
Advances in studies of the structural effects of supported Ni catalysts for CO2 hydrogenation: from nanoparticle to single atom catalyst
Zhitao
Zhang
a,
Chenyang
Shen
a,
Kaihang
Sun
a,
Xinyu
Jia
a,
Jingyun
Ye
 *b and
Chang-jun
Liu
*a
*b and
Chang-jun
Liu
*a
aSchool of Chemical Engineering and Technology, Tianjin University, Tianjin, 300072, China. E-mail: cjL@tju.edu.cn
bDepartment of Chemistry and Biomolecular Science, Clarkson University, Potsdam, New York 13699, USA. E-mail: jingyun.ye324@gmail.com
First published on 3rd February 2022
Abstract
Supported Ni catalysts are promising for CO2 hydrogenation because of their relatively cheap price with comparable activity to noble metal catalysts. The product of CO2 hydrogenation over a supported Ni catalyst can theoretically be methane, CO, methanol and formic acid. The electronic and geometric structures of a supported Ni catalyst have a significant effect on the activity and selectivity of CO2 hydrogenation. Supported single nickel atom catalysts are found to tend to form carbon monoxide, methanol and, theoretically, formic acid. The selectivity depends on the support. The supported nickel cluster on indium oxide and In2O3–ZrO2 is highly selective for methanol synthesis. Supported nickel nanoparticles are normally good catalysts for methane formation at reasonably low temperatures. However, the structural effect of the supported Ni catalyst on CO2 hydrogenation has not been well investigated. The mechanism is still in debate. To further improve the activity and stability with tunable selectivity, the structure control of the Ni catalyst is very necessary for CO2 hydrogenation, which is highly structure sensitive. In this review, recent advances in the understanding of the structural effects of supported Ni catalysts on CO2 hydrogenation are summarized, including theoretical studies, operando or in situ catalyst characterization and experimental studies. Future development is therefore finally addressed.
 Zhitao Zhang | Zhitao Zhang is currently a PhD candidate in the School of Chemical Engineering and Technology at Tianjin University, China. His research topic is the preparation and characterization of supported nickel catalysts for CO2 hydrogenation and the desulfurization of gaseous hydrocarbons. He has published two papers in Catalysis Communications and Chinese Chemical Letters and co-authored four other papers. |
 Chenyang Shen | Chenyang Shen is currently a PhD candidate in the School of Chemical Engineering and Technology at Tianjin University, China. His research topic is the theoretical and experimental studies of supported nickel and iridium catalysts for CO2 hydrogenation. He has published two papers in ACS Catalysis and Journal of Energy Chemistry and co-authored eight other papers. |
 Kaihang Sun | Kaihang Sun is currently a PhD candidate in the School of Chemical Engineering and Technology at Tianjin University, China. His research topic is the theoretical and experimental studies of supported Ni, Pt and Ag catalysts for CO2 hydrogenation. He published the first pioneering experimental work on In2O3 for CO2 hydrogenation to methanol. He published another three papers in Green Chemistry, Green Energy & Environment and Journal of Physical Chemistry C and co-authored seven other papers. |
 Xinyu Jia | Xinyu Jia, a former PhD student of Tianjin University, is currently a lecturer at the Xi'an University of Science & Technology, China. Her research topic is the experimental studies of supported nickel catalysts for CO2 hydrogenation. She invented the Ni/In2O3 catalyst for CO2 hydrogenation to methanol. She has published three papers in Applied Catalysis B, Catalysis Today and Journal of Energy Chemistry and co-authored four other papers. |
 Jingyun Ye | Jingyun Ye is currently an Assistant Professor in the Department of Chemistry of Clarkson University. She obtained her PhD degree from Tianjin University in 2014. After that, she was a postdoctoral researcher at the University of Pittsburgh and the University of Minnesota before she joined Clarkson University in 2020. She carried out the pioneering theoretical study of In2O3 for CO2 hydrogenation to methanol and initiated the new research topic on In2O3 based catalysts for CO2 hydrogenation. She is also one of the first scientists who worked on MOF based heterogenous catalysts. Her research interest is to precisely design multi-functional materials and efficient catalysts for sustainable energy conversion and storage. She has published papers in Journal of the American Chemical Society, ACS Catalysis, Applied Catalysis B: Environmental, Journal of Catalysis, ChemCatChem, Catalysis Science & Technology, ChemComm, Journal of Physical Chemistry C and others. |
 Chang-jun Liu | Chang-jun Liu is a Chang Jiang Distinguished Professor and Fellow of the Royal Society of Chemistry. His research interests include CO2 utilization, natural gas conversion, plasma nanoscience, and 3D printing. He has been in the list of highly cited Chinese authors (Chemical Engineering) by Elsevier since 2014. He served as the 2010 Program Chair of Fuel Chemistry Division of American Chemical Society and Chair of the 10th International Conference on CO2 Utilization. He is in the editorial board of Applied Catalysis B, Journal of CO2Utilization and Chinese Journal of Catalysis. He is in the advisory board of Greenhouse Gases: Science & Technology. |
1. Introduction
With the rapid development of renewable energy, the heterogeneous hydrogenation of carbon dioxide (CO2) has attracted significantly increasing attention worldwide owing to its potential in large scale utilization of carbon dioxide.1–4 CO2 hydrogenation has also been considered to be promising for energy storage.5–7 Many catalysts have been exploited and reported for CO2 hydrogenation.8–22 Among all the catalysts exploited, supported nickel catalysts are promising because of their high activity and relatively low price.8–15 In addition, supported nickel catalysts have been employed in the industrial production of syngas (CO and hydrogen) via steam reforming of methane, a reverse reaction of CO2 hydrogenation to methane, which is normally called CO2 methanation and well-known as the Sabatier reaction.3,21 Theoretically, the product of CO2 hydrogenation over a supported nickel catalyst can be methane, methanol, carbon monoxide through the reverse water gas shift (RWGS) reaction, and formic acid. Methanol, formic acid and carbon monoxide are important intermediates for syntheses of various chemicals. Methanol can be directly used as fuel. CO2 methanation has potential applications in clean fuel production, energy storage, carbon recycling and water supply for spacecraft.23 Formic acid has been used for disinfection.24 It can be used to produce hydrogen for fuel cell applications24,25 because of its high hydrogen content (53 g L−1; 4.4 wt%).24The activity, selectivity and stability of supported Ni catalysts basically depend upon the electronic and geometric structures of the catalyst. A change in the electronic structure can cause a big change in the selectivity.26–35 One can shift the selectivity of CO2 hydrogenation from selective production of methane to methanol or CO or even formic acid by the change of the catalyst structure. The electronic structure of a supported Ni catalyst can be tuned by the physical structure and components, including the supporting material, of the catalyst. The size control of nickel catalysts is an effective approach to affect the geometric and electronic structures of the catalyst. The strong nickel–support interaction with the reducible oxides can be employed to adjust the electronic structure of the supported nickel catalyst. This can not only improve the catalytic activity but can also totally change the selectivity of the product of CO2 hydrogenation.
According to a literature survey, most reported studies of CO2 hydrogenation on supported Ni catalysts are for methane synthesis. With increasing studies on nickel cluster or single nickel atom catalysts (Ni-SACs), CO2 hydrogenation to methanol or formic acid attracts more and more interest. More progress can be expected for CO2 hydrogenation to methanol or formic acid over the supported nickel catalyst. Owing to the big changes in the electronic structures from nickel nanoparticles (NPs) to Ni-SACs, CO2 hydrogenation over the supported nickel catalyst is excellent for the study of structural effects on the catalytic properties. The supported Ni-SAC has much higher accessibility of all nickel atoms to CO2 and hydrogen. It can be an excellent connection between a homogeneous catalyst, with a well-defined structure, and a heterogeneous catalyst, which can be easily separated after the reaction with low cost both in the material and reaction operation for large scale applications.
We have to acknowledge that the structural effect of Ni catalysts on CO2 hydrogenation is complex. It has not been well investigated because the structure of the nickel catalyst is subject to dynamic changes with the involvement of the oxide support, various active sites and adsorbates of different reactivities during the reaction. The reaction mechanism with structure evolution is still in debate. In this review, we attempt to summarize recent advances in the studies of structural effects of supported Ni catalysts on CO2 hydrogenation to methane, carbon monoxide and methanol. CO2 hydrogenation to formic acid on a supported nickel catalyst only showed the theoretical possibility at present. We will briefly mention it during discussions on related issues. We focus on the theoretical understanding, operando study or in situ catalyst characterization and the structure–activity relationship, via the size effect (from nickel nanoparticles to Ni-SAC), of the supported nickel catalyst for heterogeneous CO2 hydrogenation thermally. However, the discussions here are helpful for the design and preparation of supported nickel catalysts for photo-catalytic, photo-thermal-catalytic and electro-catalytic reduction of CO2. Future developments are addressed finally.
2. Reactions in CO2 hydrogenation
The characteristics of CO2 molecules and the thermodynamic aspects of CO2 hydrogenation have been well discussed in review articles.8–20 Theoretically, CO2 hydrogenation can produce carbon monoxide, methane, methanol and formic acid,10,11 as shown in Table 1. Thermodynamically, CO2 methanation (reaction (4) in Table 1) is highly favoured (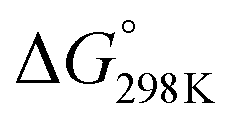 = −132.4 kJ mol−1). It has to be carried out at moderately low temperatures to limit the competitive endothermal RWGS reaction. This means that CO production from CO2 hydrogenation is normally carried out at elevated temperatures.11,36,37 A significant challenge is to improve the catalytic activity of CO2 methanation at low temperatures. CO2 hydrogenation to methanol (reaction (2) in Table 1) is also thermodynamically favoured (
= −132.4 kJ mol−1). It has to be carried out at moderately low temperatures to limit the competitive endothermal RWGS reaction. This means that CO production from CO2 hydrogenation is normally carried out at elevated temperatures.11,36,37 A significant challenge is to improve the catalytic activity of CO2 methanation at low temperatures. CO2 hydrogenation to methanol (reaction (2) in Table 1) is also thermodynamically favoured (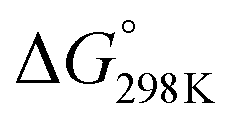 = −10.7 kJ mol−1) and exothermic. It has to be carried out at moderately low temperatures as well to avoid the reverse reaction and to inhibit the side RWGS reaction. CO2 hydrogenation to formic acid (reactions (7) and (8) in Table 1) is a little thermodynamically difficult. The catalyst for this reaction needs the aid of alkaline aqueous solution24 or unique support additives.32,33
= −10.7 kJ mol−1) and exothermic. It has to be carried out at moderately low temperatures as well to avoid the reverse reaction and to inhibit the side RWGS reaction. CO2 hydrogenation to formic acid (reactions (7) and (8) in Table 1) is a little thermodynamically difficult. The catalyst for this reaction needs the aid of alkaline aqueous solution24 or unique support additives.32,33
| No. | Reaction |
|
|
|---|---|---|---|
| 1 | CO2 + 3H2 ![[left over right harpoons]](https://www.rsc.org/images/entities/char_21cb.gif) CH3OH(g) + H2O(g) CH3OH(g) + H2O(g) |
−49.3 | +3.5 |
| 2 | CO2 + 3H2 CH3OH(l) + H2O(l) |
−137.8 | −10.7 |
| 3 | CO2 + 4H2 CH4 + 2H2O(g) |
−165.0 | −113.5 |
| 4 | CO2 + 4H2 CH4 + 2H2O(l) |
−259.9 | −132.4 |
| 5 | CO2 + H2 CO + H2O(g) |
+41.2 | +28.6 |
| 6 | CO2 + H2 CO + H2O(l) |
−6.2 | +19.2 |
| 7 | CO2 + H2 HCOOH(g) |
+43.5 | +14.9 |
| 8 | CO2 + H2 HCOOH(l) |
−31.0 | +34.3 |
| 9 | CO + 2H2 CH3OH(g) |
−90.6 | −29.1 |
| 10 | CO + 2H2 CH3OH(l) |
−131.6 | −29.9 |
| 11 | CO + 3H2 CH4 + H2O(g) |
−206.1 | −141.8 |
| 12 | CO + 3H2 CH4 + H2O(l) |
−229.7 | −165.1 |
| 13 | 2CO + 2H2 CH4 + CO2 |
−247.3 | −170.4 |
| 14 | CH4 C(s) + 2H2 |
+74.8 | +50.7 |
| 15 | 2CO C(s) + CO2 |
−172.4 | −119.7 |
| 16 | CO + H2 C(s) + H2O(g) |
−131.3 | −91.1 |
| 17 | CO + H2 C(s) + H2O(l) |
−178.7 | −100.7 |
| 18 | CO2 + 2H2 C(s) + 2H2O(g) |
−90.1 | −62.5 |
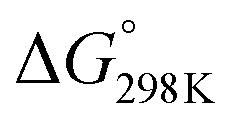
Prof. Chen and his co-workers presented a reaction network for CO2 hydrogenation to CO, CH3OH and CH4.10,15 This reaction network can be further modified based on our recent DFT studies of CO2 hydrogenation to methanol.22Fig. 1 shows the modified reaction network,22 which includes CO2 hydrogenation to formic acid via the formate route (right green path).
 | ||
| Fig. 1 The modified reaction network of CO2 hydrogenation over the supported nickel catalysts.22 (* means the adsorbed species). Copyright 2022, Elsevier. | ||
As seen in Fig. 1, a complex reaction network exists with various chemical intermediates on the catalyst. Methane, CO and methanol can be obtained from multiple routes. Two general pathways are derived from CO2 dissociation. One is the direct CO2 dissociation via the redox mechanism (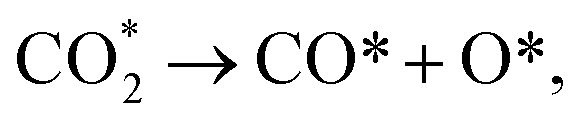 red paths). The other is the hydrogen-assisted CO2 dissociation, as shown in Fig. 1, either via formate (
red paths). The other is the hydrogen-assisted CO2 dissociation, as shown in Fig. 1, either via formate (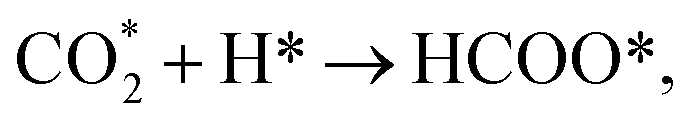 green paths) or carboxylate (
green paths) or carboxylate (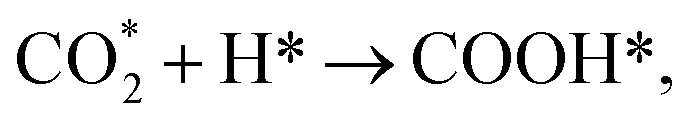 blue paths) intermediates, while H* is from H2 dissociation (H2 → H* + H*). The exact route principally depends on the catalyst structure.
blue paths) intermediates, while H* is from H2 dissociation (H2 → H* + H*). The exact route principally depends on the catalyst structure.
3. CO2 adsorption
The adsorption and activation of CO2 on the nickel surface play an important role in CO2 hydrogenation. The formation of adsorbed CO2 from gas phase CO2 can be the largest barrier for the dissociation of CO2(g).38 Two mechanisms for CO2 adsorption on the Ni catalyst exist. One is the dissociative adsorption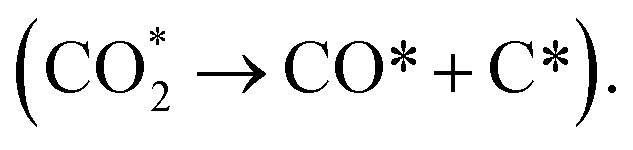 The other is the non-dissociative adsorption (CO2 + δe− → CO2δ−) to form a bent CO2δ− structural mode from the nickel to CO2 charge transfer.39 The ability of CO2 chemisorption on Ni surfaces was found in the order of Ni (211) > Ni (110) > Ni (100) > Ni (111), as shown in Table 2.40–50 The higher CO2 adsorption energies on the stepped Ni (211) was attributed to the stronger charge transfer on Ni (211) than on other low index surfaces.41 The results suggest that low coordinated Ni surfaces are more active for CO2 chemisorption with their stronger ability of electron donation. This also explains that smaller Ni clusters show stronger CO2 adsorption ability. For example, the CO2 adsorption energy on Ni13 is −1.25 eV (ref. 51) or −120.2 kJ mol−1 (ref. 51) and −100 kJ mol−1,52 while it is −58 kJ mol−1 on Ni55.52 These suggest that CO2 adsorption can be enhanced by increasing the defects (e.g., steps, edges, corners and doping with second metal atoms) of the Ni surfaces or decreasing the size of Ni nanoparticles.
The other is the non-dissociative adsorption (CO2 + δe− → CO2δ−) to form a bent CO2δ− structural mode from the nickel to CO2 charge transfer.39 The ability of CO2 chemisorption on Ni surfaces was found in the order of Ni (211) > Ni (110) > Ni (100) > Ni (111), as shown in Table 2.40–50 The higher CO2 adsorption energies on the stepped Ni (211) was attributed to the stronger charge transfer on Ni (211) than on other low index surfaces.41 The results suggest that low coordinated Ni surfaces are more active for CO2 chemisorption with their stronger ability of electron donation. This also explains that smaller Ni clusters show stronger CO2 adsorption ability. For example, the CO2 adsorption energy on Ni13 is −1.25 eV (ref. 51) or −120.2 kJ mol−1 (ref. 51) and −100 kJ mol−1,52 while it is −58 kJ mol−1 on Ni55.52 These suggest that CO2 adsorption can be enhanced by increasing the defects (e.g., steps, edges, corners and doping with second metal atoms) of the Ni surfaces or decreasing the size of Ni nanoparticles.
| E ads (eV) |
|
CO* | Ref. |
|---|---|---|---|
| a Calculated using Cambridge sequential total energy package (CASTEP). b Calculated using Spanish Initiative for Electronic Simulations with Thousands of Atoms (SIESTA). c Calculated using Material Studio (Dmol3). d Calculated using Quantum Espresso package (QE). | |||
| Ni (111) | 0.26 (q = −0.45) | −1.91 (q = −0.41) | 40 |
| −0.12 | −2.09 | 42 | |
| −0.01 | −1.93 | 43 | |
| −0.21 | 44 | ||
| −0.22 | −1.82 | 45 | |
| −0.06 | 47 | ||
| −0.24 | −2.05 | 49 | |
| −0.16 | −1.61 | 50 | |
| Ni (100) | −0.14 (q = −0.61) | −2.04 (q = −0.52) | 40 |
| −0.25 | −1.88 | 43 | |
| −0.22 | −1.81 | 45 | |
| Ni (110) | −0.42 (q = −0.67) | −1.94 (q = −0.33) | 40 |
| −0.40 | −1.87 | 45 | |
| −0.47 | 46 | ||
| −0.46 | −1.92 | 48 | |
| Ni (211) | −0.93 (q = −0.82) | 41 | |
| −0.31 | −2.09 | 42 | |
| −0.38 | −1.97 | 43 | |
| −0.35 | −1.82 | 45 | |
The support material has a significant effect on CO2 adsorption over a Ni catalyst.22,53–55 It depends on the interaction between nickel and the support. The loading of Ni clusters on yttria-stabilized zirconia (YSZ) can increase the CO2 adsorption energy from −0.37 eV on YSZ(111) to −1.16 eV on Ni4/YSZ(111).54 The nickel loading on CeO2(111) also causes an increase of the CO2 adsorption energy from −0.27 eV (CeO2(111)) to −0.79 eV (Ni/CeO2(111)).55 CO2 can be stabilized at the interface of the Ni clusters and CeO2 surface. The oxygen vacancy in CeO2 plays an important role with it.55 The oxygen vacancy has significant effects on the electronic structure and the properties of the Ni catalyst on the reducible oxide support, like CeO2 (ref. 55) and In2O3.22 The CO2 adsorption energy is −0.66 eV for Ni4/In2O3 with an oxygen vacancy, which it is −0.43 eV for the catalyst without an oxygen vacancy.22
The size of the nickel cluster on the support has a significant effect as well.54 If the nickel catalyst size is reduced to a SAC, CO2 dissociative adsorption on CeO2 supported Ni1 becomes difficult with a Gibbs free energy change of 0.76 eV, even in the presence of an oxygen vacancy.56 Alonso et al.57 found that a Ni-SAC, supported by MFI zeolite, adsorbs CO2 so strongly that CO2 cannot be dissociated to CO* and O*. It could hydrogenate CO2 to CO, via hydrogen-assisted CO2 dissociation (COOH pathway). However, there exists a high barrier (1.51 eV) rate limiting step, which suggests the high temperature needed (above 810 °C) to achieve the RWGS reaction.
Most reported studies on CO2 adsorption on nickel catalysts are based on theoretical methods. Recently, there has been some progress in the operando study of CO2 adsorption. Yuan et al.58 observed in operando the dissociation of CO2 on the Ni (111) surface at temperatures between 300 and 900 K via near ambient pressure X-ray photoelectron spectroscopy (NAP-XPS), corroborated by low energy electron diffraction (LEED) and scanning tunneling microscopy (STM) measurements. They found that CO2 quickly reacts with the clean nickel. Various Ni–O phases form even at room temperature: initially the p(2 × 2)-structured Chem–O (as shown in Fig. 2d), followed by epitaxial NiO (111) with a sodium chloride structure, and finally an oxygen enriched surface layer of Ni2O3 at the oxidative potential.58 The accumulation rate of the Ni–O phases under CO2 has a negative correlation with temperature. This suggests that the dynamic oxygen concentration is not limited by CO2 activation. The thermal decomposition of the Ni–O phases plays an important role. The Ni–O phases exhibit carbon-phobic properties due to repulsive forces to the *CO adsorbate and lower C–H bond scission rates than metallic Ni. This causes the intrinsic coke resistance.58 As shown in Fig. 2a–c, the products of the two adsorption modes (dissociative and non-dissociative) can be observed even at 300 K, except that the C 1s peak for *CO is absent.58 The reason for the absence of *CO and the presence of *CO2δ− was explained by the weakened bond between *CO and the oxidized surface.58 The content of *CO2δ− decreases with increasing temperature. This suggests its weak bonding to the surface or the dissociative adsorption as the domain mechanism.
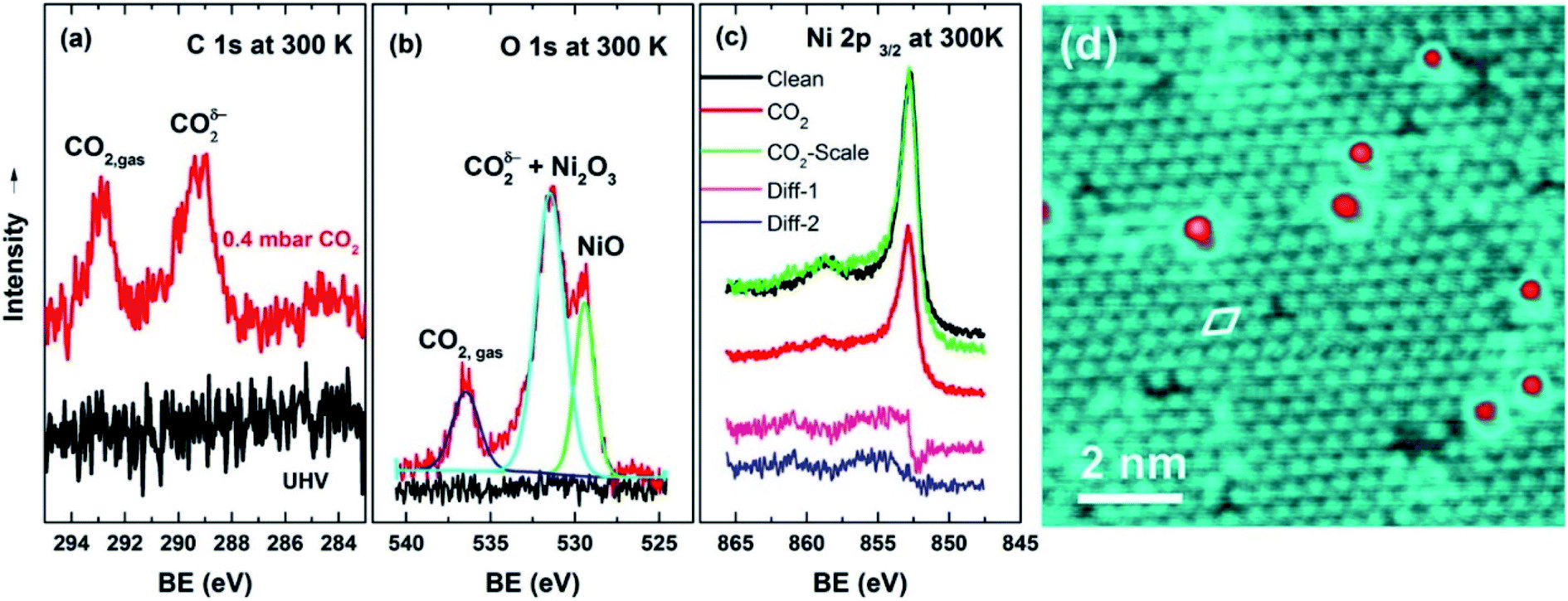 | ||
| Fig. 2 (a–c) NAP-XP spectra of C 1s, O 1s, and Ni 2p3/2 at 300 K. The spectra in black were measured in a ultra high vacuum (UHV), and those in red were measured under 0.4 mbar CO2. In (c), the green spectrum was obtained from rescaling the red one (0.4 mbar CO2) to be comparable with the black one (in UHV). The pink spectrum “Diff-1” was the difference spectrum between the green and the black, and the blue spectrum “Diff-2” was the difference spectrum with energy correction (the green shifted by 0.08 eV minus the black). (d) The STM image of a p(2 × 2) structure formed on Ni (111) after exposure to 100 L CO2 at room temperature. The unit cell is marked by a white diamond and was measured as 0.51 ± 0.03 nm. The bias voltages and tunneling currents were −0.2 V and 0.1 nA. The red dots here refer to the undesorbed *CO.58 Copyright 2016, American Chemical Society. | ||
4. CO2 hydrogenation to methane
CO2 methanation is exothermic (reactions (3) and (4) in Table 1). It is thermodynamically favoured. The supported nickel catalyst is active for CO2 methanation. Most reported Ni catalysts of CO2 hydrogenation were for methane synthesis. However, there exists a kinetic limitation at reasonably low temperatures for CO2 methanation on the supported Ni catalyst. The catalyst structure has a big effect on the activity and selectivity of the supported Ni catalyst for CO2 methanation. The mechanism for CO2 methanation on the supported Ni catalyst is still in debate, mostly because of the difficulty in the identification of the catalyst structure. The preparation of a Ni catalyst with a controllable structure remains a big challenge. Significant efforts are being made to reach the goal toward a supported nickel catalyst with a clear mechanism and enhanced low temperature activity for CO2 methanation.4.1 Theoretical studies
According to Table 2, the adsorption of CO on the Ni surface is very stable with an adsorption energy around −2.0 eV. Therefore, the reaction barrier for CO2 dissociation decides whether the reaction takes the CO (derived from direct CO2 dissociation) pathway or formate (derived from hydrogen-assisted CO2 dissociation) pathway. The corresponding reactions and their barriers are listed in Table 3.41,42,46,47,59–64 The strong binding of CO on the Ni catalyst can block the CO2 adsorption and H2 dissociation, which decreases the activity of the catalyst. In addition, the strongly bound CO species can be directly and indirectly dissociated to C species, which may result in the deactivation of the catalyst by carbon deposition over Ni particles.59 The dissociated adsorption of H2 on Ni surfaces is also important. The stronger dissociated adsorption of H2 makes the adsorption of H species compete with CO adsorption, which promotes the hydrogenation of CO or C to form methane. The weaker dissociated adsorption of H2 can facilitate the CO2 hydrogenation to form COOH* or HCOO* species. For example, the reaction barrier for CO2 dissociation on Ni (211) (Ea = 0.82 eV) is higher than that on Ni (111) (Ea = 0.57 eV) as shown in Table 3.41,42,46,47,59–64 The stronger dissociated adsorption of H2 on Ni (111) makes the adsorption of H species compete with CO adsorption, which promotes CO hydrogenation or C hydrogenation to form methane. The weaker dissociated adsorption of H2 on Ni (211) can facilitate CO2 hydrogenation to form COOH* or HCOO* species.| Surface |
|
CHO* → CO* + H* | CHO* → CH* + O* |
|
COOH* + H* → COHOH* | COHOH* + H2 → HCOH* + H2O |
|
HCOO* → CHO* + O* | CO* → C* + O* | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| a Calculated using SIESTA. b Calculated using Cambridge sequential total energy package (CASTEP). c Calculated using Simulation Tool for Atom Technology (STATE). d Calculated using DACAPO package. e Calculated based on atom superposition and electron delocalization molecular orbital (ASED-MO) theory. f Calculated using QUANTUM ESPRESSO Package (QE). | ||||||||||
| Ni (111) | 0.57(1.53) | 0.20(1.35) | 1.28(1.43) | 0.80(0.88) | 0.71(1.05) | 1.26(0.74) | 3.01(1.59) | 42 | ||
| 0.78(1.32) | 0.32(1.64) | 1.28(0.89) | 59 | |||||||
| 0.18(1.51) | 60 | |||||||||
| 0.31(1.59) | 61 | |||||||||
| 0.62 | 47 | |||||||||
| 2.97 | 62 | |||||||||
| Ni (100) | 0.53 | 63 | ||||||||
| 0.79(1.36) | 60 | |||||||||
| Ni (110) | 0.41 | 46 | ||||||||
| 0.70(0.28) | 0.34(0.27) | 0.24(1.17) | 64 | |||||||
| Ni (211) | 0.82(1.57) | 0.37(1.46) | 0.86(1.16) | 0.71(0.62) | 0.70(1.26) | 1.49(0.59) | 2.95(2.36) | 42 | ||
| 0.46(0.92) | 0.61(0.74) | 1.08(0.99) | 41 | |||||||
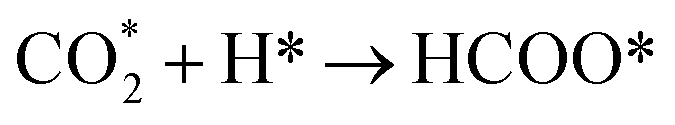
The hydrogenation of CO is favourable on low-index Ni surfaces, such as Ni (111), because the small barrier for CO2 dissociation and strong adsorption energy of CO on Ni (111) make CO the dominant species on Ni (111). As predicted by a DFT study, the chemisorption of CO2 on Ni (111) is not energetically favored.40 However, CO and atomic oxygen, products of CO2 dissociation, bind strongly to nickel. The full reaction pathway is shown in Fig. 1. Once the CO species is formed on the surface (as CO*), hydrogenation of CO to CHO* (Ea = 1.35 eV (ref. 42)) is more favourable than COH* (Ea = 1.81 eV (ref. 42)). Even COH*, formed on Ni (111), prefers to be dissociated to CO* because of the low backward reaction barrier (Ea = 0.85 eV (ref. 42)). Further dissociation of CHO* to CH* has a 1.28 eV barrier and the subsequent addition of H to CH* to form CH4 with small barriers65 (Ni (111): 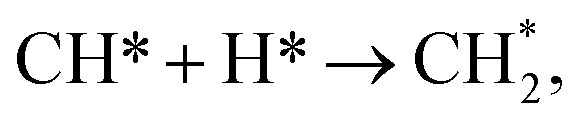 Ea = 0.52 eV (ref. 62) and
Ea = 0.52 eV (ref. 62) and 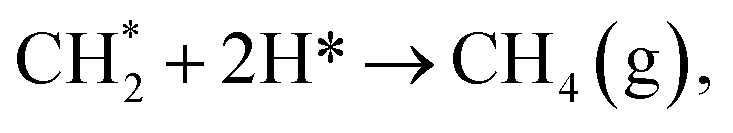 Ea = 0.50 eV (ref. 62)). Since CO2 methanation is an exothermic reaction, the heat accumulation from the reaction promotes the dissociation of CO (Ea = 3.01 eV (ref. 42)) or other intermediates, such as COH* (Ea = 2.07 eV (ref. 42)), CHO* (Ea = 1.28 eV (ref. 42)) and CH* (Ea = 1.32 eV (ref. 42)) to carbon species.
Ea = 0.50 eV (ref. 62)). Since CO2 methanation is an exothermic reaction, the heat accumulation from the reaction promotes the dissociation of CO (Ea = 3.01 eV (ref. 42)) or other intermediates, such as COH* (Ea = 2.07 eV (ref. 42)), CHO* (Ea = 1.28 eV (ref. 42)) and CH* (Ea = 1.32 eV (ref. 42)) to carbon species.
However, it is favourable for CO2 methanation over Ni (211) or Ni (110) to proceed through direct CO2 hydrogenation because of the lower barrier for CO2 hydrogenation to HCOO* (Ea = 0.70 eV (ref. 42 and 64)) than CO2 dissociation (Ea = 0.82 eV (ref. 42)) and weaker H binding energies than on Ni (111). CO2 hydrogenation to form COOH* with a barrier around 0.70 eV (ref. 42 and 64) is followed by the hydrogenation of COOH* to COHOH* (Ea = 0.34 eV (ref. 64)). Further dissociation of COHOH* to HCOH* has a 0.24 eV barrier.64 Hydrogenation of HCOH* to CH2OH* has a barrier of 1.00 eV (ref. 64) and further hydrogenation to 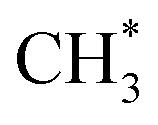 has a barrier of 1.47 eV.64 The reaction pathway through HCOO* to form CH4 presented in Fig. 1 is less favourable than through COOH* due to the higher barrier required for the hydrogenation of methoxy (CH3O*) to CH4.
has a barrier of 1.47 eV.64 The reaction pathway through HCOO* to form CH4 presented in Fig. 1 is less favourable than through COOH* due to the higher barrier required for the hydrogenation of methoxy (CH3O*) to CH4.
On the other hand, HCOO* is considered as the key intermediate for producing methanol, especially on In2O3 and Cu-based catalysts.66–71 Studies by Schatz and his co-workers72 suggested that the formation of HOCO*, HCOO*, 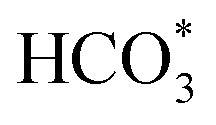 and CO* on Ni (110) is theoretically feasible during CO2 hydrogenation. The reaction temperature, CO2 coverage on the surface and the energy of the hydrogen atom determine which intermediates would be formed and which product would be obtained.72
and CO* on Ni (110) is theoretically feasible during CO2 hydrogenation. The reaction temperature, CO2 coverage on the surface and the energy of the hydrogen atom determine which intermediates would be formed and which product would be obtained.72
The support has a significant effect on CO2 methanation over the supported nickel catalyst.8–20,73,74 It has a big influence on the adsorption of the reactants, desorption, electron density of the supported Ni catalyst and Ni–support interactions. Among various supporting materials, CeO2 has been extensively investigated.10,15,73–78 CeO2 shows a better activity and a close to 100% selectivity toward CH4 than other supporting materials.77,78 The perfect CeO2 surface shows poor ability for CO2 or CO chemisorption or activation. The adsorption energies of CO2 and CO on CeO2(111) are −0.27 (ref. 55) and < −0.2 eV, respectively.79,80 CeO2 stabilizes the Ni particle and increases its activity by elongating the bond length of Ni–Ni when Ni is loaded on the CeO2 surface. CeO2 enhances the activation of CO2 at the interface of Ni particles and the CeO2 surface and promotes the adsorbed CO2 species to react with the H atoms on nickel to produce methane through the COOH reaction pathway.
The strong reducibility of CeO2 facilitates the creation of O vacancies. CeO2(110) and (111) surfaces with O vacancies favour CO oxidation but further oxidation of CO2 to carbonate is kinetically hindered due to too much electron accumulation on O vacancies.81 In addition, loading Ni clusters on the CeO2 surface was found to be favourable for the oxidization of C to CO and for further CO oxidation. CeO2 thus improves the resistance of carbon deposition from CO decomposition by facilitating the oxidization of C and CO. This leads to enhanced coke resistance for CO2 methanation.
4.2 Operando studies based on the single crystal
To study the mechanism of CO2 methanation over a supported Ni catalyst, an operando study is also important. By means of infrared-visible sum frequency generation (IR-vis SFG) vibrational spectroscopy and NAP-XPS, Roiaz et al.82 studied the hydrogenation of CO and CO2 on the Ni (110) single crystal at 10−1 mbar in situ as a function of the surface temperature in the range of 300–525 K. In addition to atomic carbon and precursors for graphenic carbon phases, five non-equivalent CO species have been distinguished, which confirms co-adsorption of H and C atoms, H-induced activation of CO, and surface reconstruction. At low temperatures, carbonate species are produced by the interaction of CO2 with atomic oxygen, which stems from the dissociation of *CO2 into *CO + *O. A metastable activated CO2− species is also detected. This species can be at the same time a precursor state toward dissociation into CO and O in the RWGS mechanism and a reactive species that undergoes direct hydrogenation conversion in the CO2 methanation process.82 This suggests that CO2 methanation occurs through the two parallel routes.Using AP-XPS, Heine et al.83 investigated the adsorption and reactions of CO2 and CO2 + H2 on the Ni (111) surface to identify the surface chemical state and the nature of the adsorbed species during the methanation reaction. In 200 mTorr CO2, they found that NiO is formed from CO2 dissociation into CO and atomic oxygen. Moreover, carbonate (CO32−) is present on the surface from further reaction of CO2 with NiO, as shown in Fig. 3. The addition of H2 into the reaction environment leads to the reduction of NiO and the disappearance of CO32−. They found that CO adsorbed on hollow sites, atomic carbon and OH species are present on the surface at temperatures > 160 °C. Their study suggested that the methanation reaction proceeds via direct dissociation of CO2. The reduction of CO is then followed to atomic carbon. The further hydrogenation of atomic carbon leads to the formation of methane. However, carbon formation on a nickel catalyst is an important but complex issue. The carbon formation of supported Ni catalysts may cause deactivation during CO2 methanation. Previous studies have confirmed that the Ni (111) facet possesses the correct symmetry and low lattice mismatch for graphene growth.84–86 This would explain the better reactivity of the catalyst with more Ni (111) for CO2 methanation87,88 since graphene like carbon tends to form methane during the hydrogenation. Hu et al.89 also found that coke is unlikely to form on Ni (111) during CO2 reforming of methane. The carbon species formed on Ni (111) have better reactivity with hydrogen and CO2.90 Patera et al.91 reported an atomic-scale description of the structure of graphene edges on Ni (111), both during and post growth, by using STM. They demonstrated that temperature acts as a control parameter driving the structure of graphene/Ni (111) edges by changing their passivation. When graphene forms above a Ni (111) substrate (i.e., at T > 300 °C), during the growth process, its edges are clean and anchored to the metal substrate. This confirms the theoretical prediction by Zhang et al.92 Growing graphene flakes are thus sealed, most probably thereby hindering the penetration of ad-species below the flakes. Upon cooling to room temperature, the growth is stopped, and supported graphene flakes are formed. Fig. 4 shows an illustrated STM image of graphene on Ni (111).93 Patera et al.94 recently reported real-time imaging of adatom promoted graphene growth on Ni (111) by means of in situ high speed STM measurements. By monitoring layer formation at the atomic scale and with a time resolution down to milliseconds, at the kink sites of the graphene edges, it was observed that single Ni atoms are involved directly in the graphene growth process. Based on the DFT study, it can be observed that reaction barriers are substantially lower for the carbon (C) atom addition driven by Ni adatoms.
 | ||
| Fig. 3 Oxidation of Ni (111) in a CO2 atmosphere. (a) C 1s, (b) O 1s, and (c) Ni 2p3/2 in 200 mTorr CO2 in the temperature range from room temperature to 200 °C. All spectra are recorded at a kinetic energy of 200 eV, except where labels indicate a more bulk-sensitive measurement at Ekin = 550 eV. Inset: molecular structure of NiCO3.83 Copyright 2016, American Chemical Society. | ||
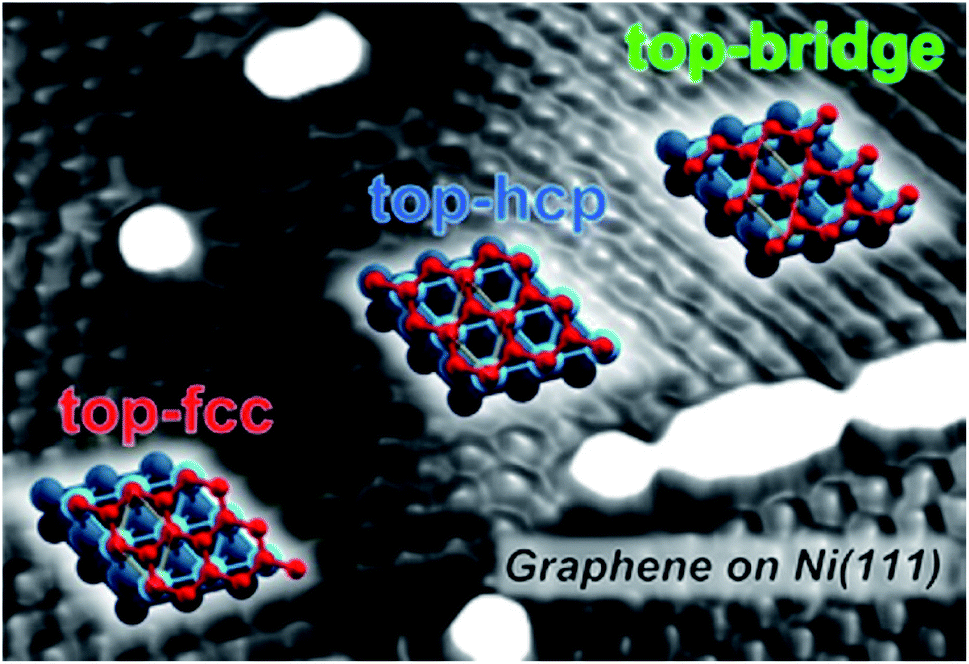 | ||
| Fig. 4 The atomic description of graphene on Ni (111).93 Copyright 2014, American Chemical Society. | ||
Methods used for graphene studies can be easily extended to the investigation of CO2 methanation on not just Ni (111). A Ni (111) substrate could enhance the reactivity of the carbon formed because of charge hybridization. In addition, the surface reactivity of the nickel catalyst can be strongly modulated by surface carbon structures.95 The content or the thickness of the formed carbon also has a significant effect for further reactions.84
4.3 Operando studies of the nickel catalysts on the porous supports
The operando studies and/or the in situ characterization of supported nickel catalysts for CO2 hydrogenation is obviously more complex and more challenging. For the investigation of the structural effect of nickel catalysts on CO2 methanation, studies based on single crystals may not be suitable because they lack the influence of electronic effects.96 Increasing publications can be found in the literature on the operando studies and/or the in situ characterization of nickel catalysts supported by a porous support, in order to understand the dynamic evolution of the catalyst structure during the reaction, to investigate the nickel–support interaction, and to ensure the optimum condition for catalyst preparation.Using operando X-ray absorption spectroscopy (XAS) analyses, Mutz et al.97 found that a fast bulk-like oxidation of the Ni particles occurred immediately after the removal of hydrogen from the gas stream, during their study on CO2 methanation over a Ni/CaO–Al2O3 catalyst with high Ni loading (23 wt%) at a H2/CO2 feed ratio of 4/1. In the following methanation step, a lower catalytic performance was observed due to residuals of partly oxidized Ni. This indicates that an efficient reactivation step is necessary after a H2 dropout to return to the initial activity. These results are important for CO2 methanation applications operating under more realistic conditions, where the fluctuating supply of hydrogen has to be considered. Consequently, oxidation of the catalyst has to be prevented or efficient reactivation procedures need to be developed.97
Vogt et al.96 applied quick operando XAS, together with operando transmission Fourier transform infrared (FT-IR) analyses, to study the size effect and thus the structure sensitivity of the SiO2 supported Ni catalysts for CO2 hydrogenation. By this study, they found that the sub-2 nm Ni particle possesses a lower d-band energy or higher electron localization, compared to large nanoparticles. This has a considerable impact on the catalytic activity.96 The Ni particles with a smaller size tend to produce CO, while the catalysts with a larger size tend to favour methane formation,96,98,99 although the formation of methane was observed for the smallest Ni clusters (∼1 nm).96 If the supported nickel catalyst further goes down to single atoms, only carbon monoxide is produced from CO2 hydrogenation.35 Moreover, the product formation rate increases by a factor of 3 at the same temperature after the particle is formed.35 The origin of the structure sensitivity of Ni/SiO2 for CO2 hydrogenation was considered to be the electronic structure of the nickel catalyst within the nickel size of 1–7 nm.96 However, if the catalyst size increases to 8–21 nm, the size effect is not the consequence of electronic effects scaling with size.99 It would be caused by the increase in the relative abundance of the active sites resulting from the change of the geometric structure.99
The operando diffuse reflectance infrared Fourier transform (DRIFT) study is very useful for the study of the structural effect of supported nickel catalysts on the adsorption, surface reaction, conversion pathway and desorption for CO2 hydrogenation to methane.35,96,99–109 With the in situ DRIFT or the operando transmission FT-IR analyses, CO2 adsorption (and related CO adsorption) on the supported nickel catalyst has been well investigated. Some of the key intermediates, as presented in Fig. 1, can be identified. Table 4 presents some illustrative identified infrared vibrational bands and the assigned types on the supported nickel catalyst during CO2 methanation.35,96,99–109 The reactive pathway(s) can be thus identified. If the catalyst takes the direct CO2 dissociative pathway, carbonyls (CO*) can be identified, followed by CO hydrogenation to form methane.106 If the catalyst takes the hydrogen-assisted CO2 dissociation pathway, oxygenate intermediates can be observed.106
| Species | Type or vibration mode | Wavenumber (cm−1) | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| TiO2 (ref. 100) | NaY101 | NaZSM-5 (ref. 101) | SiO2 (ref. 96) | MgO35 | ZrO2 (ref. 102) | Cubic ZrO2 (ref. 108) | Y2O3 (ref. 104) | CeO2 (ref. 103) | CeO2 nanoplates105 | Mesoporous CeO2 (ref. 106) | CeO2 (ref. 109) | Platelet SBA-15 (ref. 107) | ||
| CH4 gas | δ(CH) | 1305 | 1305 | 1300 | 1306 | 1300 | ||||||||
| ν(CH) | 3016 | 3015 | 3012 | 3016 | 3015 | 3000 | 3014 | 3000 | 3010 | |||||
| Subcarbonyl | Ni(CO)n (n = 2 or 3) | 2065 | 2066 | |||||||||||
| Linear CO* | ν(CO) | 2060 | 2100–1950 | 2028 | 2027 | 2040 | 2050–2020 | 2050 | 2035 | |||||
| Bridged CO* | ν(CO) | 1817 | 1903 | 1950–1750 | 1923, 1867 | 1923, 1852 | 1920 | 1930–1920 | 1912 | |||||
| Multibonded CO* | ν(CO) | 1860–1840 | ||||||||||||
| Bidentate carbonate | ν as(CO3) | 1540 | 1681 | 1552 | 1640 | 1602 | 1564 | 1580 | ||||||
| ν as(CO3) | 1523 | 1537 | 1486 | 1559 | ||||||||||
| ν s(CO3) | 1479 | 1320 | 1402 | 1290 | 1285 | |||||||||
| ν s(CO3) | 1296 | |||||||||||||
| Bicarbonate | ν as(CO3) | 1618, 1670 | 1627 | 1643 | 1650 | 1625 | 1630 | 1634 | ||||||
| ν s(CO3) | 1442 | 1430 | 1440 | 1416 | 1367 | 1438 | 1432 | |||||||
| δ(OH) | 1489 | 1250 | 1220 | 1218 | ||||||||||
| Bidentate formate | ν as(CO2) | 1589 | 1546 | 1591 | 1606–1596 | 1572 | 1578 | 1560–1588 | 1580 | 1580 | 1550 | 1647 | ||
| ν s(CO2) | 1378 | 1343 | 1356 | 1350 | 1365 | 1370 | 1371 | 1360 | 1437 | |||||
| ν(CH) | 2867 | 2850 | 2953, 2846 | 2945, 2845 | ||||||||||
| δ(CH) | 1404 | 1386 | 1386 | 1387 | ||||||||||
| Monodentate formate | ν as(CO2) | 1610 | 1610 | |||||||||||
| ν s(CO2) | 1332 | 1338–1348 | 1333 | |||||||||||
| Monodentate carbonate | ν as(CO3) | 1529 | 1521 | 1518 | 1520 | 1530 | ||||||||
| ν as(CO3) | 1435 | 1450 | 1480 | 1475 | ||||||||||
| ν s(CO3) | 1357 | 1346 | 1335 | 1436 | 1328 | 1287 | 1395 | 1390 | ||||||
| Methylene | ν(CH) | 2873 | ||||||||||||
| Methyl | ν(CH) | 2935 | ||||||||||||
| Methoxy | ν(CO) | 1735 | ||||||||||||
| Formyl | ν(CO) | 1750 | ||||||||||||
Jia et al.102 reported an operando DRIFT study of the structural effect of Ni/ZrO2 with Ni (111) as the principal exposed facet on CO2 methanation. The CO2 adsorption was firstly investigated in the temperature range of 50–350 °C. At 50 °C, bands at 1625, 1416 and 1220 cm−1 are observed. These bands are attributed to bidentate bicarbonate species. A band at 1335 cm−1, assigned to monodentate carbonates, is also observed. With the increasing temperature, the band intensities for the bicarbonates reduce monotonically. However, new bands for bidentate formates (1572 and 1356 cm−1), bidentate carbonates (1552, 1537 and 1320 cm−1) and monodentate carbonates (1521, 1450 and 1335 cm−1) present with gradually increased intensities. Meanwhile, monodentate carbonates increase more rapidly than bidentate carbonates above 200 °C. This suggests the good thermal stability of monodentate carbonates. The DRIFT studies verify that OH groups on the ZrO2 surface provide weakly basic sites for bicarbonates, whereas acid–base Zr4+–O2− pairs and O2− sites serve as moderately and strongly basic sites for bidentate and monodentate carbonates.102 The bands related to adsorbed CO (CO*) in linear and bridge forms on the Ni surface are visible in 2100–1950 cm−1 and 1950–1750 cm−1. The adsorbed CO should be formed from direct CO2 dissociation 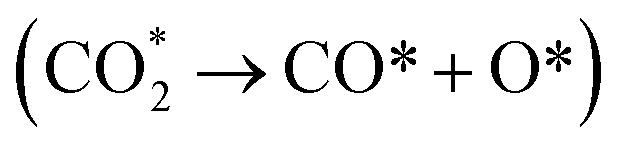 on the Ni (111) facets, followed by the reaction with OH groups towards a small amount of formates (CO* + OH* → HCOO*). The appearance of the bands for bidentate formates is good evidence for CO2 dissociation. The Ni (111) surface facilitates the dissociation of CO2 into adsorbed CO as a crucial intermediate for methane synthesis.62,83,110
on the Ni (111) facets, followed by the reaction with OH groups towards a small amount of formates (CO* + OH* → HCOO*). The appearance of the bands for bidentate formates is good evidence for CO2 dissociation. The Ni (111) surface facilitates the dissociation of CO2 into adsorbed CO as a crucial intermediate for methane synthesis.62,83,110
Operando DRIFT measurements for CO2 methanation over Ni/ZrO2 were then conducted.102 As shown in Fig. 5, at 150 °C, bicarbonates (1625, 1416 and 1220 cm−1), monodentate carbonates (1521, 1450 and 1335 cm−1) and bidentate formates (2867, 1572, 1384 and 1356 cm−1) were observed. Bidentate formates keep substantially increasing up to 200 °C, accompanied by a decrease of both bidentate bicarbonates and monodentate carbonates. However, bidentate carbonates are invisible due to their instability and rapid decomposition before hydrogenation to formates. The appearance of a shoulder band at 1610 cm−1, which is typically attributed to monodentate formates, is worth noting. It has been proposed that bidentate formates originate from hydrogenation of bidentate bicarbonates, and monodentate formates are derived from the hydrogenation of monodentate carbonates. A smaller amount of monodentate formates left on the catalyst is mainly due to their faster hydrogenation and decomposition. It is sure that Ni/ZrO2 with Ni (111) as the principal exposed facet significantly improves the activity of the (bi)carbonate hydrogenation towards bidentate and monodentate formates.102
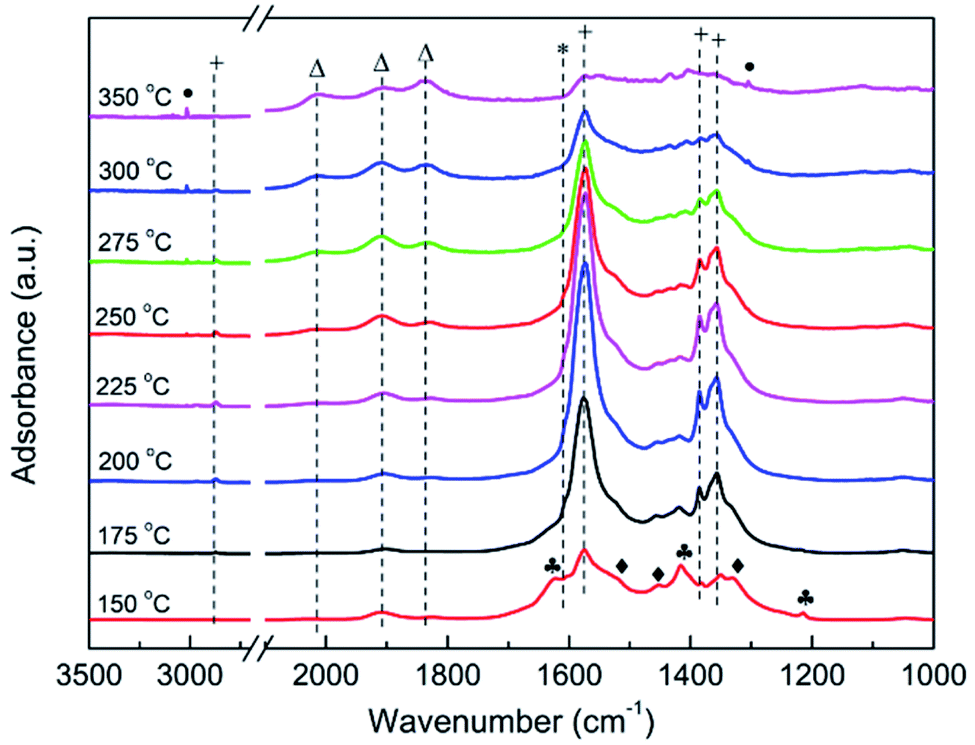 | ||
| Fig. 5 Operando DRIFT spectra during the temperature-programmed reaction of a gas mixture (40 mL min−1) containing 2.5% H2, 0.5% CO2 with argon as a carrier gas over Ni/ZrO2. The heating rate is 10 °C min−1. Symbol definition: bidentate bicarbonate (♣), monodentate carbonates (♦), bidentate formate (+), monodentate formate (*), linear CO or bridge CO (△) and gaseous CH4 (●).102 Copyright 2018, Elsevier. | ||
Operando DRIFT analyses were further conducted to study the evolution of adsorbed and gaseous species as a function of the temperature102 with the calculated relative concentrations of bidentate formates and adsorbed CO as well as gaseous methane. The quantification of adsorbed CO was made by the integration of bands in the range of 2100–1750 cm−1, while the quantifications of formates and methane were made following the band intensities at 1572 and 3015 cm−1. The highest value measured for every species was set to 100.0% in order to obtain normalized relative concentrations from 200 to 350 °C. As shown in Fig. 6, with increasing temperature from 200 to 250 °C, a gradual decrease of formates is induced from 100.0% to 70.1%, accompanied by the respective increase of adsorbed CO and methane from 20.9 to 64.6% and 1.6 to 17.2%. These confirm that the dissociation of formates into adsorbed CO is dominant for formate conversion at first.102 Above 250 °C, the methane production rate is significantly boosted by the hydrogenation steps. The adsorbed CO reaches the maximum at 275 °C, and then begins to be consumed to form methane. Considering the respective evolution of adsorbed CO and methane with the temperature, it is reasonable that methane production is mainly limited by the rate-limiting steps of CO dissociation and hydrogenation. In addition, CO2 methanation over Ni/ZrO2 with Ni (111) as the principal exposing facet prefers to proceed via the direct hydrogenation of formates (formate* → CH4(g)) and the formate dissociation followed by the hydrogenation of adsorbed CO (formate* → CO* → CH4(g)).
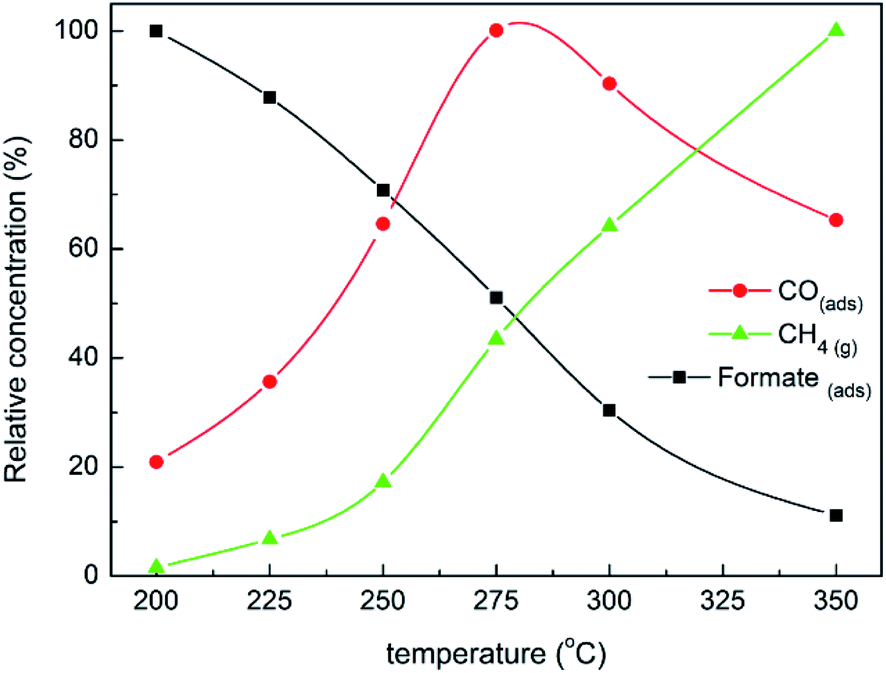 | ||
| Fig. 6 Variation of relative concentrations of CH4(g) (3015 cm−1), adsorbed CO (2100–1750 cm−1) and bidentate formates (1572 cm−1) during temperature-programmed CO2 methanation over Ni/ZrO2.102 Copyright 2018, Elsevier. | ||
4.4 Structural studies via the size effect
CO2 hydrogenation to methane over a supported nickel catalyst is structure sensitive. It is very important to prepare Ni catalysts with a controllable structure. However, obtaining a supported Ni catalyst with a desired structure remains a significant challenge. Only a few studies can be found in the literature on the preparation of catalysts with Ni (111) as the principal exposed face using specific preparation methodologies, such as cold plasma,87,88,102,111–114 external magnetic field assisted reduction115 and electrospinning,116 or using specific supports.117,118Fig. 7 and 8 show illustrative images of the supported Ni catalysts with Ni (111) as the principal exposed face.111,117 | ||
| Fig. 7 High-resolution TEM images of the Ni particles in (left) Ni/SiO2 prepared via decomposition of the nickel precursor thermally; and (right) Ni/SiO2via decomposition of the nickel precursor by energetic species of cold plasma. Both samples were reduced by flowing hydrogen at 500 °C for 2 h.111 Copyright 2013, Elsevier. | ||
 | ||
| Fig. 8 High resolution transmission electron microscope (HRTEM) images of the Nix/Mg2−xAl−MMO samples: (a and b) Ni0.4/Mg1.6Al–MMO, (c and d) Ni0.8/Mg1.2Al–MMO, (e and f) Ni/MgAl–MMO and (g and h) Ni1.6/Mg0.4Al–MMO. The insets in (b), (d), (f) and (h) show the corresponding particle-size frequency distribution histogram (400 particles analyzed).117 Copyright 2016, Royal Society of Chemistry. | ||
The studies of the structural effect of supported nickel catalysts for CO2 hydrogenation to methane are mostly via the size effect. This means that most of the reported studies affect the catalyst structure via the limited nickel nanoparticle size in empirical ways. A decrease in the size of the nickel catalyst causes an increase in the proportion of surface species, surface defects and heterogeneity of the atomic structure. This leads to improved hydrogen activation and enhanced Ni–support interactions. The decreasing particle size enhances the quantum size effects with changes in the electronic nature.119 A smaller size of the nickel catalysts results in enhanced activity for CO2 methanation. If the catalyst size goes down to a single nickel atom, the activity of the catalyst may undergo a total change because of the change in the electronic structure, induced by the quantum size effect.
The size effect is highly coupled with the support effect.120 One can even tune the product of CO2 hydrogenation from CO to methane with the change of supporting materials for the nickel catalysts of 1 to ca. 5 nm.121
The inert SiO2 is an excellent supporting material for the studies of the size effect. Vogt et al.96 applied operando transmission FT-IR spectroscopy for their study of the size effect of SiO2 supported Ni catalysts (within the nickel size of 1–7 nm) for CO2 hydrogenation. They observed that the catalyst with the highest nickel dispersion or the smallest nickel particle size exhibits relatively few bands assigned to adspecies containing CO stretching vibrations, while the catalyst with the largest size presents big peaks in this region.96 They found that the intensity of the absorption bands in the CO* stretching region during CO2 hydrogenation is positively correlated with the catalyst size.96 They suggest that this absorption intensity can be an indirect measure for the mean catalyst size.96 Gaseous CO is detected only for the smaller nickel particles (less than 1.5 nm). The amount of gaseous carbon monoxide is more or less negatively correlated with the catalyst size.96 All the CO species on the catalyst of small size are easily flushed off, while CO* presents in much more stable configurations, like bridged carbonyl or carboxylate species, on the catalyst with a larger particle size.96 Moreover, Vogt et al.96 further used isotopically labelled gas feedstocks (13CO2 and D2) for operando FT-IR measurements, to investigate the reactivity of the intermediates, such as formate and CO species. Their studies showed that no interaction occurs between the adsorbed 12C and H in the formate species and the isotopically labelled feedstocks for the catalysts of small Ni particle sizes.96 For the Ni catalysts of larger particle sizes, however, these labelled gases readily interact and shift the formate peak with both D2 and 13CO2 pulses. They found that the intermediate CO* species dominate the surface of the catalysts with larger Ni nanoparticles, while, in the case of catalysts of smaller sizes, formate species dominate the surface with detectable gaseous CO.96
Wang et al.120 carried out an operando DRIFT study of Ni/SiO2 for CO2 methanation with the nickel catalyst size of 3.5–7.5 nm. The activity for CO2 methanation quickly decreases with increasing catalyst size.120 The smaller catalysts possess higher reactivity towards CO2 methanation, whereas the larger catalysts favour the formation of CO, the major by-product. The DRIFT studies indicate that the small nickel catalysts with highly co-ordinately unsaturated surface Ni sites are active for CO2 methanation through facile adsorption and stabilization of the monodentate carbonate in the adsorption state. This facilitates the formation of monodentate formate species as well as linearly adsorbed CO species, leading to subsequent hydrogenation toward methane.120
According to the literature, the following approaches have been mostly employed to limit the size of Ni catalysts: (1) the use of promoter(s); (2) the formation of alloyed catalysts and bi-metallic catalysts; (3) modification of the surface properties of the catalyst or the supporting material; (4) the use of a supporting material with a high surface area; (5) the use of structured catalysts; (6) the use of process intensified preparation methods, such as combustion, cold plasma decomposition of the catalyst precursor, and gel or urea assisted preparation; (7) the application of specific catalyst precursors and others.
Li et al.122 developed a precise control of the growth and size of Ni nanoparticles on Al2O3 by loading of Ni2+ ions into metal–organic framework (MOF) MIL-110 (Al) via incipient wetness impregnation, followed by drying at 120 °C, as well as calcination at 600 °C in air and then hydrogen reduction at 600 or 800 °C. An up to ∼20 wt% Ni loading was applied. The particle size of Ni NPs on Al2O3 is well controlled and maintained at ∼5 nm irrespective of low- or high-temperature reduction. The formation of surface spinel species leads to a strong metal–support interaction and stabilizes the high dispersion of Ni NPs.122
The spatial distribution of the Ni nanoparticles has an effect as well.123,124 Shen et al.123 investigated the effect of micropores on the supported Ni/SiO2 catalyst for CO2 methanation. They found that there exist both ultra-small Ni nanoclusters and larger Ni nanoparticles for Ni/SiO2 with plenty of micropores. However, only large Ni nanoparticles are present on the catalyst with fewer micropores. CO2 can be activated to CO on small Ni nanoclusters during CO2 methanation, and the generated CO will be further converted to methane on larger Ni nanoparticles.
The use of high surface area porous materials for highly dispersed Ni catalysts is always attractive. Haller and his co-workers125 employed MCM-41 to load highly dispersed Ni nanoparticles on it for CO2 methanation at temperatures as low as 573 K. At a Ni loading of 3 wt%, the methane selectivity reaches almost 100%. They confirmed that the hydrogen treatment of Ni-MCM-41 induces the formation of a highly dispersed and thermally stable metallic Ni catalyst. The highest activity and selectivity were achieved on the catalyst reduced at 973 K, with which Ni is mostly reduced to Ni0 with a particle size of 1.7 nm. No serious aggregation was observed after the reaction due to the surface anchoring effect.125 Lu and his co-workers recently used metal–organic framework MOF-5 with a surface area of 2961 m2 g−1 to disperse Ni nanoparticles within it for CO2 methanation.126 A very high dispersion of Ni (41.8%) has been achieved, leading to a significant enhanced activity. CO2 conversion reaches 47.2% at 280 °C with a methane selectivity of 100% (from 200 to 320 °C). This MOF-5 supported Ni catalyst shows almost no deactivation in a long-term stability test up to 100 h.126
Chu and his co-workers127 confirmed that the basic sites of the support have a significant effect on the activity of the Ni catalyst for CO2 methanation. This was also confirmed by Muroyama et al.128 The treatment of the support can be applied towards the desired basic sites.127
The oxygen vacancy of the support significantly enhances the Ni–support interaction, stabilizes the nickel nanoparticles, promotes the formation of a Ni–support active interface and improves the adsorption of carbon dioxide and hydrogen.102,103,105,109,129–135 Obviously, the effect of the support cannot be ignored.136,137 Tsubaki and his co-workers138 recently reported the preparation of nanoparticle promoted Ni-based bimodal pore catalysts by the impregnation of nickel precursor on SiO2, Al2O3 and ZrO2 nanoparticle modified SiO2 supports. The catalysts were named Ni/SiO2–Si, Ni/SiO2–Al and Ni/SiO2–Zr. The introduction of nanoparticles into the SiO2 support increases the surface area of the catalyst and improves the dispersion of nickel. The activity for CO2 methanation is significantly enhanced. The CO2 conversion increased in the order of Ni/SiO2 (unmodified catalyst) < Ni/SiO2–Si < Ni/SiO2–Al < Ni/SiO2–Zr, corresponding to the increasing order of their surface areas. They found that the modification of ZrO2 and Al2O3 nanoparticles improved CO2 chemisorption and dissociation, and thus resulted in extremely high CH4 selectivity (about 100%) at low reaction temperatures (<450 °C).138 A strong metal–support/promoter interaction was also observed in ZrO2 and Al2O3 nanoparticle promoted catalysts, inhibiting the sintering of nickel and contributed to the high stabilities of these two catalysts in CO2 methanation.138
A strong Ni–support interaction is basically required to limit the catalyst size. Increasing investigations can be found in the use of a structured support for enhanced Ni–support interactions for CO2 methanation.139–152 Catalysts with well-defined structures can lead to higher dispersion, better reactivity, improved selectivity and enhanced stability. In some cases, the structured catalyst itself contains nickel and acts as precursors. Since CO2 methanation is an exothermal reaction, the use of a structured catalyst not only improves the activity but also results in enhanced heat and mass transfer for optimum catalyst performance. Aziz et al.145 prepared mesostructured silica nanoparticles (MSN) to obtain the Ni/MSN catalyst for CO2 methanation. They found that the catalytic performance of Ni/MSN closely depends on the structure of the catalyst, which consists of both intra- and inter-particle porosity. The presence of interparticle porosity facilitates the transport of reactant and product molecules during the reaction. The higher diffusion of CO2 in the pores of Ni/MSN increases overall reaction rates and results in higher conversion, compared to Ni/MCM-41. The internal mass transfer resistance is negligible with this Ni/MSN catalyst. The high basicity of Ni/MSN catalysts is crucial to the high activity of the reaction with enhanced CO2 adsorption. The presence of defect sites or oxygen vacancies in MSN was considered to be responsible for the formation of surface carbon species, while Ni sites dissociate hydrogen into atomic hydrogen. Carbon atoms are then combined with atomic hydrogen for the formation of methane. The Ni/MSN catalyst shows good stability with no deactivation for 200 h.145
Borgschulte et al.146 employed a nanostructured Ni@zeolite 5A catalyst for a sorption enhanced CO2 methanation with a methane yield up to 100%. Duan and his co-workers147 applied a hierarchical Al2O3 matrix for loading of high particle density nickel for CO2 methanation. A surface defect-promoted Ni catalyst with high dispersion is fabricated. It exhibits excellent activity and stability. Abundant surface vacancies generate and serve as active sites, resulting in significantly enhanced low-temperature activity. The anchoring effect from the support gives rise to high reaction stability, without sintering and/or aggregation of active species for long-term reactions.147
Li et al.148 reported a Ni–Al2O3/Ni-foam catalyst with enhanced heat transfer for CO2 methanation. This catalyst was prepared using a modified wet chemical etching method. A uniform NiO–Al2O3 composite catalyst layer (∼2 μm) is efficiently formed and firmly anchored onto the foam structure by directly immersing the Ni-foam substrate into a chemical etching solution consisting of sodium dodecyl sulphate, acetic acid, and aluminum nitrate, followed by calcination under air. A high CO2 conversion of ∼90% with a high methane selectivity (>99.9%) has been obtained and remained stable for 1200 h.148
5. CO2 hydrogenation to CO
According to Fig. 1, either direct CO2 dissociation or hydrogen-assisted CO2 dissociation can lead to the formation of CO. CO is an important intermediate for syntheses of methane and methanol. CO2 hydrogenation to CO or the RWGS reaction (reaction (5) in Table 1) is a side reaction of CO2 methanation and CO2 hydrogenation to methanol. Methane is normally the main product below 600 °C.28 A further increase in the reaction temperature causes CO formation.11,28,36,37 100% selectivity to CO can be observed at 750 °C.28 This suggests that the exothermic CO2 hydrogenation to methane dominates at temperatures below 600 °C, whereas the RWGS reaction is the predominant one at temperatures above 600 °C.28As addressed above, the size of the nickel catalyst has a significant effect on the selectivity of CO2 hydrogenation, coupled with the support effect. The small catalyst size favours the selective hydrogenation of CO2 to CO. The SiO2 supported Ni catalyst has higher activity for RWGS, compared to CeO2 and TiO2.36,120,153 The Ni catalyst that adsorbs CO strongly favours CO2 hydrogenation to methane and methanol, while the catalyst that binds CO weakly tends to formation of CO.10,13
Lin et al.34 reported a stable and active Ni/γ-Mo2N catalyst for the hydrogenation of CO2 to CO. They found that the pre-synthesized Ni particles of 4 nm are able to reverse sintering into the under-coordinated Ni species after a high temperature activation procedure driven by the intense interaction between Ni and γ-Mo2N. The high dispersion of Ni occurs after the reduction of bulk phase Ni and the removal of the surface passivated O-layer of γ-Mo2N. The obtained Ni/γ-Mo2N catalyst shows 96% CO selectivity, at temperatures between 300 and 500 °C, with high CO2 conversion.34
Based on the electronic configuration of the nickel atom, the Ni-SAC is excellent for CO2 hydrogenation to CO. It was found that a single nickel atom is only able to take part in the 2e− redox cycle. Ni clusters are needed for the full hydrogenation of CO2 with the 8e− redox cycle.35 The unique characteristics of the SAC render Ni-SAC active for electro-chemical CO2 reduction to CO.154–157 Frei and his co-workers35 confirmed that the MgO supported Ni-SAC is active for RWGS. MgO supported Ni NPs are normally highly active for CO2 methanation.158–160 In2O3 supported Ni-SACs were theoretically confirmed to be active for RWGS as well.29,161 This can be further confirmed by the effect of the reaction temperature on the stability of the supported Ni-SAC.35 It was reported that the MgO supported Ni-SAC remains stable at a reaction temperature of 300 °C, with which CO is produced through the RWGS reaction.35 Particularly, the activity of the catalyst increases when the temperature increases from 200 °C to 300 °C, as shown in Fig. 9.35 When the reaction temperature increases to 350 °C, methane and even methanol are produced with an aggregation of the Ni-SAC. As the reaction goes back to 300 °C, methane and methanol disappear. Only CO is detectable.35 This obviously suggests that CO2 hydrogenation to methane and methanol needs nickel clusters, not the Ni-SAC, with MgO as the support.35
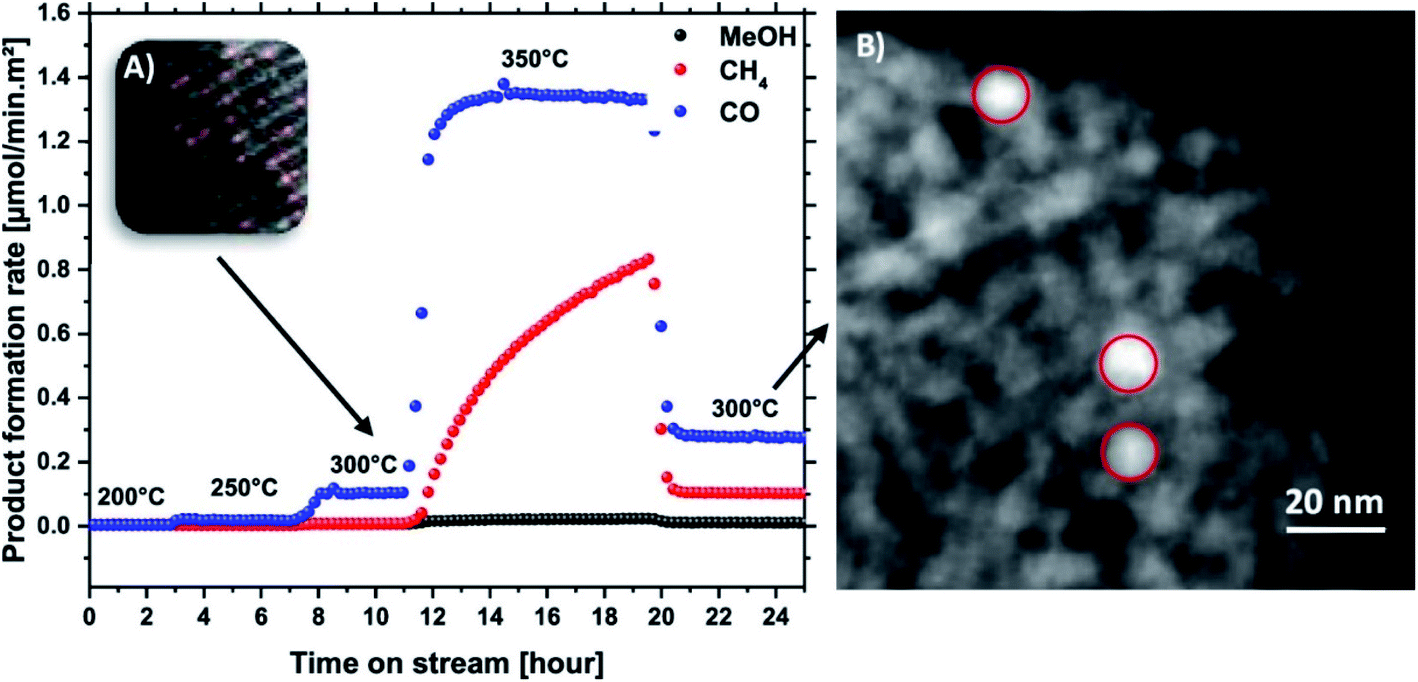 | ||
| Fig. 9 (A) Product formation rate of Ni_10 at 30 bar, and different temperatures; the upper left corner represents the sample after 300 °C measurements. (B) TEM image of the sample after testing at 350 °C.35 Copyright 2019, American Chemical Society. | ||
In situ IR analyses were employed for CO2 hydrogenation over the MgO supported Ni-SAC.35 As shown in Fig. 10, when the reaction temperature increases from 200 °C to 250 °C and then to 300 °C, a progressive decrease of the carbonate signals (1435, 1529 cm−1) from the decomposition of the surface overlayer is observed, accompanied by an increase of the bicarbonate signal (1250 cm−1), formate signals (1342, 1606 cm−1), and the appearance of methane (Fig. 10A, 1305 cm−1; Fig. 10B, 3016 cm−1). This clearly shows that, with increasing temperatures, methane starts to form, evidence of the aggregates of the Ni-SAC. In addition, a pronounced shift in the formate area from 1607 to 1596 cm−1 is observed, as shown in Fig. 10C. While values around 1600–1606 cm−1 were assigned to formate species on MgO, values around 1591 cm−1 were attributed to stretching vibrations from formate species on Ni0 nanoclusters, further supporting the correlation of cluster formation and methane production.35
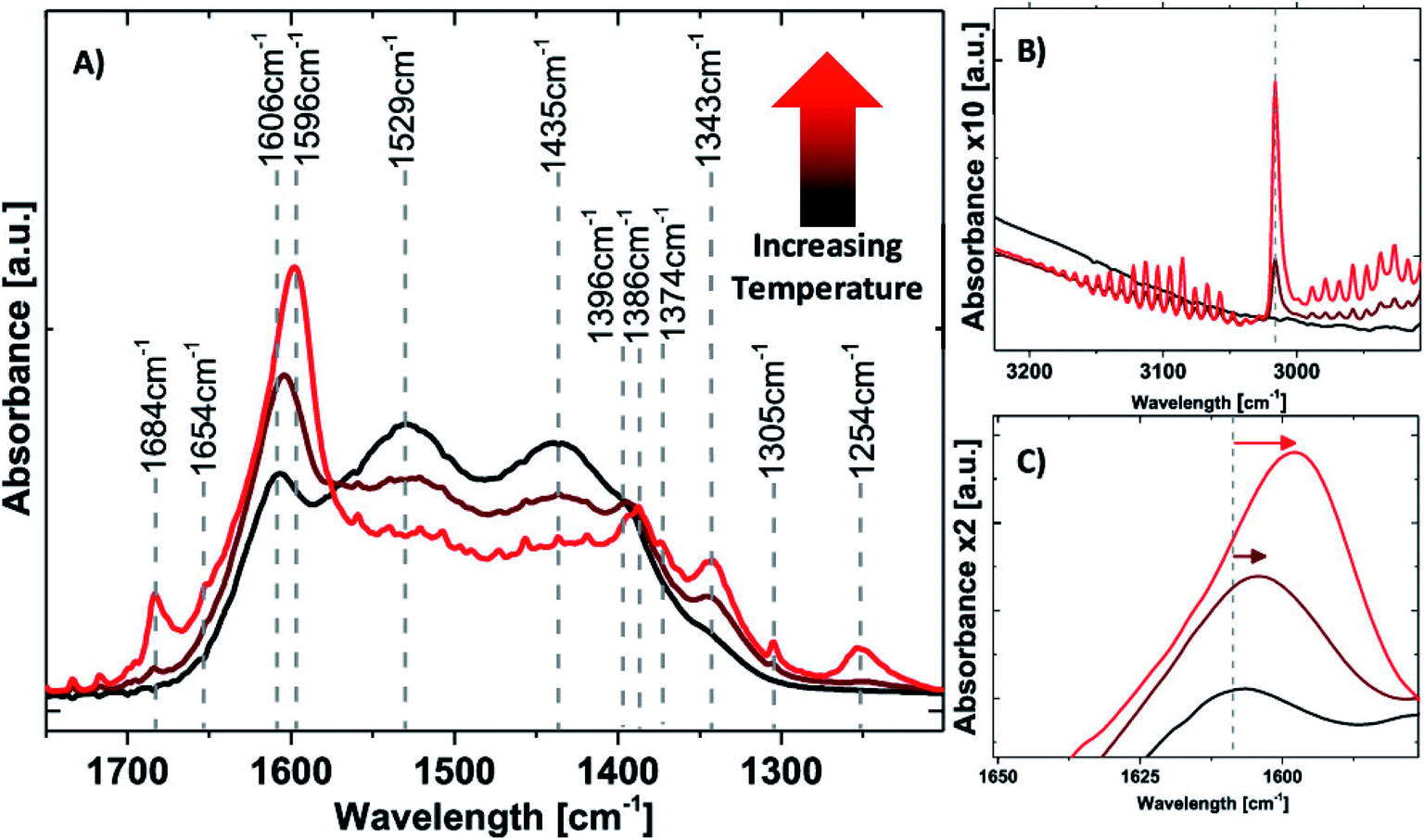 | ||
| Fig. 10 IR spectroscopy under reaction conditions (4H2/CO2); stepwise increase from 200 °C (black), to 250 °C (dark red), and to 300 °C (red): (A) zoom-in of the 1200–1700 cm−1 area; (B) zoom-in of the 2900–3200 cm−1 area; (C) zoom-in of the 1550–1650 cm−1 area.35 Copyright 2019, American Chemical Society. | ||
A limitation with the use of Ni-SACs for the RWGS reaction is the possible formation of Ni(CO)4.162 The Ni-SAC with intense Ni–support interactions is basically required for CO2 hydrogenation to avoid the formation of Ni(CO)4.
6. CO2 hydrogenation to methanol
6.1 Theoretical studies
There is increasing interest in the use of supported nickel catalysts for CO2 hydrogenation to methanol. Fig. 1 shows that either direct CO2 dissociation or hydrogen-assisted CO2 dissociation can lead to methanol synthesis from CO2 hydrogenation. Three general pathways can be considered based on the mechanism of CO2 dissociation, including the formate pathway, CO hydrogenation pathway and RWGS pathway.10,22 Peng et al.48 predicted the possibility of CO2 hydrogenation to methanol on Ni (110). They found that Ni (110) takes the formate pathway for CO2 hydrogenation to methanol. However, two routes coexist via HCOOH* and H2COO* species, as shown in Fig. 1. The hydrogenation step of CH2O* to CH3O* competes with that to CH2OH*.48 The hydrogenation of CH3O* to CH3OH* is the rate-determining step for the CH3O route on Ni (110). They also found that the presence of HCO* as a source of hydrogen facilitates the hydrogenation of CH3O*.48 This suggests the promotional effect of CO. CO can also involved in methanol synthesis.48 CO can be first hydrogenated to HCO*, which can be further hydrogenated to CH2O*, and finally to CH3OH* (through CH3O* or CH2OH*).48The formation of methanol from CO2 hydrogenation is theoretically feasible on Ni13.50 The optimized pathways for the formation of methanol and methane are 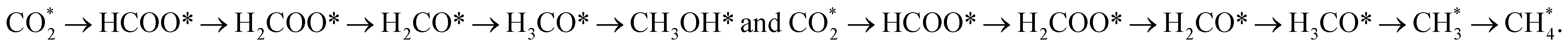 50 The hydrogenation or the dissociation of H3CO* determines the selectivity for CH3OH and CH4.50
50 The hydrogenation or the dissociation of H3CO* determines the selectivity for CH3OH and CH4.50
The oxide support plays an important role in methanol synthesis from CO2 hydrogenation, as confirmed by our recent DFT study on Ni4/In2O3.22 It was shown that the RWGS pathway is the most theoretically-favoured for CO2 hydrogenation to methanol on Ni4/In2O3.22 The complete pathway follows 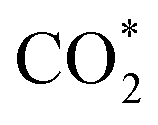 → COOH* → CO* → HCO* → H2CO* → H3CO* → H3COH*, as shown in Fig. 1. The interfacial oxygen vacancy of In2O3 serves as the active site for boosting CO2 adsorption and charge transfer between nickel species and indium oxide, which synergistically promotes the consecutive CO2 hydrogenation towards methanol.22 The dissociation of hydrogen occurs on Ni clusters in an almost barrier-less manner. The hydrogen adatoms spillover from the Ni cluster to the interfacial active site for the hydrogenation of adsorbed carbon dioxide. However, the nickel atom numbers have a big influence. Ni1/In2O3 and Ni2/In2O3 catalysts favour the RWGS reaction,29,161 leading to CO formation, because of higher energy barriers for methanol synthesis.
→ COOH* → CO* → HCO* → H2CO* → H3CO* → H3COH*, as shown in Fig. 1. The interfacial oxygen vacancy of In2O3 serves as the active site for boosting CO2 adsorption and charge transfer between nickel species and indium oxide, which synergistically promotes the consecutive CO2 hydrogenation towards methanol.22 The dissociation of hydrogen occurs on Ni clusters in an almost barrier-less manner. The hydrogen adatoms spillover from the Ni cluster to the interfacial active site for the hydrogenation of adsorbed carbon dioxide. However, the nickel atom numbers have a big influence. Ni1/In2O3 and Ni2/In2O3 catalysts favour the RWGS reaction,29,161 leading to CO formation, because of higher energy barriers for methanol synthesis.
The location of nickel in the support has an influence on the adsorption of carbon dioxide. The Ni atoms situated in the bulk do not significantly contribute to the conversion of carbon dioxide on the Ni-SAC catalyst on MgO.35 Based on the DFT calculations, as a nickel atom is located deeper in the subsurface, the contribution of Ni states to the bonding of adsorbed CO2 species is decreased.35 The location of the Ni-SAC on In2O3 has significant effects on hydrogen dissociation as well for CO2 hydrogenation.29 For the stoichiometric In2O3 surface and the In2O3(111) surface doped with a Ni single atom, H2 dissociation leads to two OH groups. This indicates a homolytic dissociation.29 The reaction over the stoichiometric surface is activated by 105 kJ mol−1 with an adsorption energy (ΔEads) of −250 kJ mol−1. Doping of a Ni single atom in the In2O3(111) surface causes a small decrease of the activation barrier to 79 kJ mol−1 with a more exothermic reaction energy (ΔEads = −300 kJ mol−1).29 If the Ni single atom is located on the In2O3(111) surface, instead of doping, H2 activation involves the single Ni atom adsorbed on top of the In2O3(111) surface. One of the H atoms remains at the Ni atom, while the other migrates to a neighbouring O atom, resulting in, respectively, Ni–H and OH fragments.29 The barrier associated with H2 dissociation is only 11 kJ mol−1.29 Similar results have been reported by Frei et al.161 for Nix–In2O3 ensembles. Different from the Ni-SAC, H2 activation over the In2O3(111) supported Ni8 cluster occurs in two steps. After adsorption of molecular H2 with ΔEads of −90 kJ mol−1, H–H bond scission causes two H atoms to be adsorbed in a bridged configuration. With respect to the gas phase, the process of H2 activation is barrier-less.29 The same result has been also obtained on a defective In2O3(111) surface supported Ni4 cluster22 for CO2 hydrogenation to methanol.
6.2 Experimental studies
A Ga2O3 supported nickel catalyst was reported to be active for CO2 hydrogenation to methanol.26 A big size (10.2 nm) of the nickel catalyst shows the highest selectivity of methanol.26 Smaller catalysts favour the RWGS reaction.26,163 However, the mechanism is not clear. Further operando studies would be needed.Jia et al.27 employed wet chemical reduction to prepare the In2O3 supported nickel catalyst with sodium borohydride (NaBH4) as a reducing agent. The obtained catalyst shows highly dispersed Ni species with intense Ni–In2O3 interactions and enhanced oxygen vacancies. The highly dispersed Ni species serve as the active sites for hydrogen activation. Abundant H adatoms are thereby generated for oxygen vacancy creation on the In2O3 surface. The enhanced surface oxygen vacancies further lead to improved CO2 conversion. As a result, an effective synergy between the active Ni sites and surface oxygen vacancies on In2O3 causes a high activity for CO2 hydrogenation to methanol. The formation of methane can be ignored even at a nickel loading up to 10 wt%. When the reaction temperature is lower than 225 °C, the selectivity of methanol is 100%. It is higher than 64% in the temperature range between 225 °C and 275 °C. The methanol selectivity is still higher than 54% at 300 °C.27 DFT calculations confirm that the electron transfer is partially from nickel to In2O3, similar to In2O3 supported gold164 and silver165 catalysts for CO2 hydrogenation to methanol. The intense Ni–In2O3 interaction causes a big change in the pathway of CO2 hydrogenation to methanol. In2O3 takes the formate pathway for methanol synthesis from CO2 hydrogenation,166 while the In2O3 supported Ni4 catalyst takes the RWGS pathway.22 Catalyst characterization suggests that no InNi alloy is formed.27 This was confirmed by Hensen and his co-workers with operando studies.29 The Ni loading has a significant effect on the activity and selectivity of Ni/In2O3 for CO2 hydrogenation. A low Ni loading leads to the formation of Ni species in the form of single atoms or very small clusters.29 These kinds of Ni species can significantly facilitate the dissociative adsorption of H2, which otherwise requires a high activation barrier on the pure In2O3 surface.29 The selective hydrogenation of CO2 to methanol is thus caused. The formation of methane and CO was observed at high Ni loadings.29
The activity of Ni/In2O3 for CO2 hydrogenation to methanol can be further improved with the use of In2O3–ZrO2 solid solution as the support.31 A highly dispersed nickel catalyst is also obtained by chemical reduction.31 A CO2 conversion of 17.9% with a methanol yield of 0.63 gMeOH gcat−1 h−1 was achieved at 300 °C and 5 MPa. Fig. 11 presents a comparison of the activities of In2O3, Ni/In2O3 and Ni/In2O3–ZrO2 with a Ni loading of 5 wt%.31 A higher Ni loading results in the formation of methane, simultaneously with methanol synthesis from CO2 hydrogenation.
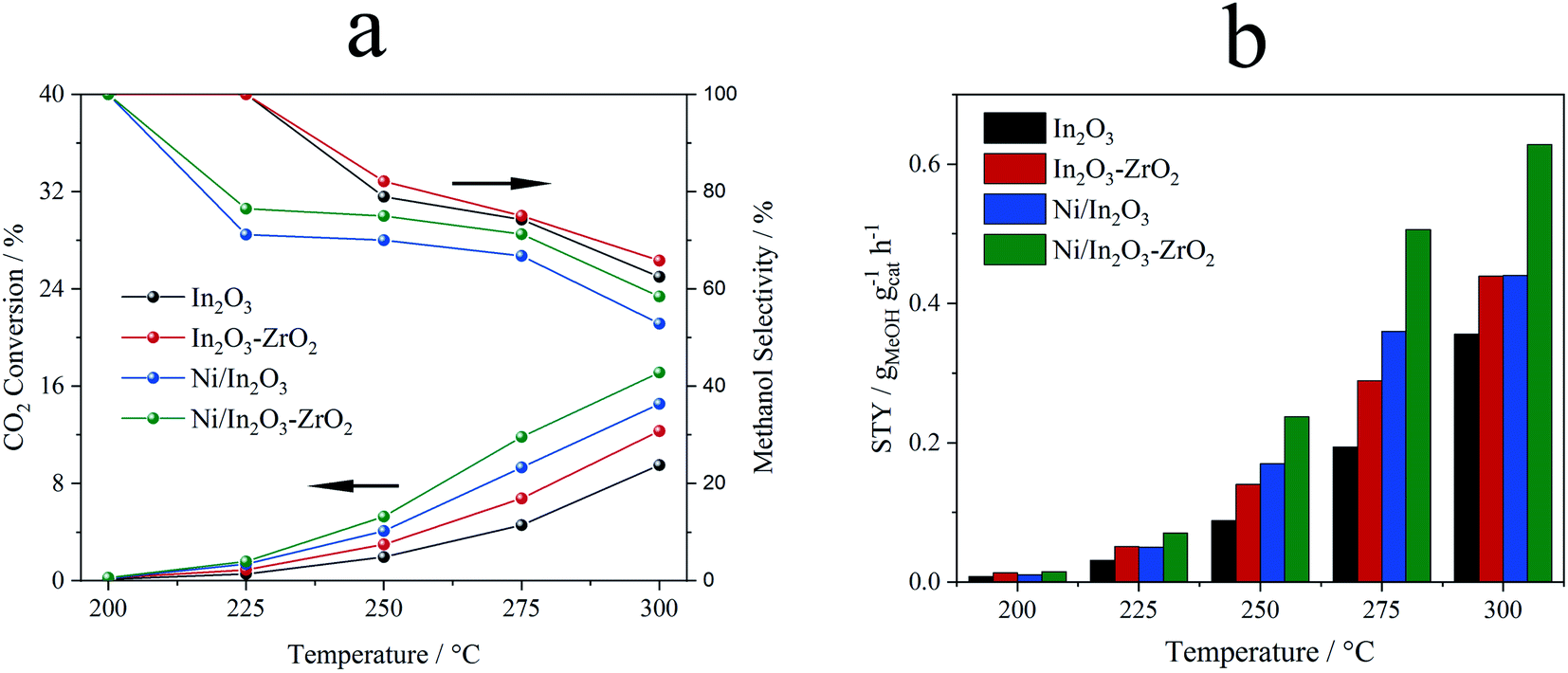 | ||
| Fig. 11 Comparison of the activities of Ni/In2O3–ZrO2, Ni/In2O3, In2O3–ZrO2 and In2O3 for CO2 hydrogenation to methanol (5 wt% Ni loading). (a) CO2 conversion and methanol selectivity and (b) STY.31 Copyright 2022, Elsevier. | ||
7. Conclusions and outlook
From the investigations reported, the following conclusions can be made:(1) The structure of the supported Ni catalyst has a significant effect on the adsorption and chemisorption of CO2 on the catalyst surface, which will further affect the activity and selectivity of the catalyst.
(2) The structural controllable preparation of a supported nickel catalyst still remains a big challenge. The studies of the structural effect on CO2 hydrogenation have mostly been via the size effect by far. A decrease in the size of a nickel catalyst causes an increase in the proportion of surface species, surface defects and heterogeneity of the atomic structure. The decreasing particle size enhances the quantum size effect with change in the electronic nature of the catalyst. This leads to improved hydrogen activation and enhanced Ni–support interactions, causing a change in the activity and selectivity for CO2 hydrogenation. If the catalyst size goes down to a single nickel atom, the activity of the catalyst may undergo a total change because of the change in the electronic structure, induced by the quantum size effect.
(3) The supported Ni-SAC is generally a good catalyst for CO2 hydrogenation to CO because of the electronic configuration of nickel and the electronic structure of the SAC. A support having intense interactions with the Ni-SAC is required for CO production in order to avoid the formation of Ni(CO)4 at low reaction temperatures. The Ni-SAC shows high activity for CO2 hydrogenation to methanol with In2O3 as the support because of the strong Ni–In2O3 interaction. The support has a significant effect on the properties of the supported Ni-SACs.
(4) The supported nickel nanoparticles are normally highly active for CO2 hydrogenation to methane at temperatures below 600 °C. The selectivity of CO will reach 100% at temperatures above 750 °C. The reaction of CO2 methanation has to compete with the side RWGS reaction. The structure of the supported nickel catalysts has a significant influence on the activity and selectivity for CO2 methanation.
(5) With some specific supports, such as Ga2O3, In2O3 and In2O3–ZrO2, the supported nickel catalyst becomes highly active towards CO2 hydrogenation to methanol at temperatures below 350 °C. This big change is caused by the intense Ni–support interaction, which changes the electronic structure of the nickel catalyst and forms an active Ni–support interface for methanol synthesis from CO2 hydrogenation. From the present understanding, it can be inferred that small nickel clusters are generally required for methanol synthesis from CO2 hydrogenation. The DFT study confirms that the Ni/In2O3 catalyst follows the RWGS pathway for CO2 hydrogenation to methanol. The reaction of CO2 hydrogenation to methanol has to compete with the side reactions of RWGS and CO2 methanation.
(6) The theoretical studies suggested that CO2 hydrogenation to formic acid is feasible over Ni-SACs with the support of unique electronic structures. Further experimental investigation is needed.
(7) Different CO2 hydrogenation pathways will be induced by different catalyst structures. Considering the challenges in the structural analyses of the catalyst, the DFT is a very useful tool for studies of the catalyst structure, nickel–support interaction, conversion pathway and others.
(8) The theoretical and experimental studies confirmed that the oxygen vacancy and basic site of the support have significant effects on the activity and reaction mechanism of nickel catalysts for CO2 hydrogenation. The strong interaction between nickel and the support helps to limit the nickel size for enhanced activity and to stabilize the catalytic activity. The intense interaction between nickel and In2O3 with surface oxygen vacancies has made the catalyst highly active for CO2 hydrogenation to methanol.
With the rapid development of renewable hydrogen technology, CO2 hydrogenation over supported nickel catalysts attracts increasing interest worldwide. The innovation of supported nickel catalysts with a clear structure for CO2 hydrogenation has become a big bottleneck. It is very necessary to conduct further fundamental studies in order to prepare nickel catalysts with controllable structures or better understand the structural effect on CO2 hydrogenation. The following future studies are therefore suggested:
(1) Because of the complex network of CO2 hydrogenation reactions, present theoretical studies have some obvious limitations. Even for the supported Ni-SAC with a well-defined structure, the reaction network is not simple. If the diffusion is further considered, the issue will be even more complex. Future studies have to consider the whole reaction network, although it is difficult at this moment. Big data technology will be useful for these important studies.
(2) The study of the supported Ni-SAC is just at the early stage. With the specific quantum size effect of the single nickel atom, we believe that the supported Ni-SAC will have more and more applications for CO2 hydrogenation, encouraged by extensive studies on the electro-chemical reduction of CO2 by Ni-SACs and the studies of In2O3 supported Ni-SACs. The investigation of supported Ni-SACs should focus on their unique properties for CO2 hydrogenation and use them to tune the activity and selectivity.
(3) Further experimental studies are needed for CO2 hydrogenation to formic acid over supported Ni-SAC catalysts. The key is to find a support that possesses unique interactions with Ni-SACs in order to stabilize the Ni-SAC, tune the electronic structure and overcome the thermodynamic obstacle.
(4) The development of in situ catalyst characterization technologies is immediately needed for the understanding of the structural effect of Ni catalysts. Operando FTIR analysis is still the most powerful tool at present for the in situ characterization of the catalyst for CO2 hydrogenation. However, IR analyses are still limited for the measurement of species with weak signals. Innovation is needed for the IR measurements of chemical intermediates during CO2 hydrogenation. NAP-XPS, STM and operando X-ray absorption spectroscopy have shown promising applications in the in situ characterization of Ni catalysts. More applications can be expected with these analyses with the development of atomic-scale observations of catalyst structures under real reaction conditions.
(5) The preparation of supported nickel catalysts with controllable structures has to be further investigated to meet the challenges in how to fabricate the desired catalyst and how to stabilize it during the reactions. This investigation should start from the very beginning of the shape and structure control of nickel nanocrystals with no supporting materials.
(6) The catalyst structure has a significant influence on CO2 hydrogenation related reactions, including those with CO, methane, various chemical intermediates and, especially, carbon species formed during the reaction. There are many opportunities in these aspects. For example, if the nickel catalyst follows the CO pathway for CO2 methanation, it must have good activity for CO methanation to obtain high activity for CO2 methanation. One can learn information on CO methanation to develop promising catalysts for CO2 methanation.
(7) The adsorbed CO species is an important intermediate for CO2 hydrogenation over the supported nickel catalyst. It was confirmed that the catalyst that adsorbs CO strongly favors the formation of methane or methanol, while the one that binds CO weakly tends to produce carbon monoxide. CO can be a probe molecule for the operando study of nickel catalysts to understand the conversion pathway of CO2 hydrogenation. Further progress can be expected.
(8) Because of the difficulty in structure identification, the present comparison of the properties of supported Ni catalysts for CO2 hydrogenation may not be accurate. For example, if one would like to study the phase structure effect, it is better to compare the experimental data with the same catalyst size. And, if one would like to study the size effect, it is better to clearly fix the catalyst phase structure. Therefore, the structure analysis of the catalyst is basic. This is a key issue related to the supported nickel catalyst for CO2 hydrogenation.
(9) Fundamental studies, including the nucleation and growth of the nickel crystal (especially on the porous supporting materials), the interaction between the nickel and the support, the preparation of the supporting material with a clear porous structure and desired phase structure, are needed. Because of the importance of defects or oxygen vacancies on the support for CO2 hydrogenation, the creation and the stability of such defects or vacancies are another significant challenge.
In summary, the studies of the structural effects of supported CO2 hydrogenation are extremely important. Multidisciplinary efforts are needed. With advances in theoretical studies, data analyses, operando studies or in situ catalyst characterization, single atom catalyst and novel catalyst preparation, we believe further progress will be made soon for the practical applications of supported nickel catalysts for CO2 hydrogenation.
Conflicts of interest
There are no conflicts to declare.References
- J. Ding, Q. Liu, R.-P. Ye, W. Gong, F. Zhang, X. He, Y. Zhang, Q. Zhong, M. D. Argyle and M. Fan, J. Mater. Chem. A, 2021, 9, 21877–21887 RSC.
- K. B. Tan, G. Zhan, D. Sun, J. Huang and Q. Li, J. Mater. Chem. A, 2021, 9, 5197–5231 RSC.
- J. Zhang, C. D. Sewell, H. Huang and Z. Lin, Adv. Energy Mater., 2021, 11, 2102767 CrossRef CAS.
- R.-P. Ye, J. Ding, W. Gong, M. D. Argyle, Q. Zhong, Y. Wang, C. K. Russell, Z. Xu, A. G. Russell, Q. Li, M. Fan and Y.-G. Yao, Nat. Commun., 2019, 10, 5698 CrossRef CAS PubMed.
- Z. Yang, Y. Qi, F. Wang, Z. Han, Y. Jiang, H. Han, J. Liu, X. Zhang and W. J. Ong, J. Mater. Chem. A, 2020, 8, 24868–24894 RSC.
- X.-Y. Meng, C. Peng, J. Jia, P. Liu, Y.-L. Men and Y.-X. Pan, J. CO2 Utiliz., 2022, 55, 101844 CrossRef CAS.
- Z.-J. Wang, H. Song, H. Liu and J. Ye, Angew. Chem., Int. Ed., 2020, 59, 8016 CrossRef CAS PubMed.
- X. Su, J. Xu, B. Liang, H. Duan, B. Hou and Y. Huang, J. Energy Chem., 2016, 25, 553–565 CrossRef.
- B. Miao, S. S. K. Ma, X. Wang, H. Su and S. H. Chan, Catal. Sci. Technol., 2016, 6, 4048–4058 RSC.
- S. Kattel, P. Liu and J. G. Chen, J. Am. Chem. Soc., 2017, 139, 9739–9754 CrossRef CAS PubMed.
- J. Ashok, S. Pati, P. Hongmanorom, Z. Tianxi, C. Junmei and S. Kawi, Catal. Today, 2020, 356, 471–489 CrossRef CAS.
- S. Rönsch, J. Schneider, S. Matthischke, M. Schlüter, M. Götz, J. Lefebvre, P. Prabhakaran and S. Bajohr, Fuel, 2016, 166, 276–296 CrossRef.
- S. De, A. Dokania, A. Ramirez and J. Gascon, ACS Catal., 2020, 10, 14147–14185 CrossRef CAS.
- X. Jiang, X. Nie, X. Guo, C. Song and J. G. Chen, Chem. Rev., 2020, 120, 7984–8034 CrossRef CAS PubMed.
- Y. Wang, L. R. Winter, J. G. Chen and B. Yan, Green Chem., 2021, 23, 249–267 RSC.
- I. Hussain, A. A. Jalil, N. S. Hassan and M. Y. S. Hamid, J. Energy Chem., 2021, 62, 377–407 CrossRef.
- F. Sha, Z. Han, S. Tang, J. Wang and C. Li, ChemSusChem, 2020, 13, 6160–6181 CAS.
- H.-Y. Kuang, Y.-X. Lin, X.-H. Li and J.-S. Chen, J. Mater. Chem. A, 2021, 9, 20857–20873 RSC.
- P. Frontera, A. Macario, M. Ferraro and P. Antonucci, Catalysts, 2017, 7, 59 CrossRef.
- Y. Li, S. H. Chan and Q. Sun, Nanoscale, 2015, 7, 8663–8683 RSC.
- J.-B. Senderens and P. Sabatier, Comptes Rendus Acad. Sci., 1902, 82, 514–516 Search PubMed.
- C. Shen, Q. Bao, W. Xue, K. Sun, Z. Zhang, X. Jia, D. Mei and C. Liu, J. Energy Chem., 2022, 65, 623–629 CrossRef.
- C. Vogt, M. Monai, G. J. Kramer and B. M. Weckhuysen, Nat. Catal., 2019, 2, 188–197 CrossRef CAS.
- G. Yang, Y. Kuwahara, K. Mori, C. Louis and H. Yamashita, Nano Res., 2021 DOI:10.1007/s12274-021-3792-2.
- J. Zhang and Y. She, Chin. J. Catal., 2020, 41, 415–425 CrossRef CAS.
- H. Choi, S. Oh, S. B. Trung Tran and J. Y. Park, J. Catal., 2019, 376, 68–76 CrossRef CAS.
- X. Jia, K. Sun, J. Wang, C. Shen and C. Liu, J. Energy Chem., 2020, 50, 409–415 CrossRef.
- I. Sancho-Sanz, S. A. Korili and A. Gil, Cat. Rev., 2021 DOI:10.1080/01614940.2021.1968197.
- J. Zhu, F. Cannizzaro, L. Liu, H. Zhang, N. Kosinov, I. A. W. Filot, J. Rabeah, A. Brückner and E. J. M. Hensen, ACS Catal., 2021, 11, 11371–11384 CrossRef CAS PubMed.
- N. H. M. Dostagir, C. Thompson, H. Kobayashi, A. M. Karim, A. Fukuoka and A. Shrotri, Catal. Sci. Technol., 2020, 10, 8196–8202 RSC.
- Z. Zhang, C. Shen, K. Sun and C. Liu, Catal. Commun., 2022, 162, 106386 CrossRef CAS.
- X. Guo, H. Zhang, H. Yang, Z. Bo, J. Yan and K. Cen, Sep. Purif. Technol., 2021, 279, 119722 CrossRef CAS.
- K. Homlamai, T. Maihom, S. Choomwattana, M. Sawangphruk and J. Limtrakul, Appl. Surf. Sci., 2020, 499, 143928 CrossRef CAS.
- L. Lin, J. Liu, X. Liu, Z. Gao, N. Rui, S. Yao, F. Zhang, M. Wang, C. Liu, L. Han, F. Yang, S. Zhang, X.-d. Wen, S. D. Senanayake, Y. Wu, X. Li, J. A. Rodriguez and D. Ma, Nat. Commun., 2021, 12, 6978 CrossRef CAS PubMed.
- M. Millet, G. Algara-Siller, S. Wrabetz, A. Mazheika, F. Girgsdies, D. Teschner, F. Seitz, A. Tarasov, S. V. Levchenko, R. Schlögl and E. Frei, J. Am. Chem. Soc., 2019, 141, 2451–2461 CrossRef CAS PubMed.
- R. V. Gonçalves, L. L. R. Vono, R. Wojcieszak, C. S. B. Dias, H. Wender, E. Teixeira-Neto and L. M. Rossi, Appl. Catal., B, 2017, 209, 240–246 CrossRef.
- Z. Zhang, X. Hu, Y. Wang, S. Hu, J. Xiang, C. Li, G. Chen, Q. Liu, T. Wei and D. Dong, Fuel, 2019, 237, 566–579 CrossRef CAS.
- A. Farjamnia and B. Jackson, J. Chem. Phys., 2017, 146, 74704 CrossRef PubMed.
- H. J. Freund and M. W. Roberts, Surf. Sci. Rep., 1996, 25, 225–273 CrossRef.
- S. Wang, D. Cao, Y. Li, J. Wang and H. Jiao, J. Phys. Chem. B, 2005, 109, 18956–18963 CrossRef CAS PubMed.
- D. Cao, Y. Li, J. Wang and H. Jiao, Surf. Sci., 2009, 603, 2991–2998 CrossRef CAS.
- R. C. Catapan, A. A. M. Oliveira, Y. Chen and D. G. Vlachos, J. Phys. Chem. C, 2012, 116, 20281–20291 CrossRef CAS.
- M. Zhou, T. N. Le, L. K. Huynh and B. Liu, Catal. Today, 2017, 280, 210–219 CrossRef CAS.
- H. Chen, M. Yang, J. Liu, G. Lu and X. Feng, Catal. Sci. Technol., 2020, 10, 5641–5647 RSC.
- B. Kreitz, G. D. Wehinger, C. F. Goldsmith and T. Turek, J. Phys. Chem. C, 2021, 125, 2984–3000 CrossRef CAS.
- G. Peng, S. J. Sibener, G. C. Schatz and M. Mavrikakis, Surf. Sci., 2012, 606, 1050–1055 CrossRef CAS.
- G. Peng, S. J. Sibener, G. C. Schatz, S. T. Ceyer and M. Mavrikakis, J. Phys. Chem. C, 2012, 116, 3001–3006 CrossRef CAS.
- G. Peng, L. Xu, V. Glezakou and M. Mavrikakis, Catal. Sci. Technol., 2021, 11, 3279–3294 RSC.
- A. L. Maulana, R. Putra, A. G. Saputro, M. K. Agusta, Nugraha and H. K. Dipojono, Phys. Chem. Chem. Phys., 2019, 21, 20276–20286 RSC.
- P. Lozano-Reis, H. Prats, P. Gamallo, F. Illas and R. Sayós, ACS Catal., 2020, 10, 8077–8089 CrossRef CAS.
- Q. Ke, L. Kang, X. Chen and Y. Wu, J. Chem. Sci., 2020, 132, 151 CrossRef CAS.
- M. Silaghi, A. Comas-Vives and C. Copéret, ACS Catal., 2016, 6, 4501–4505 CrossRef CAS.
- A. Jafarzadeh, K. M. Bal, A. Bogaerts and E. C. Neyts, J. Phys. Chem. C, 2019, 123, 6516–6525 CrossRef CAS.
- A. Cadi-Essadek, A. Roldan, X. Aparicio-Anglès and N. H. de Leeuw, J. Phys. Chem. C, 2018, 122, 19463–19472 CrossRef CAS.
- K. R. Hahn, A. P. Seitsonen, M. Iannuzzi and J. Hutter, ChemCatChem, 2015, 7, 625–634 CrossRef CAS.
- Y. Tang, Y. Wei, Z. Wang, S. Zhang, Y. Li, L. Nguyen, Y. Li, Y. Zhou, W. Shen, F. F. Tao and P. Hu, J. Am. Chem. Soc., 2019, 141, 7283–7293 CrossRef CAS PubMed.
- G. Alonso, E. López, F. Huarte-Larrañaga, R. Sayós, H. Prats and P. Gamallo, J. CO2 Utiliz., 2021, 54, 101777 CrossRef CAS.
- K. Yuan, J. Zhong, X. Zhou, L. Xu, S. L. Bergman, K. Wu, G. Q. Xu, S. L. Bernasek, H. X. Li and W. Chen, ACS Catal., 2016, 6, 4330–4339 CrossRef CAS.
- S. Wang, D. Cao, Y. Li, J. Wang and H. Jiao, J. Phys. Chem. B, 2006, 110, 9976–9983 CrossRef CAS PubMed.
- Y. Zhou, P. Lv and G. Wang, J. Mol. Catal. A-Chem., 2006, 258, 203–215 CrossRef CAS.
- I. N. Remediakis, F. Abild-Pedersen and J. K. Nørskov, J. Phys. Chem. B, 2004, 108, 14535–14540 CrossRef CAS.
- S. J. Choe, H. J. Kang, S. Kim, S. Park, D. H. Park and D. S. Huh, B. Korean Chem. Soc., 2005, 26, 1682–1688 CrossRef CAS.
- C. Liu, T. R. Cundari and A. K. Wilson, J. Phys. Chem. C, 2012, 116, 5681–5688 CrossRef CAS.
- P. Bothra, G. Periyasamy and S. K. Pati, Phys. Chem. Chem. Phys., 2013, 15, 5701 RSC.
- H. S. Bengaard, J. K. Nørskov, J. Sehested, B. S. Clausen, L. P. Nielsen, A. M. Molenbroek and J. R. Rostrup-Nielsen, J. Catal., 2002, 209, 365–384 CrossRef CAS.
- J. A. Rodriguez, J. Evans, L. Feria, A. B. Vidal, P. Liu, K. Nakamura and F. Illas, J. Catal., 2013, 307, 162–169 CrossRef CAS.
- J. Ye, C. Liu, D. Mei and Q. Ge, J. Catal., 2014, 317, 44–53 CrossRef CAS.
- J. Ye, C. Liu, D. Mei and Q. Ge, ACS Catal., 2013, 3, 1296–1306 CrossRef CAS.
- L. C. Grabow and M. Mavrikakis, ACS Catal., 2011, 1, 365–384 CrossRef CAS.
- M. Huš, D. Kopač, N. S. Štefančič, D. L. Jurković, V. D. B. C. Dasireddy and B. Likozar, Catal. Sci. Technol., 2017, 7, 5900–5913 RSC.
- N. Rui, K. Sun, C. Shen and C. Liu, J. CO2 Utiliz., 2020, 42, 101313 CrossRef CAS.
- W. Lin, K. M. Stocker and G. C. Schatz, J. Am. Chem. Soc., 2017, 139, 4663–4666 CrossRef CAS PubMed.
- X. Nie, H. Wang, W. Li, Y. Chen, X. Guo and C. Song, J. CO2 Util., 2018, 24, 99–111 CrossRef CAS.
- P. Frontera, A. Macario, G. Monforte, G. Bonura, M. Ferraro, G. Dispenza, V. Antonucci, A. S. Aricò and P. L. Antonucci, Int. J. Hydrogen Energy, 2017, 42, 26828–26842 CrossRef CAS.
- G. Varvoutis, M. Lykaki, S. Stefa, V. Binas, G. E. Marnellos and M. Konsolakis, Appl. Catal., B, 2021, 297, 120401 CrossRef CAS.
- J. W. Hu, P. Hongmanorom, P. Chirawatkul and S. Kawi, Chem. Eng. J., 2021, 426, 130864 CrossRef CAS.
- S. Tada, T. Shimizu, H. Kameyama, T. Haneda and R. Kikuchi, Int. J. Hydrogen Energy, 2012, 37, 5527–5531 CrossRef CAS.
- H. Liu, X. Zou, X. Wang, X. Lu and W. Ding, J. Nat. Gas Chem., 2012, 21, 703–707 CrossRef CAS.
- H. Chen, J. Phys. Chem. C, 2012, 116, 6239–6246 CrossRef CAS.
- M. Huang and S. Fabris, J. Phys. Chem. C, 2008, 112, 8643–8648 CrossRef CAS.
- Y. Song, L. Yin, J. Zhang, P. Hu, X. Gong and G. Lu, Surf. Sci., 2013, 618, 140–147 CrossRef CAS.
- M. Roiaz, E. Monachino, C. Dri, M. Greiner, A. Knop-Gericke, R. Schlögl, G. Comelli and E. Vesselli, J. Am. Chem. Soc., 2016, 138, 4146–4154 CrossRef CAS PubMed.
- C. Heine, B. A. J. Lechner, H. Bluhm and M. Salmeron, J. Am. Chem. Soc., 2016, 138, 13246–13252 CrossRef CAS PubMed.
- Q. Wang, L. Wei, M. Sullivan, S. Yang and Y. Chen, RSC Adv., 2013, 3, 3046–3053 RSC.
- J. Lahiri, T. Miller, L. Adamska, I. I. Oleynik and M. Batzill, Nano Lett., 2011, 11, 518–522 CrossRef CAS PubMed.
- Y. Gamo, A. Nagashima, M. Wakabayashi, M. Terai and C. Oshima, Surf. Sci., 1997, 374, 61–64 CrossRef CAS.
- Z. Fan, K. Sun, N. Rui, B. Zhao and C. Liu, J. Energy Chem., 2015, 24, 655–659 CrossRef.
- R. Zhou, N. Rui, Z. Fan and C. Liu, Int. J. Hydrogen Energy, 2016, 41, 22017–22025 CrossRef CAS.
- Z. Wang, X. M. Cao, J. Zhu and P. Hu, J. Catal., 2014, 311, 469–480 CrossRef CAS.
- A. Ambrosetti and P. L. Silvestrelli, J. Chem. Phys., 2016, 144, 111101 CrossRef PubMed.
- L. L. Patera, F. Bianchini, G. Troiano, C. Dri, C. Cepek, M. Peressi, C. Africh and G. Comelli, Nano Lett., 2015, 15, 56–62 CrossRef CAS PubMed.
- X. Zhang, L. Wang, J. Xin, B. I. Yakobson and F. Ding, J. Am. Chem. Soc., 2014, 136, 3040–3047 CrossRef CAS PubMed.
- F. Bianchini, L. L. Patera, M. Peressi, C. Africh and G. Comelli, J. Phys. Chem. Lett., 2014, 5, 467–473 CrossRef CAS PubMed.
- L. L. Patera, F. Bianchini, C. Africh, C. Dri, G. Soldano, M. M. Mariscal, M. Peressi and G. Comelli, Science, 2018, 359, 1243–1246 CrossRef CAS PubMed.
- M. Wei, Q. Fu, Y. Yang, W. Wei, E. Crumlin, H. Bluhm and X. Bao, J. Phys. Chem. C, 2015, 119, 13590–13597 CrossRef CAS.
- C. Vogt, E. Groeneveld, G. Kamsma, M. Nachtegaal, L. Lu, C. J. Kiely, P. H. Berben, F. Meirer and B. M. Weckhuysen, Nat. Catal., 2018, 1, 127–134 CrossRef CAS.
- B. Mutz, H. W. P. Carvalho, S. Mangold, W. Kleist and J. Grunwaldt, J. Catal., 2015, 327, 48–53 CrossRef CAS.
- K. Feng, J. Tian, M. Guo, Y. Wang, S. Wang, Z. Wu, J. Zhang, L. He and B. Yan, Appl. Catal., B, 2021, 292, 120191 CrossRef CAS.
- Z. Hao, J. Shen, S. Lin, X. Han, X. Chang, J. Liu, M. Li and X. Ma, Appl. Catal., B, 2021, 286, 119922 CrossRef CAS.
- Z. Yan, Q. Liu, L. Liang and J. Ouyang, J. CO2 Util., 2021, 47, 101489 CrossRef CAS.
- N. A. Sholeha, S. Mohamad, H. Bahruji, D. Prasetyoko, N. Widiastuti, N. A. Abdul Fatah, A. A. Jalil and Y. H. Taufiq-Yap, RSC Adv., 2021, 11, 16376–16387 RSC.
- X. Jia, X. Zhang, N. Rui, X. Hu and C. Liu, Appl. Catal., B, 2019, 244, 159–169 CrossRef CAS.
- N. Rui, X. Zhang, F. Zhang, Z. Liu, X. Cao, Z. Xie, R. Zou, S. D. Senanayake, Y. Yang, J. A. Rodriguez and C. Liu, Appl. Catal., B, 2021, 282, 119581 CrossRef CAS.
- Y. Li, Y. Men, S. Liu, J. Wang, K. Wang, Y. Tang, W. An, X. Pan and L. Li, Appl. Catal., B, 2021, 293, 120206 CrossRef CAS.
- Y. Du, C. Qin, Y. Xu, D. Xu, J. Bai, G. Ma and M. Ding, Chem. Eng. J., 2021, 418, 129402 CrossRef CAS.
- P. Hongmanorom, J. Ashok, P. Chirawatkul and S. Kawi, Appl. Catal., B, 2021, 297, 120454 CrossRef CAS.
- M. Liu, H. Chen, C. Chen, J. Wu, H. Wu and C. Yang, Nanoscale, 2019, 11, 20741–20753 RSC.
- X. Xu, Y. Tong, J. Huang, J. Zhu, X. Fang, J. Xu and X. Wang, Fuel, 2021, 283, 118867 CrossRef CAS.
- S. Tada, H. Nagase, N. Fujiwara and R. Kikuchi, Energ. Fuel., 2021, 35, 5241–5251 CrossRef CAS.
- J. Ren, H. Guo, J. Yang, Z. Qin, J. Lin and Z. Li, Appl. Surf. Sci., 2015, 351, 504–516 CrossRef CAS.
- X. Yan, Y. Liu, B. Zhao, Z. Wang, Y. Wang and C. Liu, Int. J. Hydrogen Energy, 2013, 38, 2283–2291 CrossRef CAS.
- X. Zhu, P. Huo, Y. Zhang, D. Cheng and C. Liu, Appl. Catal., B, 2008, 81, 132–140 CrossRef CAS.
- Y. Dai, R. Zou, T. Ba, J. Zhang and C. Liu, J. CO2 Util., 2021, 51, 101647 CrossRef CAS.
- J. Zhang, X. Jia and C. Liu, RSC Adv., 2022, 12, 721–727 RSC.
- P. Lan, C. Wang and C. Chen, J. Taiwan Inst. Chem. E., 2020, 116, 188–196 CrossRef CAS.
- L. Liu, S. Wang, Y. Guo, B. Wang, P. Rukundo, S. Wen and Z. Wang, Int. J. Hydrogen Energy, 2016, 41, 17361–17369 CrossRef CAS.
- J. Liu, W. Bing, X. Xue, F. Wang, B. Wang, S. He, Y. Zhang and M. Wei, Catal. Sci. Technol., 2016, 6, 3976–3983 RSC.
- X. Li, D. Li, H. Tian, L. Zeng, Z. Zhao and J. Gong, Appl. Catal., B, 2017, 202, 683–694 CrossRef CAS.
- F. Yang, D. Deng, X. Pan, Q. Fu and X. Bao, Natl. Sci. Rev., 2015, 2, 183–201 CrossRef CAS.
- K. Wang, Y. Men, S. Liu, J. Wang, Y. Li, Y. Tang, Z. Li, W. An, X. Pan and L. Li, Fuel, 2021, 304, 121388 CrossRef CAS.
- C. Vogt, M. Monai, E. B. Sterk, J. Palle, A. E. M. Melcherts, B. Zijlstra, E. Groeneveld, P. H. Berben, J. M. Boereboom, E. J. M. Hensen, F. Meirer, I. A. W. Filot and B. M. Weckhuysen, Nat. Commun., 2019, 10, 5330 CrossRef PubMed.
- S. Li, Q. Wang, J. Lu, X. Deng, S. Bi, Z. Song, C. Guo, R. Li and X. Yan, Cryst. Eng. Comm., 2019, 21, 6709–6718 RSC.
- L. Shen, M. Zhu and J. Xu, Greenhouse Gas. Sci. Technol., 2021, 11, 1213–1221 CAS.
- D. Beierlein, D. Häussermann, M. Pfeifer, T. Schwarz, K. Stöwe, Y. Traa and E. Klemm, Appl. Catal., B, 2019, 247, 200–219 CrossRef CAS.
- G. Du, S. Lim, Y. Yang, C. Wang, L. Pfefferle and G. L. Haller, J. Catal., 2007, 249, 370–379 CrossRef CAS.
- W. Zhen, B. Li, G. Lu and J. Ma, Chem. Commun., 2015, 51, 1728–1731 RSC.
- M. Chen, Z. Guo, J. Zheng, F. Jing and W. Chu, J. Energy Chem., 2016, 25, 1070–1077 CrossRef.
- H. Muroyama, Y. Tsuda, T. Asakoshi, H. Masitah, T. Okanishi, T. Matsui and K. Eguchi, J. Catal., 2016, 343, 178–184 CrossRef CAS.
- P. Unwiset, K. C. Chanapattharapol, P. Kidkhunthod, Y. Poo-arporn and B. Ohtani, Chem. Eng. Sci., 2020, 228, 115955 CrossRef CAS.
- Y. Quan, N. Zhang, Z. Zhang, Y. Han, J. Zhao and J. Ren, Int. J. Hydrogen Energy, 2021, 46, 14395–14406 CrossRef CAS.
- M. Zhu, P. Tian, X. Cao, J. Chen, T. Pu, B. Shi, J. Xu, J. Moon, Z. Wu and Y. Han, Appl. Catal., B, 2021, 282, 119561 CrossRef CAS.
- G. I. Siakavelas, N. D. Charisiou, S. AlKhoori, A. A. AlKhoori, V. Sebastian, S. J. Hinder, M. A. Baker, I. V. Yentekakis, K. Polychronopoulou and M. A. Goula, Appl. Catal., B, 2021, 282, 119562 CrossRef CAS.
- Y. Zhang, T. Zhang, F. Wang, Q. Zhu and Q. Liu, Greenhouse Gas. Sci. Technol., 2021, 11, 1222–1233 CAS.
- S. Lin, Z. Hao, J. Shen, X. Chang, S. Huang, M. Li and X. Ma, J. Energy Chem., 2021, 59, 334–342 CrossRef.
- R. Tang, N. Ullah, Y. Hui, X. Li and Z. Li, Mol. Catal., 2021, 508, 111602 CrossRef CAS.
- P. Strucks, L. Failing and S. Kaluza, Chem.-Ing.-Tech., 2021, 93, 1–12 CrossRef.
- L. Shen, J. Xu, M. Zhu and Y. Han, ACS Catal., 2020, 10, 14581–14591 CrossRef CAS.
- P. Zhu, Q. Chen, Y. Yoneyama and N. Tsubaki, RSC Adv., 2014, 4, 64617–64624 RSC.
- X. Fang, L. Xia, S. Li, Z. Hong, M. Yang, X. Xu, J. Xu and X. Wang, Fuel, 2021, 293, 120460 CrossRef CAS.
- H. S. Lim, G. Kim, Y. Kim, M. Lee, D. Kang, H. Lee and J. W. Lee, Chem. Eng. J., 2021, 412, 127557 CrossRef CAS.
- H. Tian, X. Li, L. Zeng and J. Gong, ACS Catal., 2015, 5, 4959–4977 CrossRef CAS.
- J. Zhang and F. Li, Appl. Catal., B, 2015, 176–177, 513–521 CrossRef CAS.
- L. Xu, Z. Miao, H. Song, W. Chen and L. Chou, Catal. Sci. Technol., 2014, 4, 1759–1770 RSC.
- Z. Li, L. Mo, Y. Kathiraser and S. Kawi, ACS Catal., 2014, 4, 1526–1536 CrossRef CAS.
- M. A. A. Aziz, A. A. Jalil, S. Triwahyono, R. R. Mukti, Y. H. Taufiq-Yap and M. R. Sazegar, Appl. Catal., B, 2014, 147, 359–368 CrossRef CAS.
- A. Borgschulte, N. Gallandat, B. Probst, R. Suter, E. Callini, D. Ferri, Y. Arroyo, R. Erni, H. Geerlings and A. Zuttel, Phys. Chem. Chem. Phys., 2013, 15, 9620–9625 RSC.
- S. He, C. Li, H. Chen, D. Su, B. Zhang, X. Cao, B. Wang, M. Wei, D. G. Evans and X. Duan, Chem. Mater., 2013, 25, 1040–1046 CrossRef CAS.
- Y. Li, Q. Zhang, R. Chai, G. Zhao, Y. Liu, Y. Lu and F. Cao, AIChE J., 2015, 61, 4323–4331 CrossRef CAS.
- A. Vita, C. Italiano, L. Pino, P. Frontera, M. Ferraro and V. Antonucci, Appl. Catal., B, 2018, 226, 384–395 CrossRef CAS.
- L. Xu, F. Wang, M. Chen, D. Nie, X. Lian, Z. Lu, H. Chen, K. Zhang and P. Ge, Int. J. Hydrogen Energy, 2017, 42, 15523–15539 CrossRef CAS.
- G. Zhou, H. Liu, K. Cui, H. Xie, Z. Jiao, G. Zhang, K. Xiong and X. Zheng, Int. J. Hydrogen Energy, 2017, 42, 16108–16117 CrossRef CAS.
- Q. Zhang, R. Xu, N. Liu, C. Dai, G. Yu, N. Wang and B. Chen, Appl. Surf. Sci., 2022, 579, 152204 CrossRef CAS.
- M. Li, H. Amari and A. C. van Veen, Appl. Catal., B, 2018, 239, 27–35 CrossRef CAS.
- J. Zhang, H. Zhang, Y. Wu, C. Liu, Y. Huang, W. Zhou and B. Zhang, J. Mater. Chem. A, 2022 10.1039/d1ta07910g.
- Y. Huang, F. Rehman, M. Tamtaji, X. Li, Y. Huang, T. Zhang and Z. Luo, J. Mater. Chem. A, 2022 10.1039/d1ta08337f.
- L. Wang, W. Chen, D. Zhang, Y. Du, R. Amal, S. Qiao, J. Wu and Z. Yin, Chem. Soc. Rev., 2019, 48, 5310–5349 RSC.
- T. Cui, L. Li, C. Ye, X. Li, C. Liu, S. Zhu, W. Chen and D. Wang, Adv. Funct. Mater., 2021, 2108381 CrossRef.
- J. Huang, X. Li, X. Wang, X. Fang, H. Wang and X. Xu, J. CO2 Utiliz., 2019, 33, 55–63 CrossRef CAS.
- G. Baldauf-Sommerbauer, S. Lux, W. Aniser, B. Bitschnau, I. Letofsky-Papst and M. Siebenhofer, J. CO2 Utiliz., 2018, 23, 1–9 CrossRef CAS.
- W. K. Fan and M. Tahir, J. Environ. Chem. Eng., 2021, 9, 105460 CrossRef CAS.
- M. S. Frei, C. Mondelli, R. Garcia-Muelas, J. Morales-Vidal, M. Philipp, O. V. Safonova, N. Lopez, J. A. Stewart, D. C. Ferre and J. Perez-Ramirez, Nat. Commun., 2021, 12, 1960 CrossRef CAS PubMed.
- A. M. Abdel-Mageed and S. Wohlrab, Catalysts, 2022, 12, 16 CrossRef CAS.
- J. Gong, M. Chu, W. Guan, Y. Liu, Q. Zhong, M. Cao and Y. Xu, Ind. Eng. Chem. Res., 2021, 60, 9448–9455 CrossRef CAS.
- N. Rui, F. Zhang, K. Sun, Z. Liu, W. Xu, E. Stavitski, S. D. Senanayake, J. A. Rodriguez and C. Liu, ACS Catal., 2020, 10, 11307–11317 CrossRef CAS.
- K. Sun, Z. Zhang, C. Shen, N. Rui and C. Liu, Green Energy Environ., 2021 DOI:10.1016/j.gee.2021.05.004.
- J. Ye, C. Liu, D. Mei and Q. Ge, ACS Catal., 2013, 3, 1296–1306 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2022 |