
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGreen synthesis of zirconium MOF-808 for simultaneous phosphate recovery and organophosphorus pesticide detoxification in wastewater†
Lydia
González
a,
Rodrigo
Gil-San-Millán
 b,
Jorge A. R.
Navarro
a,
Carmen R.
Maldonado
a,
Elisa
Barea
*a and
Francisco J.
Carmona
*a
b,
Jorge A. R.
Navarro
a,
Carmen R.
Maldonado
a,
Elisa
Barea
*a and
Francisco J.
Carmona
*a
aDepartamento de Química Inorgánica, Universidad de Granada, Av. Fuentenueva S/N, 18071, Granada, Spain. E-mail: fjcarmona@ugr.es
bFaculty of Chemistry, University of Wroclaw, Fryderyka Joliot-Curie 14, F, 50-383, Wroclaw, Poland
First published on 22nd April 2022
Abstract
Agricultural wastewater posseses serious threats to the environment and human health. In this work, we report a green microwave assisted synthesis of MOF-808 with tuned particle size. These materials have been proved to simultaneously degrade a toxic pesticide and capture phosphate from water over several cycles. The adsorbed phosphate can be further recovered, favoring a P-circular economy.
 Francisco J. Carmona | Dr Francisco J. Carmona obtained his PhD in Chemistry in 2017 at the University of Granada (Spain). Later, he worked as a postdoctoral researcher at Insubria University (Italy, 2017–2019) and Kyoto University (Japan, 2019–2020). In 2020, he received a Marie Sklodowska-Curie postdoctoral fellowship to join the Department of Inorganic Chemistry at the University of Granada (Spain). His current research is focused on the green preparation of metal–organic frameworks (MOFs) and polyhedra (MOPs) with potential application in biomedicine and environmental remediation. |
Due to the continuous growth of the world population and the negative effects derived from global climate change, agrochemicals are essential to maintain the current crop yields and ensure food security.1 However, their low efficiency together with their indiscriminate use implies severe threats to the environment and human health.2,3 In this context, phosphorus-based compounds are extensively employed in modern agriculture and manufactured from phosphoric rock, which is a non-renewable resource. In addition, less than 30% of the phosphorus applied to soils reaches the food chain, with the main P-losses occurring in croplands. Consequently, the P-compounds, which contaminate fresh waters, contribute to eutrophication and decimate biodiversity, which also entails a significant societal cost.4 On the other hand, organophosphorus pesticides (OPs), widely used in agriculture, are highly toxic compounds due to their ability to inhibit the activity of acetylcholinesterase, an enzyme present in several animals, including humans. Indeed, OP poisoning causes 110
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 deaths per year globally.5,6 For these reasons, the design of new strategies to decontaminate polluted wastewater and recover phosphate from it is of paramount importance and may contribute to the much-needed P circular economy.
000 deaths per year globally.5,6 For these reasons, the design of new strategies to decontaminate polluted wastewater and recover phosphate from it is of paramount importance and may contribute to the much-needed P circular economy.
In this regard, Zr-based metal–organic frameworks (Zr-MOFs) are robust and non-toxic crystalline porous materials composed of Zr6O4(OH)4 clusters connected by organic spacers. Their elevated porosity, together with the Pearson hard-acid nature of Zr(IV) sites and concomitant strong affinity for phosphate compounds, has led to Zr-MOFs being considered as promising materials towards the decontamination and recovery of P-polluted wastewater. Indeed, some Zr-MOFs have recently been proved to efficiently capture phosphate or pesticides. However, most of these examples are limited to the removal of a single pollutant,7–10 far from the real conditions of application. Only one very recent study, of our research group, reported the simultaneous capture and selective recovery of both P-pollutants by Zr-MOF NU-1000.11 On the other hand, certain Zr-MOFs can hydrolytically degrade OPs in buffered media containing organic amines (e.g. N-ethylmorpholine).12,13 Nevertheless, the catalytic activity of these materials decreases dramatically in unbuffered media such as wastewater, suggesting the role of these molecules as co-catalysts (i.e. t1/2 < 1 min in N-ethylmorpholine solution and t1/2 = 94 min in Milli-Q water for Tabun degradation by MOF-808).14,15 Finally, it is important to highlight that Zr-MOF materials are usually prepared through long solvothermal synthesis using toxic N,N-dimethylformamide (DMF). Given that DMF is considered as a substance of very high concern by the European Chemicals Agency,16 the development of environmentally friendly methods to synthesize Zr-MOFs is urgently needed in order to facilitate their industrial manufacture and application. In this regard, it should be noted that green synthesis of Zr-UiO-66 (ref. 17) and related MOF-808(Ce)18 and MOF-808(Hf)19 has been recently reported. However, in neither case has particle size control been carried out. To the best of our knowledge, only one example of particle size control in MOF-808(Zr) has recently been achieved in the absence of DMF, although isopropanol solvent was used for the preparation of the Zr6 oxocluster.20
Taking into account this background, in this work, we report a water-based microwave assisted synthetic method to prepare porous [Zr6O4(OH)4(trimesate)2(formate)6] (MOF-808) materials (pore sizes: 1.8 and 2.2 nm)21 with crystal size growth control. We also demonstrate the ability of these as-synthesized MOF-808 materials to simultaneously capture phosphate ions and hydrolytically degrade the OP pesticide dimethyl-4-nitrophenylphosphate (methyl paraoxon, MP) in unbuffered aqueous solution over several cycles. Moreover, the captured phosphate ions can be easily recovered, regenerating the adsorbent, and favouring a P-circular economy (Scheme 1).
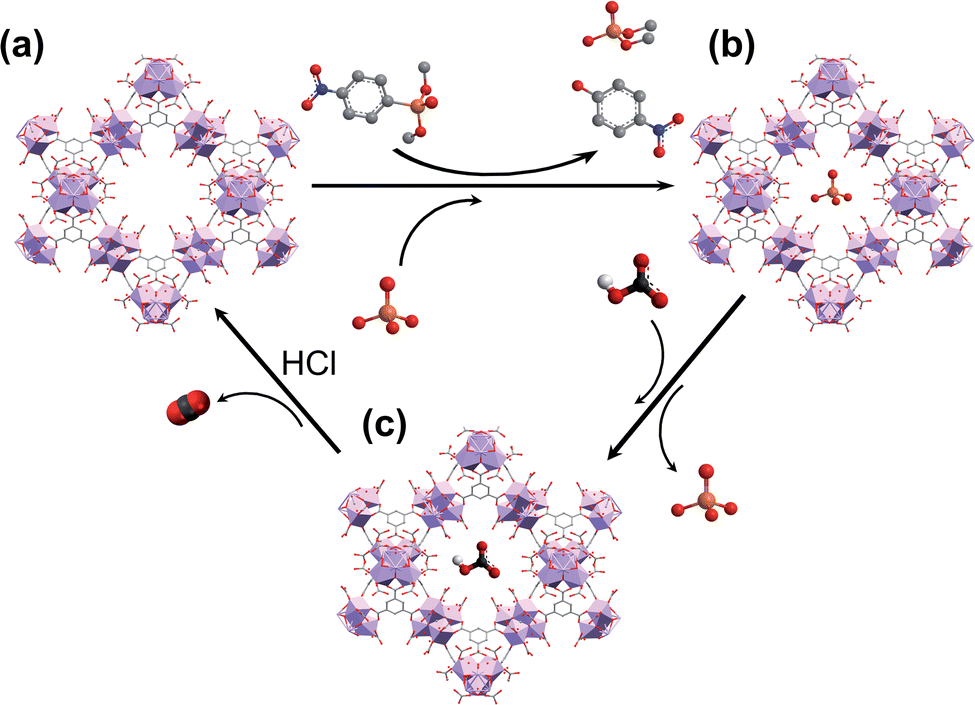 | ||
| Scheme 1 Schematic representation of (a) simultaneous phosphate removal and pesticide degradation, (b) recovery of phosphate by hydrogen carbonate treatment, and (c) regeneration of MOF-808 with hydrochloric acid. | ||
Firstly, three MOF-808 materials, with particle size ranging from 345 to 570 nm, were prepared by water-based microwave (MW) assisted synthesis using different heating ramps. It should be highlighted that this environmentally friendly approach allowed the reaction time to be significantly shortened (from days to a few hours) in comparison with typical solvothermal methods, which employ a two-day synthesis and toxic DMF as solvent.21,22 The designed synthetic procedure encompasses four different steps: (i) a heating ramp of 15, 60 and 120 min up to 95 °C for MOF-808_15, MOF-808_60 and MOF-808_120, respectively; (ii) a 1 h dwelling period at 95 °C; (iii) a fast cooling-step of 2 min down to room temperature and (iv) a washing process using water as solvent (Fig. 1a). Noteworthily, using this water-based and faster synthetic method our MOF-808 materials were recovered with similar reaction yields (61–75%) to those obtained in traditional DMF-based solvothermal synthesis (70%).21 Once MOF-808_15, MOF-808_60 and MOF-808_120 were isolated, their purity and crystallinity were firstly confirmed by powder X-ray diffraction analysis (Fig. 1b). In addition, the N2 adsorption studies proved the permanent porosity of the three materials after thermal activation (Fig. 1c). The calculated BET surface areas for MOF-808_15 (1970 m2 g−1), MOF-808_60 (2050 m2 g−1) and MOF-808_120 (1960 m2 g−1) were in good agreement with the value previously reported in the solvothermal synthesis of MOF-808 (2060 m2 g−1),21 corroborating the high quality of the synthesized materials. Likewise, these results confirmed the efficiency of the water washing process, avoiding the typical solvent exchange with DMF for several days.21 Scanning electron microscopy (SEM) was used in order to explore the impact of MW heating ramp speed on MOF-808 crystal growth. The results reveal that MOF-808_15, MOF-808_60 and MOF-808_120 were fairly homogeneous with increasing particle sizes of 345 ± 25 nm, 485 ± 30 nm and 570 ± 30 nm, respectively (Fig. 1d). These results indicate that the slower the heating-ramp, the larger the crystals. For comparative studies, three control materials, denoted as MOF-808_15_c, MOF-808_60_c and MOF-808_120_c, were also synthesized using the technically fastest heating ramp available (1 min) followed by increasing dwelling times at 95 °C of 75 min, 120 min and 180 min, respectively. Thus, the overall reaction time of each control material was the same as that of the corresponding counterpart, allowing us to analyse the effect of the heating ramp speed on the properties of the final materials. Noteworthily, all control materials showed more polydisperse crystal size distribution (Fig. S1 and Table S1†). In addition, the powder X-ray diffractograms suggested the co-existence of an amorphous phase together with MOF-808 microcrystals, especially in the case of MOF-808_120_c (Fig. S2†). Indeed, this material showed a very low N2 adsorption capacity at 77 K (BET surface area = 710 m2 g−1). Significant porosity drops were also found for MOF-808_60_c (BET surface area = 1235 m2 g−1) and MOF-808_15_c (BET surface area = 1540 m2 g−1) (Fig. S3 and Table S1†). These results are in agreement with previous reports for microwave assisted MOF-808 synthesis without a controlled heating ramp.23
 | ||
| Fig. 1 (a) Controlled-heating ramp used in the preparation of MOF-808 (continuous lines) and control materials (dashed lines). (b) Powder X-ray diffractograms, (c) N2 adsorption at 77 K and (d) SEM images of MOF-808_15 (red), MOF-808_60 (blue) and MOF-808_120 (green). Scale bar: 1 μm. | ||
Once MOF-808_15, MOF-808_60 and MOF-808_120 materials were fully characterized, we proceeded to assess their behaviour towards the adsorption of phosphate ions and the degradation of the organophosphorus pesticide methyl paraoxon (MP). Firstly, experimental data confirmed the incorporation of phosphate from aqueous solutions in all MOF-808 structures. The corresponding solid–liquid adsorption isotherms at 25 °C could be successfully fitted to a Langmuir model with maximum adsorption capacities of 1.60 (MOF-808_15), 1.26 (MOF-808_60) and 0.77 (MOF-808_120) mol mol−1 (Fig. S4†). Since the three samples showed similar porosity, the difference in maximum phosphate loading capacities could be attributed to a hindered diffusion of phosphate in the inner pore structure. Consequently, the adsorbate loading inversely correlates with MOF particle size. In addition, an aqueous solution of phosphate (0.08 mM, 1 mL, pH = 6.9) was exposed to an equimolar amount of MOF-808 materials (0.08 μmol) to evaluate their adsorption kinetics. A pseudo-second order kinetic model was the best to fit the experimental data (Fig. 2a and Table S2†), demonstrating in all cases a fast diffusion and high affinity of phosphate anions for the pore structure (MOF-808_15: t1/2 = 20.9 min; MOF-808_60: t1/2 = 24.8 min and MOF-808_120: t1/2 = 26.2 min). A nearly complete uptake of this pollutant was reached after 24 hours for MOF-808_15 (98.0 ± 2.3%) and MOF-808_60 (96.1 ± 0.9%), while a lower payload was observed for MOF-808_120 (79.9 ± 5.1%). These results are in agreement with the solid–liquid adsorption isotherms (Fig. S4†). In addition, we evaluated the ability of MOF-808 materials to catalyse the hydrolysis of toxic methyl paraoxon (oral rat LD50 = 3.27 mg kg−1)24 into 4-nitrophenol (oral rat LD50 = 667 mg kg−1)25 and dimethylphosphate (oral rat LD50 = 3283 mg kg−1),26 in unbuffered aqueous solution. All assayed MOF-808 materials lead to a significant hydrolysis of the ester P–O bond after 24 hours (MOF-808_15: 92.8 ± 0.9%; MOF-808_60: 91.1 ± 6.4%; MOF-808_120: 86.0 ± 5.8%) (Fig. 2b). The degradation profiles can be fitted to a second-order kinetics model, with half-life times of 113 min, 137 min and 231 min, for MOF-808_15, MOF-808_60 and MOF-808_120, respectively (Table S3†). These results demonstrate the detrimental impact of crystal size on catalytic activity, with the MOF-808_120 material exhibiting the worst performance in the assayed series. These results are in line with previously published studies on MP hydrolysis by MOF-808 in pure water (t1/2 = 300 min).15
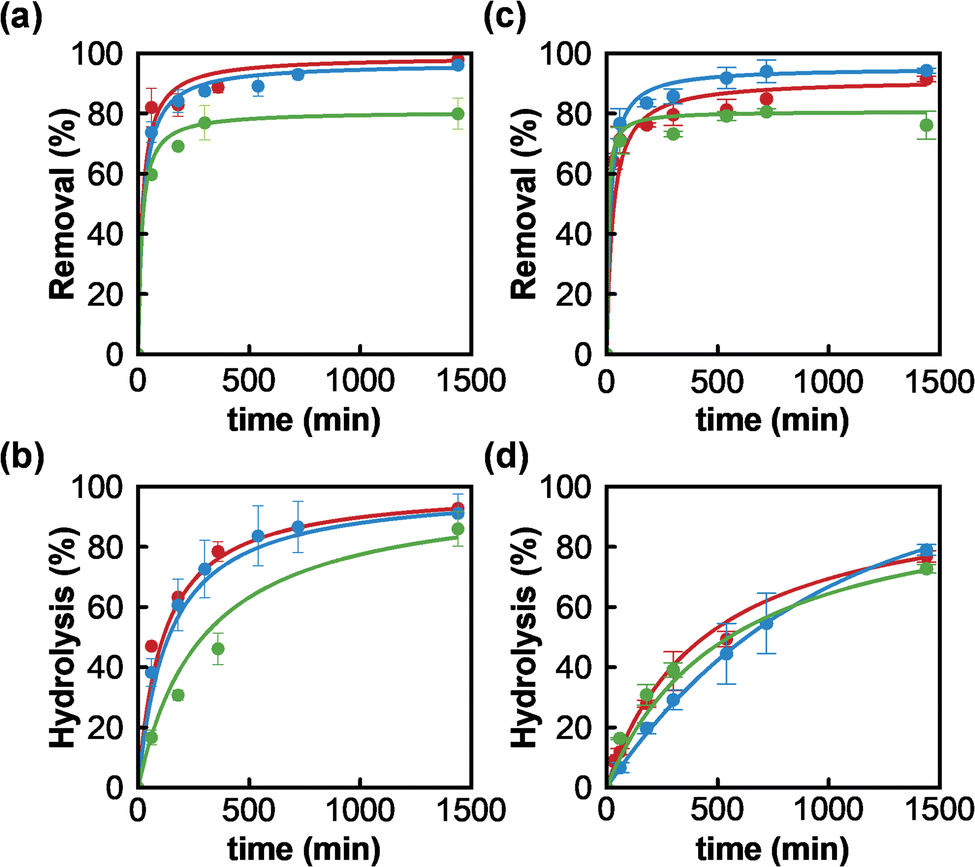 | ||
| Fig. 2 (a) Adsorption kinetics of phosphate. (b) Degradation kinetics of methyl paraoxon. (c) Adsorption kinetics of phosphate in the presence of methyl paraoxon. (d) Degradation kinetics of methyl paraoxon in the presence of phosphate. Experimental conditions: unbuffered aqueous solutions at 25 °C (1 mL), 0.08 μmol of MOF-808, [MP] = 0.08 mM, and [PO43−] = 0.08 mM (MOF-808_15: red, MOF-808_60: blue and MOF-808_120: green). | ||
Interestingly, these porous materials were also able to simultaneously degrade methyl paraoxon and capture phosphate ions from water. It should be noted that phosphate adsorption kinetic profiles and uptakes, after 24 hours, were similar to the ones reported in the absence of pesticide (MOF-808_15: 91.5 ± 0.9%; MOF-808_60: 94.3 ± 0.8%; MOF-808_120: 76.1 ± 4.3%) (Fig. 2c and Table S2†), suggesting a negligible interference effect of pesticide on phosphate adsorption. We also evaluated the impact of the presence of phosphate ions on pesticide degradation. An important degradation took place after 24 hours, although it was somehow 10–15% smaller than the MP conversion observed in the absence of phosphate (MOF-808_15: 76.8 ± 1.9%; MOF-808_60: 79.0 ± 1.8%; MOF-808_120: 72.7 ± 1.4%). Similarly, the degradation rate was slowed down, with half-life times of 440 min, 390 min and 552 min, for MOF-808_15, MOF-808_60 and MOF-808_120, respectively (Table S3†). According to this, phosphate ions seem to be responsible for an important catalyst poisoning, with a relatively larger impact on the activity of the smaller particles, which may point to active sites being mainly located at the particles' external surface.
In order to prove this hypothesis, we carried out UV-vis studies which revealed that pesticide concentration in the supernatants decreased at the same rate as the concentration of p-nitrophenol (one of the degradation products) increased. Interestingly, an isosbestic point was observed at 297 nm, pointing out that neither methyl paraoxon nor p-nitrophenol is incorporated into the porous matrixes (Fig. 3b). Furthermore, suspensions of MOF-808_60 (5.6 μmol) and methyl paraoxon (5.6 mM) were incubated in 1 mL of D2O for 24 h at room temperature in the presence and absence of phosphate (5.6 mM). The supernatants were analysed by means of 1H and 31P NMR spectroscopy using glyphosate as an internal reference. Both the pesticide and its degradation products (p-nitrophenol and dimethylphosphate) were successfully identified in 1H and 31P spectra. In particular, the 1H NMR spectra confirm that 62.0 and 48.0% of methyl paraoxon was hydrolysed after 24 h in the absence and presence of phosphate, respectively. These conversion values were appreciably lower than those obtained in the batch experiments, which could be attributed to kinetic issues related to the use of deuterated water. Noteworthily, the comparison of the integrated H signals of non-degraded methyl paraoxon and p-nitrophenol with the internal reference (glyphosate) indicated that neither the pesticide nor p-nitrophenol was incorporated into the porous matrix (Fig. 3c, S5 and S6†). Conversely, dimethylphosphate (the other degradation product) was partially encapsulated inside the MOF (76.6% in the absence of phosphate vs. 44.4% in the presence of this anion) (Fig. S5 and S6†). Despite this fact, it seems that dimethylphosphate does not really compete with phosphate during the adsorption process, as corroborated by the previously discussed kinetic studies where phosphate uptake after 24 h was not affected by the presence of methyl paraoxon (see above). 31P spectra further supported these experimental results, confirming the generation and partial encapsulation of dimethylphosphate molecules as well as the almost complete encapsulation of phosphate ions after 24 h of incubation (Fig. S7 and S8†). In summary, pesticide degradation seems to take place on the particle surface and, consequently, the high surface area of the pores is available for the adsorption of phosphate ions. In contrast, methyl paraoxon molecules compete with phosphate ions for the active sites on the external particle surface, explaining the drop of about 10–15% in the hydrolytic efficiency when both P-pollutants are present.
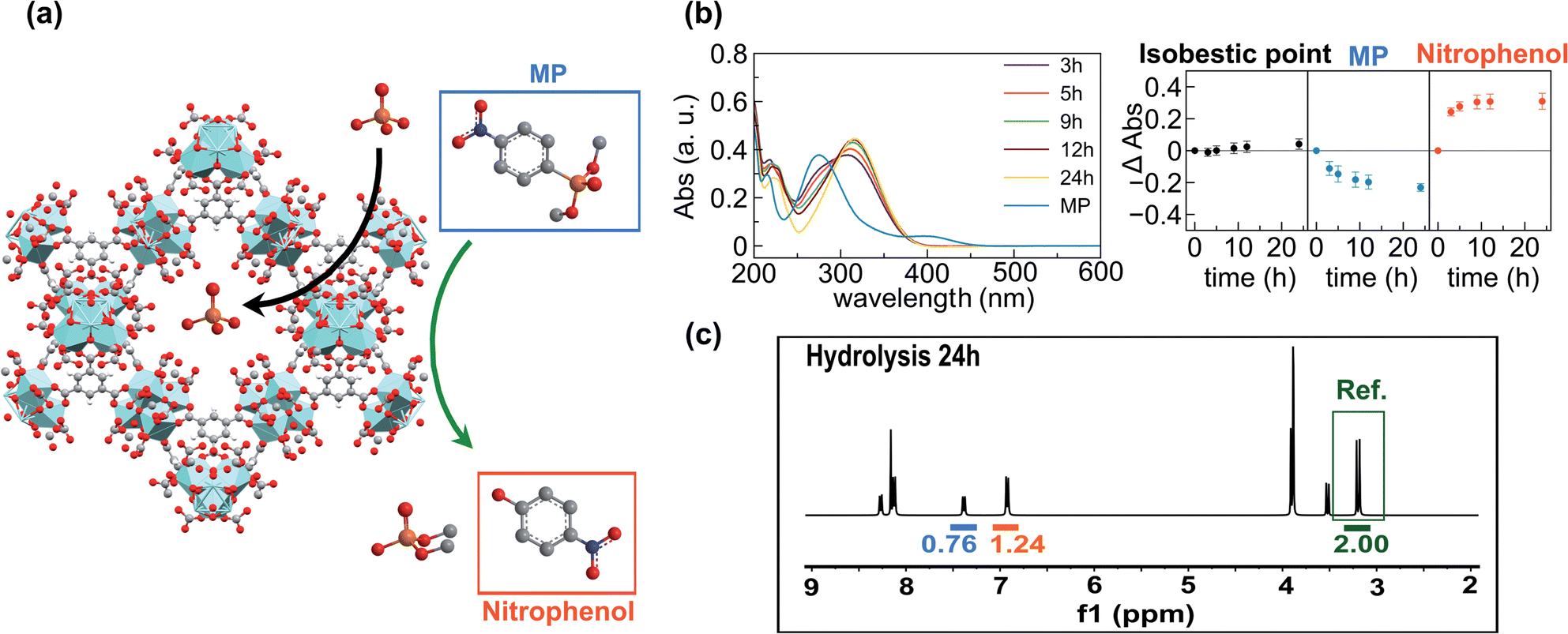 | ||
| Fig. 3 (a) Schematic representation of phosphate encapsulation into the cavities of MOF-808 and degradation of methyl paraoxon taking place on the MOF surface. (b) UV-Vis spectra showing the degradation of methyl paraoxon by MOF-808_60. On the right, evolution of absorbance at 297 nm (isosbestic point, black), 274 nm (methyl paraoxon, blue) and 316 nm (p-nitrophenol, orange). (c) 1H NMR of the supernatant after the hydrolysis of methyl paraoxon by MOF-808_60 after 24 h. The concentration of the internal reference (glyphosate) is the same as that of the total concentration of unreacted methyl paraoxon and generated p-nitrophenol, ruling out the encapsulation of the pesticide. | ||
To further explore the suitability of these materials as decontaminating agents, recyclability studies were carried out with MOF-808_60. After a first incubation of MOF-808_60 (0.08 μmol) with equimolecular amounts of methyl paraoxon and phosphate (0.08 mM, 1 mL), the solid was filtered off and treated with a highly concentrated hydrogencarbonate solution (4 mM, 1 mL, pH = 8.3, 5 h) recovering a substantial amount of the previously adsorbed phosphate (79.2 ± 3.0%) (Fig. 4). Then, and in order to decompose the entrapped hydrogencarbonate anions, we proceeded to regenerate the MOF-808_60 matrix by suspending the solid with diluted hydrochloric acid (4 mM, 1 mL, 5 h). The recyclability was further evaluated over two additional cycles. The results revealed that the material maintained similar performances to the ones observed, in the first cycle, towards phosphate adsorption, pesticide degradation and phosphate recovery. A drop of only 10% occurred between the second and third cycles for methyl paraoxon hydrolysis (Fig. 4). To prove the impact of all these processes (catalysis and adsorption, subsequent desorption of phosphate and matrix regeneration) on the structural integrity of the dual material, both the MOF_808_60 solid and eluted solutions were analysed by XRPD and ICP-MS, respectively. Although a certain loss of crystallinity was observed after the different treatments (Fig. S9†), neither Zr nor trimesate linker leaching was observed, ruling out ligand exchange. Nevertheless, a partial displacement of the –COO– sites of BTC linkers cannot be ruled out, as previously observed by Zhang, Chen et al.27 In any case, this observed amorphization does not affect the final performances of our material, as demonstrated by its good recyclability.
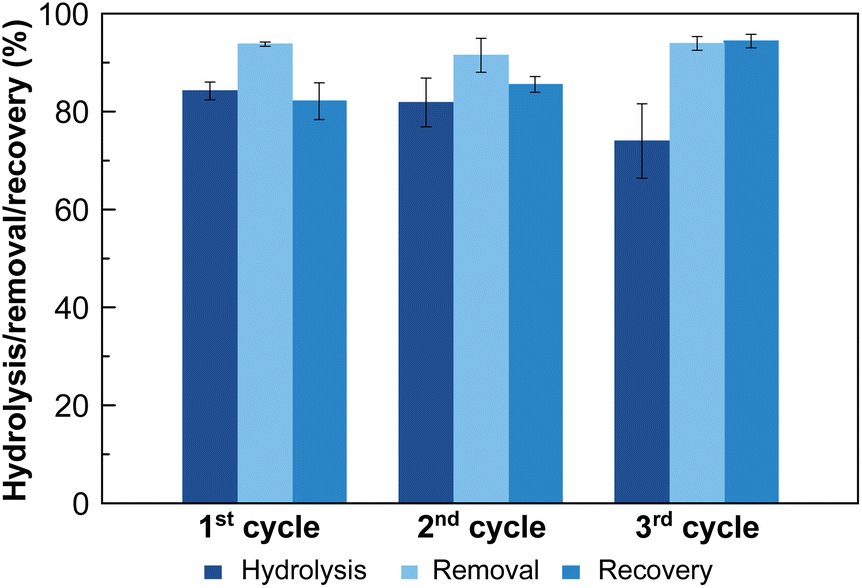 | ||
| Fig. 4 Recyclability studies of MOF-808_60 along three successive cycles: (i) simultaneous hydrolysis of methyl paraoxon and removal of phosphate and (ii) subsequent recovery of phosphate by exchange with hydrogencarbonate anions. After each cycle, MOF-808_60 was regenerated by treatment with an aqueous solution of hydrochloric acid (2.5 mM). | ||
Furthermore, the ability of MOF-808_60 to both decompose methyl paraoxon and capture phosphate in the presence of several interferents typically found in water, such as chloride, sulphate, nitrate and hydrogencarbonate ions (interference:pesticide:phosphate ratio 5:1:1), was investigated (Fig. S10†). Regarding pesticide hydrolysis, only the presence of sulphate and nitrate seemed to slightly decrease the total amount of degraded methyl paraoxon after 24 h (80.3 ± 1.5 and 71.2 ± 2.1%, respectively). In contrast, the phosphate adsorption capacity of MOF-808_60 remained unaltered in all cases except when hydrogencarbonate was used as an interferent (phosphate uptake dropped by up to 55.9 ± 5.7%). Notwithstanding, the hydrogencarbonate to phosphate molar ratio used in these assays is much larger than that typically found in wastewaters (1.5:1).28
Conclusions
We have demonstrated the feasibility of preparing high quality MOF-808 materials through a fast and environmentally friendly water-based microwave reaction. By modulating the heating ramp, highly crystalline and porous materials with homogeneous and tuned crystal size were obtained. Moreover, these matrixes have been proved to exhibit a good performance toward the capture of phosphate ions or the degradation of toxic methyl paraoxon pesticide in water. In mixtures containing both pollutants, these MOFs were also able to simultaneously decompose methyl paraoxon and capture phosphate ions without a significant detriment to the individual performance. UV-Vis and NMR spectroscopy analysis have demonstrated that while phosphate ions can be adsorbed inside the pores, pesticide degradation takes place mainly on the external particle surface. Furthermore, a strategy has been designed to regenerate the adsorbent and recover the trapped phosphate ions which may contribute to the P circular economy. Specifically, MOF-808_60 has been proved to exhibit good reusability maintaining a good performance over at least three degradation–adsorption–desorption cycles. These results represent a step forward in the application of MOF materials in wastewater treatment.Author contributions
Lydia González: investigation, visualization. Rodrigo Gil-San Millán: investigation, methodology. Jorge A. R. Navarro: supervision, funding acquisition, writing – review & editing. Carmen R. Maldonado: project administration, supervision, funding acquisition, writing – original draft, writing – review & editing. Elisa Barea: project administration, supervision, funding acquisition, writing – original draft, writing – review & editing. Francisco J. Carmona: conceptualization, investigation, methodology, funding acquisition, writing – original draft, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
MCIN/AEI/10.13039/501100011033 (Project PID2020-113608RB-I00), FEDER/Junta de Andalucía-Consejería de Economía y Conocimiento (Project B-FQM-364-UGR18) and FEDER/Junta de Andalucía-Consejería de Transformación Económica, Industria, Conocimiento y Universidades (Project P20_00672 and P18-RT-612) and the University of Granada (Project PPJIA2021.20) are acknowledged for funding. F. J. C. is thankful for the financial support provided by the Marie Skłodowska-Curie Individual Fellowship (H2020-MSCA-IF-2019-EF-ST-888972-PSustMOF) within the European Union H2020 programme and EU FEDER. L. G. thanks MCIN for her contract (Garantía Juvenil en I+D+i, PEJ2018-004022-A). Funding for open access charge: Universidad de Granada/CBUA.Notes and references
- FAO, IFAD, UNICEF, WFP and WHO, The State of Food Security and Nutrition in the World 2021., FAO, Rome, 2021 Search PubMed.
- D. W. Sun, L. Huang, H. Pu and J. Ma, Chem. Soc. Rev., 2021, 50, 1070–1110 RSC.
- D. Tilman, K. G. Cassman, P. A. Matson, R. Naylor and S. Polasky, Nature, 2002, 418, 671–677 CrossRef CAS PubMed.
- W. J. Brownlie, M. A. Sutton, D. S. Reay, K. V. Heal, L. Hermann, C. Kabbe and B. M. Spears, Nat. Food, 2021, 2, 71–74 CrossRef CAS.
- A. Mojiri, J. L. Zhou, B. Robinson, A. Ohashi, N. Ozaki, T. Kindaichi, H. Farraji and M. Vakili, Chemosphere, 2020, 253, 126646 CrossRef CAS PubMed.
- W. Boedeker, M. Watts, P. Clausing and E. Marquez, BMC Public Health, 2020, 20, 1–19 CrossRef PubMed.
- X. Min, X. Wu, P. Shao, Z. Ren, L. Ding and X. Luo, Chem. Eng. J., 2019, 358, 321–330 CrossRef CAS.
- Y. Gu, D. Xie, Y. Ma, W. Qin, H. Zhang, G. Wang, Y. Zhang and H. Zhao, ACS Appl. Mater. Interfaces, 2017, 9, 32151–32160 CrossRef CAS PubMed.
- Q. Yang, J. Wang, X. Chen, W. Yang, H. Pei, N. Hu, Z. Li, Y. Suo, T. Li and J. Wang, J. Mater. Chem. A, 2018, 6, 2184–2192 RSC.
- X. Zhu, B. Li, J. Yang, Y. Li, W. Zhao, J. Shi and J. Gu, ACS Appl. Mater. Interfaces, 2015, 7, 223–231 CrossRef CAS.
- L. González, F. J. Carmona, N. M. Padial, J. A. R. Navarro, E. Barea and C. R. Maldonado, Mater. Today Chem., 2021, 22, 100596 CrossRef.
- J. E. Mondloch, M. J. Katz, W. C. Isley, P. Ghosh, P. Liao, W. Bury, G. W. Wagner, M. G. Hall, J. B. Decoste, G. W. Peterson, R. Q. Snurr, C. J. Cramer, J. T. Hupp and O. K. Farha, Nat. Mater., 2015, 14, 512–516 CrossRef CAS PubMed.
- M. J. Katz, J. E. Mondloch, R. K. Totten, J. K. Park, S. T. Nguyen, O. K. Farha and J. T. Hupp, Angew. Chem., Int. Ed., 2014, 53, 497–501 CrossRef CAS PubMed.
- M. C. De Koning, M. Van Grol and T. Breijaert, Inorg. Chem., 2017, 56, 11804–11809 CrossRef CAS.
- S. J. Garibay, O. K. Farha and J. B. Decoste, Chem. Commun., 2019, 55, 7005–7008 RSC.
- E. European Chemical Agency, Substance Infocard (DMF), (accessed 22 February, 2022), https://echa.europa.eu/es/substance-information/-/substanceinfo/100.000.617 Search PubMed.
- K. Užarević, T. C. Wang, S. Y. Moon, A. M. Fidelli, J. T. Hupp, O. K. Farha and T. Friščić, Chem. Commun., 2016, 52, 2133–2136 RSC.
- J. M. Yassin, A. M. Taddesse and M. Sánchez-Sánchez, Microporous Mesoporous Mater., 2021, 324, 111303 CrossRef CAS.
- Z. Hu, T. Kundu, Y. Wang, Y. Sun, K. Zeng and D. Zhao, ACS Sustainable Chem. Eng., 2020, 8, 17042–17053 CrossRef CAS.
- S. Dai, C. Simms, I. Dovgaliuk, G. Patriarche, A. Tissot, T. N. Parac-Vogt and C. Serre, Chem. Mater., 2021, 33, 7057–7066 CrossRef CAS.
- H. Furukawa, F. Gándara, Y. B. Zhang, J. Jiang, W. L. Queen, M. R. Hudson and O. M. Yaghi, J. Am. Chem. Soc., 2014, 136, 4369–4381 CrossRef CAS PubMed.
- R. Gil-San-Millan, E. López-Maya, A. E. Platero-Prats, V. Torres-Pérez, P. Delgado, A. W. Augustyniak, M. K. Kim, H. W. Lee, S. G. Ryu and J. A. R. Navarro, J. Am. Chem. Soc., 2019, 141, 11801–11805 CrossRef CAS PubMed.
- H. Reinsch, S. Waitschat, S. M. Chavan, K. P. Lillerud and N. Stock, Eur. J. Inorg. Chem., 2016, 2016, 4490–4498 CrossRef CAS.
- Supelco, Saf. data sheet Paraoxon-methyl, (accessed 6 April, 2022), https://www.sigmaaldrich.com/ES/en/sds/sial/46192 Search PubMed.
- Sigma-Aldrich, Saf. data sheet 4-Nitrophenol, Safety data sheet of 4-Nitrophenol, (accessed 6 April, 2022) Search PubMed.
- USA.Gov, Dimethyl hydrogen phosphate, PubChem Database, (accessed 6 April, 2022) Search PubMed.
- W. Zhang, A. Bu, Q. Ji, L. Min, S. Zhao, Y. Wang and J. Chen, ACS Appl. Mater. Interfaces, 2019, 11, 33931–33940 CrossRef CAS PubMed.
- M. B. Pescod, Wastewater treatment and use in agriculture - FAO irrigation and drainage, 1992, vol. 47 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details of experimental procedures, synthesis of compounds, PXRD diffractograms, adsorption isotherms and kinetics, 1H and 31P NMR spectra, and recyclability and interference studies, including Fig. S1–S10 and Tables S1–S3. See https://doi.org/10.1039/d2ta02074b |
| This journal is © The Royal Society of Chemistry 2022 |