, H. Kondoh and K.-i. Shimizu, ACS Catal., 2020, 10, 1010–1023 CrossRef CAS.
- H. Wang, J. Dong, L. F. Allard, S. Lee, S. Oh, J. Wang, W. Li, M. Shen and M. Yang, Appl. Catal., B, 2019, 244, 327–339 CrossRef CAS.
- C. Dong and D. Ma, Sci. China: Chem., 2020, 63, 1584–1585 CrossRef CAS.
- Y. Xiong, W. Sun, Y. Han, P. Xin, X. Zheng, W. Yan, J. Dong, J. Zhang, D. Wang and Y. Li, Nano Res., 2021, 14, 2418–2423 CrossRef CAS.
- Y. Xiong, W. Sun, P. Xin, W. Chen, X. Zheng, W. Yan, L. Zheng, J. Dong, J. Zhang, D. Wang and Y. Li, Adv. Mater., 2020, 32, 2000896 CrossRef CAS.
- X. He, Y. Deng, Y. Zhang, Q. He, D. Xiao, M. Peng, Y. Zhao, H. Zhang, R. Luo, T. Gan, H. Ji and D. Ma, Cell Rep. Phys. Sci., 2020, 1, 100004 CrossRef.
- J. Wang, Z. Li, Y. Wu and Y. Li, Adv. Mater., 2018, 30, 1801649 CrossRef.
- L. H. Vieira, J. M. Assaf and E. M. Assaf, Mater. Lett., 2020, 277, 128354 CrossRef CAS.
- Y. Chen, S. Ji, W. Sun, W. Chen, J. Dong, J. Wen, J. Zhang, Z. Li, L. Zheng, C. Chen, Q. Peng, D. Wang and Y. Li, J. Am. Chem. Soc., 2018, 140, 7407–7410 CrossRef PubMed.
- X.-L. Wang, X.-P. Fu, W.-W. Wang, C. Ma, R. Si and C.-J. Jia, J. Phys. Chem. C, 2019, 123, 9001–9012 CrossRef CAS.
- X. I. Pcrcira-Hcrnandcz, A. DeLaRiva, V. Muravev, D. Kunwar, H. Xiong, B. Sudduth, M. Engelhard, L. Kovarlk, E. J. M. Hcnscn, Y. Wang and A. K. Datye, Nat. Commun., 2019, 10, 1358 CrossRef.
- M. J. Islam, M. G. Mesa, A. Osatiashtiani, M. J. Taylor, J. C. Manayil, C. M. A. Parlett, M. A. Isaacs and G. Kyriakou, Appl. Catal., B, 2020, 273, 119062 CrossRef CAS.
- S. Tian, W. Gong, W. Chen, N. Lin, Y. Zhu, Q. Feng, Q. Xu, Q. Fu, C. Chen, J. Luo, W. Yan, H. Zhao, D. Wang and Y. Li, ACS Catal., 2019, 9, 5223–5230 CrossRef CAS.
- K. Mou, Z. Chen, X. Zhang, M. Jiao, X. Zhang, X. Ge, W. Zhang and L. Liu, Small, 2019, 15, 1903668 CrossRef CAS PubMed.
- D. Ji, L. Fan, L. Li, S. Peng, D. Yu, J. Song, S. Ramakrishna and S. Guo, Adv. Mater., 2019, 31, 1808267 CrossRef.
- J. Wan, W. Chen, C. Jia, L. Zheng, J. Dong, X. Zheng, Y. Wang, W. Yan, C. Chen, Q. Peng, D. Wang and Y. Li, Adv. Mater., 2018, 30, 1808267 Search PubMed.
- J. Kim, C.-W. Roh, S. K. Sahoo, S. Yang, J. Bae, J. W. Han and H. Lee, Adv. Energy Mater., 2018, 8, 1701476 CrossRef.
- X. Shi, X. Wang, X. Shang, X. Zou, W. Ding and X. Lu, ChemCatChem, 2017, 9, 3743–3751 CrossRef CAS.
- J. Chen, Y. Wanyan, J. Zeng, H. Fang, Z. Li, Y. Dong, R. Qin, C. Wu, D. Liu, M. Wang, Q. Kuang, Z. Xie and L. Zheng, ACS Sustainable Chem. Eng., 2018, 6, 14054–14062 CrossRef CAS.
- J. Zhang, X. Wu, W.-C. Cheong, W. Chen, R. Lin, J. Li, L. Zheng, W. Yan, L. Gu, C. Chen, Q. Peng, D. Wang and Y. Li, Nat. Commun., 2018, 9, 1002 CrossRef PubMed.
- H. Wang, J. Shen, J. Huang, T. Xu, J. Zhu, Y. Zhu and C. Li, Nanoscale, 2017, 9, 16817–16825 RSC.
- X. Wang, X.-P. Fu, W.-Z. Yu, C. Ma, C.-J. Jia and R. Si, Inorg. Chem. Front., 2017, 4, 2059–2067 RSC.
- Z. Bo, L. R. McCullough, S. Dull, M. A. Ardagh, J. Wang and J. Notestein, J. Chem. Phys., 2019, 151, 214703 CrossRef PubMed.
- L. DeRita, S. Dai, K. Lopez-Zepeda, N. Pham, G. W. Graham, X. Pan and P. Christopher, J. Am. Chem. Soc., 2017, 139, 14150–14165 CrossRef CAS PubMed.
- M. Kottwitz, Y. Li, R. M. Palomino, Z. Liu, G. Wang, Q. Wu, J. Huang, J. Timoshenko, S. D. Senanayake, M. Balasubramanian, D. Lu, R. G. Nuzzo and A. I. Frenkel, ACS Catal., 2019, 9, 8738–8748 CrossRef CAS.
- J. Lu, J. W. Elam and P. C. Stair, Surf. Sci. Rep., 2016, 71, 410–472 CrossRef CAS.
- H. Wang and J. Lu, Acta Phys.-Chim. Sin., 2018, 34, 1334–1357 CAS.
- J. Lu, J. W. Elam and P. C. Stair, Acc. Chem. Res., 2013, 46, 1806–1815 CrossRef CAS.
- P. Z. Chen, T. P. Zhou, L. L. Xing, K. Xu, Y. Tong, H. Xie, L. D. Zhang, W. S. Yan, W. S. Chu, C. Z. Wu and Y. Xie, Angew. Chem., Int. Ed., 2017, 56, 610–614 CrossRef CAS PubMed.
- B. Y. Xia, H. B. Wu, N. Li, Y. Yan, X. W. Lou and X. Wang, Angew. Chem., Int. Ed., 2015, 54, 3797–3801 CrossRef CAS PubMed.
- S. Sun, G. Zhang, N. Gauquelin, N. Chen, J. Zhou, S. Yang, W. Chen, X. Meng, D. Geng, M. N. Banis, R. Li, S. Ye, S. Knights, G. A. Botton, T.-K. Sham and X. Sun, Sci. Rep., 2013, 3, 1775 CrossRef.
- H. Shang, W. Sun, R. Sui, J. Pei, L. Zheng, J. Dong, Z. Jiang, D. Zhou, Z. Zhuang, W. Chen, J. Zhang, D. Wang and Y. Li, Nano Lett., 2020, 20, 5443–5450 CrossRef CAS.
- C. Wang, H. Tissot, J. H. Stenlid, S. Kaya and J. Weissenrieder, J. Phys. Chem. Lett., 2019, 10, 7318–7323 CrossRef CAS.
- U. Heiz, A. Sanchez, S. Abbet and W. D. Schneider, J. Am. Chem. Soc., 1999, 121, 3214–3217 CrossRef CAS.
- S. Abbet, A. Sanchez, U. Heiz, W. D. Schneider, A. M. Ferrari, G. Pacchioni and N. Rosch, J. Am. Chem. Soc., 2000, 122, 3453–3457 CrossRef CAS.
- S. Proch, M. Wirth, H. S. White and S. L. Anderson, J. Am. Chem. Soc., 2013, 135, 3073–3086 CrossRef CAS PubMed.
- J. Liu, B. R. Bunes, L. Zang and C. Wang, Environ. Chem. Lett., 2018, 16, 477–505 CrossRef CAS.
- S. Wei, A. Li, J.-C. Liu, Z. Li, W. Chen, Y. Gong, Q. Zhang, W.-C. Cheong, Y. Wang, L. Zheng, H. Xiao, C. Chen, D. Wang, Q. Peng, L. Gu, X. Han, J. Li and Y. Li, Nat. Nanotechnol., 2018, 13, 856–861 CrossRef CAS.
- Z. Huang, X. Gu, Q. Cao, P. Hu, J. Hao, J. Li and X. Tang, Angew. Chem., Int. Ed., 2012, 51, 4198–4203 CrossRef CAS PubMed.
- J. Yu, A. Wang, W. Yu, X. Liu, X. Li, H. Liu, Y. Hu, Y. Wu and W. Zhou, Appl. Catal., B, 2020, 277, 119236 CrossRef CAS.
- K. Liu, X. Zhao, G. Ren, T. Yang, Y. Ren, A. F. Lee, Y. Su, X. Pan, J. Zhang, Z. Chen, J. Yang, X. Liu, T. Zhou, W. Xi, J. Luo, C. Zeng, H. Matsumoto, W. Liu, Q. Jiang, K. Wilson, A. Wang, B. Qiao, W. Li and T. Zhang, Nat. Commun., 2020, 11, 1263 CrossRef CAS PubMed.
- A. Han, J. Zhang, W. Sun, W. Chen, S. Zhang, Y. Han, Q. Feng, L. Zheng, L. Gu, C. Chen, Q. Peng, D. Wang and Y. Li, Nat. Commun., 2019, 10, 3787 CrossRef.
- M. Flytzani-Stephanopoulos, Acc. Chem. Res., 2014, 47, 783–792 CrossRef CAS.
- Q. Fu, W. L. Deng, H. Saltsburg and M. Flytzani-Stephanopoulos, Appl. Catal., B, 2005, 56, 57–68 CrossRef CAS.
- L. Wang, W. Zhang, S. Wang, Z. Gao, Z. Luo, X. Wang, R. Zeng, A. Li, H. Li, M. Wang, X. Zheng, J. Zhu, W. Zhang, C. Ma, R. Si and J. Zeng, Nat. Commun., 2016, 7, 14036 CrossRef CAS PubMed.
- H. Xu, T. Liu, S. Bai, L. Li, Y. Zhu, J. Wang, S. Yang, Y. Li, Q. Shao and X. Huang, Nano Lett., 2020, 20, 5482–5489 CrossRef CAS PubMed.
- X. Ge, P. Zhou, Q. Zhang, Z. Xia, S. Chen, P. Gao, Z. Zhang, L. Gu and S. Guo, Angew. Chem., Int. Ed., 2020, 59, 232–236 CrossRef CAS PubMed.
- A. V. Crewe, J. Wall and J. Lanomore, Science, 1970, 168, 1338–1340 CrossRef CAS PubMed.
- S. Duan, R. Wang and J. Liu, Nanotechnology, 2018, 29, 204002 CrossRef PubMed.
- J. Liu, Chin. J. Catal., 2017, 38, 1460–1472 CrossRef CAS.
- C. Wang, X.-K. Gu, H. Yan, Y. Lin, J. Li, D. Liu, W.-X. Li and J. Lu, ACS Catal., 2017, 7, 887–891 CrossRef CAS.
- S. B. Rice, J. Y. Koo, M. M. Disko and M. M. J. Treacy, Ultramicroscopy, 1990, 34, 108–118 CrossRef CAS.
- T. Sun, Y. Li, T. Cui, L. Xu, Y. G. Wang, W. Chen, P. Zhang, T. Zheng, X. Fu, S. Zhang, Z. Zhang, D. Wang and Y. Li, Nano Lett., 2020, 20, 6206–6214 CrossRef CAS PubMed.
- H. Li, L. Wang, Y. Dai, Z. Pu, Z. Lao, Y. Chen, M. Wang, X. Zheng, J. Zhu, W. Zhang, R. Si, C. Ma and J. Zeng, Nat. Nanotechnol., 2018, 13, 411–417 CrossRef CAS PubMed.
- Z. Jiang, X. Feng, J. Deng, C. He, M. Douthwaite, Y. Yu, J. Liu, Z. Hao and Z. Zhao, Adv. Funct. Mater., 2019, 29, 1902041 CrossRef.
- M. D. Marcinkowski, M. T. Darby, J. Liu, J. M. Wimble, F. R. Lucci, S. Lee, A. Michaelides, M. Flytzani-Stephanopoulos, M. Stamatakis and E. C. H. Sykes, Nat. Chem., 2018, 10, 325–332 CrossRef CAS PubMed.
- S. Koust, L. Arnarson, P. G. Moses, Z. Li, I. Beinik, J. V. Lauritsen and S. Wendt, Phys. Chem. Chem. Phys., 2017, 19, 9424–9431 RSC.
- A. Mellor, D. Humphrey, C. M. Yim, C. L. Pang, H. Idriss and G. Thornton, J. Phys. Chem. C, 2017, 121, 24721–24725 CrossRef CAS PubMed.
- C. Ye, N. Zhang, D. Wang and Y. Li, Chem. Commun., 2020, 56, 7687–7697 RSC.
- B. C. Gates, Chem. Rev., 1995, 95, 511–522 CrossRef CAS.
- G. V. Korshin, A. I. Frenkel and E. A. Stern, Environ. Sci. Technol., 1998, 32, 2699–2705 CrossRef CAS.
- R. B. Greegor and F. W. Lytle, J. Catal., 1980, 63, 476–486 CrossRef CAS.
- F. de Groot, Chem. Rev., 2001, 101, 1779–1808 CrossRef CAS PubMed.
- H. Zhang, G. Liu, L. Shi and J. Ye, Adv. Energy Mater., 2018, 8, 1701343 CrossRef.
- O. Alexeev and B. C. Gates, Top. Catal., 2000, 10, 273–293 CrossRef CAS.
- J. J. Rehr and R. C. Albers, Rev. Mod. Phys., 2000, 72, 621–654 CrossRef CAS.
- Q. Wang, X. Huang, Z. L. Zhao, M. Wang, B. Xiang, J. Li, Z. Feng, H. Xu and M. Gu, J. Am. Chem. Soc., 2020, 142, 7425–7433 CrossRef CAS PubMed.
- B. B. Sarma, P. N. Plessow, G. Agostini, P. Concepcion, N. Pfaender, L. Kang, F. R. Wang, F. Studt and G. Prieto, J. Am. Chem. Soc., 2020, 142, 14890–14902 CrossRef CAS PubMed.
- N. C. Nelson, L. Chen, D. Meira, L. Kovarik and J. Szanyi, Angew. Chem., Int. Ed., 2020, 59, 17657–17663 CrossRef CAS PubMed.
- L. Yuan, S.-F. Hung, Z.-R. Tang, H. M. Chen, Y. Xiong and Y.-J. Xu, ACS Catal., 2019, 9, 4824–4833 CrossRef CAS.
- L. Ye, X. Duan, S. Wu, T.-S. Wu, Y. Zhao, A. W. Robertson, H.-L. Chou, J. Zheng, T. Ayvali, S. Day, C. Tang, Y.-L. Soo, Y. Yuan and S. C. E. Tsang, Nat. Commun., 2019, 10, 914 CrossRef PubMed.
- K. Siegbahn, C. Nordling, A. Fahlman, K. Hamrin, J. Hedman, R. Nordberg, C. Johansson, T. Bergmark, S.-E. Karlsson, I. Lindgren and B. Lindberg, Nova Acta Regiae Soc. Sci. Ups. 20.1-282, Almqvist and Wiksells, 1967 Search PubMed.
- R. W. Joyner, M. W. Roberts and K. Yates, Surf. Sci., 1979, 87, 501–509 CrossRef CAS.
- Q. Shang, N. Tang, H. Qi, S. Chen, G. Xu, C. Wu, X. Pan, X. Wang and Y. Cong, Chin. J. Catal., 2020, 41, 1812–1817 CrossRef CAS.
- Y. Yao, S. Hu, W. Chen, Z. Q. Huang, W. Wei, T. Yao, R. Liu, K. Zang, X. Wang, G. Wu, W. Yuan, T. Yuan, B. Zhu, W. Liu, Z. Li, D. He, Z. Xue, Y. Wang, X. Zheng, J. Dong, C. R. Chang, Y. Chen, X. Hong, J. Luo, S. Wei, W. X. Li, P. Strasser, Y. Wu and Y. Li, Nat. Catal., 2019, 2, 304–313 CrossRef CAS.
- G. Giannakakis, A. Trimpalis, J. Shan, Z. Qi, S. Cao, J. Liu, J. Ye, J. Biener and M. Flytzani-Stephanopoulos, Top. Catal., 2018, 61, 475–486 CrossRef CAS.
- G. X. Pei, X. Y. Liu, X. Yang, L. Zhang, A. Wang, L. Li, H. Wang, X. Wang and T. Zhang, ACS Catal., 2017, 7, 1491–1500 CrossRef CAS.
- J. Liu, J. Shan, F. R. Lucci, S. Cao, E. C. H. Sykes and M. Flytzani-Stephanopoulos, Catal. Sci. Technol., 2017, 7, 4276–4284 RSC.
- L. Zhang, A. Wang, J. T. Miller, X. Liu, X. Yang, W. Wang, L. Li, Y. Huang, C.-Y. Mou and T. Zhang, ACS Catal., 2014, 4, 1546–1553 CrossRef CAS.
- F. Xing, J. Jeon, T. Toyao, K.-i. Shimizu and S. Furukawa, Chem. Sci., 2019, 10, 8292–8298 RSC.
- C. Lamberti, A. Zecchina, E. Groppo and S. Bordiga, Chem. Soc. Rev., 2010, 39, 4951–5001 RSC.
- J. Ryczkowski, Catal. Today, 2001, 68, 263–381 CrossRef CAS.
- C. Asokan, L. DeRita and P. Christopher, Chin. J. Catal., 2017, 38, 1473–1480 CrossRef CAS.
- F. Yang, S. Ding, H. Song and N. Yan, Sci. China Mater., 2020, 63, 982–992 CrossRef CAS.
- M.-M. Millet, G. Algara-Siller, S. Wrabetz, A. Mazheika, F. Girgsdies, D. Teschner, F. Seitz, A. Tarasov, S. V. Leychenko, R. Schloegl and E. Frei, J. Am. Chem. Soc., 2019, 141, 2451–2461 CrossRef CAS PubMed.
- Y. Lu, C.-T. Kuo, L. Kovarik, A. S. Hoffman, A. Boubnov, D. M. Driscoll, J. R. Morris, S. R. Bare and A. M. Karim, J. Catal., 2019, 378, 121–130 CrossRef CAS.
- A. J. Foster and R. F. Lobo, Chem. Soc. Rev., 2010, 39, 4783–4793 RSC.
- M. J. Huelsey, B. Zhang, Z. Ma, H. Asakura, D. A. Do, W. Chen, T. Tanaka, P. Zhang, Z. Wu and N. Yan, Nat. Commun., 2019, 10, 1330 CrossRef PubMed.
- C. Schilling, M. Ziemba, C. Hess and M. Veronica Ganduglia-Pirovano, J. Catal., 2020, 383, 264–272 CrossRef CAS.
- D. B. Burueva, L. M. Kovtunova, V. I. Bukhtiyarov, K. V. Kovtunov and I. V. Koptyug, Chem.–Eur. J., 2019, 25, 1420–1431 CrossRef CAS PubMed.
- W. Zhang, S. Xu, X. Han and X. Bao, Chem. Soc. Rev., 2012, 41, 192–210 RSC.
- A. Brueckner, Chem. Soc. Rev., 2010, 39, 4673–4684 RSC.
- K. Dyrek and M. Che, Chem. Rev., 1997, 97, 305–331 CrossRef CAS PubMed.
- Q. Shen, C. Cao, R. Huang, L. Zhu, X. Zhou, Q. Zhang, L. Gu and W. Song, Angew. Chem., Int. Ed., 2020, 59, 1216–1219 CrossRef CAS PubMed.
- S. Hejazi, S. Mohajernia, B. Osuagwu, G. Zoppellaro, P. Andryskova, O. Tomanec, S. Kment, R. Zboril and P. Schmuki, Adv. Mater., 2020, 32, 1908505 CrossRef CAS PubMed.
- S. M. Heald, J. Synchrotron Radiat., 2015, 22, 436–445 CrossRef CAS PubMed.
- J. Zhang, C. Liu and B. Zhang, Small Methods, 2019, 3, 1800481 CrossRef.
- Z. Xu, Y. Zhang, L. Qin, Q. Meng, Z. Xue, L. Qiu, G. Zhang, X. Guo and Q. Li, Small, 2020, 16, 2002071 CrossRef CAS PubMed.
- Y. Lou, Y. Cai, W. Hu, L. Wang, Q. Dai, W. Zhan, Y. Guo, P. Hu, X.-M. Cao, J. Liu and Y. Guo, ACS Catal., 2020, 10, 6094–6101 CrossRef CAS.
- B. Qiao, J.-X. Liang, A. Wang, C.-Q. Xu, J. Li, T. Zhang and J. J. Liu, Nano Res., 2015, 8, 2913–2924 CrossRef CAS.
- Z. Jakub, J. Hulva, P. T. P. Ryan, D. A. A. Duncan, D. J. J. Payne, R. Bliem, M. Ulreich, P. Hofegger, F. Kraushofer, M. Meier, M. Schmid, U. Diebold and G. S. S. Parkinson, Nanoscale, 2020, 12, 5866–5875 RSC.
- W. Chen, Y. Ma, F. Li, L. Pan, W. Gao, Q. Xiang, W. Shang, C. Song, P. Tao, H. Zhu, X. Pan, T. Deng and J. Wu, Adv. Funct. Mater., 2019, 29, 1904278 CrossRef CAS.
- Q. Fu, H. Saltsburg and M. Flytzani-Stephanopoulos, Science, 2003, 301, 935–938 CrossRef CAS PubMed.
- S. Yao, X. Zhang, W. Zhou, R. Gao, W. Xu, Y. Ye, L. Lin, X. Wen, P. Liu, B. Chen, E. Crumlin, J. Guo, Z. Zuo, W. Li, J. Xie, L. Lu, C. J. Kiely, L. Gu, C. Shi, J. A. Rodriguez and D. Ma, Science, 2017, 357, 389–393 CrossRef CAS PubMed.
- Y. Xiang, J. He, N. Sun, Y. Fan, L. Yang, C. Fang and L. Kuai, Microporous Mesoporous Mater., 2020, 308, 110507 CrossRef CAS.
- S. C. Ammal and A. Heyden, ACS Catal., 2019, 9, 7721–7740 CrossRef CAS.
- X. Sun, J. Lin, Y. Zhou, L. Li, Y. Su, X. Wang and T. Zhang, AIChE J., 2017, 63, 4022–4031 CrossRef CAS.
- H. Guan, J. Lin, B. Qiao, S. Miao, A.-Q. Wang, X. Wang and T. Zhang, AIChE J., 2017, 63, 2081–2088 CrossRef CAS.
- M. Yan, X. Ma, Y. Yang, X. Wang, W. C. Cheong, Z. Chen, X. Xu, Y. Huang, S. Wang, C. Lian and Y. Li, Nano Lett., 2018, 18, 6017–6021 CrossRef CAS PubMed.
- S. C. Ammal and A. Heyden, ACS Catal., 2017, 7, 301–309 CrossRef CAS.
- M. Xing, L. Guo and Z. Hao, Int. J. Quantum Chem., 2018, 118, e25767 CrossRef.
- J. Shan, M. Li, L. F. Allard, S. Lee and M. Flytzani-Stephanopoulos, Nature, 2017, 551, 605–608 CrossRef CAS PubMed.
- D. Bajec, A. Kostyniuk, A. Pohar and B. Likozar, Chem. Eng. J., 2020, 396, 125182 CrossRef CAS.
- S. Bai, F. Liu, B. Huang, F. Li, H. Lin, T. Wu, M. Sun, J. Wu, Q. Shao, Y. Xu and X. Huang, Nat. Commun., 2020, 11, 954 CrossRef CAS PubMed.
- K. Harrath, X. Yu, H. Xiao and J. Li, ACS Catal., 2019, 9, 8903–8909 CrossRef CAS.
- Y. Kwon, T. Y. Kim, G. Kwon, J. Yi and H. Lee, J. Am. Chem. Soc., 2017, 139, 17694–17699 CrossRef CAS PubMed.
- X. Guo, G. Fang, G. Li, H. Ma, H. Fan, L. Yu, C. Ma, X. Wu, D. Deng, M. Wei, D. Tan, R. Si, S. Zhang, J. Li, L. Sun, Z. Tang, X. Pan and X. Bao, Science, 2014, 344, 616–619 CrossRef CAS PubMed.
- Y. Tang, Y. Wei, Z. Wang, S. Zhang, Y. Li, L. Nguyen, Y. Li, Y. Zhou, W. Shen, F. F. Tao and P. Hu, J. Am. Chem. Soc., 2019, 141, 7283–7293 CrossRef CAS PubMed.
- M. Akri, S. Zhao, X. Li, K. Zang, A. F. Lee, M. A. Isaacs, W. Xi, Y. Gangarajula, J. Luo, Y. Ren, Y.-T. Cui, L. Li, Y. Su, X. Pan, W. Wen, Y. Pan, K. Wilson, L. Li, B. Qiao, H. Ishii, Y.-F. Liao, A. Wang, X. Wang and T. Zhang, Nat. Commun., 2019, 10, 5181 CrossRef PubMed.
- Z. Zuo, S. Liu, Z. Wang, C. Liu, W. Huang, J. Huang and P. Liu, ACS Catal., 2018, 8, 9821–9835 CrossRef CAS.
- W.-J. Jang, J.-O. Shim, H.-M. Kim, S.-Y. Yoo and H.-S. Roh, Catal. Today, 2019, 324, 15–26 CrossRef CAS.
- D. Pakhare and J. Spivey, Chem. Soc. Rev., 2014, 43, 7813–7837 RSC.
- S. Li, J. Yang, C. Song, Q. Zhu, D. Xiao and D. Ma, Adv. Mater., 2019, 31, 1901796 CrossRef CAS PubMed.
- X. Meng, X. Cui, N. P. Rajan, L. Yu, D. Deng and X. Bao, Chem, 2019, 5, 2296–2325 CAS.
- P. Schwach, X. Pan and X. Bao, Chem. Rev., 2017, 117, 8497–8520 CrossRef CAS PubMed.
- D. A. Bulushev and J. R. H. Ross, Catal. Rev.: Sci. Eng., 2018, 60, 566–593 CrossRef CAS.
- X. Ye, C. Yang, X. Pan, J. Ma, Y. Zhang, Y. Ren, X. Liu, L. Li and Y. Huang, J. Am. Chem. Soc., 2020, 142, 19001–19005 CrossRef CAS PubMed.
- C. Rivera-Carcamo, C. Scarfiello, A. B. Garcia, Y. Tison, H. Martinez, W. Baaziz, O. Ersen, C. Le Berre and P. Serp, Adv. Mater. Interfaces, 2020, 2001777, DOI:10.1002/admi.202001777.
- M. Xia, J. Ding, X. Du, R. Shang and Q. Zhong, J. Alloys Compd., 2019, 777, 406–414 CrossRef CAS.
- Y. Wang, H. Arandiyan, J. Scott, K.-F. Aguey-Zinsou and R. Amal, ACS Appl. Energy Mater., 2018, 1, 6781–6789 CrossRef CAS.
- Y. Guo, S. Mei, K. Yuan, D.-J. Wang, H.-C. Liu, C.-H. Yan and Y.-W. Zhang, ACS Catal., 2018, 8, 6203–6215 CrossRef CAS.
- F. J. Caparros, L. Soler, M. D. Rossell, I. Angurell, L. Piccolo, O. Rossell and J. Llorca, ChemCatChem, 2018, 10, 2365–2369 CrossRef CAS.
- S. Ni, H. Zhang, Y. Zhao, X. Li, Y. Sun, J. Qian, Q. Xu, P. Gao, D. Wu, K. Kato, M. Yamauchi and Y. Sun, Chem. Eng. J., 2019, 366, 631–638 CrossRef CAS.
- W. K. Xiang, Y. H. Zhao, Z. Jiang, X. P. Li, H. Zhang, Y. Sun, Z. J. Ning, F. P. Du, P. Gao, J. Qian, K. Kato, M. Yamauchi and Y. H. Sun, J. Mater. Chem. A, 2018, 6, 23366–23377 RSC.
- Y. Pan, K. Sun, S. Liu, X. Cao, K. Wu, W. C. Cheong, Z. Chen, Y. Wang, Y. Li, Y. Liu, D. Wang, Q. Peng, C. Chen and Y. Li, J. Am. Chem. Soc., 2018, 140, 2610–2618 CrossRef CAS PubMed.
- Z. Hu, X. Li, S. Zhang, Q. Li, J. Fan, X. Qu and K. Lv, Small, 2020, 16, 2004583 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2022 |