
Mechanistic understanding and design of non-noble metal-based single-atom catalysts supported on two-dimensional materials for CO2 electroreduction
Ya
Huang
 a,
Faisal
Rehman
a,
Mohsen
Tamtaji
a,
Xuning
Li
b,
Yanqiang
Huang
b,
Tao
Zhang
*b and
Zhengtang
Luo
*a
a,
Faisal
Rehman
a,
Mohsen
Tamtaji
a,
Xuning
Li
b,
Yanqiang
Huang
b,
Tao
Zhang
*b and
Zhengtang
Luo
*a
aDepartment of Chemical and Biological Engineering, Guangdong-Hong Kong-Macao Joint Laboratory for Intelligent Micro-Nano Optoelectronic Technology, William Mong Institute of Nano Science and Technology, Hong Kong Branch of Chinese National Engineering Research Center for Tissue Restoration and Reconstruction, The Hong Kong University of Science and Technology, Clear Water Bay, Kowloon, Hong Kong 999077, P. R. China. E-mail: keztluo@ust.hk
bState Key Laboratory of Catalysis, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, Liaoning, China. E-mail: taozhang@dicp.ac.cn
First published on 26th November 2021
Abstract
Single-atom catalysts (SACs), which are low-cost, contain earth-abundant metals, and feature two-dimensional material supports, have shown great potential for us in a wide range of electrochemical reactions, including the CO2 reduction reaction (CO2RR) to convert CO2 to valuable chemicals and fuels. In recent years, substantial advances have been achieved in the preparation methodologies, with improved catalytic performances, but the underlying structure–activity relationship from a general perspective remains elusive. In particular, it is urgent to summarize the progress made on SACs with diatom metal centers toward efficient CO2RR. Based on the recent progress in this area, this review synopsizes the fundamental understandings of non-noble metal-based SACs for CO2RR using selected examples. We also highlight the representative atomic structures of active sites from the latest progress, including M–N–C, heteroatom co-doping, vacancy/edge defects and bimetallic SACs, with the aim of elucidating the nature of active sites on various 2D substrates. Moreover, we summarize the spectroscopic and computational studies to verify the atomic-level regulation of the geometric and electronic properties of SACs. We anticipate that this review will deepen the mechanistic understanding of the structure–performance relationship and inspire future studies on SACs for CO2RR.
1. Introduction
The environmental issues brought about by the tremendous consumption of fossil fuels has led to the exploration of renewable energy.1–3 According to a recent report, the global average concentration of CO2 in the atmosphere reached a recorded level of over 415 ppm in the first half of 2021 and this trend is expected to continue in the next 20 years if no counteraction is taken.4 The urgent need for clean energy technologies in reducing atmospheric CO2 concentration presents a great research challenge and opportunity.5 In recent decades, tremendous efforts have been made in the field of electrochemical CO2 reduction reaction (CO2RR), to mitigate global warming and tackle the worldwide energy shortage.6–9 Ultimately, carbon neutrality can be achieved by converting CO2 into storable valuable fuels, such as methane, methanol, formate, ethanol, etc.10,11 Since the implementation of the CO2RR mainly relies on high-performance electrocatalysts, the development of highly efficient and scalable catalysts is the key. Significant insights have been obtained from extensive studies on bulk or nanoscale electrocatalysts over the past few years.12,13 However, two issues are notably worthy of being considered for these categories: (1) low atomic utilization, where only a small fraction of metal atoms are electrochemically active during electrolysis, and (2) relatively poor selectivity due to the high kinetic barriers of CO2 activation and the competing hydrogen evolution reaction (HER).14–16 The emerging single-atom catalysts (SACs), with adjustable geometric and electronic structures of their catalytic sites, have been considered as ideal candidate to address this dilemma.SACs are catalysts with atomically isolated metal active sites stabilized by a support, which were first introduced by Zhang, Li, Liu, and co-workers in 2011.17 This work put forward the first example of Pt SACs dispersed on FeOx for CO oxidation and demonstrated the catalytic mechanism through density functional theory (DFT) studies. After this, SACs as a new class of heterogeneous catalysts rose to be a research hotspot, with substantial work being carried out. The interest in SACs towards the CO2RR arises from three main aspects: (1) their extremely high atom efficiency, where every single atom is exposed as a reactive site.18 (2) The low coordination state of the metal single atom directly affects the electronic and geometric properties and therefore leads to different catalytic activities.19–21 Compared with non-SACs, atomically dispersed 3d nonprecious metals (especially Fe, Co, and Ni) exhibit better selectivity towards CO2RR over the HER.22–24 (3) SAC coordination into a graphitic framework can be easily simulated using DFT calculation models. Such simplicity of active regions endows them to be ideal platforms to study catalytic mechanism.25
Research on CO2 electro-conversion has made a significant breakthrough recently and has been categorized according to various factors. For instance, a series of SAC fabrication methods, substrate materials, the types of central metals, and the preferred products have been systematically summarized in previous reviews.26–30 Despite numerous progress reports on SACs for electrochemical reactions, the catalytic design strategies and experimental performance have been substantially focused upon, while the additional insights into the spectroscopies and underlying relationship among different atomic structures for the CO2RR have been rarely systematically reviewed. To narrow the scope, this review discusses nonprecious metal-based SACs, especially those featuring 3d transition metals. Non-noble metal single atoms have been widely studied as carbon-based low-cost SACs show great possibility to replace the noble metal versions and be scaled up to an industrial level in the future.28,31,32 Among them, Cu has been found to be a unique metal that can reduce CO2 to C2 or C2+ products, which plays a decisive role in this field and requires more attention.11,33–35
As shown in the workflow chart (Fig. 1), this review begins with fundamental knowledge on the CO2RR selected from recent representative works and factors that can impact the selectivity of SACs. In-depth insights into a variety of molecular models with diverse coordination structures extracted from carbon-based SACs, involving M–N–C systems, heteroatom co-doping, vacancy/edge defects and bimetallic models are discussed. Special attention is paid to the recent development of advanced spectroscopies that can be used to identify the atomic structures of SACs and interpret their evolution during electrolysis. Furthermore, we present a computational part, which focuses on DFT and machine learning (ML), providing atomic-level insight into SACs and reaction mechanisms. Finally, major challenges and perspectives on non-noble metal-based SACs are discussed. We hope this review can pave the way towards the rational design of SACs and provide fundamental guidance for the future research on the CO2RR. The abbreviations using it in the review are listed in the Table 1.
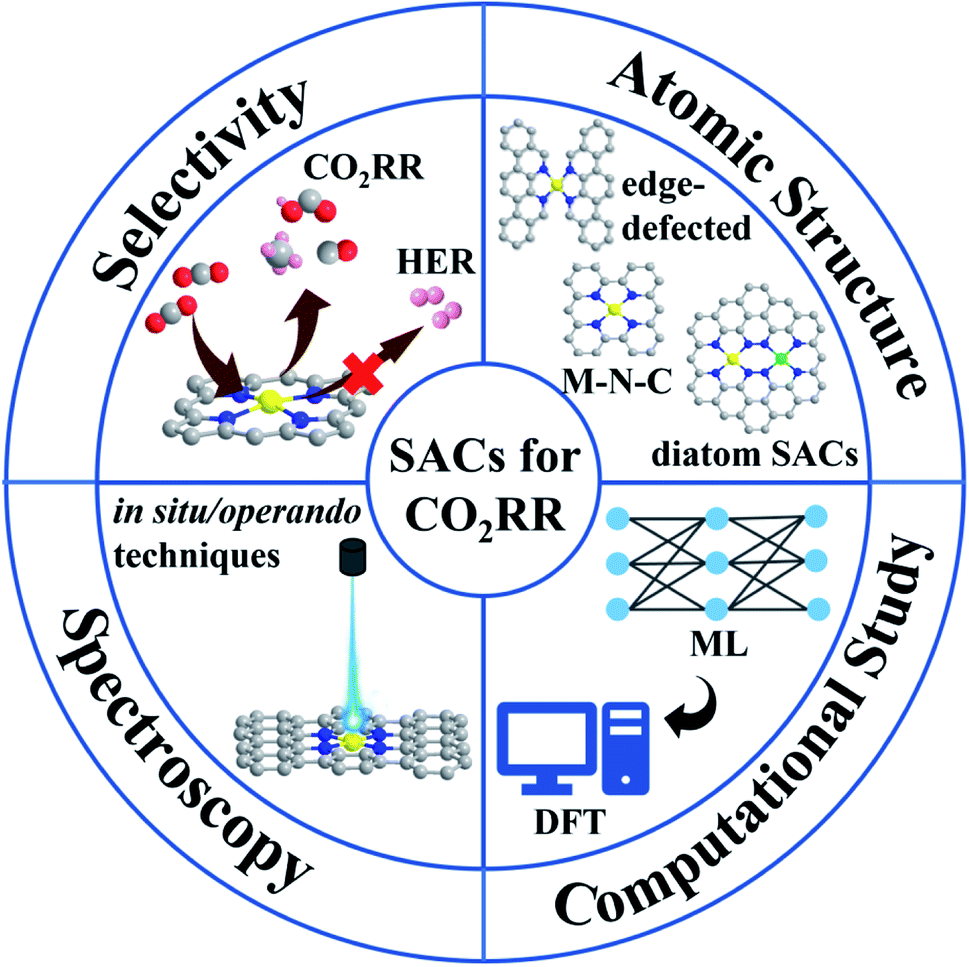 | ||
| Fig. 1 Workflow chart of this review. The review follows four steps, including the selectivity of SACs, atomic structural tuning, advanced spectroscopy, and computational study. | ||
2. Fundamentals of the CO2RR on SACs
The mechanism pathways of the CO2RR are complex due to multi-electron transfer leading to various possible products.14 This process starts with CO2 adsorption on the active sites, followed by the step-wise transfer of protons (H+) and/or electrons (e−) and generation of diverse intermediates and products.36 As illustrated in Scheme 1a, the CO2 molecule is first activated to *COOH via a one proton-coupled electron transfer (PCET) process.37 This step is critical for the determination of the selectivity of SACs due to the existing competition of *H adsorption. It should be noted that the species illustrated in Scheme 1 represent the product formation based on the number of electrons/protons transferred. Two-electron transfer is commonly observed for SACs, with CO or HCOOH being released as final products. And deeply reduced C1 products like methanol and methane require six and eight H+/e− transfer, respectively. Scheme 1b illustrates the proposed pathways toward C2 products, where the OC–CO dimerization is the rate-determining step (RDS).37 An alternative pathway toward C2 products is through the coupling of *CHO/*COH and *CO, which has been theoretically proved on diatom SACs supported by 2D graphene nitrene.38 All subsequent evolutions of species come from these C–C coupling processes. Notably, stabilized C2 intermediates can be transferred to n-propanol via CO insertion,34 but it is very difficult for this to take place on SACs, therefore this pathway will not be discussed in detail in this review.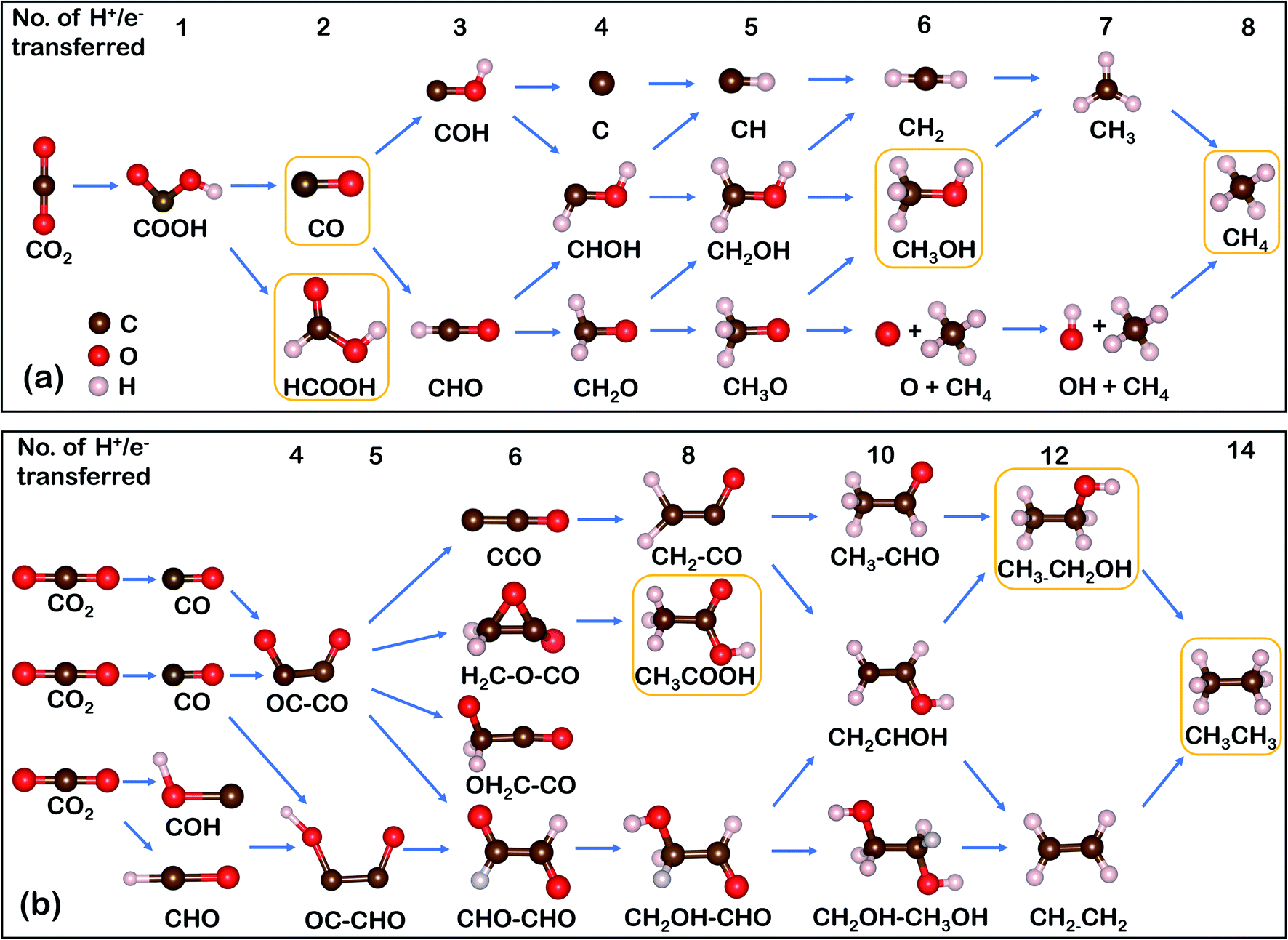 | ||
| Scheme 1 Overview of possible CO2RR roadmaps. The product formation of CO2RR on SACs toward various (a) C1 and (b) C2 products (main products are boxed) with the number of H+/e− transferred. | ||
| 1E_M | First ionization energy | MOFs | Metal–organic frameworks |
| ATR-IR | Attenuated total reflection-infrared | MS | Intermedium-spin |
| C2N | Nitrogen-doped graphene | OCV | Open-circuit voltage |
| CO2RR | CO2 reduction reaction | OER | Oxygen evolution reaction |
| COFs | Covalent organic frameworks | Pc | Phthalocyanine |
| DFT | Density functional theory | PCET | Proton-coupled electron transfer |
| DOS | Density of state | RDS | Rate-determining step |
| E_M | Pauling-electronegativity | RHE | Reversible hydrogen electrode |
| EXAFS | Extended X-ray absorption fine structure | SACs | Single-atom catalysts |
| FE | Faradaic efficiency | TCNQ | Tetracyanoquinodimethane |
| FT | Fourier transform | TM | 3d transition metal |
| g-C3N4 | Graphitic carbon nitride | TOF | Turnover frequency |
| GQDs | Graphene quantum dots | TPP | Porphyrin |
| HER | Hydrogen evolution reaction | WT | Wavelet transform |
| HS | High-spin | XANES | X-ray absorption near-edge structure |
| j CO | CO partial current density | XAS | X-ray absorption spectroscopy |
| LS | Low-spin | XGBR | Extreme gradient boosting regression |
| M_cov | Covalent radius | ΔG | Gibbs free energy |
| MEA | Membrane electrode assembly | η | Overpotentials |
| ML | Machine learning |
In contrast to bulk and nanostructured non-noble metals, which have been developed on an industrial-scale and exhibit high current densities toward C2 products, the majority of SACs for the CO2RR are still at the stage of laboratory studies.39,40 The most commonly used H-type electrolyzer for the CO2RR, constructed from two gas-tight half-cell chambers separated by an ion-exchange membrane, is illustrated in Fig. 2a. Furthermore, the half-cell reactions in the CO2RR with their reduction potentials of the main products at pH 7 are listed in Table 2.41 Most commonly, the CO2RR on SACs proceeds through a two-electron transfer process and generates CO or formate, balanced with oxygen evolution reaction (OER) as the anodic reaction.10 However, the selectivity of the CO2RR over carbon-based SACs requires specific attention because several factors are believed to influence the catalytic performance and reduced products. In this section, selected fundamental factors and how they impact CO2 activation and intermediate stabilization will be discussed.
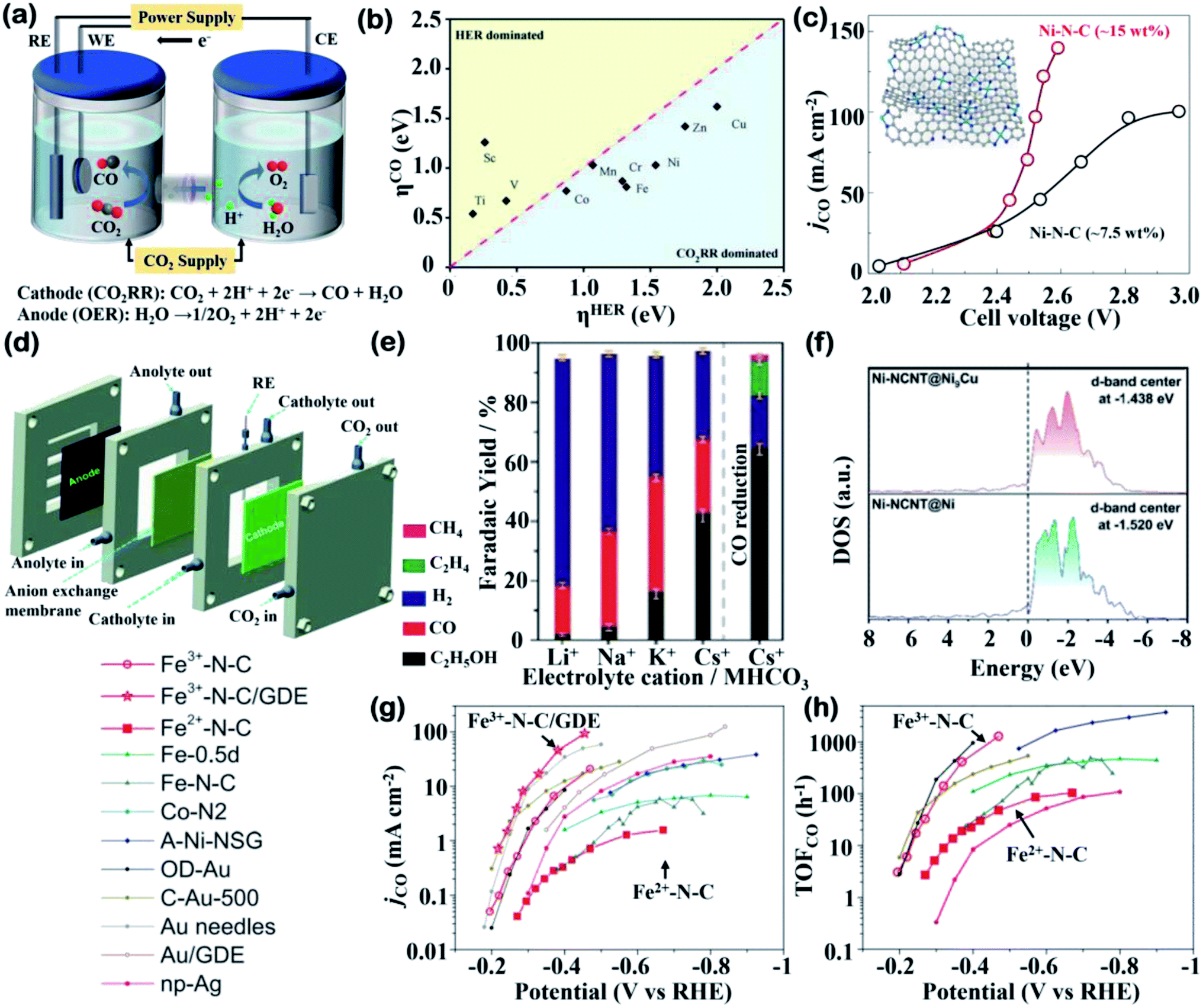 | ||
| Fig. 2 Cell configurations and electrochemical data. (a) A CO2 reduction H-cell with electrode reaction toward CO. (b) Overpotential for CO2RR against that for HER on 3d transition metal SACs. Reproduced with permission from ref. 19, copyright 2021, Wiley-VCH. (c) The partial current densities of CO (jCO) of nearly 7.5 wt% and 15 wt% Ni–N–C catalysts under different cell voltages. Inset: illustration of SACs prepared with the crosslinking and then self-assembled GQDs. Reproduced with permission from ref. 62, copyright 2019, Springer Nature Publication. (d) Schematic illustration of the flow cell configuration for CO2RR. Reproduced with permission from ref. 65, copyright 2021, Royal Society of Chemistry. (e) faradaic yields (FY) of CO2RR at −1.2 V vs. RHE in 0.1 M various cationic (Li+, Na+, K+ and Cs+) electrolytes. FY of CO reduction at −1.2 V vs. RHE in 0.1 M CsHCO3 was reported on the right-hand side. Reproduced with permission from ref. 69, copyright 2019, Wiley-VCH. (f) Calculated DOS for Ni d-band center in Ni-NCNT@Ni and Ni-NCNT@Ni9Cu. Reproduced with permission from ref. 76, copyright 2020, Wiley-VCH. (g and h) Comparison of (g) jCO and (h) TOF toward CO of Fe3+–N–C in an H-cell (red circles) and on a GDE (red stars) and of Fe2+–N–C (red squares) as well as other reported catalysts. Reproduced with permission from ref. 77, copyright 2019, Science. | ||
| Product | Half-cell reaction | E 0 (V vs. RHE) |
|---|---|---|
| Hydrogen | 2H+ + 2e− → H2 | 0 |
| Carbon monoxide | CO2 + 2H+ + 2e− → CO + H2O | −0.11 |
| Formate/formic acid | CO2 + 2H+ + 2e− → HCOOH | −0.21 |
| Methanol | CO2 + 6H+ + 6e− → CH3OH + H2O | 0.03 |
| Methane | CO2 + 8H+ + 8e− → CH4 + 2H2O | 0.17 |
| Acetate/acetic acid | 2CO2 + 8H+ + 8e− → CH3COOH + 2H2O | −0.26 |
| Ethylene | 2CO2 + 12H+ + 12e− → C2H4 + 4H2O | 0.07 |
| Ethanol | 2CO2 + 12H+ + 12e− → CH3CH2OH + 3H2O | 0.09 |
2.1 Metal center selection
A big challenge associated with the CO2RR is the competitive HER in aqueous electrolytes. From a thermodynamic perspective, the reduction potential from CO2 to CO is slightly different from the HER (E0(CO2/CO) = −0.11 V vs. RHE).41 However, the high kinetic barrier of CO2 activation leads to a sluggish reaction rate, which requires careful selection of metal centers and improved catalyst designs.42 Due to the reduction process mainly occurring at metal sites, the type of the metal center directly leads to distinct electronic properties and further affects the reaction pathways. By using density functional theory (DFT) calculations, SACs embedded in porphyrin-like supports with M–N4–C active sites have been extensively investigated to predict efficient non-noble metals for the CO2RR.43,44 DFT calculations have revealed that the overpotentials (η) for the CO2RR and HER at the atomic 3d transition metal (TM) sites and it has been found that the CO2RR is favorable over the HER on most SACs (Fig. 2b). A new descriptor has also been established associated with the charge state of metal atoms to reassess the CO2RR activity to exclude the HER influence, which provides rational guidance towards metal center selection.19 The distinct catalytic performance of SACs compared to metal nanocrystals may be attributed to isolated M–N–C sites suppressing the *H adsorption and therefore alleviating the HER competition.44,45 For example, an investigation among various M–N–C catalysts demonstrated that Fe and Co exhibit the best catalytic efficiency at low and high potentials, respectively, consistent with the experimental observation that the CO2RR activities of Fe and Ni SACs could rival those of noble metal-based (Ag or Au) catalysts.46 It can be explained by the changes in the Gibbs free energies at the RDS that Fe and Co–M–C catalysts are both favored in the first PCET step under low overpotentials, but Co suffers from a high energy barrier in CO formation. On the other hand, Ni-based catalysts with weak binding of COOH* required large overpotentials and their weak *H adsorption under a large potential suppresses the HER.46 Similar results from another study proved a volcano relationship between the CO2RR performance and a family of M–N–C catalysts, and demonstrated that Fe and Co SACs are located at the summit of the volcano under different overpotentials.472.2 Loading and distribution
Despite the metal center being used to guide DFT calculations, the loading and distribution of SACs play a key role in enhancing selectivity and deeper reducing of CO2 toward C2 products. Due to the high surface energy of isolated metal atoms, substrates such as carbon-based supports, metal alloys or metal oxides are essential to stabilize single atoms and prevent their aggregation.18,42,48–50 Low single-atom densities (typically less than 5 wt%) hamper electrocatalytic rates and catalyst usage for the CO2RR. Therefore, huge efforts have been devoted to the design and synthesis of supports for higher loading of SACs.The commonly used strategies include metal ion/molecule-seeding pyrolysis,51,52 polymer-assisted pyrolysis,53 metal–organic framework (MOF)-derived pyrolysis,54,55 and covalent organic frameworks (COF)-supported methods.56,57 The seeding strategy, polymer-assisted pyrolysis, and MOF-derived pyrolysis are widely applied due to their universal and facile operation for most metals, as summarized in many previous reviews.41,45,49,58,59 On the other hand, the recent emerging pyrolysis-free COF-supported SACs are prepared via a mild and well-defined synthetic method with better control of the structure of the active sites. Covalently constructed using diverse organic modules, COFs act as a versatile platform for stabilizing single metal atoms with single distribution. Besides this, the N-containing organic building blocks in COF structures are CO2-philic and can facilitate CO2 adsorption during electrocatalysis, while the S-containing aromatic heterocycles are electron-rich and can serve as electron donors.57,59,60 For instance, stable metal–porphyrin–tetrathiafulvalene COFs (M-TTCOFs) have been developed for the CO2RR and a faradaic efficiency (FE) of 91.3% was achieved for CO at −0.7 V vs. RHE, as well as high cycling stability for over 40 h. Exfoliated COFs have a high number of active sites, which hence boost the FE to almost 100% at −0.8 V vs. RHE. This high efficiency may be ascribed to the synergistic combination of electron-migrating porphyrinic ligands and electron-donating tetrathiafulvalene.56 This donor–acceptor conjugation is a promising approach by which to design polycrystalline COFs that exhibit a high electron transfer rate. Another strategy involves the incorporation of S-containing aromatic heterocycles with porphyrinic electron acceptors to prepare COF-supported Co-SACs. The catalysts exhibit outstanding electron conduction (1.38 × 10−8 S m−1) and carrier mobility (0.18 cm2 V−1 s−1) and a FECO of 91.4% was achieved at −0.6 V vs. RHE.61 However, the biggest challenges for COF-supported SACs are their relatively high cost and complicated preparation processes.
In recent work, low-cost ultrahigh loading SACs were synthesized via a facile route, in which amine-functionalized graphene quantum dots (GQDs) were used as a support on which to anchor metal atoms, achieving a record high content at 40 wt% or 3.8 at%. Using the general synthetic protocol, these mass-produced GQDs could be interwoven into a carbon matrix and applied in a flow cell reactor, featuring an anion membrane electrode assembly (MEA) with a Ni-SAC. The CO2RR results (Fig. 2c) demonstrated that ∼15 wt% Ni-SACs reached the highest CO partial current densities, representing a 2.5-fold improvement over ∼7.5 wt% counterparts at a lower applied voltage of 2.55 V.62 This example clearly shows that high loading is a prerequisite for the development of SACs toward their future industrialization.
2.3 Electrolyzer and electrolyte
The poor solubility of CO2 (ca. 34 mM) in aqueous electrolytes leads to CO2 diffusion limitations in H-type cells, which may not affect the catalytic performance at a moderate overpotential but hinder the development of high-loading SACs at a relatively high overpotential.63,64 To enhance the kinetics of the CO2RR, a flow cell can operate as a promising electrolyzer. In a very recent study, an amino-modification strategy was used to facilitate the current density of M–N–C (M = Ni, Fe and Zn) SACs for the CO2RR. The high loading of Ni–N4/C–NH2 (5.51 wt%) allowed for amplifying to a gas-fed flow cell, as shown in Fig. 2d.65 Constructed in a gas diffusion electrode, this catalyst achieved a CO partial current density (jCO) of 447.6 mA cm−2 with almost 90% FECO at 1.0 V, where the remarkable jCO in the flow cell was 7-fold that in an H-type cell, indicating the superiority of flow cell configuration.65 Despite the exciting breakthrough in practical application, the poor long-term stability in this work caused by carbon paper used in the gas diffusion electrodes calls for the development of devices with better operation. The MEA cell designed was a good example as it realized 20 h of continuous operation on Ni-SACs at a high current density (85 mA cm−2) with the FECO maintained at ∼100%.Furthermore, another insightful direction toward industrial scale-up is the type of electrolyte used in the CO2RR system, which is of great significance, but has always been ignored. The concentration, pH and cationic species of electrolytes influence the product composition, which has been widely studied for bulk or nanostructured inexpensive metals (especially Cu).66,67 In the very few studies that have involved SACs, an atomically dispersed Cu catalyst was shown to reduce CO2 to ethanol with a high FE of 55% under optimized electrolyte conditions (0.1 M CsHCO3). Normally, Cu-SACs rarely generate multi-carbon products such as CH3CH2OH or C2H4 due to the isolated active sites are hardly affordable for C–C coupling, which is the only route to form C2 intermediates.68 The appearance and then disappearance of the Cu–Cu peak in operando XAS analysis reveals that single metal sites are converted into Cu nanoparticles at −1.2 V and reversibly recover into SACs after electrolysis. Thus, CO2 is catalyzed by temporarily aggregated Cu atoms and the synergistic effect boosts ethanol production. In addition, the influence of electrolyte cations has been investigated, where 0.1 M CsHCO3 demonstrates the best selectivity toward ethanol and suppresses activity toward the HER (Fig. 2e) due to the formation of a large cation stabilized active Cu(I) species.69 Overall, more efforts are needed to be put in toward this crucial research direction of cell configuration development, as well as electrolyte improvement.
2.4 Cu-based SACs
Cu-based SACs are a class of electrocatalysts that have great potential to obtain more favorable products. The unique properties of Cu for the CO2RR have prompted researchers to conduct deeper investigations on C2 and C2+ products, as valuable chemicals. In contrast, SACs with other metal centers normally generate CO or formate as products, while in several cases, highly reduced chemicals such as methanol can be produced.70–72 A recent report demonstrated that 4.9%mol Cu–Nx–C SACs obtained at 800 °C favored C2H4 formation, while a lower concentration of Cu atoms (2.4%mol) was inclined to form CH4.73 Besides the importance of the implementation of high-loading SACs being reaffirmed, the preferential products prove that the adjacent Cu active sites can work synergistically when they are close enough to one another. This enhanced result was supported by DFT calculations that showed that the porous C2N layer supported Cu dimers exhibit excellent activity for the CO2RR, with methane and ethylene being the main products.74 A groundbreaking study demonstrated that Cu–pyrrolic-N4 active sites could help to stabilize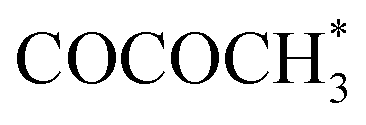 intermediates for acetone production. The proposed reaction pathway for acetone investigated via DFT calculations and experiments showed that the synergy between Cu and coordinated pyrrolic N led to further C–C coupling toward a C3 species.75 Copper doped into other metal SACs can also improve their CO2RR selectivity. For example, by introducing Cu into Ni-SACs to form a Ni9Cu outer layer, the adsorption energy of H* can be tuned, thus contributing toward the suppression of the HER.76 The change in the electronic structure from the difference in the electronic density of state (DOS) was explored, as shown in Fig. 2f. The Cu addition upshifts the d-band center of Ni, which coincides well with the experimental results that Ni–Ni9Cu exhibits a high FECO of 97%.
intermediates for acetone production. The proposed reaction pathway for acetone investigated via DFT calculations and experiments showed that the synergy between Cu and coordinated pyrrolic N led to further C–C coupling toward a C3 species.75 Copper doped into other metal SACs can also improve their CO2RR selectivity. For example, by introducing Cu into Ni-SACs to form a Ni9Cu outer layer, the adsorption energy of H* can be tuned, thus contributing toward the suppression of the HER.76 The change in the electronic structure from the difference in the electronic density of state (DOS) was explored, as shown in Fig. 2f. The Cu addition upshifts the d-band center of Ni, which coincides well with the experimental results that Ni–Ni9Cu exhibits a high FECO of 97%.
2.5 Metal valence
Besides its determining role in center metal selection, valence is a non-negligible factor in the electronic structure of SACs. Two valence states of Fe-SACs on carbon supports were used to explore how valence state affects the catalytic efficiency. As the valence may change during the electrocatalysis, operando X-ray absorption spectroscopy was used to confirm the oxidation state of the Fe ions. The results showed that Fe maintained a +3 valence during the CO2RR, which exhibits better stability, lower onset potential, higher jCO, and a higher turnover frequency (TOF) than its Fe2+ counterpart (Fig. 2g and h).77 This superior ability can be attributed to the Fe3+ ions being stabilized via coupling with pyrrolic N, resulting in a Fe3+/2+ state and a lowering of the CO2RR potential. However, results from another study presented that Fe+ coordinated with four pyrrolic nitrogen atoms was identified as the reactive center during the CO2RR. The monovalent Fe species emerged when potential was applied to catalysts with a relative content of ∼17.1% (−0.9 V vs. RHE) and disappeared after electrolysis.78 These inconsistent results may originate from the complexity of the geometric and electronic structure of the single-Fe-atom sites, which require advanced operando characterization, and this will be discussed in detail in the section on spectroscopy.3. Insights into the atomic structures of active sites
SACs can be considered as a conceptual bridge between heterogeneous and homogeneous catalysts due to their unique geometry configuration, high stability, designable active species, and the ability to clarify the structure–activity relationship at the atomic and molecular levels.79 However, it should be noted that the catalytic sites never involve only single metal atoms. The adjacent atoms are non-trivial in that they can influence the CO2RR performance, as the doping elements can tune the coordination environment and the carbon-based framework supports the basic conductive function. Until now, carbon-based non-noble metal-based SACs have been extensively reported and have provided an opportunity to clarify the mechanism of inter-atomic interactions. Insights gained from these studies are based on simplified molecular models. In this section, the regulation of coordination structures, including coordination numbers, heteroatom types and vacancy/edge defected designs, will be discussed. Moreover, we will focus on the recent progress on diatomic models toward C2 products in the fourth section.3.1 M–N–C structure
Fig. 3 illustrates all the possible M–N–C structures with different coordination numbers and configurations of N atoms. Nitrogen is a low-cost, earth-abundant element, which creates an electron-rich environment to anchor metal atoms. Different coordination environments regulate the electronic properties of single metal atoms. A series of carbon-based SACs with coordination structures of isolated M–N–C as typical catalytically active sites have been widely studied. Plenty of studies have explored how the different coordination numbers on graphene supports affect the CO2RR performance. Representative models of the active sites are shown in Fig. 3. In general, the coordination number can be regulated by the pyrosis temperature but it is uncontrollable to obtain the same active motifs.25,32,80,81 On the other hand, molecules like porphyrin (TPP)82 or phthalocyanine (Pc)83 as well as periodic N-containing substrates like graphitic carbon nitride (g-C3N4)84 or holey nitrogen-doped graphene (C2N)85 are usually used to help develop SACs with uniformity.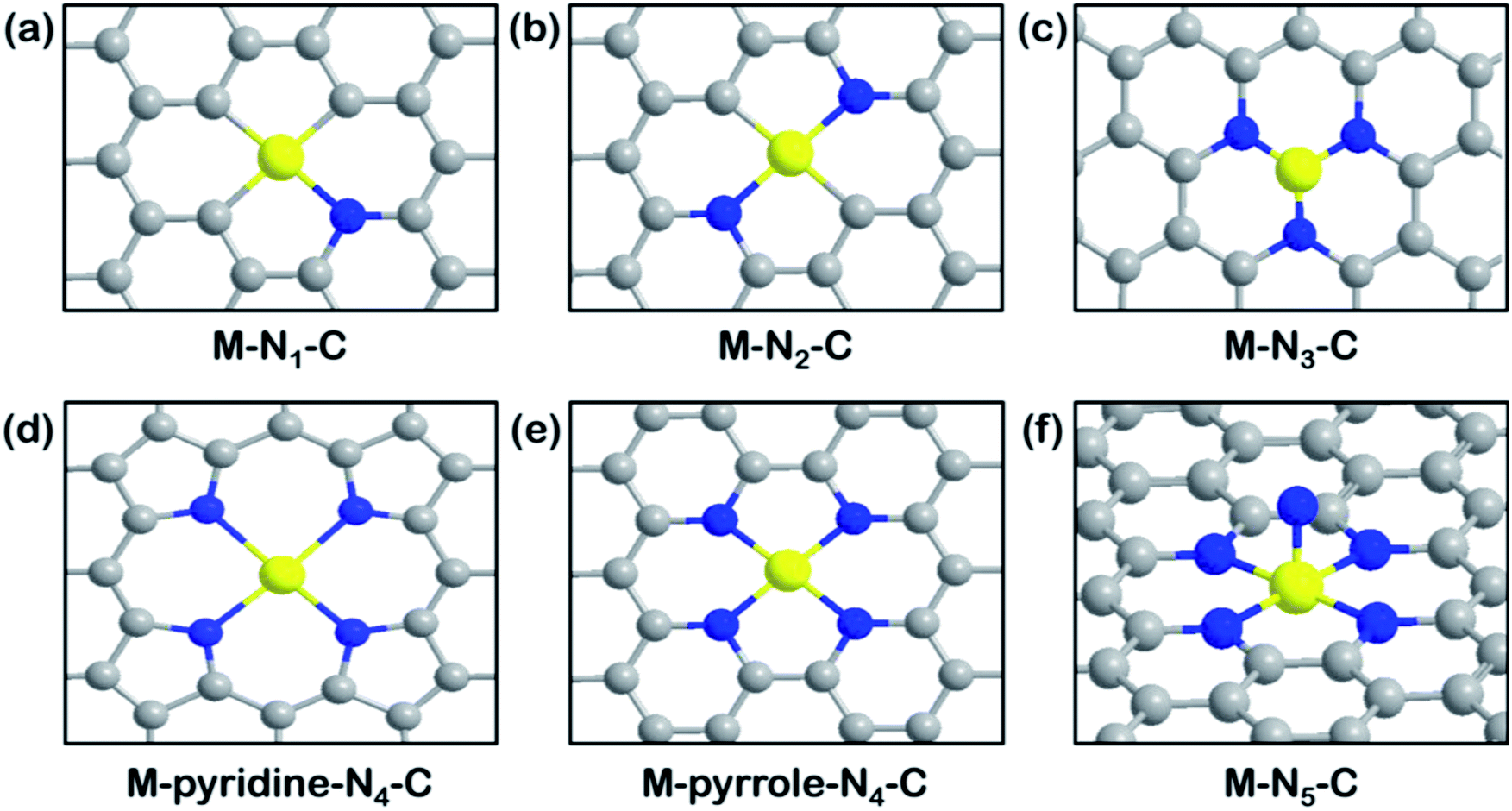 | ||
| Fig. 3 Overview of M–N–C structures. Schematic illustration of (a) M–N1–C. Reproduced with permission from ref. 86, copyright 2020, American Chemical Society. (b) M–N2–C. Reproduced with permission from ref. 88, copyright 2019, Wiley-VCH. (c) M–N3–C. Reproduced with permission from ref. 81, copyright 2018, Wiley-VCH. (d) M–pyridine-N4–C. Reproduced with permission from ref. 91, copyright 2020, Springer Nature Publication. (e) M–pyrrole-N4–C. Reproduced with permission from ref. 77, copyright 2019, Science. (f) M–N5–C molecular models. Reproduced with permission from ref. 93, copyright 2019, Wiley-VCH. (M: yellow; C: grey; N: blue). | ||
Despite a diversity of synthesis strategies, controversies about the most active M–N–C site still exist. DFT calculation results have predicted that a single Ni metal atom anchored with three carbon and one nitrogen (Fig. 3a) exhibits the best catalytic activity and selectivity toward the CO2RR.86 The computational results demonstrate that the introduction of one nitrogen (1N) decreases the CO* desorption barrier and yields better CO productivity on a Ni–C3N1 site than a Ni–C4 site. Meanwhile, the high charge capacity of the 1N site allows for the carrying of more charge to facilitate the reduction process. In comparison, a lower work function at the charge-neutral state restricts the charge capacity of 4N counterparts when they are charged to the same final Fermi level. Nonetheless, other studies from an experimental perspective reached different conclusions. SACs with different coordination numbers (M–N2, M–N3, M–N4, M–N5) on carbon-based supports have been evaluated according to several aspects. Frequently mentioned is coordination unsaturated M–Nx, which is more reactive than M–N4 motifs during the CO2RR due to the suppressed HER activity and lower free energy of the formation of the *COOH intermediate.87 For example, a graphene-supported Cu–N2 site (Fig. 3b) has shown superior activity than Cu–N4 sites, reaching a high FECO of 81% at −0.5 V vs. RHE.88 This result can be rationalized by geometry structure differences, in that the bond lengths of Cu–N2 sites are shorter than those of Cu–N4, which is beneficial toward promoting electron transfer from the central metal to the activated *CO2. Another example compared the activity of Co–Nx–C (x = 2, 3, 4) SACs pyrolyzed at different temperatures and Co–N2–C was found to exhibit the best CO2RR performance, with a record TOF value of 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 200 h−1.80 The characterization of three samples with different coordination numbers was presented in detail and the N2 coordination state was reconfirmed upon NH3 treatment. The M–N3–C sites (Fig. 3c) also show advantages for improved CO2RR performance. Ni–N3–C exhibits much lower free energy for the first electronic step (activation of CO2 to *COOH) than Ni–N4–C, from computational investigations, making it optimal to serve as the active sites.81 In recent work, Mn–N3–C3N4 on carbon nanotubes (CNTs) was exploited for the CO2RR and was found to exhibit better performance (FECO of 98.8% and jCO of 14 mA cm−2 at an overpotential of 0.44 V) than previously reported graphene-supported Mn–N4 SACs.89In situ XAS results indicated that the electron redistribution of the Mn active site after CO2 adsorption increased the Mn oxidation state, which then returned to its original state after one CO2 reduction cycle, indicating that the CO2RR takes place at the Mn–N3 sites. The calculated local density of states on Mn–N3–C3N4 exhibit a closer distance between the d-band center and the Fermi level than Mn–N4–C models, which is favorable toward CO2 binding and activation.
200 h−1.80 The characterization of three samples with different coordination numbers was presented in detail and the N2 coordination state was reconfirmed upon NH3 treatment. The M–N3–C sites (Fig. 3c) also show advantages for improved CO2RR performance. Ni–N3–C exhibits much lower free energy for the first electronic step (activation of CO2 to *COOH) than Ni–N4–C, from computational investigations, making it optimal to serve as the active sites.81 In recent work, Mn–N3–C3N4 on carbon nanotubes (CNTs) was exploited for the CO2RR and was found to exhibit better performance (FECO of 98.8% and jCO of 14 mA cm−2 at an overpotential of 0.44 V) than previously reported graphene-supported Mn–N4 SACs.89In situ XAS results indicated that the electron redistribution of the Mn active site after CO2 adsorption increased the Mn oxidation state, which then returned to its original state after one CO2 reduction cycle, indicating that the CO2RR takes place at the Mn–N3 sites. The calculated local density of states on Mn–N3–C3N4 exhibit a closer distance between the d-band center and the Fermi level than Mn–N4–C models, which is favorable toward CO2 binding and activation.
However, based on different calculation models and imposed forces, along with different rate-determining steps, the most reactive sites have been identified as M–N4 or M–N5 in some published studies.90 For instance, we reported that Ni–N4 exhibits the best catalytic activity for the CO2RR using grand canonical potential kinetics (GCP-K) calculation methodology, which was found to be in good agreement with experimental results showing that FECO reached ∼100% and jCO was 40 mA cm−2 at −1.05 V vs. RHE.91 The HER performance was shown to be correlated to coordinated C numbers from three aspects (FECO, TOF, and Tafel slope) and we found that the Ni–N4 sites are superior to Ni–N2 and Ni–N3. A similar conclusion has been drawn by other research teams, in that Co–N4 SACs exhibit higher FECO and jCO values than coordination unsaturated Co–N4−x over a full range of applied potential from −0.5 to −1.0 V vs. RHE.92 On the other hand, it is critical to distinguish between M–N4 motifs constructed using different N-containing ligands (Fig. 3d and e), as pyrrolic N ligands might stabilize Fe3+ for faster CO2 adsorption, whereas pyridinic N ligands exhibit the opposite effect in that Fe3+ ions are reduced to Fe2+ ions at −0.1 to −0.2 V vs. RHE.77
Axial coordination of addition N to the M–N4 plane to form M–N5 sites has been demonstrated to be an effective way to promote the CO2RR. As shown in Fig. 3f, the axial ligand tunes the electronic properties of the central metal, especially the valence state. Fe–N5 SACs have been prepared by dispersing Fe–N5 on N-doped graphene and XAS tests were used to identify that the Fe–N5 sites exhibit higher oxidation states than typical Fe–N4 sites.93 A subsequent computational investigation determined that boosting d electron transfer from Fe to the additional N led to a weaker binding strength of *CO, thus facilitating the final CO production. Another related study reported that the valence of central Co in a five coordinated N species was higher than that of a Co–N4 analog.94 The coordination affinity is beneficial to the rapid formation of *COOH as well as CO desorption. All these studies emphasize the influence of different coordination numbers on the catalytic performance of M–N–C toward the CO2RR.
3.2 Heteroatom co-doping
Fig. 4 illustrates an overview of representative engineering models, such as heteroatom co-doping and vacancy or edge defects. Due to the difference in electronegativity of common non-metals (like N, O, P, S), it is feasible to break the symmetry of electronic distribution by modulating the types of coordination atoms among catalytic centers.95 Atomic-level tuning is sensible for SACs as electronic redistribution is effective, while precise regulation is quite challenging. In recent work, Cu–N2O2 (Fig. 4a) SACs were prepared with the help of copper disodium EDTA on carbon dots and the resulting catalysts exhibited extraordinary selectivity toward CH4.96 The central metal experienced an oxidation state change from Cu2+ to Cu+/Cu0 due to the strong covalent interactions between Cu2+ ions and the O-containing ligand, which decreased the Cu 2p binding energy. The less positive charge density of the Cu atoms in Cu–N2O2 sites rationalized the lower energy barriers of all of the CO2RR intermediates and the higher adsorption energy for *H than typical Cu–N4 sites. Apart from the chelate-assisted method, well-defined heteroatom co-doping can also be achieved by modifying the molecular structure directly. By breaking the perfect D4h symmetry of tetraphenylporphyrin (N4-TPP) to obtain 21-oxatetraphenylporphyrin (N3O-TPP), the structural uniformity of N3O has been shown to became an ideal framework for the anchoring of single Ni atoms.97 DFT calculations revealed that the polarization of O-doping lowered the valency of the Ni center and led to a stronger Ni–C bond to stabilize *COOH intermediates. S substitution in the porphyrinic framework produced a similar effect, reported in another theoretical study.98 By replacing two opposable pyrrole N atoms with S atoms, the electronic optimization of the Fe center showed better intramolecular electron transfer ability than Fe–N4. Moreover, N, S co-coordination resulted in a weaker CO binding energy for efficient CO production and a decreased limiting potential that surpasses those of most reported SACs. Other co-doping elements, such as P, S in Co–N3, for electronic density modulation of Co-SACs has also proven the importance of heteroatoms.99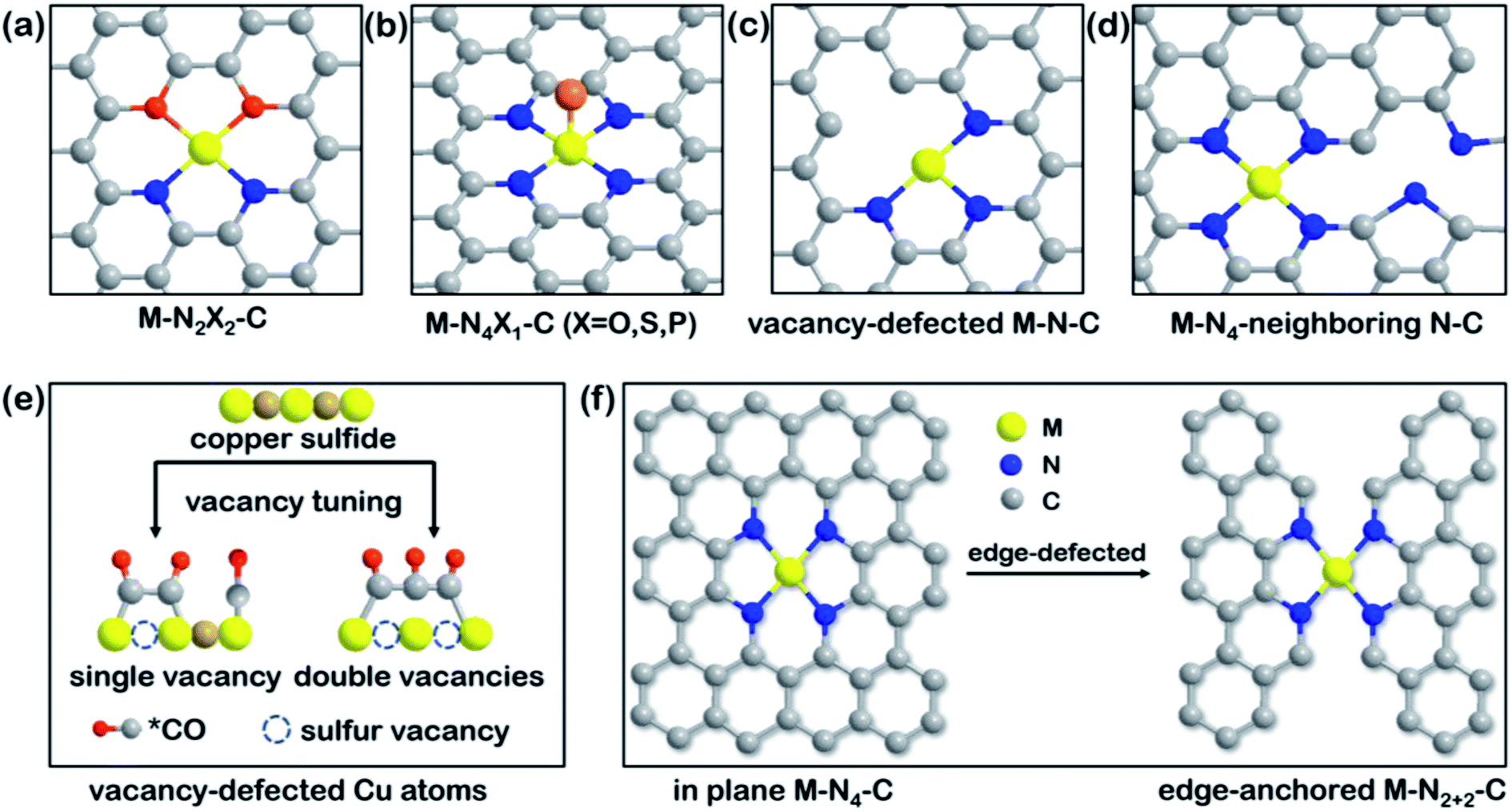 | ||
| Fig. 4 Overview of model engineering of SACs. Schematic illustration of (a) N-heteroatom co-doped M–N2X2–C. Reproduced with permission from ref. 96, copyright 2021, Springer Nature Publication. (b) Axial-coordinated M–N4X1–C. Reproduced with permission from ref. 100, copyright 2021, Wiley-VCH. (c) Vacancy-defected M–N–C. Reproduced with permission from ref. 104, copyright 2020, Wiley-VCH. (d) Non-coordinated M–N4-neighboring N–C models. Reproduced with permission from ref. 105, Copyright 2021, Elsevier. (e) Mechanism of C3 product formation by tuning from a single sulfur vacancy to double vacancies in copper sulfide. Reproduced with permission from ref. 106, copyright 2021, Springer Nature Publication. (f) Edge-defected design of in-plane M–N4–C and edge-anchored M–N2+2–C. Reproduced with permission from ref. 107, copyright 2019, Royal Society of Chemistry. Model engineering such as heteroatom co-doping or creating vacancies/edges introduces diverse electronic states for SACs. | ||
The axial coordination of heteroatoms results in a similar atomic structure to M–N5, as depicted in Fig. 4b, but leads to different CO2RR performance. A rarely reported metal, Cd, was used to prepare Cd–N4S1 SACs for the CO2RR, the catalytic activities of which were compared with that of Cd–N5.100 The straightforward comparison of axial atoms showed that even though both active sites effectively boost CO2RR, the high spin density and charge delocalization of S remarkably decrease the highest free energy barrier on Cd–N4S1 compared with that on Cd–N5. Despite the Cd atom preferring to form five coordination structures due to its large atomic radius, the enhancement of the added axial heteroatom has also worked in other systems. For example, diphenyl sulfide was used as an axial ligand to promote interfacial electron communication, resulting in a 1.5 times increase in the TOF (2.4 s−1) of Co–N4S1 at −0.87 V compared with that of pristine Co–N4.101 In another study, O coordinated with Fe–N4 from an out-of-plane position and was shown to maintain excellent stability at low overpotentials. DFT calculations showed that *CO could be desorbed from the Fe–N4O motifs easier than from Fe–N4 and Fe–N5 sites.102 Accordingly, regulated coordination models based on other elements are conducive to the CO2RR and should be considered for improving electrocatalysts.
3.3 Vacancy/edge defects
As is known, defect structures are appropriate anchoring sites for single metal atoms.41,103 Strategies to create and control vacancy or edge defects around metal centers has attracted increasing attention. Pyrolysis, as the most used method to prepare SACs, can also be applied to create controllable vacancies in carbon substrates. For instance, vacancy-defected Ni–N3–V SACs (Fig. 4c) could be obtained by taking advantage of the easy evaporation of coordinated oxygen at high temperatures.104 Vacancy defects provide moderate free energies for CO2 activation and CO desorption in comparison to defect-free Ni–N3, as shown in Fig. 3c. Thus, the manageable structure of Ni–N3–V exhibits the best catalytic performance, with a high jCO (65 mA cm−2) value at −0.9 V vs. RHE. The vacancy beyond the adjacent atoms also modulates the catalytic activity, so that the non-coordinated N in vacant carbon support can tune the electronic properties of the M–N4 motif, as illustrated in Fig. 4d.105 The authors of the study demonstrated a surprising discovery that the Ni atom in the Ni–N4 species was no longer the active site, with the four adjacent N atoms being responsible for catalyzing the CO2RR. Due to the pyrrolic-N in the neighboring Ni–N4 site inducing electron transfer from Ni 4s orbitals to adjacent N 2s orbitals, the electron-rich N facilitated the *COOH formation and then boosted the CO2RR performance. This latest study presents the impact that vacancy adjustment has on preferred electrocatalytic products by tuning the sulfur vacancies of copper sulfide (Fig. 4e).106 It was found that the single vacancy could not support CO–OCCO coupling due to the strong electrostatic repulsion of three *CO intermediates. In contrast, the double adjacent co-planar vacancies enabled the formation of a polyline OC–COCO trimer with a decreased adsorption energy of −0.23 eV. The advantages like the reduced steric hindrance and enriched negative charge density of double vacancies promote CO2-to-C3H7OH conversion. Although this example is not about SACs, the significance of vacancies should be understood and more SACs models with multiple controllable vacancies are encouraged to be developed in the future.Edge defect engineering regulates the electronic structure of the active sites indirectly. Two different structural models, in-plane Ni–N4–C10 and edge-anchored Ni–N2+2–C8, were explored and their CO2RR performance compared (Fig. 4f). Although their coordination numbers of N were the same, the Ni–N2+2–C8 motif with edge defects contained a dangling bond for faster dissociation of the C–O bond in *COOH while Ni–N4–C10 did not, according to DFT calculations.107 However, the difference between these two structures was not well revealed in the experiments. In another study, edge-located unsaturated N was shown to play a role in anchoring single Ni atoms and increasing electron density, resulting in an excellent TOF, comparable to those of noble metal-based catalysts, such as those containing Ag.108 Aberration-corrected scanning TEM confirmed that Ni single atoms were primarily anchored on the edge of defective graphene. Moreover, DFT demonstrated that the in-plane Ni–N3 site was unfavorable for CO desorption, while the edge-anchored analog with intermediate adsorption strength exhibited the optimal catalytic activity based on the Sabatier principle.
3.4 Bimetallic SACs
Although traditional SACs have been shown to exhibit great performance in the electrochemical CO2RR, the isolated metal atoms work independently with each other. However, the electronic nature of each non-noble metal limits further improvement in terms of higher efficiency and deep reduction products. For instance, Fe-SACs exhibit superior onset potential for CO2-to-CO reduction but are burdened by the strong binding energy of CO, while Ni-SACs show high current density but suffer from the sluggish kinetics of the first PCET process.109,110 Researchers have further discovered that neighboring single atoms could affect CO2RR performance when they are close enough, as they optimize the adsorption energy of intermediates or even lead to the formation of a C2 product.73 Accordingly, bimetallic SACs with tunable collocation and molecular models have emerged as a new frontier in electrocatalysis.Fig. 5 illustrates two dual-atom catalyst models and the latest work on bimetallic SACs for the CO2RR. The proposed structures of bimetallic SACs in recent work, M1M2–N8–C and M1M2–N6–C, are shown in Fig. 5a and b. M1M2–N8–C is composed of two adjacent but separate M–N4-type SACs with two different metal centers. Recently, NiSn atomic pair catalysts supported on N-doped carbon nanosheets with an M1M2–N8–C structure were utilized for the CO2RR.111 The separate configurations of Ni–N4 and Sn–N4 were proven by XPS and XAFS analysis, as no Ni–Sn bond was observed. The excellent overall FE and TOF for CO2-to-formate transition could be attributed to the cooperation of dual metal centers, in which Ni–N4 favors CO2 adsorption and Sn sites boost *OCHO intermediate formation. The detached bimetallic SACs provide a promising method to realize syngas production with a controllable CO/H2 ratio for typical downstream thermochemical reactions. In another study, CoNi–N8–C dual-atom SACs for syngas evolution were demonstrated to exhibit a high total current (>74 mA cm−2) with a tunable CO/H2 ratio of 0.23–2.26 by controlling the ratio between Co and Ni.112 DFT revealed that Co–N4 and Ni–N4 sites selectively promoted the HER and CO2RR, respectively, and the reaction pathways on coexisting CoNi dual atoms were similar to those of single metal sites. Thus, the regulation of product composition was able to be achieved by tuning the Co/Ni ratio. Similarly, a synergistic effect was observed on Ni paired with Fe in an M1M2–N8–C model.113 The introduction of Ni changed the energy barrier for the CO2RR reaction and increased the stability of Fe-SACs due to strong CO* adsorption. Moreover, differing the configuration of bimetallic centers changes the rate-determining step, as evidenced by the calculated DOS after COOH* and CO* adsorption.
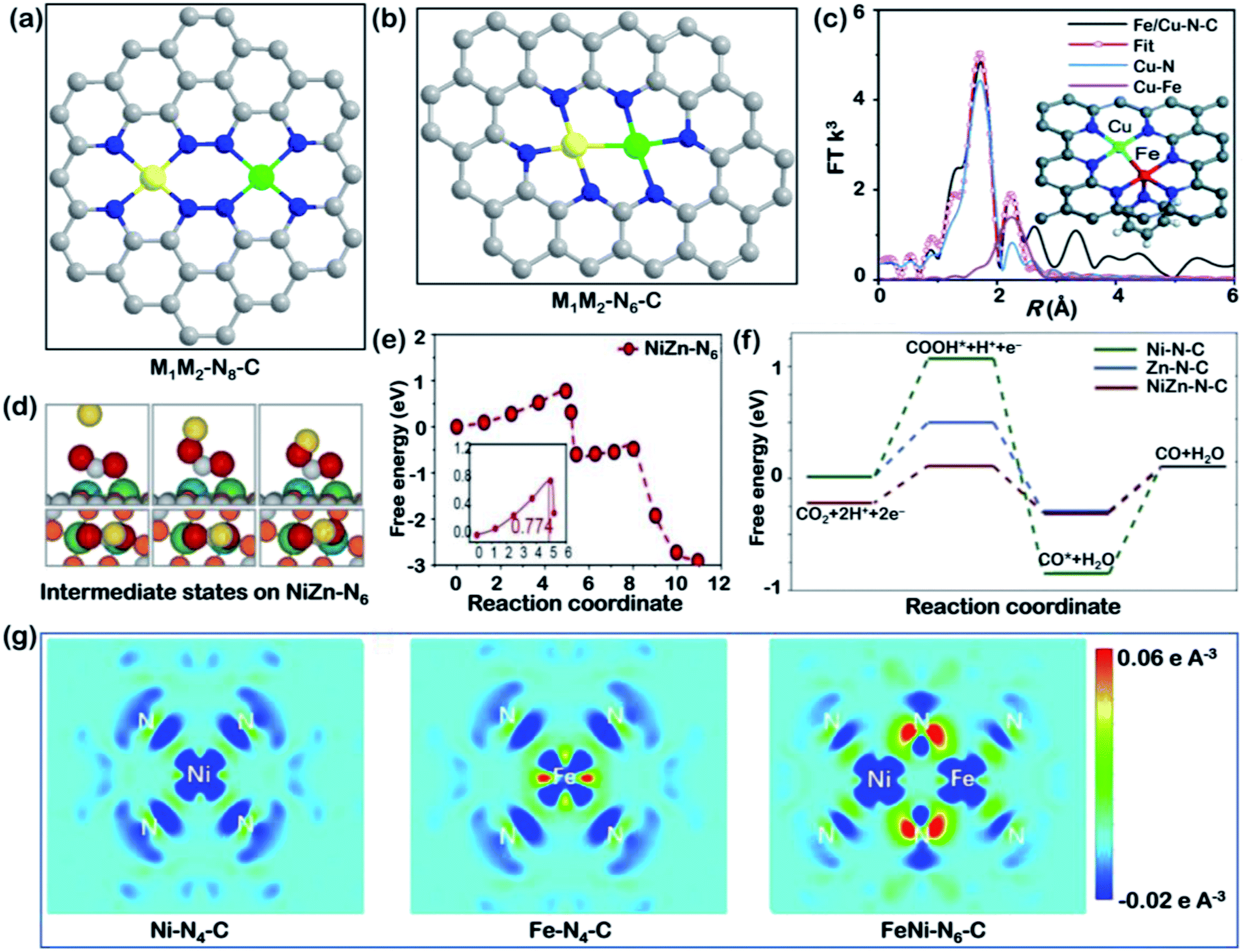 | ||
| Fig. 5 Bimetallic SACs. Schematic illustration of (a) M1M2–N8–C and (b) M1M2–N6–C models (M1: yellow; M2: green; C: grey; N: blue). (c) EXAFS fitting for FeCu–N6–C in R space; inset: a proposed model for FeCu–N6–C, where black, blue, red, and indigo represent C, N, Fe, and Cu atoms, respectively. Reproduced with permission from ref. 115, copyright 2021, Royal Society of Chemistry. (d) Diagrams of the atomic structure and energy changes of the starting, intermediate, and final states of the CO2RR, and (e) CI–NEB-calculated *COOH on NiZn–N6–C reaction kinetics, and (f) the free energy of each intermediate state on the metal atom sites in Zn–N–C, Ni–N–C, and ZnNi–N–C. Reproduced with permission from ref. 117, copyright 2021, Wiley-VCH. (g) The differential charge density maps of Ni–N4–C, Fe–N4–C, and NiFe–N6–C. From aquamarine to orange indicates the transition from electron depletion to accumulation. Reproduced with permission from ref. 118, copyright 2021, Springer Nature Publication. | ||
On the other hand, the M1M2–N6–C structure contains direct metal–metal coupling within the diatomic molecular model (Fig. 5b). A series of DFT simulations were conducted for this dual atom structure supported on graphene.114 Based on multiple combinations of transition metals with different electronegativity, Cu/Mn, Ni/Mn and Ni/Fe were presented that showed superior activity for the CO2RR compared with Au (211). These three diatomic catalysts break the scaling relationship between COOH* and CO* adsorption strength and serve as a guide to design optimal bimetallic SACs. In particular, a unique seven coordinated structure, N4Fe–CuN3, was reported by He and coworkers very recently, as illustrated in the inset model in Fig. 5c.115 The fitting results of EXAFS spectra (Fig. 5c) suggested the existence of Cu–Fe coordination. The diatomic catalyst exhibited a synergistic effect and showed a much higher TOF than Fe–N4 and Cu–N4 over a wide potential range of −0.4 to −1.1 V vs. RHE. Its enhanced catalytic activity arose from the closer distance between the d-band center of N4Fe–CuN3 to the Fermi level than those of Cu- and Fe-SACs. Except for this example, most heteronuclear systems leads to bimetallic SACs with six coordinated nitrogen atoms.116 The latest reported work involved the study of atomically dispersed Ni–Zn catalysts with an M1M2–N6–C structure from both kinetical and thermodynamic perspectives.117 In terms of kinetics, the authors utilized the climbing image nudged elastic band (CI-NEB) calculation to study the reaction process, with the starting, intermediate, and final states, as depicted in Fig. 5d.117 The calculated free energy of *COOH revealed a reduced barrier on the Ni–Zn bimetallic SACs (Fig. 5e). In terms of thermodynamics, ZnNi–N–C was shown to be more selective toward the CO2RR than Zn–N–C and Ni–N–C due to its lower free energy for COOH* formation (Fig. 5f). A decreased gap between the d-band center of the Ni 3d orbitals and the Fermi level was also observed in this work. The activation energy of CO2 hydrogenation on bimetallic sites was significantly lower than on the other two single-atom configurations, explaining the efficient CO2RR pathway on ZnNi–N6–C sites. For another NiFe–N6–C catalyst, the electronic structures of single-atom and diatom catalysts were visually expressed using differential charge density maps (Fig. 5g).118 These maps indicate that, in a bimetallic catalyst, more electrons are delocalized around the coordinating N between Ni and Fe with electrons partially transferred from an Fe atom. The electron redistribution leads to a higher oxidation state of Fe and therefore enhances CO2 activation and CO desorption.
Besides the dual-atom sites built in a relatively fixed atomic structure of active sites, several bimetallic SACs comprise two types of SACs. Isolated Zn and Co atoms coordinated on N-doped carbon black were prepared via pyrolysis and achieved a FECO of 93.2% and jCO of 26 mA cm−2 at −0.5 V vs. RHE.119 The electronic interaction between neighboring Zn/Co active sites was demonstrated by XANES spectra analysis, but the complex and uncertain diatomic structure may lead to less persuasive simulation results. Another Zn/Co tandem catalyst demonstrated good CO2RR performance beyond two-electron transfer.120 Experiments and calculation results revealed a two-step mechanism in which CO2-to-CO transfer preferentially takes place on the Co atom first and then CO diffuses to a Zn site and is further reduced to CH4. The high FE toward CH4 (15%) can be attributed to the Co site enhancing the adsorbed *H over the coordinated N of Zn–N4.
Despite the various molecular models based on single atoms or dual atoms that have been investigated, factors beyond the metal-containing active sites can also determine the reaction pathways. For example, the immobilization of Co phthalocyanine (CoPc) onto a conductive CNT led to a six-electron transfer process from CO2 to methanol in comparison to a two-electron reduction to CO for homogeneous CoPc.71 It was observed that the close contact of molecular CoPc with the conductive CNT/electrode partially contributed to the high FE toward methanol due to the high degree of electronic communication. The electrocatalytic nanocarbon thus plays a significant role that should not be ignored.121 Furthermore, SACs derived from MOFs or COFs can adsorb CO2 gas due to their porous nature, which affects the reaction results.58 To sum up, research on active molecular models is very important, but this should not be the sole point by which to evaluate a catalytic system. Standards and protocols for experimental and theoretical data acquisition are highly recommended to identify the intrinsic mechanism of SACs toward the CO2RR.
4. Insights from spectroscopy
The atom-level identification of SAC structures is difficult but essential for further mechanism investigation. Widely used microscopies such as scanning transmission electron microscopy (STEM) can be used to visually determine surface morphology, but they cannot provide detail on the explicit structural and electronic properties of SACs.23 The molecular models and proposed reaction pathways based on theoretical calculations require experimental evidence for confirmation. The urgent demand for advanced spectroscopic characterization has brought about breakthroughs in the field of the CO2RR.4.1 XAS
X-ray absorption spectroscopy (XAS), including X-ray absorption near-edge structure (XANES) and extended X-ray absorption fine structure (EXAFS), is a remarkably useful technique in characterizing SACs. An XANES spectrum can be used to determine the chemical valence state and electronic configuration of the target element, while pre-edge features can offer information such as bond angles.122,123 EXAFS can be used to probe the local structure and give us valuable spatial insight into the geometric structure and coordination environment.122 Specifically, fitting the results from Fourier-transform EXAFS (FT-EXAFS) can help to infer the interatomic distances between the absorber and scattering atoms and coordination numbers.121,122Fig. 6a shows the different valence states of two Cu–N–C catalysts prepared at different pyrolysis temperatures and a comparison of their Cu K-edge XANES spectra with reference samples.73 It can be seen that the main peak of the Cu-SACs shift toward the middle energy region between the Cu2O and CuO references, indicating that the Cu atoms are in higher oxidation states of Cu(I) or Cu(II). The atomic structure investigated by FT-EXAFS confirmed the presence of Cu–N bonds and the existence of Cu–Cu bonds in the annealed samples, suggesting that the Cu atoms are isolated and coordinated to N atoms (Fig. 6b). Li and coworkers explored antimony (Sb) single atoms dispersed on N-doped carbon for the CO2RR, which can efficiently produce formate as a product. N K-edge soft XAS measurements were used to identify the electronic structure of the N and C species surrounding the Sb atoms, revealing the existence of pyridinic N, pyrrolic N and graphitic N.124,125 The ultra-high sensitivity of XAS makes it a useful tool with which to figure out the electronic state of target metal atoms and ligand species.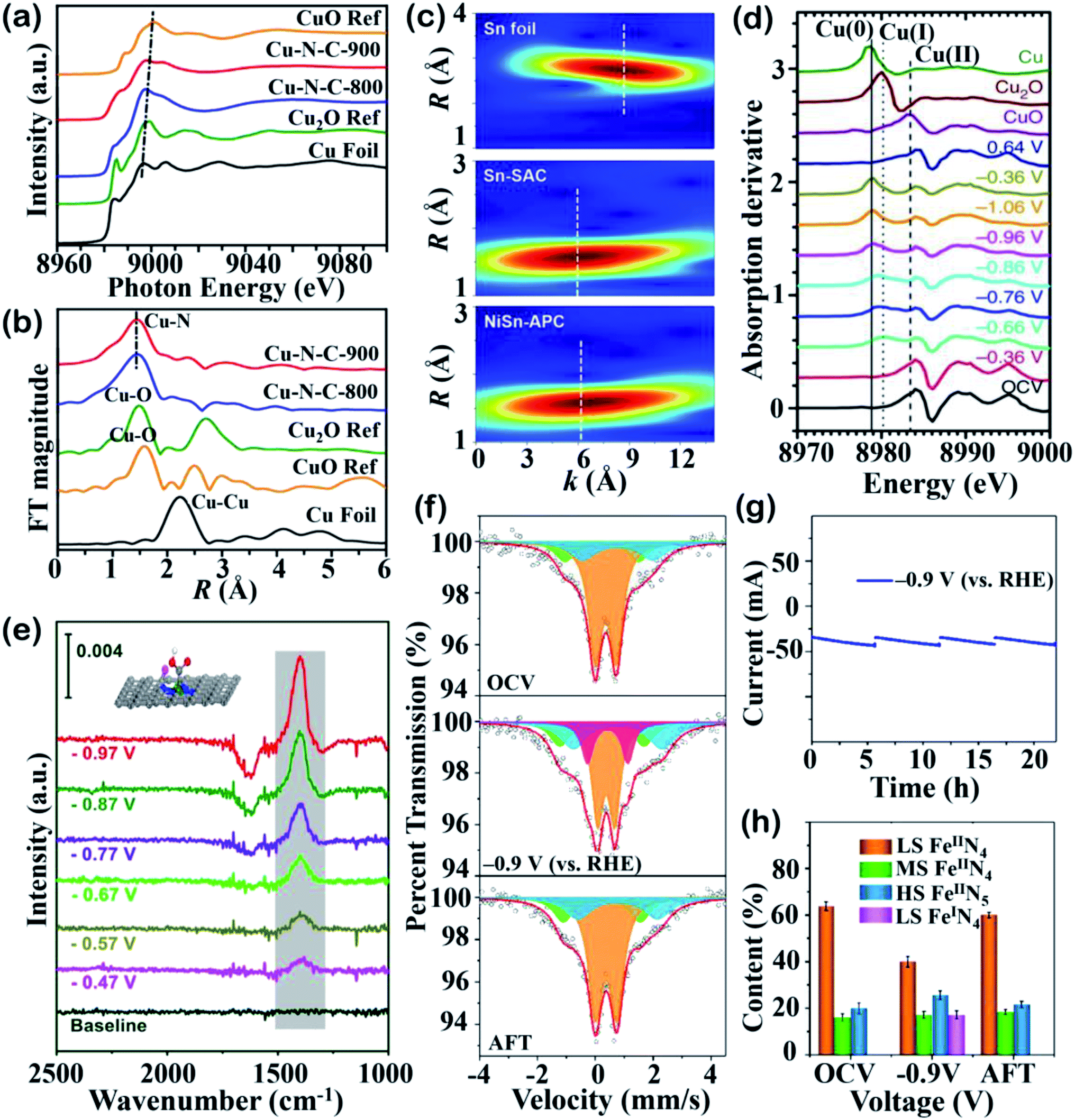 | ||
| Fig. 6 Spectroscopic studies. (a) XANES spectra and (b) FT-EXAFS spectra. Reproduced with permission from ref. 73, copyright 2020, American Chemical Society. (c) EXAFS wavelet-transform (WT) images of Sn foil, Sn-SAC and NiSn-APC. Reproduced with permission from ref. 111, copyright 2021, Wiley-VCH. (d) First-order derivatives of the Cu K-edge in situ XANES spectra for Cu(II) phthalocyanine under electrocatalytic reaction conditions. Reproduced with permission from ref. 128, copyright 2018, Springer Nature Publication. (e) In situ ATR-IR spectra of Ni–N4 SACs under increasing applied potentials in 0.5 M KHCO3. Reproduced with permission from ref. 53, copyright 2021, Elsevier. (f) Operando57Fe Mössbauer spectra of 57Fe-enriched Fe–NC–S collected under OCV, −0.9 V (vs. RHE), and after reaction. The orange, green, blue, and purple doublets could be assigned to LS Fe2+ in FeIIN4, MS Fe2+ in FeIIN4, HS Fe2+ in N–FeIIN4, and LS Fe2+ in FeIN4, respectively. (g) Current–time response and (h) contents of different Fe moieties derived from Fig. 5f. Reproduced with permission from ref. 78, copyright 2021, American Chemical Society. Advanced spectroscopic techniques help to probe the atomic structure under ex situ/in situ conditions and provide experimental proof. | ||
Wavelet-transform (WT)-EXAFS can be used to discriminate the diverse back-scatterers in the overlapped signals and allow us to interpret EXAFS spectra visually.126 As shown in Fig. 6c, the WT-EXAFS images of NiSn diatomic catalysts and Sn-SACs exhibit a similar maximum intensity at 6 Å−1, which can be assigned to Sn–N contributions.111 The maximum intensity of Sn–Sn located at 8.6 Å−1, as displayed for the Sn foil, suggests that Sn in both the NiSn catalyst and Sn-SACs exist in the form of being independently dispersed. In another study, a similar method was used to recognize the Cu single-atom dispersity of Cu–N–C by comparing the highest WT positions of CuPc and Cu foil.127
The active centers under electrolysis are not always the same structure as what is detected by ex situ spectroscopies, as the isolated sites may go through geometric or electronic evolution. The identification of real active sites under working conditions has encouraged researchers to focus on in situ and operando techniques. The abovementioned example by Fontecave and colleagues reported that the Cu–N4 sites were reconstructed into small Cu nanoparticles to enhance CO2RR toward deeply reduced products.69 This geometric change was monitored by operando XAS and explained why their Cu-SACs could produce chemicals beyond CO. In situ XANES spectra can also help researchers to explore the evolution of different Cu oxidated species during the CO2RR. As shown in Fig. 6d, the appearance of Cu(I) and Cu(0) peaks under increased reduction potentials indicates that Cu changes oxidation state during working status while in open-circuit voltage (OCV), where Cu(II) was the dominant spectral feature.128 This transition was found to be reversible upon switching the potential back to 0.64 V, and the XANES spectrum was almost back to that of the OCV state. Combined in situ XAS and electrochemical results give us valuable insight to link the CO2RR performance to realistic catalytic sites.
4.2 Optical spectroscopies
In situ attenuated total reflection-infrared (ATR-IR) spectroscopy was introduced to characterize the reaction intermediates, and the latest work used this technique to monitor *COOH adsorbed on active sites via the vibration of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching (Fig. 6e).53 The increasing peak intensity at 1400 cm−1 achieved a maximum at −0.97 V, indicating that *COOH formation could be regarded as RDS for the CO2RR on fluorine-doped Ni-SACs. Operando Raman spectroscopy has also been used to detect the oxidation states of active moieties during the CO2RR process. Actually, the aqueous electrolyte in the CO2RR system is more suitable for operando Raman spectroscopy rather than operando IR spectroscopy due to the low scattering of water in Raman spectra.129 By utilizing operando Raman spectroscopy, Liu and coworkers found that the vibration peak of Ni–N (at ∼244 cm−1) shifted to a lower wavenumber when the applied cathodic potential was above 0.57 V, implying a weaker interaction between the Ni atom and coordinated N atoms.130 This redshift could be attributed to the in situ-formed Ni+ from Ni2+ reduction via the addition of an electron to a Ni 3d orbital. The monovalent Ni unraveled by operando Raman spectroscopy provided experimental proof for the proposed reaction mechanisms of the CO2RR.
O stretching (Fig. 6e).53 The increasing peak intensity at 1400 cm−1 achieved a maximum at −0.97 V, indicating that *COOH formation could be regarded as RDS for the CO2RR on fluorine-doped Ni-SACs. Operando Raman spectroscopy has also been used to detect the oxidation states of active moieties during the CO2RR process. Actually, the aqueous electrolyte in the CO2RR system is more suitable for operando Raman spectroscopy rather than operando IR spectroscopy due to the low scattering of water in Raman spectra.129 By utilizing operando Raman spectroscopy, Liu and coworkers found that the vibration peak of Ni–N (at ∼244 cm−1) shifted to a lower wavenumber when the applied cathodic potential was above 0.57 V, implying a weaker interaction between the Ni atom and coordinated N atoms.130 This redshift could be attributed to the in situ-formed Ni+ from Ni2+ reduction via the addition of an electron to a Ni 3d orbital. The monovalent Ni unraveled by operando Raman spectroscopy provided experimental proof for the proposed reaction mechanisms of the CO2RR.
4.3 Mössbauer spectroscopy
Mössbauer spectroscopy is a precise technology that is used to probe the elemental spin, oxidation states and geometric symmetry of Mössbauer isotopes such as 57Fe, 119Sn, etc.129 In the field of SACs for the CO2RR, Mössbauer measurements have been extensively used to identify Fe species with different coordination and electronic structures. For instance, in the work done by Li and coworkers, the doublets (D1 and D2) fitted in the Mössbauer spectra are in accordance with Fe–Nx moieties wherein the metal center is in low-spin (LS) and intermedium-spin (MS) states, respectively.123 Fontecave and colleagues exploited a series of Fe–N–C catalysts for the CO2RR and utilized 57Fe Mössbauer spectroscopy to distinguish its components, such as α-Fe and iron carbide.131 In composition analysis by Mössbauer spectroscopy, the isolated Fe–N4 was proven to be the active site for CO2-to-CO conversion. Operando Mössbauer spectroscopy characterization can therefore provide quantitative information on the geometric structure and electronic environment of real active sites under reaction conditions. The development of operando Mössbauer spectroscopy on the laboratory scale was recently achieved using a specially designed H-cell to prevent spectral interference. Steady current–time responses during operando Mössbauer spectroscopy measurements were obtained and the spectra were fitted with multiple doublets, as shown in Fig. 6f and g.78 The three doublets in orange, green, and blue represent LS Fe2+ in FeIIN4, MS Fe2+ in FeIIN4, and high-spin (HS) Fe2+ in N–FeIIN4, respectively. When applying a voltage of −0.9 V vs. RHE., the appearance of a new purple doublet could be assigned to LS Fe2+ in FeIN4, which arose due to the decreased LS content, suggesting that LS Fe2+ in FeIIN4 sites experiences dynamic evolution during the electrolysis. The relative content of the four Fe species in Fig. 6h implies the in situ generation of LS FeIN4 as a highly active site for the electrochemical CO2RR.5. Insights from computational studies
5.1 DFT studies on the CO2RR
The electrochemical performance of SACs for the CO2RR has been determined experimentally, but it remains challenging to analyze the features that determine the catalytic activity. In this regard, theoretical studies could give more insights into the reaction mechanism. Specifically, using DFT, the reaction cycle and energy barriers for each elementary step can be estimated accurately (Scheme 1). Furthermore, theoretical calculations can help to reveal the electronic structure of the active sites to determine structure–performance relationships.Uniformly-dispersed transition metal-based SACs provide a perfect platform for acquiring a clear theoretical comprehension of the CO2RR process. For example, the CO2RR process by DFT calculation was investigated using a series of transition-metal based SACs, M/SV–graphene and M/DV–graphene.132 By comparing the free energies of the first protonation step with that of the *H adsorption process, most of the SACs exhibited preferential selectivity for the CO2RR over the competitive HER. The results indicate that the efficiency and selectivity for the CO2RR products can be correlated to the valence electrons, where the reduction ability decreases with decreased energy at the d-level. As re-summarized in Fig. 7a, CH4 is preferentially produced by Fe/SV and Co/SV, while the main product for Ni/DV, Cu/DV, and noble-metal (such as Pt and Pd) based catalysts is CH3OH.132 A similar result on the correlation between efficiency and selectivity for the CO2RR according to the elemental properties of metals as a function of their group number has also been reported in another study.133
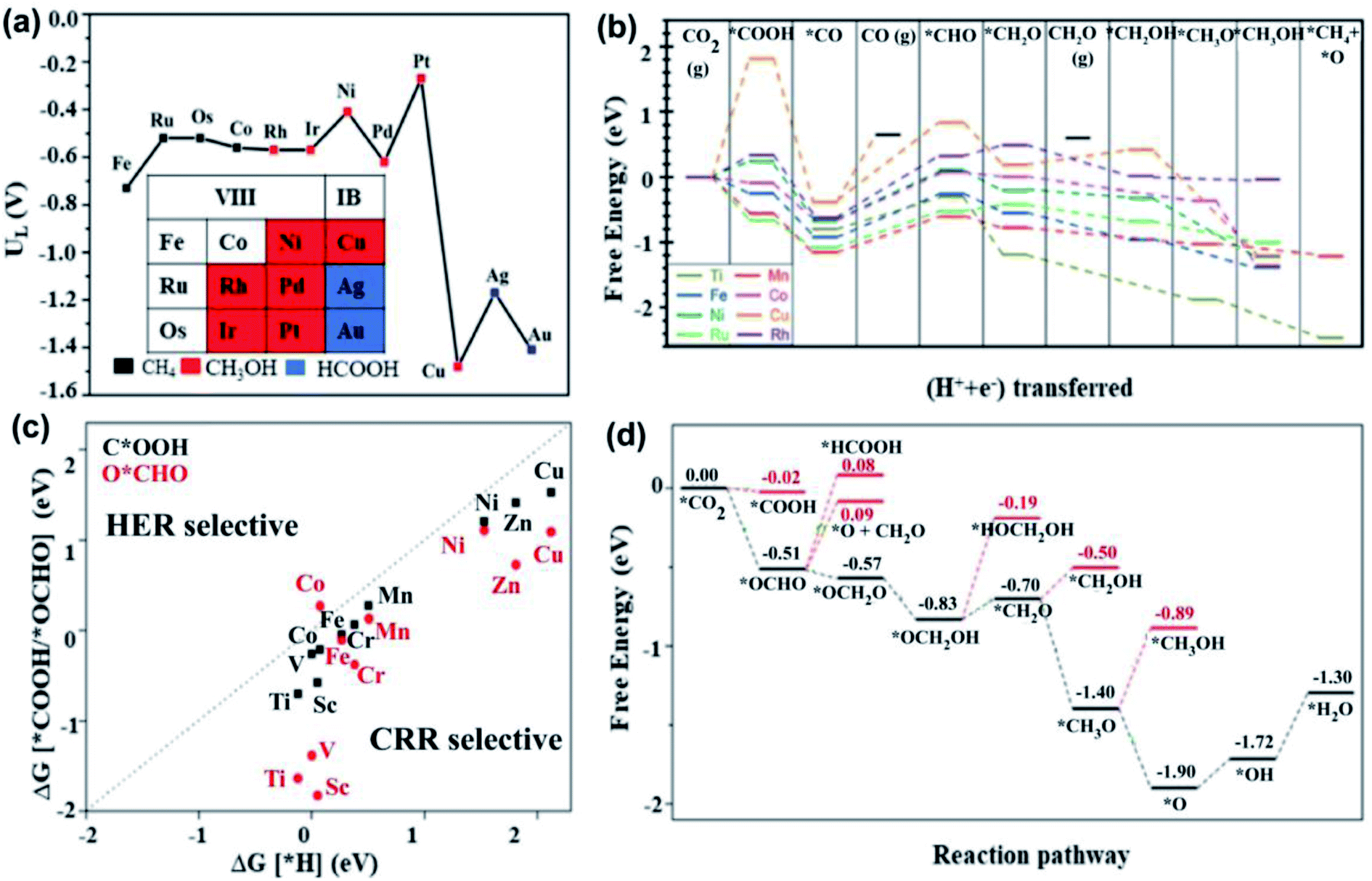 | ||
| Fig. 7 DFT analyses. (a) Limiting potentials and product selectivity of the CO2RR on various TM-based SACs anchored on graphene. The results indicate that a Ru SAC has a minimum limiting potential of −0.52 V for CH4 production, while Pt is more prominent for the CH3OH synthesis, and Ag, and Au preferentially produce formic acid. Reproduced with permission from ref. 133, copyright 2017, Royal Society of Chemistry. (b) Free energy profile for CO2RR to various products on various TM@C2N active sites. CH4 is only produced by Ti and Mn-based SACs. Reproduced with permission from ref. 138, copyright 2018, Royal Society of Chemistry. (c) Comparison of selectivity of first protonation step of CO2RR and the competitive HER. It shows that all the studied transition metals except for Co preferentially participate in the CO2RR over the HER. Reproduced with permission from ref. 146, copyright 2018, Royal Society of Chemistry. (d) The free energy profile of the CO2RR for the Mo-MOF electrocatalyst that shows preferable CH4 production, while the formation of other products exhibit high energy barriers, as highlighted by the red lines. Reproduced with permission from ref. 145, copyright 2020, Elsevier. The last protonation step to produce H2O is the RDS, which required a limiting potential of 0.42 eV. | ||
Transition metals are regarded as promising candidates because of their many d-orbital electrons and the variation valence states. The electrochemical activity and selectivity of SACs toward the CO2RR depends on the central metal atom and its coordination environment. More precisely, the activity is related to the electronic structure, which can be modulate through the coordination environment and valence states. In such a study, the CO2RR performance of metal-doped nitrogen carbon based SACs was correlated with an intrinsic descriptor, based on the electronic states of the transition metals. The intrinsic descriptor Φ constitutes the number of valence electrons, the electronegativity of the transition metal and the radius of the transition metal.19 The theoretical calculations predicted that Co-based SACs could achieve high electrochemical performance for the CO2RR. Various products can be visualized for the electrochemical CO2RR, but CO (as a simple one due to the two-electron transfer process) is used to access the electrochemical performance of the CO2RR. CO formation is hindered by the weak adsorption of CO2 and desorption of the CO from the active surface, which is limited by the strong adsorption of CO. In such a case, Ni atoms coordinated to a graphene support as an active site were shown to form an electronic structure that facilitates the CO formation in the CO2RR while simultaneously hindering the HER.134 In another investigation, TM–Nx–C moieties with double vacancies were thoroughly studied for the electrochemical reduction of CO2 to CO. A scaling relationship was observed between the adsorption energy of *COOH and *CO to measure the CO2RR activity for CO formation. According to thermodynamics analysis, Ni–N1, Pd–N1, Pt–N1, Co–N4, and Rh–N4 are the most prominent active sites for the CO formation because these systems show moderate binding strength toward *COOH and *CO, which facilitates CO formation. While, Fe–N4 was predicted to be more favorable for the further reduction of CO to CH3OH and CH4 at a relatively low electrode potential of between −0.7 and 0.9 V.135 In another study, Co-doped graphene altered with pyridinic nitrogen atoms was studied for the conversion of CO2 to HCOOH. The electrocatalytic performance and electronic structure of Co SACs anchored on monovacancy (Co–N3) and divacancy (Co–N4) structures were compared towards the CO2RR. The theoretical results revealed that the Co atom supported by a monovacancy (Co–N3) exhibits superior performance in terms of CO2 activation and product selectivity. The conversion of CO2 to *HCOO requires an energy barrier of 0.31 eV, indicating that this reaction can easily accelerate under ambient conditions, while the second hydrogenation step to form the required product requires an overpotential of 0.51 eV.136
Regular and planar carbon materials, besides graphene, as substrates of SACs are highly desired for DFT calculations. For instance, Wang and coworkers studied transition metals anchored on a C2N monolayer for the CO2RR and found that the N6 moiety serves as an active site for CO2 activation and then protonation to form different products.137 It was predicted that C2N-supported SACs are more selective for CO2 reduction over the HER and that CO2-to-*COOH conversion is the RDS. According to the DFT calculated free energies of each intermediate, Ti and Mn-based SACs favor methane production, while Fe, Co, Ni, and Ru show high selectivity toward methanol over a potential range of 0.5–0.8 eV, with the detailed pathways shown in Fig. 7b. In search of a more stable and conductive substrate, graphyne as a two-dimensional carbon planar materials with high stability and plasticity is a good candidate on which to stabilize single metal atoms. DFT was employed to assess the performance of SACs embedded in graphyne for the CO2RR.138 Based on reaction barriers, it was discovered that Cr–G was the most efficient catalyst for the CO2RR, with the HER being suppressed during electrocatalysis. In the optimal pathway, the Cr–G catalyst most probably generates a final product of CH4, and the RDS is *CH3 to CH4 conversion, which only requires a limiting potential of −0.29 eV.138 Graphdiyne, a new 2D planar carbon allotrope, formed from sp–sp2 hybridized carbon atoms, has received a huge amount of attention due to its unique characteristics and wide applications in catalysis and other fields.139 In a theoretical study, the electrochemical performance of alkali metals supported on a graphdiyne substrate was evaluated for the reduction of CO2 to HCOOH. The results indicate that the electrocatalysts exhibit excellent stability and can be realized experimentally. Among the studied alkali metals, Na and Li had the capacity to strongly adsorb CO2 and activate it. Based on adsorption free energy, the binding between *OCHO and alkali metals is stronger than that between *COOH and alkali metals. This indicates that *HCCOH is preferentially formed via the CO2RR, and Li@GDY required a limiting potential of −0.56 V, while Na@GDY required a lower overpotential of −0.16 V.140 Fe anchored on graphdiyne has been shown to achieve high selectivity and activity in the CO2RR towards CH4 production. The reduction of CO to *COH is the rate determining step, which requires a limiting potential of −0.33 V.141
g-C3N4 has been used to reveal the mechanistic aspects of the CO2RR by embedding it with a variety of transition metals.142 Based on the adsorption energies of *CO and *H, a descriptor-based picture was built for the product distribution, where seven metals, Pt, Ru, Co, Ni, Pd, Fe, and Cu, were predicted to have the capacity to generate products toward C2H4. Notably, in terms of the rich vacancy/edge sites and N-atom contents of g-C3N4, the atomic model in the calculation was constructed as a unique M–N6 structure, which may contribute toward the results that seven metal-based SACs generate materials beyond C1 products.142 A series of transition metals supported on graphitic carbon nitride have been investigated for the CO2RR using DFT. Mn–C3N was shown to be the most prominent electrocatalyst due to its high activity and selectivity toward HCOOH formation. The HER was successfully suppressed on this surface and required a very low overpotential of 0.04 V, while a kinetic energy barrier of 0.75 eV was required for the first protonation step.143 While, in another study, TM-doped g-C3N4 was theoretically evaluated for the reduction of CO2 to C1 products, i.e. CH3OH and CH4. Ni/g-C3N4 tends to convert CO2 to CH3OH with a limiting potential of 0.72 V, while Fe/g-C3N4 and Co/g-C3N4 preferably produce CH4 with limiting potentials of 0.67 and 0.81 V, respectively.144 In an interesting study, a novel 2D MOF was utilized as a support for SACs for use in the CO2RR.145 Among the studied transition metals, a Mo-based MOF was determined to be the most efficient electrocatalyst due to its performance during the adsorption and transformation of CO2. The low energy barrier of 0.47 eV for Mo-SACs was even lower than that of a (211) Cu surface, and the proposed optimal mechanism on the Mo-SACs was *CO → *COOH → *HCOOH → *CHO → *CH2O → *CH2OH → *CH3 → *CH4, as shown in Fig. 7d.145
Adding metal atoms into coordination networks with unsaturated active sites that act as Lewis-acid sites can produce active materials. Phthalocyanine (Pc) has been predicted to be an efficient candidate due to its good binding energy toward independent metal atoms uniformly dispersed on a monolayer. As shown in Fig. 7c, a Pc monolayer was shown to support TM-SACs and it was found that most metals favored the first protonation step of the CO2RR rather than the competitive HER.146 Co–Pc was predicted to be the most efficient electrocatalyst for HCHO, with an overpotential of 0.36 V, while Cr, Mn, and Zn-based SACs were dominated by HCOOH products according to their calculated Gibbs free energies. Among all these studied metals, Mn–Pc exhibited the lowest overpotential of 0.017 V, while Fe–Pc showed the highest overpotential of 0.819 V toward the CO2RR. In another case, coordinated multi-dentate ligands were shown to make structures flexible and ideal for applications. For instance, tetracyanoquinodimethane (TCNQ) was explored as a coordination molecule to be doped with various single metal atoms for the CO2RR.147 HCOOH was predicted to be the primary product for V, Cr, Mn, Ni and Cu-based SACs, while Co and Fe-based electrocatalysts favored four-electron transferred HCHO as the main product. The modification of the coordinated environment of the active metal center alters the d-electron configuration and is used to tune the adsorption energies of the reactants and reaction intermediates for better electrochemical performance. In such a DFT study, the effect of coordination engineering of a Co–N4 porphyrin toward the CO2RR was studied by replacing N with C or O.148 By evaluating 24 different Co-centered coordination sites, the Co–O3N1 porphyrin was predicted to be the most efficient for the electrochemical conversion of CO2 to CO. Detailed analysis of the electronic structure revealed that the lack of π-bonding in Co–O bonds compared to Co–N and Co–C bonds was responsible for the enhanced catalytic activity.148 In another research study, N and S coordination effects were studied for the CO2RR by comparing Fe–N2S2 with an Fe–N4 porphyrin. The results indicated that the combination of N and S provides a more stable structure than N coordination alone. The addition of S atoms forms a non-coplanar porphyrin framework, and more electrons accumulate on the Fe–S bond, promoting more vital interaction between the Fe and S atoms. The additional dz2 orbitals of Fe efficiently tune the adsorption of CO2 and intermediates and accelerate the overall kinetics of the CO2RR. The S addition in coordination effectively decreases the limiting potential, and the Fe–N2S2 electrocatalyst shows a limiting potential of −0.38 eV for CO2 conversion to HCOOH.149 A similar study revealed that Ni-SACs with two C, one N, and one S coordination environment were estimated to be the most efficient catalyst for the CO2RR, with a theoretically calculated onset potential of 0.67 eV.150
5.2 Machine learning-accelerated design of SACs
DFT calculations have been extensively applied for the discovery and design of SACs for the CO2RR through the prediction of their activity, stability, and structure sensitivity.151,152 However, DFT calculations are time- and resource-consuming for the high-throughput screening of SACs in the vast parameter space. In this regard, machine learning (ML) as a strong and supportive tool can guide DFT calculations to accelerate and simplify the rational design of SACs via the establishment of accurate and deep structure–activity relationships coupled with feature importance analysis.153–156 For example, neural network algorithms were developed to calculate the adsorption energies of CO* and the formation energies of HOCO* in order to identify the performances of surface sites on Au nanoparticles as well as de-alloyed Au surfaces for the CO2RR.157 Similarly, ML was applied to DFT-calculated data to discover active bimetallic facets for the CO2RR and the results revealed that most facets of nickel gallium bimetallics lead to similar activity on Ni surfaces.155 Moreover, structure–activity relationships were established for predicting CO and H adsorption energies based on structural properties using active learning across intermediates. In fact, an automated screening approach through integration and optimization of ML was presented to guide DFT calculations for predicting catalytic activity. The feasibility of this approach was demonstrated by screening various alloys combining 31 elements, which resulted in 131 candidate surfaces across 54 alloys being identified for the CO2RR and the identification of 258 surfaces across 54 alloys for the CO2RR.158 Similarly, active learning was then used to accelerate the screening of CO adsorption energy on Cu-based components.159 In addition, extreme gradient boosting regression (XGBR) was implemented as a supervised ML algorithm to screen ΔGCO* and ΔGH* of 1060 SACs embedded in metal-non-metal co-doped graphene for the CO2RR, as shown in the inset of Fig. 8a.160 Based on feature importance analysis, the Pauling-electronegativity (E_M), covalent radius (M_cov), and first ionization energy of the SACs (1E_M) are the most important parameters on ΔGCO* (Fig. 8a). Comparison of predicted values using the XGBR model and DFT calculations, shown in Fig. 8b, indicated that ML was able to accurately predict ΔGCO*. Therefore, the XGBR model was used for the accelerated high-throughput screening of ΔGCO* in the design of SACs, leading to a huge decrease in the number of DFT calculations, as depicted in Fig. 8c.160 Additionally, atomic properties were used to predict the catalytic activity of SACs and dual-atom catalysts for the CO2RR. Based on results from the XGBR algorithm, Ag–MoPc was revealed as an excellent electrocatalyst, with a limiting potential of −0.33 V.161 Subsequently, the data from the abovementioned work was used as an example to evaluate the efficiency of a DFT–ML hybrid program for catalysis programming.162 Obviously, ML has been successfully applied to accelerate the design and discovery of SACs for the CO2RR. However, this technique is still in its infancy and there is thus a need for further optimization and development of materials simulations. | ||
| Fig. 8 Machine Learning for the CO2RR. (a) Feature importance analysis for ΔGCO* prediction using ML. Pauling electronegativity (E_M), covalent radius (M_cov), and first ionization energy of transition metal atoms as SACs (1E_M) are the most important parameters. The inset shows the structures of SACs used for the CO2RR. Green, gray, and blue spheres indicate nonmetal, carbon, and transition metal atoms, respectively. (b) Comparison of ML- and DFT-predicted ΔGCO*. (c) High-throughput screening of ΔGCO* using XGBR models. Reproduced with permission from ref. 160, copyright 2020, American Chemical Society. These figures indicate that ML can accurately predict ΔGCO* for the better design of SACs. | ||
6. Summary and perspective
As discussed in this review, the fundamental knowledge for the CO2RR on non-noble metal-based SACs on 2D substrates is introduced based on recent studies. The decisive factors for efficient CO2 transformation, including central metal selection, metal valence regulation, SAC loading and distribution, cell configurations, electrolytes, etc. were discussed and Cu-based SACs for C2/C2+ products were emphasized. The proposed atomic structures of the active sites on carbon-based materials that can be used to intuitively compare mechanistic investigations between various studies and the rational design of optimum catalytic sites were systematically summarized. In addition, the application of state-of-art spectroscopies, especially in situ or operando techniques, in CO2RR studies presents an exciting approach to gain insights from structure–activity relationships. Furthermore, computational inspiration of the novel theoretical understanding of the catalytic mechanisms and ML as a rising research direction toward accelerating catalyst screening by integrating DFT calculations were discussed.Despite the fast development in SAC designs and mechanism studies laying the foundation for future research on the CO2RR, there are still multiple challenges that remain in the field:
(1) Rational designs of electrodes and electrolyzers that enable the CO2RR to be endured at a high current density and relatively large applied potential. To date, a few studies have reported the large-scale preparation of SACs and measured their CO2RR performance in flow cell or MEA-cell electrolyzers.62,65,163 It shows great promise for future that researchers can start with these prototypes and subsequently expand SACs to an industrial scale. Accordingly, the exploration of scalable SACs, robust electrodes that can integrate SACs readily, electrolyzer configuration and even electrolytes are further needed.
(2) Enriching the designs of SACs with reduced CO2RR products. An isolated active site is difficult to afford C–C coupling, not to mention the restrictions on metal selection. Many existing reports have achieved nearly 100% CO2-to-CO conversion on nonprecious SACs but hardly reduced CO2 to C2/C2+ products. Hence, fabricating diatom or even multi-atom-based SACs is highly encouraged.90 As tunable adsorption and desorption energies can be realized on neighboring dual metal atoms, there are new opportunities to promote C–C coupling and find optimal metal pairs for obtaining target products.
(3) Proper monitoring of the dynamic structural evolution of active sites. Further development of spectroscopic and microscopic techniques, especially in situ/operando techniques, to offer more real-time information during electrocatalysis is significant to identify the geometric and electronic structures of the real active moieties and make molecular models more plausible.
(4) Although remarkable progress has been made in developing SACs, product selectivity, energy efficiency, reaction kinetics, and electrocatalyst stability under harsh conditions are still challenging for practical realization. Theoretical calculations play an essential role in predicting the active catalysts, reaction mechanism, and electronic structure of the active site to understand the catalyst activity. However, only a few mechanistic studies based on DFT calculations on SACs are available regarding product selectivity and reaction kinetics. Usually, the reaction conditions are ignored in predicting prominent electrocatalysts and free energy profiles via DFT calculations. Therefore, to gain fundamental insights into the reaction energies and product selectivity, it is highly desirable to include environmental factors such as solvation effects (implicit or explicit), pH, and electric field in DFT calculations.
(5) ML in combination with DFT has been recently implemented in the design and discovery of SACs for the CO2RR. The accuracy and predictive power depends strongly on the input features, ML algorithms, and the quality as well as size of training and test database.29 Therefore, more studies and efforts are still required to use universal descriptors as input features, use a general ML algorithm, and increase the input database.164 Moreover, the descriptors obtained from ML can be helpful for constructing volcano plots to understand the origin of SAC activity. However, due to the diversity of reaction intermediates, the complexity of the CO2RR, and lack of appropriate descriptors as input features to accelerate the rational design of SACs, the application of ML algorithms in the reaction mechanism of the CO2RR is still in its early stage.91,165 Active ML is also highly desired for the future investigation of catalytic activity, metal–support interactions, and manipulation of the coordination structure of highly selective and stable SACs.
Overall, mechanistic understanding from the integration of experimental and computational insights can help to guide the design of SACs toward high activity and selectivity. We believe a more thorough investigation of nonprecious metal-based SACs for the CO2RR will be achieved in the future.
Author contributions
Y. H., F. R. and M. T. participated in the organization, writing and editing of this manuscript. X. L., Y. Q. H., T. Z. and Z. L. inspired this review and assisted in the editorial process.Conflicts of interest
The authors declare no competing interests.Acknowledgements
Z. L. acknowledges support from the NSFC-RGC Joint Research Scheme (N_HKUST607/17), the IER foundation (HT-JD-CXY-201907), the “International science and technology cooperation projects” of the Science and Technological Bureau of Guangzhou Huangpu District (2019GH06), the Guangdong Science and Technology Department (Project#: 2020A0505090003), and the Research Fund of Guangdong-Hong Kong-Macao Joint Laboratory for Intelligent Micro-Nano Optoelectronic Technology (No. 2020B1212030010).References
- S. Chu, Y. Cui and N. Liu, Nat. Mater., 2016, 16, 16–22 CrossRef PubMed
.
- E. V. Kondratenko, G. Mul, J. Baltrusaitis, G. O. Larrazábal and J. Pérez-Ramírez, Energy Environ. Sci., 2013, 6, 3112–3135 RSC
- Z. Yang, J. Zhang, M. C. Kintner-Meyer, X. Lu, D. Choi, J. P. Lemmon and J. Liu, Chem. Rev., 2011, 111, 3577–3613 CrossRef CAS PubMed
- WMO, Greenhouse Gas Concentrations in the Atmosphere WMO-Global Atmosphere Watch, 2021, https://library.wmo.int/doc_num.php?explnum_id=10794 Search PubMed.
- A. Majumdar and J. Deutch, Joule, 2018, 2, 805–809 CrossRef
- D. Wakerley, S. Lamaison, F. Ozanam, N. Menguy, D. Mercier, P. Marcus, M. Fontecave and V. Mougel, Nat. Mater., 2019, 18, 1222–1227 CrossRef CAS PubMed
- X. Li, J. Lin, L. Li, Y. Huang, X. Pan, S. E. Collins, Y. Ren, Y. Su, L. Kang and X. Liu, Angew. Chem., Int. Ed., 2020, 59, 19983–19989 CrossRef CAS PubMed
- J. E. Huang, F. Li, A. Ozden, A. S. Rasouli, F. P. G. de Arquer, S. Liu, S. Zhang, M. Luo, X. Wang and Y. Lum, Science, 2021, 372, 1074–1078 CrossRef CAS PubMed
- L. Zhang, Z. Wei, S. Thanneeru, M. Meng, M. Kruzyk, G. Ung, B. Liu and J. He, Angew. Chem., Int. Ed., 2019, 58, 15834–15840 CrossRef CAS PubMed
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Norskov, T. F. Jaramillo and I. Chorkendorff, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS PubMed
- C. G. Morales-Guio, E. R. Cave, S. A. Nitopi, J. T. Feaster, L. Wang, K. P. Kuhl, A. Jackson, N. C. Johnson, D. N. Abram, T. Hatsukade, C. Hahn and T. F. Jaramillo, Nat. Catal., 2018, 1, 764–771 CrossRef CAS
- R. Francke, B. Schille and M. Roemelt, Chem. Rev., 2018, 118, 4631–4701 CrossRef CAS PubMed
- L. Wang, W. Chen, D. Zhang, Y. Du, R. Amal, S. Qiao, J. Wu and Z. Yin, Chem. Soc. Rev., 2019, 48, 5310–5349 RSC
- K. P. Kuhl, E. R. Cave, D. N. Abram and T. F. Jaramillo, Energy Environ. Sci., 2012, 5, 7050–7059 RSC
- J. T. Feaster, C. Shi, E. R. Cave, T. Hatsukade, D. N. Abram, K. P. Kuhl, C. Hahn, J. K. Nørskov and T. F. Jaramillo, ACS Catal., 2017, 7, 4822–4827 CrossRef CAS
- J. J. Leung, J. A. Vigil, J. Warnan, E. Edwardes Moore and E. Reisner, Angew. Chem., Int. Ed., 2019, 58, 7697–7701 CrossRef CAS PubMed
- B. Qiao, A. Wang, X. Yang, L. F. Allard, Z. Jiang, Y. Cui, J. Liu, J. Li and T. Zhang, Nat. Chem., 2011, 3, 634–641 CrossRef CAS PubMed
- X.-F. Yang, A. Wang, B. Qiao, J. Li, J. Liu and T. Zhang, Acc. Chem. Res., 2013, 46, 1740–1748 CrossRef CAS PubMed
- L. Gong, D. Zhang, C. Y. Lin, Y. Zhu, Y. Shen, J. Zhang, X. Han, L. Zhang and Z. Xia, Adv. Energy Mater., 2019, 9, 1902625 CrossRef CAS
- S. Tian, M. Hu, Q. Xu, W. Gong, W. Chen, J. Yang, Y. Zhu, C. Chen, J. He and Q. Liu, Sci. China Mater., 2021, 64, 642–650 CrossRef CAS
- J. Mao, C.-T. He, J. Pei, Y. Liu, J. Li, W. Chen, D. He, D. Wang and Y. Li, Nano Lett., 2020, 20, 3442–3448 CrossRef CAS PubMed
- S. Back, J. Lim, N. Y. Kim, Y. H. Kim and Y. Jung, Chem. Sci., 2017, 8, 1090–1096 RSC
- K. Jiang, S. Siahrostami, T. Zheng, Y. Hu, S. Hwang, E. Stavitski, Y. Peng, J. Dynes, M. Gangisetty, D. Su, K. Attenkofer and H. Wang, Energy Environ. Sci., 2018, 11, 893–903 RSC
- T. Sheng and S.-G. Sun, Chem. Phys. Lett., 2017, 688, 37–42 CrossRef CAS
- X. Song, H. Zhang, Y. Yang, B. Zhang, M. Zuo, X. Cao, J. Sun, C. Lin, X. Li and Z. Jiang, Adv. Sci., 2018, 5, 1800177 CrossRef PubMed
- C. Xu, A. Vasileff, Y. Zheng and S. Z. Qiao, Adv. Mater. Interfaces, 2020, 8, 2001904 CrossRef
- M. Jia, Q. Fan, S. Liu, J. Qiu and Z. Sun, Curr. Opin. Green Sustain., 2019, 16, 1–6 CrossRef
- T. Wang, Q. Zhao, Y. Fu, C. Lei, B. Yang, Z. Li, L. Lei, G. Wu and Y. Hou, Small Methods, 2019, 3, 1900210 CrossRef CAS
- S. Zhu, E. P. Delmo, T. Li, X. Qin, J. Tian, L. Zhang and M. Shao, Adv. Mater., 2021, e2005484, DOI:10.1002/adma.202005484
- X. Li, X. Yang, Y. Huang, T. Zhang and B. Liu, Adv. Mater., 2019, 31, 1902031 CrossRef CAS PubMed
- Y. Cheng, S. Zhao, B. Johannessen, J. P. Veder, M. Saunders, M. R. Rowles, M. Cheng, C. Liu, M. F. Chisholm and R. De Marco, Adv. Mater., 2018, 30, 1706287 CrossRef PubMed
- C. Lu, J. Yang, S. Wei, S. Bi, Y. Xia, M. Chen, Y. Hou, M. Qiu, C. Yuan, Y. Su, F. Zhang, H. Liang and X. Zhuang, Adv. Funct. Mater., 2019, 29, 1806884 CrossRef
- A. Bagger, W. Ju, A. S. Varela, P. Strasser and J. Rossmeisl, ChemPhysChem, 2017, 18, 3266–3273 CrossRef CAS PubMed
- L. Fan, C. Xia, F. Yang, J. Wang, H. Wang and Y. Lu, Sci. Adv., 2020, 6, eaay3111 CrossRef CAS PubMed
- P. P. Yang, X. L. Zhang, F. Y. Gao, Y. R. Zheng, Z. Z. Niu, X. Yu, R. Liu, Z. Z. Wu, S. Qin, L. P. Chi, Y. Duan, T. Ma, X. S. Zheng, J. F. Zhu, H. J. Wang, M. R. Gao and S. H. Yu, J. Am. Chem. Soc., 2020, 142, 6400–6408 CrossRef CAS PubMed
- R. Kortlever, J. Shen, K. J. P. Schouten, F. Calle-Vallejo and M. T. Koper, J. Phys. Chem. Lett., 2015, 6, 4073–4082 CrossRef CAS PubMed
- A. D. Handoko, F. Wei, Jenndy, B. S. Yeo and Z. W. Seh, Nat. Catal., 2018, 1, 922–934 CrossRef CAS
- S. Chen, H. Yuan, S. I. Morozov, L. Ge, L. Li, L. Xu and W. A. Goddard III, J. Phys. Chem. Lett., 2020, 11, 2541–2549 CrossRef CAS PubMed
- M. G. Kibria, J. P. Edwards, C. M. Gabardo, C. T. Dinh, A. Seifitokaldani, D. Sinton and E. H. Sargent, Adv. Mater., 2019, 31, 1807166 CrossRef PubMed
- P. De Luna, R. Quintero-Bermudez, C.-T. Dinh, M. B. Ross, O. S. Bushuyev, P. Todorović, T. Regier, S. O. Kelley, P. Yang and E. H. Sargent, Nat. Catal., 2018, 1, 103–110 CrossRef CAS
- S. G. Han, D. D. Ma and Q. L. Zhu, Small Methods, 2021, 5, 2100102 CrossRef CAS
- A. Vasileff, C. Xu, Y. Jiao, Y. Zheng and S.-Z. Qiao, Chem, 2018, 4, 1809–1831 CAS
- Z. Wang, J. Zhao and Q. Cai, Phys. Chem. Chem. Phys., 2017, 19, 23113–23121 RSC
- A. Bagger, W. Ju, A. S. Varela, P. Strasser and J. Rossmeisl, Catal. Today, 2017, 288, 74–78 CrossRef CAS
- M. Li, H. Wang, W. Luo, P. C. Sherrell, J. Chen and J. Yang, Adv. Mater., 2020, 32, e2001848 CrossRef PubMed
- W. Ju, A. Bagger, G. P. Hao, A. S. Varela, I. Sinev, V. Bon, B. Roldan Cuenya, S. Kaskel, J. Rossmeisl and P. Strasser, Nat. Commun., 2017, 8, 1–9 CrossRef CAS PubMed
- J. Li, P. Pršlja, T. Shinagawa, A. J. Martin Fernandez, F. Krumeich, K. Artyushkova, P. Atanassov, A. Zitolo, Y. Zhou and R. García-Muelas, ACS Catal., 2019, 9, 10426–10439 CrossRef CAS
- Q. Fan, P. Hou, C. Choi, T. S. Wu, S. Hong, F. Li, Y. L. Soo, P. Kang, Y. Jung and Z. Sun, Adv. Energy Mater., 2019, 10, 1903068 CrossRef
- R. Sun, Y. Liao, S.-T. Bai, M. Zheng, C. Zhou, T. Zhang and B. F. Sels, Energy Environ. Sci., 2021, 14, 1247–1285 RSC
- S. Back and Y. Jung, ACS Energy Lett., 2017, 2, 969–975 CrossRef CAS
- S. Zhao, G. Chen, G. Zhou, L. C. Yin, J. P. Veder, B. Johannessen, M. Saunders, S. Z. Yang, R. De Marco, C. Liu and S. P. Jiang, Adv. Funct. Mater., 2019, 30, 1906157 CrossRef
- C. Gao, S. Chen, Y. Wang, J. Wang, X. Zheng, J. Zhu, L. Song, W. Zhang and Y. Xiong, Adv. Mater., 2018, 30, 1704624 CrossRef PubMed
- S.-G. Han, D.-D. Ma, S.-H. Zhou, K. Zhang, W.-B. Wei, Y. Du, X.-T. Wu, Q. Xu, R. Zou and Q.-L. Zhu, Appl. Catal. B, 2021, 283, 119591 CrossRef CAS
- Y. Qu, Z. Li, W. Chen, Y. Lin, T. Yuan, Z. Yang, C. Zhao, J. Wang, C. Zhao, X. Wang, F. Zhou, Z. Zhuang, Y. Wu and Y. Li, Nat. Catal., 2018, 1, 781–786 CrossRef CAS
- L. Jiao, R. Zhang, G. Wan, W. Yang, X. Wan, H. Zhou, J. Shui, S. H. Yu and H. L. Jiang, Nat. Commun., 2020, 11, 1–7 Search PubMed
- H. J. Zhu, M. Lu, Y. R. Wang, S. J. Yao, M. Zhang, Y. H. Kan, J. Liu, Y. Chen, S. L. Li and Y. Q. Lan, Nat. Commun., 2020, 11, 1–10 Search PubMed
- Q. Cao, L.-L. Zhang, C. Zhou, J.-H. He, A. Marcomini and J.-M. Lu, Appl. Catal. B, 2021, 294, 120238 CrossRef CAS
- L. Zou, Y. S. Wei, C. C. Hou, C. Li and Q. Xu, Small, 2021, 17, e2004809 CrossRef PubMed
- K. Huang, J.-Y. Zhang, F. Liu and S. Dai, ACS Catal., 2018, 8, 9079–9102 CrossRef CAS
- S. B. Peh and D. Zhao, Science, 2020, 369, 372–373 CrossRef CAS PubMed
- Q. Wu, M. J. Mao, Q. J. Wu, J. Liang, Y. B. Huang and R. Cao, Small, 2021, 17, 2004933 CrossRef CAS PubMed
- C. Xia, Y. Qiu, Y. Xia, P. Zhu, G. King, X. Zhang, Z. Wu, J. Y. T. Kim, D. A. Cullen, D. Zheng, P. Li, M. Shakouri, E. Heredia, P. Cui, H. N. Alshareef, Y. Hu and H. Wang, Nat. Chem., 2021, 13, 887–894 CrossRef CAS PubMed
- M. R. Singh, E. L. Clark and A. T. Bell, Phys. Chem. Chem. Phys., 2015, 17, 18924–18936 RSC
- D. Higgins, C. Hahn, C. Xiang, T. F. Jaramillo and A. Z. Weber, ACS Energy Lett., 2018, 4, 317–324 CrossRef
- Z. Chen, X. Zhang, W. Liu, M. Jiao, K. Mou, X. Zhang and L. Liu, Energy Environ. Sci., 2021, 14, 2349–2356 RSC
- H. Jiang, L. Wang, B. Gao, Y. Li, Y. Guo, M. Zhuo, K. Sun, B. Lu, M. Jia, X. Yu, H. Wang and Y. Li, Chem. Eng. J., 2021, 422, 129923 CrossRef CAS
- D. Gao, R. M. Arán-Ais, H. S. Jeon and B. Roldan Cuenya, Nat. Catal., 2019, 2, 198–210 CrossRef CAS
- T. Zheng, K. Jiang and H. Wang, Adv. Mater., 2018, 30, 1802066 CrossRef PubMed
- D. Karapinar, N. T. Huan, N. Ranjbar Sahraie, J. Li, D. Wakerley, N. Touati, S. Zanna, D. Taverna, L. H. Galvão Tizei and A. Zitolo, Angew. Chem., Int. Ed., 2019, 58, 15098–15103 CrossRef CAS PubMed
- E. Boutin, M. Wang, J. C. Lin, M. Mesnage, D. Mendoza, B. Lassalle-Kaiser, C. Hahn, T. F. Jaramillo and M. Robert, Angew. Chem., Int. Ed., 2019, 58, 16172–16176 CrossRef CAS PubMed
- Y. Wu, Z. Jiang, X. Lu, Y. Liang and H. Wang, Nature, 2019, 575, 639–642 CrossRef CAS PubMed
- H. Yang, Y. Wu, G. Li, Q. Lin, Q. Hu, Q. Zhang, J. Liu and C. He, J. Am. Chem. Soc., 2019, 141, 12717–12723 CrossRef CAS PubMed
- A. Guan, Z. Chen, Y. Quan, C. Peng, Z. Wang, T.-K. Sham, C. Yang, Y. Ji, L. Qian, X. Xu and G. Zheng, ACS Energy Lett., 2020, 5, 1044–1053 CrossRef CAS
- J. Zhao, J. Zhao, F. Li and Z. Chen, J. Phys. Chem. C, 2018, 122, 19712–19721 CrossRef CAS
- K. Zhao, X. Nie, H. Wang, S. Chen, X. Quan, H. Yu, W. Choi, G. Zhang, B. Kim and J. G. Chen, Nat. Commun., 2020, 11, 1–10 Search PubMed
- T. Zhang, X. Han, H. Yang, A. Han, E. Hu, Y. Li, X. q. Yang, L. Wang, J. Liu and B. Liu, Angew. Chem., Int. Ed., 2020, 132, 12153–12159 CrossRef
- J. Gu, C.-S. Hsu, L. Bai, H. M. Chen and X. Hu, Science, 2019, 364, 1091–1094 CrossRef CAS PubMed
- X. Li, Y. Zeng, C.-W. Tung, Y.-R. Lu, S. Baskaran, S.-F. Hung, S. Wang, C.-Q. Xu, J. Wang, T.-S. Chan, H. M. Chen, J. Jiang, Q. Yu, Y. Huang, J. Li, T. Zhang and B. Liu, ACS Catal., 2021, 11, 7292–7301 CrossRef CAS
- A. Wang, J. Li and T. Zhang, Nat. Rev. Chem., 2018, 2, 65–81 CrossRef CAS
- X. Wang, Z. Chen, X. Zhao, T. Yao, W. Chen, R. You, C. Zhao, G. Wu, J. Wang, W. Huang, J. Yang, X. Hong, S. Wei, Y. Wu and Y. Li, Angew. Chem., Int. Ed., 2018, 57, 1944–1948 CrossRef CAS PubMed
- J. Yang, Z. Qiu, C. Zhao, W. Wei, W. Chen, Z. Li, Y. Qu, J. Dong, J. Luo, Z. Li and Y. Wu, Angew. Chem., Int. Ed., 2018, 57, 14095–14100 CrossRef CAS PubMed
- S. Feng, W. Zheng, J. Zhu, Z. Li, B. Yang, Z. Wen, J. Lu, L. Lei, S. Wang and Y. Hou, Appl. Catal. B., 2020, 270 Search PubMed
- X. Zhang, Y. Wang, M. Gu, M. Wang, Z. Zhang, W. Pan, Z. Jiang, H. Zheng, M. Lucero, H. Wang, G. E. Sterbinsky, Q. Ma, Y.-G. Wang, Z. Feng, J. Li, H. Dai and Y. Liang, Nat. Energy, 2020, 5, 684–692 CrossRef CAS
- S. Tian, Q. Fu, W. Chen, Q. Feng, Z. Chen, J. Zhang, W.-C. Cheong, R. Yu, L. Gu and J. Dong, Nat. Commun., 2018, 9, 1–7 CrossRef PubMed
- J. Mahmood, F. Li, S. M. Jung, M. S. Okyay, I. Ahmad, S. J. Kim, N. Park, H. Y. Jeong and J. B. Baek, Nat. Nanotechnol., 2017, 12, 441–446 CrossRef CAS PubMed
- X. Zhao and Y. Liu, J. Am. Chem. Soc., 2020, 142, 5773–5777 CrossRef CAS PubMed
- C. Yan, H. Li, Y. Ye, H. Wu, F. Cai, R. Si, J. Xiao, S. Miao, S. Xie, F. Yang, Y. Li, G. Wang and X. Bao, Energy Environ. Sci., 2018, 11, 1204–1210 RSC
- W. Zheng, J. Yang, H. Chen, Y. Hou, Q. Wang, M. Gu, F. He, Y. Xia, Z. Xia, Z. Li, B. Yang, L. Lei, C. Yuan, Q. He, M. Qiu and X. Feng, Adv. Funct. Mater., 2019, 30, 1907658 CrossRef
- J. Feng, H. Gao, L. Zheng, Z. Chen, S. Zeng, C. Jiang, H. Dong, L. Liu, S. Zhang and X. Zhang, Nat. Commun., 2020, 11, 4341 CrossRef PubMed
- Q. Qu, S. Ji, Y. Chen, D. Wang and Y. Li, Chem. Sci., 2021, 12, 4201–4215 RSC
- M. D. Hossain, Y. Huang, T. H. Yu, W. A. Goddard III and Z. Luo, Nat. Commun., 2020, 11, 2256 CrossRef CAS PubMed
- Z. Geng, Y. Cao, W. Chen, X. Kong, Y. Liu, T. Yao and Y. Lin, Appl. Catal. B, 2019, 240, 234–240 CrossRef CAS
- H. Zhang, J. Li, S. Xi, Y. Du, X. Hai, J. Wang, H. Xu, G. Wu, J. Zhang, J. Lu and J. Wang, Angew. Chem., Int. Ed., 2019, 58, 14871–14876 CrossRef CAS PubMed
- Y. Pan, R. Lin, Y. Chen, S. Liu, W. Zhu, X. Cao, W. Chen, K. Wu, W. C. Cheong, Y. Wang, L. Zheng, J. Luo, Y. Lin, Y. Liu, C. Liu, J. Li, Q. Lu, X. Chen, D. Wang, Q. Peng, C. Chen and Y. Li, J. Am. Chem. Soc., 2018, 140, 4218–4221 CrossRef CAS PubMed
- E. Jung, H. Shin, B.-H. Lee, V. Efremov, S. Lee, H. S. Lee, J. Kim, W. H. Antink, S. Park and K.-S. Lee, Nat. mater., 2020, 19, 436–442 CrossRef CAS PubMed
- Y. Cai, J. Fu, Y. Zhou, Y. C. Chang, Q. Min, J. J. Zhu, Y. Lin and W. Zhu, Nat. Commun., 2021, 12, 1–9 CrossRef PubMed
- H. Kim, D. Shin, W. Yang, D. H. Won, H. S. Oh, M. W. Chung, D. Jeong, S. H. Kim, K. H. Chae, J. Y. Ryu, J. Lee, S. J. Cho, J. Seo, H. Kim and C. H. Choi, J. Am. Chem. Soc., 2021, 143, 925–933 CrossRef CAS PubMed
- S. Cao, S. Wei, X. Wei, S. Zhou, H. Chen, Y. Hu, Z. Wang, S. Liu, W. Guo and X. Lu, Small, 2021, 17, e2100949 CrossRef PubMed
- Y. Chen, R. Gao, S. Ji, H. Li, K. Tang, P. Jiang, H. Hu, Z. Zhang, H. Hao, Q. Qu, X. Liang, W. Chen, J. Dong, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2021, 60, 3212–3221 CrossRef CAS PubMed
- Y. Wu, C. Chen, X. Yan, X. Sun, Q. Zhu, P. Li, Y. Li, S. Liu, J. Ma, Y. Huang and B. Han, Angew. Chem., Int. Ed., 2021, 113, 20971–20978 CrossRef
- J. Wang, X. Huang, S. Xi, H. Xu and X. Wang, Angew. Chem., Int. Ed., 2020, 59, 19162–19167 CrossRef CAS PubMed
- X. Wang, Y. Pan, H. Ning, H. Wang, D. Guo, W. Wang, Z. Yang, Q. Zhao, B. Zhang, L. Zheng, J. Zhang and M. Wu, Appl. Catal. B, 2020, 266, 118630 CrossRef CAS
- K. Mou, Z. Chen, X. Zhang, M. Jiao, X. Zhang, X. Ge, W. Zhang and L. Liu, Small, 2019, 15, e1903668 CrossRef PubMed
- X. Rong, H. J. Wang, X. L. Lu, R. Si and T. B. Lu, Angew. Chem., Int. Ed., 2020, 59, 1961–1965 CrossRef CAS PubMed
- X. Wang, S. Ding, T. Yue, Y. Zhu, M. Fang, X. Li, G. Xiao, Y. Zhu and L. Dai, Nano Energy, 2021, 82, 105689 CrossRef CAS
- C. Peng, G. Luo, J. Zhang, M. Chen, Z. Wang, T. K. Sham, L. Zhang, Y. Li and G. Zheng, Nat. Commun., 2021, 12, 1580 CrossRef CAS PubMed
- F. Pan, H. Zhang, Z. Liu, D. Cullen, K. Liu, K. More, G. Wu, G. Wang and Y. Li, J. Mater. Chem. A, 2019, 7, 26231–26237 RSC
- Y. Cheng, S. Zhao, H. Li, S. He, J.-P. Veder, B. Johannessen, J. Xiao, S. Lu, J. Pan, M. F. Chisholm, S.-Z. Yang, C. Liu, J. G. Chen and S. P. Jiang, Appl. Catal. B, 2019, 243, 294–303 CrossRef CAS
- Z. Zhang, J. Xiao, X. J. Chen, S. Yu, L. Yu, R. Si, Y. Wang, S. Wang, X. Meng and Y. Wang, Angew. Chem., Int. Ed., 2018, 57, 16339–16342 CrossRef CAS PubMed
- W. Ju, A. Bagger, G.-P. Hao, A. S. Varela, I. Sinev, V. Bon, B. R. Cuenya, S. Kaskel, J. Rossmeisl and P. Strasser, Nat. Commun., 2017, 8, 1–9 CrossRef CAS PubMed
- W. Xie, H. Li, G. Cui, J. Li, Y. Song, S. Li, X. Zhang, J. Y. Lee, M. Shao and M. Wei, Angew. Chem., Int. Ed., 2021, 60, 7382–7388 CrossRef CAS PubMed
- Q. He, D. Liu, J. H. Lee, Y. Liu, Z. Xie, S. Hwang, S. Kattel, L. Song and J. G. Chen, Angew. Chem., Int. Ed., 2020, 59, 3033–3037 CrossRef CAS PubMed
- X. Yang, T. Tat, A. Libanori, J. Cheng, X. Xuan, N. Liu, X. Yang, J. Zhou, A. Nashalian and J. Chen, Mater. Today, 2021, 45, 54–61 CrossRef CAS
- G. Luo, Y. Jing and Y. Li, J. Mater. Chem. A, 2020, 8, 15809–15815 RSC
- M. Feng, X. Wu, H. Cheng, Z. Fan, X. Li, F. Cui, S. Fan, Y. Dai, G. Lei and G. He, J. Mater. Chem. A, 2021, 9, 23817–23827 RSC
- W. Ren, X. Tan, W. Yang, C. Jia, S. Xu, K. Wang, S. C. Smith and C. Zhao, Angew. Chem., Int. Ed., 2019, 58, 6972–6976 CrossRef CAS PubMed
- Y. Li, B. Wei, M. Zhu, J. Chen, Q. Jiang, B. Yang, Y. Hou, L. Lei, Z. Li, R. Zhang and Y. Lu, Adv. Mater., 2021, 33, 2102212 CrossRef CAS PubMed
- Z. Zeng, L. Y. Gan, H. Bin Yang, X. Su, J. Gao, W. Liu, H. Matsumoto, J. Gong, J. Zhang, W. Cai, Z. Zhang, Y. Yan, B. Liu and P. Chen, Nat. Commun., 2021, 12, 4088 CrossRef CAS PubMed
- W. Zhu, L. Zhang, S. Liu, A. Li, X. Yuan, C. Hu, G. Zhang, W. Deng, K. Zang, J. Luo, Y. Zhu, M. Gu, Z. J. Zhao and J. Gong, Angew. Chem., Int. Ed., 2020, 59, 12664–12668 CrossRef CAS PubMed
- L. Lin, T. Liu, J. Xiao, H. Li, P. Wei, D. Gao, B. Nan, R. Si, G. Wang and X. Bao, Angew. Chem., Int. Ed., 2020, 59, 22408–22413 CrossRef CAS PubMed
- E. J. Askins, M. R. Zoric, M. Li, Z. Luo, K. Amine and K. D. Glusac, Nat. Commun., 2021, 12, 3288 CrossRef CAS PubMed
- A. Roe, D. Schneider, R. Mayer, J. Pyrz, J. Widom and L. Que Jr, J. Am. Chem. Soc., 1984, 106, 1676–1681 CrossRef CAS
- L. Fang, S. Seifert, R. E. Winans and T. Li, Small Methods, 2021, 5, 2001194 CrossRef CAS
- Z. Jiang, T. Wang, J. Pei, H. Shang, D. Zhou, H. Li, J. Dong, Y. Wang, R. Cao and Z. Zhuang, Energy Environ. Sci., 2020, 13, 2856–2863 RSC
- P. Chen, N. Zhang, S. Wang, T. Zhou, Y. Tong, C. Ao, W. Yan, L. Zhang, W. Chu and C. Wu, Proc. Natl. Acad. Sci. U.S.A., 2019, 116, 6635–6640 CrossRef CAS PubMed
- J. Timoshenko and A. Kuzmin, Comput. Phys. Commun., 2009, 180, 920–925 CrossRef CAS
- S. Chen, Y. Li, Z. Bu, F. Yang, J. Luo, Q. An, Z. Zeng, J. Wang and S. Deng, J. Mater. Chem. A, 2021, 9, 1705–1712 RSC
- Z. Weng, Y. Wu, M. Wang, J. Jiang, K. Yang, S. Huo, X. F. Wang, Q. Ma, G. W. Brudvig, V. S. Batista, Y. Liang, Z. Feng and H. Wang, Nat. Commun., 2018, 9, 415 CrossRef PubMed
- J. Li and J. Gong, Energy Environ. Sci., 2020, 13, 3748–3779 RSC
- S. Liu, H. B. Yang, S. F. Hung, J. Ding, W. Cai, L. Liu, J. Gao, X. Li, X. Ren, Z. Kuang, Y. Huang, T. Zhang and B. Liu, Angew. Chem., Int. Ed., 2020, 59, 798–803 CrossRef CAS PubMed
- T. N. Huan, N. Ranjbar, G. Rousse, M. Sougrati, A. Zitolo, V. Mougel, F. Jaouen and M. Fontecave, ACS Catal., 2017, 7, 1520–1525 CrossRef CAS
- S. Back, J. Lim, N.-Y. Kim, Y.-H. Kim and Y. Jung, Chem. Sci., 2017, 8, 1090–1096 RSC
- H. He and Y. Jagvaral, Phys. Chem. Chem. Phys., 2017, 19, 11436–11446 RSC
- K. Jiang, S. Siahrostami, T. Zheng, Y. Hu, S. Hwang, E. Stavitski, Y. Peng, J. Dynes, M. Gangisetty and D. Su, Energy Environ. Sci., 2018, 11, 893–903 RSC
- C. Guo, T. Zhang, X. Liang, X. Deng, W. Guo, Z. Wang, X. Lu and C.-M. L. Wu, Appl. Surf. Sci., 2020, 533, 147466 CrossRef CAS
- M. D. Esrafili and B. Nejadebrahimi, Appl. Surf. Sci., 2019, 475, 363–371 CrossRef CAS
- X. Cui, W. An, X. Liu, H. Wang, Y. Men and J. Wang, Nanoscale, 2018, 10, 15262–15272 RSC
- L. Fu, R. Wang, C. Zhao, J. Huo, C. He, K.-H. Kim and W. Zhang, Chem. Eng. J., 2021, 414, 128857 CrossRef CAS
- X. Gao, H. Liu, D. Wang and J. Zhang, Chem. Soc. Rev., 2019, 48, 908–936 RSC
- Z. Feng, G. Su, H. Ding, Y. Ma, Y. Li, Y. Tang and X. Dai, Mol. Catal., 2020, 494, 111142 CrossRef CAS
- T. He, L. Zhang, G. Kour and A. Du, J. CO2 Util., 2020, 37, 272–277 CrossRef CAS
- S. Fu, X. Liu, J. Ran and Y. Jiao, Catal. Today, 2021 DOI:10.1016/j.cattod.2021.06.013
- Y. Meng, Y. Gao, K. Li, H. Tang, Y. Wang and Z. Wu, Appl. Surf. Sci., 2021, 542, 148568 CrossRef CAS
- C. Ao, B. Feng, S. Qian, L. Wang, W. Zhao, Y. Zhai and L. Zhang, J. CO2 Util., 2020, 36, 116–123 CrossRef CAS
- Q. Cui, G. Qin, W. Wang, K. Geethalakshmi, A. Du and Q. Sun, Appl. Surf. Sci., 2020, 500, 143993 CrossRef CAS
- J.-H. Liu, L.-M. Yang and E. Ganz, ACS Sustain. Chem. Eng., 2018, 6, 15494–15502 CrossRef CAS
- J.-H. Liu, L.-M. Yang and E. Ganz, J. Mater. Chem. A, 2019, 7, 3805–3814 RSC
- H. Zhou, X. Zou, X. Wu, X. Yang and J. Li, J. Phys. Chem. Lett., 2019, 10, 6551–6557 CrossRef CAS PubMed
- S. Cao, S. Wei, X. Wei, S. Zhou, H. Chen, Y. Hu, Z. Wang, S. Liu, W. Guo and X. Lu, Small, 2021, 2100949, DOI:10.1002/smll.202100949
- Q. Yuan, Y. Li, P. Yu, B. Ma, L. Xu, Q. Sun, H. Yang, M. Xie and T. Cheng, J. Exp. Nanosci., 2021, 16, 256–265 CrossRef CAS
- K. K. Rao, Q. K. Do, K. Pham, D. Maiti and L. C. Grabow, Top. Catal., 2020, 63, 728–741 CrossRef CAS
- D. Gao, T. Liu, G. Wang and X. Bao, ACS Energy Lett., 2021, 6, 713–727 CrossRef CAS
- Z.-K. Han, D. Sarker, R. Ouyang, A. Mazheika, Y. Gao and S. Levchenko, Nat. Commun., 2021, 12, 1–9 CrossRef PubMed
- X. Ma, Z. Li, L. E. Achenie and H. Xin, J. Phys. Chem. Lett., 2015, 6, 3528–3533 CrossRef CAS PubMed
- Z. W. Ulissi, M. T. Tang, J. Xiao, X. Liu, D. A. Torelli, M. Karamad, K. Cummins, C. Hahn, N. S. Lewis and T. F. Jaramillo, ACS Catal., 2017, 7, 6600–6608 CrossRef CAS
- J. R. Kitchin, Nat. Catal., 2018, 1, 230–232 CrossRef
- Y. Chen, Y. Huang, T. Cheng and W. A. Goddard III, J. Am. Chem. Soc., 2019, 141, 11651–11657 CrossRef CAS PubMed
- K. Tran and Z. W. Ulissi, Nat. Catal., 2018, 1, 696–703 CrossRef CAS
- M. Zhong, K. Tran, Y. Min, C. Wang, Z. Wang, C.-T. Dinh, P. De Luna, Z. Yu, A. S. Rasouli and P. Brodersen, Nature, 2020, 581, 178–183 CrossRef CAS PubMed
- A. Chen, X. Zhang, L. Chen, S. Yao and Z. Zhou, J. Phys. Chem. C, 2020, 124, 22471–22478 CrossRef CAS
- X. Wan, Z. Zhang, H. Niu, Y. Yin, C. Kuai, J. Wang, C. Shao and Y. Guo, J. Phys. Chem. Lett., 2021, 12, 6111–6118 CrossRef CAS PubMed
- X. Wan, Z. Zhang, W. Yu and Y. Guo, Materials Reports: Energy, 2021, 1, 100046 CrossRef
- T. Zheng, K. Jiang, N. Ta, Y. Hu, J. Zeng, J. Liu and H. Wang, Joule, 2019, 3, 265–278 CrossRef CAS
- M. G. Kibria, J. P. Edwards, C. M. Gabardo, C. T. Dinh, A. Seifitokaldani, D. Sinton and E. H. J. A. M. Sargent, Adv. Mater., 2019, 31, 1807166 CrossRef PubMed
- T. Cheng, H. Xiao and W. A. Goddard III, J. Am. Chem. Soc., 2016, 138, 13802–13805 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2022 |