
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Catalytic conversion of chitin as a nitrogen-containing biomass
Hirokazu
Kobayashi
 *a,
Takuya
Sagawa
b and
Atsushi
Fukuoka
c
*a,
Takuya
Sagawa
b and
Atsushi
Fukuoka
c
aKomaba Institute for Science, The University of Tokyo, 3-8-1 Komaba, Meguro-ku, Tokyo 153-8902, Japan. E-mail: kobayashi-hi@g.ecc.u-tokyo.ac.jp
bDepartment of Industrial Chemistry, Faculty of Engineering, Tokyo University of Science, 6-3-1 Niijuku, Katsushika-ku, Tokyo 125-8585, Japan
cInstitute for Catalysis, Hokkaido University, Kita 21 Nishi 10, Kita-ku, Sapporo, Hokkaido 001-0021, Japan
First published on 25th April 2023
Abstract
The conversion of chitin enables the utilisation of naturally-fixed nitrogen in addition to carbon toward establishing a sustainable carbon and nitrogen cycle. Chitin is an abundant biomass, 100 Gt per year, but most chitin-containing waste is discarded due to its recalcitrant properties. This feature article summarises the challenges and our work on chitin conversion to N-acetylglucosamine and oligomers with fascinating applications. Afterwards, we introduce recent progress on the chemical transformation of N-acetylglucosamine, followed by a discussion of future perspectives based on current status and findings.
 Hirokazu Kobayashi | Hirokazu Kobayashi received his PhD degree from Tokyo Institute of Technology in 2009. He moved to the Catalysis Research Centre, which became the Institute for Catalysis in 2015, at Hokkaido University as an Assistant Professor in 2009. He was appointed an Associate Professor in Komaba Institute for Science, The University of Tokyo in 2022. He is studying the catalytic conversion of natural resources such as biomass and alkanes to establish a sustainable society. |
 Takuya Sagawa | Takuya Sagawa received his PhD in Engineering from Tokyo University of Science under the guidance of Prof. T. Gunji in 2018. Then he joined the group of Prof. A. Fukuoka at Hokkaido University as a postdoctoral researcher in 2018. Then he moved to the group of Prof. M. Hashizume at Tokyo University of Science in 2020 and has been an assistant professor there. His research interests are in the conversion of chitin-derived amide sugar alcohol and the fabrication of composite materials composed of polysaccharides. |
 Atsushi Fukuoka | Atsushi Fukuoka is a Professor at the Institute for Catalysis (ICAT) at Hokkaido University. He studied homogeneous catalysis and received a PhD from The University of Tokyo in 1989. He then joined the Research Institute for Catalysis and Catalysis Research Centre (RIC and CRC, now ICAT) and started research on heterogeneous catalysis. He served as the Director of CRC from 2010 to 2014 and is currently appointed as an Advisor to the President of Hokkaido University. His current research interests are in catalytic biomass conversion and freshness preserving catalysts. |
Introduction
The basic concept of biomass utilisation is to establish a sustainable energy and material cycle. Current societies rely on fossil resources, but these fuels cannot regenerate within a reasonable timespan. Human activity required 6.4 × 1017 kJ of energy and released 36 Gt of CO2 in 2021, ca. 20-fold larger than the amounts in 1900. The increased amount of CO2 is causing the greenhouse effect and ocean acidification.1 Meanwhile, plants fix solar energy as organic materials using atmospheric CO2, and the net primary production (NPP) is about 1017 carbon-g per year, accumulating 4 × 1018 kJ of energy.2 Accordingly, biomass energy is several times larger than our demand. The utilisation of biomass, together with other renewable energy and material cycle systems, including artificial photosynthesis and chemical recycling of plastics,3 will become an indispensable technology for building a sustainable society.Biomass conversion has mainly dealt with lignocellulose, starch and lipids, which are all composed of C, H and O atoms.4,5 Recently, artificially introducing N atoms to biomass-derived molecules has attracted significant interest as organic nitrogen compounds show a wide variety of functions.6–8 In this sense, chitin and chitosan, which are the most abundant biomass (annually ca. 100 Gt in total) next to lignocellulose,9 originally contain N atoms. Chitin and chitosan are both copolymers consisting of N-acetylglucosamine (NAG) and glucosamine units linked by β-1,4-glycosidic bonds (Fig. 1). Chitin contains more than 40 mol% NAG, and the polymers having lower ratios of NAG are called chitosan. Therefore, the conversion of chitin and chitosan enables the utilisation of naturally-fixed nitrogen to produce functional chemicals (Fig. 2).10,11 In terms of ‘planetary boundaries’, the utilisation of artificially-fixed nitrogen has exceeded the capacity of the Earth.12 Thus, the nitrogen cycle is important in addition to carbon, and a potential solution is to use naturally-fixed nitrogen with high efficiency. Proteins also consist of nitrogen-containing monomers, but the structures are often complicated. The simple structures of chitin and chitosan may facilitate their utilisation in chemical industries.
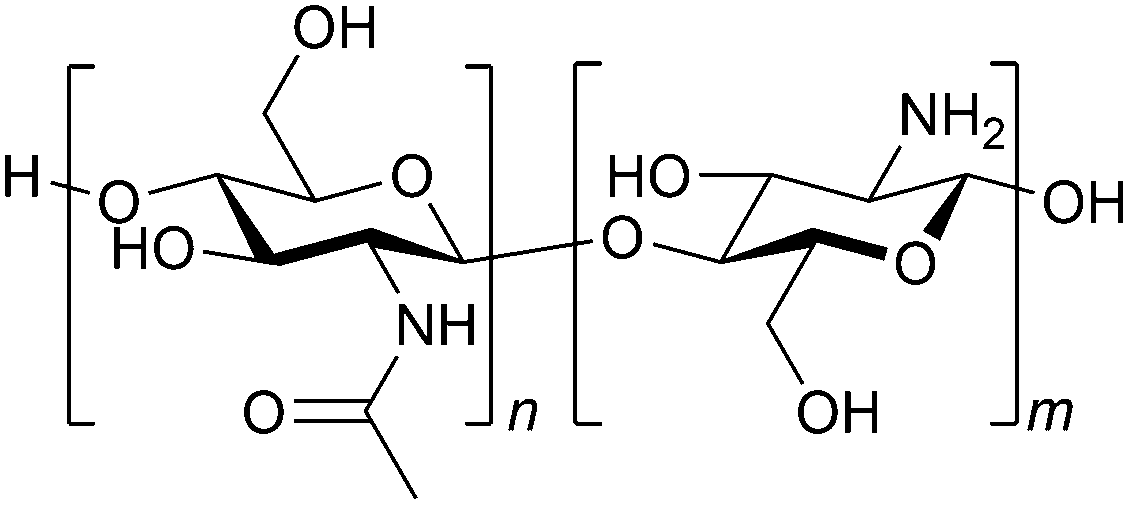 | ||
| Fig. 1 Structures of chitin and chitosan. NAG and glucosamine units are randomly mixed. Chitin: n/(n + m) > 40%, chitosan: n/(n + m) ≤ 40% as a typical standard. | ||
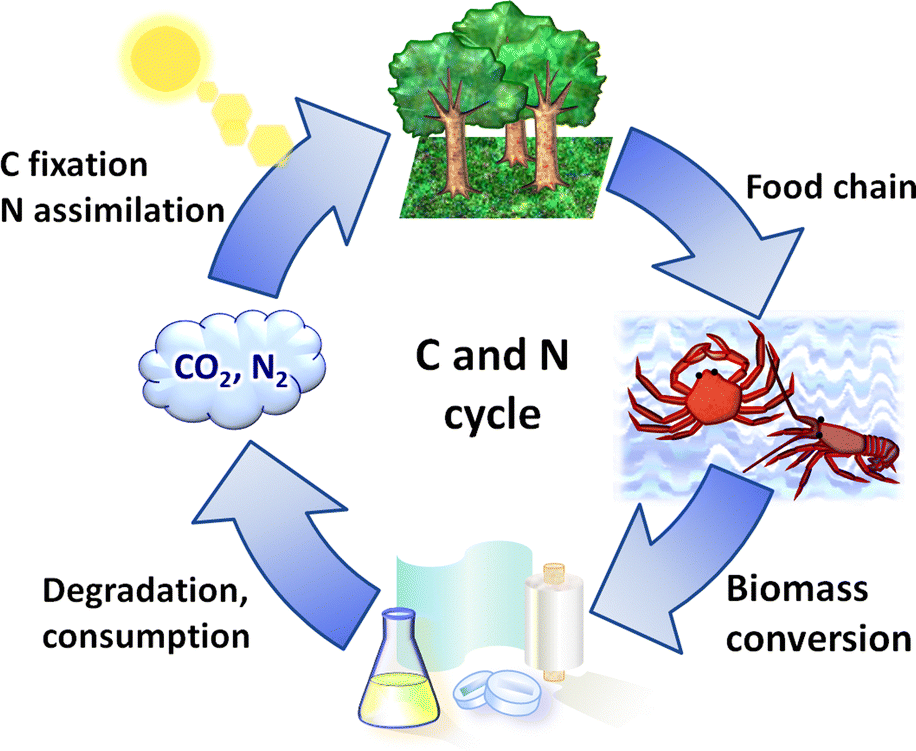 | ||
| Fig. 2 Nitrogen-containing biorefinery. | ||
While chitin and chitosan are attractive resources, they are resistant to degradation reactions, as described in detail in the following section. The development of new catalytic systems is crucial to achieving efficient utilisation of nitrogen-containing biomass.13–15 The selective scission of rigid chemical bonds within biomass molecules is especially important. Herein, this article summarises our recent work on the catalytic conversion of chitin to value-added organonitrogen compounds. We have preferred chitin over chitosan because the protection of reactive amino groups by acetyl groups is beneficial for selective chemical transformation. We show that acid catalysts including mineral acids and activated carbons hydrolyse chitin in the presence of mechanical milling. The section also discusses the reaction mechanisms and a significant application of chitin-oligosaccharides, the intermediates of hydrolysis. Afterwards, the authors introduce the conversion of NAG to useful derivatives that contain N atoms and its challenges, followed by concluding remarks.
Hydrolysis of chitin
Manufacture of NAG
Chitin contains many inter- and intra-molecular hydrogen bonds, which makes the polymer recalcitrant and insoluble in water. This nature is similar to that of cellulose, but there is a major difference between the two compounds. Cellulose forms hydrogen bonds to make two-dimensional sheets and they stack by dispersion forces, while chitin contains additional strong hydrogen bonds using amide groups (Fig. 3) to produce a three-dimensional hydrogen bond nework.16 Even after the ball-milling treatment, chitin preserves most of the hydrogen bonds.17 The hydrolysis of cellulose is a difficult subject,18 but that of chitin requires even harsher reaction conditions. Moreover, chitin has amide bonds in addition to glycosidic bonds, and both can be hydrolysed. The selective scission of the glycosidic linkage is needed to synthesise NAG. Accordingly, the hydrolysis of chitin is a challenge.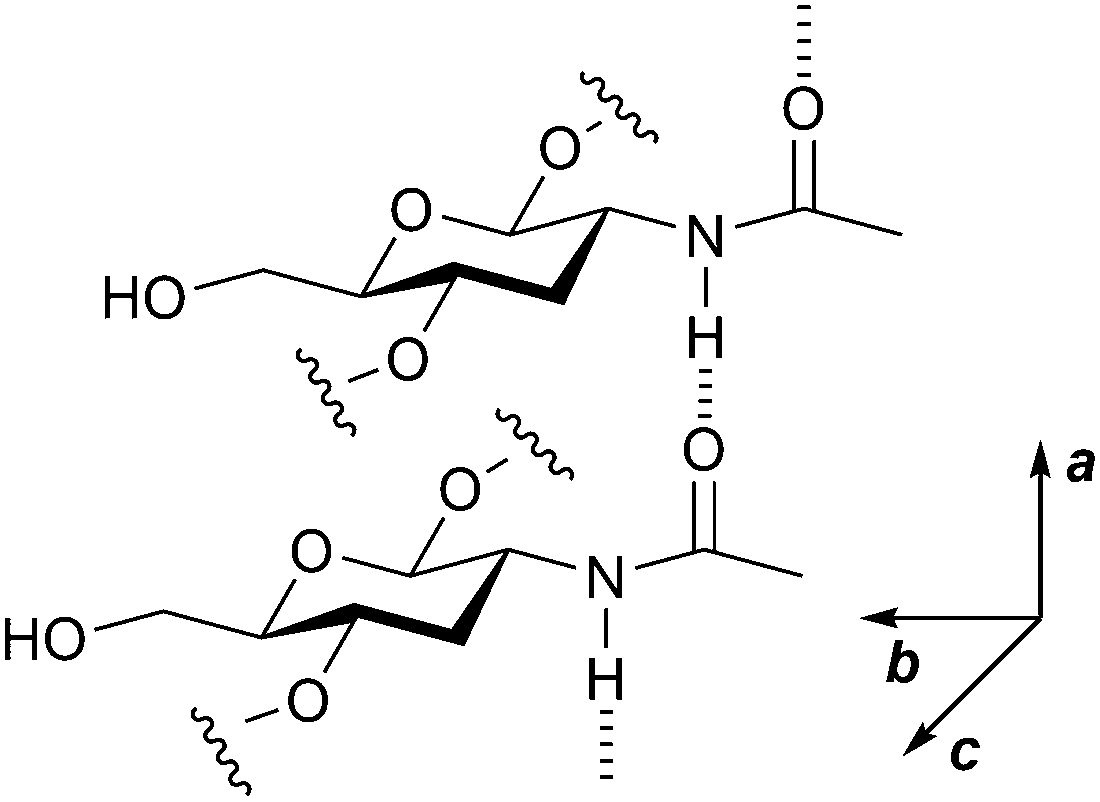 | ||
| Fig. 3 Hydrogen bonds made by amide groups in chitin. Amide groups form hydrogen bonds along the a-axis direction. The image omits some functional groups for better visibility. | ||
To manufacture NAG from chitin, typical industrial processes use concentrated HCl and enzymes.19,20 A high concentration of HCl is needed to accelerate the hydrolysis of glycosidic bonds while limiting deacetylation.21–23 The turnover numbers of acids are far less than 1 and this process produces a large amount of neutralisation waste. The hydrolysis of glycosidic bonds is an SN1 reaction, and the bond cleavage after the pre-equilibrium of protonation is the rate-determining step (Fig. 4). Therefore, the hydrolysis rate relies strongly on the activity of H3O+. In contrast, the hydrolysis of amide bonds under acidic conditions is an SN2 reaction. The carbonyl oxygen in the amide structure is basic, easily undergoing protonation. Our density functional theory (DFT) calculations estimate that the proton affinities (−ΔH° at 298 K) of the O atoms in amide and glycosidic bonds are 885 and 811 kJ mol−1, respectively. Moreover, the rate-determining step is the addition of water.24 Thus, the acid strength only slightly affects the reaction rate and the decrease in water activity at a high acid concentration slows down the reaction.21,24 Perhaps for a similar reason, pure formic acid selectively converts chitin to NAG formates with the retention of acetyl groups.25 In contrast, a mixture of aprotic polar solvents and water accelerates deacetylation,26 which is likely to be due to the increased nucleophilicity of water molecules by hindering hydrogen bonds (H2O⋯H2O). The activity and nucleophilicity of water are major factors in determining the hydrolysis selectivity. Another method to synthesise NAG is acetylating glucosamine using acetic anhydride.27 Water and methanol solvents allow the acetylation of amino groups while inhibiting the esterification of hydroxy groups. However, the direct synthesis of NAG from chitin would be better. These factors have pushed the price of NAG to over 50 GBP per kilogram. An efficient process for hydrolysing chitin to NAG is essential to achieve a chitin-based biorefinery.
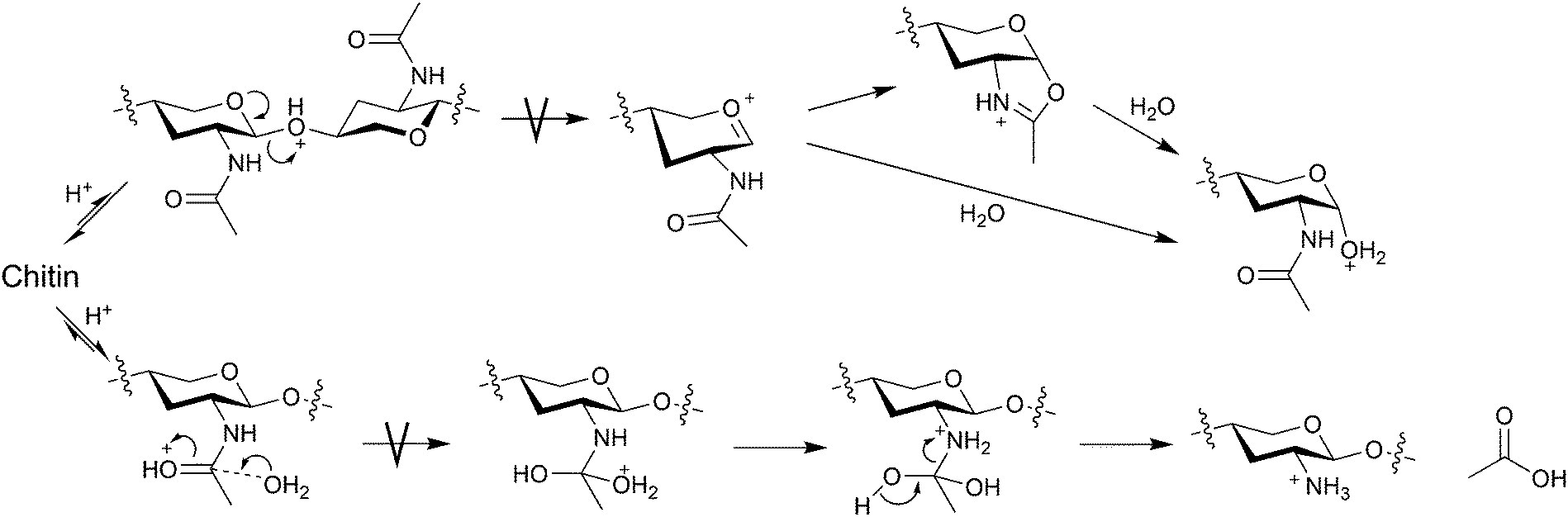 | ||
| Fig. 4 Hydrolysis mechanisms of glycosidic and amide bonds. The image omits some functional groups for better visibility. | ||
Hydrolysis of chitin by mineral acids under ball-milling conditions
Dilute mineral acids are ineffective for the selective hydrolysis of chitin as described above. Among other types of catalyst, carbon materials with weak acid sites characteristically hydrolyse cellulose in water,18,28 but this system does not function for chitin as it is. Chitin is more recalcitrant than cellulose, requiring us to add a new idea for achieving its hydrolysis.Previously, several groups have reported that acid catalysts hydrolyse cellulose to oligomers during the milling process.29–33 We expected that the mechanical forces of ball-milling will activate the recalcitrant chitin, thereby allowing the hydrolysis of chitin with a small amount of acid.34 Indeed, the effect of milling was obvious; a catalytic amount of H2SO4 quantitatively hydrolysed chitin to soluble compounds under dry planetary milling (Fig. 5). In detail, we added H2SO4 to chitin powder (substrate/catalyst ratio (S/C) = 8.1 mol-NAG unit/mol-H2SO4) dispersed in diethylether, and the sample was dried by a rotary evaporator. The resulting powder was milled using alumina balls and an alumina pot at 500 rpm for 6 h. The temperature inside the pot was at most 60 °C during milling. No solvent was used for this reaction, but physisorbed water on the sample (2–5 wt%) worked as a reagent for hydrolysis. 1H nuclear magnetic resonance (NMR) spectroscopy showed that the major products were oligomers of NAG with β-1,4-glycosidic bonds and 1,6-bonds including anhydride structures. Small quantities of unidentified products were also included. Surprisingly, acetic acid (AcOH) was not detected in the product mixture, suggesting no hydrolysis of amide bonds for the following reasons: first, low water content is beneficial to reducing deacetylation (Fig. 4). Second, the mechanical forces selectively accelerate the hydrolysis of glycosidic bonds (see below). Therefore, the reaction cleaved glycosidic bonds while preserving amide bonds.
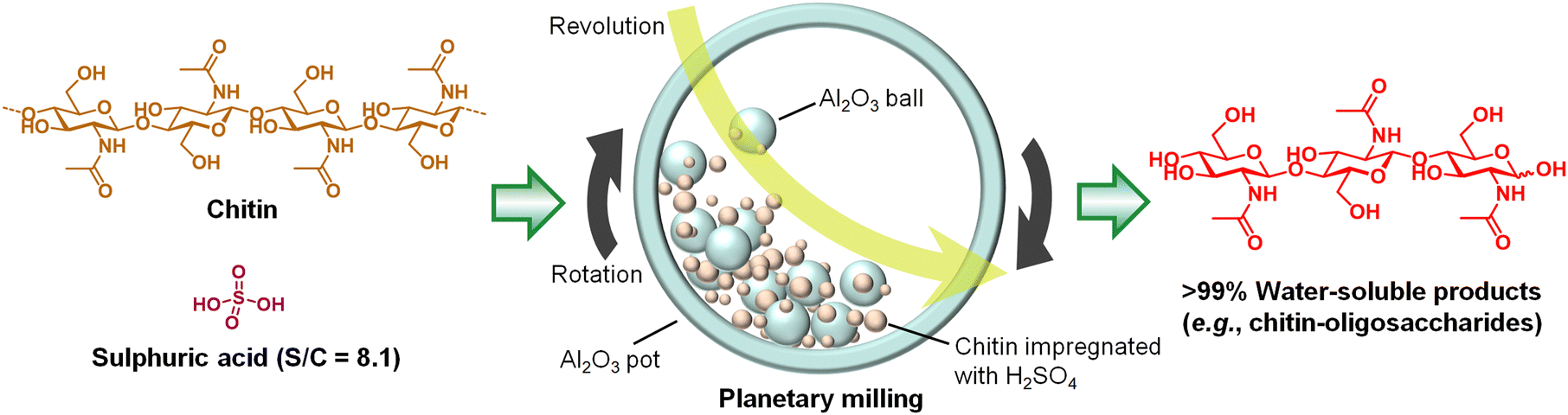 | ||
| Fig. 5 Hydrolysis of chitin by H2SO4 in the mechanochemical method. | ||
The solid sample produced by milling with H2SO4 underwent further hydrolysis in water solvent at 190 °C, giving NAG in 32% yield, based on original chitin.34 The yield increased to 61% at an S/C of 3.4 at 175 °C (Fig. 6).35 The turnover number of H2SO4 for the formation of NAG was 2.1, which was a catalytic reaction.
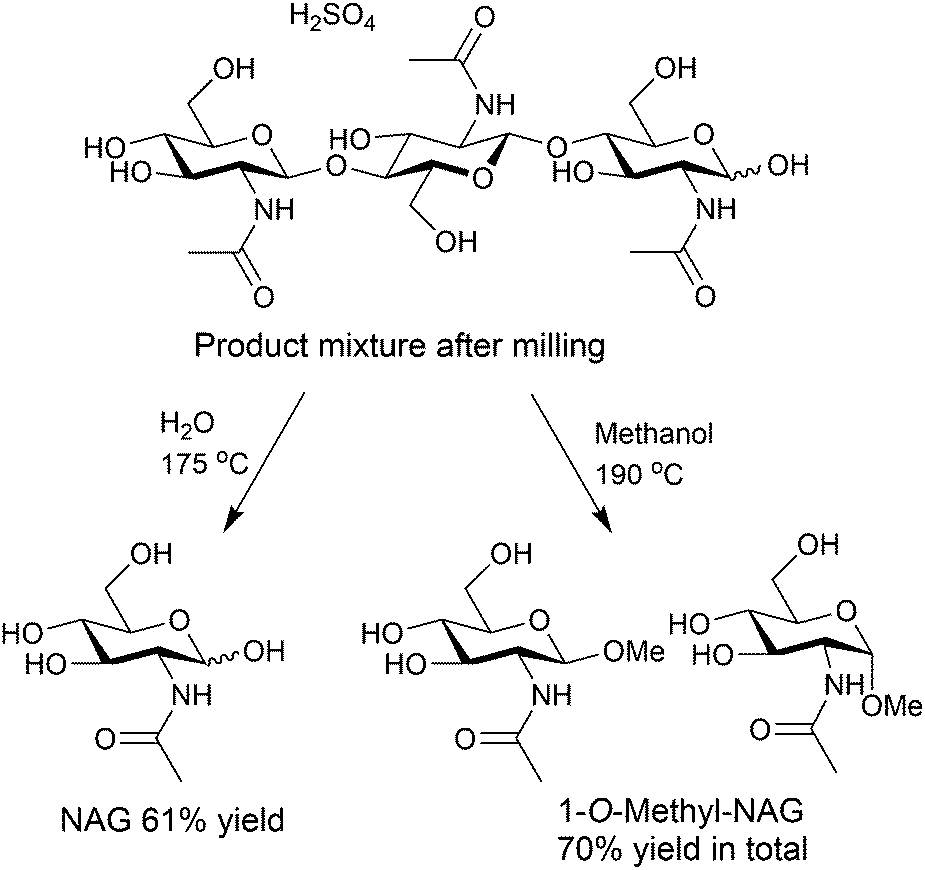 | ||
| Fig. 6 Conversion of the oligomers produced by a mechanochemical reaction to monomers. | ||
Instead of hydrolysis, alcoholysis is also possible for degrading the oligomer intermediates (Fig. 6). Changing the solvent to methanol, we synthesised 1-O-methylated NAG in up to 70% yield.34 NAG is an unstable compound, but alcoholysis can protect the reactive hemiacetal groups, decreasing the decomposition of products.36
Mechanistic study of mechanocatalytic hydrolysis
To clarify the effect of milling, we designed several hydrolysis experiments and measured the solubilisation ratios of the products by preparing a 1 wt% suspension in water at room temperature.34 Initially, we confirmed that chitin contains no soluble products. In the second experiment, chitin was milled for 6 h, and the product showed a solubilisation ratio of 5.2%. Accordingly, the milling treatment causes a very limited degree of hydrolysis in the absence of a catalyst. For the next control experiment, chitin was milled, impregnated with H2SO4 and aged for 6 h, which showed a solubilisation ratio of only 7.8%. H2SO4 alone does not hydrolyse chitin. By altering the order, chitin was impregnated with H2SO4 and milled for 6 h, giving >99% solubilisation ratio. These results show that H2SO4 hydrolyses chitin under milling conditions.Acid strength dominates the solubilisation ratio after the milling process.35 Chitin was impregnated with several acids with different pKa values, and the samples were milled for 6 h. HClO4 (pKa = −10) and H2SO4 (pKa = −3) gave completely soluble products. CH3SO3H (pKa = −2) and C3F7CO2H (pKa = 0.4) showed ca. 90% solubilisation, followed by H3PO4 (pKa = 2.1) (44% solubilisation) and AcOH (pKa = 4.7) (2.8% solubilisation). These results suggest that the protonation of glycosidic bonds is an essential step for hydrolysis under our milling conditions.
In the planetary milling process, alumina balls accelerated by the centrifugal force hit the pot wall. This event provides mechanical forces and heat at the collision point. We performed control experiments to reveal which factor is predominant in facilitating the hydrolysis of chitin (Fig. 7).37 A planetary milling of H3PO4-impregnated chitin afforded a pale yellow product containing NAG and oligomers consisting of two to six NAG units in 40% yield in total. This reaction produced no AcOH. In control experiments, chitin was milled, impregnated with H3PO4 and aged with no solvent at a high temperature in the range of 60–120 °C for a designated time. The reaction temperature was decided by the following assumption; the temperature of the bulk phase was at most 60 °C during the milling and the increase in the temperature at the local area by ball collision was estimated to be roughly as high as 50 °C. The optimum reaction produced NAG and oligomers in 33% yield. However, the product was dark brown and contained AcOH in 24 mol% yield based on the number of NAG units in chitin. Accordingly, the effect of milling cannot be ascribed to local heat but to mechanical forces. This is a mechanocatalytic reaction.
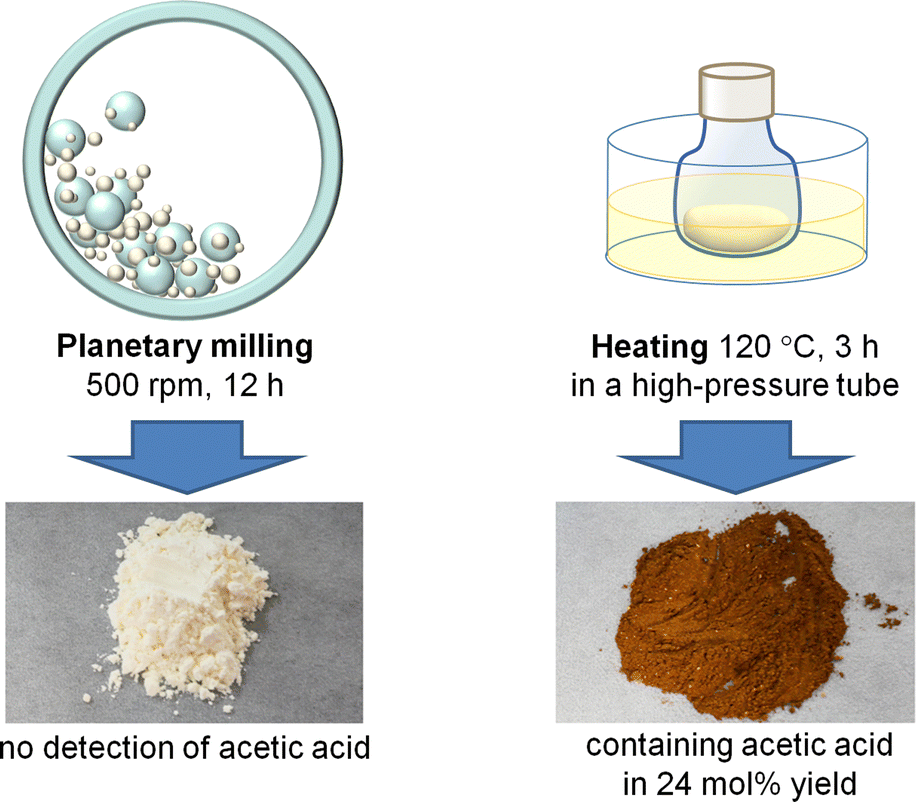 | ||
| Fig. 7 Comparison of milling and heating reactions. Chitin was impregnated with H3PO4 (S/C = 5.0) before the treatments. | ||
We speculated that mechanical forces easily work along the polymer chain of a chitin molecule, while they are rarely available within side-chains, namely amide groups.34 However, it was unclear how great forces are applied to chitin during milling and if they are large enough to activate glycosidic bonds in chitin. These questions are also applicable to cellulose.29–33 In a simulation of milling, Rinaldi et al. and Takagaki et al., respectively, estimated the relationship between the total milling energy and hydrolysis rate, which showed good correlation.32,38 To consider the effect of mechanical forces, we solved the motion equation of an alumina ball during planetary milling with a rough approximation, which indicated that the alumina ball hits the pot wall at a velocity of 4 m s−1.37 Based on the collision velocity, finite element simulations estimated that up to 7 GPa of compressive stress and 1.5 GPa of tensile stress are imposed on chitin and cellulose. Therefore, the compressive stress is larger, except when stress concentration occurs. Multiplying by the molecular cross-sectional area, we can estimate that the maximum tensile and compressive stresses correspond to 0.5 and 2.4 nN of forces, respectively, on one chitin or cellulose molecule. These values are similar to the connecting forces of covalent bonds, as evaluated by pulling polymer molecules with atomic force microscopes.39
DFT calculations were employed to estimate the effects of forces on chemical bonds. Amirjalayer et al. showed that tensile forces decrease the activation energy of hydrolysing glycosidic bonds.40 Using the B3LYP functional41,42 within DFT, we demonstrated that the conformation of the tetrahydropyran ring of NAG changes to increase the electron density on C–O σ* bonds in the glycosidic bonds under a tensile-force field.43 This facilitates an elimination reaction to cleave the glycosidic bonds in a syn form between an O lone pair and C–O bond (Fig. 8). This type of reaction is rare as the syn orientation provides larger steric repulsion due to the eclipsed conformation and a smaller overlap of orbitals than the anti forms. This indicates that forces could alter reaction mechanisms from the usual thermal reactions. In addition, our calculations showed that the tensile forces do not accelerate the hydrolysis of amide.43 The rate-determining step of acid-catalysed amide hydrolysis is the addition of water (Fig. 4), which is not related to the tensile forces on the amide group. This feature is different from alkaline hydrolysis under a mechanochemical condition that cleaves amide bonds,44 where the scission of C–N bonds is the rate-determining step for amide hydrolysis.45
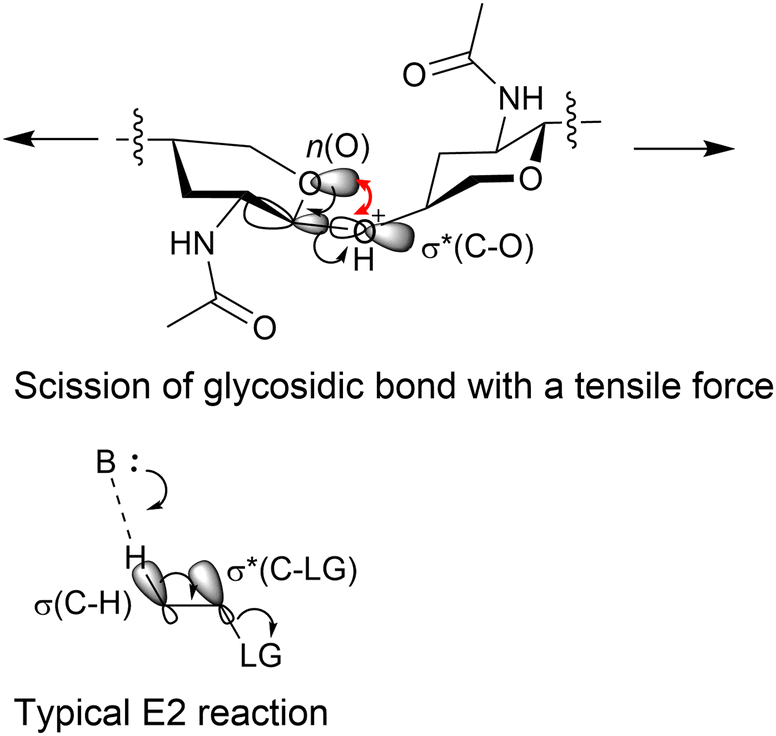 | ||
| Fig. 8 Reaction mechanisms of bond cleavage. The image omits some functional groups of chitin for better visibility. In the syn orientation (top), the red arrow highlights opposite signs between the n(O) and the OH side of σ*(C–O) orbitals. In contrast, the anti orientation (bottom) does not have such a repulsion. | ||
We have also demonstrated that not only tensile forces but also compressive ones activate glycosidic bonds in hydrolysis.37 Cellobiose was employed as a small model to reproduce the effects of forces, which significantly reduced the calculation time. In the DFT calculations at the B3LYP/6-31+G(d,p) level, the conformation of H3PO4-coordinated cellobiose was initially optimised, and then the distance between two oxygen atoms at the tips of the molecule was changed to provide tensile or compressive forces. Then, we simulated hydrolysis reactions in the force-affected molecules (Fig. 9). This procedure, called the COGEF method,46,47 slightly changes the intensity of forces during reactions but easily evaluates the effect of forces. Note that the protonation of cellobiose is simpler than the coordination of H3PO4, but cellobiose protonated at the glycosidic bond underwent bond scission with only 7.4 kJ mol−1 of activation energy. The naked proton is too active and it does not reproduce actual experiments. In the absence of forces, the glycosidic bond of H3PO4-coordinated cellobiose was cleaved at an activation energy of 120 kJ mol−1. An anti-periplanar conformation appeared in the transition state, which is similar to typical E2 reactions. Stretching the cellobiose molecule by 7.5%, corresponding to ca. 3 nN of pulling force, decreased the energy barrier to 93 kJ mol−1. The energy needed can be further decreased to 76 kJ mol−1 at 9.4% elongation of the molecule. A syn conformation was found during the bond cleavage, as described above. Meanwhile, compressing by 7.5%, ca. 1.2 nN of force, also lowered the required energy to 102 kJ mol−1. The force stabilised the twisted boat structure, giving an anti-periplanar conformation for the bond scission. It is easy to assume that a tensile force activates a chemical bond, but our calculations show that a compressive force also works to some extent. Moreover, our classical mechanics calculations have indicated that the maximum tensile and compressive forces are 0.5 and 2.4 nN, as described above. Taking this fact into account, the compressive forces produced by planetary milling are large enough to activate glycosidic bonds, whereas tensile forces can do so only when there is force concentration. Thus, we propose that compressive forces are as important as tensile forces in the mechanocatalytic reaction.
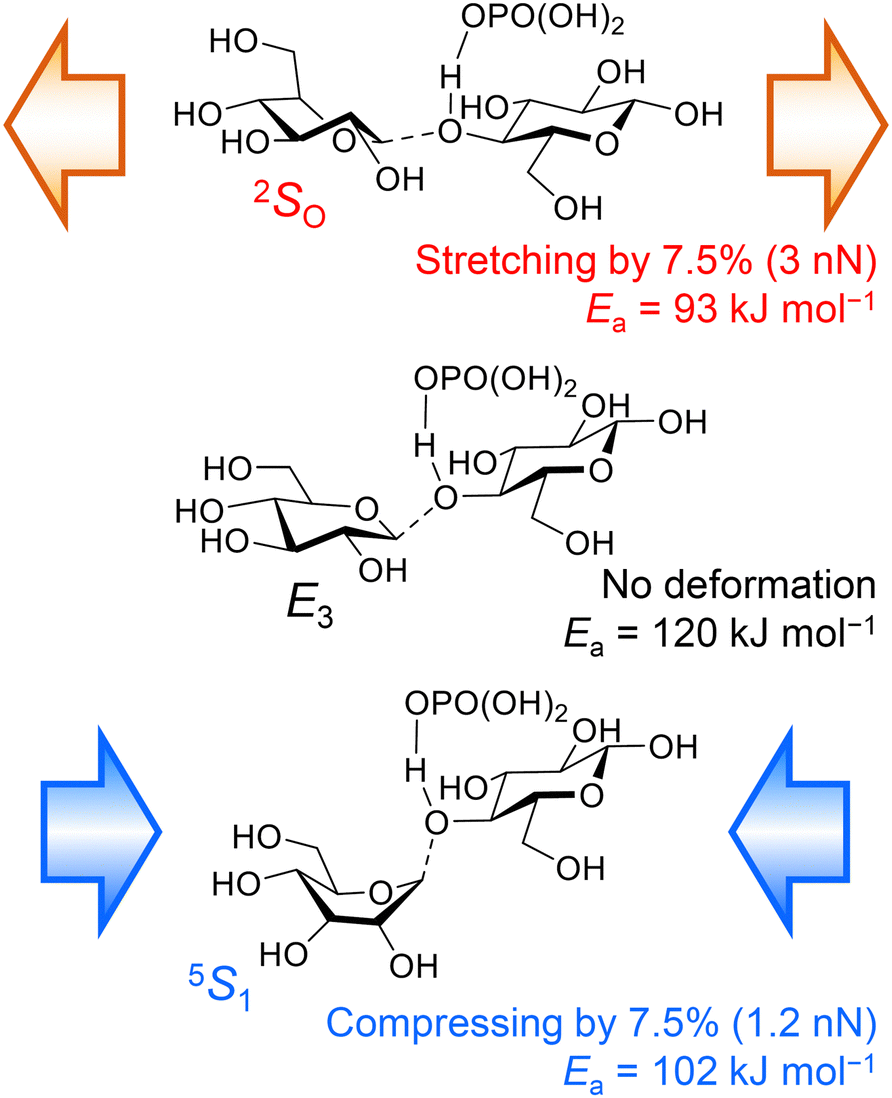 | ||
| Fig. 9 Effects of forces on the cleavage of glycosidic bonds. The Ea value is not an activation energy under a constant force but under a constant distance between the tips of the molecule. | ||
DFT is designed to calculate the ground states of molecular systems.48 However, in the presence of tensile forces, chemical bonds are stretched in the homolysis manner, which may mix excited states. To evaluate the accuracy of the DFT calculations, we compared B3LYP and the full configuration interaction (full CI) with the aug-cc-pV5Z basis set49,50 and the closed shell system in terms of the energy needed to stretch H2.37 Surprisingly, their difference was less than 0.5 kJ mol−1 up to 12% elongation. Stretching the CO molecule also gave similar energies between B3LYP and the complete active-space second-order perturbation theory (CASPT2; ten electrons, eight orbitals)51 up to 25% elongation. Hence, the calculation method is reliable over this range.
Catalysis of oxygenated carbon in the hydrolysis
Mineral acids are active for the mechanocatalytic hydrolysis of chitin, but enabling the use of solid catalysts may expand practical applications. Kerton et al. reported that kaolinite, an acidic layered clay mineral, can accelerate the hydrolysis of chitin in the milling process.52 Later, we found that carbons with weak acid sites are active and selective catalysts for converting chitin to chitin-oligosaccharides under planetary milling conditions.53 This is different from the conventional aqueous-phase reaction, where they do not hydrolyse milled chitin, as described above.In our work, a carbon-based catalyst was prepared by the air oxidation54,55 of a commercial activated carbon (BA) at 425 °C. The recovery of the catalyst, which was named AC-Air, was about 50 wt%, and the remaining part was burnt out. AC-Air had 0.90 mmol g−1 of carboxylic groups and 0.84 mmol g−1 of phenolic hydroxy groups. Chitin was treated with AC-Air in a planetary mill (Fritsch P-6) at 500 rpm for 12 h. The product showed a 34% solubilisation ratio and contained water-soluble oligosaccharides in 32% yield (>90% selectivity) (Fig. 10). In contrast, the reaction in the absence of a catalyst gave an 8% solubilisation ratio and produced oligosaccharides in 1.2% yield. Accordingly, the catalyst elevated the yield of oligosaccharides by 27 times, compared to the non-catalytic reaction. This enhancement is remarkable as kaolinite increased the yield by only three times in previous work.52 In the presence of AC-Air, extending the milling time to 48 h increased the solubilisation ratio to 72% and the yield of oligosaccharides to 66%. Accordingly, this reaction system provides more than 90% selectivity for oligosaccharides among soluble products (66/72 = 92%).
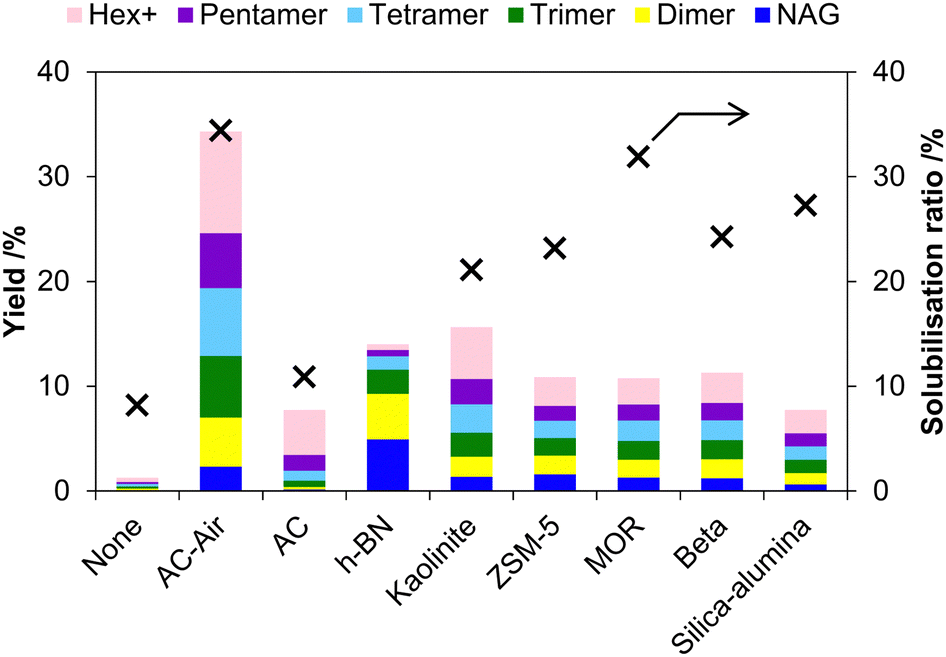 | ||
| Fig. 10 Screening of solid catalysts in the mechanochemical hydrolysis of chitin. Hex+ indicates the total yield of hexamer and larger oligomers. Yield is based on moles of carbon. | ||
1H NMR analysis showed a high purity of chitin-oligosaccharides in the water-soluble product after 48 h of milling with AC-Air. We focused on the peaks of C1–H (Fig. 11) as they specifically appear in a low magnetic field region. The major peaks were assignable to chitin-oligosaccharides, namely β-1,4-oligomers of NAG. Additionally, anhydrides were found, but the peak area was only about 5% among all C1–H peaks. In contrast, when we used H2SO4 as a catalyst, the product gave many peaks in the C1–H region, suggesting the presence of oligomers with different structures.
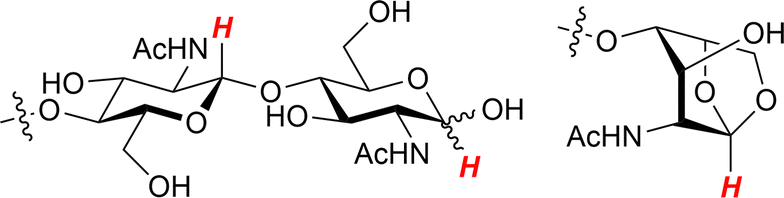 | ||
| Fig. 11 C1–H (highlighted in red) in oligomers and anhydrides. | ||
The selective production of chitin-oligosaccharides has a fascinating application in agriculture. Recently, it has been a challenge to decrease the use of agrichemicals to retain high productivity while preserving the natural environment. To overcome this problem, biostimulants that bring out the abilities of plants themselves will be an alternative to conventional agrichemicals,56 and the market will grow up to 6–8 billion GBP per year by 2030. In this sense, chitin-oligosaccharides can activate plant immune systems.57,58 If plants are infected with fungi, oligosaccharides present on the microbe cells are released (Fig. 12). Plants detect the fragments as pathogen-associated molecular patterns (PAMPs) and express immune-related genes. Relatively large but water-soluble chitin-oligosaccharides work as PAMPs so that plants become more resistant to microbes. Additionally, oligosaccharide mixtures containing chitin-oligosaccharides assist the growth of plants, possibly increasing the yield of crops.59
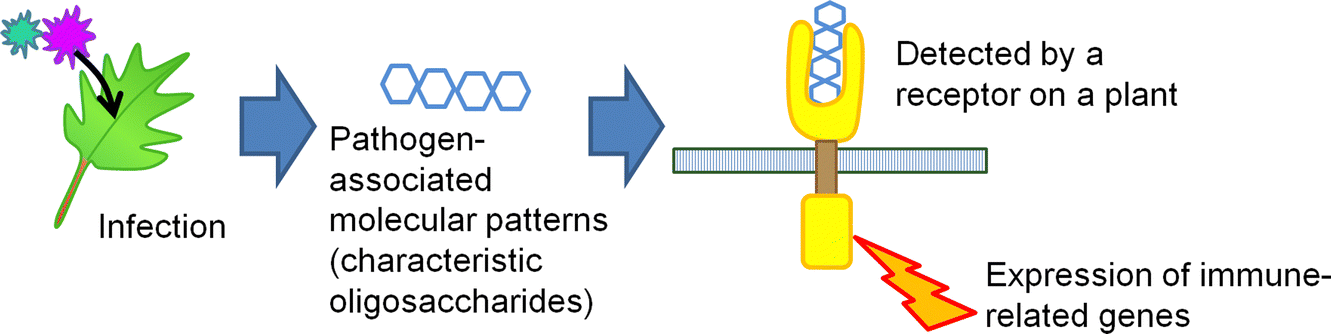 | ||
| Fig. 12 Schematic of PAMP recognition. | ||
Taking into account the application of chitin-oligosaccharides, the distribution of polymerisation degree is of interest. We changed the reaction time from 12 h to 48 h using AC-Air. Major products were trimer to hexamer without dependence on the reaction time, and the successive hydrolysis was limited. This result is noteworthy, because small oligomers are usually more reactive than large molecules, giving monomers as the predominant product and only a small amount of oligomers.60,61 As a representative example, a trial using 11 M HCl to convert chitin to oligosaccharides produced NAG as a major product and the yield of oligomers decreased with increasing polymerisation degree.62 Our Monte-Carlo simulations indicated that AC-Air cleaves glycosidic bonds almost with the same probability regardless of the polymerisation degree. Therefore, for example, a decamer (with nine glycosidic bonds) undergoes hydrolysis roughly nine times faster than a dimer (one glycosidic bond). These kinetics allow for the accumulation of a water-soluble but not too small range of chitin-oligosaccharides, namely bioactive ones. Carbons can easily adsorb large polysaccharides,63 and the homogeneously adsorbed polymeric substrates have the chance to receive mechanical forces. This system is suited for synthesising chitin-oligosaccharides working as PAMPs.
We further elucidated the cause of the high activity of AC-Air by examining related acid catalysts (Fig. 10). While AC-Air gave 32% yield of oligosaccharides after 12 h of milling, pristine activated carbon (AC) was inactive (oligosaccharides 6.2% yield), suggesting that the oxygenated functional groups with weak acidity are the active sites. Hexagonal boron nitride, with a similar structure to graphite and very low acid strength,64,65 gave 9.1% yield of oligosaccharides. Kaolinite was the second most active (14% yield) among the catalysts we tested. Proton-type zeolites (ZSM-5, beta and MOR) and silica-alumina produced oligosaccharides in 7–10% yields. A sulfonic acid resin, Amberlyst, almost completely converted chitin to soluble products. However, Amberlyst became soluble in water after the reaction, suggesting that this resin was virtually similar to H2SO4 in this system. Accordingly, a solid catalyst should have many acid sites but could be a weak acid such as carboxylic acid. Moreover, layered materials, especially carbons, are better than typical three-dimensionally bonded oxides. In the mechanocatalytic hydrolysis of cellulose, Takagaki et al. have shown the activity of HNbMoO6,32 which is also a layered material. We guess that the thin-layer structure is useful to contact with chitin particles at the molecular level due to its flexibility. In addition, a carbon material has polycyclic aromatics as the basal plane, and they attract chitin by CH–π interactions (Fig. 13).65–67 This is different from the fact that oxides apply electrostatic interactions. These characteristics make the air-oxidised carbon more active for the hydrolysis of chitin.
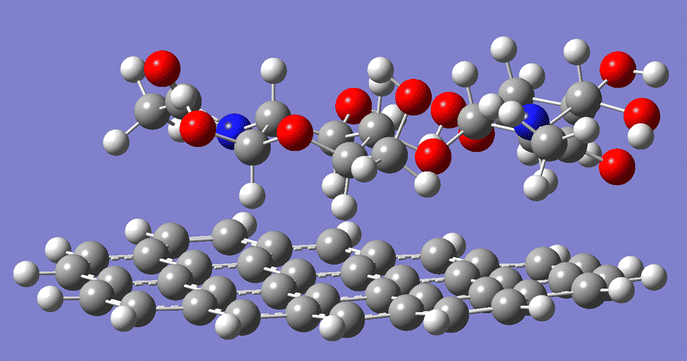 | ||
| Fig. 13 Adsorption structure of N,N′-diacetylchitobiose on carbon. Optimised at B3LYP-D3/6-31G(d,p). Many C–H groups of the sugar are directed toward aromatic rings, and the carbon is slightly bent to fit the sugar molecule. | ||
Conversion of NAG
Potential derivatives of NAG
NAG is a potential feedstock to produce a variety of chemicals (Fig. 14). Thermal decomposition of NAG or chitin produces many compounds, such as pyridine, pyrazine, chromogens, 3-acetamido-5-acetylfuran (3A5AF) and 3-acetamidofuran.68–71 The use of NaOH can change the product distribution, giving a small amount of pyrrole.72 An important and characteristic product among the derivatives is 3A5AF. This compound can be a precursor to proximicins, which are potential anticancer medicines.73 The oxidation of 3A5AF may produce 3-acetamido-5-carboxyfuran (3A5CF), with one carboxy and one amide group, likely to be a monomer of polyamide. Reacting 3A5AF with a methyl Grignard reagent, followed by dehydration, leads to vinyl compounds.74 Its radical polymerisation perhaps produces side-chain polymers. Meanwhile, the hydrogenation of NAG selectively produces 2-acetamido-2-deoxysorbitol (ADS).75,76 This compound is transformed into 2-acetamido-2-deoxyisosorbide (ADI), a promising precursor to polymers.77 An oxazolidinone with 5-(S) configuration, a potential precursor to antibacterial agents, is also synthesised from ADS.78 Combining a retroaldol reaction and hydrogenation converts NAG to acetyl monoethanolamine, and its oxidation provides an amino acid in a protected form.79 The oxidation of NAG also produces an amino acid, specifically N-acetylglucosaminic acid.80 Supported Au catalysts convert NAG and related amino sugars such as mannosamine to corresponding amino acids. The system is applicable to chitosan for synthesising glucosaminic acid.81 As most studies on NAG conversion have been started in recent years, further efforts will expand the potential derivatives and applications. The following sections focus on 3A5AF and ADI as their applications are promising.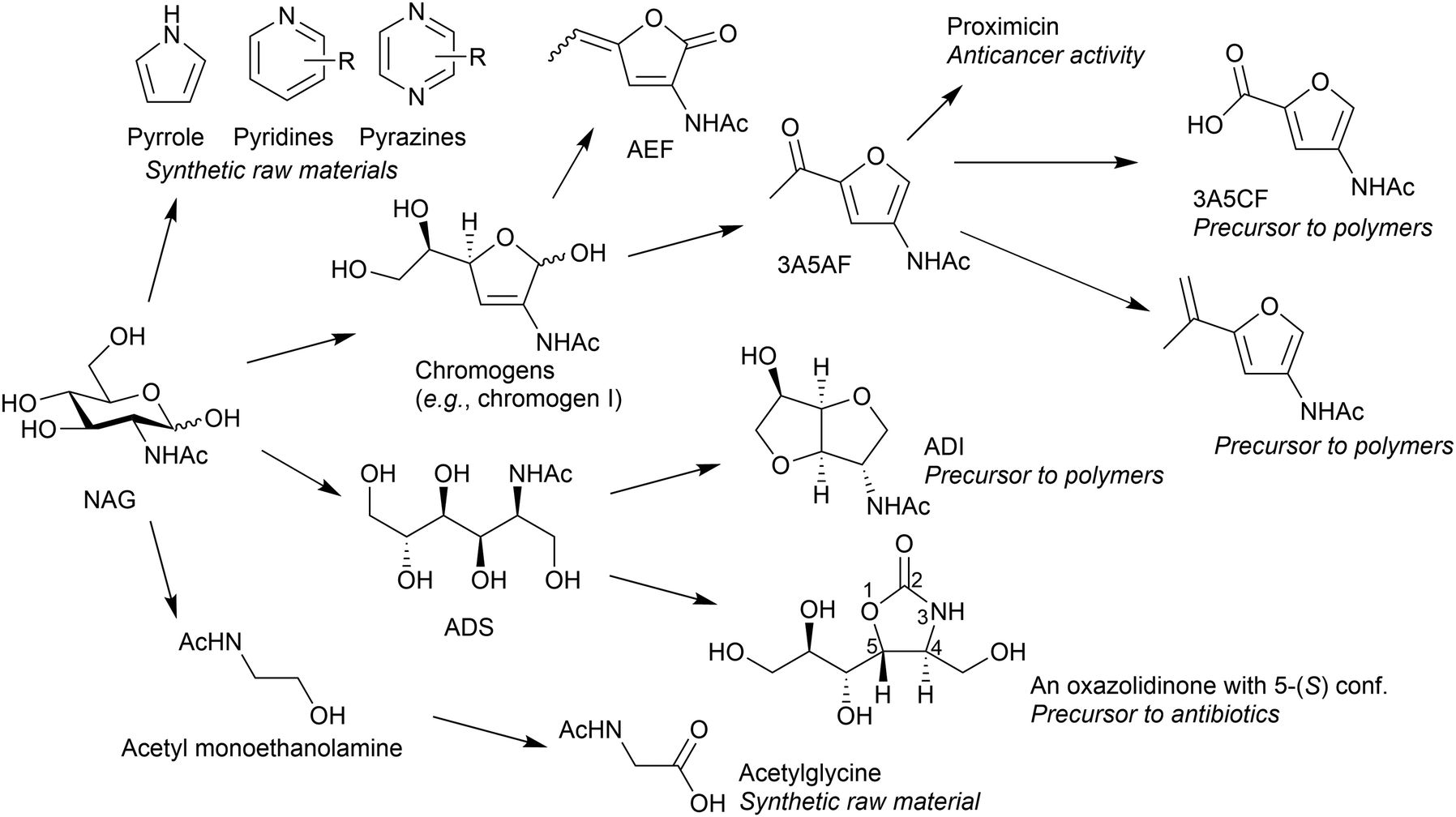 | ||
| Fig. 14 Conversion of NAG to organonitrogen compounds and their potential applications. | ||
Conversion of NAG to 3A5AF
The furanic compound 3A5AF was found in a small quantity in the pyrolysis products of NAG and chitin.68,69 Kerton et al. reported a liquid-phase conversion of NAG to 3A5AF in up to 60% yield in the presence of 1-butyl-3-methylimidazolium chloride ([BMIM]Cl) and B(OH)3 under microwave irradiation (Table 1, entry 1).82 The ionic liquid can be replaced with dimethylacetamide (DMAc) and NaCl (entry 2).83 Afterwards, Yan and co-workers found that B(OH)3 and NaCl produce 3A5AF in 7.5% yield from chitin in N-methylpyrrolidone (NMP) (entry 3).84 The use of ball-milled chitin improves the yield of 3A5AF to 29% (entry 4).85 They also clarified that Cl− significantly increases 3A5AF yield, by comparing several anions.86| Entry | Substrate | Catalyst and additive (mol%) | Solvent | T/°C | Yield of 3A5AF/% | Ref. |
|---|---|---|---|---|---|---|
| a Microwave heating. | ||||||
| 1 | NAG | B(OH)3 (200) | [BMIM]Cl | 180a | 60 | 82 |
| 2 | NAG | B(OH)3 (100), NaCl (400) | DMAc | 220a | 58 | 83 |
| 3 | Chitin | B(OH)3 (400), NaCl (200) | NMP | 215 | 7.5 | 84 |
| 4 | Ball-milled chitin | B(OH)3 (400), HCl (100) | [BMIM]Cl | 180 | 29 | 85 |
| 5 | NAG | AlCl3·6H2O (100) | DMF | 120 | 30 | 93 |
For the reaction mechanism, it is well known that a similar dehydration reaction of glucose produces 5-hydroxymethyl-2-furaldehyde (5-HMF). However, the positions of the functional groups in 3A5AF and 5-HMF are different (Fig. 15). Glucose initially isomerises to fructose, which is a ketose with a furanose form, followed by dehydration (Fig. 15b). However, NAG cannot isomerise to the form corresponding to fructose due to a large disadvantage in energy (+73 kJ mol−1, predicted with DFT calculations).79 Therefore, NAG directly undergoes dehydration possibly in the linear form, giving functional groups at 3- and 5-positions (Fig. 15(a)), although the detailed reaction mechanism is under discussion.82,86–88 Boron compounds may shift the equilibrium among the pyranose, furanose and linear forms by producing esters with NAG toward accelerating dehydration. Cl− might change the hydrogen bonding structures of sugars. Note that the deacetylation of NAG enables the formation of 5-HMF, because a double bond (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) can be produced in the resulting structure.89–92 Accordingly, the amide group plays a predominant role in the NAG transformation.
N) can be produced in the resulting structure.89–92 Accordingly, the amide group plays a predominant role in the NAG transformation.
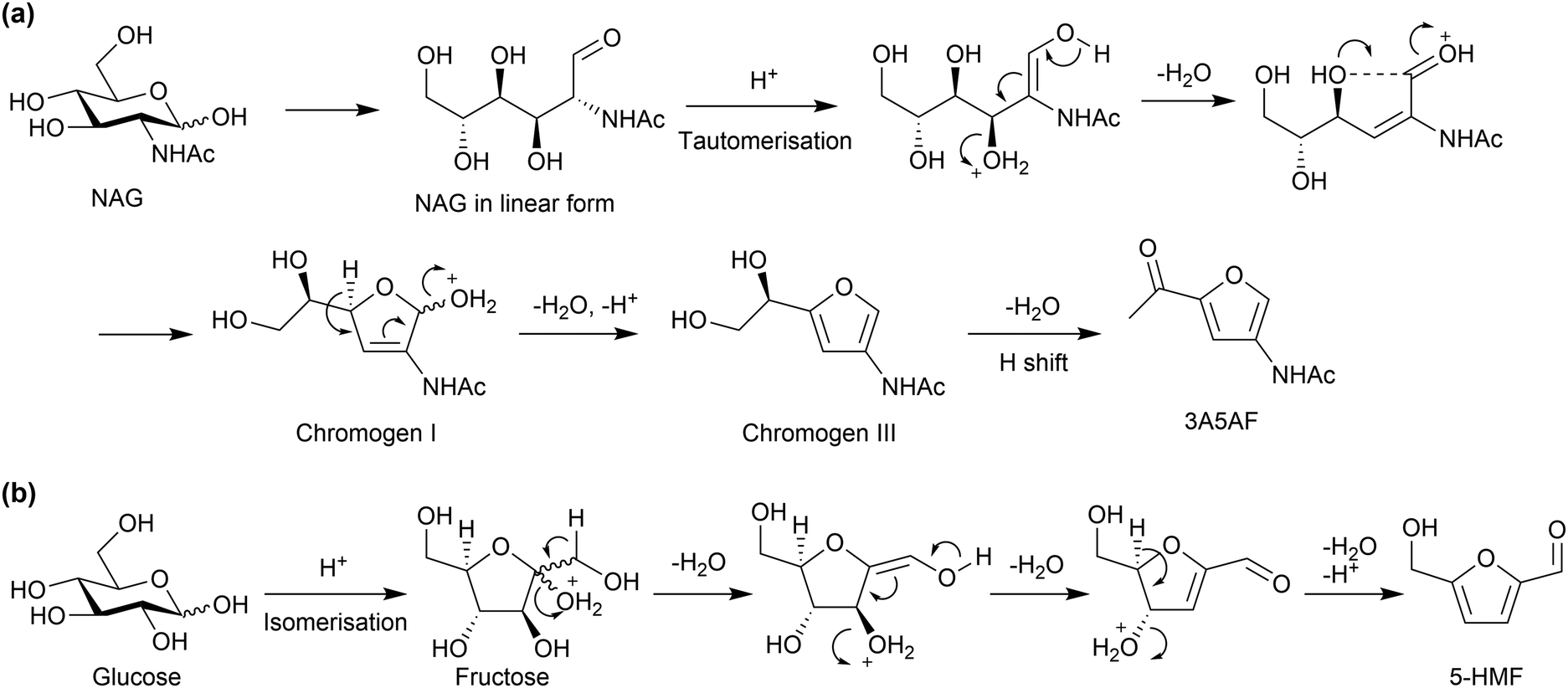 | ||
| Fig. 15 Plausible reaction mechanisms to produce (a) 3A5AF and (b) 5-HMF. The dehydration reactions have derivatives in the mechanisms. | ||
The above-mentioned systems use B(OH)3, but its removal is sometimes problematic as B(OH)3 easily produces adducts with sugar compounds. In addition, these reactions need high temperatures, often with unconventional methods. Decreasing the reaction temperature to 140 °C dropped the yield of 3A5AF to 1% despite the almost complete conversion of NAG in the presence of NaCl and B(OH)3 in DMAc.93 One probable reason is the low solubility of NaCl in the aprotic solvents.94 Rapid heating to a high temperature dissolves the salt to accelerate the desired reaction before side-reactions.
Accordingly, we need an easy method that many researchers can use to synthesise and isolate 3A5AF for further studies.
Previous studies suggest that the conversion of NAG to 3A5AF needs aprotic amide solvents and chloride ions. B(OH)3 could be a chelating agent or a Lewis acid. Based on these assumptions, we found that AlCl3·6H2O transforms NAG into 3A5AF in 30% yield in dimethylformamide (DMF) at 120 °C by conventional oil-bath heating without adding boron compounds (Table 1, entry 5).93 Moreover, this reaction can be performed at a 100 mL scale with 5% concentration of NAG, giving 0.70 g of 3A5AF (18% isolated yield) with more than 98% purity after silica gel chromatography. This reaction also generated 3-acetamido-5-ethylidene-2(5H)-furanone (AEF), with a sweet scent similar to dairy products, as a side product. The yield of AEF increased to 7% by using Al(OTf)3 as a catalyst. This compound could also be interesting.
In summary, 3A5AF is available at a laboratory scale, but the efficiency is not very high. We should further improve the synthetic method to make the compound more useful and easily accessible.
The oxidation of an acetyl group in 3A5AF produces 3A5CF, with one carboxy and one amide group. This compound is likely to be a promising precursor to polyamides. However, the conversion of 3A5AF to 3A5CF is a challenge. Currently, the only way to achieve this transformation is to use the iodoform reaction with a stoichiometric amount of iodine (Fig. 16(a)).73 In other oxidation reactions, the Baeyer–Villiger oxidation of 3A5AF should produce a predominant isomer (Fig. 16(b)), while radical reactions such as autoxidation probably attack the furan ring due to the low aromatic stabilisation.95,96 It is desirable to develop an efficient catalytic method to produce 3A5CF by overcoming these problems.
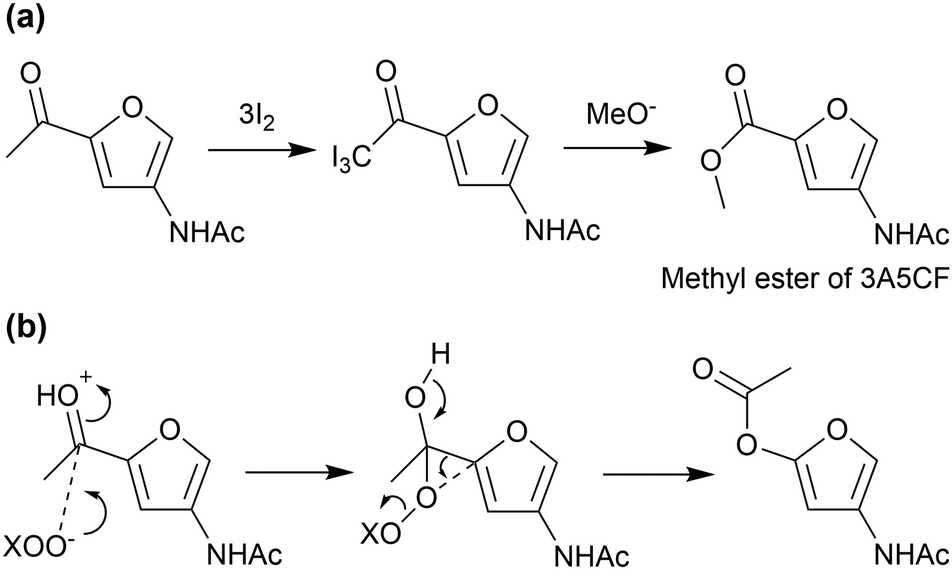 | ||
| Fig. 16 (a) Iodoform reaction and (b) Baeyer–Villiger oxidation of 3A5AF. | ||
Synthesis of ADI
This condensed five-membered ring compound has one amide and one hydroxy group (Fig. 17(a)). Although the application has not yet been developed, the structure is attractive as a feedstock for polymer synthesis. Isosorbide, with the same skeleton as ADI and two hydroxy groups, is a precursor to engineering plastics (Fig. 17(b)).97 Indeed, the isosorbide-based polycarbonate named Durabio shows excellent scratch resistance, high transparency with remarkable chromogenic property and high thermal resistance, and is thus applied in automotive parts (e.g., Mazda CX-5) and mobile phones.98 ADI-based plastics may also exhibit remarkable properties. ADI may be synthesised from NAG in a similar manner to the glucose-to-isosorbide system, and we reported the synthesis of ADI from NAG for the first time.77 The production of an ADS intermediate has been established at a laboratory scale as follows: NAG (4 wt% concentration) was hydrogenated over Ru/C under 4 MPa of H2 in water at 80 °C, and the product mixture was filtrated and evaporated. The resulting condensed material was dissolved in methanol at 60 °C and recrystallised at −30 °C, giving 85% isolated yield of ADS. The concentration of NAG can be increased to improve the productivity. The produced ADS was converted to ADI by a CF3SO3H catalyst (S/C = 2) at 150 °C, which afforded 33% yield of ADI.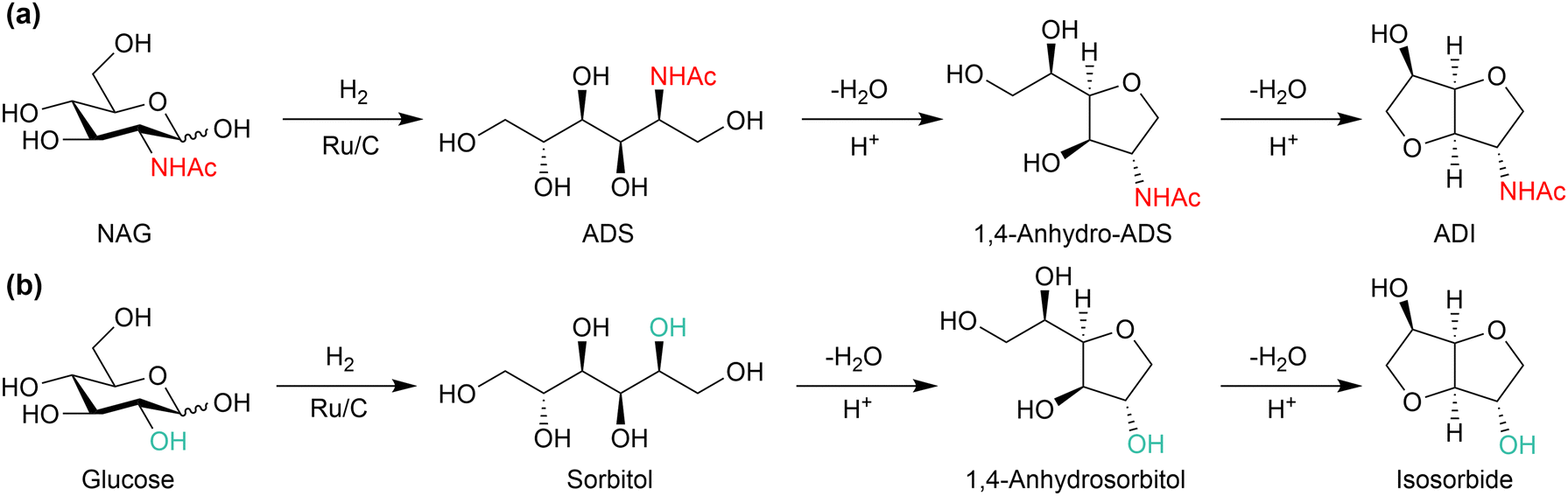 | ||
| Fig. 17 Similarity between (a) conversion of NAG to ADI and (b) that of glucose to isosorbide. | ||
The conversion of ADS needs a large amount of CF3SO3H (S/C = 2), unlike sorbitol undergoing dehydration with a small amount of H2SO4 or H-beta zeolite.99,100 A major reason is the high basicity of the amide group in ADS.76 The carbonyl oxygen traps protons, which increases the energy for protonating hydroxy groups for the dehydration reaction. Our DFT calculations have shown that the second dehydration step of ADS is the rate-determining step, and the activation energy is 58 kJ mol−1 higher than the corresponding reaction of sorbitol. To decrease the influence of proton trapping, a large amount of strong acid was needed. The addition of lanthanoid triflate slightly increases the yield of ADI to 40%, perhaps by coordinating with the amide group of the substrate to hamper proton trapping.101
We explored the possibility of replacing strong acids with weak acids and unexpectedly found that H3PO3 accelerates the conversion of ADS to ADI.102 Other acids with similar pKa values (H3PO3 1.3) showed much lower activity for this reaction. NMR analysis indicated the presence of phosphite esters of ADS and anhydro-ADS that are possible intermediates. To clarify the role of the esters, cyclisation reactions of the esters to produce ADI were simulated by DFT calculations. The second cyclisation reaction gave 82 kJ mol−1 of activation energy for the phosphite ester system (Fig. 18 phosphite mechanism), which was 36 kJ mol−1 lower than that in the absence of the ester (Fig. 18 acid mechanism). H3PO3 easily produces an ester with anhydro-ADS, and its PO group receives a proton with a significantly lower energy than a hydroxy group for producing ADI. Although the yield of ADI has been at most 25%, this result indicates that the phosphite mechanism could overcome the proton trapping problem. Nozaki et al. have proposed that Al(PO3)3 produces esters with phenolic hydroxy groups, which facilitates the hydrodeoxygenation of the phenolic compounds over a Pt/Al(PO3)3 catalyst.103 The utilisation of esters is an interesting approach to activate hydroxy groups.
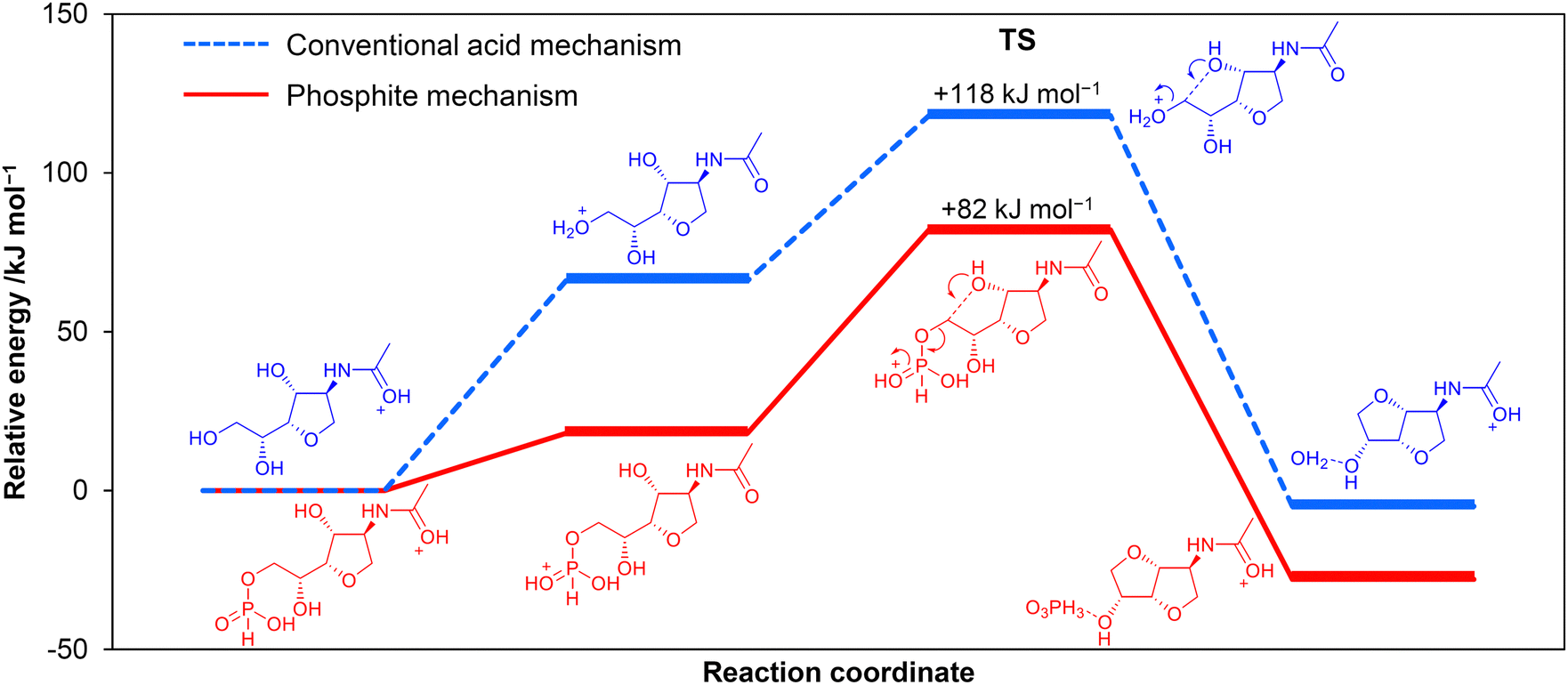 | ||
| Fig. 18 Energy profiles for the cyclisation reactions to produce ADI, determined by DFT calculations. Red: phosphite mechanism. Blue: conventional acid mechanism. Calculation level: B3LYP/6-31+G(d,p) with solvent effects of DMF by SMD. The zero-point vibration energy was included. The energy required to transfer a proton from amide to PO (phosphite mechanism) or hydroxy (acid mechanism) groups dominates the activation energy. | ||
Conclusions and outlook
Chitin is a simple molecule consisting of nitrogen-containing sugars, and therefore it is a fascinating feedstock for producing organonitrogen compounds. However, the strong hydrogen bonds and the presence of multiple reactive groups in chitin have hampered its selective and efficient hydrolysis to NAG. We found that mechanical forces applied in planetary milling selectively activate glycosidic bonds so that acids can cleave the bonds while preserving amide bonds. Our quantitative analysis of mechanical forces indicated that tensile and compressive forces produced in the planetary mill both contribute to dissociating glycosidic bonds. Moreover, the forces could change the reaction mechanism. This mechanochemical system can utilise solid catalysts and a carbon-based weak acid is especially effective. The catalyst generates chitin-oligosaccharides in a high yield, and the product can not only be a source of NAG but also a biostimulant that assists the growth of plants with a smaller amount of agrichemicals. The latter application provides a high value to chitin-oligosaccharides so it makes the implementation much easier than usual bulk chemical productions. The limitation of the current hydrolysis system is that it consumes a considerable amount of energy in the milling process during the long reaction time. Although the use of renewable energy may alleviate this issue, we hope that the development of new catalysts and mechanochemical systems based on our mechanistic studies will improve energy efficiency. In addition, the use of deep eutectic solvents or highly concentrated salt solutions has recently emerged as a potential method to activate chitin.104–107 They would also contribute to the selective extraction of chitin from biomass, because the current extraction method uses a large amount of NaOH and HCl.10,108 These technologies based on solid science will expand the possibility of chitin conversion. Moreover, as mentioned in a previous section, chitin is similar to but more recalcitrant than cellulose, so the chemistry established here may also be useful in cellulose conversion.The synthesis of NAG derivatives is an emerging topic and has not been very efficient, except for ADS. The latest studies have clarified that we should pay attention to the different reactivity of the amide group from a hydroxy group (glucose). Amide has much higher basicity and the imine isomer (CN–CO) corresponding to fructose is not produced due to a large energy disadvantage. Therefore, the dehydration of NAG provides a furan compound functionalised at the 3- and 5-positions (3A5AF), which is not observed in the glucose conversion. The dehydration of ADS requires a large amount of super-strong acid due to proton trapping by the amide group. We introduced the idea that H3PO3 can relieve this problem by producing esters with the substrate and easy protonation of the PO moiety instead of OH groups. A new strategy like this is necessary to control the reactivity of NAG or its derivatives.
Regarding 3A5AF, we propose that the development of a Lewis acid catalyst is needed to improve the synthetic efficiency. Previous work used B(OH)3 and Cl− with S/C ratios less than 1 to accomplish a good yield (up to 60%). Instead, we found that AlCl3·6H2O works for this reaction at a larger S/C (1.0) and a lower temperature (120 °C), which allowed us to increase the concentration of NAG up to 10 wt% and to isolate 3A5AF in a gram scale. However, the yield of 3A5AF was up to 30%. A new catalyst should be tolerant to amide and selectively activate OH groups for dehydration and H shift (Fig. 15), which further improves efficiency. After the synthesis of 3A5AF, its conversion to 3A5CF may open the door to bio-based polymers. The only method currently available is the iodoform reaction, and hence creating a new oxidation system is a fascinating subject.
We hope that the integration of these studies will achieve the practical utilisation of chitin toward establishing a sustainable carbon and nitrogen cycle.
Author contributions
HK: writing – original draft, visualisation. TS: writing – review & editing. AF: writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Japan Society for the Promotion of Science KAKENHI (21H01973, 18H01781, 26709060, 19K15608), by Japan Science and Technology Agency PRESTO (Grant Number JPMJPR22N5) and by JACI Prize for Encouraging Young Researcher from Japan Association for Chemical Innovation. The authors would like to thank Mr Shogo Ito, Dr Mizuho Yabushita, Mr Kyoichi Kuroki and Dr Kota Techikawara for the development of mechanocatalytic hydrolysis, Prof. Jun-ya Hasegawa and Dr Danjo De Chavez for DFT calculations in a force field, Mr Makoto Saito for the fruitful discussion on the product application, Mr Yusuke Suzuki for studying carbon systems, Dr Daniele Padovan for the synthesis and reactivity assessment of 3A5AF, Dr Cheng Yang for the study of H3PO3 system and all the other collaborators for their contributions.References
- S. C. Doney, D. S. Busch, S. R. Cooley and K. J. Kroeker, Ann. Rev. Environ. Resour., 2020, 45, 83–112 CrossRef.
- R. J. Geider, E. H. Delucia, P. G. Falkowski, A. C. Finzi, J. P. Grime, J. Grace, T. M. Kana, J. La Roche, S. P. Long, B. A. Osborne, T. Platt, I. C. Prentice, J. A. Raven, W. H. Schlesinger, V. Smetacek, V. Stuart, S. Sathyendranath, R. B. Thomas, T. C. Vogelmann, P. Williams and F. I. Woodward, Global Change Biol., 2001, 7, 849–882 CrossRef.
- K. Lee, Y. Jing, Y. Wang and N. Yan, Nat. Rev. Chem., 2022, 6, 635–652 CrossRef PubMed.
- A. Shrotri, H. Kobayashi and A. Fukuoka, Acc. Chem. Res., 2018, 51, 761–768 CrossRef CAS PubMed.
- R. Rinaldi, R. Jastrzebski, M. T. Clough, J. Ralph, M. Kennema, P. C. A. Bruijnincx and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2016, 55, 8164–8215 CrossRef CAS PubMed.
- X. Chen, S. Song, H. Li, G. Gözaydın and N. Yan, Acc. Chem. Res., 2021, 54, 1711–1722 CrossRef CAS PubMed.
- G. Liang, A. Wang, L. Li, G. Xu, N. Yan and T. Zhang, Angew. Chem., Int. Ed., 2017, 56, 3050–3054 CrossRef CAS PubMed.
- W. Deng, Y. Wang, S. Zhang and N. Yan, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 5093–5098 CrossRef CAS.
- K. Kurita, Mar. Biotechnol., 2006, 8, 203–226 CrossRef CAS.
- N. Yan and X. Chen, Nature, 2015, 524, 155–157 CrossRef CAS PubMed.
- A. Shrotri, H. Kobayashi and A. Fukuoka, Adv. Catal., 2017, 60, 59–123 CAS.
- J. Rockström, M. Klum, P. Miller and J. Lokrantz, Big World Small Planet Abundance Within Planetary Boundaries, Yale University Press, 2015 Search PubMed.
- P. Sudarsanam, R. Zhong, S. Van den Bosch, S. M. Coman, V. I. Parvulescu and B. F. Sels, Chem. Soc. Rev., 2018, 47, 8349–8402 RSC.
- X. Chen, H. Yang and N. Yan, Chem. – Eur. J., 2016, 22, 13402–13421 CrossRef CAS PubMed.
- J. Dai, F. Li and X. Fu, ChemSusChem, 2020, 13, 6498–6508 CrossRef CAS PubMed.
- P. Sikorski, R. Hori and M. Wada, Biomacromolecules, 2009, 10, 1100–1105 CrossRef CAS PubMed.
- M. Osada, C. Miura, Y. S. Nakagawa, M. Kaihara, M. Nikaido and K. Totani, Carbohydr. Polym., 2013, 92, 1573–1578 CrossRef CAS PubMed.
- H. Kobayashi and A. Fukuoka, Bull. Chem. Soc. Jpn., 2018, 91, 29–43 CrossRef CAS.
- J.-K. Chen, C.-R. Shen and C.-L. Liu, Mar. Drugs, 2010, 8, 2493–2516 CrossRef CAS PubMed.
- M. Ishikawa, F. Nanjo and K. Sakai, Pat., JP1822027 (publication number H05-033037), 1994 Search PubMed.
- A. Einbu and K. M. Vårum, Biomacromolecules, 2007, 8, 309–314 CrossRef CAS PubMed.
- A. Einbu and K. M. Vårum, Biomacromolecules, 2008, 9, 1870–1875 CrossRef CAS PubMed.
- M. Z. Abidin, M. P. Junqueira-Gonçalves, V. V. Khutoryanskiy and K. Niranjan, J. Chem. Technol. Biotechnol., 2017, 92, 2787–2798 CrossRef CAS.
- M. L. Bender, Chem. Rev., 1960, 60, 53–113 CrossRef CAS.
- J. Zhang and N. Yan, Green Chem., 2016, 18, 5050–5058 RSC.
- J. Zhang and N. Yan, ChemCatChem, 2017, 9, 2790–2796 CrossRef CAS.
- S. Roseman and J. Ludowieg, J. Am. Chem. Soc., 1954, 76, 301–302 CrossRef CAS.
- A. Shrotri, H. Kobayashi and A. Fukuoka, Acc. Chem. Res., 2018, 51, 761–768 CrossRef CAS PubMed.
- S. M. Hick, C. Griebel, D. T. Restrepo, J. H. Truitt, E. J. Buker, C. Bylda and R. G. Blair, Green Chem., 2010, 12, 468–474 RSC.
- N. Meine, R. Rinaldi and F. Schüth, ChemSusChem, 2012, 5, 1449–1454 CrossRef CAS PubMed.
- A. Shrotri, L. K. Lambert, A. Tanksale and J. Beltramini, Green Chem., 2013, 15, 2761–2768 RSC.
- S. Furusato, A. Takagaki, S. Hayashi, A. Miyazato, R. Kikuchi and S. T. Oyama, ChemSusChem, 2018, 11, 888–896 CrossRef CAS PubMed.
- P. Dornath, H. J. Cho, A. Paulsen, P. Dauenhauer and W. Fan, Green Chem., 2015, 17, 769–775 RSC.
- M. Yabushita, H. Kobayashi, K. Kuroki, S. Ito and A. Fukuoka, ChemSusChem, 2015, 8, 3760–3763 CrossRef CAS PubMed.
- H. Kobayashi, K. Techikawara and A. Fukuoka, Green Chem., 2017, 19, 3350–3356 RSC.
- Y. Pierson, X. Chen, F. D. Bobbink, J. Zhang and N. Yan, ACS Sustainable Chem. Eng., 2014, 2, 2081–2089 CrossRef CAS.
- H. Kobayashi, Y. Suzuki, T. Sagawa, K. Kuroki, J. Hasegawa and A. Fukuoka, Phys. Chem. Chem. Phys., 2021, 23, 15908–15916 RSC.
- M. Kessler, R. T. Woodward, N. Wong and R. Rinaldi, ChemSusChem, 2018, 11, 552–561 CrossRef CAS PubMed.
- M. K. Beyer and H. Clausen-Schaumann, Chem. Rev., 2005, 105, 2921–2948 CrossRef CAS PubMed.
- S. Amirjalayer, H. Fuchs and D. Marx, Angew. Chem., Int. Ed., 2019, 58, 5232–5235 CrossRef CAS PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- D. De Chavez, H. Kobayashi, A. Fukuoka and J. Hasegawa, J. Phys. Chem. A, 2021, 125, 187–197 CrossRef CAS PubMed.
- X. Chen, H. Yang, Z. Zhong and N. Yan, Green Chem., 2017, 19, 2783–2792 RSC.
- C. O’Connor, Q. Rev., Chem. Soc., 1970, 24, 553–564 RSC.
- M. K. Beyer, J. Chem. Phys., 2000, 112, 7307–7312 CrossRef CAS.
- I. M. Klein, C. C. Husic, D. P. Kovács, N. J. Choquette and M. J. Robb, J. Am. Chem. Soc., 2020, 142, 16364–16381 CrossRef CAS PubMed.
- P. Hohenberg and W. Kohn, Phys. Rev., 1964, 136, B864–B871 CrossRef.
- T. H. Dunning Jr., J. Chem. Phys., 1989, 90, 1007–1023 CrossRef.
- R. A. Kendall and T. H. Dunning Jr., J. Chem. Phys., 1992, 96, 6796–6806 CrossRef CAS.
- J. J. W. McDouall, K. Peasley and M. A. Robb, Chem. Phys. Lett., 1988, 148, 183–189 CrossRef CAS.
- G. Margoutidis, V. H. Parsons, C. S. Bottaro, N. Yan and F. M. Kerton, ACS Sustainable Chem. Eng., 2018, 6, 1662–1669 CrossRef CAS.
- H. Kobayashi, Y. Suzuki, T. Sagawa, M. Saito and A. Fukuoka, Angew. Chem., Int. Ed., 2023, 62, e202214229 CAS.
- H. Kobayashi, H. Kaiki, A. Shrotri, K. Techikawara and A. Fukuoka, Chem. Sci., 2016, 7, 692–696 RSC.
- A. Shrotri, H. Kobayashi and A. Fukuoka, ChemSusChem, 2016, 9, 1299–1303 CrossRef CAS PubMed.
- O. I. Yakhin, A. A. Lubyanov, I. A. Yakhin and P. H. Brown, Front. Plant Sci., 2017, 7, 2049 Search PubMed.
- H. Kaku, Y. Nishizawa, N. Ishii-Minami, C. Akimoto-Tomiyama, N. Dohmae, K. Takio, E. Minami and N. Shibuya, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 11086–11091 CrossRef CAS PubMed.
- M. Hayafune, R. Berisio, R. Marchetti and N. Shibuya, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, E404–E413 CrossRef CAS PubMed.
- J. He, W. Han, J. Wang, Y. Qian, M. Saito, W. Bai, J. Song and G. Lv, J. Plant Growth Regul., 2022, 41, 535–545 CrossRef CAS.
- M. Yabushita, H. Kobayashi, K. Hara and A. Fukuoka, Catal. Sci. Technol., 2014, 4, 2312–2317 RSC.
- P. Chen, A. Shrotri and A. Fukuoka, Catal. Sci. Technol., 2020, 10, 4593–4601 RSC.
- J. A. Rupley, Biochim. Biophys. Acta, 1964, 83, 245–255 CAS.
- P.-W. Chung, M. Yabushita, A. T. To, Y. Bae, J. Jankolovits, H. Kobayashi, A. Fukuoka and A. Katz, ACS Catal., 2015, 5, 6422–6425 CrossRef CAS.
- S. Torii, K. Jimura, S. Hayashi, R. Kikuchi and A. Takagaki, J. Catal., 2017, 335, 176–184 CrossRef.
- H. Kobayashi and A. Fukuoka, J. Phys. Chem. C, 2017, 121, 17332–17338 CrossRef CAS.
- P.-W. Chung, A. Charmot, O. M. Gazit and A. Katz, Langmuir, 2012, 28, 15222–15232 CrossRef CAS PubMed.
- M. Yabushita, H. Kobayashi, J. Hasegawa, K. Hara and A. Fukuoka, ChemSusChem, 2014, 7, 1443–1450 CrossRef CAS PubMed.
- R. A. Franich and S. J. Goodin, J. Anal. Appl. Pyrol., 1984, 7, 91–100 CrossRef CAS.
- J. Chen, M. Wang and C.-T. Ho, J. Agric. Food Chem., 1998, 46, 3207–3209 CrossRef CAS.
- L. Zeng, C. Qin, L. Wang and W. Li, Carbohydr. Polym., 2011, 83, 1553–1557 CrossRef CAS.
- M. Osada, K. Kikuta, K. Yoshida, M. Ogata and T. Usui, Green Chem., 2013, 15, 2960–2966 RSC.
- X. Gao, X. Chen, J. Zhang, W. Guo, F. Jin and N. Yan, ACS Sustainable Chem. Eng., 2016, 4, 3912–3920 CrossRef CAS.
- A. D. Sadiq, X. Chen, N. Yan and J. Sperry, ChemSusChem, 2018, 11, 532–535 CrossRef CAS PubMed.
- Y. Liu, C. N. Rowley and F. M. Kerton, ChemPhysChem, 2014, 15, 4087–4094 CrossRef CAS PubMed.
- F. D. Bobbink, J. Zhang, Y. Pierson, X. Chen and N. Yan, Green Chem., 2015, 17, 1024–1031 RSC.
- H. Kobayashi, K. Techikawara and A. Fukuoka, Green Chem., 2017, 19, 3350–3356 RSC.
- T. Sagawa, H. Kobayashi, C. Murata, Y. Shichibu, K. Konishi and A. Fukuoka, ACS Sustainable Chem. Eng., 2019, 7, 14883–14888 CrossRef CAS.
- T. Sagawa, H. Kobayashi, C. Murata, Y. Shichibu, K. Konishi, M. Hashizume and A. Fukuoka, Bull. Chem. Soc. Jpn., 2022, 95, 1054–1059 CrossRef CAS.
- K. Techikawara, H. Kobayashi and A. Fukuoka, ACS Sustainable Chem. Eng., 2018, 6, 12411–12418 CrossRef CAS.
- Y. Ohmi, S. Nishimura and K. Ebitani, ChemSusChem, 2013, 6, 2259–2262 CrossRef CAS PubMed.
- J. Dai, G. Gözaydın., C. Hu and N. Yan, ACS Sustainable Chem. Eng., 2019, 7, 12399–12407 CAS.
- M. W. Drover, K. W. Omari, J. N. Murphy and F. M. Kerton, RSC Adv., 2012, 2, 4642–4644 RSC.
- K. W. Omari, L. Dodot and F. M. Kerton, ChemSusChem, 2012, 5, 1767–1772 CrossRef CAS PubMed.
- X. Chen, S. L. Chew, F. M. Kerton and N. Yan, Green Chem., 2014, 16, 2204–2212 RSC.
- X. Chen, Y. Gao, L. Wang, H. Chen and N. Yan, ChemPlusChem, 2015, 80, 1565–1572 CrossRef CAS PubMed.
- X. Chen, Y. Liu, F. M. Kerton and N. Yan, RSC Adv., 2015, 5, 20073–20080 RSC.
- M. Osada, S. Shoji, S. Suenaga and M. Ogata, Fuel Proc. Technol., 2019, 195, 106154 CrossRef CAS.
- C. H. M. van der Loo, M. L. G. Borst, K. Pouwer and A. J. Minnaard, Org. Biomol. Chem., 2021, 19, 10105–10111 RSC.
- K. W. Omari, J. E. Besaw and F. M. Kerton, Green Chem., 2012, 14, 1480–1487 RSC.
- Y. Wang, C. M. Pedersen, T. Deng, Y. Qiao and X. Hou, Bioresour. Technol., 2013, 143, 384–390 CrossRef CAS PubMed.
- T.-W. Tzeng, P. Bhaumik and P.-W. Chung, Mol. Catal., 2019, 479, 110627 CrossRef.
- X. Cai, Z. Wang, Y. Ye, D. Wang, Z. Zhang, Z. Zheng, Y. Liu and S. Li, Renewable Sustainable Energy Rev., 2021, 150, 111452 CrossRef CAS.
- D. Padovan, H. Kobayashi and A. Fukuoka, ChemSusChem, 2020, 13, 3594–3598 CrossRef CAS PubMed.
- D. Pabsch, P. Figiel, G. Sadowski and C. Held, J. Chem. Eng. Data, 2022, 67, 2706–2718 CrossRef CAS.
- C. W. Bird, Tetrahedron, 1992, 48, 335–340 CrossRef CAS.
- K. E. Horner and P. B. Karadakov, J. Org. Chem., 2013, 78, 8037–8043 CrossRef CAS PubMed.
- D. J. Saxon, A. M. Luke, H. Sajjad, W. B. Tolman and T. M. Reineke, Prog. Polym. Sci., 2020, 101, 101196 CrossRef CAS.
- T. Setoyama, Catal. Surv. Asia, 2014, 18, 183–192 CrossRef CAS.
- H. Kobayashi, H. Yokoyama, B. Feng and A. Fukuoka, Green Chem., 2015, 17, 2732–2735 RSC.
- R. Otomo, T. Yokoi and T. Tatsumi, Appl. Catal., A, 2015, 505, 28–35 CrossRef CAS.
- T. Sagawa, H. Kobayashi and A. Fukuoka, Mol. Catal., 2020, 498, 111282 CrossRef CAS.
- C. Yang, T. Sagawa, A. Fukuoka and H. Kobayashi, Green Chem., 2021, 23, 7228–7234 RSC.
- X. Jin, R. Tsukimura, T. Aihara, H. Miura, T. Shishido and K. Nozaki, Nat. Catal., 2021, 4, 312–321 CrossRef CAS.
- N. Özel and M. Elibol, Carbohydr. Polym., 2021, 262, 117942 CrossRef PubMed.
- G. Gözaydın, S. Song and N. Yan, Green Chem., 2020, 22, 5096–5104 RSC.
- G. Gözaydın, Q. Sun, M. Oh, S. Lee, M. Choi, Y. Liu and N. Yan, ACS Sustainable Chem. Eng., 2023, 11, 2511–2519 CrossRef.
- Y. Wang, J. Kou, X. Wang and X. Chen, Green Chem., 2023, 25, 2596–2607 RSC.
- J. Wang, C. Teng and L. Yan, Green Chem., 2022, 24, 552–564 RSC.
| This journal is © The Royal Society of Chemistry 2023 |