
Enhanced hydrogen evolution activity of CsPbBr3 nanocrystals achieved by dimensionality change†
Qing
Guo
 *a,
Jin-Dan
Zhang
a,
Ya-Jing
Chen
b,
Ke-Yuan
Zhang
a,
Li-Na
Guo
a,
Qi-Chao
Shan
a,
Jun-Lin
Lu
a,
Xin-Hua
Duan
*a and
Li-Zhu
Wu
b
*a,
Jin-Dan
Zhang
a,
Ya-Jing
Chen
b,
Ke-Yuan
Zhang
a,
Li-Na
Guo
a,
Qi-Chao
Shan
a,
Jun-Lin
Lu
a,
Xin-Hua
Duan
*a and
Li-Zhu
Wu
b
aXi’an Key Laboratory of Sustainable Energy Material Chemistry, School of Chemistry, Xi’an Jiaotong University, Xi’an 710049, P. R. China. E-mail: guoqing92@xjtu.edu.cn; duanxh@xjtu.edu.cn
bKey Laboratory of Photochemical Conversion and Optoelectronic Materials, Technical Institute of Physics and Chemistry, Chinese Academy of Sciences, Beijing 100190, P. R. China
First published on 7th March 2023
Abstract
Dimensionality plays a vital role at the nanoscale in tuning the electronical and photophysical properties and surface features of perovskite nanocrystals. Here, 3D and 1D all-inorganic CsPbBr3 nanocrystals were chosen as model materials to systemically reveal the dimensionality-dependent effect in photocatalytic H2 evolution. In terms of facilitating photoinduced electron–hole pair separation and charge transfer, as well as inducing proton reduction potential with the presence of fewer Br vacancies, 1D CsPbBr3 nanorods gave about a 5-fold improvement for solar H2 evolution.
On account of its high-energy density, environmental benignity, storability and transportability, hydrogen (H2), generated from a photocatalytic or photoelectrocatalytic technique by utilizing inexhaustible solar energy is regarded as one of the most ideal solar fuels,1–4 especially for semiconductor-based photocatalysis.5–7 Recently, halide perovskites, which are mostly applied in photovoltaic devices, have attracted great interest in the field of photocatalytic H2 evolution,8–11 owing to their superior optical and electronic properties,12–14 since the first report of three-dimensional (3D) methylammonium lead iodide (MAPbI3) as a photocatalyst for H2 evolution from hydriodic acid (HI) splitting under solar light irradiation.15 Inspired by this work, various halide perovskites, either all-inorganic16 or organic–inorganic hybrids,17–19 stabilized in concentrated haloid acid, have acted as photocatalysts to realize H2 production from haloid acid splitting. Composition and structural modification were focused on to improve photocatalytic efficiency by promoting charge carrier separation and surface reaction.20–23
At the nanoscale, dimensionality plays a vital role in tuning the electronical and photophysical properties of perovskite nanomaterials, as well as the surface features, which are another key factors, due to the high surface-to-volume ratio.24 For example, compared to 2D and 0D nanostructures, 1D nanowires/nanorods exhibit an intrinsically higher aspect ratio and a novel enclosing surface, resulting in a higher degree of anisotropy, leading to reduced grain boundaries and a direct carrier transportation pathway in one dimension.25 To the best of our knowledge, exploration of the effect of dimensionality in photocatalysis, especially for H2 evolution, has rarely been studied.
In this context, all-inorganic CsPbBr3 nanocrystals were chosen as model materials. 3D nanocubes and 1D nanorods have been extensively studied to reveal the dimensionality-dependent effect in photocatalytic H2 evolution. As shown in Scheme S1b (ESI†), the ultrasmall diameter of 1D nanorods maintains the quantum size effects of the exciton in the radial direction. Moreover, a 1D nanostructure provides fast carrier transport in the axial direction to achieve long-distance separation.26 Under visible light irradiation, an almost 5-fold enhancement in the H2 generation rate was observed for 1D nanorods when using Pt as a cocatalyst, compared to that of 3D nanocubes. Mechanistic insights revealed that the superior H2 evolution activity of 1D nanorods benefited from facilitated photogenerated charge carrier separation and transfer, as well as induced proton reduction potential with fewer Br vacancies on the surface.
In order to accurately control the shape and size of the materials, CsPbBr3 nanocubes (denoted 3D NCs) were synthesized via a traditional hot injection method at high temperature.27,28 Through the oriented attachment mechanism, the initially formed nanocubes could gradually transform into nanorods during the synthesis process.29 Hence, 1D nanorods (denoted 1D NRs) could be obtained by prolonging the reaction time (see details in ESI†). The narrow peaks of the prepared materials in the X-ray diffraction (XRD) pattern (Fig. S1, ESI†) matched well with the standard pattern of CsPbBr3 (PDF No. 18-0364),27 verifying the successful formation of orthorhombic phase CsPbBr3. Transmission electron microscopy (TEM) images confirmed that the obtained 3D NCs and 1D NRs acquired the shape of a cube (Fig. 1a) and a rod (Fig. 1d), respectively. The enlarged TEM images show a size of about 13–14 nm (Fig. S2, ESI†) and a diameter of about 12–13 nm (Fig. S4, ESI†), with a clear lattice distance of ∼5.88 Å (Fig. 1b and Fig. S3, ESI†) and ∼5.98 Å (Fig. 1e and Fig. S5, ESI†) for 3D NCs and 1D NRs, respectively, corresponding to the (001) plane of orthorhombic phase CsPbBr3 with a lattice constant of 5.87 Å. Moreover, energy-dispersive X-ray (EDX) analysis (Fig. S6, ESI†) and the full X-ray photoelectron spectroscopy (XPS) spectrum (Fig. S7, ESI†) revealed the coexistence of Cs, Pb and Br elements, and the corresponding elemental mapping (Fig. 1c and f) showed a uniform distribution of the three elements in the nanocrystals, all of which indicated the successful preparation of 3D NCs and 1D NRs.
 | ||
| Fig. 1 (a) TEM image, (b) high-resolution TEM image and (c) corresponding elemental mapping of Cs, Pb and Br elements, respectively, over 3D NCs. (d) TEM image, (e) high-resolution TEM image and (f) corresponding elemental mapping of Cs, Pb and Br elements, respectively, over 1D NRs. | ||
In addition, the first absorption peaks centered at ∼506 nm (Fig. S8, ESI†) and approximate positions of the photoluminescence (PL) peak (Fig. S9, ESI†) indicated the size of 3D NCs was similar to the diameter of the 1D NRs, because of the quantum confinement effects of 1D NRs in the radial direction. Moreover, higher PL intensity of 1D NRs mainly attributed to inhibited nonradiative recombination of electron–hole pairs on surface defects, implied that fewer defects existed on 1D NRs, which might be favorable for H2 evolution.
To evaluate the performance of the prepared CsPbBr3 samples as photocatalysts for H2 generation, post-synthetic treatment of CsPbBr3 is in high demand, because of the poor electrical conductivity of long-chain organic ligands capped on the obtained nanocrystal surfaces. Herein, a facile ligand exchange was performed using tetrafluoroborate salt (NH4BF4) as a treatment agent to remove the long-chain ligands (see details in ESI†).27 The Fourier transform-infrared (FT-IR) spectra show (Fig. S10, ESI†) a lower density of organic ligands on the CsPbBr3 surface, suggesting the effective removal of the long-chain ligands from the surface by NH4BF4.
The activities of photocatalytic H2 evolution over the as-prepared CsPbBr3 samples were evaluated in saturated HBr aqueous solution with the addition of H3PO2 under visible-light irradiation (λ ≥ 400 nm) (see details in ESI†).15 Pt as cocatalyst was deposited through an in situ photoreduction method. As displayed in Fig. 2a, CsPbBr3, HBr, H3PO2, cocatalyst and visible light were all necessary for efficient solar H2 evolution. Excitingly, after ligand exchange, the H2 evolution rate of the 3D NCs could be improved almost 3.4-fold (Fig. 2b). This can be attributed to the ionic ligand being able to facilitate charge separation and transfer as well as proton adsorption. Based on this, CsPbBr3 nanocrystals used for photocatalytic experiments were capped with ionic ligands of BF4− unless otherwise noted. After that, photocatalytic experiments were carried out with 3D NCs and 1D NRs as photocatalysts, respectively, under identical conditions to evaluate the effect of dimensionality on H2 evolution activity. Obviously, 1D NRs showed a much higher H2 evolution rate than 3D NCs (Fig. 2c). And a normalized H2 generation rate with BET surface area gave about a 4-fold improvement for 1D NRs (Table S1, ESI†).
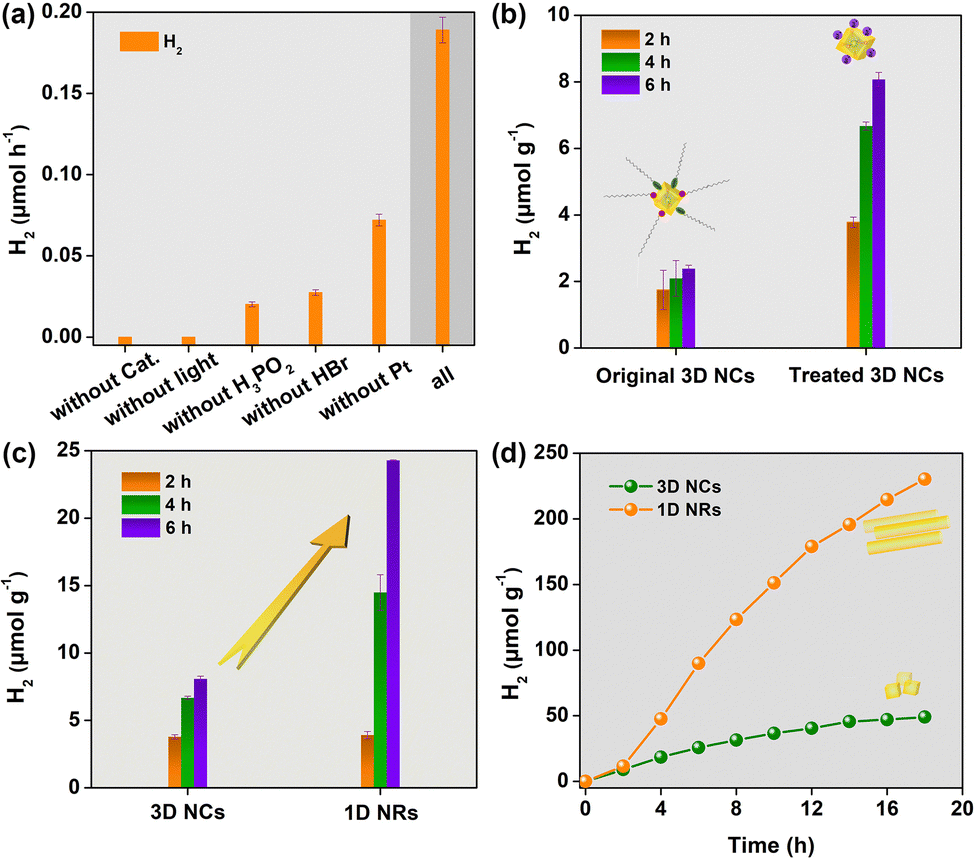 | ||
| Fig. 2 (a) Control experiments of solar H2 evolution in 2.0 h of irradiation over 3D NCs after ligand exchange (λ ≥ 400 nm). (b) Comparison of solar H2 evolution between 3D NCs before and after ligand exchange under identical conditions. (c) Solar H2 evolution with 3D NCs and 1D NRs capped with BF4−. (d) Solar H2 production under optimal conditions with long irradiation time. Error bars represent mean ± standard deviation (s.d.) of at least three independent experiments. | ||
In order to further improve the efficiency of solar H2 evolution, the mass ratio of Pt to CsPbBr3 was optimized (Fig. S11, ESI†), and 1.0 wt% was chosen as the optimal mass ratio for the photocatalytic reaction from the perspective of economics. Under optimal conditions, 1D NRs showed superior H2 evolution activity compared to 3D NCs even with long irradiation time. As shown from the kinetics curves of H2 evolution (Fig. 2d), the photocatalytic activity was dramatically boosted after the induction period of 2 h, mainly attributed to the formation of Pt cocatalyst in solution. After 18 h of irradiation, 1D NRs exhibited an H2 evolution rate of ∼240 μmol g−1, which was an almost 5-fold enhancement, further indicating the superior activity of 1D NRs towards photocatalytic H2 evolution. The results demonstrated that tuning the dimensionality of perovskite nanocrystals plays a vital role in enhanced photocatalytic H2 evolution. In addition, even with 44 h of irradiation under AM 1.5, H2 was sustainably evolved (Fig. S12, ESI†). A comparison of the crystalline structure (Fig. S13, ESI†), morphology and elemental distribution (Fig. S14, ESI†) shows no obvious changes between fresh and recycled 1D NRs, further signifying the excellent stability of the 1D NRs. Subsequently, the apparent quantum yield (AQY) was measured to be ∼0.04% (Fig. S15, ESI†). In further, the wavelength-dependent activity of 1D NRs for H2 production was surveyed (Fig. S16, ESI†), which shown analogous trends to the UV-vis absorption spectra of CsPbBr3 (Fig. S8, ESI†), implying CsPbBr3 is an active photocatalyst.
Then the effect of reaction time during the preparation of NRs on H2 evolution was investigated, which was varied from 90 min to 70 min (denoted 1D NRs-70 min), 110 min (denoted 1D NRs-110 min) and 130 min (denoted 1D NRs-130 min) (Fig. S17 and S18, ESI†). As shown in Fig. S19 (ESI†), 1D NRs exhibited the highest H2 evolution activity compared to 1D NRs-70 min, 1D NRs-110 min or 1D NRs-130 min. This indicated that the time for nanorod growth was critical for photocatalytic activity, which was optimal when the initially formed nanocubes were completely transformed to nanorods. Moreover, the photocatalytic activity of 1D NRs was compared with other reported all-inorganic and organic–inorganic perovskites (Fig. S20, ESI†) under identical conditions (Table S2, ESI†), which further suggested the superior activity of 1D NRs in our work.
To gain insight into the mechanism of the outstanding photocatalytic performance of 1D NRs, the driving force of the photocatalytic reaction was first investigated. In this regard, valence-band (VB) XPS analysis (Fig. S21, ESI†) was utilized to reveal the VB potentials of 3D NC and 1D NR materials, in which the band positions were determined to be ∼ + 1.49 V and ∼ + 1.36 V versus the normal hydrogen electrode (NHE) for 3D NCs and 1D NRs, respectively. The band gaps of 3D NCs and 1D NRs were calculated to be ∼2.36 eV and ∼2.37 eV, respectively, using Tauc plots (Fig. S22, ESI†). Accordingly, the conduction-band (CB) positions were estimated to be ∼−0.87 V and ∼−1.01 V vs. NHE, respectively, and the corresponding band diagram has been depicted (Fig. S23, ESI†). Thus, the driving force (0.87 eV and 1.01 eV) for H2 generation is both thermodynamically feasible and sufficient for 3D NCs and 1D NRs.
Subsequently, steady-state and time-resolved spectroscopic techniques were conducted to investigate photoexcited charge separation behavior over CsPbBr3 capped with BF4−. As shown in Fig. 3a, the PL intensity of 1D NRs at ∼525 nm was higher than that of 3D NCs. This demonstrated that the recombination of photogenerated electron–hole pairs through a nonradiative pathway on surface defects is suppressed over 1D NRs, owing to a lower amount of surface defects. Moreover, the PL lifetime was prolonged from the 79.9 ns of 3D NCs to the 125.6 ns of 1D NRs, further indicating fewer defects on the surface of 1D NRs (Fig. 3b and Table S3, ESI†). The surface features will be investigated in detail through XPS analysis; see later in the text.
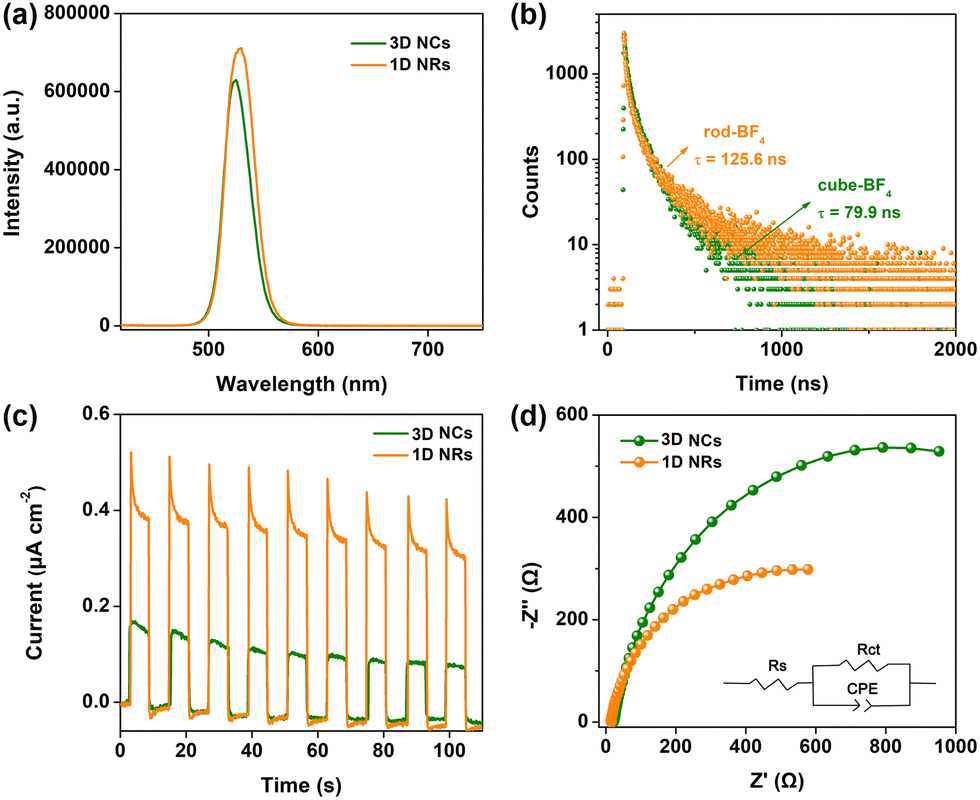 | ||
| Fig. 3 (a) PL spectra and (b) time-resolved PL spectra of 3D NCs and 1D NRs powered counterparts (excitation: 405 nm laser). (c) Transient photocurrent responses and (e) EIS Nyquist plots of 3D NC and 1D NR electrodes under the same conditions (inset is the fitted equivalent circuit). | ||
In further, a facile interfacial charge transfer process was unveiled by photoelectrochemical (PEC) measurements. The experimental details for preparing the electrodes are provided in ESI.† As shown in Fig. 3c, a 1D NR electrode exhibited much improved transient photocurrent response, compared with the 3D NC counterpart under identical conditions, implying the facilitated charge transfer kinetics in 1D NRs, owing to the improved charge separation.30 Electrochemical impedance spectroscopy (EIS) was also measured to monitor the interfacial charge-transfer properties. A smaller arc radius for a 1D NR electrode than for a 3D NC electrode was observed in the EIS Nyquist plots (Fig. 3d), suggesting much lower interfacial charge transfer resistance. The equivalent circuit was further fitted to reveal charge transfer resistance (Table S4, ESI†), in which the 1D NR electrode exhibited a lower charge transfer resistance (Rct) of 778 Ω than that of 3D NCs with an Rct of 1456 Ω, further confirming the favorable interfacial charge transfer.31 Moreover, the polarization curves of 3D NC and 1D NR electrodes (Fig. S24, ESI†) suggested that dimensionality tuning from 3D NCs to 1D NRs could decrease the overpotential and promote proton reduction to H2, leading to the boosted photocatalytic H2 production activity.
To further investigate the surface states of the obtained CsPbBr3 samples, XPS spectroscopy was conducted. Full XPS spectrum analysis showed that 3D NCs and 1D NRs had a Pb/Br ratio of 1.351/1 and 1.350/1, respectively, suggesting that both samples possessed Br vacancies. Then, high-resolution XPS signals of Pb 4f were fitted with Gaussian functions. As shown in Fig. S25 (ESI†), two strong peaks at ∼142.78 eV and ∼137.9 eV are indexed to Pb(II) species coordinated with Br−. Another two peaks at ∼142.28 eV and ∼137.36 eV are attributed to Pb (δ+) species coordinated with BF4−. We calculated the percentages of Pb (δ+) species in CsPbBr3 according to the integral areas (Table S5, ESI†). It was found that the atomic ratios of Pb (δ+) species were 46.3% and 41.3% over 3D NCs and 1D NRs, respectively, confirming that 1D NCs possessed fewer Br vacancies, which was further verified by the surface charge as revealed by zeta-potential (Table S6, ESI†). The zeta potentials of 3D NCs and 1D NRs were −10.0 and −3.42 mV, respectively, which not only illustrated their surface negative charge27 but also demonstrated decreased negative charge with reducing concentration of Br vacancies, since they acted as electron traps.32 This was consistent with the above spectroscopic results.
Based on that, we utilized density functional theory (DFT) calculations to investigate the ability of CsPbBr3 for proton reduction (see details in ESI†).  values of CsPbBr3 with and without Br vacancies were compared. Fig. S26 (ESI†) shows the corresponding structural models of H atom adsorption. Notably, the
values of CsPbBr3 with and without Br vacancies were compared. Fig. S26 (ESI†) shows the corresponding structural models of H atom adsorption. Notably, the 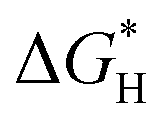 value of CsPbBr3 with Br vacancies was dramatically reduced to 0.21 eV (Fig. S27, ESI†), suggesting the potential of CsPbBr3 with Br vacancies for H2 evolution. Thus, Br vacancies are necessary for proton reduction, while large amounts of Br vacancies have a negative effect on photogenerated electron–hole separation, which is consistent with the above spectroscopic results. We came to the conclusion that the superior H2 generation activity of 1D NRs benefited from the induced proton reduction potential with the presence of Br vacancies, as well as the facilitated charge carriers kinetics with 1D nanostructures.
value of CsPbBr3 with Br vacancies was dramatically reduced to 0.21 eV (Fig. S27, ESI†), suggesting the potential of CsPbBr3 with Br vacancies for H2 evolution. Thus, Br vacancies are necessary for proton reduction, while large amounts of Br vacancies have a negative effect on photogenerated electron–hole separation, which is consistent with the above spectroscopic results. We came to the conclusion that the superior H2 generation activity of 1D NRs benefited from the induced proton reduction potential with the presence of Br vacancies, as well as the facilitated charge carriers kinetics with 1D nanostructures.
In summary, we have systematically demonstrated that dimensionality played an important role for CsPbBr3 nanocrystals towards solar H2 evolution activity. Under optimal conditions, it can be improved to give a five-fold enhancement after dimensionality tuning from 3D to 1D nanostructures. By a combination of theoretical and experimental investigations, the superior activity was revealed, which was the result of the promoted charge carrier kinetics, as well as the proton reduction ability induced by fewer Br vacancies present on 1D NRs. This work provides new insights for tuning photocatalytic H2 evolution performance under mild conditions.
This work was financially supported by the Natural Science Foundation of China (No. 21971201, 22171220), the Natural Science Basic Research Plan in Shanxi Province of China (2021JM-003), the Fundamental Research Funds of the Center Universities (No. xtr072022003, 11913222000020), and the China Postdoctoral Science Foundation Funded Project (2019M663659). We thank Prof. Fei Liang at Shandong University for the DFT calculations.
Conflicts of interest
There are no conflicts to declare.Notes and references
- M. Xiao, M. Hao, M. Lyu, E. G. Moore, C. Zhang, B. Luo, J. Hou, J. Lipton-Duffin and L. Wang, Adv. Funct. Mater., 2019, 29, 1905683 CrossRef CAS
.
- Y. Wu, Q. Wu, Q. Zhang, Z. Lou, K. Liu, Y. Ma, Z. Wang, Z. Zheng, H. Cheng, Y. Liu, Y. Dai, B. Huang and P. Wang, Energy Environ. Sci., 2022, 15, 1271 RSC
- H. Zhang, Z. Yang, W. Yu, H. Wang, W. Ma, X. Zong and C. Li, Adv. Energy Mater., 2018, 8, 1800795 CrossRef
- M. Crespo-Quesada, L. M. Pazos-Outon, J. Warnan, M. F. Kuehnel, R. H. Friend and E. Reisner, Nat. Commun., 2016, 7, 12555 CrossRef CAS PubMed
- Y.-J. Gao, X.-B. Li, X.-Z. Wang, N.-J. Zhao, Y. Zhao, Y. Wang, Z.-K. Xin, J.-P. Zhang, T. Zhang, C.-H. Tung and L.-Z. Wu, Matter, 2020, 3, 571 CrossRef
- G. Zhang, Z. A. Lan and X. Wang, Chem. Sci., 2017, 8, 5261 RSC
- J. He, Z. Yi, Q. Chen, Z. Li, J. Hu and M. Zhu, Chem. Commun., 2022, 58, 10723 RSC
- Y. Wu, P. Wang, X. Zhu, Q. Zhang, Z. Wang, Y. Liu, G. Zou, Y. Dai, M. H. Whangbo and B. Huang, Adv. Mater., 2018, 30, 1704342 CrossRef PubMed
- P. Zhou, H. Chen, Y. Chao, Q. Zhang, W. Zhang, F. Lv, L. Gu, Q. Zhao, N. Wang, J. Wang and S. Guo, Nat. Commun., 2021, 12, 4412 CrossRef CAS PubMed
- X. Zhao, S. Chen, H. Yin, S. Jiang, K. Zhao, J. Kang, P. F. Liu, L. Jiang, Z. Zhu, D. Cui, P. Liu, X. Han, H. G. Yang and H. Zhao, Matter, 2020, 3, 935 CrossRef
- Y. Zhao, Q. Zeng, Y. Yu, T. Feng, Y. Zhao, Z. Wang, Y. Li, C. Liu, J. Liu, H. Wei, S. Zhu, Z. Kang, H. Zhang and B. Yang, Mater. Horiz., 2020, 7, 2719 RSC
- T. Krishnamoorthy, H. Ding, C. Yan, W. L. Leong, T. Baikie, Z. Y. Zhang, M. Sherburne, S. Li, M. Asta, N. Mathews and S. G. Mhaisalkar, J. Mater. Chem. A, 2015, 3, 23829 RSC
- F. Wei, Z. Deng, S. Sun, F. Xie, G. Kieslich, D. M. Evans, M. A. Carpenter, P. D. Bristowe and A. K. Cheetham, Mater. Horiz., 2016, 3, 328 RSC
- Y.-F. Xu, M.-Z. Yang, B.-X. Chen, X.-D. Wang, H.-Y. Chen, D.-B. Kuang and C.-Y. Su, J. Am.
Chem. Soc., 2017, 139, 5660 CrossRef CAS PubMed
- S. Park, W. J. Chang, C. W. Lee, S. Park, H.-Y. Ahn and K. T. Nam, Nat. Energy, 2016, 2, 16185 CrossRef
- M. V. Pavliuk, M. Abdellah and J. Sá, Mater. Today Commun., 2018, 16, 90 CrossRef CAS
- W. Li, F. Wang, Z. Zhang and S. Min, Chem. Commun., 2021, 57, 7774 RSC
- F. Wang, X. Liu, Z. Zhang and S. Min, Chem. Commun., 2020, 56, 3281 RSC
- W. Li, F. Wang, Z. Zhang, X. Ma and S. Min, Sustainable Energy Fuels, 2022, 6, 76 RSC
- H. Wang, X. Wang, R. Chen, H. Zhang, X. Wang, J. Wang, J. Zhang, L. Mu, K. Wu, F. Fan, X. Zong and C. Li, ACS Energy Lett., 2018, 4, 40 CrossRef
- H. Wang, H. Zhang, J. Wang, Y. Gao, F. Fan, K. Wu, X. Zong and C. Li, Angew. Chem., Int. Ed., 2021, 60, 7376 CrossRef CAS PubMed
- Y. Wu, P. Wang, Z. Guan, J. Liu, Z. Wang, Z. Zheng, S. Jin, Y. Dai, M.-H. Whangbo and B. Huang, ACS Catal., 2018, 8, 10349 CrossRef CAS
- Z. Guan, Y. Wu, P. Wang, Q. Zhang, Z. Wang, Z. Zheng, Y. Liu, Y. Dai, M.-H. Whangbo and B. Huang, Appl. Catal., B, 2019, 245, 522 CrossRef CAS
- Z. Liu, H. Yang, J. Wang, Y. Yuan, K. Hills-Kimball, T. Cai, P. Wang, A. Tang and O. Chen, Nano Lett., 2021, 21, 1620 CrossRef CAS PubMed
- S. Pan, J. Li, Z. Wen, R. Lu, Q. Zhang, H. Jin, L. Zhang, Y. Chen and S. Wang, Adv. Energy Mater., 2021, 11, 202004002 Search PubMed
- Z. K. Xin, Y. J. Gao, Y. Gao, H. W. Song, J. Zhao, F. Fan, A. D. Xia, X. B. Li, C. H. Tung and L. Z. Wu, Adv. Mater., 2022, 34, e2106662 CrossRef PubMed
- J.-C. Wang, N. Li, A. M. Idris, J. Wang, X. Du, Z. Pan and Z. Li, Sol. RRL, 2021, 5, 2100154 CrossRef CAS
- L. Protesescu, S. Yakunin, M. I. Bodnarchuk, F. Krieg, R. Caputo, C. H. Hendon, R. X. Yang, A. Walsh and M. V. Kovalenko, Nano Lett., 2015, 15, 3692 CrossRef CAS PubMed
- D. Zhang, S. W. Eaton, Y. Yu, L. Dou and P. Yang, J. Am. Chem. Soc., 2015, 137, 9230 CrossRef CAS PubMed
- C. Cai, Y. Teng, J. H. Wu, J. Y. Li, H. Y. Chen, J. H. Chen and D. B. Kuang, Adv. Funct. Mater., 2020, 30, 2001478 CrossRef CAS
- Q. Guo, F. Liang, X.-Y. Gao, Q.-C. Gan, X.-B. Li, J. Li, Z.-S. Lin, C.-H. Tung and L.-Z. Wu, ACS Catal., 2018, 8, 5890 CrossRef CAS
- Z. Ni, C. Bao, Y. Liu, Q. Jiang, W.-Q. Wu, S. Chen, X. Dai, B. Chen, B. Hartweg, Z. Yu, Z. Holman and J. Huang, Science, 2020, 367, 1352 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details. See DOI: https://doi.org/10.1039/d2cc06731e |
| This journal is © The Royal Society of Chemistry 2023 |