
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRing expansion reactions of P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h2_char_e001.gif) O-containing molecules†
O-containing molecules†
Zhongzhen
Yang
 ,
Jerry K. F.
Tam‡
,
Jack M.
Wootton‡
,
Jason M.
Lynam
* and
William P.
Unsworth
*
,
Jerry K. F.
Tam‡
,
Jack M.
Wootton‡
,
Jason M.
Lynam
* and
William P.
Unsworth
*
Department of Chemistry, University of York, Heslington, York YO10 5DD, UK. E-mail: william.unsworth@york.ac.uk
First published on 30th May 2023
Abstract
A series of ring expansion reactions of P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O-containing molecules have been developed for the synthesis of medium-sized ring cyclic phosphonate esters and phosphonamidates. The reactivity trends initially appear to be counter-intuitive, compared with more well established ring expansion reactions of lactam derivatives, but are explained by considering the differences in heteroatom bonding to P and C respectively.
O-containing molecules have been developed for the synthesis of medium-sized ring cyclic phosphonate esters and phosphonamidates. The reactivity trends initially appear to be counter-intuitive, compared with more well established ring expansion reactions of lactam derivatives, but are explained by considering the differences in heteroatom bonding to P and C respectively.
Molecules containing P
O bonds (e.g. DNA, RNA and ATP) are essential to all life on earth.1 Organophosphorus compounds are also important in medicinal chemistry and agrochemistry, with various biologically active PO-containing molecules known (e.g.1–3, Scheme 1A).2 Their potential to be used as therapeutic and crop protection agents has therefore been well studied, often using prodrug approaches.3
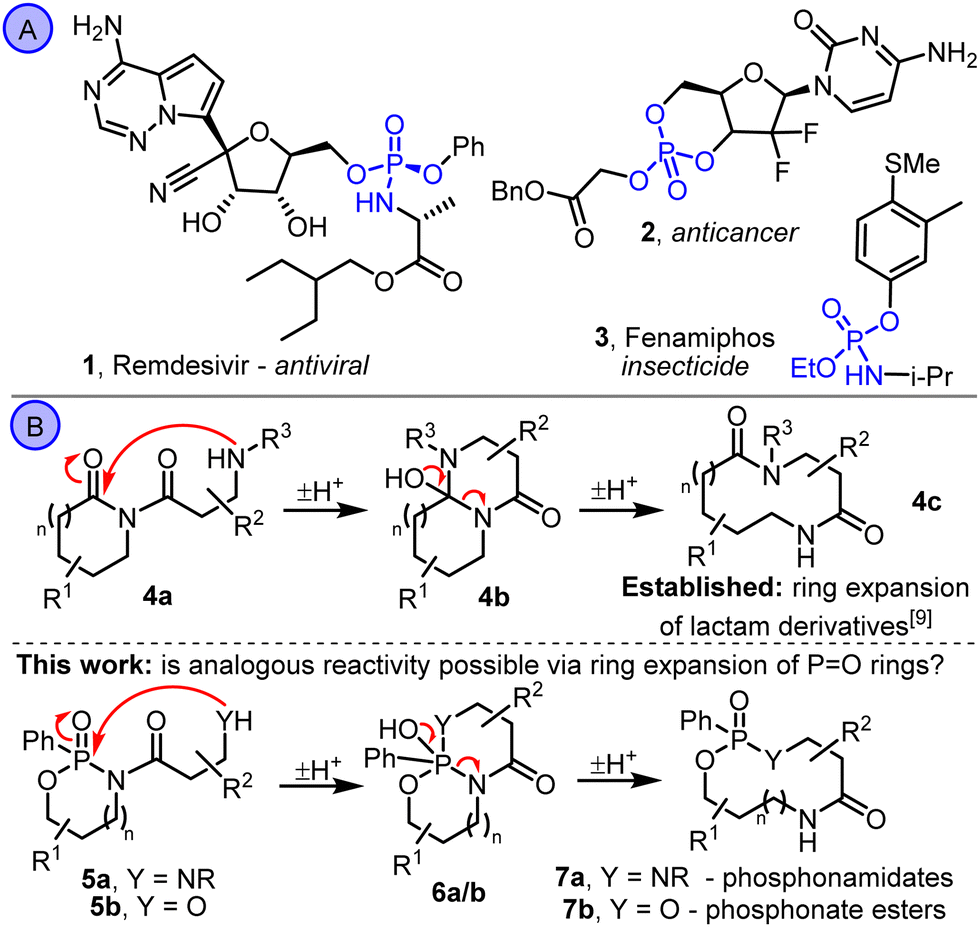 | ||
| Scheme 1 (A) Bioactive PO containing small organic molecules. (B) Ring expansion reactions of lactams vs cyclic phosphonamidates. | ||
Cyclic PO-containing molecules are routinely used in prodrug-based medicinal chemistry studies, but almost always as 5- or 6-membered ring derivatives (e.g. the recently reported anti-tumor candidate 2).4 In view of this, and interest in medium-sized rings and macrocycles in medicinal chemistry more generally,5 our aim in this study was to develop new methods to synthesise PO-containing medium-sized rings using ring expansion reactions (Scheme 1B).6,7 Synthetic methods to make medium-sized ring PO compounds are rare,8 and to the best of our knowledge there are no published examples that make use of ring expansion reactions. We therefore set out to explore whether strategies similar to those able to promote the ring expansion of lactam derivatives (e.g.4a → 4b → 4c, Scheme 1B)9,10 can be applied to phosphonamidate derivatives of the type 5. By testing amine (5a) and alcohol (5b) tethered substrates, a reactivity trend was revealed that contrasts that seen in the established lactam ring expansions; the more nucleophilic amine derivatives 5a rearrange less easily (or not at all), while less nucleophilic alcohol derivatives 5b rearrange well to form cyclic phosphonate esters 7b. Calculated Gibbs free energy data for the isomeric intermediates 5, 6 and 7 indicate that while both reaction series are exergonic, there is a much stronger thermodynamic force for ring expansion, and a lower kinetic barrier, in alcohol derivatives 5b compared with the analogous amine substrates 5a.
Synthetic studies started by exploring amine-based substrates of the type 5a; our previous work showed that related ring expansions are generally faster, more exergonic and higher yielding using amine side chains compared with alcohol-11 or thiol-based systems.12 A Conjugate Addition/Ring Expansion (CARE)13 cascade was devised, with phosphonamidate derivative 9 reacted with different nucleophilic primary amines (Scheme 2A). The first part of the CARE cascade reaction proceeded as expected, with amine conjugate addition taking place in each case to form amines 10a–f in good yields.14 However, no evidence of rearrangement to ring expanded products 11a–f was obtained under any of the basic reaction conditions screened (see ESI,† Table S2). Similar results were obtained starting from phosphoramidate derivative 12, with conjugate addition products (13a,b) isolated. Aniline derivative 16 was also prepared via reduction of nitrobenzene 15 but this substrate also failed to undergo ring expansion (Scheme 2B).
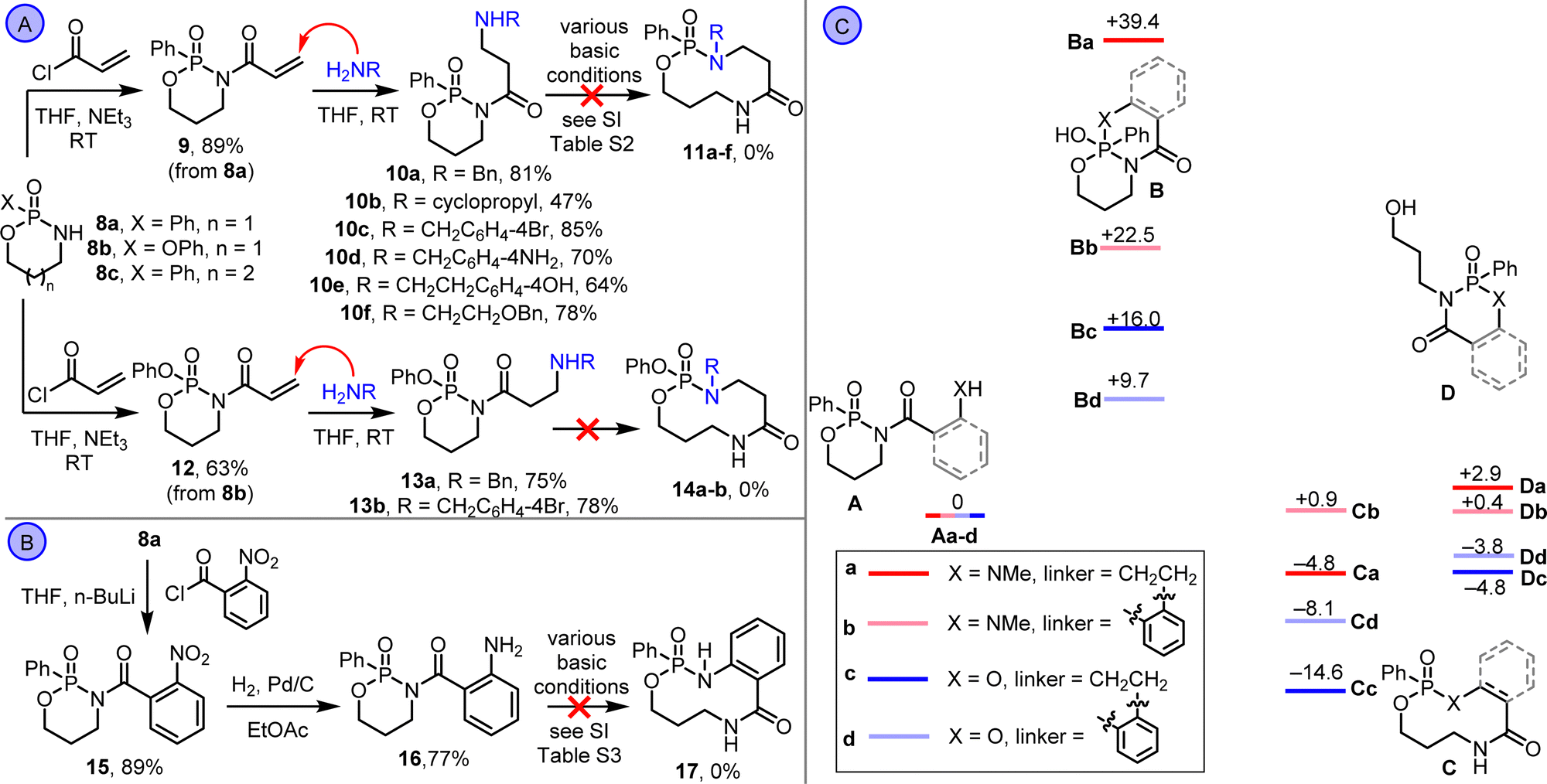 | ||
| Scheme 2 (A) Unsuccessful ring expansion of N-acyl phosphonamidates with tethered aliphatic amines. (B) Unsuccessful ring expansion of 16. (C) Relative energies of isomeric species in ring expansion of N-acyl phosphonamidates using a DFT/B3LYP/6-31G* approach. ΔG°rel values at 298 K are given in kcal mol−1. | ||
At this point, with the planned ring expansion reactions not proceeding as hoped, their viability was assessed using Density Functional Theory (DFT), using a method that was established and benchmarked for lactam ring expansion reactions in our previous work (Scheme 2C).15 Thus, the ground state energy of the free amine isomer (A), ring-closed (B) and ring-expanded isomer (C) were calculated for a representative aliphatic amine (analogous to compounds 10a–f, labelled with ‘a’ and highlighted in red) and aniline systems (compound 16, labelled with ‘b’ and highlighted in pink). The energy of a fourth isomer (D), accessible via an alternative fragmentation of the endocyclic P–O bond, was also calculated, in addition to analogous calculations for aliphatic alcohol (labelled with ‘c’, highlighted in dark blue) and phenol (labelled with ‘a’, highlighted in pale blue) for comparison. The energies are in kcal/mol and relative to reference states A.
Several observations emerge from the calculations. First, ring expanded isomer (C) was calculated to be the lowest energy in three out of the four series, and borderline for the aniline system (Ab, Cb, Db all have similar energy). Notably, the unwanted isomer D was significantly higher in energy than C in three out of four cases. The isomer with the highest calculated energy in all systems was ring-closed isomer B, with this especially marked for the aliphatic amine series (isomer Ba). With the caveat that these data are calculated for intermediates and not transition states, this may be indicative of a high kinetic barrier to cyclisation being the reason for the failure of 10a–f and 16 to rearrange. Finally, the energies of states B–D were all significantly lower relative to the reference state for the analogous alcohol and phenol systems compared to the analogous amines; this is best visualised in Scheme 2C by comparing the alcohol states depicted in dark/pale blue (c and d) to the amines depicted in red/pink (a and b).
These data provide three key learnings that informed subsequent synthetic studies: (1) the ring expansion reactions are thermodynamically viable based on the calculated energy of states Ca–d; (2) there appears to be a significant kinetic barrier to ring expansion, in contrast to our previous work on lactam systems which are under thermodynamic control;15 (3) alcohol-based substrates should work better than the analogous amines, based on their calculated thermodynamic profiles. At this point, we switched attention to alcohol-based substrates, starting with protected-phenol derivative 18a, which was synthesised via the N-acylation of phosphonamidate 8a (Scheme 3A). Hydrogenolysis of 18a followed, to form phenol 19a, which rearranged spontaneously in situ. However, rather than rearrange via ring expansion, we instead isolated product 20a, via fragmentation of the endocyclic P–O bond. This observation was surprising, considering that this isomer was calculated to be higher in energy than ring expanded product 21a (compare Cd and Dd in Scheme 2C). But pleasingly, stirring 20a with triethylamine in chloroform at RT promoted further rearrangement into the thermodynamic product 21a, which was isolated in 84% yield. The same sequence (N-acylation, hydrogenolysis and ring expansion under basic conditions) was also used to form medium-sized ring phosphonate esters 21b–g (Scheme 3A).16 The structure of compound 21d was confirmed by X-ray crystallography.17
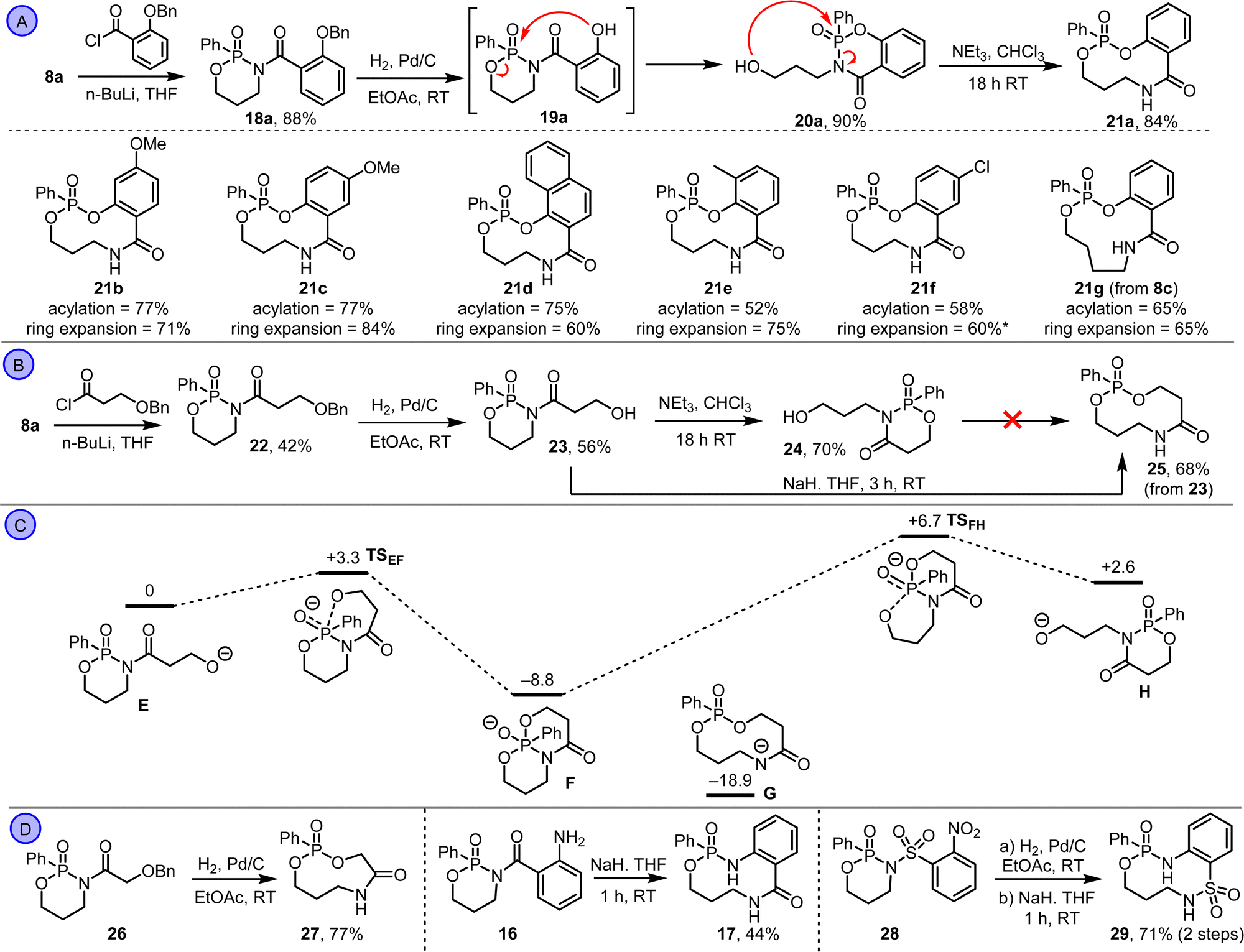 | ||
| Scheme 3 (A) The ring expansion of N-acyl phosphonamidates with tethered phenols. (B) The ring expansion of N-acyl phosphonamidates 23. (C) Relative energies of isomeric species in an anionic ring expansion manifold, using a DFT/B3LYP/6-31+G* approach. ΔG°rel values at 298 K are given in kcal mol−1. (D) Other ring expansion reactions of PO containing molecules. *Contaminated with ≈10% 21a, presumably as a result of hydrogenolysis of the C–Cl bond. | ||
Attention then turned to aliphatic alcohol derivative 22 (Scheme 3B). Hydrogenolysis of 22 was performed as before, but there was no evidence of spontaneous rearrangement, with alcohol 23 the only product isolated. This suggests a higher kinetic barrier compared to the phenol systems, which aligns with the calculated energies for Bc and Bd in Scheme 2C. In an attempt to overcome the kinetic barrier, alcohol 23 was reacted with triethylamine in chloroform at RT; this did promote rearrangement, but again led to the formation of unwanted isomer 24. Sodium hydride was therefore tested as base in place of triethylamine, and pleasingly this enabled the smooth conversion of 23 into ring-expanded product 25 in 68% yield. It is likely that the use of this stronger base enables an anionic reaction manifold to be accessed that allows the kinetic barrier to ring expansion to be overcome; notably, in the case of the phenol substrates (e.g.19a) their lower pKa presumably enables a similar anionic pathway to be accessed when using triethylamine. Calculations performed for the aliphatic alcohol system (Scheme 3C) reinforce the notion that accessing an anionic pathway is important, with the five-coordinate phosphorus intermediate F calculated to be lower in energy than its corresponding precursor E, in stark contrast to the neutral pathway (Scheme 2C). A transition state (TSEF) for the conversion of E into F was found at just 3.3 kcal mol−1, consistent with a facile reaction at RT, and a low energy transition state (TSFH) linking isomers F and H was also found, indicating that the conversion of H back into F is also viable if any H forms. We were unable to find a transition state linking F to the ring expanded isomer G, but notably G was comfortably the lowest energy isomer on the potential energy surface, in line with the formation of 25 as the reaction product.18
We ended by testing two new reaction systems and re-visiting one that had previously failed. First, hydrogenolysis of 26 enabled its direct conversion into 9-membered ring phosphonate ester 27 in 77% yield. In this 3-atom ring expansion, the fact that it proceeds via 5-membered ring cyclisation as opposed to 6-, likely leads to a lower kinetic barrier and hence precludes the need to add base to promote ring expansion. We also found that by using sodium hydride as base, aniline 16 could be converted into 17 in 44% yield. Notably this reaction failed using the less basic condition tested previously and confirms that ring expansion via amine nucleophiles is viable provided the kinetic barrier can be overcome. Finally, an alternative aniline-based ring expansion was achieved successfully from sulfonamide derivative 28; nitro reduction followed by treatment with sodium hydride in THF promoted its conversion into 10-memebered ring phosphonamidate 29 in 71% yield over two steps.
In summary, ring expansion reactions of PO-containing starting materials have been developed, allowing access to medium-sized ring cyclic phosphonate ester and phosphonamidates. Compared to more well-established ring expansion reactions at CO bonds (e.g. lactam derivatives 4, Scheme 1B), two key differences emerged. First is the greater reactivity of alcohol-tethered systems than the analogous amines. This contrasts to reactivity at CO, where amines generally react faster and in higher yields, and is likely due to the change in relative bond strengths on switching from C to P, in particular the high P–O bond strength. The second key difference is the higher kinetic barriers, which in most cases can be overcome by using either a more acidic substrate (e.g. phenol 19a) or more basic reaction conditions to access an anionic rearrangement pathway. An advantage to the higher kinetic barriers is the ability to isolate isomeric species (e.g.23, 24 and 25, in Scheme 3C) in high yields under appropriate kinetically controlled conditions.
The synthetic chemistry featured in the manuscript was done by Z. Y., with guidance from W. P. U. The computational chemistry was done by J. K. F. T. and J. M. W., with guidance from J. M. L. The manuscript was written through contributions from all authors. The project was directed and managed by W. P. U. and J. M. L. The authors are grateful for funding from the China Scholarship Council (Z. Y.), the University of York Wild Fund (J. K. F. T.), GSK (J. M. W.) and the Royal Society (J. M. L.) for an Industry Fellowship (INF\R1\221057).
Conflicts of interest
There are no conflicts to declare.Notes and references
- F. H. Westheimer, Science, 1987, 235, 1173–1178 CrossRef CAS PubMed.
- (a) M. O. Fabrício, RSC Adv., 2014, 4, 18998–19012 RSC; (b) Y.-Y. Zhu, T. Zhang, L. Zhou and S.-D. Yang, Green Chem., 2021, 23, 9417–9421 RSC; (c) F. Bouchareb and M. Berredjem, Phosphorus, Sulfur Silicon Relat. Elem., 2022, 197, 711–731 CrossRef CAS.
- (a) K.-M. Huttunen, N. Mähönen, J. Leppänen, J. Vepsäläinen, R. O. Juvonen, H. Raunio, H. Kumpulainen, T. Järvinen and J. Rautio, Pharm. Res., 2007, 24, 679–687 CrossRef CAS PubMed; (b) M. Slusarczyk, M. Seppi and F. Pertusati, Antiviral Chem. Chemother., 2018, 26, 1–31 Search PubMed; (c) R. L. Mackman, ACS Med. Chem. Lett., 2022, 13, 338–347 CrossRef CAS PubMed; (d) M. Krećmerová, P. Mayer, R. Rais and B. S. Slusher, Front. Chem., 2022, 10, 889737 CrossRef PubMed.
- L. Zhang, K. Qi, J. Xu, Y. Xing, X. Wang, L. Tong, Z. He, W. Xu, X. Li and Y. Jiang, J. Med. Chem., 2023, 66, 4150–4166 CrossRef CAS PubMed.
- For medium-sized rings and macrocycles in medicinal chemistry, see: (a) F. Kopp, C. F. Stratton, L. B. Akella and D. S. Tan, Nat. Chem. Biol., 2012, 8, 358 CrossRef CAS PubMed; (b) C. Zhao, Z. Ye, Z.-X. Ma, S. A. Wildman, S. A. Blaszczyk, L. Hu, I. A. Guizei and W. Tang, Nat. Commun., 2019, 10, 4015 CrossRef PubMed; (c) E. M. Driggers, S. P. Hale, J. Lee and N. K. Terrett, Nat. Rev. Drug Discovery, 2008, 7, 608 CrossRef CAS PubMed; (d) E. Marsault and M. L. Peterson, J. Med. Chem., 2011, 54, 1961 CrossRef CAS PubMed; (e) F. Giordanetto and J. Kihlberg, J. Med. Chem., 2014, 57, 278 CrossRef CAS PubMed; (f) A. K. Yudin, Chem. Sci., 2015, 6, 30 RSC.
- For reviews of ring expansion chemistry, see: (a) M. Hesse, Ring Enlargement in Organic Chemistry, Wiley-VCH, Weinheim, 1991 Search PubMed; (b) W. P. Unsworth and J. R. Donald, Chem. – Eur. J., 2017, 23, 8780 CrossRef PubMed; (c) K. Prantz and J. Mulzer, Chem. Rev., 2010, 110, 3741 CrossRef CAS PubMed; (d) T. C. Stephens and W. P. Unsworth, Synlett, 2020, 133 CAS; (e) A. K. Clarke and W. P. Unsworth, Chem. Sci., 2020, 11, 2876 RSC; (f) B. Biletskyi, P. Colonna, K. Masson, J.-L. Parrain, L. Commeiras and G. Chouraqui, Chem. Soc. Rev., 2021, 50, 7513–7538 RSC.
- For selected recent examples relevant to this study, see: (a) L. Li, Z.-L. Li, F.-L. Wang, Z. Guo, Y.-F. Cheng, N. Wang, X.-W. Dong, C. Fang, J. Liu, C. Hou, B. Tan and X.-Y. Liu, Nat. Commun., 2016, 7, 13852, DOI:10.1038/ncomms13852; (b) R. Costil, Q. Lefebvre and J. Clayden, Angew. Chem., Int. Ed., 2017, 56, 14602 CrossRef CAS PubMed; (c) A. Lawer, J. A. Rossi-Ashton, T. C. Stephens, B. J. Challis, R. G. Epton, J. M. Lynam and W. P. Unsworth, Angew. Chem., Int. Ed., 2019, 58, 13942 CrossRef CAS PubMed.
- (a) A.-M. Caminade and J. P. Majoral, Chem. Rev., 1994, 94, 1183–1213 CrossRef CAS; (b) K. C. Majumdar, R. K. Nandi and S. Ganai, Tetrahedron Lett., 2014, 55, 1247–1250 CrossRef CAS.
- For relevant lactam ring expansion methods, see: (a) T. C. Stephens, M. Lodi, A. Steer, Y. Lin, M. Gill and W. P. Unsworth, Chem. – Eur. J., 2017, 23, 13314 CrossRef CAS PubMed; (b) R. Mendoza-Sanchez, V. B. Corless, Q. N. N. Nguyen, M. Bergeron-Brlek, J. Frost, S. Adachi, D. J. Tantillo and A. K. Yudin, Chem. – Eur. J., 2017, 23, 13319 CrossRef CAS PubMed; (c) J. Shang, V. J. Thombare, C. L. Charron, U. Wille and C. Hutton, Chem. – Eur. J., 2021, 26, 1620–1625 CrossRef PubMed; (d) Z. Yang, C. R. B. Swanson and W. P. Unsworth, Synlett DOI:10.1055/a-1932-9717.
- For conceptually related ring expansion reactions of non-lactam substrates, see: (a) C. Kitsiou, J. J. Hindes, P. I’Anson, P. Jackson, T. C. Wilson, E. K. Daly, H. R. Felstead, P. Hearnshaw and W. P. Unsworth, Angew. Chem., Int. Ed., 2015, 54, 15794 CrossRef CAS PubMed; (b) L. G. Baud, M. A. Manning, H. L. Arkless, T. C. Stephens and W. P. Unsworth, Chem. – Eur. J., 2017, 23, 2225 CrossRef CAS PubMed; (c) D. R. Loya, A. Jean, M. Cormier, C. Fressigné, S. Nejrotti, J. Blanchet, J. Maddaluno and M. De Paolis, Chem. – Eur. J., 2018, 24, 2080 CrossRef CAS PubMed.
- T. C. Stephens, A. Lawer, T. French and W. P. Unsworth, Chem. – Eur. J., 2018, 24, 13947–13953 CrossRef CAS PubMed.
- K. Y. Palate, R. G. Epton, A. C. Whitwood, J. M. Lynam and W. P. Unsworth, Org. Biomol. Chem., 2021, 19, 1404–1411 RSC.
- (a) K. Y. Palate, Z. Yang, A. C. Whitwood and W. P. Unsworth, RSC Chem. Biol., 2022, 3, 334–340 RSC; (b) Z. Yang, I. Zalessky, R. G. Epton, A. C. Whitwood, J. M. Lynam and W. P. Unsworth, Angew. Chem., Int. Ed., 2023, e202217178 CAS.
- The same reaction was also attempted using NH3 (delivered as a 7 N solution in methanol) but failed, with only unreacted starting material 19a recovered.
- A. Lawer, R. G. Epton, T. C. Stephens, K. Y. Palate, M. Lodi, E. Marotte, K. J. Lamb, J. K. Sangha, J. Lynam and W. P. Unsworth, Chem. – Eur. J., 2020, 26, 12674–12683 CrossRef CAS PubMed See also ref. 11, 13a and 13b.
- The analogous thiol-based system (c.f. 18a but OBn = SBn) was also tested, but the benzyl cleavage step failed (see ESI†).
- CCDC 2260255† (21d) contains the crystallographic data see: www.ccdc.cam.ac.uk/data_request/cif.
- For all novel products synthesised in this manuscript, the reliable assignment of isomeric species was aided by the observation of 13C–31P coupling in their 13C NMR spectra. This allowed the proximity of different carbon atoms to phosphorus to be easily observed and hence provide a simple method to map the progress of the rearrangement (e.g. to distinguish 23, 24 and 25). See ESI,† Tables S6 and S7 for additional detail.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2260255. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3cc02087h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |