
Stability of single-atom catalysts for electrocatalysis
Hao
Hu†
a,
Jiale
Wang†
a,
Peng
Tao
 a,
Chengyi
Song
a,
Wen
Shang
a,
Tao
Deng
ab and
Jianbo
Wu
*abcd
a,
Chengyi
Song
a,
Wen
Shang
a,
Tao
Deng
ab and
Jianbo
Wu
*abcd
aState Key Laboratory of Metal Matrix Composites, School of Materials Science and Engineering, Shanghai Jiao Tong University, 800 Dongchuan Rd, Shanghai 200240, China. E-mail: jianbowu@sjtu.edu.cn
bCenter of Hydrogen Science, Shanghai Jiao Tong University, Shanghai 200240, China
cMaterials Genome Initiative Center, Shanghai Jiao Tong University, Shanghai 200240, China
dFuture Material Innovation Center, Zhangjiang Institute for Advanced Study, Shanghai Jiao Tong University, Shanghai 200240, People's Republic of China
First published on 16th December 2021
Abstract
Single-atom catalysts (SACs) have recently attracted significant attention due to their maximum atom utilization and high efficiency in a series of electrocatalytic reactions. However, the atomically dispersed metal atoms have intrinsically extreme mobility due to their high surface energy. Besides, the harsh reaction conditions of electrocatalysis also challenged the catalytic stability of SACs. The excellent electrocatalytic performance of SACs always degrades under long-term operating conditions. Most previous studies of SACs have focused more on the activity and selectivity of SACs in electrocatalysis, while catalytic stability has received little attention as a more critical factor limiting their large-scale industrial application. In this review, we provide an overview of the recent advances in SACs in terms of selecting the metal and support materials, synthetic strategies, and electrocatalytic applications with a focus on catalytic stability. A deep understanding of the instability behaviors of SACs under different electrocatalytic conditions contributes to the design of effective synthetic strategies to further optimize their catalytic stability, which is particularly discussed. Finally, we present the challenges and prospects for the future development of stable SACs in electrocatalysis.
 Hao Hu | Hao Hu received his B.S. and M.S. degrees from the Faculty of Materials Science and Chemistry, China University of Geosciences, Wuhan in 2017 and 2020, respectively. Then he joined the School of Materials Science and Engineering at Shanghai Jiao Tong University to pursue his PhD degree under the direction of Professor Jianbo Wu. His current research focuses on the design of Pt-based electrocatalysts for fuel cells utilizing in situ TEM characterization. |
 Jiale Wang | Jiale Wang received his B.S. degree from the College of Materials Science and Engineering, Hunan University in 2019. Then he joined the School of Materials Science and Engineering at Shanghai Jiao Tong University to pursue his M.S. degree under the direction of Professor Jianbo Wu. His current research focuses on the design and preparation of CO2 reduction electrocatalysts. |
 Jianbo Wu | Dr Jianbo Wu has served as a tenured professor in the School of Materials Science and Engineering (MS&E) at Shanghai Jiao Tong University since 2014. He received his B.S. and M.S. degrees from Zhejiang University, China in 2007, his PhD degree in Chemical Engineering from the University of Rochester, USA in 2012, and two years' postdoctoral training in the Department of MS&E at the University of Illinois. His current research focuses on shape-controlled nanocrystals, fuel cells, electrocatalysis, functional materials for energy and environmental applications, and in situ TEM characterization to understand the catalyst formation and degradation during catalysis. |
1. Introduction
To meet the growing energy demand and solve the worldwide environmental issue, developing electrocatalytic technologies, such as water electrolysis,1 fuel cells,2 and CO2 reduction,3 has been identified as one of the most promising and renewable approaches to ensure sustainable and environmentally friendly energy conversion. For these electrocatalytic reactions, the design and development of economic and efficient electrocatalysts are crucial to achieving high efficiency and stable energy conversion. Although noble metals like Pt,4 Pd,5 Ru,6 Ir,7 and their derived materials have exhibited excellent properties in a variety of electrocatalytic reactions, the disadvantages of high cost and low natural abundance hindered their large-scale practical applications.8 The catalytic activity of the electrocatalysts typically depends on two aspects, including the intrinsic activity of the individual active site and the number of active sites.9 Numerous efforts have been made to increase the surface area by reducing the size of metallic nanomaterials to nanoclusters and single atoms,10,11 of which a larger number of active sites are adequately exposed and hence improve the electrocatalytic performance.12–14 Single-atom catalysts (SACs) have gained considerable attention as superior electrocatalysts due to their maximum atom utilization and the improved intrinsic catalytic activity since Zhang's group reported the fabrication of isolated Pt atoms anchored on iron oxide (Pt1/FeOx) and exhibited excellent performance for CO oxidation in 2011.15 Moreover, SACs with the individual metal atoms dispersed on a substrate generally display many superior properties, such as quantum size effects, unsaturated coordination configuration, and strong metal–support interactions (SMSI).16 In the last decade, SACs have achieved rapid development, and numerous studies have focused on the activity and selectivity of SACs in electrocatalysis. However, the stability of SACs in the catalytic reactions has been given considerably less attention in comparison to activity and selectivity issues. The single metallic atoms are likely to migrate and aggregate under synthetic and catalytic conditions due to their extremely high surface energy. Besides, owing to the harsh reaction conditions of different electrocatalyses, such as strong acid and alkaline solutions,17 high and low working potentials,18,19 and the gas environment,20,21 the dissolution, peeling off, and poisoning of SACs frequently occur during the catalytic reaction and lead to the degradation of the catalysts, hence hindering their long-term durability.22 Therefore, synthesizing SACs with excellent catalytic stability is crucial for their large-scale industrial applications.To suppress the instability behavior of SACs during the catalytic reactions, a comprehensive understanding of the coordination configuration and the electronic structure is significant, which is generally associated with the coactions between the isolated metallic atoms and support. The concept of strong metal–support interactions (SMSI) was initially demonstrated by Garten et al. in 1978, indicating that the adsorption capacity for H and CO of precious metals supported on TiO2 refers to the chemical bonds between metallic species and the relevant interface.23 Guided by SMSI, on the one hand, building strong bonds between the metal atoms and the support can inhibit the mobility of individual metal atoms; on the other hand, regulating the local electronic structure of the electroactive sites via charge transfer between metal and coordination atoms can also effectively increase the resistance of SACs to harsh conditions.24 Therefore, the rational design of the synthetic strategy is critical to improving their resistance to harsh catalytic conditions. Up to now, the major synthetic strategies have included spatial confinement,25 defect engineering,26,27 and coordination environment design strategies.28 SACs with single atoms stably dispersed on the support can be easily achieved with these synthetic strategies. However, owing to the different operating conditions of different electrocatalytic reactions, the mechanism of catalyst degradation also differs. Therefore, fully understanding the instability behaviors of SACs under different electrocatalytic conditions is essential for designing effective synthetic strategies to further optimize their catalytic stability. However, as far as we know, a dedicated and comprehensive overview of the stability of SACs in electrocatalytic applications is rarely reported.
In this review, we first focus on selecting the metal and support materials of SACs for electrocatalysis. Subsequently, the main synthetic strategies to enhance the stability of SACs in both synthetic and catalytic processes are summarized. Furthermore, the catalytic stability of SACs under different harsh catalytic conditions is discussed in depth. Finally, the current challenges and the prospects for the development of stable SACs in electrocatalysis are proposed (Fig. 1).
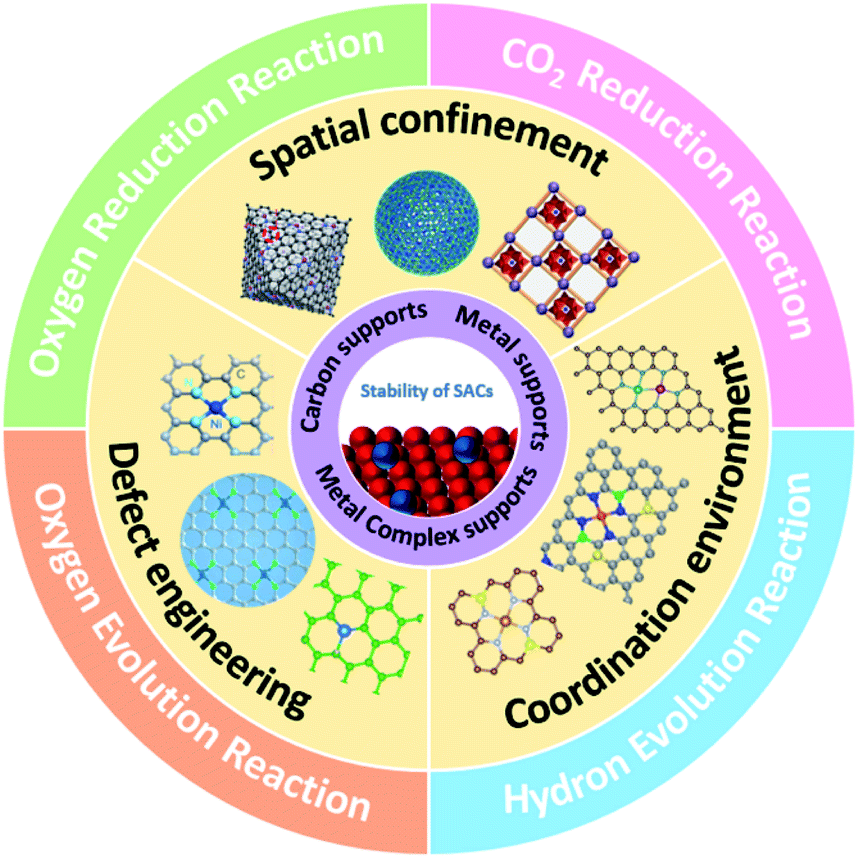 | ||
| Fig. 1 Schematic diagram of the stability of SACs, including metals and supports, synthetic strategies, and electrocatalytic stability.29–36,123 | ||
2. Selection of metals and supports
Seeking a suitable metal and support material for the fabrication of SACs is a prerequisite for achieving excellent performance in electrocatalysis.37 However, the single metal atoms tend to aggregate easily due to their high surface energy during the preparation process. Besides, SACs are also likely to suffer from adverse structural evolution during the catalytic reactions leading to degradation. Therefore, designing and synthesizing stable SACs have become a significant challenge to achieve excellent electrocatalytic performance. Up to now, constructing SMSI has been the central principle for the synthesis of stable SACs, as strong surface interactions can efficiently avoid aggregation while modifying the electronic structure of the electroactive sites to further improve the catalytic stability via electronic metal–support interactions (EMSI). In general, the interaction can be regulated by modifying the supports such as defect construction and the introduction of a functional group to obtain more anchoring sites since the properties of supports determine the coordination configuration, chemical bonding, and electronic structure that influence the activity and stability of SACs. Therefore, the deliberate selection of a suitable combination of metal and support materials is the basis for the preparation of stable SACs. Numerous types of support material have been investigated.38–40 Based on MSI, we will review the recent progress in the rational selection of metal and support materials for stable SACs.2.1. Selection of metals
First, the selection of supported metal atoms is essential to all the electrocatalytic reactions, as the metal atoms usually are part of the active sites assisting in the catalysis. In general, transition metal elements are primarily selected in electrocatalysis and have been widely investigated.41 This is mainly due to the alternative d orbitals of the transition metals that allow easy access to obtain the optimal electronic structure favorable for the reactions. In addition, transition metals have many other unique properties, such as high melting/boiling point, high rigidity, excellent electrical/thermal conductivity, and multiple redox potentials, which also contribute to their excellent performance. However, owing to the large surface energy and low-coordination environment of single metals, SACs always aggregate over time, which leads to the loss of activity and stability. Gates et al. prepared a sequence of MgO-supported catalysts with different metal nanoclusters, including Ir, Pt, and Au.42 Through the same treatment in H2 and an electron beam, most of the Ir species became clusters with a diameter of approximately 1 nm, while the Pt and Au nanoparticles had diameters ranging from 2 to 5 nm. The results demonstrated that different metals have different resistances to sintering, and Ir is probably an inherently sinter-resistant metal compared to the others. Therefore, understanding the fundamental properties of transition metal elements helps investigate the stability mechanism during the preparation and under the reaction conditions, and thus in designing stable catalysts. For SACs, the appropriate selection of single-atom metals will facilitate the fabrication of stable SACs by adjusting the coordination number and the strength of the chemical bonds between the single metal and support. Zheng et al. designed a sequence of 3d-transition metals (Cr, Mn, Fe, Co, Ni, Cu, and Zn) supported on graphitic carbon nitride (g-C3N4) SACs.43 By calculating the binding energies of the g-C3N4 support with different metals, Fe, Ni, and Co possess the highest M–N2 binding energies, indicating their highest structural stabilities. Therefore, the design of stable SACs in electrocatalysis requires the rational selection of metals.2.2. Selection of supports
Except for selecting the supported atom, the selection of support materials is much more essential for stabilizing isolated metallic atoms on a substrate and the resistance to unstable behaviors in electrocatalysis. Ideal supports could offer abundant immobilizing sites to firmly anchor the isolated atoms and construct strong bonding to prevent the aggregation of isolated metallic atoms during preparation. More importantly, a suitable support can efficiently enhance the stability of SACs under harsh catalytic conditions. The properties of support materials determine the coordination number, steric environment, and chemical bonding, which result in different electronic and morphological structures of SACs. Therefore, rational selection and design of supports are significant for synthesizing stable SACs in electrocatalytic reactions. Until now, many kinds of support have been reported, including carbon, metals, and metal (hydr)oxides/nitrides/carbides/sulfides.Zhang and co-workers successfully synthesized almost all the transition metal atoms on a conductive carbon support with a metal loading of over 1.8 wt% via the low-temperature pyrolysis method (Fig. 2a).72 The X-ray absorption near-edge structure spectroscopy (XANES) spectra and X-ray absorption fine structure (EXAFS) suggested that the as-prepared Ni-SACs contained Ni2+, corresponding to Ni–N4 coordination. The modified carbon support tightly bound the metal cations and prevented the agglomeration of metal atoms into clusters and nanoparticles. Recently, an emerging class of porous carbon-based materials, MOFs, with ultra-high surface area, flexible structural stability, and well-defined ordered structures, have been considered the most promising candidates for the SAC substrate. MOFs have numerous anchoring sites, defects, and porosities to stabilize the single metal atoms. In general, utilizing MOFs as precursors for MOF-derived SACs improves not only the loading of metals in SACs but also the stability of SACs due to the formation of stable metal–C, O, N, and S coordination bonds. Since MOFs were firstly reported as an efficient support to stabilize single Pt atoms by Jiang and co-workers,76 numerous studies have been widely performed in the last ten years. For instance, Li's group developed an effective method to synthesize Ru-SACs with Ru species anchored on MOFs.51 The spatial separation of Ru single atoms was precisely controlled by cages of ZIF-8 by the formation of strong bonding between Ru and ZIF-8 after thermal reduction treatment to prevent active species from migrating and aggregating, thus contributing to the excellent stability and recyclability of Ru-SACs.
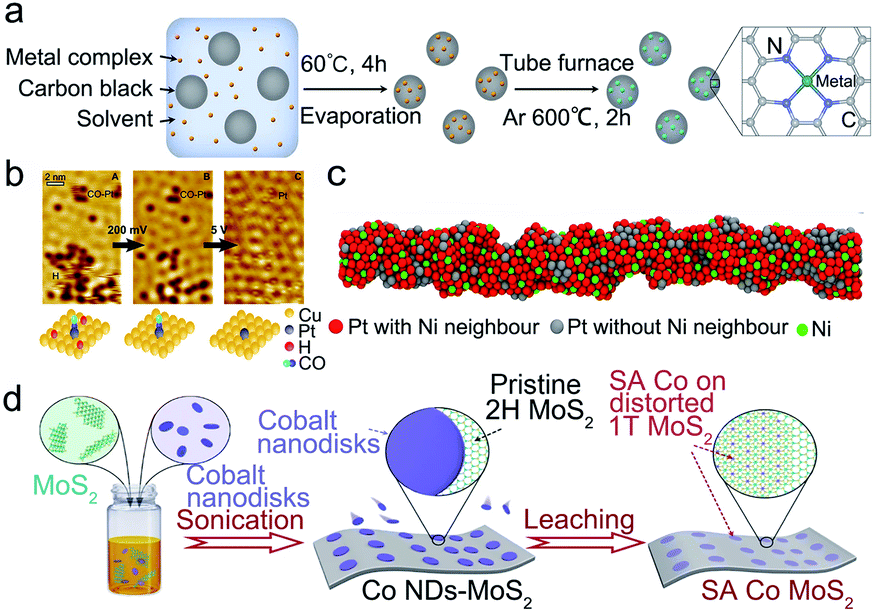 | ||
| Fig. 2 Selection of the support for SACs. (a) The synthetic process of M-SACs. Reproduced with permission from ref. 72. Copyright Springer Nature 2019. (b) STM images of H and CO on a Pt–Cu (111) surface. Reproduced with permission from ref. 63. Copyright 2016 American Chemical Society. (c) Schematic illustration of SA Ni–PtNWs. Reproduced with permission from ref. 60. Copyright 2019 Springer Nature. (d) Schematic illustration of the synthetic procedure for SA Co-D 1T MoS2. Reproduced with permission from ref. 71. Copyright Springer Nature 2019. | ||
The key to achieving a stable isolated atom on a metal support is enhancing the bonding strength between the metal and support. To suppress migration and agglomeration of isolated metal atoms to form clusters and nanoparticles, the bonding energy between the support and single atoms should be larger than that of the single atoms themselves.55 Metal atoms like Pt, Pd, Ni, Cu, and Au are the popular active elements in SACs for electrocatalysis.54,56 Michaelides et al. have studied the stability of SACs with different alloy combinations via the DFT calculation method.57 They showed that the Gibbs free energy of aggregation (ΔGagg) of PdAu, PtCu, NiAu, and PdCu is positive, which means that they could form the SACs. But the ΔGagg of NiCu is near zero, indicating the existence of single metal atoms and clusters. It provided an effective approach to design stable SACs. Subsequently, Liu et al. successfully synthesized Pt SACs on a Cu support with a Pt/Cu ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :125 without forming metal clusters (Fig. 2b).58 Besides, Zhang's group selected Pd and Cu supports to synthesize Pd SACs.59 Due to the electron transfer between Cu and Pd, the as-prepared Pd SACs exhibited a high ethylene selectivity and especially showed excellent durability with stable operation for 24 hours. To lower the catalytic cost and scarcity of catalysts, Duan et al. anchored single Ni atoms on Pt nanowires via an electrochemical dealloying method (Fig. 2c).60 The synthesized single Ni modified Pt nanowires exhibited enhanced durability compared to pure Pt nanowires and a commercial Pt/C catalyst for the HER. The enhanced stability may be attributed to multipoint line contacts between the nanowires and the carbon carrier, thus preventing physical movement and aggregation.
:125 without forming metal clusters (Fig. 2b).58 Besides, Zhang's group selected Pd and Cu supports to synthesize Pd SACs.59 Due to the electron transfer between Cu and Pd, the as-prepared Pd SACs exhibited a high ethylene selectivity and especially showed excellent durability with stable operation for 24 hours. To lower the catalytic cost and scarcity of catalysts, Duan et al. anchored single Ni atoms on Pt nanowires via an electrochemical dealloying method (Fig. 2c).60 The synthesized single Ni modified Pt nanowires exhibited enhanced durability compared to pure Pt nanowires and a commercial Pt/C catalyst for the HER. The enhanced stability may be attributed to multipoint line contacts between the nanowires and the carbon carrier, thus preventing physical movement and aggregation.
As one of the metal (hydr)oxides, Fe (hydr)oxide is a typical 3d transition metal-based oxide that could stabilize the isolated metallic atoms by the intense metal–metal bonding.62 For instance, Zhang et al. first successfully synthesized a Pt1/FeOx SAC with a Pt loading of 0.06 wt% by a co-precipitation method.15 Afterwards, owing to the large surface area (290 m2 g−1) of the FeOx support and strong interactions between Pt and FeOx, they increased the Pt content in Pt/FeOx (0.17 wt%) without detecting Pt aggregation. Besides, Au is easily aggregated in the high-temperature synthetic process. A good approach to this issue is to utilize the interaction between Au and an appropriate support to select a suitable support.63 Liu and co-workers found that Au dispersed on FeOx has larger resistance to sintering than Au nanostructures; thus they successfully synthesized a Au1/FeOx SAC by anchoring single Au atoms on FeOx.63 The formation of a strong covalent bond between Au and the FeOx support resulted in the excellent stability of Au1/FeOx SACs for over 100 hours. In addition, CeO2 and Al2O3 have also been reported as effective support materials for stabilizing single metal atoms.64–66
Except for metal (hydr)oxides, metal nitrides/carbide/sulfides are also frequently chosen as the support materials. DFT calculations have demonstrated that single Pt atoms can be stably fixed on a TiN support.67 Lee et al. successfully synthesized single Pt atoms supported on TiN via an incipient wetness impregnation method,68 which showed high mass activity and unique selectivity in selective electrochemical reactions. Significantly, the corrosion resistance of TiN enhanced the durability of Pt SACs on TiN. WxC, as a popular metal carbide support, has been demonstrated to be an excellent support for Pt SACs via DFT calculations.69 By comparing the stabilities of Pt atoms on different WxC low-index facets, they discovered that the WxC (100) surface could strongly adsorb Pt atoms and resist clustering. Besides, MoC could also stabilize the isolated Pt atoms due to the intense interactions between isolated Pt atoms and MoC after a high-temperature activation.70 In addition, Liu's group designed an interface Co SAC supported on distorted 1T MoS2.71 Due to the lattice mismatch and the covalent bonding between Co and S, the isolated Co atoms could be firmly anchored on the support and exhibited long-term durability for over 10 days (Fig. 2d).
3. Synthetic strategies of stable SACs
Due to the large surface energy of single metal atoms, they spontaneously tend to migrate and aggregate under synthesis.62 Therefore, the rational design of synthetic strategies to stably anchor the atomically dispersed metal atoms on the support plays a key role in achieving remarkable electrocatalytic performance. Besides, the instability behaviors of SACs under harsh electrocatalytic conditions could also be suppressed by the reasonable synthetic strategy.73 Therefore, a deep understanding of synthetic strategies is essential to the design and fabrication of stable SACs. At present, constructing strong bonding and regulating the electronic interaction between single metal atoms and the support are the most effective strategies to develop stable SACs, which could be divided mainly into the spatial confinement, defect engineering, and coordination environment design strategies. Herein, the recent advances in synthetic strategies for stable SACs will be discussed.743.1. Spatial confinement strategy
The spatial confinement strategy was considered an efficient method to synthesize stable SACs.25 Using porous materials, the isolated atoms could be confined in molecular-scale cages to avoid migration and agglomeration of target metal atoms into nanoparticles.75 The synthetic strategy was applied to the preparation of numerous stable SACs due to its irreplaceable advantages in predictable anchor sites, locations, and extremely durable structures under harsh conditions. Moreover, the rational spatial confinement strategy can efficiently avoid the structural and compositional evolution, thus causing instability during the catalytic reaction. Specific substrates with a porous structure can provide strong metal–support interactions to protect the isolated atoms from migration, agglomeration, and peeling off. Meanwhile, the electronic interaction between isolated metal sites and coordination atoms can regulate the electronic structure of electroactive species to enhance the resistance to dissolution and poisoning which frequently occur in catalytic reactions.76Up to now, porous carbon-based materials like zeolites, MOFs, and COFs have been widely used as templates in the spatial confinement strategy for the preparation of stable SACs.77 In these materials, MOF-derived SACs have achieved great progress in the past few years. MOFs are constructed with metal ions as connection points and organic ligands as supports to form three-dimensional networks. Furthermore, due to the features of large specific area, adjustable aperture size, high porosity, and diverse structure configurations, MOFs could enable the stabilization and homogeneity of single metal atoms on the anchor sites and make it possible to achieve high loading SACs. Therefore, MOFs are considered as the ideal support materials for single metal atoms.
Jiang et al. firstly proposed the successful synthesis of single Pt atoms confined in a MOF with Pt loading of 0.07 and 0.29 wt%.76 The EXAFS spectra showed the prominent peak at about 1.5 Å attributed to Pt–N, confirming that the single-atom Pt was stabilized by nitrogen atoms. Afterward, W-SACs with a higher metal content (1.21 wt%) were also reported.78 The W-SACs were synthesized by confining the W atoms in the N-doped carbon matrix derived from a MOF (UiO-66-NH2). As illustrated in Fig. 3a and b, metal precursor WCl5 was encapsulated in the skeleton of the MOF, and a further pyrolysis process was performed to form isolated W atoms stabilized by the N-doped carbon frame. In the atom trapping process, the uncoordinated amine groups in the MOF are vital in limiting the aggregation of W species. The W-SACs showed excellent HER stability in both alkaline and acidic media with no appreciable activity degradation after 10000 CV cycles, mainly due to the unique structure of the W1N1C3 moiety resulting in the strong interaction between the isolated W atoms and the N doped carbon substrate. Recently, Liu et al. introduced a modified spatial confinement strategy for the fabrication of SACs, which is suitable for various MOF systems (Fig. 3c).31 Typically, classical Keggin-type polyoxometalates (POMs) were used to immobilize single Pt atoms in the square-planar sites on the surface of monodisperse POMs in different MOFs, and the subsequent pyrolysis caused the formed isolated Pt atoms to be stabilized by oxygen species in the support. Further characterization proved the isolated Pt coordinated with four bridging O atoms, implying the intense interactions between isolated Pt atoms and the substrate. For the high loading of MOF-derived SACs, Jiang's group designed a high Fe content (3.46 wt%) implanted N-doped porous carbon.79 During the pyrolysis as illustrated in Fig. 3d, Fe atoms were spatially isolated and firmly immobilized by nitrogen atoms in porphyrin linkers which was the essential protective barrier to avoid the aggregation of Fe atoms. Furthermore, the FeSA–N–C showed excellent durability and methanol tolerance, shown by the LSV curves after 20000 CV cycles and methanol addition.
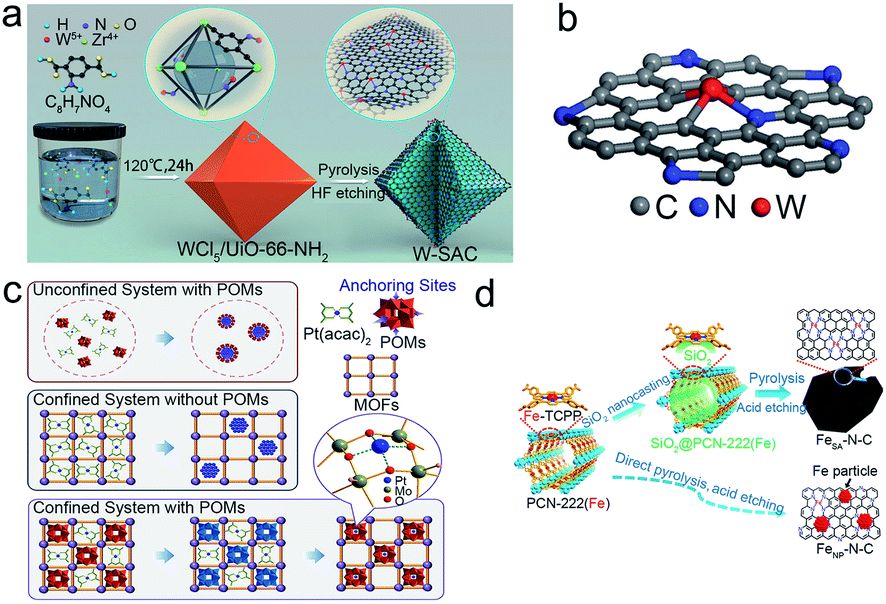 | ||
| Fig. 3 Spatial confinement strategies for the preparation of stable SACs. (a) The schematic diagram for the preparation, and (b) atomic model of the W-SAC. Reproduced with permission from ref. 78. Copyright John Wiley and Sons 2018. (c) Schematic diagram of the preparation of different Pt nanomaterials in an unconfined/confined system and with/without POMs condition. Reproduced with permission from ref. 31. Springer Nature 2021. (d) Schematic diagram of the nanocasting-assisted preparation of FeSA–N–C. Reproduced with permission from ref. 79. Copyright Springer Nature 2020. | ||
3.2. Defect engineering
The defect engineering strategy is also an effective strategy to stabilize the SACs.27 The precursors are firmly captured by the defect sites against sintering via the strong bonding and electronic interactions between isolated metal atoms and defective sites.26 Moreover, the adverse structural evolution during the electrocatalytic reactions could be inhibited by the regulation of the local electronic structure and coordination environment. Till now, defects like intrinsic defects, heteroatoms, and anion/cation vacancies have been reported to synthesize and stabilize SACs.Due to the high surface energy and high activity of isolated atoms, many studies have been devoted to avoiding aggregation in both the synthetic and catalytic processes. For example, Wang et al. applied intrinsic defects to construct a thermodynamically stable structure of SACs (Fig. 4a and b).36 By means of DFT calculations, they calculated the binding energy of FeN4S2 to be −10.57 eV while that of FeN3S1 is only −5.01 eV, which means that FeN4S4 is more stable than FeN3S1. Similarly, they further showed that CoN3S1 and NiN3S1 are thermodynamically more stable than CoN4S2 and NiN4S, respectively. Thus, the defective atoms play a key role in the durability of SACs, which could inhibit the tendency to aggregate. In addition, adverse structural evolution, such as dissolution and oxidation, is also a key issue in the stability of SACs. Pt-free catalysts like Ni-based catalysts suffer from low stability under acidic conditions.33 Chen's group utilized the orbital charge transfer between carbons and doped Ni to enhance the durability of Ni SACs.33 The overlap of the partial density of states (pDOS) projected onto the Ni atom and the three surrounding C atoms indicated a strong C–Ni chemical binding, which improved the resistance of Ni SACs to dissolution in acidic solution (Fig. 4c and d). Therefore, the Ni SACs showed superior HER cycling stability in 0.5 M H2SO4 solutions. Except for the intrinsic defects, the external introduction of defects could also contribute to stabilizing the isolated metal atoms. Oxygen vacancy in the metal oxide support is a superior medium to form a stable configuration of SACs. Li and co-workers introduced oxygen vacancies in TiO2 nanosheets by a thermal treatment method (Fig. 4e).80 Through the DFT calculations, isolated Au atoms could form a stable three-center Ti–Au–T structure on the defective TiO2 (001) surface, while it could only constitute a four-center Ti–Au–O–Ti structure when immobilized on the pristine TiO2 (001). The binding energy of the Ti–Au–T structure is smaller than that of Ti–Au–O–Ti, demonstrating the better durability of Ti–Au–Ti than Ti–Au–O–Ti.
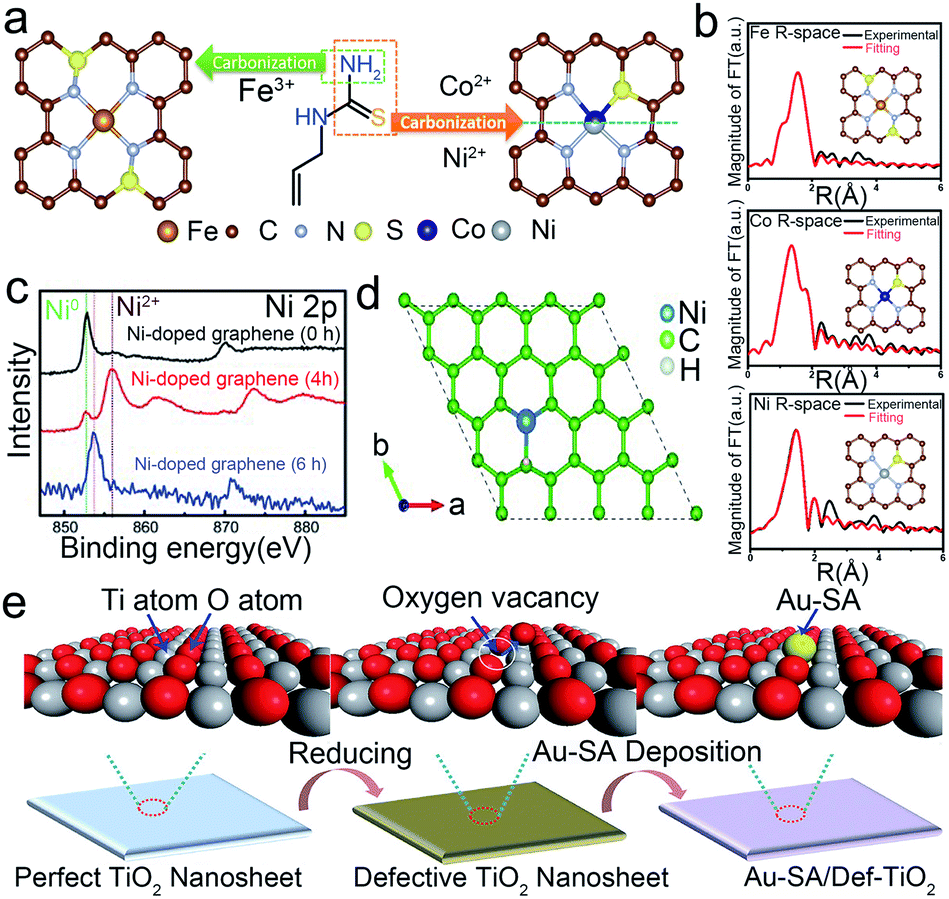 | ||
| Fig. 4 Defect engineering for the preparation of stable SACs. (a) The schematic illustration of the formation, (b) EXAFS data of Fe–SAs/NSC, Co–SAs/NSC and Ni–SAs/NSC. Reproduced with permission from ref. 36. Copyright American Chemical Society 2019. (c) Ni 2p XPS data of pristine graphene@np-Ni and Ni/np-G for 3 h and 6 h Ni dissolution, respectively. (d) H adsorption sites and configuration of the nickel doped graphene. Reproduced with permission from ref. 33. Copyright John Wiley and Sons 2015. (e) Synthetic process of a single Au atom on defective TiO2 with oxygen vacancies. Reproduced with permission from ref. 80. Copyright John Wiley and Sons 2018. | ||
3.3. Coordination environment
The isolated atoms in SACs are generally anchored on a support which contains unsaturated coordination atoms. Numerous studies have demonstrated that the stability of SACs is sensitive to the local coordination configuration, and thus constructing an appropriate coordination environment of SACs is essential to bind metal atoms while hindering the migration and aggregation of SACs.28 Till now, the coordination atoms have included O, N, and Se, and dual atoms.81 Optimizing the coordination configuration (including the coordination numbers and coordination atoms) of electroactive sites is a promising and essential approach to adjust the bond strength and the electronic structure, thus enhancing the stability of SACs.82O is the common coordination atom in SACs. Ren et al. have designed variable valence state Pt SACs on Fe2O3 by a controllable rapid thermal treatment method (Fig. 5a).83 It was found that the coordination number of Pt to O and the oxidation state of Pt decreased with increasing the treatment temperature (Fig. 5b). Based on this conclusion, the optimized Pt1/Fe2O3-600 catalyst remained in the isolated state without any aggregation during the catalytic reaction, indicating that the isolated Pt atoms were stably anchored. Besides, the N atom has also been utilized as a coordination agent for single metal atoms. Co SACs could be designed in Co–N2, Co–N3, and Co–N4 coordination structures by tuning the pyrolysis temperature (Fig. 5c).84 The results showed that the Co–N2 structure possesses the highest catalytic stability with negligible decay in current density and almost invariable CO faradaic efficiency during a 60 h operation. The TEM images showed that the morphology and the atomic dispersion of Co atoms were well preserved. This excellent catalytic stability was attributed to the lower oxidation state and binding energy of CO–N2. In addition, by substituting the Co atom with Pt, the coordination number of Se could increase in the np-Co0.85Se catalyst.85 The corresponding XPS and XAS experiments illustrated that Pt atoms were strongly anchored by six Se atoms, forming a stable octahedral structure (Fig. 5d). It presented an excellent stability without showing obvious structural changes after 40 hours of long-term stability testing in the acid electrolyte. Except for controlling the coordinate number, changing the coordinate atom species like forming dual metallic SACs is also an effective approach to stabilize SACs. Sun's group found that Pt–Ru dual metal atom catalysts with 6H on N-doped carbon nanotubes (NCNTs) possessed excellent stability.86 To confirm the stability of Pt–Ru with 6H on NCNTs, they performed first-principles molecular dynamics calculations on the Pt–Ru SACs at 300 K for 5 ps. The results indicated that no obvious structural change was observed in Pt–Ru with 6H.
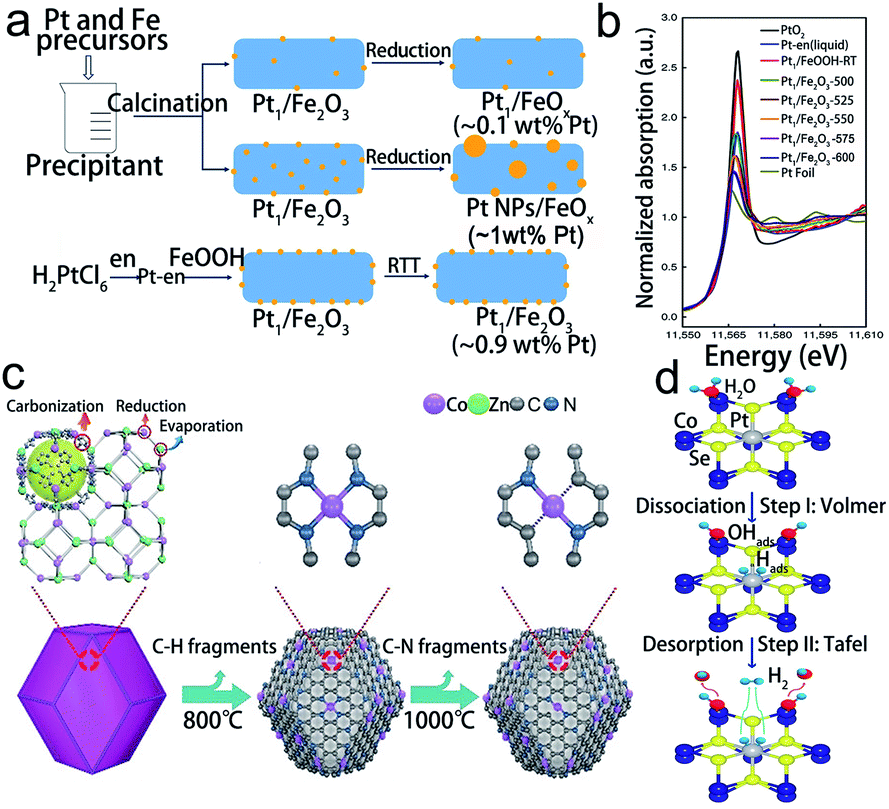 | ||
| Fig. 5 Coordination environment design for the synthesis of stable SACs. (a) Schematic diagram of different synthetic strategies, (b) the normalized XANES data of Pt1/Fe2O3 catalysts. Reproduced with permission from ref. 83. Copyright Springer Nature 2019. (c) The formation process of Co–N4 and Co–N2. Reproduced with permission from ref. 84 Copyright John Wiley and Sons 2018. (d) HER mechanism illustration with the assistance of in situ and operando XAS technologies of Pt/np-Co0.85Se. Reproduced with permission from ref. 85. Copyright Springer Nature 2019. | ||
4. Stability of SACs in electrocatalytic applications
As novel electrocatalysts, SACs possess unique properties, including maximized atom utilization and highly active sites. The excellent electrocatalytic activity of SACs has been widely confirmed by numerous relevant studies, while the stability of SACs during the catalytic process was frequently neglected. In addition to the instability of the single metal atoms in the synthetic process, the instability of SACs under actual reaction conditions is more significant for achieving industrial applications. Owing to the large surface energy and the low coordination number of single metal atoms, they intrinsically possess a high degree of mobility. Besides, the varying external potential and the strong acidic and alkaline environments always promote adverse structural evolution, such as migration, aggregation, dissolution, and peeling off, resulting in the degradation of SACs. Currently, the main principle to enhance the catalytic stability under harsh conditions is to construct strong bonding and tune the electronic structure of active sites via MSI. Guided by this, representative studies, as well as recent advances in the catalytic stability of SACs, are summarized in this section.4.1. ORR
The oxygen reduction reaction (ORR) generally occurs at the cathode of electrochemical energy conversion devices through a two-electron (2e−) or four-electron (4e−) pathway.87 Herein, we mainly discuss the 4e− ORR for its wide application in electrocatalysis, such as fuel cells and metal–air batteries. The wide range of external potential, as well as the acidic and alkaline electrolytes in the ORR, frequently promotes the oxidation and reduction of the metal and support, resulting in a variety of structural evolution, such as migration, aggregation, dissolution, and peeling off.88 Moreover, CO poisoning also leads to the degradation of electrocatalysts.89 Therefore, it places strict requirements on the stability of SACs under harsh reaction conditions.During the ORR process, isolated metal atoms tend to move and subsequently aggregate due to the high surface energy. When the bonding between the metal and support is not strong enough, the single metal atoms will aggregate rapidly into clusters or nanoparticles, resulting in the loss of ECSA and performance degradation. Therefore, SMSI are critical for the excellent durability of SACs. The main approach is to steadily immobilize the single metal atoms on the support to form strong bonding. Meanwhile, establishing the stable chemical bond between the metal and anchoring site is also an efficient approach to suppress the migration and aggregation of metal atoms. For example, Song et al. synthesized MOF-derived Pt SACs via the spatial confinement strategy, which exhibited excellent durability during the 10000 cycle ADT (Fig. 6a and b).90 The HADDF-STEM image indicated that most Pt species maintained the isolated single atoms and no crystalline Pt particles could be detected, which was because the MOF-derived N-doped carbon support with abundant N-anchoring sites can strongly interact with the Pt atoms to avoid Pt atoms from moving and gathering. Besides, Li's group designed and synthesized a MOF-derived Co1–N3PS/HC catalyst. They firstly constructed a Co–N4 structure, and then two heteroatoms were introduced into Co–N4 to form CoN4@P2 and CoN4@S2 structures. By comparing the doping energy in CoN4@P2 and CoN4@S2, they found that P was more stable than S. Thus, based on CoN4@P2, they further introduced 2 S atoms to form CoN4@P2S2 and CoN3S@P2S. Interestingly, they found that these two structures (CoN4@P2S2 and CoN3S@P2S) would distort to form the more stable CoN3PS@PS structure.91 Guided by DFT calculations and the optimized structure, they synthesized a Co1–N3PS/HC catalyst which exhibited excellent acidic stability compared to Co1–N4/HC and NPS/HC catalysts. Furthermore, the HAADF-STEM image revealed that the structure and atomic-level dispersion of Co species in Co1–N3PS/HC are well preserved after the ADT (Fig. 6c), indicating the remarkable capability to suppress the migration and aggregation. It was attributed to the unique atomic configuration of single Co atoms with coordination atoms.
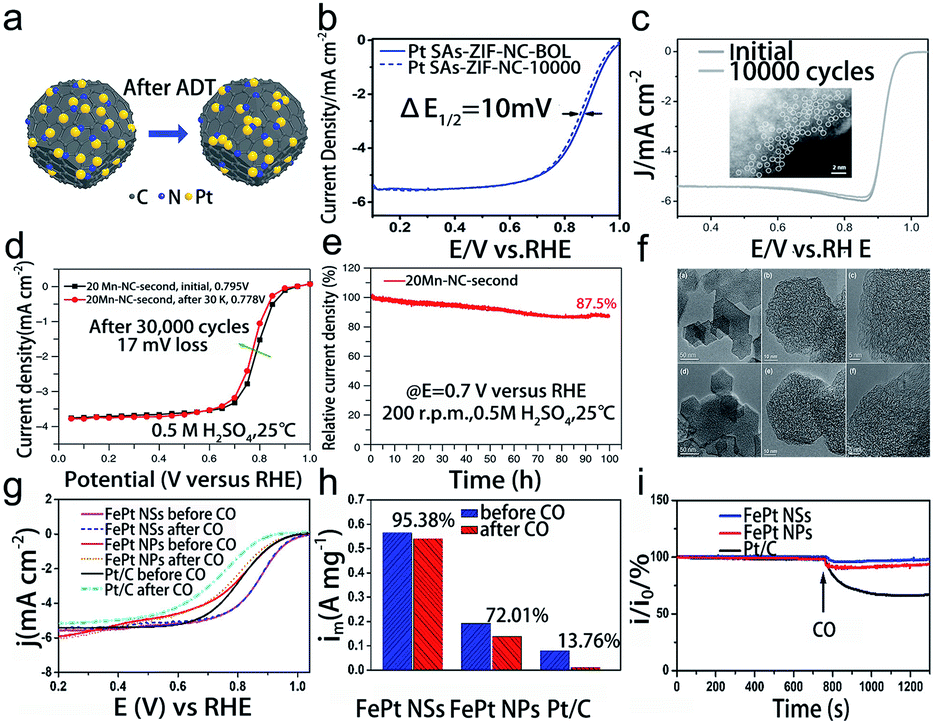 | ||
| Fig. 6 Electrocatalytic stability of SACs in the oxygen reduction reaction. (a) Schematic diagram after the stability test, (b) the oxygen reduction reaction polarization data before BOL and after the stability test for Pt SAs-ZIF-NC. Reproduced with permission from ref. 90. Copyright John Wiley and Sons 2020. (c) ORR polarization curves before and after the durability test. The inset image is the HAADF-STEM image of Co1–N3PS/HC after the stability test. Reproduced with permission from ref. 91. Copyright Elsevier 2015. (d) Steady-state oxygen reduction reaction polarization data before and after durability measurements, (e) i–t curves at 0.7 V, (f) HRTEM images before (top) and after (bottom) stability tests for 20Mn–NC-second. Reproduced with permission from ref. 93. Copyright Springer Nature 2018. (g) Oxygen reduction reaction polarization data for various catalysts before and after CO-tolerance tests. (h) The comparison of mass activities at 0.9 V before and after CO-tolerance tests, (i) CO-poisoning effect for the three catalysts in the presence of oxygen. Reproduced with permission from ref. 89. Copyright American Chemical Society 2018. | ||
The dissolution and subsequent redeposition of metal atoms are the major issues for the degradation of these materials in the ORR, especially in acid environments. The anchored single atoms are easily leached during the catalytic reaction due to the breaking of the chemical bonds between metallic species and the surrounding ligands in the support with the continuous high potential cycles.92 Moreover, the saturated oxygen environment further accelerates the oxidation and dissolution of the catalysts. O2, as the reactant of the ORR, is adsorbed in large quantities on the surface of the catalyst, resulting in the monoatomic metals being more susceptible to oxidation and dissolution. Such dissolution decreases the ECSA and utilization of the metal, thus leading to the degradation of SACs. Moreover, support materials such as carbon-based materials are unstable and easily oxidized in an oxidizing environment, which also promotes the dissolution of the metal sites.93 Regulating the electronic structure of electroactive sites through EMSI between the anchored single metal and ligand was considered efficient to suppress the dissolution. For instance, Holby and co-workers investigated the dissolution of Fe at a given potential in an acid ORR environment by DFT calculation.18 They calculated the stability of FeN4, OO–FeN4, and HO–FeN4vs. dissolution as a function of pH and potential. Combining the stability diagrams, they found that the relative stability plots for ligated FeN4 structures (OO–FeN4C138 with O2 binding and HO–FeN4C138 with OH binding) are more stable than ligand-free FeN4 structures, especially in low pH situations. They speculated that the free radicals formed in the ORR system would attack the C/N support matrix or Fe directly and cause the activity and stability to drop, and with the existence of protective ligands, these free radicals may interact with ligands and leave the FeN4. Thus, they indicated that the existence of ligands could help to stabilize Fe against dissolution, and without the interaction with the ligand or ORR intermediate, the FeN4 structure will be thermodynamically driven to dissolve. Moreover, Li et al. synthesized Mn–NC SACs through the carbonization of ZIF-8 and further acid leaching.93 The as-prepared catalyst exhibited excellent cycling stability in acid solution, as demonstrated by a loss of only 17 mV in E1/2 after 30000 cycles shown in Fig. 6d, and remarkable durability, maintaining 88% of the initial current density and exhibiting an E1/2 loss of 29 mV after 100 hours (Fig. 6e). Further characterization confirmed that the atomic Mn sites are in the oxidation state coordinating with four N atoms. The DFT calculation of the possible MnNxCy configurations proved that the MnN4 structure is the most stable site for the ORR in acids, which may be attributed to the formation of strong coordination bonds between the Mn and pyridinic-N via EMSI. Meanwhile, the corrosion resistance of carbon-based supports in SACs was further evaluated by cycling tests at a high potential range (1.0–1.5 V). After 5000 cycles, the 20Mn–NC-second and 30Mn–NC-second catalysts exhibited fewer capacitance increments than the 0Mn–NC-second and the 20Fe–NC-second catalyst, indicating the enhanced carbon stability of the as-prepared SACs. As illustrated in Fig. 6f, the TEM images after 5000 cycles showed no change in morphology, further proving the stability of carbon under the reaction conditions, which is attributed to the relatively high degree of graphitization that enhanced the corrosion resistance of the carbon.
As Pt-based catalysts have been widely investigated to show remarkable performance in the ORR, another long-standing problem regarding the CO tolerance for the ORR emerged as a major limitation to the actual utilization of fuel cells. Due to the strong adsorption of precious metals to CO, these ORR catalysts are easily poisoned by the presence of CO in the electrocatalytic reactions. Even the presence of 0.001 vol% CO impurities in oxygen and fuel would lead to loss of activity. The main principle to solve this issue is also to modify the electronic structure of electroactive sites that weaken the Pt–CO bond. For instance, Wu et al. synthesized neighboring dispersed Pt atoms anchored on ultrathin FePt nanosheets for the ORR with a loading of 6.7 wt%.89 The FePt NSs exhibited excellent durability performance with a retention of 73.9% mass activity after a 20000 cycle durability test, and especially strong CO tolerance (Fig. 6g–i). Further characterization indicated that the isolated Pt atoms have an oxidation valence between Pt0 and Pt4+ with the coordination of Pt–O and Pt–Fe bonds. The electronic interaction between Pt and FePt NSs effectively downshifted the Pt d band, which weakened the interaction between Pt atoms and the CO p orbital and thus decreased the adsorption of CO on the Pt atom surface. The DFT calculation revealed that the adsorption energy of the CO species on the FePt surface is smaller than that on a pure Pt surface, further indicating that the adsorption of CO on the FePt surface was weakened. But the FePt surface shows many OH species close to the Fe, thus promoting the elimination of CO by electro-oxidation. Except for the CO poisoning, methanol poisoning is also a key factor in catalyst deactivation. Li et al. designed Cu SACs with the single Cu atoms immobilized on the ultrathin nitrogenated 2D carbon matrix with a high loading of 20.9 wt%. The doped N atoms firmly coordinated with Cu atoms with the Cu–N2 and Cu–N4 structures during the synthetic process, which not only formed the strong bonding but also regulated the electronic structure. The prepared Cu–N–C catalyst exhibited superb resistance to methanol poisoning, as evidenced by the absence of obvious current loss after 2 mL methanol was added, and it also showed superior CO tolerance.94
4.2. OER
The oxygen evolution reaction (OER) is a half reaction in multiple electrochemical techniques,95 such as water analysis and rechargeable metal–air batteries. In general, the OER is limited by sluggish kinetics96 and requires a very high working potential,97 thus raising the need for high-performance and stable catalysts under harsh conditions. Although non-monatomic metallic catalysts such as Ir or Ru show remarkable OER activity, they suffer from low stability in catalytic environments. For instance, Ru is likely to react with oxygen in the lattice to form unstable RuO4, and the further escape of the RuO4 intermediate from the electrode will lead to the dissolution of the catalyst.98,99 The instability issues of the Ir electrode are very similar to those of Ru. The IrO3 intermediate formed in the OER will further react to give IrO42−, thus resulting in the transient dissolution and the peeling off of metals into the electrolyte and the degradation of catalysts.100 Due to the large surface energy of single metal atoms, the stability of SACs is relatively poor. Hence, a reasonable approach to suppress the dissolution of metals is anchoring the single metal atoms in a conductive support to construct a robust structure and enable precise control over the electronic structure of electroactive metal atoms associated with the coordination environment. For example, Cao et al. synthesized a single Ru atom confined in an acid-resistant N–C support as an efficient and stable catalyst for the OER in acidic solutions.101 In a chronoamperometry test at a high potential of 1.5 V, negligible degradation (5%) of Ru–N–C was detected after the 30 h operation, while the Ru dissolution ratio was only 5% shown in Fig. 7b, indicating the excellent stability of the Ru SACs. Besides, the as-prepared catalyst can operate continuously for 24 h without obvious performance degradation (Fig. 7c). The XAFS spectrum confirmed that Ru atoms mostly coordinated with four N atoms to form the Ru1–N4 structural configuration (Fig. 7a). To investigate the structural evolution of Ru–N–C during the OER, the operando XAFS was utilized to show that the average chemical valence state of Ru increased slightly between +3 and +4 valence states, and there was a slight constriction in Ru–N bonds under the working conditions, which further immobilized the Ru atom on the support surface, preventing dissolution under the working potentials, thus enhancing the stability of Ru SACs. To engineer the electronic structure of Ru atoms, Li's group reported a series of Ru SACs by utilizing different PtCu alloys as supports. The prepared catalysts exhibited great resistance to oxidation and subsequent dissolution of Ru1 (Fig. 7d).102 The DFT calculation implied that the Pt–Cu alloys could provide electrons to the reaction intermediates and hence avoided the over-oxidation and subsequent dissolution of Ru1.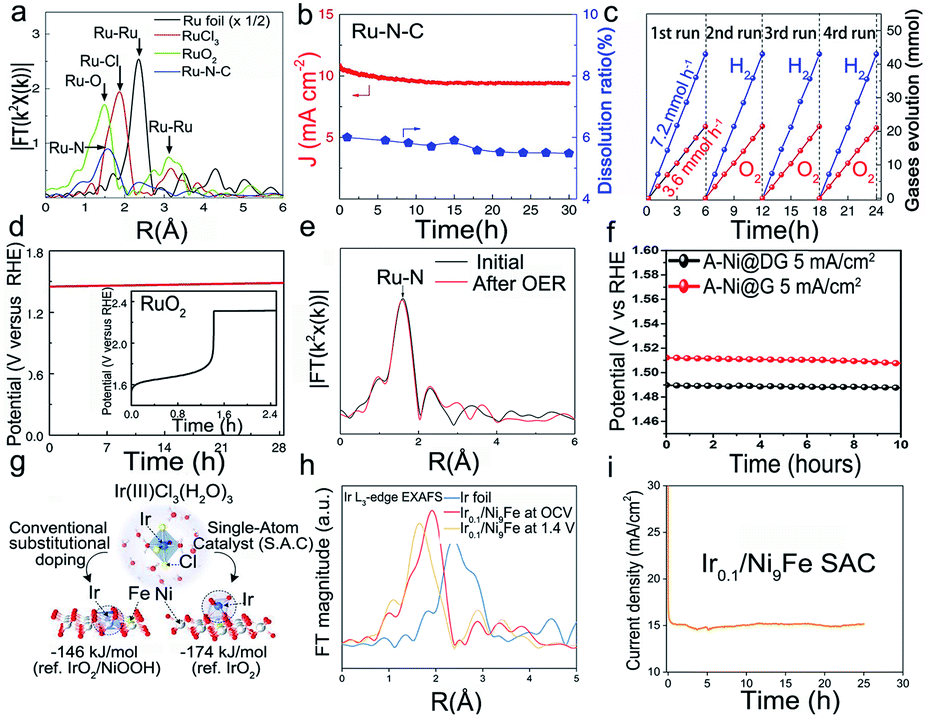 | ||
| Fig. 7 Electrocatalytic stability of SACs in the oxygen evolution reaction. (a) Fourier transform data for different catalysts, (b) Data of current density and Ru dissolution mass ratio with time for Ru–N–C (at 1.49 V vs. RHE) in 0.5 M H2SO4, (c) Time distributions of O2 and H2 evolution in overall water splitting. Reproduced with permission from ref. 101. Copyright Springer Nature 2019. (d) Chronoamperometry data for Ru1–Pt3Cu and RuO2 at 10 mA cm−2. Reproduced with permission from ref. 102. Copyright Springer Nature 2019. (e) The EXAFS data of Ru–N–C before and after the electrochemical test. Reproduced with permission from ref. 101. Copyright Springer Nature 2019. (f) The durability of A-Ni@DG and A-Ni@G at 5 mA cm−2 in 1 M KOH. Reproduced with permission from ref. 103. Copyright Elsevier 2018. (g) DFT calculation of the preferred Ir atom embedded in the NiFeOOH layered structure under operating conditions. (h) EXAFS data for Ir foil and Ir0.1/Ni9Fe. (i) Durability test of the Ir0.1/Ni9Fe SAC at 1.43 V (vs. RHE) for 100 hours. Reproduced with permission from ref. 104. Copyright PNAS 2021. | ||
Moreover, the single atom agglomeration also deactivates the SACs. For this purpose, Cao et al. also tested the morphology and structure of Ru–N–C before and after long-term operation, as detected by TEM, XRD, and XAFS (Fig. 7e).101 No obvious change was observed, indicating the good resistance of the Ru–N–C catalyst to agglomeration. Zhang et al. reported graphene defects to trap isolated Ni SACs through a defect engineering strategy.103 The carbon defects efficiently trapped atomic Ni atoms in electrochemical reactions as anchoring sites. The relevant experiment indicated that the unique vacancy and coordination environment could tune the electronic structure and strong charge transfer between the single Ni atoms and the carbon atoms, thus improving its stability in both acidic and alkaline solutions (Fig. 7f). Recently, Cui and co-workers reported Ir SACs in which Ir single-atom sites are immobilized on NiFe oxyhydroxides through an in situ reduction method (Fig. 7g). The synthesized Ir SACs exhibited superior OER performance, especially long-term durability for 100 h of operation in 1 M KOH solution (Fig. 7i). The in situ EXAFS spectra shown in Fig. 7h indicated that Ir retained its atomic dispersion under catalytic conditions.104
4.3. HER
As one of two half-reactions in water splitting, the hydrogen evolution reaction (HER) is regarded as one of the promising methods to produce hydrogen.105 HER generally occurs at a relatively low potential, so the oxidation of active sites may not happen as easily as in the ORR and OER. Besides, the HER process usually happens under acid/alkaline conditions, which may cause structural instability such as aggregation, dissolution, and electronic structure change, thus reducing the instability of SACs during the catalytic reaction.Due to the high activity of isolated metal atoms and harsh conditions (commonly in acid or alkaline solution), SACs, especially Pt-free catalysts, would aggregate and dissolve, making them suffer from low stability. Designing a stable structure and stabilizing the components are essential to improve the stability of SACs. Chen and co-workers have devised a single Ni-doped nanoporous graphene catalyst that exhibited an excellent stability of more than 120 hours, and the overpotential was about 45 mV in 0.5 M H2SO4 (Fig. 8a–c).33 As evidenced by the overlapped projected density of states (pDOS) projection, the charge transfer between nickel and the neighboring carbon atoms indicated the strong bonding of nickel and carbon atoms, thus resulting in the excellent stability of Ni SACs in acidic solutions. But compared to non-monatomic Ni, it indicated that once the residual Ni metallic particle existed in Ni-doped graphene, the stability would decrease, which may be attributed to the dissolution of metallic Ni0.33 Recently, some studies have reported that the doping of transition metal phosphides (TMPs) with appropriate elements can improve stability.106 For instance, Guan and co-workers have constructed a W-doped CoP nanoarray (W–CoP) catalyst. In order to investigate the stability of W–CoP, they performed the HER tests in different pH solutions, and the results showed that W–CoP has an excellent stability.107 The following SEM image after HER measurements indicated that W–CoP could maintain the initial morphological structure.
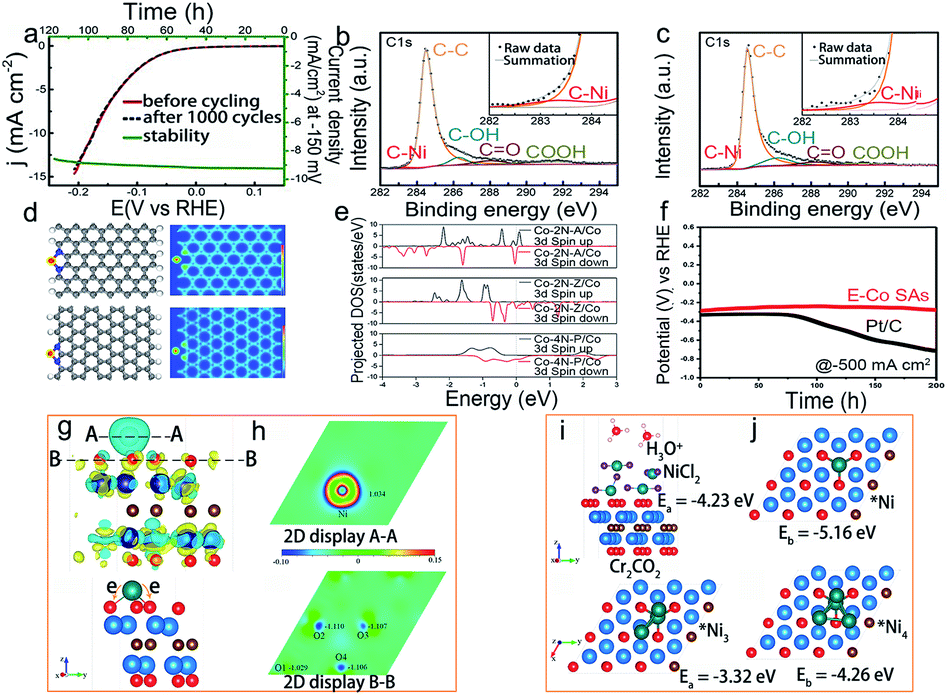 | ||
| Fig. 8 Electrocatalytic stability of SACs in the hydrogen evolution reaction. (a) Stability test of nickel doped graphene at 150 mV for 120 hours. XPS spectra of Ni-doped graphene (b) before and (c) after 1000 cycle measurement. Reproduced with permission from ref. 33. Copyright John Wiley and Sons 2015. (d) Structure and electron localization function for Co–2N-A and Co–2N-Z, respectively, (e) pDOS of Co in Co–2N-A, Co–2N-Z and Co–4N–P, respectively, and (f) stability test of E-Co SAs and Pt/C at −500 mA cm−2 for 200 h. Reproduced with permission from ref. 112. Copyright John Wiley and Sons 2021. (g) Schematic illustration of charge density on the Ni/Cr2CO2 surface, (h) charge transfer in Ni/Cr2CO2, and the adsorption structures and energy of (i) top: NiCl2, and bottom: Ni3 cluster, and (j) top: Ni single atom, and bottom: Ni4 clusters on the Cr2CO2 surface. Reproduced with permission from ref. 110. Copyright American Chemical Society 2019. | ||
Much attention has also been paid to control the electronic structure of SACs in electrocatalysis. Recently, defect engineering was utilized to design stable HER SACs through vacancies or dopants. Mu et al. designed an isolated MoRu pair (rGO–MoO3−x–MoRu) catalyst, which could be firmly stabilized by graphene and MoO3−x.108 The existence of ion holes distoring the neighboring atoms and the obvious electron delocalization guaranteed the stability of rGO–MoO3−x–MoRu, which eventually exhibited stability for more than 35 hours in both acid and alkaline solutions. Because O with fewer bound Ti is more stable and the formation of vacancies is beneficial to localize the structure of TiOx and promote the local electron transfer. Wang et al. designed a single atom doped TiO2 (TM1/TiO2) and controlled the charge transfer of lattice oxygen in TM1/TiO2 for HER application.109 Owing to the promising chemical stability of MXenes, Zhang's group110 used Cr2CO2 MXenes to design a single Ni catalyst. According to AIMD simulations at 600 K, Ni atoms anchored on Cr2CO2 would not aggregate into clusters, and using CI-NEB technology, they further indicated that the diffusion of Ni was difficult on anchor sites. Both AIMD simulations and CI-NEB results showed that Cr2CO2 MXenes are very stable (Fig. 8g–j). Similarly, some researchers have proposed a Co replaced MXene catalyst. The EXAFS results confirmed that Co atoms occupied Mo sites in the Mo2CTx. After the stability test, the oxidation state and morphology of Mo2CTx: Co were negligibly changed, indicating its stable electronic structure and morphology.111 For Co SACs, Lee et al.112 discovered that the zigzag and armchair edge sites are beneficial in stabilizing the Co sites (E-Co SAs) by DFT calculations. The unique and optimized electronic structures of E-Co SAs promoted the durability to 480 h under alkaline conditions (Fig. 8d–f). Moreover, Pt is typically a thermodynamically stable element compared to other transition metals due to its higher redox potential in electrocatalysis. However, several adverse structural evolutions may also occur after the long-term stability test; thus, some efforts have been devoted to solving this issue. For example, the isolated Pt SACs on the NiO/Ni support (PtSA–NiO/Ni) were reported as stable SACs for the HER in alkaline solutions. After a long HER test in 1 M KOH, no obvious changes in morphology and elemental distribution were detected. The Pt atoms remained dispersed on an atomic level which may be attributed to the existence of the charge delocalization of Pt toward O atoms and adjacent Ni atoms.113
4.4. CO2RR
The carbon dioxide reduction reaction (CO2RR) is a promising approach to convert carbon into value-added fuels and chemicals.3 Different from the ORR, OER, and HER, it usually occurs under neutral conditions (such as CO2 saturated 0.1 M KHCO3; pH is about 6.8) with a relatively more negative potential.114 Actually, single-atom metals are not in the 0 valent state due to the coordinated binding with ligands, and thus they are likely reduced to a metallic state at a negative potential.115 Recently, SACs have shown remarkable specific activity and selectivity for the CO2RR. However, they still suffer from insufficient stability. The instability behaviors of SACs such as aggregation, peeling off, and the change in electronic structure generally lead to the decrease in the current density and FE, which results in catalyst degradation in the CO2RR.Typically, because of the high surface energy in SACs, the isolated metal atoms are likely to aggregate during catalytic reactions. Constructing stable bonding between the single metal atoms and support is a valid solution. Liu and co-workers have calculated the binding energies between the different transition metal-tetracyanoquinodimethanes (TM-TCNQs) and the support. It was proved that the weak binding energy could induce the agglomeration of single metal atoms.116 Thus, the strong binding between the metal and support plays a key role in suppressing the aggregation issue. Liang et al. have developed a catalyst of Ni phthalocyanine molecules supported on carbon nanotubes (NiPc–OMe MDE).117 The catalyst exhibited excellent durability without aggregation issues (Fig. 9a and b). Compared to electron-withdrawing CN groups (NiPc MDE and NiPc–CN MDE), the enhanced stability in the electron-donating OMe group (NiPc–OMe MDE) may be attributed to the strong Ni–N bonds and accelerated CO desorption process (Fig. 9c). Besides, due to the strong bonding strength of Zn–N4 (Fig. 9d and e), SA–Zn/MNC showed a remarkable stability (Fig. 9f) with no obvious change in performance and morphology after 30 hours of operation.38 In addition, the instability of SACs is also reflected in CO tolerance.89 Some computational studies have indicated that the instability of FePc/CNT may be attributed to the strong binding ability with CO.118 For example, the FECO of FePc/CNT was larger than 98% from −0.44 to −0.52 V; however, the durability of the catalyst was poor, as evidenced by the change in potential from −0.58 to −0.78 V after 40 min catalytic reaction at −4.0 mA cm−2.119
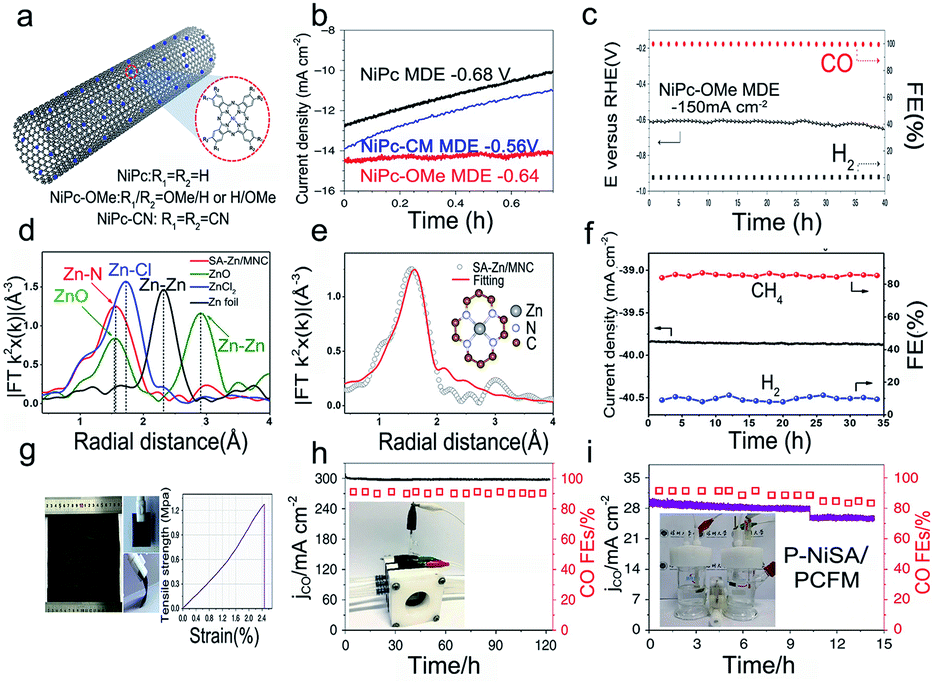 | ||
| Fig. 9 Electrocatalytic stability of SACs in the CO2 reduction reaction. (a) Schematic illustration of the NiPc MDEs, (b) The stability of the NiPc–OMe MDE, NiPc MDE, and NiPc–CN MDE via chronoamperometry, respectively. (c) Long-term durability test of the NiPc–OMe MDE. Reproduced with permission from ref. 117. Copyright Springer Nature 2020. (d) FT-EXAFS spectra, and (e) the Zn K-edge plot of SA–Zn/MNC. The inset image illustrates the atomic structure of Zn–N4. (f) Durability measurement of the ERC for SA–Zn/MNC at −1.8 V (vs. SCE). Reproduced with permission from ref. 38. Copyright American Chemical Society 2020. (g) Digital images and stress–strain plot of the NiSA/PCFM membrane. (h) Long-term durability measurements in the GDE cell and (i) H cell at −1.0 V (vs. RHE). Reproduced with permission from ref. 121. Copyright Springer Nature 2020. | ||
Except for the aggregation issue, other instability behaviors should also be considered, such as the change in electronic configuration of the electroactive sites and peeling off of isolated metal atoms. Cao et al. designed a Ni–phthalocyanine-based COF (NiPc-COF) catalyst.120 The Ni SACs can maintain their CO2RR activity for more than 10 hours. After the long-term operation, there was no change in the crystallinity and morphology of NiPc-COF and valence state of Ni. The above results fully manifested the high stability of NiPc-COF anchored by pyrazine units. Although lots of SACs have exhibited excellent CO2RR performances, they are still a long way from the large-scale practical applications due to the poor long-term stability, especially at high current density. He et al. reported single Ni atom adorned carbon membranes (NiSA/PCFM), which showed a large current density of 308 mA cm−2. Significantly, the activity and selectivity of the NiSA/PCFM membrane were almost unchanged for about 120 hours (retaining 95% of the original value in Fig. 9g–i).121 In addition, they utilized the integrated carbon fiber membrane to synthesize SACs via the electrospinning method, which exhibited sufficient mechanical strength and flexibility. Compared to the facile coating method, the integrated SACs exhibited greater mechanical stability, enough to prevent the single metal atom from peeling off from the support, especially at large current density. In addition, due to the relatively negative reduction potential in the CO2RR, SACs are easily reduced to metallic clusters. For instance, the structure of Cu–Nx could be reduced to Cu nanoparticles.115 Compared to Cu–Nx in Cu1–N/CNT, FECO in Ni1–N/CNT could reach nearly 100%, and Ni1–N/CNT exhibited an outstanding stability at a full-cell voltage of 2.8 V. Interestingly, the bond length between isolated atoms and anchored sites might also play an important role in stability. Xie and co-workers constructed a relatively shorter Sn–N bond of Sn SACs, which maintained its structure during a 200 h durability test.122
5. Conclusions and prospects
Developing renewable energy techniques to meet energy requirements urgently requires the extensive development of low-cost, high-performance, and robust electrocatalysts. Downsizing the nanoparticle to the atomic level to increase the utilization of metal atoms can impart unique and attractive properties to electrocatalysts. For SACs in practical electrocatalytic applications, the high performance of SACs containing a large number of active sites has been proven, while the catalytic stability under the harsh reaction conditions has rarely been investigated. Therefore, a deep understanding of instability behaviors of SACs in different electrocatalytic reactions can guide the design of rational synthesis strategies to enhance catalytic stability. Up to now, the main principle for improving the stability of SACs has been to build strong bonds between the metal atoms and support to stabilize the single metal atoms. Meanwhile, the appropriate electron transfer between isolated metal atoms and the coordination ligands could also suppress the adverse structural evolution of SACs, which leads to the degradation of SACs. Herein, we summarized the recent advances in stable SACs in terms of metal and support materials, synthetic strategies, and catalytic stability in electrocatalysis.Although SACs have achieved attractive advances, there are still many challenges, including the following.
(1) The common problem is the focus on increasing the metal loading through controllable and facile synthetic methods. In general, the high performance of SACs depends on the high content of individual metal atoms to ensure plenty of active sites. However, the individual metal atoms are likely to aggregate due to their high surface energy, which also frequently occurs in the catalytic reactions, which result in the deactivation of SACs. To prevent aggregation of single metal atoms, current studies mainly focus on preparing SACs with low metal loading via rational synthetic methods, which hinders the development of SACs in large-scale applications. As a result, for practical applications, it is essential to explore facile, economical, and scalable synthetic strategies for the synthesis of SACs with a high loading amount.
(2) Investigating the structural and compositional evolution of SACs during the synthetic and electrocatalytic process is crucial for the design of stable SACs. An in-depth understanding of the evolution of SACs requires advanced in situ characterization technologies with a higher spatial and temporal resolution. In recent decades, in situ TEM has been rapidly developed due to the advances in nanofabrication technology, which offers a platform for investigating the structures and compositions of materials. In situ TEM characterization of SACs may be a convincing approach to offer a clear view of the evolution of SACs during the reaction, improving the understanding of the instability mechanism of SACs. Besides, the theoretical calculation is also an effective way to predict the dynamic behavior and possible structural evolution during catalysis. The binding energy between the metal atoms and support can be calculated to reflect the MSI, which is helpful for the design of stable SACs.
(3) Understanding the interaction between the single metal atoms and the support is beneficial for optimizing the activity and durability of SACs. In supported metal catalysts, all the active metal atoms generally directly interact with the support and coordinate with the surface ligands. Therefore, the properties of the support materials play a key role in determining the activity and stability of the SACs. SMSI can stabilize the single metal atoms by anchoring metal atoms on the support. Meanwhile, EMSI will efficiently tune the electronic structure of the electroactive sites to enhance the stability of SACs in electrocatalysis. However, the understanding of the MSI remains in its infancy owing to the challenge in exploring their effects from inner reaction parameters guided by their natures. The investigation of SMSI is critical to revealing their catalytic and stabilization mechanisms.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was sponsored by the Thousand Talents Program for Distinguished Young Scholars from the Chinese government, the National Key R&D Program of China (No. 2017YFB0406000), the National Natural Science Foundation of China (21875137, 51521004, and 51420105009), the Innovation Program of Shanghai Municipal Education Commission (Project No. 2019-01-07-00-02-E00069), the 111 Project (Project No. B16032), and the fund from the Center of Hydrogen Science and Joint Research Center for Clean Energy Materials at Shanghai Jiao Tong University.Notes and references
- X. Zou and Y. Zhang, Chem. Soc. Rev., 2015, 44, 5148–5180 RSC.
- J. Wu and H. Yang, Acc. Chem. Res., 2013, 46, 1848–1857 CrossRef CAS PubMed.
- L. Wang, W. Chen, D. Zhang, Y. Du, R. Amal, S. Qiao, J. Wu and Z. Yin, Chem. Soc. Rev., 2019, 48, 5310–5349 RSC.
- J. N. Tiwari, S. Sultan, C. W. Myung, T. Yoon, N. Li, M. Ha, A. M. Harzandi, H. J. Park, D. Y. Kim, S. S. Chandrasekaran, W. G. Lee, V. Vij, H. Kang, T. J. Shin, H. S. Shin, G. Lee, Z. Lee and K. S. Kim, Nat. Energy, 2018, 3, 773–782 CrossRef CAS.
- H. Liao, C. Wei, J. Wang, A. Fisher, T. Sritharan, Z. Feng and Z. J. Xu, Adv. Energy Mater., 2017, 7, 1701129 CrossRef.
- Y.-H. Fang and Z.-P. Liu, J. Am. Chem. Soc., 2010, 132, 18214–18222 CrossRef CAS PubMed.
- F. Li, G.-F. Han, H.-J. Noh, J.-P. Jeon, I. Ahmad, S. Chen, C. Yang, Y. Bu, Z. Fu, Y. Lu and J.-B. Baek, Nat. Commun., 2019, 10, 4060 CrossRef PubMed.
- B. Wu and N. Zheng, Nano Today, 2013, 8, 168–197 CrossRef CAS.
- X. Huang, Z. Zhao, L. Cao, Y. Chen, E. Zhu, Z. Lin, M. Li, A. Yan, A. Zettl, Y. M. Wang, X. Duan, T. Mueller and Y. Huang, Science, 2015, 348, 1230–1234 CrossRef CAS PubMed.
- S. Yang, R. Du, Y. Yu, Z. Zhang and F. Wang, Nano Energy, 2020, 77, 105057 CrossRef CAS.
- D. Wu, X. Wang, L. Shi, K. Jiang, M. Wang, C. Lu, Z. Chen, P. Liu, J. Zhang, D. Tranca, Y. Hou, Y. Chen and X. Zhuang, J. Mater. Chem. A, 2020, 8, 21661–21667 RSC.
- H. Mistry, R. Reske, Z. Zeng, Z.-J. Zhao, J. Greeley, P. Strasser and B. R. Cuenya, J. Am. Chem. Soc., 2014, 136, 16473–16476 CrossRef CAS PubMed.
- X. Li, X. Yang, Y. Huang, T. Zhang and B. Liu, Adv. Mater., 2019, 31, 1902031 CrossRef CAS PubMed.
- L. Liu, H. Su, F. Tang, X. Zhao and Q. Liu, Nano Energy, 2018, 46, 110–116 CrossRef CAS.
- B. Qiao, A. Wang, X. Yang, L. F. Allard, Z. Jiang, Y. Cui, J. Liu, J. Li and T. Zhang, Nat. Chem., 2011, 3, 634–641 CrossRef CAS PubMed.
- C. Zhu, S. Fu, Q. Shi, D. Du and Y. Lin, Angew. Chem., Int. Ed. Engl., 2017, 56, 13944–13960 CrossRef CAS PubMed.
- X. Li, A. E. Surkus, J. Rabeah, M. Anwar, S. Dastigir, H. Junge, A. Bruckner and M. Beller, Angew. Chem., Int. Ed. Engl., 2020, 59, 15849–15854 CrossRef CAS PubMed.
- E. F. Holby, G. Wang and P. Zelenay, ACS Catal., 2020, 10, 14527–14539 CrossRef CAS.
- J. Wang, S. Qi and M. Zhao, J. Phys. Chem. C, 2020, 124, 17675–17683 CrossRef CAS.
- R. Bliem, J. van der Hoeven, A. Zavodny, O. Gamba, J. Pavelec, P. E. de Jongh, M. Schmid, U. Diebold and G. S. Parkinson, Angew. Chem., Int. Ed. Engl., 2015, 54, 13999–14002 CrossRef CAS PubMed.
- G. S. Parkinson, Z. Novotny, G. Argentero, M. Schmid, J. Pavelec, R. Kosak, P. Blaha and U. Diebold, Nat. Mater., 2013, 12, 724–728 CrossRef CAS PubMed.
- R. Qin, P. Liu, G. Fu and N. Zheng, Small Methods, 2018, 2, 1700286 CrossRef.
- S. F. SJ Tauster and R. L. Garten, J. Am. Chem. Soc., 1978, 100, 170–175 CrossRef.
- J. Li, Q. Guan, H. Wu, W. Liu, Y. Lin, Z. Sun, X. Ye, X. Zheng, H. Pan, J. Zhu, S. Chen, W. Zhang, S. Wei and J. Lu, J. Am. Chem. Soc., 2019, 141, 14515–14519 CrossRef CAS PubMed.
- A. Han, B. Wang, A. Kumar, Y. Qin, J. Jin, X. Wang, C. Yang, B. Dong, Y. Jia, J. Liu and X. Sun, Small Methods, 2019, 3, 1800471 CrossRef.
- K. Qi, M. Chhowalla and D. Voiry, Mater. Today, 2020, 40, 173–192 CrossRef CAS.
- Z. Geng, Y. Liu, X. Kong, P. Li, K. Li, Z. Liu, J. Du, M. Shu, R. Si and J. Zeng, Adv. Mater., 2018, 30, 1803498 CrossRef PubMed.
- P. Liu, Y. Zhao, R. Qin, S. Mo, G. Chen, L. Gu, D. M. Chevrier, P. Zhang, Q. Guo, D. Zang, B. Wu, G. Fu and N. Zheng, Science, 2016, 352, 797–800 CrossRef CAS PubMed.
- X. Wang, W. Chen, L. Zhang, T. Yao, W. Liu, Y. Lin, H. Ju, J. Dong, L. Zheng, W. Yan, X. Zheng, Z. Li, X. Wang, J. Yang, D. He, Y. Wang, Z. Deng, Y. Wu and Y. Li, J. Am. Chem. Soc., 2017, 139, 9419–9422 CrossRef CAS PubMed.
- Y. Zuo, T. Li, N. Zhang, T. Jing, D. Rao, P. Schmuki, Š. Kment, R. Zbořil and Y. Chai, ACS Nano, 2021, 15, 7790–7798 CrossRef CAS PubMed.
- Y. Liu, X. Wu, Z. Li, J. Zhang, S. X. Liu, S. Liu, L. Gu, L. R. Zheng, J. Li, D. Wang and Y. Li, Nat. Commun., 2021, 12, 4205 CrossRef CAS PubMed.
- L. Zhang, D. Liu, Z. Muhammad, F. Wan, W. Xie, Y. Wang, L. Song, Z. Niu and J. Chen, Adv. Mater., 2019, 31, 1903955 CrossRef CAS PubMed.
- H. J. Qiu, Y. Ito, W. Cong, Y. Tan, P. Liu, A. Hirata, T. Fujita, Z. Tang and M. Chen, Angew. Chem., Int. Ed., 2015, 54, 14031–14035 CrossRef CAS PubMed.
- Z. Lu, B. Wang, Y. Hu, W. Liu, Y. Zhao, R. Yang, Z. Li, J. Luo, B. Chi and Z. Jiang, Angew. Chem., Int. Ed., 2019, 2648–2652 CrossRef.
- Y. Zhu, J. Sokolowski, X. Song, Y. He, Y. Mei and G. Wu, Adv. Energy Mater., 2020, 10, 1902844 CrossRef CAS.
- J. Zhang, Y. Zhao, C. Chen, Y.-C. Huang, C.-L. Dong, C.-J. Chen, R.-S. Liu, C. Wang, K. Yan, Y. Li and G. Wang, J. Am. Chem. Soc., 2019, 141, 20118–20126 CrossRef CAS PubMed.
- J. Zhang, C. Liu and B. Zhang, Small Methods, 2019, 3, 1800481 CrossRef.
- L. Han, S. Song, M. Liu, S. Yao, Z. Liang, H. Cheng, Z. Ren, W. Liu, R. Lin, G. Qi, X. Liu, Q. Wu, J. Luo and H. L. Xin, J. Am. Chem. Soc., 2020, 142, 12563–12567 CrossRef CAS PubMed.
- F. D. Speck, M. T. Y. Paul, F. Ruiz-Zepeda, M. Gatalo, H. Kim, H. C. Kwon, K. J. J. Mayrhofer, M. Choi, C. H. Choi, N. Hodnik and S. Cherevko, J. Am. Chem. Soc., 2020, 142, 15496–15504 CrossRef CAS PubMed.
- R. Lang, W. Xi, J. C. Liu, Y. T. Cui, T. Li, A. F. Lee, F. Chen, Y. Chen, L. Li, L. Li, J. Lin, S. Miao, X. Liu, A. Q. Wang, X. Wang, J. Luo, B. Qiao, J. Li and T. Zhang, Nat. Commun., 2019, 10, 234 CrossRef PubMed.
- H. Sun, Z. Yan, F. Liu, W. Xu, F. Cheng and J. Chen, Adv. Mater., 2020, 32, 1806326 CrossRef CAS PubMed.
- C. Aydin, J. Lu, N. D. Browning and B. C. Gates, Angew. Chem., Int. Ed. Engl., 2012, 51, 5929–5934 CrossRef CAS PubMed.
- Y. Zheng, Y. Jiao, Y. Zhu, Q. Cai, A. Vasileff, L. H. Li, Y. Han, Y. Chen and S.-Z. Qiao, J. Am. Chem. Soc., 2017, 139, 3336–3339 CrossRef CAS PubMed.
- C. Lu, R. Fang and X. Chen, Adv. Mater., 2020, 32, 1906548 CrossRef CAS PubMed.
- Q. Jiang, J. Zhang, H. Huang, Y. Wu and Z. Ao, J. Mater. Chem. A, 2020, 8, 287–295 RSC.
- S. Wei, Y. Wang, W. Chen, Z. Li, W.-C. Cheong, Q. Zhang, Y. Gong, L. Gu, C. Chen, D. Wang, Q. Peng and Y. Li, Chem. Sci., 2020, 11, 786–790 RSC.
- Z. Song, L. Zhang, K. Doyle-Davis, X. Fu, J.-L. Luo and X. Sun, Adv. Energy Mater., 2020, 10, 2001561 CrossRef CAS.
- M. Wang, H. Ji, S. Liu, H. Sun, J. Liu, C. Yan and T. Qian, Chem. Eng. J., 2020, 393, 124702 CrossRef CAS.
- H. Zhong, Q. Zhang, J. Wang, X. Zhang, X. Wei, Z. Wu, K. Li, F. Meng, D. Bao and J. Yan, ACS Catal., 2018, 8, 3965–3970 CrossRef CAS.
- C. H. Choi, C. Baldizzone, J.-P. Grote, A. K. Schuppert, F. Jaouen and K. J. J. Mayrhofer, Angew. Chem., Int. Ed., 2015, 54, 12753–12757 CrossRef CAS PubMed.
- S. Ji, Y. Chen, S. Zhao, W. Chen, L. Shi, Y. Wang, J. Dong, Z. Li, F. Li, C. Chen, Q. Peng, J. Li, D. Wang and Y. Li, Angew. Chem., Int. Ed. Engl., 2019, 58, 4271–4275 CrossRef CAS PubMed.
- G. Giannakakis, M. Flytzani-Stephanopoulos and E. C. H. Sykes, Acc. Chem. Res., 2019, 52, 237–247 CrossRef CAS PubMed.
- W. Chen, Y. Ma, F. Li, L. Pan, W. Gao, Q. Xiang, W. Shang, C. Song, P. Tao, H. Zhu, X. Pan, T. Deng and J. Wu, Adv. Funct. Mater., 2019, 29, 1904278 CrossRef CAS.
- M. T. Greiner, T. E. Jones, S. Beeg, L. Zwiener, M. Scherzer, F. Girgsdies, S. Piccinin, M. Armbrüster, A. Knop-Gericke and R. Schlögl, Nat. Chem., 2018, 10, 1008–1015 CrossRef CAS PubMed.
- L. Zhang, Y. Ren, W. Liu, A. Wang and T. Zhang, Natl. Sci. Rev., 2018, 5, 653–672 CrossRef CAS.
- G. Kyriakou, M. B. Boucher, A. D. Jewell, E. A. Lewis, T. J. Lawton, A. B. ber, H. L. Tierney, M. Flytzani-Stephanopoulos and E. Sykes, Science, 2012, 335, 1209 CrossRef CAS PubMed.
- M. T. Darby, E. C. H. Sykes, A. Michaelides and M. Stamatakis, Top. Catal., 2018, 61, 428–438 CrossRef CAS PubMed.
- J. Liu, F. R. Lucci, M. Yang, S. Lee, M. D. Marcinkowski, A. J. Therrien, C. T. Williams, E. C. H. Sykes and M. Flytzani-Stephanopoulos, J. Am. Chem. Soc., 2016, 138, 6396–6399 CrossRef CAS PubMed.
- G. X. Pei, X. Y. Liu, X. Yang, L. Zhang, A. Wang, L. Li, H. Wang, X. Wang and T. Zhang, ACS Catal., 2017, 7, 1491–1500 CrossRef CAS.
- M. Li, K. Duanmu, C. Wan, T. Cheng, L. Zhang, S. Dai, W. Chen, Z. Zhao, P. Li, H. Fei, Y. Zhu, R. Yu, J. Luo, K. Zang, Z. Lin, M. Ding, J. Huang, H. Sun, J. Guo, X. Pan, W. A. Goddard, P. Sautet, Y. Huang and X. Duan, Nat. Catal., 2019, 2, 495–503 CrossRef CAS.
- R. Shen, W. Chen, Q. Peng, S. Lu, L. Zheng, X. Cao, Y. Wang, W. Zhu, J. Zhang, Z. Zhuang, C. Chen, D. Wang and Y. Li, Chem, 2019, 5, 2099–2110 CAS.
- A. Wang, J. Li and T. Zhang, Nat. Rev. Chem., 2018, 2, 65–81 CrossRef CAS.
- B. Qiao, J.-X. Liang, A. Wang, C.-Q. Xu, J. Li, T. Zhang and J. J. Liu, Nano Res., 2015, 8, 2913–2924 CrossRef CAS.
- Y. Lu, S. Zhou, C.-T. Kuo, D. Kunwar, C. Thompson, A. S. Hoffman, A. Boubnov, S. Lin, A. K. Datye, H. Guo and A. M. Karim, ACS Catal., 2021, 11, 8701–8715 CrossRef CAS.
- E. J. Peterson, A. T. DeLaRiva, S. Lin, R. S. Johnson, H. Guo, J. T. Miller, J. Hun Kwak, C. H. F. Peden, B. Kiefer, L. F. Allard, F. H. Ribeiro and A. K. Datye, Nat. Commun., 2014, 5, 4885 CrossRef CAS PubMed.
- Z. Zhang, Y. Zhu, H. Asakura, B. Zhang, J. Zhang, M. Zhou, Y. Han, T. Tanaka, A. Wang, T. Zhang and N. Yan, Nat. Commun., 2017, 8, 16100 CrossRef CAS PubMed.
- R. Q. Zhang, C. E. Kim, B. D. Yu, C. Stampfl and A. Soon, Phys. Chem. Chem. Phys., 2013, 15, 19450–19456 RSC.
- S. Yang, J. Kim, Y. J. Tak, A. Soon and H. Lee, Angew. Chem., Int. Ed., 2016, 55, 2058–2062 CrossRef CAS PubMed.
- C. K. Poh, S. H. Lim, J. Lin and Y. P. Feng, J. Phys. Chem. C, 2014, 118, 13525–13538 CrossRef CAS.
- L. Lin, W. Zhou, R. Gao, S. Yao, X. Zhang, W. Xu, S. Zheng, Z. Jiang, Q. Yu, Y.-W. Li, C. Shi, X.-D. Wen and D. Ma, Nature, 2017, 544, 80–83 CrossRef CAS PubMed.
- K. Qi, X. Cui, L. Gu, S. Yu, X. Fan, M. Luo, S. Xu, N. Li, L. Zheng, Q. Zhang, J. Ma, Y. Gong, F. Lv, K. Wang, H. Huang, W. Zhang, S. Guo, W. Zheng and P. Liu, Nat. Commun., 2019, 10, 5231 CrossRef PubMed.
- H. Yang, L. Shang, Q. Zhang, R. Shi, G. I. N. Waterhouse, L. Gu and T. Zhang, Nat. Commun., 2019, 10, 4585 CrossRef PubMed.
- J. Li, L. Zhang, K. Doyle-Davis, R. Li and X. Sun, Carbon Energy, 2020, 2, 488–520 CrossRef CAS.
- J. Yang, W. Li, D. Wang and Y. Li, Adv. Mater., 2020, 32, 2003300 CrossRef CAS PubMed.
- C. Zhao, X. Dai, T. Yao, W. Chen, X. Wang, J. Wang, J. Yang, S. Wei, Y. Wu and Y. Li, J. Am. Chem. Soc., 2017, 139, 8078–8081 CrossRef CAS PubMed.
- X. Fang, Q. Shang, Y. Wang, L. Jiao, T. Yao, Y. Li, Q. Zhang, Y. Luo and H.-L. Jiang, Adv. Mater., 2018, 30, 1705112 CrossRef PubMed.
- E. Zhang, T. Wang, K. Yu, J. Liu, W. Chen, A. Li, H. Rong, R. Lin, S. Ji, X. Zheng, Y. Wang, L. Zheng, C. Chen, D. Wang, J. Zhang and Y. Li, J. Am. Chem. Soc., 2019, 141, 16569–16573 CrossRef CAS PubMed.
- W. Chen, J. Pei, C.-T. He, J. Wan, H. Ren, Y. Wang, J. Dong, K. Wu, W.-C. Cheong, J. Mao, X. Zheng, W. Yan, Z. Zhuang, C. Chen, Q. Peng, D. Wang and Y. Li, Adv. Mater., 2018, 30, 1800396 CrossRef PubMed.
- L. Jiao, R. Zhang, G. Wan, W. Yang, X. Wan, H. Zhou, J. Shui, S.-H. Yu and H.-L. Jiang, Nat. Commun., 2020, 11, 2831 CrossRef CAS PubMed.
- J. Wan, W. Chen, C. Jia, L. Zheng, J. Dong, X. Zheng, Y. Wang, W. Yan, C. Chen, Q. Peng, D. Wang and Y. Li, Adv. Mater., 2018, 30, 1705369 CrossRef PubMed.
- H.-J. Zhu, M. Lu, Y.-R. Wang, S.-J. Yao, M. Zhang, Y.-H. Kan, J. Liu, Y. Chen, S.-L. Li and Y.-Q. Lan, Nat. Commun., 2020, 11, 497 CrossRef CAS PubMed.
- J. Zhang, H. Yang and B. Liu, Adv. Energy Mater., 2021, 11, 2002473 CrossRef CAS.
- Y. Ren, Y. Tang, L. Zhang, X. Liu, L. Li, S. Miao, D. Sheng Su, A. Wang, J. Li and T. Zhang, Nat. Commun., 2019, 10, 4500 CrossRef PubMed.
- X. Wang, Z. Chen, X. Zhao, T. Yao, W. Chen, R. You, C. Zhao, G. Wu, J. Wang and W. Huang, Angew. Chem., Int. Ed., 2018, 1962–1966 CrossRef.
- K. Jiang, B. Liu, M. Luo, S. Ning, M. Peng, Y. Zhao, Y.-R. Lu, T.-S. Chan, F. M. F. de Groot and Y. Tan, Nat. Commun., 2019, 10, 1743 CrossRef PubMed.
- L. Zhang, R. Si, H. Liu, N. Chen, Q. Wang, K. Adair, Z. Wang, J. Chen, Z. Song, J. Li, M. N. Banis, R. Li, T.-K. Sham, M. Gu, L.-M. Liu, G. A. Botton and X. Sun, Nat. Commun., 2019, 10, 4936 CrossRef PubMed.
- H. A. Gasteiger, S. S. Kocha, B. Sompalli and F. T. Wagner, Appl. Catal., B, 2005, 56, 9–35 CrossRef CAS.
- F. T. Wagner, B. Lakshmanan and M. F. Mathias, J. Phys. Chem. Lett., 2010, 1, 2204–2219 CrossRef CAS.
- W. Chen, W. Gao, P. Tu, T. Robert, Y. Ma, H. Shan, X. Gu, W. Shang, P. Tao, C. Song, T. Deng, H. Zhu, X. Pan, H. Yang and J. Wu, Nano Lett., 2018, 18, 5905–5912 CrossRef CAS PubMed.
- Z. Song, Y.-N. Zhu, H. Liu, M. N. Banis, L. Zhang, J. Li, K. Doyle-Davis, R. Li, T.-K. Sham, L. Yang, A. Young, G. A. Botton, L.-M. Liu and X. Sun, Small, 2020, 16, 2003096 CrossRef CAS PubMed.
- Y. Chen, R. Gao, S. Ji, H. Li, K. Tang, P. Jiang, H. Hu, Z. Zhang, H. Hao, Q. Qu, X. Liang, W. Chen, J. Dong, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2021, 60, 3212–3221 CrossRef CAS PubMed.
- D. Banham, S. Ye, K. Pei, J.-i. Ozaki, T. Kishimoto and Y. Imashiro, J. Power Sources, 2015, 285, 334–348 CrossRef CAS.
- J. Li, M. Chen, D. A. Cullen, S. Hwang, M. Wang, B. Li, K. Liu, S. Karakalos, M. Lucero, H. Zhang, C. Lei, H. Xu, G. E. Sterbinsky, Z. Feng, D. Su, K. L. More, G. Wang, Z. Wang and G. Wu, Nat. Catal., 2018, 1, 935–945 CrossRef CAS.
- F. Li, G.-F. Han, H.-J. Noh, S.-J. Kim, Y. Lu, H. Y. Jeong, Z. Fu and J.-B. Baek, Energy Environ. Sci., 2018, 11, 2263–2269 RSC.
- Q. Zhang, H. Zhong, F. Meng, D. Bao, X. Zhang and X. Wei, Nano Res., 2018, 11, 1294–1300 CrossRef CAS.
- H. Zhong, J. Wang, F. Meng and X. Zhang, Angew. Chem., Int. Ed., 2016, 55, 10091–10095 CrossRef.
- J. Wang, H.-x. Zhong, Y.-l. Qin and X.-b. Zhang, Angew. Chem., Int. Ed., 2013, 52, 5248–5253 CrossRef CAS PubMed.
- S. Cherevko, Curr. Opin. Electrochem., 2018, 8, 118–125 CrossRef CAS.
- T. Reier, M. Oezaslan and P. Strasser, ACS Catal., 2012, 2, 1765–1772 CrossRef CAS.
- P. Jovanovič, N. Hodnik, F. Ruiz-Zepeda, I. Arčon, B. Jozinović, M. Zorko, M. Bele, M. Šala, V. S. Šelih, S. Hočevar and M. Gaberšček, J. Am. Chem. Soc., 2017, 139, 12837–12846 CrossRef PubMed.
- L. Cao, Q. Luo, J. Chen, L. Wang, Y. Lin, H. Wang, X. Liu, X. Shen, W. Zhang, W. Liu, Z. Qi, Z. Jiang, J. Yang and T. Yao, Nat. Commun., 2019, 10, 4849 CrossRef PubMed.
- Y. Yao, S. Hu, W. Chen, Z.-Q. Huang, W. Wei, T. Yao, R. Liu, K. Zang, X. Wang, G. Wu, W. Yuan, T. Yuan, B. Zhu, W. Liu, Z. Li, D. He, Z. Xue, Y. Wang, X. Zheng, J. Dong, C.-R. Chang, Y. Chen, X. Hong, J. Luo, S. Wei, W.-X. Li, P. Strasser, Y. Wu and Y. Li, Nat. Catal., 2019, 2, 304–313 CrossRef CAS.
- L. Zhang, Y. Jia, G. Gao, X. Yan, N. Chen, J. Chen, M. T. Soo, B. Wood, D. Yang, A. Du and X. Yao, Chem, 2018, 4, 285–297 CAS.
- X. Zheng, J. Tang, A. Gallo, J. A. Garrido Torres, X. Yu, C. J. Athanitis, E. M. Been, P. Ercius, H. Mao, S. C. Fakra, C. Song, R. C. Davis, J. A. Reimer, J. Vinson, M. Bajdich and Y. Cui, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2101817118 CrossRef CAS PubMed.
- Y. Wu, F. Li, W. Chen, Q. Xiang, Y. Ma, H. Zhu, P. Tao, C. Song, W. Shang, T. Deng and J. Wu, Adv. Mater., 2018, 30, 1803151 CrossRef PubMed.
- T. Liu, X. Ma, D. Liu, S. Hao, G. Du, Y. Ma, A. M. Asiri, X. Sun and L. Chen, ACS Catal., 2017, 7, 98–102 CrossRef CAS.
- J. Wu, N. Han, S. Ning, T. Chen, C. Zhu, C. Pan, H. Wu, S. J. Pennycook and C. Guan, ACS Sustainable Chem. Eng., 2020, 8, 14825–14832 CrossRef CAS.
- S. Liu, C. Chen, Y. Zhang, Q. Zheng, S. Zhang, X. Mu, C. Chen, J. Ma and S. Mu, J. Mater. Chem. A, 2019, 7, 14466–14472 RSC.
- D. Yi, F. Lu, F. Zhang, S. Liu, B. Zhou, D. Gao, X. Wang and J. Yao, Angew. Chem., Int. Ed., 2020, 59, 15855–15859 CrossRef CAS PubMed.
- Y. Cheng, J. Dai, Y. Song and Y. Zhang, ACS Appl. Energy Mater., 2019, 2, 6851–6859 CrossRef CAS.
- D. A. Kuznetsov, Z. Chen, P. V. Kumar, A. Tsoukalou, A. Kierzkowska, P. M. Abdala, O. V. Safonova, A. Fedorov and C. R. Müller, J. Am. Chem. Soc., 2019, 141, 17809–17816 CrossRef CAS PubMed.
- X. Liu, L. Zheng, C. Han, H. Zong, G. Yang, S. Lin, A. Kumar, A. R. Jadhav, N. Q. Tran, Y. Hwang, J. Lee, S. Vasimalla, Z. Chen, S.-G. Kim and H. Lee, Adv. Funct. Mater., 2021, 31, 2100547 CrossRef CAS.
- K. L. Zhou, Z. Wang, C. B. Han, X. Ke, C. Wang, Y. Jin, Q. Zhang, J. Liu, H. Wang and H. Yan, Nat. Commun., 2021, 12, 3783 CrossRef CAS PubMed.
- Q. Xiang, F. Li, J. Wang, W. Chen, Q. Miao, Q. Zhang, P. Tao, C. Song, W. Shang, H. Zhu, T. Deng and J. Wu, ACS Appl. Mater. Interfaces, 2021, 13, 10837–10844 CrossRef CAS PubMed.
- S. Jin, Y. Ni, Z. Hao, K. Zhang, Y. Lu, Z. Yan, Y. Wei, Y.-R. Lu, T.-S. Chan and J. Chen, Angew. Chem., Int. Ed., 2020, 59, 21885–21889 CrossRef CAS PubMed.
- J.-H. Liu, L.-M. Yang and E. Ganz, J. Mater. Chem. A, 2019, 7, 3805–3814 RSC.
- X. Zhang, Y. Wang, M. Gu, M. Wang, Z. Zhang, W. Pan, Z. Jiang, H. Zheng, M. Lucero, H. Wang, G. E. Sterbinsky, Q. Ma, Y.-G. Wang, Z. Feng, J. Li, H. Dai and Y. Liang, Nat. Energy, 2020, 5, 684–692 CrossRef CAS.
- W. Ju, A. Bagger, G.-P. Hao, A. S. Varela, I. Sinev, V. Bon, B. Roldan Cuenya, S. Kaskel, J. Rossmeisl and P. Strasser, Nat. Commun., 2017, 8, 944 CrossRef PubMed.
- Z. Jiang, Y. Wang, X. Zhang, H. Zheng, X. Wang and Y. Liang, Nano Res., 2019, 12, 2330–2334 CrossRef CAS.
- M.-D. Zhang, D.-H. Si, J.-D. Yi, S.-S. Zhao, Y.-B. Huang and R. Cao, Small, 2020, 16, 2005254 CrossRef CAS PubMed.
- H. Yang, Q. Lin, C. Zhang, X. Yu, Z. Cheng, G. Li, Q. Hu, X. Ren, Q. Zhang, J. Liu and C. He, Nat. Commun., 2020, 11, 593 CrossRef CAS PubMed.
- X. Zu, X. Li, W. Liu, Y. Sun, J. Xu, T. Yao, W. Yan, S. Gao, C. Wang, S. Wei and Y. Xie, Adv. Mater., 2019, 31, 1808135 CrossRef PubMed.
- J. He, N. Li, Z. -G, M. Zhong, Z. -X. Fu, M. Liu, J. -C. Yin, Z. Shen, W. Li, J. Zhang, Z. Chang and X. -H. Bu, Adv. Funct. Mater., 2021, 31, 2103597 CrossRef CAS.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |