
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Immobilization strategies for porphyrin-based molecular catalysts for the electroreduction of CO2
Maryam
Abdinejad
 *a,
Keith
Tang
b,
Caitlin
Dao
b,
Saeed
Saedy
a and
Tom
Burdyny
*a
*a,
Keith
Tang
b,
Caitlin
Dao
b,
Saeed
Saedy
a and
Tom
Burdyny
*a
aDepartment of Chemical Engineering, Delft University of Technology, Van der Maasweg 9, 2629 HZ Delft, The Netherlands. E-mail: M.Abdinejad@tudelft.nl; T.E.Burdyny@TUDelft.nl
bDepartment of Physical and Environmental Sciences, University of Toronto Scarborough, 1265 Military Trail, Toronto, ON M1C 1A4, Canada
First published on 17th March 2022
Abstract
The ever-growing level of carbon dioxide (CO2) in our atmosphere, is at once a threat and an opportunity. The development of sustainable and cost-effective pathways to convert CO2 to value-added chemicals is central to reducing its atmospheric presence. Electrochemical CO2 reduction reactions (CO2RRs) driven by renewable electricity are among the most promising techniques to utilize this abundant resource; however, in order to reach a system viable for industrial implementation, continued improvements to the design of electrocatalysts is essential to improve the economic prospects of the technology. This review summarizes recent developments in heterogeneous porphyrin-based electrocatalysts for CO2 capture and conversion. We specifically discuss the various chemical modifications necessary for different immobilization strategies, and how these choices influence catalytic properties. Although a variety of molecular catalysts have been proposed for CO2RRs, the stability and tunability of porphyrin-based catalysts make their use particularly promising in this field. We discuss the current challenges facing CO2RRs using these catalysts and our own solutions that have been pursued to address these hurdles.
Introduction
Carbon dioxide (CO2) conversion technology is emerging as a promising tool to aid in the quest to lower CO2 emissions.1,2 Current advances have successfully converted CO2 to small C1 building block chemicals: CO,3 CH4,4 formaldehyde5 and formic acid;6–8 high energy dense liquid fuels: methanol (MeOH),9 ethylene (CH2CH2),10 ethanol,11 petrochemical polymers,12 and hydrogels.13 The abundance and cost efficiency of CO2 as a resource, makes its conversion economically viable as a competitor to traditional methods of manufacturing (e.g., carbonylation14 and the methanol to olefin (MTO) process15–17). Research efforts to further utilize captured CO2 as a raw material for the production of higher value compounds and chemical feedstocks have intensified in recent years with the advent of efficient electrolyzer technologies and heterogenization techniques. Electrochemical (EC) and photoelectrochemical (PEC) CO2 reduction technologies are the leading approaches to achieve CO2 reduction.18,19Electrocatalysts are instrumental to CO2RR due to their contributions to overcoming kinetic energy barriers and in mediating Proton Coupled Electron Transfers (PCETs).20–22 Benchmarks for potential commercial implementation stipulate the necessity of high current densities (j > 200 mA cm−2), long operation capacities, high selectivities (>90%), and low overpotentials.23 Although the catalytic capabilities of proposed systems have improved considerably, their current state remains insufficient for industrial/commercial application. Compared to the initial performance of catalysts, their long-term stability needs to be considered. Based on the techno-economic analysis, the stability of electrocatalysts for CO2RR should be at least 4000 hours.24,25 The stability of the catalytic system depends on multiple factors including catalyst's structure, catalyst immobilization including chemical and physical including covalent and non-covalent bonding, support material, catalyst's loading, catalyst surface morphology and the type of metal centre in the case of metallo-porphyrins.26,27
Advances in electrochemical CO2 reduction reactions (CO2RRs) offer a realistic pathway to utilization of CO2 as an abundant and inexpensive source for C1 building blocks.28 Until recently, solid state electrocatalysts had been leading the field in terms of conversion efficiency (current density). Often composed of heavy metals such as Pt, Pd, Au, Ag, Cu, etc., yet costly to implement and maintain.29–32 Although solid state catalysts have proven themselves capable of reducing CO2 to energy dense compounds like MeOH, ethylene, and ethanol; there is much to be desired for their selectivity for the reduction products they produce.33
On the other hand, molecular catalysts34,35 are favoured for their high selectivity and are capable of converting CO2 to CO,3 formaldehyde,36 formic acid,37 oxalic acid/oxalate,38 cyclic carbonates,39etc. with selectivities at near unity. Macrocyclic tetrapyrrolic ligands such as porphyrins and phthalocyanines are used as molecular catalysts,40 and often incorporate earth-abundant metals such as Fe,41 Cu,42,43 Co,44–46 Ni,47 Zn48etc., which are popular due to their high stability and facile tunability.49–53 Additionally, the highly conjugated system of porphyrin-based molecules result in a number of invaluable properties, such as enhanced electronic conductivity and π–π stacking capabilities.54–57
Although there exist several overarching reviews of CO2 electrocatalysts including even more specific reviews on porphyrin/phthalocyanine catalysts,50,51 a coverage of more recent developments in the field is required. Herein, major advances involving the functionalization of porphyrin and phthalocyanine catalysts for electrochemical CO2RR will be recounted in detail with a focus on recent advances in heterogeneous electrocatalysts and the effects of various immobilization strategies on catalytic performance.
Mechanistic pathways of CO2RR
The identity of the metal centre plays a significant role in the activity and selectivity of the catalyst. In the first step of the CO2RR mechanism, the electrophilic C atom is activated by nucleophilic attack from an electron-rich metal centre. The initial binding of CO2 requires the C–O σ* (LUMO) and degenerate C–O π* (LUMO+1) orbitals on the C atom be filled with electrons from the metal center.57,58 To satisfy this condition, an M+1,0 centre with a d8 configuration in a square-pyramidal ligand field is best for binding CO2via its filled dz2 (σ) and dxz/yz (π back-bonding) orbitals. For this reason, Fe, Co and Ni are hypothesized to be the best metals for catalysis due to their d8 electron configuration. Product selectivity is then influenced by the ability of CO to remain adsorbed to the metal for further reduction or desorption, leading to the release of CO.59 It has been proposed that Fe, Co, and Ni contain doubly occupied dz2 orbitals that would repel the lone pair of electrons on CO after CO2 reduction, releasing CO as the major product.60 However, if the CO remains bound to the metal via σ bonding, further reduction can take place, producing CH4. In metals having outermost s or p electrons, the electron transfer happens at the more localized, lower energy orbital, which is not strong enough to reduce CO, leading to the production of [CO2] followed by further reduction to formic acid depending on the availability of the proton source.The prediction from the molecular orbital theory closely resembles to the qualitative results observed in the literature. For example, the effect of the metal centre on CO2RR has been studied in extensively with 17 different metallophthalocyanines (MPcs) using gas diffusion electrodes (GDEs).61 Co, Ni, Fe, and Pd, belonging to group VIII of the transition elements, generate CO as the main CO2RR product. Co and Ni in particular, were most impressive with current efficiencies of 98 and 100% respectively (between −1.0 and −1.75 V (vs. RHE)). Sn, Pb, In, Zn, and Al produce formic acid as the main product, with Zn also being able to generate CO to a comparable extent. Cu, Ga, and Ti are unique in being the only metals that give methane as the main product, with current efficiencies being as large as 30–40%, while methane production for other metals is almost negligible. Lastly, V, Mn, Mg, Pt, and H show poor activity for CO2RR, with competing hydrogen evolution at current efficiencies of 90–100%. Although the selectivity of these metals towards one product is favourable, the activity must also be considered. CoPc and FePc showed higher current densities at lower applied potentials, while NiPc, although having a high selectivity for CO, requires much more energy to drive similar CO current densities. The high activity and selectivity of Co porphyrins towards CO makes the use of Co porphyrins one of the most promising catalysts for CO2RR, and therefore, has been subject to many detailed investigations.62,63 Although, closely following behind are NiPc and FePc, which are also promising catalysts if activity could be increased in the case of NiPc, and selectivity is enhanced in the case of FePc.64
Incorporation of a metal active canter to porphyrin/phthalocyanine-based catalysts promotes a cooperative effect between the catalyst metal site and the metal electrode. For instance, combining CoPc and Fe single-atom sites, showed that the free energy decreased in the activation and desorption steps of CO2RR (Fig. 1a–c).65 In this case, CoPc molecules reduced the adsorption energy of *CO and H*, without weakening the formation activity of *COOH. Therefore, combining CoPc and Fe–NC enhances the CO2RR activity on the Co centre while reducing the adsorption of CO on the Fe site.66
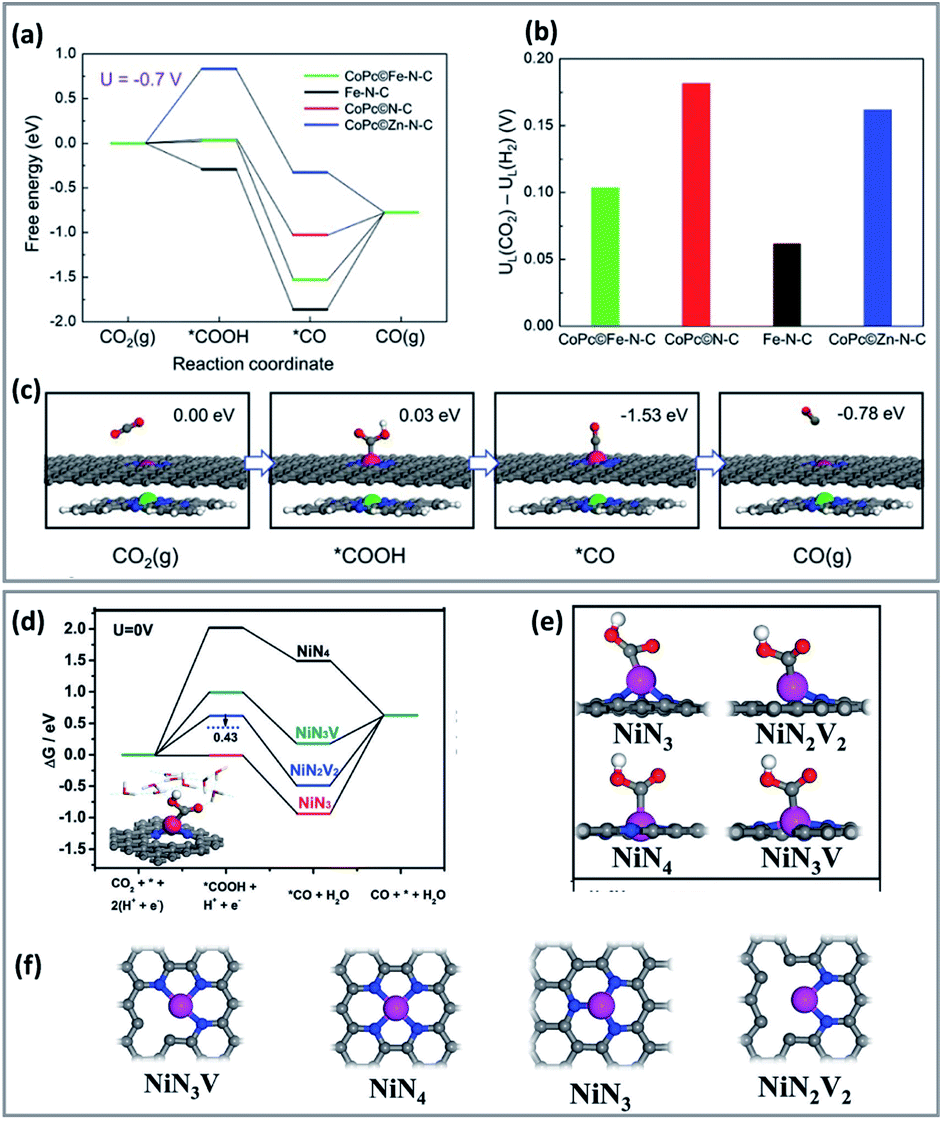 | ||
| Fig. 1 Computational results of CO2RR process on different catalysts. (a) Free energy diagram for the CO2RR to CO at U = −0.7 V vs. RHE on the Fe site in CoP/Fe–N–C, Fe site in Fe–N–C, Co site in CoP/N–C, and Zn site in CoP/Zn–N–C, respectively. (b) The differences in limiting potentials for CO2RR (UL (CO2)) and HER (UL (H2)) on the active sites. (c) Schematic atomic structure of CO2RR process on the Fe site in CoPc/Fe–N–C with the free energies at U = −0.7 V versus RHE. DFT calculations (copyright 2019 Wiley-VCH).65 (d) Free energy diagrams solvation effect corrections for the CO2RR and the HER. (e and f) Optimized atomic structures of different Ni–N structures with Ni atoms coordinated with 4 N atoms (NiN4), 3 N atoms (NiN3 and NiN3V), 2 N atoms (NiN2V2) (copyright 2012 Science).67 | ||
In another study, theoretical calculations indicate that a lower coordination number could change the electronic structure of the active site and increase the success of CO2RR over HER (Fig. 1d–f).67 The free energy of *COOH of coordinated, unsaturated NiN3, NiN3V, and NiN2V2 is lower than that of saturated NiN4. The *H blocking was also relatively weak in the case of NiN3V and NiN2V2, and for NiN2V2, resulting in high product selectivity.
The effect of the ligand can drastically affect the activity of these catalysts. Strategies for improving homogeneous catalysts include introducing functional groups that serve as local proton sources,68,69 hydrogen bond donors,70 or cationic moieties in the second sphere environment,71,72 all of which have resulted in increased CO2RR rates in part due to the stabilization of the formed CO2 intermediate by the substituents on the porphyrin ligand. A classic strategy of improving performance was pioneered by Savéant's group on Fe porphyrins through structural modification of the porphyrin ligand by incorporating substituents that can induce through-structure electronic effects.73 Introducing electron-withdrawing groups such as fluorine atoms has been shown to decrease overpotential by lowering electron density near the metal active site, making it is easier to inject an electron into the catalyst. However, this may also subsequently decrease catalytic activity by decreasing the nucleophilicity of the metal and its ability to bind to CO2. On the other hand, the introduction of methoxy substituents increases catalytic activity by increasing the propensity of the metal center to bind to CO2via inductive electron-donating effects of the ligand. Careful balance of electron-donating (lowering overpotential) and electron-withdrawing (increasing TOF) is required in ligand design and an optimal push–pull system is needed to achieve the ideal molecular catalyst.74
In the case of heterogeneous catalysts however, the effect of electron withdrawing (i.e. F and CN)75,76 and electron-donating (i.e. octaalkoxyl)77 substituents show little improvement for CO2RR in terms of the desired electronic effect. However, these substituents are crucial in reducing aggregation and reducing π–π stacking interactions that lead to improved catalytic activity. Ligand modification within the context of heterogenized molecular catalysts presumably affects other factors in the immobilized catalyst system including electron transfer between the catalyst and electrode, the ability for CO2 to coordinate with the catalyst, the desorption rate of the reduced products, and solvation energies.26 These findings emphasize the need to not screen heterogeneous molecular catalysts by the same criteria as homogeneous catalysts alone.
Homogeneous vs. heterogeneous electrocatalysts
Molecular catalysts can be applied in two general categories: as either homogeneous or heterogeneous systems.78 Whereas heterogeneous catalysts exist in a separate physical phase from the reactant (CO2), homogeneous systems operate in the same phase as the reactant. Homogeneous studies are a convenient way to assess the initial CO2 reduction ability of novel molecular catalysts. Oftentimes, only those that show promise under these conditions are further investigated with more vigorous heterogeneous studies. Hu et al.79 make a compelling argument for a reassessment of this method of screening, that can sometimes allow promising but underperforming molecules to slip through the cracks. Their report of cobalt tetraphenylporphyrin (CoTPP) immobilized onto carbon nanotubes (CNTs) illustrates how CoTPP, a catalyst whose activity is traditionally eclipsed by iron tetraphenylporphyrin (FeTPP) in homogeneous conditions, performs significantly better when immobilized onto a conductive CNT support in aqueous media (FEco = 83%; j = −0.59 mA cm−2 at −1.15 V vs. SCE) than an analogous FeTPP–CNT (FEco = 64%; j = −0.9 mA cm−2).80 They propose a new, simple deposition method consisting of sonicating the dissolved catalysts and CNTs, drop casting the solution, and drying as a means to quickly screen new molecular catalysts.Comprehensive studies comparing identical catalysts in homogeneous and heterogeneous environments widely demonstrate an overall enhancement to catalytic performance upon immobilization onto electron conductive supports.76,79 Systematic studies show that a significant enhancement in catalytic reactivity was achieved through immobilization of Fe-TPP-dimers onto CNTs in aqueous solution (TOF = 10 s−1; FECO = ∼90%) compared to their homogeneous analogues in DMF (TOF = 0.11 s−1; FECO = 48% at −1.33 V vs. RHE).81 We have also shown that heterogeneous pyridine–porphyrin complexes exhibit higher catalytic activity and product selectivity (FEtotal > 92% and j = −30 mA cm−2 at −0.6 V vs. RHE) compared to their homogeneous counterparts (FEtotal = 76% and j = −1.34 mA cm−2 at −1.4 V vs. RHE).74
Heterogeneous immobilization of molecular catalysts onto conductive solid supports is advantageous in several ways: (1) unlike in homogeneous systems, immobilized catalysts are locally bound to the electrode, the source of reductive capability, guaranteeing a high degree of catalytic site exposure.82,83 This serves to streamline the pathway of electron transfer from the electrode to the catalytically active site to CO2; (2) moreover, the solid support is often chosen by virtue of its exceptional electrical conductivity, further ensuring efficient electron transfer processes;84,85 (3) most organic/inorganic molecules are limited by their solubility in aqueous solvents. Heterogeneous systems enable molecular catalysts to overcome such limitations, freeing them to operate in proton-rich aqueous solutions, which serve a dual purpose in being more green.69,86 These strategies have proven to be a promising approach to efficiently enhancing catalytic activity. In the previous study of pyridine–porphyrin complexes, we demonstrate that even with a lower catalyst load concentration, the performance of heterogeneous molecules on CNTs is superior to that of its homogeneous analog.74
Converting CO2 into value-added materials is thought to occur via several mechanistic pathways. After capturing CO2, an initial proton coupled electron transfer (PCET) process forms intermediates such as *COOH and *OCHO. Among various carbonaceous products, CO and formic acid are considered pivotal C1 building blocks for C2+ products. The formation of C2+ products is much more challenging due to the number of reaction steps and intermediates required to form the C–C bond. This difficulty is also due to the linear relationship between the binding energies of individual reaction intermediates and their activation energies (kinetic barrier).87,88 Given the competition of C–C coupling with H–H and C–H bond formation,89 strategies that improve CO* dimerization to OC–CO* is key to the production of C2+ products. General strategies to achieve this include manipulating CO* binding strength through catalytic design, increasing CO* coverage, controlling CO* adsorption energetics,87 and re-adsorbing electrogenerated CO.90
To achieve C–C bond formation, the adsorbed *CO species may interact with each other via the Langmuir–Hinshelwood (LH) step through surface-bound species and a species in solution described in the Eley–Rideal (ER) step.91 In the case of metallo-porphyrins, the metal active site needs to bind to the *CO intermediates strongly enough to facilitate C–C coupling, but not too much as to significantly increase the energy barriers. Fundamental theoretical studies are valuable when designing catalysts.92 Li et al.93 demonstrated the potential of molecule-enhanced surfaces and how the CO2 to CO conversion efficiency of 5,10,15,20-tetraphenyl-21H,23H-porphine iron(III) chloride (FeTPP[Cl]) contributes to enhanced C2 production on a Cu electrode. They were able to utilize immobilized FeTPP[Cl] to create a localized concentration of ·CO, which serves as a key intermediate for the Cu active sites in the production of ethanol. By showing that the binding energy of CO to FeTPP[Cl] was 0.2 eV weaker than that of the Cu (111) substrate, the authors hypothesized that the CO produced by FeTPP[Cl] was readily spilling over onto the Cu active sites.
Noncovalent electrode immobilization by adsorption
Non-covalent immobilization relies on the π–π interactions from the conjugated aromatic system that exists on aromatic macrocycles to bind to carbon surfaces.76,94,95 Porphyrins and phthalocyanines being aromatic macrocycles are good candidates for surface immobilization due to their strong π–π interactions (Table 1). These interactions lead to improved electron transport rates due to the closer proximity of the catalyst to the electrode and the potential for improved electron conductivity from the in-plane π–π stacking. Coverage of the electrode surface with molecular catalysts may minimize its contact with water and reduce the opportunity for HERs.96 This method has been used for different applications such as water oxidation97 and proton reduction98 in addition to electrocatalytic reduction of CO2 to CO.76,79,99| Catalyst | V vs. RHE | j (mA cm−2) | FE% (CO) | TOF (s−1) | Ref. |
|---|---|---|---|---|---|
| Fe-TPP | −0.8 | 7.8 | 54% | 0.02 | 86 |
| Fe-TPP–NH2 | −0.8 | 12.9 | 79% | 0.05 | 86 |
| Fe-TPP–adj(NH2)2 | −0.8 | 8.0 | 70% | 0.03 | 86 |
| Fe-Tetra-Py | −0.7 | 19.6 | 37 | 0.9 | 74 |
| Fe-Cis-Py | −0.7 | 30.4 | 67 | 3.49 | 74 |
| Fe-Tri-OMe-Py | −0.7 | −23 | 50 | 1.49 | 74 |
| Fe-TPP-dimer | −0.8 | 16 | 89 | 10.2 | 81 |
| CoPc–CNT | −0.63 | ∼15 | 98 | 4.1 | 76 |
| CoP–pc | −0.61 | 18 | 90 | 1.4 | 100 |
| COF-367-Co | −0.67 | 3.3 | 90 | 0.53 | 50 |
| CoPc–P4VP | −0.73 | 2.0 | 89 | 4.8 | 101 |
| CoPc2 | −0.67 | 18.1 | 93 | 6.8 | 95 |
| Co-TPP | −0.8 | 0.9 | 70 | 2.75 | 79 |
| Fe-CATpyr | −0.59 | 0.24 | 93 | 0.04 | 102 |
| CATCO2H | −1.35 | 0.4 | 80 | 0.05 | 103 |
| CoFPC | −0.9 | 6.0 | 88 | 2.05 | 75 |
| CoTAP | −0.8 | 2.5 | 86 | 2 | 104 |
| Co-TPP | −0.9 | 0.8 | 52 | 4.5 | 105 |
| FePGH | −0.4 | 2.8 | 96 | 0.8 | 106 |
| NiPor-CTF | −0.9 | 42 | 97 | 0.47 | 107 |
| N–CoMe2Pc/NRGO | −0.8 | 8.7 | 90 | 1 | 108 |
| FePc-Gr75 | −0.6 | 1.7 | 90 | — | 109 |
| CoPc/Znln2S4 | −0.83 | 8.1 | 92.6 | — | 110 |
| PCN-222(Fe)/C | −0.6 | 1.2 | 91 | 1.2 | 111 |
| Ni-TPP–NItBu | −0.5 | 22 | 94 | 7.2 | 47 |
| Co-qpyCOOH/CNT | −0.65 | 6.7 | 100 | 0.28 | 112 |
| CoTPyPP/CNT | −0.6 | 7.5 | 95 | 2.1 | 54 |
The support material, surface functionality, morphology, and conductivity of the electrode are necessary for CO2RR and have been shown to enhance the catalytic efficiency, catalyst regeneration, and product separation.113 The support material and its interaction with the molecular catalyst directly affect the electron transfer, transport of species, the strength of catalyst bonding to the surface, and durability of catalyst; it also may alter the CO2RR mechanism.114 Highly conductive support ensures suitable electron transfer and reduces the ohmic resistance of the electrode, making high current densities possible.76,79 Carbon-based materials such as CNTs, carbon black (CB), carbon paper (CP), graphene derivatives, etc. are of particular interest for CO2RR due to their high stability and conductive surface area (Fig. 2).76,79,94,97,98,102,115–117 In another study, it has been reported that the CoPc catalysts immobilized on CNTs reveal an exceptional CO activity compared to CoPc immobilized onto other carbon-based materials such as reduced graphene oxide, carbon fiber paper, and CB.76
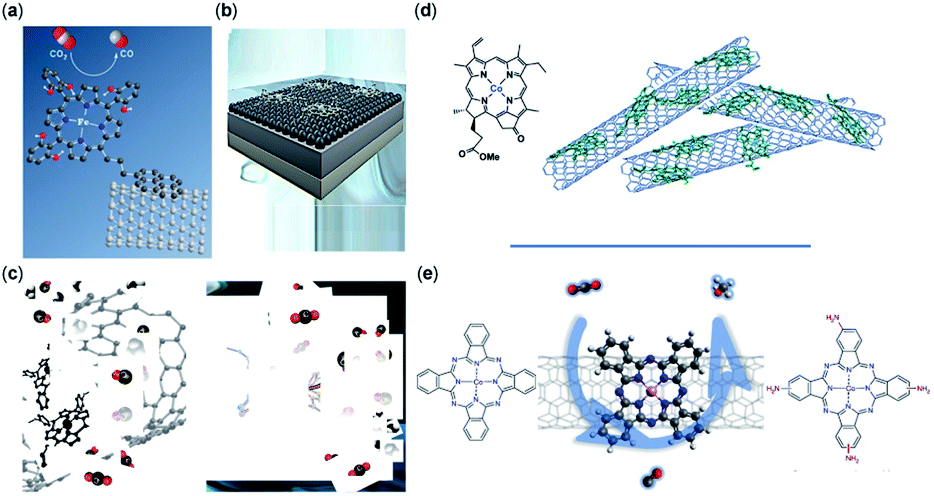 | ||
| Fig. 2 Non-covalent immobilization of porphyrin/phthalocyanine CO2RR catalysts onto carbon-based electrodes representative of the catalysts reported in (a) an iron triphenylporphyrin bearing 6 pendant –OH groups on the ortho positions of the phenyl rings immobilized on CNTs with a pyrene group (copyright 2016, Royal Society of Chemistry);102 (b) iron tetraphenylporphyrin immobilized in a flow cell (copyright 2020 Elsevier);86 (c) iron porphyrin–pyridine derivatives immobilized on CNTs (copyright 2020 American Chemical Society);74 (d) cobalt chlorin adsorbed on MWCNTs on a glassy carbon electrode (copyright Royal Society of Chemistry);116 and (e) cobalt phthalocyanine immobilized on CNTs (copyright 2019, Nature Publishing Group).117 | ||
Several techniques may be used to achieve noncovalent hybridization, such as dip coating and drop-casting. These methods involve dissolving the catalyst and immersing the carbon-based support material in a suitable solvent such as DMF, followed by deposition of the mixture onto the desired surface. Suspension methods ensure a homogeneous dispersion of the catalyst throughout the solid support and minimize the chance of unfavourable molecular aggregation, which can inhibit electron delivery. Shen et al.3 propose a detailed mechanistic scheme for CO2 electroreduction to CO and CH4 with CoTPP immobilized onto pyrolytic graphite (PG). Their work emphasizes the importance of pH in facilitating the initial electron transfer that activates CO2, by demonstrating the pH dependency of CH4 production, as well as in minimizing H2 evolution, which is predominantly produced at low pH (pH = 1). They also identify the CO2 radical anion (CO2˙−) as the key reaction intermediate in CO production. Although the formation of CO2˙− typically occurs at very negative potentials, a key strategy in successful catalytic systems lies in stabilizing its coordination to the catalyst. Using a narrow pH range (pH 1–3), they identified conflicting reaction pathways for the reaction products, where CO production is catalysed at pH = 3, and CH4 production is catalysed at pH = 1. They achieved 60% FECO at pH = 3, pressure = 10 atm, at −0.6 V vs. RHE and traces of CH4 (∼2.4% FECH4) at higher overpotentials (−0.8 V vs. RHE).
The surface morphology and graphitic degree of different materials should be considered when choosing a solid support. Wang et al.76 compared the catalytic activity of CoPc catalysts immobilized directly onto several carbon materials including CNTs, carbon fiber paper, reduced graphene oxide, and CB. Compared to CNTs, these other materials were found to have less than a third of the current density, ∼10% lower FEs, and inferior catalytic stability. The morphology of the immobilized CoPc/CNT can be visualized with transmission electron microscopy (TEM) (Fig. 3a and b). Aoi et al.116 found that a significant decline in FECO selectivity of a cobalt-porphyrin chlorin complex occurred when a graphene oxide matrix was used compared to when the same catalysts were deposited onto multi-wall CNTs (MWCNTs) in similar conditions. This decline in selectivity was attributed to the higher graphitic degree of CNTs, which resulted in increased π–π interactions between the molecular catalyst and the carbon support.79 In their study, Hu et al. noted a higher level of catalyst detachment occurring with a CB scaffold during electrolysis, whereas a comparable CNT support was more stable.
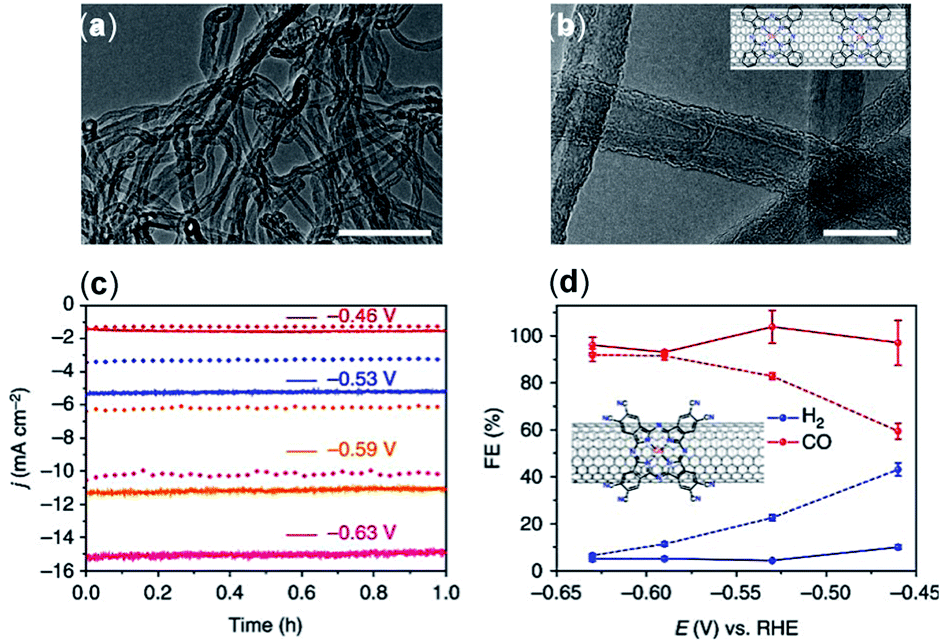 | ||
| Fig. 3 (a and b) TEM images of the CoPc/CNT (6%) hybrid. Inset in (b) shows a schematic representation of the CoPc/CNT hybrid. (c) Representative chronoamperograms of CO2 electroreduction catalyzed by the CoPc/CNT (2.5%) hybrid for 1 h at various potentials in 0.1 M KHCO3 aqueous solution; (d) FE of reduction products at different potentials for CoPc–CN/CNT (solid line) and CoPc/CNT (dotted line).76 Copyright 2017, Nature Publishing Group. | ||
In another recent report, an enhancement in electrochemical CO2RR of free base phthalocyanines was reported using N-doped carbon materials (N-Cmat).118 It was demonstrated that reduction of CO2 to CO occurred with the pyridinic N's as opposed to the pyrrolic N's. Introduction of Co nanoparticles, Co@Pc/C, led to CO production with a FECO of 84% and current density of 28 mA cm−2 at −0.9 V (Fig. 4).
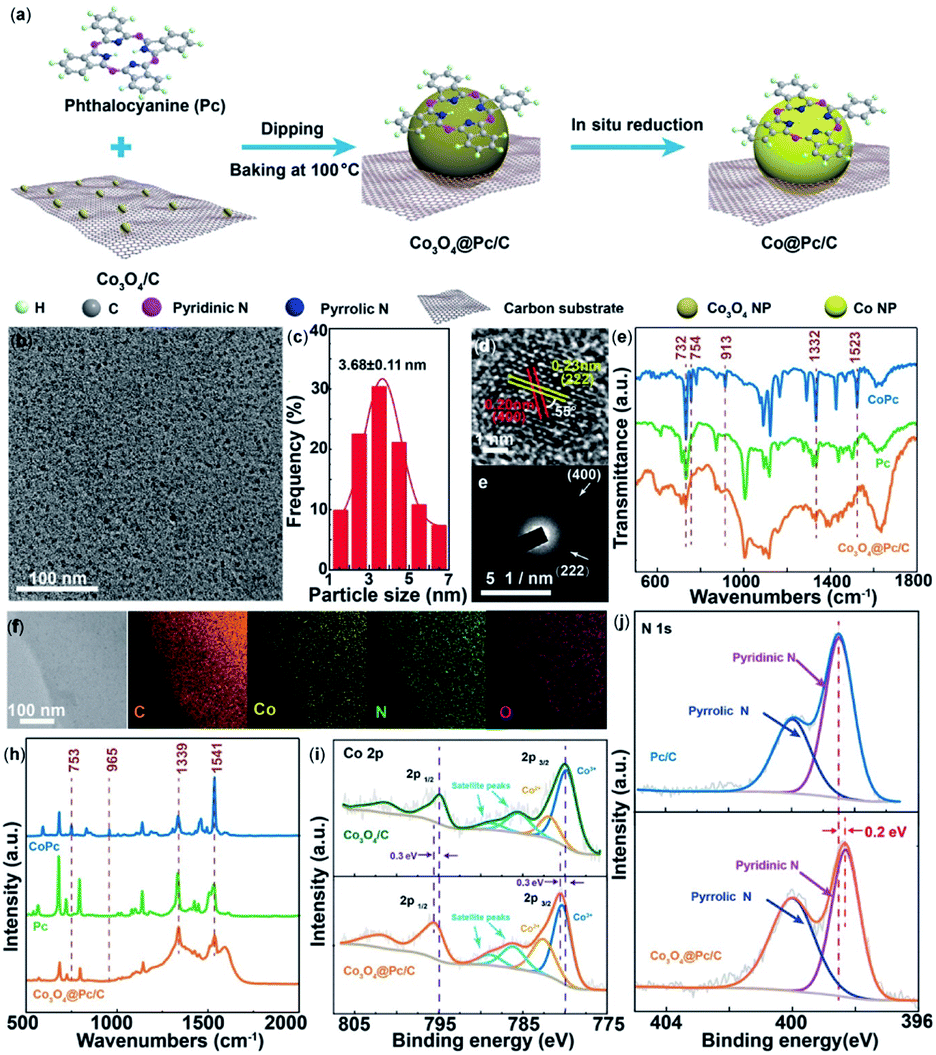 | ||
| Fig. 4 (a) An illustration of the synthetic procedure for Co@Pc/C; (b) TEM image; (c) size distribution histogram; (d) high-magnification TEM image; and the corresponding (e) fast Fourier-transform (FFT) pattern; (f) STEM-EDS elemental mapping images; and (h) Raman spectra of Co3O4@Pc/C alongside commercial CoPc and Pc; (i) high-resolution Co 2p XPS spectra of Co3O4@Pc/C and Co3O4/C. (j) High-resolution N 1s XPS spectra of Co3O4@Pc/C and Pc/C.118 Copyright 2020 Wiley-VCH. | ||
Other studies of immobilized CoII-2,3-naphthalocyanine (NapCo) complexes onto doped graphene in aqueous solution find that the electronic transfer processes between the catalyst and the conductive surface are improved through axial Co–O coordination to the terminal sulfoxide groups, resulting in a 3-fold increase to the TOF and a FECO of up to 97% (Fig. 5).119
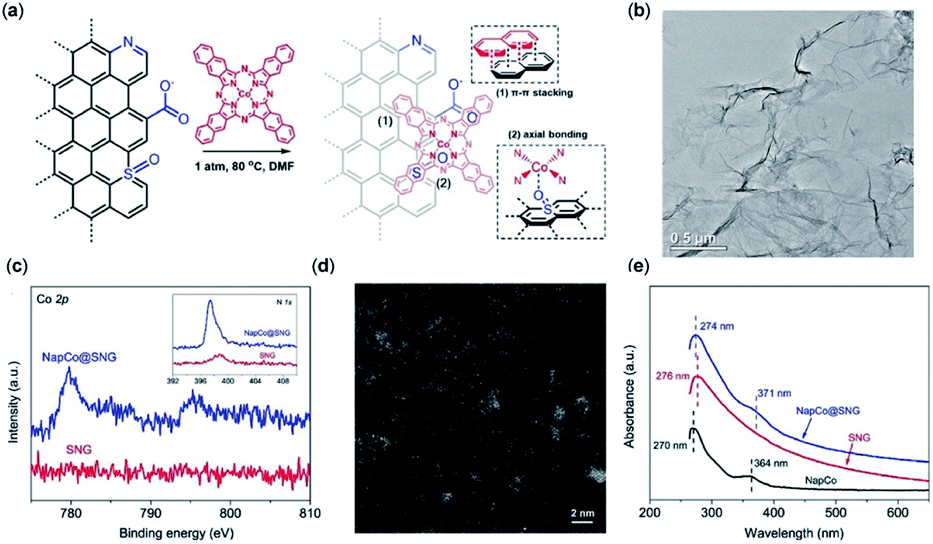 | ||
| Fig. 5 (a) Heterogenization of NapCo onto graphene through π–π stacking and heteroatom coordination; (b) bright-field TEM image of NapCo@S/N/O heteroatoms doped graphene (SNG); (c) XPS surveys of Co 2p core electron levels of SNG and NapCo@SNG. Inset: the N 1s core electron levels of SNG and NapCo@SNG; (d) HAADF-STEM images of NapCo@SNG where the Co atoms are indicated by the bright spots; (e) UV/Vis spectra of NapCo before and after immobilizing onto SNG.119 Copyright 2019 Wiley-VCH. | ||
Deposition of porphyrin molecules onto hydrophobic substrates such as polytetrafluoroethylene (PTFE) and Nafion has also seen success.120,121 The hydrophobic microenvironment of the polymer significantly enhances CO2 gas diffusion and mass transport, increasing the local concentration of CO2 on the electrode for CO2RR.122 Nafion is another example of a tetrafluoroethylene based polymer that possesses additional ionic properties due to its sulfonic acid groups which facilitates proton transfer for CO2 reduction. It was shown to work synergistically with carbon-based materials such as CNTs, demonstrating a ∼10 fold current enhancement for the reduction of CO2 to CO at −1.4 V vs. Ag/AgCl (pH 7).123 However, CO2 permeability through the Nafion membrane remains limited, resulting in lower FE and current density when used for CO2 reduction to formate.124
Early studies of covalent modification of an electrode surface with metalloporphyrins was reported by Aramata et al.125 in which Co-5,10,15,20-tetrakis(4-carboxylphenyl)porphyrin (CoTCPP) was fixed to a glassy carbon electrode functionalized with 4-aminopyridine groups via coordination of the Co centre with pyridine. The modified electrode demonstrated a FECO of 50% at −1.2 V vs. SCE in a CO2-saturated standard phosphate buffer solution (pH 6.8). Even after prolonged potentiostatic electrolysis under above conditions, the electrode remained stable, with no decrease in current density for more than 4 h. The authors attribute this improvement in catalytic activity to the increased electron density on the central Co(II) ion after axial coordination to the electron-donating pyridine moiety, thereby stabilizing the binding of CO2 on the opposite coordination site.
A later study utilizes a similar strategy to immobilize Co phthalocyanine (CoPc) onto polymeric films composed of pyridines (poly-4-vinylpyridine or P4VP) via a coordination bond.126 The CoPc–P4VP films display a FECO of ∼90%, with a TOF of 4.8 s−1 at −0.75 vs. RHE, which is drastically improved over the CoPc alone, adsorbed onto an edge-plane graphite (EPG) electrode. The latter only displays a 36% FECO along with a TOF of 0.6 s−1. In addition to the increase in dz2 orbital energy from the axial coordination, the authors hypothesize the improvement in catalytic activity to be from the encapsulation of the porphyrin catalyst inside the polymer film. This leads to higher CO2 solubility in the otherwise hydrophobic membrane due to basic pyridine sites and the second sphere hydrogen bond/proton network provided by the ionizable pyridine groups.
Covalent modification of electrode
Covalent immobilization establishes a direct bond between the molecular catalyst and the electrode surface (Table 2). This is beneficial in a number of ways. For one, the bond connecting the electrode to the catalyst layer can lead to heightened electron conductivity, and by extension more efficient use of energy (lower potentials).127 Secondly, covalent immobilization is a more robust alternative to non-covalent approaches which can show signs of catalyst displacement after several hours of operation.108,115,128 Here, the ligand groups of the porphyrin must be functionalized in a way that both allows for covalent binding to a surface, without destabilizing the molecule, while also remaining active for CO2RR.112,129 Such an approach provides the opportunity for long-term stability and predictable catalyst orientations, but leads to a high degree of constraints, generally adding complexity to the synthetic approach required.Covalent attachment of an electrocatalyst to a solid support has been shown to improve catalytic performance as demonstrated by Y.-F. Han et al.131 in which protoporphyrin IX cobalt chloride (CoPPCl) was covalently linked to hydroxyl-functionalized carbon nanotubes (CNT–OH). The grafted catalyst was synthesized by refluxing CoPPCl with CNT–OH (3.06 wt% hydroxyls) in ethanol with triethylamine, generating a covalent bond between the hydroxyl O atom and the Co center, and resulting in the functionalized material CoPP@CNT. The CoPP@CNT composite and Nafion were suspended in ethanol and drop cast onto carbon paper reaching a catalyst loading of 60 μg cm−2. The catalytic performance was then evaluated in a low-volume two-compartment cell with a CO2-saturated 0.5 M NaHCO3 electrolyte. The FECO of the CoPP@CNT composite ranges from 90% at −0.65 V to 80% at −0.5 V vs. RHE, with TOFCO varying from 0.34 s−1 to 2.1 s−1 respectively.
Although the CoPP@CNT composite showed negligible current decay over time, electrodes that were prepared by non-covalent attachment (physically mixed samples) of CoPPCl/CNT–OH with various CoPPCl loadings (CoPPCl/CNT–OH weight ratios of 4.4 × 10−4 to 5.6 × 10−1) at −0.55 vs. RHE showed a 20% decrease in the current density after a 1 hour electrolysis. Not only does covalent grafting improve catalyst stability, but the current density is also enhanced; physically mixed CoPPCl/CNT–OH showed a 50% lower current density at −0.55 vs. RHE (1 mA cm−2) compared to the covalently grafted CoPP@CNT value (2.1 mA cm2). The authors attribute this decrease in current density to catalyst aggregation which is a consequence of non-covalent grafting. The formation of aggregates blocks available active sites on the catalyst and hinder efficient electron transfer, especially at higher catalyst loadings, resulting in lower current densities. Through covalent grafting, the number of immobilized catalysts on the electrode can be optimized, while maintaining a high level of dispersion such that all grafted Co porphyrins are catalytically active.
Molecules with amine/amide-derived functionalized groups (e.g. amines, pyridine linkers) are well positioned for covalent anchoring to a surface through their monodentate axial ligands.34,104 Recent approaches have pioneered new techniques whereby a similar effect can be accomplished with organic molecules interfaced with solid supports.85,133,134 Marianov et al.105 have successfully demonstrated direct attachment of porphyrin derivatives (CoTPP-cov) through a aniline-mediated linkage onto glassy carbon (Fig. 6a). In these conditions a higher current density (4.7 mA cm−2) was observed compared to their unlinked counterparts (1.4 mA cm−2) (Fig. 6b). A positive correlation between the current density and catalyst loading concentration and active surface area was also shown (Fig. 6b and c).
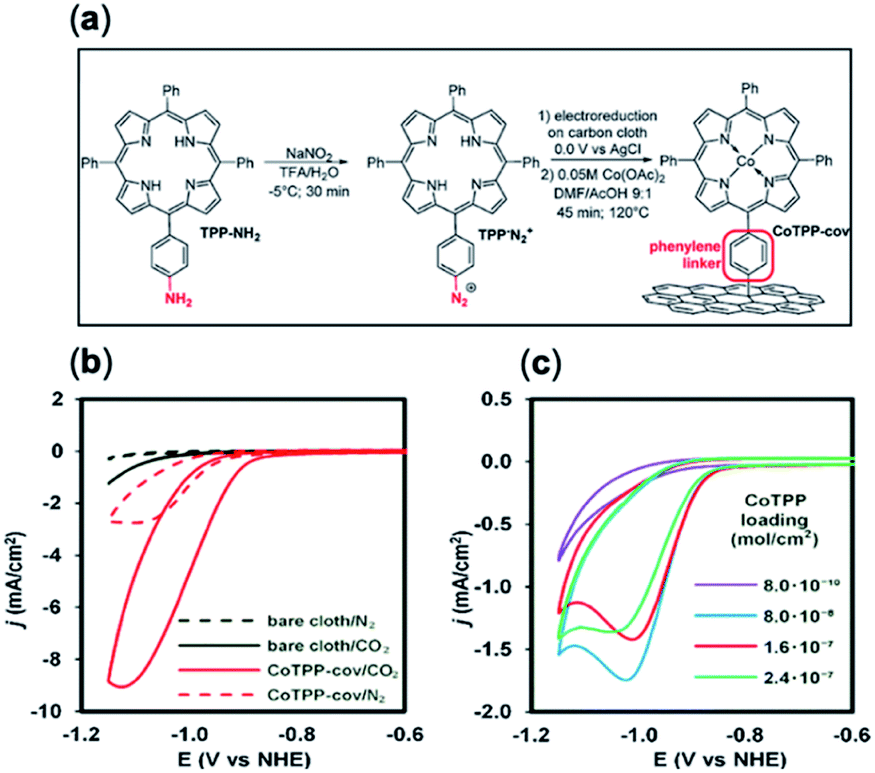 | ||
| Fig. 6 (a) Preparation of covalently immobilized Co tetraphenylporphyrin (CoTPP-cov); (b) cyclic voltammetry (CVs) of CoTPP-cov in N2- and CO2-purged aqueous solution, CVs of bare carbon cloth are shown for clarity; (c) CV traces of CoTPP-noncov with a variable amount of noncovalently immobilized CoTPP in CO2-saturated solution. Conditions: electrolyte: 0.5 M KHCO3 in all cases, potential scan rate is 100 mV s−1.105 Copyright 2019 Elsevier B.V. | ||
Lessons from homogeneous electrocatalysts have been incorporated into the design of a number of heterogeneous systems. For example, electron donating groups are known to increase the partial negative charge on the metal centre via inductive effect resulting in higher CO2-to-metal binding energy and enhanced CO2RR. Covalent immobilization of an iron tetraphenylporphyrin with six pendant –OH groups in the ortho positions and one carboxylic acid group, resulted in a high FE of 92%.130 Jiang et al.104 covalently grafted cobalt tetrakis-(4-aminophenyl)porphyrin (CoTAP) bearing 4 electron-donating amino groups onto a carboxylic acid functionalized CNT via an amide linkage. This strategy resulted in an unprecedented ∼100% FECO at overpotentials of 550 mV and a TOFCO of 6.0 s−1, while the non-covalent grafted electrode demonstrated a more moderate FECO of 85% and TOFCO of 2.3 s−1. In comparison to their previous work with covalent and non-covalent grafted cobalt tetraphenylporphyrin (CoTPP), a much lower FECO was observed in both electrodes; 67% (TOFCO 8.3 s−1) for covalent and 52% (TOFCO 4.5 s−1) for non-covalent. The authors rationalize this improvement of catalytic activity for several reasons; the presence of electron donating amino groups improves the intrinsic catalytic activity of each individual catalyst, furthermore the amide bond acts as a molecular wire that enhances electron transfer from the CNT to the catalytically active Co centre. Direct covalent connection of the CoTAP to the surface of CNT improves overall reaction rate due to faster electron migration and the diffusion of CO2 towards the active centres is no longer hindered by the layers of CoTAP aggregates.
Electrode immobilization by electropolymerization
Electrode surface immobilization via electropolymerization involves a monomer unit consisting of the molecular catalyst and a reactive moiety that undergoes polymerization upon oxidation, which then propagates onto the electrode surface. The oxidation of the monomer can be initiated by chemical means, however electrochemical oxidation grants control of film thickness, the possibility for in situ characterization during polymer growth, the lack of complicated purification steps, and most importantly is devoid of toxic oxidants, making this immobilization technique essentially ‘green’.The polymerization of the film is generally achieved by voltammetrically cycling the monomer in solution at an appropriate potential range and at a controlled sweep rate. Care must be taken to determine the optimal potential for deposition of these films as many have found that they can undergo oxidative degradation at more positive potentials, having negative consequences for the catalytic properties of the film. This technique is demonstrated in one study where the authors use a thiophene ((T)–3,4-ethylenedioxythiophene or EDOT) moiety attached to CoTPP via a flexible 1,3-aminothiopropylene spacer, which was electropolymerized into polythiophene on indium-tin-oxide (ITO)-coated glass and carbon paper substrates.135 At −0.66 V vs. RHE, the Co-porphyrin-based polymer demonstrated a FECO of 66%, as well as a TOF and TON of 1.6 s−1 and 5.7 × 103 respectively, after 1 hour. The polymer film is highly stable and demonstrated a relatively constant current density of 0.936 mA cm−2 and FECO of 36% over the course of a 6 hour controlled potential electrolysis (CPE).
Metal–organic frameworks (MOFs)136,137 and covalent organic frameworks (COFs)138 introduce more structure and conformation to the aforementioned covalent strategies. Due to the breadth of this field, the topic of (MOFs) and (COFs) is not covered in this review.
Conclusions and future prospects
As described in this review, electrocatalytic reduction of CO2 into fuels and higher value chemicals has become increasingly viable with the advent of recent methodical and technological advances. In order for CO2 electroreduction to be industrially viable, electrocatalysts need to perform with both high activity and high selectivity. The use of metalloporphyrins as molecular catalysts has achieved unprecedented results for the reduction of CO2 due to their favourable structural and electronic properties. Namely, their structural tuneability enables one to benefit from a wide range of immobilization techniques unavailable to other species. Furthermore, their highly conjugated system allows for enhanced electron conductivity and the ability to tune the electronic structure of the catalytic metal centre. These advantages are further accentuated though immobilization onto heterogeneous electrodes. A number of porphyrin catalysts and their electrocatalytic propensity for CO2 electroreduction in heterogeneous systems have been reported herein.The catalytic activity of these catalysts is strongly dependent on their structural properties and the immobilization technique chosen. Although the goal behind these immobilization techniques is to reduce catalyst aggregation and improve electron transfer from the electrode to the catalyst, the structural complexity of porphyrin molecules coupled with the particular constraints of synthesizing immobilization-compatible molecules hinders rapid development. Advances in structural design allow successful molecules to form stable interactions with the electrode to prevent dissociation, resulting in longer operation capacities.
Despite the variety of optimized heterogeneous molecular catalysts reported so far, there are still limitations which need to be addressed. For commercial electrochemical CO2 conversion, it is crucial to achieve a high selectivity of reduction products while ensuring long-term stability of the molecular catalysts. Promising strides in understanding multi-step reaction mechanisms that use molecular catalysts to localize reaction intermediates for reducing CO2 to complex C2 products is underway.
Conflicts of interest
There are no conflicts to declare.References
- S. Kumar, M. Y. Wani, C. T. Arranja, J. d. A. e Silva, B. Avula and A. J. F. N. Sobral, J. Mater. Chem. A, 2015, 3, 19615–19637 RSC.
- P. Falkowski, Science, 2000, 290, 291–296 CrossRef CAS PubMed.
- J. Shen, R. Kortlever, R. Kas, Y. Y. Birdja, O. Diaz-Morales, Y. Kwon, I. Ledezma-Yanez, K. J. P. Schouten, G. Mul and M. T. M. Koper, Nat. Commun., 2015, 6, 8177 CrossRef PubMed.
- K. W. Frese and S. Leach, J. Electrochem. Soc., 1985, 132, 259–260 CrossRef CAS.
- K. Nakata, T. Ozaki, C. Terashima, A. Fujishima and Y. Einaga, Angew. Chem., Int. Ed., 2014, 53, 871–874 CrossRef CAS PubMed.
- W. Zhang, Y. Hu, L. Ma, G. Zhu, Y. Wang, X. Xue, R. Chen, S. Yang and Z. Jin, Adv. Sci., 2018, 5, 1700275 CrossRef PubMed.
- M. Abdinejad, C. Ferrag, M. N. Hossain, M. Noroozifar, K. Kerman and H. B. Kraatz, J. Mater. Chem. A, 2021, 9, 12870–12877 RSC.
- M. Abdinejad, M. K. Motlagh, M. Noroozifar and H. B. Kraatz, Materials Advances, 2022, 3, 1224–1230 RSC.
- S. Kar, R. Sen, A. Goeppert and G. K. S. Prakash, J. Am. Chem. Soc., 2018, 140, 1580–1583 CrossRef CAS PubMed.
- D. Kim, C. S. Kley, Y. Li and P. Yang, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 10560–10565 CrossRef CAS PubMed.
- Y. C. Li, Z. Wang, T. Yuan, D.-H. Nam, M. Luo, J. Wicks, B. Chen, J. Li, F. Li, F. P. G. de Arquer, Y. Wang, C.-T. Dinh, O. Voznyy, D. Sinton and E. H. Sargent, J. Am. Chem. Soc., 2019, 141, 8584–8591 CrossRef CAS PubMed.
- L. Guo, J. Sun, X. Ji, J. Wei, Z. Wen, R. Yao, H. Xu and Q. Ge, Commun. Chem., 2018, 1, 11 CrossRef.
- C. Ferrag, M. Abdinejad and K. Kerman, Can. J. Chem., 2019, 1, 1–8 Search PubMed.
- A. Brennführer, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2009, 48, 4114–4133 CrossRef PubMed.
- H. Schulz, Appl. Catal., A, 1999, 186, 3–12 CrossRef CAS.
- O. S. Bushuyev, P. De Luna, C. T. Dinh, L. Tao, G. Saur, J. van de Lagemaat, S. O. Kelley and E. H. Sargent, Joule, 2018, 2, 825–832 CrossRef CAS.
- M. Bertau, H. Offermanns, L. Plass, F. Schmidt and H. J. Wernicke, Methanol: The Basic Chemical and Energy Feedstock of the Future: Asinger's Vision Today, 2014 Search PubMed.
- J. A. Herron, J. Kim, A. A. Upadhye, G. W. Huber and C. T. Maravelias, Energy Environ. Sci., 2015, 8, 126–157 RSC.
- W. A. Smith, T. Burdyny, D. A. Vermaas and H. Geerlings, Joule, 2019, 3, 1822–1834 CrossRef CAS.
- J. C. Calabrese, T. Herskovitz and J. B. Kinney, J. Am. Chem. Soc., 1983, 105, 5914–5915 CrossRef CAS.
- M. Aresta, C. F. Nobile, V. G. Albano, E. Forni and M. Manassero, J. Chem. Soc., Chem. Commun., 1975, 636–637 RSC.
- R. Lin, J. Guo, X. Li, P. Patel and A. Seifitokaldani, Catalysts, 2020, 10, 473 CrossRef CAS.
- S. Verma, B. Kim, H.-R. “Molly” Jhong, S. Ma and P. J. A. Kenis, ChemSusChem, 2016, 9, 1972–1979 CrossRef CAS PubMed.
- M. Jouny, W. Luc and F. Jiao, Ind. Eng. Chem. Res., 2018, 57, 2165–2177 CrossRef CAS.
- T. Burdyny and W. A. Smith, Energy Environ. Sci., 2019, 12, 1442–1453 RSC.
- X.-M. Hu, S. U. Pedersen and K. Daasbjerg, Curr. Opin. Electrochem., 2019, 15, 148–154 CrossRef CAS.
- W. Choi, D. H. Won and Y. J. Hwang, J. Mater. Chem. A, 2020, 8, 15341–15357 RSC.
- J. Qiao, Y. Liu, F. Hong and J. Zhang, Chem. Soc. Rev., 2014, 43, 631–675 RSC.
- K. P. Kuhl, E. R. Cave, D. N. Abram and T. F. Jaramillo, Energy Environ. Sci., 2012, 5, 7050–7059 RSC.
- N. J. Firet, T. Burdyny, N. T. Nesbitt, S. Chandrashekar, A. Longo and W. A. Smith, Catal. Sci. Technol., 2020, 10, 5870–5885 RSC.
- Y. Ling, Q. Ma, Y. Yu and B. Zhang, Trans. Tianjin Univ., 2021, 27, 180–200 CrossRef CAS.
- M. Abdinejad, Z. Mirza, X. Zhang and H.-B. Kraatz, ACS Sustainable Chem. Eng., 2020, 8, 1715–1720 CrossRef CAS.
- Z. Tang, E. Nishiwaki, K. E. Fritz, T. Hanrath and J. Suntivich, ACS Appl. Mater. Interfaces, 2021, 13, 14050–14055 CrossRef CAS PubMed.
- M. Abdinejad, M. N. Hossain and H.-B. Kraatz, RSC Adv., 2020, 10, 38013–38023 RSC.
- E. Boutin, L. Merakeb, B. Ma, B. Boudy, M. Wang, J. Bonin, E. Anxolabéhère-Mallart and M. Robert, Chem. Soc. Rev., 2020, 49, 5772–5809 RSC.
- M. Rauch, Z. Strater and G. Parkin, J. Am. Chem. Soc., 2019, 141, 17754–17762 CrossRef CAS PubMed.
- M. Isaacs, F. Armijo, G. Ramírez, E. Trollund, S. R. Biaggio, J. Costamagna and M. J. Aguirre, J. Mol. Catal. A: Chem., 2005, 229, 249–257 CrossRef CAS.
- S. Ikeda, T. Takagi and K. Ito, Bull. Chem. Soc. Jpn., 1987, 60, 2517–2522 CrossRef CAS.
- Z. Dai, Q. Sun, X. Liu, C. Bian, Q. Wu, S. Pan, L. Wang, X. Meng, F. Deng and F.-S. Xiao, J. Catal., 2016, 338, 202–209 CrossRef CAS.
- M. Hammouche, D. Lexa, M. Momenteau and J. M. Saveant, J. Am. Chem. Soc., 1991, 113, 8455–8466 CrossRef CAS.
- X. Yang, Q.-X. Li, S.-Y. Chi, H.-F. Li, Y.-B. Huang and R. Cao, SmartMat, 2022 DOI:10.1002/smm2.1086.
- Z. Weng, J. Jiang, Y. Wu, Z. Wu, X. Guo, K. L. Materna, W. Liu, V. S. Batista, G. W. Brudvig and H. Wang, J. Am. Chem. Soc., 2016, 138, 8076–8079 CrossRef CAS PubMed.
- K. Kosugi, H. Kashima, M. Kondo and S. Masaoka, Chem. Commun., 2022, 58, 2975–2978 RSC.
- C. L. Yao, J. C. Li, W. Gao and Q. Jiang, Phys. Chem. Chem. Phys., 2017, 19, 15067–15072 RSC.
- M. Usman, M. Humayun, M. D. Garba, L. Ullah, Z. Zeb, A. Helal, M. H. Suliman, B. Y. Alfaifi, N. Iqbal, M. Abdinejad, A. A. Tahir and H. Ullah, Nanomaterials, 2021, 11, 2029 CrossRef CAS PubMed.
- H. Tian, K. Wang, Z. Shui, M. Ali Raza, H. Xiao, M. Que, L. Zhu and X. Chen, Mater. Lett., 2022, 310, 131482 CrossRef CAS.
- M. Abdinejad, L. F. B. Wilm, F. Dielmann and H. B. Kraatz, ACS Sustainable Chem. Eng., 2021, 9, 521–530 CrossRef CAS.
- H. Yang, D. Yang, Y. Zhou and X. Wang, J. Am. Chem. Soc., 2021, 143, 13721–13730 CrossRef CAS PubMed.
- B. J. Fisher and R. Eisenberg, J. Am. Chem. Soc., 1980, 102, 7361–7363 CrossRef CAS.
- S. Lin, C. S. Diercks, Y.-B. Zhang, N. Kornienko, E. M. Nichols, Y. Zhao, A. R. Paris, D. Kim, P. Yang, O. M. Yaghi and C. J. Chang, Science, 2015, 349, 1208–1213 CrossRef CAS PubMed.
- R. B. Ambre, Q. Daniel, T. Fan, H. Chen, B. Zhang, L. Wang, M. S. G. Ahlquist, L. Duan and L. Sun, Chem. Commun., 2016, 52, 14478–14481 RSC.
- I. Hod, M. D. Sampson, P. Deria, C. P. Kubiak, O. K. Farha and J. T. Hupp, ACS Catal., 2015, 5, 6302–6309 CrossRef CAS.
- Z. N. Zahran, E. A. Mohamed and Y. Naruta, Sci. Rep., 2016, 6, 24533 CrossRef CAS PubMed.
- S. Dou, L. Sun, S. Xi, X. Li, T. Su, H. J. Fan and X. Wang, ChemSusChem, 2021, 14, 2126–2132 CrossRef CAS PubMed.
- Y.-R. Wang, M. Liu, G.-K. Gao, Y.-L. Yang, R.-X. Yang, H.-M. Ding, Y. Chen, S.-L. Li and Y.-Q. Lan, Angew. Chem., Int. Ed., 2021, 60, 21952–21958 CrossRef CAS PubMed.
- X. Zhang, M. C. Wasson, M. Shayan, E. K. Berdichevsky, J. Ricardo-Noordberg, Z. Singh, E. K. Papazyan, A. J. Castro, P. Marino, Z. Ajoyan, Z. Chen, T. Islamoglu, A. J. Howarth, Y. Liu, M. B. Majewski, M. J. Katz, J. E. Mondloch and O. K. Farha, Coord. Chem. Rev., 2021, 429, 213615 CrossRef CAS PubMed.
- P. Saha, S. Amanullah and A. Dey, Acc. Chem. Res., 2022, 55, 134–144 CrossRef PubMed.
- G. F. Manbeck and E. Fujita, J. Porphyrins Phthalocyanines, 2015, 19, 45–64 CrossRef CAS.
- L. Feng, K.-Y. Wang, E. Joseph and H.-C. Zhou, Trends Chem., 2020, 2, 555–568 CrossRef CAS.
- N. Furuya and S. Koide, Electrochim. Acta, 1991, 36, 1309–1313 CrossRef CAS.
- N. Furuya and K. Matsui, J. Electroanal. Chem. Interfacial Electrochem., 1989, 271, 181–191 CrossRef CAS.
- H.-J. Zhu, M. Lu, Y.-R. Wang, S.-J. Yao, M. Zhang, Y.-H. Kan, J. Liu, Y. Chen, S.-L. Li and Y.-Q. Lan, Nat. Commun., 2020, 11, 497 CrossRef CAS PubMed.
- J. Shen, M. J. Kolb, A. J. Göttle and M. T. M. Koper, J. Phys. Chem. C, 2016, 120, 15714–15721 CrossRef CAS.
- X. Zhang, Y. Wang, M. Gu, M. Wang, Z. Zhang, W. Pan, Z. Jiang, H. Zheng, M. Lucero, H. Wang, G. E. Sterbinsky, Q. Ma, Y.-G. Wang, Z. Feng, J. Li, H. Dai and Y. Liang, Nat. Energy, 2020, 5, 684–692 CrossRef CAS.
- L. Lin, H. Li, C. Yan, H. Li, R. Si, M. Li, J. Xiao, G. Wang and X. Bao, Adv. Mater., 2019, 31, 1903470 CrossRef CAS PubMed.
- W. Ren, X. Tan, W. Yang, C. Jia, S. Xu, K. Wang, S. C. Smith and C. Zhao, Angew. Chem., Int. Ed., 2019, 58, 6972–6976 CrossRef CAS PubMed.
- C. Yan, H. Li, Y. Ye, H. Wu, F. Cai, R. Si, J. Xiao, S. Miao, S. Xie, F. Yang, Y. Li, G. Wang and X. Bao, Energy Environ. Sci., 2018, 11, 1204–1210 RSC.
- C. Cyrille, D. Samuel, R. Marc and S. Jean-Michel, Science, 2012, 338, 90–94 CrossRef PubMed.
- M. Abdinejad, A. Seifitokaldani, C. Dao, E. H. Sargent, X. A. Zhang and H. B. Kraatz, ACS Appl. Energy Mater., 2019, 2, 1330–1335 CrossRef CAS.
- C. G. Margarit, C. Schnedermann, N. G. Asimow and D. G. Nocera, Organometallics, 2019, 38, 1219–1223 CrossRef CAS.
- I. Azcarate, C. Costentin, M. Robert and J.-M. Savéant, J. Am. Chem. Soc., 2016, 138, 16639–16644 CrossRef CAS PubMed.
- A. Khadhraoui, P. Gotico, B. Boitrel, W. Leibl, Z. Halime and A. Aukauloo, Chem. Commun., 2018, 54, 11630–11633 RSC.
- C. Costentin, M. Robert and J.-M. Savéant, Acc. Chem. Res., 2015, 48, 2996–3006 CrossRef CAS PubMed.
- M. Abdinejad, C. Dao, B. Deng, F. Dinic, O. Voznyy, X. Zhang and H.-B. Kraatz, ACS Sustainable Chem. Eng., 2020, 8, 9549–9557 CrossRef CAS.
- N. Morlanés, K. Takanabe and V. Rodionov, ACS Catal., 2016, 6, 3092–3095 CrossRef.
- X. Zhang, Z. Wu, X. Zhang, L. Li, Y. Li, H. Xu, X. Li, X. Yu, Z. Zhang, Y. Liang and H. Wang, Nat. Commun., 2017, 8, 14675 CrossRef PubMed.
- J. Choi, P. Wagner, S. Gambhir, R. Jalili, D. R. MacFarlane, G. G. Wallace and D. L. Officer, ACS Energy Lett., 2019, 4, 666–672 CrossRef CAS.
- S. Zhang, Q. Fan, R. Xia and T. J. Meyer, Acc. Chem. Res., 2020, 53, 255–264 CrossRef CAS PubMed.
- X. M. Hu, M. H. Rønne, S. U. Pedersen, T. Skrydstrup and K. Daasbjerg, Angew. Chem., Int. Ed., 2017, 56, 6468–6472 CrossRef CAS PubMed.
- I. Bhugun, D. Lexa and J. M. Savéant, J. Am. Chem. Soc., 1996, 118, 1769–1776 CrossRef CAS.
- M. Abdinejad, C. Dao, B. Deng, M. E. Sweeney, F. Dielmann, X. Zhang and H. B. Kraatz, ChemistrySelect, 2020, 5, 979–984 CrossRef CAS.
- L. Sun, V. Reddu, A. C. Fisher and X. Wang, Energy Environ. Sci., 2020, 13, 374–403 RSC.
- G. Zhao, X. Huang, X. Wang and X. Wang, J. Mater. Chem. A, 2017, 5, 21625–21649 RSC.
- S. Meshitsuka, M. Ichikawa and K. Tamaru, J. Chem. Soc., Chem. Commun., 1974, 158–159 RSC.
- C. Copéret, F. Allouche, K. W. Chan, M. P. Conley, M. F. Delley, A. Fedorov, I. B. Moroz, V. Mougel, M. Pucino, K. Searles, K. Yamamoto and P. A. Zhizhko, Angew. Chem., Int. Ed., 2018, 57, 6398–6440 CrossRef PubMed.
- M. Abdinejad, C. Dao, X. Zhang and H. B. Kraatz, J. Energy Chem., 2021, 58, 162–169 CrossRef.
- Y. Zheng, A. Vasileff, X. Zhou, Y. Jiao, M. Jaroniec and S.-Z. Qiao, J. Am. Chem. Soc., 2019, 141, 7646–7659 CrossRef CAS PubMed.
- X. Liu, J. Xiao, H. Peng, X. Hong, K. Chan and J. K. Nørskov, Nat. Commun., 2017, 8, 15438 CrossRef CAS PubMed.
- W. Zhang, Z. Jin and Z. Chen, Adv. Sci., 2022, 2105204 CrossRef PubMed.
- J. H. Montoya, C. Shi, K. Chan and J. K. Nørskov, J. Phys. Chem. Lett., 2015, 6, 2032–2037 CrossRef CAS PubMed.
- R. J. Davis and M. E. Davis, Fundamentals of Chemical Reaction Engineering, McGraw-Hill Higher Education, New York, 1st edn, 2003 Search PubMed.
- K. D. Yang, C. W. Lee, K. Jin, S. W. Im and K. T. Nam, J. Phys. Chem. Lett., 2017, 8, 538–545 CrossRef CAS PubMed.
- F. Li, Y. C. Li, Z. Wang, J. Li, D.-H. Nam, Y. Lum, M. Luo, X. Wang, A. Ozden, S.-F. Hung, B. Chen, Y. Wang, J. Wicks, Y. Xu, Y. Li, C. M. Gabardo, C.-T. Dinh, Y. Wang, T.-T. Zhuang, D. Sinton and E. H. Sargent, Nat. Catal., 2020, 3, 75–82 CrossRef CAS.
- D. Tasis, N. Tagmatarchis, A. Bianco and M. Prato, Chem. Rev., 2006, 106, 1105–1136 CrossRef CAS PubMed.
- M. Wang, K. Torbensen, D. Salvatore, S. Ren, D. Joulié, F. Dumoulin, D. Mendoza, B. Lassalle-Kaiser, U. Işci, C. P. Berlinguette and M. Robert, Nat. Commun., 2019, 10, 3602 CrossRef PubMed.
- M. N. Hossain, P. Prslja, C. Flox, N. Muthuswamy, J. Sainio, A. M. Kannan, M. Suominen, N. Lopez and T. Kallio, Appl. Catal., B, 2022, 304, 120863 CrossRef CAS.
- F. Li, B. Zhang, X. Li, Y. Jiang, L. Chen, Y. Li and L. Sun, Angew. Chem., Int. Ed., 2011, 50, 12276–12279 CrossRef CAS PubMed.
- P. D. Tran, A. Le Goff, J. Heidkamp, B. Jousselme, N. Guillet, S. Palacin, H. Dau, M. Fontecave and V. Artero, Angew. Chem., Int. Ed., 2011, 50, 1371–1374 CrossRef CAS PubMed.
- J. D. Blakemore, A. Gupta, J. J. Warren, B. S. Brunschwig and H. B. Gray, J. Am. Chem. Soc., 2013, 135, 18288–18291 CrossRef CAS PubMed.
- N. Han, Y. Wang, L. Ma, J. Wen, J. Li, H. Zheng, K. Nie, X. Wang, F. Zhao, Y. Li, J. Fan, J. Zhong, T. Wu, D. J. Miller, J. Lu, S.-T. Lee and Y. Li, Chem, 2017, 3, 652–664 CAS.
- W. W. Kramer and C. C. L. McCrory, Chem. Sci., 2016, 7, 2506–2515 RSC.
- A. Maurin and M. Robert, J. Am. Chem. Soc., 2016, 138, 2492–2495 CrossRef CAS PubMed.
- A. Maurin and M. Robert, Chem. Commun., 2016, 52, 12084–12087 RSC.
- S. Gu, A. N. Marianov and Y. Jiang, Appl. Catal., B, 2022, 300, 120750 CrossRef CAS.
- A. N. Marianov and Y. Jiang, Appl. Catal., B, 2019, 244, 881–888 CrossRef CAS.
- J. Choi, J. Kim, P. Wagner, S. Gambhir, R. Jalili, S. Byun, S. Sayyar, Y. M. Lee, D. R. MacFarlane, G. G. Wallace and D. L. Officer, Energy Environ. Sci., 2019, 12, 747–755 RSC.
- C. Lu, J. Yang, S. Wei, S. Bi, Y. Xia, M. Chen, Y. Hou, M. Qiu, C. Yuan, Y. Su, F. Zhang, H. Liang and X. Zhuang, Adv. Funct. Mater., 2019, 29, 1806884 CrossRef.
- M. Li, C. Yan, R. Ramachandran, Y. Lan, H. Dai, H. Shan, X. Meng, D. Cui, F. Wang and Z.-X. Xu, Chem. Eng. J., 2022, 430, 133050 CrossRef CAS.
- X. Li, G. Chai, X. Xu, J. Liu, Z. Zhong, A. Cao, Z. Tao, W. You and L. Kang, Carbon, 2020, 167, 658–667 CrossRef CAS.
- C. Chen, X. Sun, D. Yang, L. Lu, H. Wu, L. Zheng, P. An, J. Zhang and B. Han, Chem. Sci., 2019, 10, 1659–1663 RSC.
- B.-X. Dong, S.-L. Qian, F.-Y. Bu, Y.-C. Wu, L.-G. Feng, Y.-L. Teng, W.-L. Liu and Z.-W. Li, ACS Appl. Energy Mater., 2018, 1, 4662–4669 CrossRef CAS.
- P. B. Pati, R. Wang, E. Boutin, S. Diring, S. Jobic, N. Barreau, F. Odobel and M. Robert, Nat. Commun., 2020, 11, 3499 CrossRef CAS PubMed.
- S. Messias, M. Nunes da Ponte and A. S. Reis-Machado, React. Chem. Eng., 2019, 4, 1982–1990 RSC.
- X. Lu, Y. Wu, X. Yuan, L. Huang, Z. Wu, J. Xuan, Y. Wang and H. Wang, ACS Energy Lett., 2018, 3, 2527–2532 CrossRef CAS.
- P. Kang, S. Zhang, T. J. Meyer and M. Brookhart, Angew. Chem., Int. Ed., 2014, 53, 8709–8713 CrossRef CAS PubMed.
- S. Aoi, K. Mase, K. Ohkubo and S. Fukuzumi, Chem. Commun., 2015, 51, 10226–10228 RSC.
- Y. Wu, Z. Jiang, X. Lu, Y. Liang and H. Wang, Nature, 2019, 575, 639–642 CrossRef CAS PubMed.
- C. He, Y. Zhang, Y. Zhang, L. Zhao, L.-P. Yuan, J. Zhang, J. Ma and J.-S. Hu, Angew. Chem., Int. Ed., 2020, 59, 4914–4919 CrossRef CAS PubMed.
- J. Wang, X. Huang, S. Xi, J.-M. Lee, C. Wang, Y. Du and X. Wang, Angew. Chem., Int. Ed., 2019, 58, 13532–13539 CrossRef CAS PubMed.
- F. Li, Y. C. Li, Z. Wang, J. Li, D.-H. Nam, Y. Lum, M. Luo, X. Wang, A. Ozden, S.-F. Hung, B. Chen, Y. Wang, J. Wicks, Y. Xu, Y. Li, C. M. Gabardo, C.-T. Dinh, Y. Wang, T.-T. Zhuang, D. Sinton and E. H. Sargent, Nat. Catal., 2020, 3, 75–82 CrossRef CAS.
- N. Corbin, J. Zeng, K. Williams and K. Manthiram, Nano Res., 2019, 12, 2093–2125 CrossRef CAS.
- Z. Xing, L. Hu, D. S. Ripatti, X. Hu and X. Feng, Nat. Commun., 2021, 12, 136 CrossRef CAS PubMed.
- J. J. Walsh, G. Neri, C. L. Smith and A. J. Cowan, Chem. Commun., 2014, 50, 12698–12701 RSC.
- Y. Y. Birdja, R. E. Vos, T. A. Wezendonk, L. Jiang, F. Kapteijn and M. T. M. Koper, ACS Catal., 2018, 8, 4420–4428 CrossRef CAS PubMed.
- T. Atoguchi, A. Aramata, A. Kazusaka and M. Enyo, J. Chem. Soc., Chem. Commun., 1991, 156–157 RSC.
- Y. Liu and C. C. L. McCrory, Nat. Commun., 2019, 10, 1683 CrossRef PubMed.
- M. Abdinejad, I. Santos da Silva and H. B. Kraatz, J. Mater. Chem. A, 2021, 9, 9791–9797 RSC.
- F. Greenwell, G. Neri, V. Piercy and A. J. Cowan, Electrochim. Acta, 2021, 392, 139015 CrossRef CAS.
- H. Dong, M. Lu, Y. Wang, H.-L. Tang, D. Wu, X. Sun and F.-M. Zhang, Appl. Catal., B, 2022, 303, 120897 CrossRef CAS.
- A. Maurin and M. Robert, Chem. Commun., 2016, 52, 12084–12087 RSC.
- M. Zhu, J. Chen, L. Huang, R. Ye, J. Xu and Y.-F. Han, Angew. Chem., Int. Ed., 2019, 58, 6595–6599 CrossRef CAS PubMed.
- Y. Lu, J. Zhang, W. Wei, D.-D. Ma, X.-T. Wu and Q.-L. Zhu, ACS Appl. Mater. Interfaces, 2020, 12, 37986–37992 CrossRef CAS PubMed.
- C. Copéret, F. Allouche, K. W. Chan, M. P. Conley, M. F. Delley, A. Fedorov, I. B. Moroz, V. Mougel, M. Pucino, K. Searles, K. Yamamoto and P. A. Zhizhko, Angew. Chem., Int. Ed., 2018, 57, 6398–6440 CrossRef PubMed.
- C. M. Lieber and N. S. Lewis, J. Am. Chem. Soc., 1984, 106, 5033–5034 CrossRef CAS.
- S. Watpathomsub, J. Luangchaiyaporn, N. S. Sariciftci and P. Thamyongkit, New J. Chem., 2020, 44, 12486–12495 RSC.
- N. Kajal, V. Singh, R. Gupta and S. Gautam, Environ. Res., 2022, 204, 112320 CrossRef CAS PubMed.
- H. Zhong, M. Wang, G. Chen, R. Dong and X. Feng, ACS Nano, 2022, 16(2), 1759–1780 CrossRef CAS PubMed.
- Y. Shan, L. Chen, H. Pang and Q. Xu, Small Struct., 2021, 2, 2000078 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2022 |