DOI:
10.1039/D2TA00328G
(Paper)
J. Mater. Chem. A, 2022,
10, 12378-12390
Solvent polarity tuning to enhance the crystallinity of 2D-covalent organic frameworks for visible-light-driven hydrogen generation†
Received
13th January 2022
, Accepted 17th March 2022
First published on 17th March 2022
Abstract
The fabrication of crystalline COF materials with predictable structures and desirable features is one of the crystal engineering strategic goals. Many synthetic factors play an invisible role in the crystallization of COFs, and the nature of the solvent is one of the most influential. Herein, we synthesized two series of benzothiadiazole-based COFs using three pairs of solvent systems of different polarities to explore the effect on the COF synthesis product crystallization, as well as the photocatalytic performance of H2 evolution under visible light irradiation. The results showed that the crystallinity of the COFs increases with increasing solvent polarity. This work showed that increasing the solvent polarity not only enhanced the crystallinity, but also enhanced the thermal stability and gave highly crystalline COFs with a shorter reaction time (30 min). More interestingly, the amorphous materials generated by the use of less solvent polarity (dioxane) can be transferred into crystalline COFs by increasing the polarity of the solvent with butanol or ethanol. The high crystallinity COFs exhibited excellent hydrogen evolution rates. Therefore, this work demonstrates that the choice of solvent is very important.
Introduction
Covalent organic frameworks (COFs) are a new type of porous crystalline material with interesting periodic structures and versatile functions, built from organic linkers by forming a reversible covalent bond.1,2 COFs provide in situ control of their building blocks in two and three dimensions (2D and 3D). This control allows fine-tuning of the chemical and physical properties of the network, as well as fine-tuning of the highly arranged structures. Since Yaghi reported the first 2D COF in 2005,3 pioneering works have attracted increased study attention due to their potential applications in gas storage, batteries and optoelectronics, environmental remediation, sensing, drug delivery, catalysis, and so on.4,5 Recently, 2D COFs have been revealed as a potential type of excellent photocatalyst for H2 generation under visible light due to their unique properties, such as prominent visible light absorbance, large surface areas, tunable bandgaps, and long-range ordered structures beside π–π stacked architectures, which can improve their charge-carrier mobilities, charge separation, and optical properties.6–9 A number of conjugated organic photocatalysts, including linear polymers,10–13 polymer dots,14,15 hydrophilic polymers, and conjugated microporous polymers (CMPs),16–21 have been used for photocatalytic hydrogen generation from water under visible light. However, COF materials are distinguished from polymer photocatalysts by (a) an ideally one-step reaction, (b) limited solvent wastage, (c) high reaction yields, (d) a cheap and available co-catalyst, (e) high crystallinity, and (f) efficient charge transfer.22,23 In spite of that, thermodynamically, the COF formation by covalent binding organic building blocks is expected to be unfavorable.24 In many cases, crosslinked amorphous polymers or poorly defined solids are often obtained through the linkage of organic building blocks with strong covalent bonds. Although several building blocks have been developed, it remains a challenge to build COFs with high porosity and crystallization. It is critical to find proper conditions for the formation of a crystal structure. The solvothermal method is the most commonly used for the synthesis of COFs, but this method requires prolonged reaction time for 3–7 days at high pressures and temperatures.22,25,26 Thus, an appropriate pressure and reaction temperature are required for further optimization, and appropriate solvent combinations and ratios are critical in achieving a balance between framework development and crystallization. Smith and co-workers reported that the crystallinity of imine-linked 2D COFs is mostly affected by the acetic acid concentration and water in the reaction mixture. They observed that in the absence of acetic acid, no solid precipitate is generated during the synthesis of the TAPB-PDA COF. The absence of water led to the rapid precipitation of amorphous materials.27 The co-catalyst is another factor used in condensation reactions. For example, metal triflates, rather than acetic acid, were used as co-catalysts for the formation of imine-linked COFs by Matsumoto and co-workers. They found that quantitative reaction yields of highly crystalline COFs were formed using a catalytic amount of Sc(OTf)3 performed in short reaction times (10–30 min) at room temperature.28 One of the most important factors affecting the 2D COF synthesis with the help of a binary solvent mixture was generally used to enhance the solubility and crystallinity of COFs in the presence of an acidic catalyst. For example, common co-solvent mixtures, such as mesitylene/dioxane,29,30 DMF/NaCN,31o-dichlorobenzene/n-butanol,32 and dioxane/ethanol,33 have been employed to construct imine-linked COFs. Feng and co-workers reported that the crystallinity of the porphyrin boronic ester-linked COF produced was highly dependent on the dioxane:mesitylene mixture composition. With a mesitylene:dioxane ratio of 19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, the maximum long-range order was achieved, whereas mesitylene alone resulted in a weakly crystalline material and a dioxane concentration of 20% already resulted in an amorphous product.34 Chen et al. developed [4 + 4] COFs with different topologies (kgm and sql), pore shapes and sizes from the same A2B2 monomers by changing the reaction solvent.35 Nevertheless, several researchers have attempted to study the possibility of altering one of the factors used in imine-linked 2D COF formation to adjust the synthesis conditions to obtain a crystalline product. Because polymer crystallinity is an important feature in many applications, including photocatalysis, several of the reported studies attempted to improve COF crystallinity as shown in Table S1.† However, it should be noted that the impact of the solvent polarity on the crystallinity of the product of a COF synthesis has not been reported in detail so far.
:1, the maximum long-range order was achieved, whereas mesitylene alone resulted in a weakly crystalline material and a dioxane concentration of 20% already resulted in an amorphous product.34 Chen et al. developed [4 + 4] COFs with different topologies (kgm and sql), pore shapes and sizes from the same A2B2 monomers by changing the reaction solvent.35 Nevertheless, several researchers have attempted to study the possibility of altering one of the factors used in imine-linked 2D COF formation to adjust the synthesis conditions to obtain a crystalline product. Because polymer crystallinity is an important feature in many applications, including photocatalysis, several of the reported studies attempted to improve COF crystallinity as shown in Table S1.† However, it should be noted that the impact of the solvent polarity on the crystallinity of the product of a COF synthesis has not been reported in detail so far.
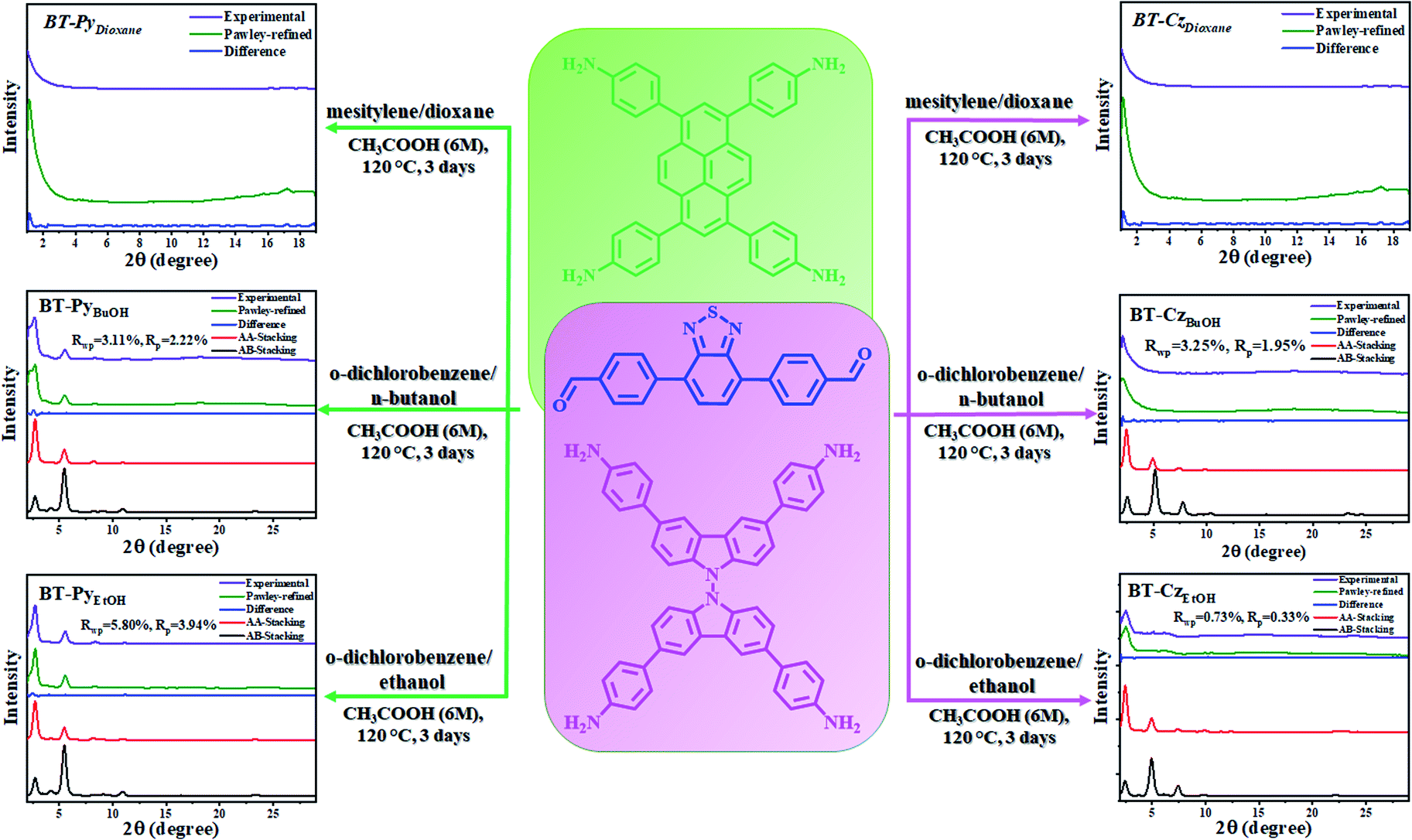
Herein, to confirm and understand this effect, we selected and synthesized two benzothiadiazole (BT)-based COFs with pyrene (Py) and carbazole (Cz) using three pairs of solvent systems (mesitylene/p-dioxane, o-dichlorobenzene/n-butanol, and o-dichlorobenzene/ethanol) (Scheme 1) and studied the effect on the crystallinity and stacking behaviors of 2D COF formation. The resulting products were denoted as BT-PySolvent and BT-CzSolvent COFs, respectively, and utilized for photocatalytic H2 evolution under visible light irradiation. Indeed, a high crystallinity was obtained from the two different COFs when using highly polar solvents (butanol and ethanol), which was experimentally confirmed from the powder X-ray diffraction (PXRD) patterns and surface areas.
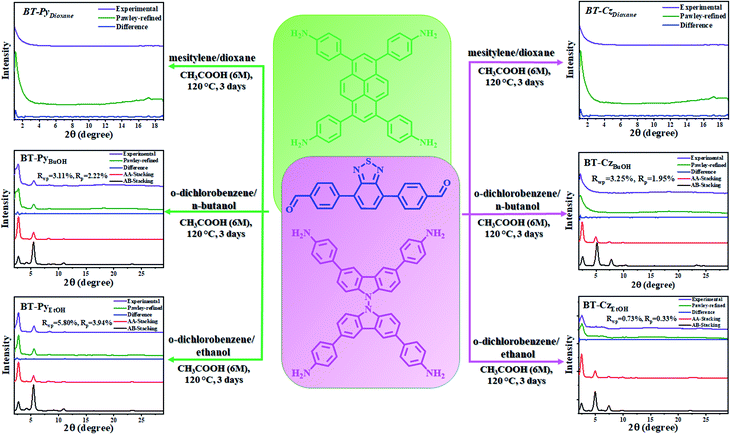 |
| | Scheme 1 Schematic diagram of the structure and preparation of BT–PySolvent and BT–CzSolvent COFs in different solvents, and the experimentally observed PXRD patterns. | |
Results and discussion
COF synthesis and characterization
The two series of BT-Pysolvent and BT-Czsolvent 2D COFs were synthesized using a condensation reaction of the corresponding diphenyl benzothiadiazole dialdehyde with pyrene and carbazole tetraamines in three pairs of mixed solvents with different solvent polarity in aqueous acetic acid at 120 °C for three days, respectively (Scheme 1).
The detailed procedure for the synthesis of these two compounds and the corresponding starting materials, as well as the characterization of these species, are described in the experimental section and ESI (Schemes S1–S5 and Fig. S1–S6).† Firstly, we synthesized two benzothiadiazole-(BT)-based COFs using pyrene (Py) and carbazole (Cz) using six different solvent systems (mesitylene/p-dioxane, o-DCB/p-dioxane, o-DCB/CHCl3, o-DCB/n-BuOH, o-DCB/EtOH, and o-DCB/MeOH) to investigate the influence on the crystallinity of 2D COF formation. All solvent systems show that when the polarity of the solvent rises, the crystallinity increases (Fig. S7†). The BT-Py COF, as demonstrated in Fig. S7,† in o-DCB/p-dioxane exhibits some crystallinity compared to that in mesitylene/p-dioxane, and we attributed this to the fact that the polarity of o-DCB is higher than that of mesitylene. Since the mixtures of solvents mesitylene/p-dioxane, o-DCB/n-BuOH and o-DCB/EtOH are the most prevalent in the synthesis of COFs, we focused our research on these solvents. A PXRD experiment was carried out to determine the crystalline nature of the as-prepared COFs (Fig. S7†). Furthermore, two different models were constructed, fully eclipsed AA stacking and staggered AB stacking (Fig. 1 and S8†). The Materials Studio 2017 software was then used to simulate a detailed crystal model. The experimental PXRD patterns agreed well with the patterns simulated from an AA-eclipsed layer stacking model. Moreover, the Pawley refinement for these COFs gave PXRD patterns that were in good agreement with the experimentally observed data, as indicated by the negligible difference (see Section 2.2.3 for more details). The construction of a periodic porous framework for the BT-Py COF was confirmed using HR-TEM. Fig. 1e shows the lattice fringes, which suggests an orderly alignment with a high degree of crystallinity. Fourier transform infrared (FT-IR) spectroscopy, solid-state 13C-nuclear magnetic resonance (13C-NMR) spectroscopy, and X-ray photoelectron spectroscopy (XPS) were used to assess the chemical structure of these COFs. The FT-IR spectra reveal a stretching vibration band at around 1625 cm−1 assigned to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond, indicating the formation of an imine linkage (Fig. S10†). Furthermore, their 13C-NMR spectra showed a signal at 158.1 and 159.6 ppm for the BT-PySolvent and BT-CzSolvent COFs, respectively (Fig. 2a and b), confirming again the formation of an imine bond. A full assignment of all resonances for other 13C-NMR spectra of COFs is shown in Fig. 2a and b. Nitrogen sorption isotherms were collected at 77 K to examine the porosity of the COFs. As shown in Fig. 2c, the isotherms of the BT-PySolvent COFs are typical type-IV isotherms with a steep increase at low relative pressures, indicating the mesoporous nature. The Brunauer–Emmett–Teller (BET) surface areas were computed to be 455, 1669, and 842 m2 g−1 for BT-PyDioxane, BT-PyBuOH, and BT-PyEtOH, respectively, and 57.9, 64.7, and 100 m2 g−1 for BT-CzDioxane, BT-CzBuOH, and BT-CzEtOH, respectively (Table S1†). The pore-size distribution of the synthesized COFs calculated using density functional theory (DFT) displayed a prominent peak at around 3.22 nm and 2.33 nm, which is in good agreement with the calculated pore size of 3.21 nm and 2.90 nm for BT-PySolvent and BT-CzSolvent COFs, respectively (Fig. 2d and S11b†). The morphological features of the as-prepared COFs were characterized using field-emission scanning electron microscopy (FE-SEM) (Fig. 4a–c and S12a–c†) and transmission electron microscopy (TEM) (Fig. 4d–f and S12d–f†). SEM images revealed that BT-PyDioxane exhibited spherical-like aggregated particles, while BT-PyBuOH presents particles with a rod morphology. The TEM images showed that all BT-Py COFs acquired a rod morphology. This indicates that the shape is strongly dependent on the nature of the solvent. The morphological difference between the BT-PyEtOH COF and the BT-PyBuOH COF could explain the significantly lower BET surface area of the BT-PyEtOH COF. Although the two COFs have good crystallinity BT-PyEtOH has a hollow tube structure as shown in Fig. 4f, and hollow structures have large free voids in the core, as these voids are very large in size (about 123.2 nm wide) for the adsorption of N2 gas molecules with a small kinetic diameter (3.64 Å) and cross-sectional area (0.162 nm2).29,36,37 On the other hand, the BT-Cz COFs exhibited low BET surface area compared to the BT-Py COFs, as seen from the nitrogen sorption analysis (Fig. 2c and S11†). The low sorption capacity and thus possibly obstructed pores of the COFs may either be attributed to their low crystallinity and the ill-defined stacking in the material, or due to residuals present in the pores, or both.38,39 Furthermore, the thermal stability of the COFs was revealed by thermogravimetric analysis (TGA) (Fig. S10†). The TGA experiments were performed using a constant flow of nitrogen gas in the range of 30–800 °C by heating the samples at a rate of 10 °C min−1. The TGA result indicated that the crystal COFs have higher thermally stability with a value of Td10 of approximately 550 and 516 °C for BT-PyBuOH and BT-CzEtOH, respectively, and displayed only less than 30% weight loss even when heated at 800 °C (Table S4†). On the other hand, the analogous amorphous COFs (BT-PyDioxane and BT-CzDioxane) displayed lower thermal stability as shown in Fig. S10.† Thus, increasing the crystallinity increased the thermal stability of the corresponding COFs. The high stability makes these COFs good candidates for potential applications in photocatalysis.18 To further investigate the formation of the COFs, the XPS spectra of the BT-Py and BT-Cz COFs were used to confirm the formation of the imine linkage, as shown in Fig. 3 and S11.† The peak at 287.1 eV in the C 1s spectrum and the signal at 398.53 eV in the N 1s spectrum are assigned to the formation of an imine (C=N) linkage. The morphological features of the as-prepared COFs were characterized using field-emission scanning electron microscopy (FE-SEM) (Fig. 4a–c and S12a–c†) and transmission electron microscopy (TEM) (Fig. 4d–f and S12d–f†). SEM images revealed that BT-PyDioxane exhibited spherical-like aggregated particles, while BT-PyBuOH presented particles with a rod morphology. TEM images showed that all of the BT-Py COFs acquired a rod morphology. This indicates that the shape is strongly dependent on the nature of the solvent.
N bond, indicating the formation of an imine linkage (Fig. S10†). Furthermore, their 13C-NMR spectra showed a signal at 158.1 and 159.6 ppm for the BT-PySolvent and BT-CzSolvent COFs, respectively (Fig. 2a and b), confirming again the formation of an imine bond. A full assignment of all resonances for other 13C-NMR spectra of COFs is shown in Fig. 2a and b. Nitrogen sorption isotherms were collected at 77 K to examine the porosity of the COFs. As shown in Fig. 2c, the isotherms of the BT-PySolvent COFs are typical type-IV isotherms with a steep increase at low relative pressures, indicating the mesoporous nature. The Brunauer–Emmett–Teller (BET) surface areas were computed to be 455, 1669, and 842 m2 g−1 for BT-PyDioxane, BT-PyBuOH, and BT-PyEtOH, respectively, and 57.9, 64.7, and 100 m2 g−1 for BT-CzDioxane, BT-CzBuOH, and BT-CzEtOH, respectively (Table S1†). The pore-size distribution of the synthesized COFs calculated using density functional theory (DFT) displayed a prominent peak at around 3.22 nm and 2.33 nm, which is in good agreement with the calculated pore size of 3.21 nm and 2.90 nm for BT-PySolvent and BT-CzSolvent COFs, respectively (Fig. 2d and S11b†). The morphological features of the as-prepared COFs were characterized using field-emission scanning electron microscopy (FE-SEM) (Fig. 4a–c and S12a–c†) and transmission electron microscopy (TEM) (Fig. 4d–f and S12d–f†). SEM images revealed that BT-PyDioxane exhibited spherical-like aggregated particles, while BT-PyBuOH presents particles with a rod morphology. The TEM images showed that all BT-Py COFs acquired a rod morphology. This indicates that the shape is strongly dependent on the nature of the solvent. The morphological difference between the BT-PyEtOH COF and the BT-PyBuOH COF could explain the significantly lower BET surface area of the BT-PyEtOH COF. Although the two COFs have good crystallinity BT-PyEtOH has a hollow tube structure as shown in Fig. 4f, and hollow structures have large free voids in the core, as these voids are very large in size (about 123.2 nm wide) for the adsorption of N2 gas molecules with a small kinetic diameter (3.64 Å) and cross-sectional area (0.162 nm2).29,36,37 On the other hand, the BT-Cz COFs exhibited low BET surface area compared to the BT-Py COFs, as seen from the nitrogen sorption analysis (Fig. 2c and S11†). The low sorption capacity and thus possibly obstructed pores of the COFs may either be attributed to their low crystallinity and the ill-defined stacking in the material, or due to residuals present in the pores, or both.38,39 Furthermore, the thermal stability of the COFs was revealed by thermogravimetric analysis (TGA) (Fig. S10†). The TGA experiments were performed using a constant flow of nitrogen gas in the range of 30–800 °C by heating the samples at a rate of 10 °C min−1. The TGA result indicated that the crystal COFs have higher thermally stability with a value of Td10 of approximately 550 and 516 °C for BT-PyBuOH and BT-CzEtOH, respectively, and displayed only less than 30% weight loss even when heated at 800 °C (Table S4†). On the other hand, the analogous amorphous COFs (BT-PyDioxane and BT-CzDioxane) displayed lower thermal stability as shown in Fig. S10.† Thus, increasing the crystallinity increased the thermal stability of the corresponding COFs. The high stability makes these COFs good candidates for potential applications in photocatalysis.18 To further investigate the formation of the COFs, the XPS spectra of the BT-Py and BT-Cz COFs were used to confirm the formation of the imine linkage, as shown in Fig. 3 and S11.† The peak at 287.1 eV in the C 1s spectrum and the signal at 398.53 eV in the N 1s spectrum are assigned to the formation of an imine (C=N) linkage. The morphological features of the as-prepared COFs were characterized using field-emission scanning electron microscopy (FE-SEM) (Fig. 4a–c and S12a–c†) and transmission electron microscopy (TEM) (Fig. 4d–f and S12d–f†). SEM images revealed that BT-PyDioxane exhibited spherical-like aggregated particles, while BT-PyBuOH presented particles with a rod morphology. TEM images showed that all of the BT-Py COFs acquired a rod morphology. This indicates that the shape is strongly dependent on the nature of the solvent.
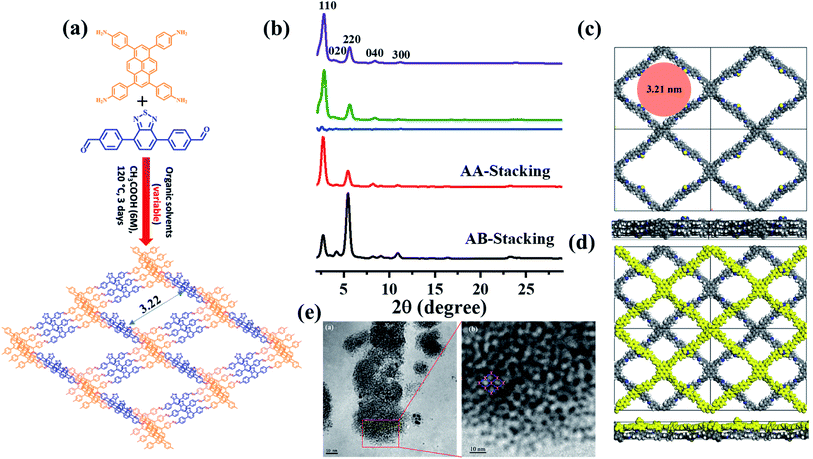 |
| | Fig. 1 (a) The synthesis of BT-PySolvent COF; (b) experimental PXRD patterns of BT-PySolvent (purple), Pawley refinement (green), their difference (blue), simulated profiles using AA-stacking (red), and AB-stacking (black) modes; unit cells of the (c) AA-stacking and (d) AB-stacking models; (e) high resolution TEM image of BT-Py COF. | |
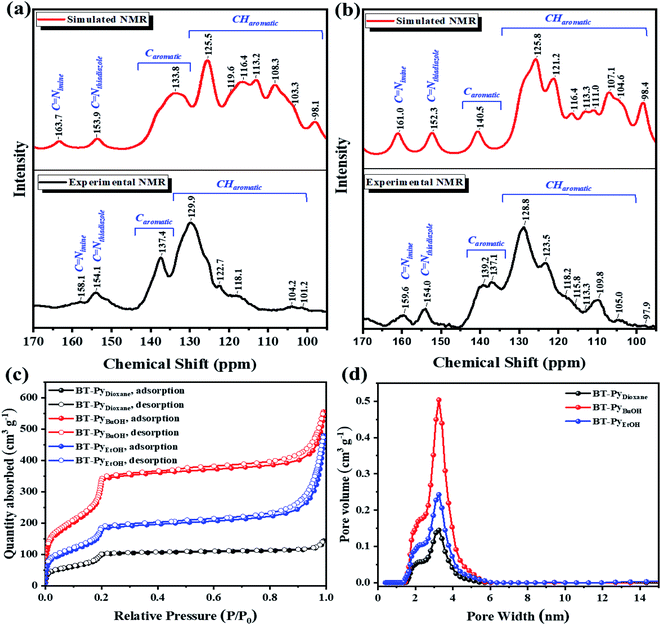 |
| | Fig. 2 Simulated and experimental 13C-NMR spectra of the (a) BT-PySolvent COFs and (b) BT-CzSolvent COFs. (c) N2 adsorption (solid symbols) and desorption (open symbols) isotherms of the BT-PySolvent COFs at 77 K. (d) The pore-size distribution of the BT-PySolvent COFs. | |
 |
| | Fig. 3 FE-SEM images of (a) BT-PyDioxane, (b) BT-PyBuOH, and (c) BT-PyEtOH. TEM images of (d) BT-PyDioxane, (e) BT-PyBuOH, and (f) BT-PyEtOH. | |
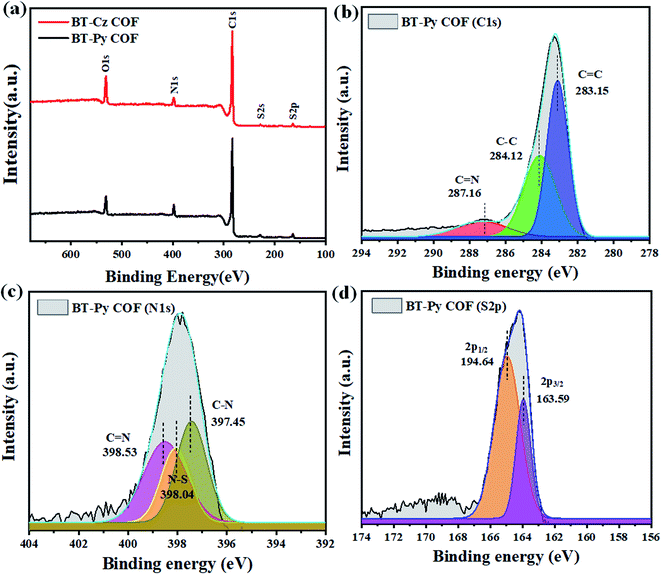 |
| | Fig. 4 (a) XPS spectra of the COFs, and high-resolutions of the (b) C 1s, (c) N 1s, and (d) S 2p peaks in the BT-Py COF. | |
Computational study
Combining experimental and computer simulation methods is an effective method for predicting and interpreting the structure of large molecules;40 see Section 3 in the ESI† for more details. It is worth noting, though, that the DFT calculations are performed for a free molecule in a vacuum, whereas the experiments are performed on solid samples that are subjected to various intra- and intermolecular interactions.
Vibrational spectral analysis (IR spectra)
The chosen characteristic vibrational band values as well as the observed values are presented in Tables S2 and S3.† The B3LYP predicted wavenumbers using the 6–31 G(d) basis set agree well with the observed IR bands of the BT-PySolvent and BT-CzSolvent COFs (Fig. S8†). In the FT-IR spectra recorded for the COFs, the vibrational stretching absorbance that appears between 1623–1626 cm−1 is due to the CN of the imine group, in agreement with the IR bands at 1646 and 1647 cm−1 for the BT-PySolvent and BT-CzSolvent COFs. The medium stretching band observed at 1523 cm−1 describes the endocyclic CN group of the thiadiazol ring.41 In addition, the CC stretching vibrations occur in the region 1430–1625 cm−1.42 In our work, this peak occurs in the range of 1482–1523 cm−1 and is calculated at 1495–1500 cm−1. The C–N vibrational stretching calculated at 1235 cm−1 was almost identical to the band for the BT-PySolvent and BT-CzSolvent COFs at 1235 and 1229 cm−1, respectively. The estimated N–N stretching of the BT-CzSolvent COFs at 1461 cm−1 corresponds to the observed IR vibrational mode at 1455–1485 cm−1.
13C NMR spectral analysis
The theoretical and experimental 13C NMR results of the BT-PySolvent and BT-CzSolvent COFs are shown in Fig. 2a and b. According to the 13C NMR spectrum of the BT-PySolvent and BT-CzSolvent COFs, the two signals at 158.1/159.6 and 154.1/154.0 ppm are assigned to the C atoms of the imine group (CN) and thiadiazol ring (CN), respectively, due to the coordination to electronegative N atoms, which agrees with the B3LYP calculated values at 163.7/161.0 and 153.9/152.3 ppm.43 The aromatic carbons of the BT-PySolvent COFs give 13C NMR signals at 137.4–101.2 ppm, and these signals are calculated at 133.8–98.1 ppm. Similarly, the BT-CzSolvent COF aromatic C atoms were observed at 139.2–97.9 ppm, which agreed with the estimated values at 140.5–98.4 ppm.
Powder X-ray diffraction data and structure simulation
The PXRD analyses of the BT-PySolvent and BT-CzSolvent COFs revealed that they were crystalline in n-butanol and ethanol (high polarity), but amorphous in dioxane (low polarity). The crystal structure was built and optimized in Materials Studio (MS) software, and the MS Reflex Plus module was used to calculate the PXRD pattern, which closely matched the experimentally observed pattern, as evidenced by very slight differences (Fig. S15†). Pawley refinement was carried out for the simulated structures and produced good agreement factors for the BT-PySolvent COFs (BT-PyBuOH: Rwp = 3.11%, Rp = 2.22%; BT-PyEtOH: Rwp = 5.80%, Rp = 3.94%) and BT-Czsolvent COFs (BT-CzBuOH: Rwp = 3.25%, Rp = 1.95%; BT-CzEtOH: Rwp = 0.73%, Rp = 0.33%). The theoretical XRD patterns obtained from the AA stacking models were highly consistent with those of the experimental ones (red curves), but the AB stacking models were considerably different (black curves). To learn more about their 2D layer conformations and unit cell parameters, fully eclipsed AA layer stacking (Fig. 1c and S8†) and staggered AB layer stacking models (Fig. 1d and S8†) of the BT-PySolvent and BT-CzSolvent COFs were constructed. The simulated structure with the AA stacking mode reveals that the BT-PySolvent and BT-CzSolvent COFs have theoretical pore sizes of 3.21 and 2.90 nm, respectively (Fig. S8a and b†), which is consistent with the experimental results (Fig. 2d). The final lattice parameters were attained by the full profile Pawley-refined fitting from the AA stacking model of the BT-PySolvent and BT-CzSolvent COFs (Tables S9–S12†).
Electronic properties
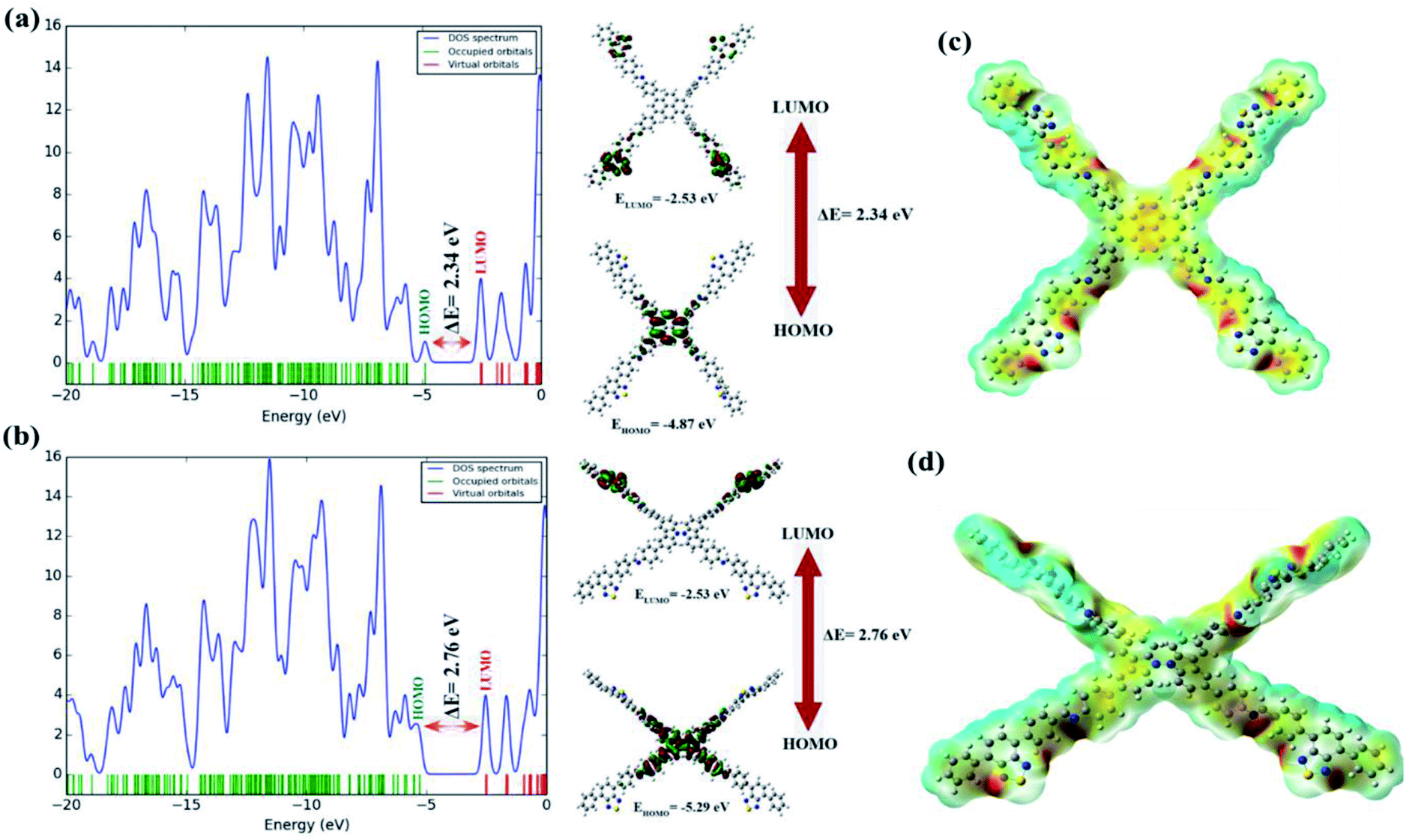
The HOMO and LUMO, as well as their energy eigenvalues, play an important role in defining the molecule's chemical reactivity descriptors. The total energy, HOMO and LUMO energies, energy gap (ΔE), absolute electronegativity (χ), ionization potential (I), electron affinity (A), absolute hardness (ɳ) and softness (S) for the title COFs were calculated at the DFT/B3LYP level in conjunction with the 6-31G(d) basis set, and the obtained results are given in Table S7†. Some important molecular orbitals (i.e., HOMO−2, HOMO−1, HOMO, LUMO, LUMO+1 and LUMO+2) of the COFs have been described as shown in Fig. S16a and b.† The molecular orbital figures reflect that the HOMO is mainly localized over the Py and Cz molecules connected to the BT in the BT-Py and BT-Cz COFs, respectively. On the other hand, the LUMO is almost spread over the BT linkers in both the BT-Py and BT-Cz COFs. The HOMO/LUMO energies for the BT-Py and BT-Cz COFs were calculated to be −4.87/−2.53 eV and −5.29/−2.53 eV, respectively. The calculated values show that the energy gap of BT-Py (2.34 eV) is lower than that of BT-Cz (2.76 eV) and is in good agreement with those obtained experimentally (Fig. 5a and b). The chemical hardness of the BT-Cz COF is found to be 1.38 eV and the value decreases to 1.17 eV in the BT-Py COF due to replacing carbazole with pyrene. The chemical reactivity descriptors show that the BT-Py COFs are more chemically reactive than the BT-Cz COFs. The frontier molecular orbital (FMO) distribution was confirmed by the density of states (DOS) spectrum performed by the Gauss Sum 3.0 program44 based on the results obtained from Gaussian 16 revision A.03 software and shown in Fig. 5.
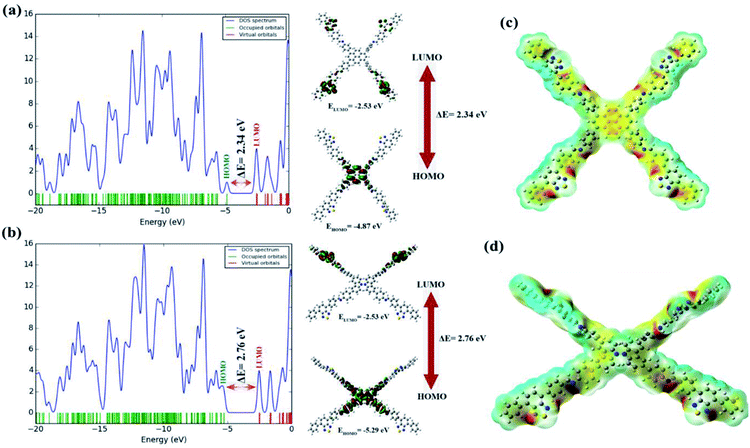 |
| | Fig. 5 HOMO–LUMO gap and DOS of the (a) BT-Py COF and (b) BT-Cz COF. The electrostatic potential map on the molecular surface for the (c) BT-Py COF and (d) BT-Cz COF with their optimized structures. | |
The DOS plot, which is a population study per orbital, clearly reveals the composition of molecular orbitals in a specific energy range (Fig. 5). The molecular electrostatic potential (MEP) is related to the electronic density around the molecule and is a very powerful descriptor for determining potential interaction sites as well as hydrogen-bonding interactions.45 MEP can predict electrophilic and nucleophilic sites in target molecules to determine the reactivity of an investigated system. The positive region represented by blue color of the MEP is a nucleophilic site, while the negative region represented by red color is associated with an electrophilic site. The potential increases in the order red < orange < yellow < green < blue. The MEP surfaces for the BT-Py and BT-Cz COFs were calculated using the B3LYP level and the 6-31G(d) basis set and are shown in Fig. 5c and d. The MEP shows that the most negative potential is found around N atoms. When it comes to the positive potential, the BT linker proton carries the most positive potential. All the other protons have a negligible positive potential.
Effect of solvent on the crystallization formation of the COFs
Solvent combinations are essential to achieve a balance between the formation and crystallization of the framework.46 When a mixture of solvents is used in the synthesis of COFs, usually one is polar and the other one is non-polar, because polar-nonpolar interfacial growth enables the spatially confined growth of COFs.47 In order to explore the role of solvent polarity in the growth of the COFs, three pairs of solvent systems of different polarities (mesitylene/p-dioxane, o-dichlorobenzene/n-butanol, and o-dichlorobenzene/ethanol), as shown in Table S2,† were used for monitoring the progress of the porous crystalline network. Thus, the condensation reactions were performed at 120 °C for 72 h. PXRD measurements were used to monitor their crystallinity. The crystallinity obtained from these three types of solvothermal solvent systems had nearly identical FT-IR and 13C NMR spectra, but differed in their PXRD patterns, indicating that they have different framework structures with the same chemical composition. As shown in Fig. S17,† amorphous materials of BT-PyDioxane and BT-CzDioxane were obtained when a solvent of low polarity (mesitylene/p-dioxane) was used. The low polarity of the solvent leads to rapid precipitation of amorphous materials. On the other hand, as the polarity of the solvent increases, the crystallization of the COFs increases. Therefore, highly crystalline COFs were obtained using the o-dichlorobenzene/ethanol system with higher solvent polarity as shown in Fig. S17.† Thus, it can be speculated that the observed crystallization of the COFs may be related to the solvents used during the reaction. In general, crystallization starts with nucleation followed by growth.48 The impact of the solvent on the crystallization kinetics may also be represented by way of its impact on nucleation and growth. In general, the solvent has two effects on the crystal nucleation and growth rate. On the one hand, the solute molecules in the solution are bound to the molecules of the solvent, which are called solvated. Solvated solute molecule desolvation must precede their integration into the crystal lattice during the nucleation and crystal growth step. On the other hand, the solvent molecules are adsorbed on the surface of a cluster of nuclei or a growing crystal surface. Both facets can be evaluated during the solvent–solute interaction, which incorporates van der Waals force and hydrogen bonding.49 The hydrogen bond donor ability is the main factor affecting crystal formation, playing a major role in determining the nucleation process.50 We can see that the hydrogen bond donor ability is increased according to the following order EtOH > BuOH > dioxane. Therefore, the higher the polarity or the hydrogen bond donor ability of the solvent, the more easily COFs crystallize, as shown in Fig. S17.† This is because the values of interaction energy (Ea) decrease with the increase of the polarity of the solvents, which enhances the crystal growth rate.51 To shed more light on the growth of the BT-Pysolvent COFs, their growth was conducted in o-dichlorobenzene/n-butanol, and o-dichlorobenzene/ethanol at different times for monitoring the evolution of the porous crystalline network as shown in Fig. 6. Thus, the condensation reactions were carried out at 0.5, 1.0, 3.0, 5.0, 12.0, 24.0 and 72.0 h. PXRD measurements were used to monitor their crystallinity. As shown in Fig. 6a, there are no peaks for BT-PyBuOH COF at 0.5 h. The characteristic peak (2θ = 2.32°) for BT-PyBuOH COF became visible after 1.0 h and a highly crystalline BT-PyBuOH COF sample could be obtained after 5 h, and the intensity of the diffraction peak did not exhibit further enhancement after 5.0 h. On the other hand, the characteristic peak (2θ = 2.32°) of BT-PyEtOH COF becomes visible rapidly at 0.5 h and a highly crystalline BT-PyEtOH COF sample can be obtained after 1.0 h as shown in Fig. 6b. In addition, the condensation reactions for the BT-PyBuOH and BT-PyEtOH COFs were carried out at 90 °C and 120 °C. PXRD measurements were used to monitor their crystallinity as shown in Fig. S18.† The BT-PyBuOH COF showed low crystallization at low temperature (90 °C); on the other hand, the BT-PyEtOH COF showed high crystallization at both temperatures, and this corresponds to the lower activation energy of the reaction with increasing solvent polarity. This means that the higher the polarity of the solvent or the hydrogen bonding of the solvent, the more easily COFs crystallize.
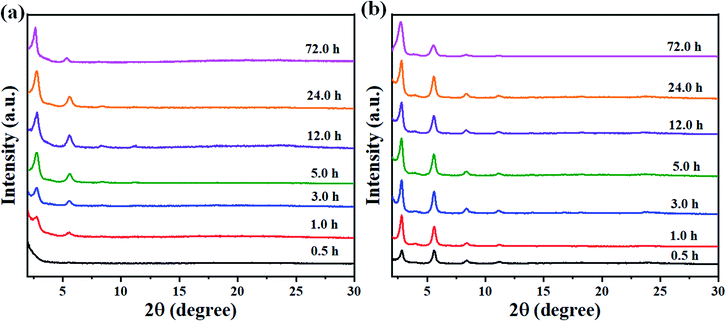 |
| | Fig. 6 Experimentally observed PXRD patterns of (a) BT-PyBuOH COF and (b) BT-CzEtOH COF at different reaction times. | |
Transforming amorphous or CMP materials into crystalline COFs
Motivated by the self-healing ability of dynamic covalent bonds, we hypothesized a disorder-to-order transition scenario from amorphous organic networks to crystallized COFs by changing the polarity of the solvent. Using mesitylene/dioxane, we synthesized the COF in the same way as stated previously at 120 °C for 72 h, yielding amorphous materials as shown in Fig. 7. Then, we split the amorphous material into two tubes, one with o-dichlorobenzene/n-butanol and the other with o-dichlorobenzene/ethanol, and performed the reaction under identical conditions as given in the section on COF synthesis in the ESI† at 120 °C for 72 h. As shown in Fig. 7, in the case of dioxane of low polarity, we obtained amorphous organic networks, and the amorphous material is transferred to a crystallized COF, and the crystallinity increases with increasing polarity of the solvent, which clearly indicates that the polarity of the solvent has a significant impact on the synthesis of 2D COFs.
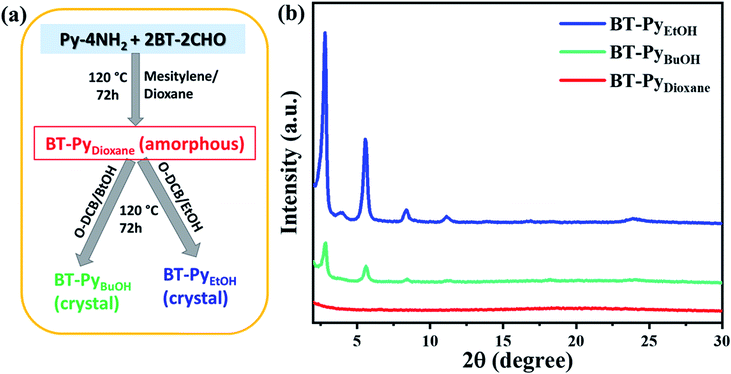 |
| | Fig. 7 (a) Schematic synthesis and (b) experimentally observed PXRD patterns of transforming amorphous material BT-PyDioxane COF into crystalline BT-PyBuOH COF and BT-PyEtOH COF. | |
Optical and electronic properties
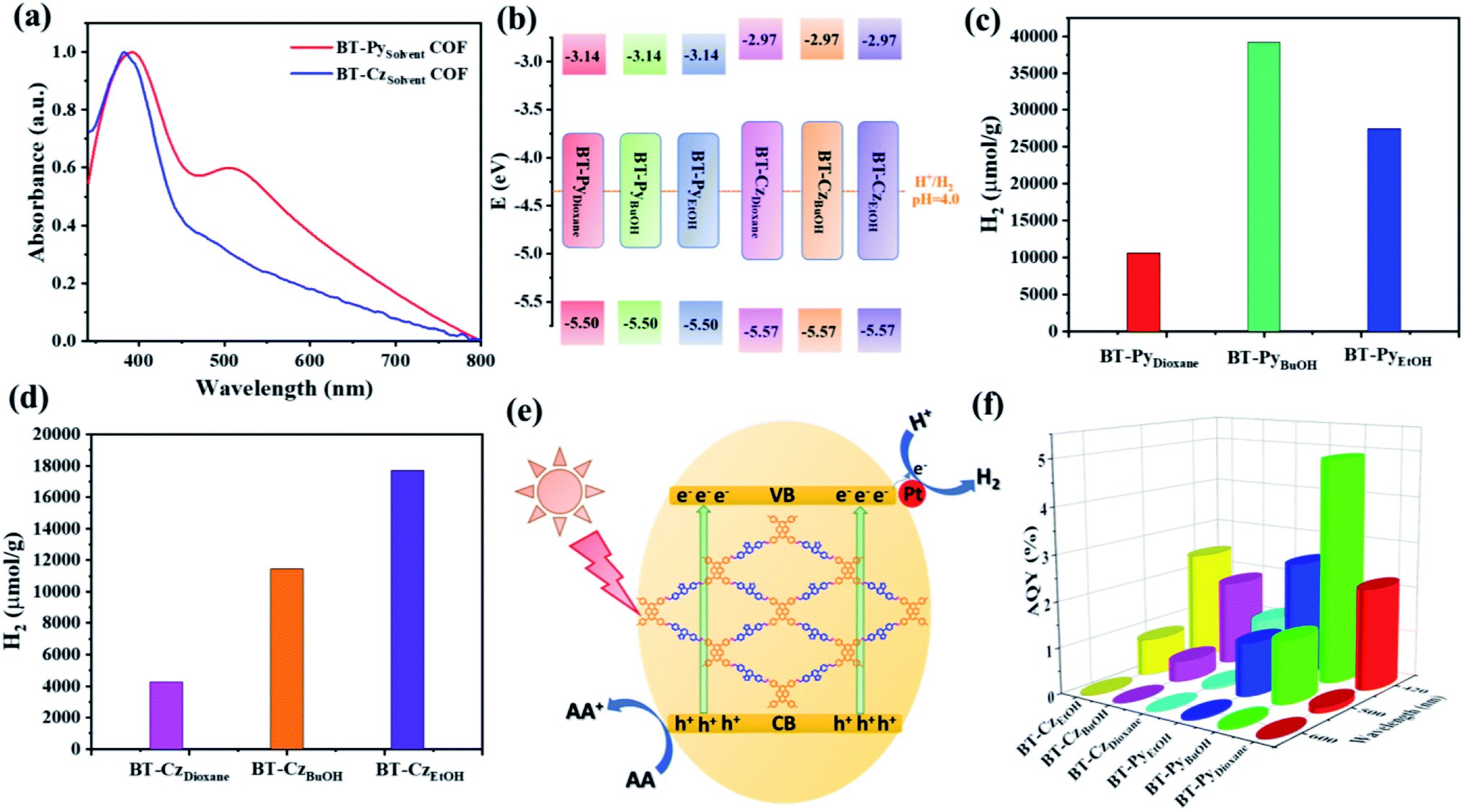
The UV-vis diffuse reflection absorption spectra of BT-Py and BT-Cz COFs show absorption peaks in both the UV and visible regions (Fig. 8a). Nevertheless, the frameworks with a pyrene moiety exhibited redshifts in the visible light region in comparison with the BT-Cz COF, which is an indication that the pyrene moiety is favorable for enhancing visible light absorption due to extended π-conjugation. Accordingly, from the Tauc plot spectra, the optical band gap energies (Eg) for BT-Py COF and BT-Cz COF were estimated to be 2.36 and 2.60 eV, respectively (Table 1). Ultraviolet photoelectron spectroscopy (UPS) is used to determine the energy levels of the VBs of the BT-PySolvent and BT-CzSolvent COFs as shown in Table 1, and Fig. 8b, S17 and S18†.
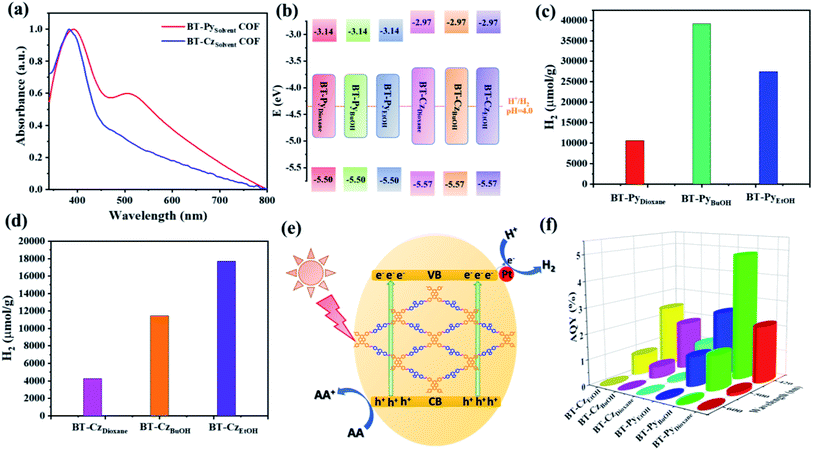 |
| | Fig. 8 (a) UV-vis diffuse absorption spectra of the BT-Py and BT-Cz COFs. (b) Calculated VB/CB energy levels of the COFs. Time course of the produced H2 of the (c) BT-PySolvent COFs and (d) BT-CzSolvent COFs for 4 h under irradiation with visible light (λ = 380–780 nm). (e) Schematic diagram of the photocatalytic H2 reduction over the COFs under visible light irradiation. (f) Wavelength dependence of AQY on the H2 evolution of the COFs. | |
Table 1 Photophysical properties and H2 evolution rates of the COFs
| Polymer |
VB/CB (eV)a,b |
Bandgap (eV)c |
Lifetime [ns] |
HER (μmol g−1 h−1)d |
AQY (%) |
| 420 nm |
500 nm |
|
VB calculated using ultraviolet photoelectron spectroscopy.
CB = VB + Eg.
Calculated from Tauc plots.
Conditions: 1 mg of COFs in 10 mL of a mixture of water/MeOH (2:1)/0.1 M AA and 2% Pt as a co-catalyst, measured under 350 W Xe light (AM 1.5; λ = 380–780 nm; 1000 W m−2).
|
| BT-PyDioxane |
−5.50/−3.14 |
2.36 |
4.42 |
3037 |
2.20 |
0.12 |
| BT-PyBuOH |
−5.50/−3.14 |
2.36 |
4.74 |
11285 |
4.88 |
1.36 |
| BT-PyEtOH |
−5.50/−3.14 |
2.36 |
4.62 |
8137 |
2.52 |
1.15 |
| BT-CzDioxane |
−5.57/−2.97 |
2.60 |
4.29 |
1198 |
1.12 |
0.06 |
| BT-CzBuOH |
−5.57/−2.97 |
2.60 |
4.33 |
3485 |
1.86 |
0.45 |
| BT-CzEtOH |
−5.57/−2.97 |
2.60 |
4.64 |
4546 |
2.41 |
0.79 |
Photocatalytic hydrogen evolution efficacy of the COFs
The photocatalytic hydrogen evolution experiments were carried out under visible light irradiation (λ = 380– 780 nm) at ambient temperature. The evolved gas (500 μL) from the photoreactor was periodically monitored every hour and analyzed by gas chromatography (GC). Two series of BT-PySolvent and BT-CzSolvent COFs were investigated for photocatalytic hydrogen evolution from water in the presence of ascorbic acid (AA) as a sacrificial electron donor (SED) and methanol was added to improve the dispersion of the photocatalysts in water. H2PtCl6 was added for the in situ formation of the platinum (Pt) co-catalyst to enhance the photocatalytic hydrogen evolution. Firstly, we investigated potential sacrificial donors, such as triethylamine (TEA), triethanolamine (TEOA), and AA, in an aqueous solution for the BT-PySolvent and BT-CzSolvent COFs by measuring the hydrogen evolution rate (HER) for 4 h under visible-light irradiation in the presence of the Pt cocatalysts (Fig. S21 and S22†). The hydrogen evolution rate produced from the photocatalytic reaction is the highest with AA as the SED. Therefore, ascorbic acid was used in the additional optimization tests, as reported in the ESI† (Fig. S23–S25†). As shown in Fig. 8c, the HERs of the BT-PyDioxane, BT-PyBuOH, and BT-PyEtOH COFs are 3037, 11285, and 8137 μmol g−1 h−1, respectively. Fig. 8c and d demonstrate that increasing the crystallinity of the COFs enhances the photocatalytic HER. This is because crystalline materials have a long-range order, which facilitates charge transfer, prevents charge carrier recombination, and reduces charge trapping at defect sites.52
The crystalline BT-PyBuOH COF displayed a 3.66-fold increase in the HER compared to its corresponding amorphous BT-PyDioxane analogue. Tan and his group obtained the same results, in which they reported that the high crystallinity CTF-COFs showed higher photocatalytic activity compared to their low crystallinity CTF-COF analogues.53 The HER of the crystalline CTF-HUST-C1 was 4.75 times higher than that of its low-crystalline analogue, CTF-HUST-1. Similarly, employing Pt as a co-catalyst and AA as a SED, the crystalline FS-COF showed a 9-fold higher H2 production than its amorphous counterpart.54 Furthermore, we summarized the HER performances of various COF-based photocatalysts under visible-light irradiation in the presence of a Pt cocatalyst and sacrificial solution conditions in Table S13.† Among them, our designed COFs achieved the best photocatalytic efficiencies under the optimized photocatalytic conditions. On the other hand, the photocatalytic efficiency of BT-PyBuOH COF is higher than that of BT-PyEtOH COF, even though the crystallization of both is very close to each other. This is due to the BET surface area of BT-PyBuOH COF (1669 m2 g−1) being substantially higher than that of BT-PyEtOH COF (800 m2 g−1), resulting in more active sites, where high BET surface areas are advantageous for enhancing the catalyst dispersity and interaction with the reactants, thus giving more efficient hydrogen production. BT-PyBuOH not only improves the hydrogen production rate, but also has a good photocatalytic stability under optimal conditions as shown in Fig. S26.† The predicted mechanism of the basic principle of photocatalytic H2 production on a COF photocatalyst is depicted in Fig. 8e. The apparent quantum yields (AQYs) for the hydrogen evolution were measured under the illumination of a 300 W Xe lamp with different bandpass filters (420, 500, and 600 nm) (Fig. 8f). The AQY of the BT-PyBuOH COF gives the highest value up to 4.88% at 420 nm, which is consistent with the photocatalytic efficiency. The reasons for the enhanced photocatalytic performances of these COFs were further investigated. The generation of photoinduced electron–hole pairs, as well as separation and migration, is considered to be the basic process of photocatalytic reactions. In general, PL spectra can reflect the separation efficiency of photogenerated electron–hole pairs of photocatalysts,55 and thus the steady-state PL spectra were measured as shown in Fig. 9a and b. The PL intensity decreases with an increase in the crystallinity of the COFs. This is an indication that higher crystallinity could improve the charge migration efficiency. As shown in Fig. 9c and d, the time-resolved photoluminescence decay spectra in tetrahydrofuran solution without adding a Pt co-catalyst were recorded at room temperature. Thus, the obtained long excited fluorescence lifetimes are 4.42, 4.74, 4.62, 4.29, 4.33, and 4.64 ns, for BT-PyDioxane, BT-PyBuOH, BT-PyEtOH, BT-CzDioxane, BT-CzBuOH, and BT-CzEtOH, respectively, which were consistent with the photocatalytic hydrogen evolution efficiency. Furthermore, transient photocurrent measurements and electrochemical impedance spectroscopy (EIS) were employed to investigate the charge separation and migration behaviours of the different COFs as shown in Fig. 9e–h. The markedly increased photocurrent of BT-PyBuOH, BT-PyEtOH, and BT-CzEtOH further proves the inhibition of charge recombination, in good agreement with the EIS data. The efficient charge transfer for BT-PyBuOH, BT-PyEtOH, and BT-CzEtOH should originate from the high crystallinity, which could synergistically improve charge transfer, resulting in high photocatalytic efficiency. Finally, the control experiments showed that the kinetic curve of the photocatalytic process was increased under a light-on condition and decreased under a light-off condition, confirming that our system was a photocatalytic reaction (Fig. S27†). It is important to note that no H2 was detected in the absence of AA or photocatalyst, validating the photoactive function of our COFs.
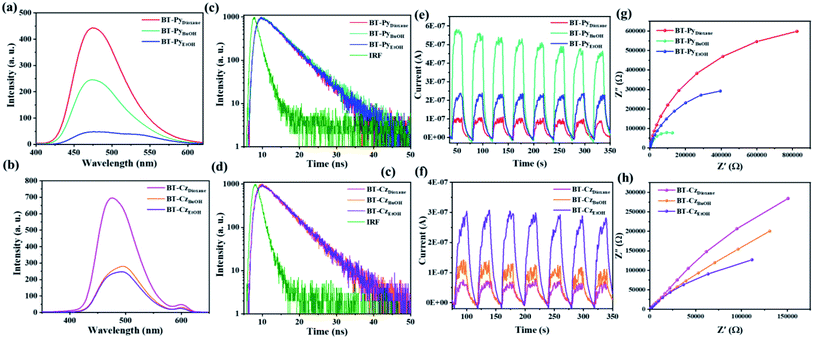 |
| | Fig. 9 PL spectra of the (a) BT-PySolvent COFs and (b) BT-CzSolvent COFs. Time-resolved fluorescence decay in DMF of the (c) BT-PySolvent COFs and (d) BT-CzSolvent COFs (IRF: instrument response factor). Transient photocurrent measurements of the (e) BT-PySolvent COFs and (f) BT-CzSolvent COFs under visible light irradiation (LED lamp, λ > 420 nm). Nyquist plots of the (g) BT-PySolvent COFs and (h) BT-CzSolvent COFs. | |
Conclusions
In summary, we have successfully studied the effect of organic solvents of different polarities on the synthesis of two-dimensional imine-linked 2D COFs. We found that the polarity of the solvent plays a vital role in the nucleation and crystal growth. The use of a highly polar organic solvent as the reaction medium greatly facilitates the preparation of crystalline COFs as elucidated by PXRD. Therefore, the higher the polarity or the hydrogen bond donor ability of the solvent, the more easily the COFs crystallize. This is because the values of interaction energy (Ea) decrease with an increase in the polarity of the solvents, which enhances the crystal growth rate. The structure of the synthesized COFs has been illustrated by several characterizations, as well as the interpretation of the structure of the COFs by the combination of experimental and computer simulation methods. The photocatalytic applications of the synthesized COFs for hydrogen evolution from water have been investigated. The BT-PySolvent COFs show significantly higher photocatalytic H2 evolution compared to the BT-CzSolvent COFs. The high crystallinity COFs exhibited excellent hydrogen evolution rates up to 11.28 mmol h−1 g−1 with apparent quantum yields of 4.88% at 420 nm for BT-PyBuOH under visible-light-driven conditions.
Author contributions
Conceptualization: Ho-Hsiu Chou, Ahmed M. Elewa Data curation: Ahmed M. Elewa Formal analysis: Ahmed M. Elewa Funding acquisition: Ho-Hsiu Chou Investigation: Ho-Hsiu Chou, Ahmed M. Elewa Methodology: Ahmed M. Elewa, Ahmed F. M. EL-Mahdy, Ahmed E. Hassan, Zhenhai Wen, Jayachandran Jayakumar, Tsung-Lin Lee, Li-Yu Ting, Islam M. A. Mekhemer, Tse-Fu Huang, Mohamed Hammad Elsayed, Chih-Li Chang, Wei-Cheng Lin Project administration: Ho-Hsiu Chou, Ahmed M. Elewa Resources: Ho-Hsiu Chou Software: Ahmed M. Elewa, Ahmed E. Hassan, Zhenhai Wen Supervision: Ho-Hsiu Chou Validation: Ho-Hsiu Chou Visualization: Ho-Hsiu Chou, Ahmed M. Elewa Writing – original draft: Ahmed M. Elewa Writing – review & editing: Ho-Hsiu Chou, Ahmed M. Elewa, Ahmed F. M. EL-Mahdy
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors gratefully acknowledge the financial support of the Ministry of Science and Technology of Taiwan (MOST 110-2636-E-007-020 and MOST 111-2628-E-007-009-) and thank the National Center for High-Performance Computing of Taiwan for providing the computing time. The authors appreciate the Precision Instrument Support Center of National Tsing Hua University for providing the analysis and measurement facilities.
References
- M. S. Lohse and T. Bein, Adv. Funct. Mater., 2018, 28, 1705553 CrossRef
 .
.
- Y. Li, Q. Chen, T. Xu, Z. Xie, J. Liu, X. Yu, S. Ma, T. Qin and L. Chen, J. Am. Chem. Soc., 2019, 141, 13822–13828 CrossRef CAS PubMed .
- A. P. Cote, A. I. Benin, N. W. Ockwig, M. O'Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166–1170 CrossRef CAS PubMed .
- P. Dong, Y. Wang, A. Zhang, T. Cheng, X. Xi and J. Zhang, ACS Catal., 2021, 11, 13266–13279 CrossRef CAS .
- S. Mondal, B. Mohanty, M. Nurhuda, S. Dalapati, R. Jana, M. Addicoat, A. Datta, B. K. Jena and A. Bhaumik, ACS Catal., 2020, 10, 5623–5630 CrossRef CAS .
- S. Ghosh, A. Nakada, M. A. Springer, T. Kawaguchi, K. Suzuki, H. Kaji, I. Baburin, A. Kuc, T. Heine and H. Suzuki, J. Am. Chem. Soc., 2020, 142, 9752–9762 CAS .
- Y. Wang, A. Vogel, M. Sachs, R. S. Sprick, L. Wilbraham, S. J. Moniz, R. Godin, M. A. Zwijnenburg, J. R. Durrant and A. I. Cooper, Nat. Energy, 2019, 4, 746–760 CrossRef CAS .
- W. Li, X. Huang, T. Zeng, Y. A. Liu, W. Hu, H. Yang, Y.-B. Zhang and K. Wen, Angew. Chem., Int. Ed., 2021, 60, 1869–1874 CrossRef CAS PubMed .
- J. Yang, F. Kang, X. Wang and Q. Zhang, Mater. Horiz., 2022, 9, 121–146 RSC .
- W.-C. Lin, M. H. Elsayed, J. Jayakumar, L.-Y. Ting, C.-L. Chang, A. M. Elewa, W.-S. Wang, C.-C. Chung, C.-Y. Lu and H.-H. Chou, Int. J. Hydrogen Energy, 2020, 45, 32072–32081 CrossRef CAS .
- W.-H. Wang, L.-Y. Ting, J. Jayakumar, C.-L. Chang, W.-C. Lin, C.-C. Chung, M. H. Elsayed, C.-Y. Lu, A. M. Elewa and H.-H. Chou, Sustainable Energy Fuels, 2020, 4, 5264–5270 RSC .
- W.-C. Lin, J. Jayakumar, C.-L. Chang, L.-Y. Ting, T.-F. Huang, M. H. Elsayed, A. M. Elewa, Y.-T. Lin, J.-J. Liu, Y.-T. Tseng and H.-H. Chou, J. Mater. Chem. A, 2022, 10, 6641–6648 RSC .
- M.-J. Yu, C.-L. Chang, H.-Y. Lan, Z.-Y. Chiao, Y.-C. Chen, H. W. Howard Lee, Y.-C. Chang, S.-W. Chang, T. Tanaka, V. Tung, H.-H. Chou and Y.-J. Lu, ACS Photonics, 2021, 8, 3125–3132 CrossRef CAS .
- M. H. Elsayed, J. Jayakumar, M. Abdellah, T. H. Mansoure, K. Zheng, A. M. Elewa, C.-L. Chang, L.-Y. Ting, W.-C. Lin and H.-h. Yu, Appl. Catal., B, 2021, 283, 119659 CrossRef CAS .
- A. M. Elewa, J. Jayakumar, Y.-W. Huang, M. H. Elsayed, C.-L. Chang, L.-Y. Ting, W.-C. Lin, C.-C. Chueh and H.-H. Chou, J. Environ. Chem. Eng., 2022, 10, 106927 CrossRef CAS .
- T.-L. Lee, A. Elewa, M. G. Kotp, H.-H. Chou and A. F. EL-Mahdy, Chem. Commun., 2021, 57, 11968–11971 RSC .
- M. G. Mohamed, M. H. Elsayed, A. M. Elewa, A. F. EL-Mahdy, C.-H. Yang, A. A. Mohammed, H.-H. Chou and S.-W. Kuo, Catal. Sci. Technol., 2021, 11, 2229–2241 RSC .
- A. M. Elewa, A. F. EL-Mahdy, M. H. Elsayed, M. G. Mohamed, S.-W. Kuo and H.-H. Chou, Chem. Eng. J., 2021, 421, 129825 CrossRef CAS .
- A. M. Elewa, M. H. Elsayed, A. F. El-Mahdy, C.-L. Chang, L.-Y. Ting, W.-C. Lin, C.-Y. Lu and H.-H. Chou, Appl. Catal., B, 2021, 285, 119802 CrossRef CAS .
- M. G. Kotp, A. M. Elewa, A. F. M. El-Mahdy, H.-H. Chou and S.-W. Kuo, ACS Appl. Energy Mater., 2021, 4(11), 13140–13151 CrossRef CAS .
- J. Byun and K. A. Zhang, Mater. Horiz., 2020, 7, 15–31 RSC .
- X. Ma and T. F. Scott, Commun. Chem., 2018, 1, 1–15 CrossRef .
- Y. Song, Q. Sun, B. Aguila and S. Ma, Adv. Sci., 2019, 6, 1801410 CrossRef PubMed .
- L. Deng, J. Zhang and Y. Gao, Mesoporous Mater., 2018, 1–27 Search PubMed .
- X. Li, C. Yang, B. Sun, S. Cai, Z. Chen, Y. Lv, J. Zhang and Y. Liu, J. Mater. Chem. A, 2020, 8, 16045–16060 RSC .
- H. R. Abuzeid, A. F. EL-Mahdy and S.-W. Kuo, Giant, 2021, 100054 CrossRef CAS .
- B. J. Smith, A. C. Overholts, N. Hwang and W. R. Dichtel, Chem. Commun., 2016, 52, 3690–3693 RSC .
- M. Matsumoto, R. R. Dasari, W. Ji, C. H. Feriante, T. C. Parker, S. R. Marder and W. R. Dichtel, J. Am. Chem. Soc., 2017, 139, 4999–5002 CrossRef CAS PubMed .
- A. F. El-Mahdy, C. Young, J. Kim, J. You, Y. Yamauchi and S.-W. Kuo, ACS Appl. Mater. Interfaces, 2019, 11, 9343–9354 CrossRef CAS PubMed .
- V. S. Vyas, F. Haase, L. Stegbauer, G. Savasci, F. Podjaski, C. Ochsenfeld and B. V. Lotsch, Nat. commun., 2015, 6, 1–9 Search PubMed .
- A. F. El-Mahdy, Y. H. Hung, T. H. Mansoure, H. H. Yu, T. Chen and S. W. Kuo, Chem.–Asian J., 2019, 14, 1429–1435 CrossRef CAS PubMed .
- A. F. EL-Mahdy, A. M. Elewa, S. W. Huang, H. H. Chou and S. W. Kuo, Adv. Opt. Mater., 2020, 8, 2000641 CrossRef CAS .
- L.-H. Li, X.-L. Feng, X.-H. Cui, Y.-X. Ma, S.-Y. Ding and W. Wang, J. Am. Chem. Soc., 2017, 139, 6042–6045 CrossRef CAS PubMed .
- X. Feng, L. Chen, Y. Dong and D. Jiang, Chem. Commun., 2011, 47, 1979–1981 RSC .
- Y. Li, L. Guo, Y. Lv, Z. Zhao, Y. Ma, W. Chen, G. Xing, D. Jiang and L. Chen, Angew. Chem., Int. Ed., 2021, 60, 5363–5369 CrossRef CAS PubMed .
- P. Pachfule, S. Kandmabeth, A. Mallick and R. Banerjee, Chem. Commun., 2015, 51, 11717–11720 RSC .
- A. F. El-Mahdy, M.-Y. Lai and S.-W. Kuo, J. Mater. Chem. C, 2020, 8, 9520–9528 RSC .
- F. Haase, T. Banerjee, G. Savasci, C. Ochsenfeld and B. V. Lotsch, Faraday Discuss., 2017, 201, 247–264 RSC .
- H.-L. Qian, C.-X. Yang and X.-P. Yan, Nat. commun., 2016, 7, 1–7 Search PubMed .
-
T. Schlick, Molecular Modeling and Simulation: an Interdisciplinary Guide, Springer, 2010 Search PubMed .
- T. A. Mohamed, A. E. Hassan, I. A. Shaaban, A. M. Abuelela and W. M. Zoghaib, J. Mol. Struct., 2017, 1130, 434–441 CrossRef CAS .
- A. Teimouri, A. N. Chermahini, K. Taban and H. A. Dabbagh, Spectrochim. Acta - A: Mol. Biomol. Spectrosc., 2009, 72, 369–377 CrossRef PubMed .
- R. K. Dani, M. K. Bharty, S. K. Kushawaha, S. Paswan, O. Prakash, R. K. Singh and N. K. Singh, J. Mol. Struct., 2013, 1054–1055, 251–261 CrossRef CAS .
- N. M. O'boyle, A. L. Tenderholt and K. M. J. Langner, J. Comput. Chem., 2008, 29, 839–845 CrossRef PubMed .
-
E. Scrocco and J. Tomasi, in Advances in Quantum Chemistry, ed. P.-O. Löwdin, Academic Press, 1978, vol. 11, pp. 115–193 Search PubMed .
- J. You, Y. Zhao, L. Wang and W. Bao, J. Cleaner Prod., 2021, 125822 CrossRef CAS .
- S. Kim and H. C. Choi, ACS omega, 2019, 5, 948–958 CrossRef PubMed .
- D. Erdemir, A. Y. Lee and A. S. Myerson, Acc. Chem. Res., 2009, 42, 621–629 CrossRef CAS PubMed .
- W. Du, Q. Yin, H. Hao, Y. Bao, X. Zhang, J. Huang, X. Li, C. Xie and J. Gong, Ind. Eng. Chem., 2014, 53, 5652–5659 CrossRef CAS .
- W. Du, Q. Yin, J. Gong, Y. Bao, X. Zhang, X. Sun, S. Ding, C. Xie, M. Zhang and H. Hao, Cryst. Growth Des., 2014, 14, 4519–4525 CrossRef CAS .
- C.-l. Wu, S.-h. Zhang, R.-j. Gou, G. Han and S.-f. Zhu, Comput. Theor. Chem., 2018, 1127, 22–30 CrossRef CAS .
- T. Banerjee, K. Gottschling, G. k. Savasci, C. Ochsenfeld and B. V. Lotsch, ACS Energy Lett., 2018, 3, 400–409 CrossRef CAS PubMed .
- M. Liu, Q. Huang, S. Wang, Z. Li, B. Li, S. Jin and B. Tan, Angew. Chem., Int. Ed., 2018, 57, 11968–11972 CrossRef CAS PubMed .
- X. Wang, L. Chen, S. Y. Chong, M. A. Little, Y. Wu, W.-H. Zhu, R. Clowes, Y. Yan, M. A. Zwijnenburg, R. S. Sprick and A. I. Cooper, Nat. Chem., 2018, 10, 1180–1189 CrossRef CAS PubMed .
- Y. Zhao, Q. Zeng, Y. Yu, T. Feng, Y. Zhao, Z. Wang, Y. Li, C. Liu, J. Liu and H. Wei, Mater. Horiz., 2020, 7, 2719–2725 RSC .
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d2ta00328g |
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 ab,
Ahmed F. M.
EL-Mahdy
ab,
Ahmed F. M.
EL-Mahdy