
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceComputation meets experiment: identification of highly efficient fibrillating peptides†
Lorenzo
Sori‡
 a,
Andrea
Pizzi‡
*a,
Greta
Bergamaschi
b,
Alessandro
Gori
b,
Alfonso
Gautieri
c,
Nicola
Demitri
d,
Monica
Soncini
c and
Pierangelo
Metrangolo
*a
a,
Andrea
Pizzi‡
*a,
Greta
Bergamaschi
b,
Alessandro
Gori
b,
Alfonso
Gautieri
c,
Nicola
Demitri
d,
Monica
Soncini
c and
Pierangelo
Metrangolo
*a
aLaboratory of Supramolecular and BioNano Materials (SupraBioNanoLab), Department of Chemistry, Materials, and Chemical Engineering “Giulio Natta”, Politecnico di Milano, Via Luigi Mancinelli 7, 20131 Milan, Italy. E-mail: andrea.pizzi@polimi.it; pierangelo.metrangolo@polimi.it
bIstituto di Scienze e Tecnologie Chimiche – National Research Council of Italy (SCITEC-CNR), 20131 Milan, Italy
cDepartment of Electronics, Information and Bioengineering, Politecnico di Milano, 20131 Milan, Italy
dElettra – Sincrotrone Trieste, S.S. 14 Km 163.5 in Area Science Park, 34149 Basovizza – Trieste, Italy
First published on 4th July 2023
Abstract
Self-assembling peptides are of huge interest for biological, medical and nanotechnological applications. The enormous chemical variety that is available from the 20 amino acids offers potentially unlimited peptide sequences, but it is currently an issue to predict their supramolecular behavior in a reliable and cheap way. Herein we report a computational method to screen and forecast the aqueous self-assembly propensity of amyloidogenic pentapeptides. This method was found also as an interesting tool to predict peptide crystallinity, which may be of interest for the development of peptide based drugs.
Introduction
Self-assembly processes lead molecular entities to organize themselves in particularly stable and functional supramolecular structures, moving from an original state of disorder to a subsequent one of order.1 These are essential events that regulate various biological processes, which are mainly driven by noncovalent interactions.2 The folding of nucleic acids and proteins is undoubtedly the best-known self-assembly phenomenon.3 In some specific conditions the resulting supramolecular structure does not exert a specific function; this is the case of amyloid fibers, which occur as a consequence of aberrant protein misfolding and aggregation and are considered the hallmark of several debilitating diseases like Alzheimer's (AD), Parkinson's (PD), Huntington's (HD) and type-II diabetes (T2D).4,5 Amyloids have a particularly defined hierarchical structure (Fig. 1) where the lamination of β-sheet layers forms the cross-β architecture, i.e. the core motif of supramolecular subunits (protofilaments) that group laterally and compose the finished fiber. | ||
| Fig. 1 Schematic representation of the structure of an amyloid fibril, formed by several protofilaments showing a cross-β structure. | ||
In addition to the interest closely related to these pathologies, the processes driving the formation of amyloids (fibrillation) are the subject of intense research in the fields of biomaterials and bioinspired materials, due to their intriguing mechanical properties.
In this context the study of self-assembling peptides to prepare functional nanostructures and materials6 is bringing new potential and opportunities to the fields of medicine and nanotechnology.7
The huge combinatorial possibilities offered by the chemical variety of natural and customized amino acids lead to almost unlimited peptide sequences, hence a wide range of achievable supramolecular morphologies. Unfortunately, the high potential of a system is strictly connected to its complexity. Indeed, the prediction and the control of the self-assembly of peptide-based systems are the factors that hamper their full exploitation.8
The issues related to the control of the self-assembly process can be addressed through the use of solvents,9 or by means of chemical switches.10 Computations represent a relatively new approach for the prediction of the supramolecular behavior of a peptide. In this regard, a recent study by Frederix et al. shows how simulations based on parameters such as hydrophobicity and aggregation propensity (AP) offer a reliable model to forecast the amyloidogenic propensity of tripeptides.11
Some of us reported that halogenation can be exploited as an effective single-point modification12 to amplify the self-assembly properties of some peptide sequences13 derived from amyloid beta (Aβ)14 and human calcitonin (hCT),15 whose fibrillation may be related to the occurrence of AD and medullary thyroid carcinoma, respectively. Very recently, bromination of tyrosine residues16 was proved as an efficient strategy to promote the self-assembly and the consequent emergence of elastomeric properties in a short peptide derived from resilin.17
In this view, with the purpose of expanding the available tools to strengthen peptide fibrillation, possibly alternative to halogenation, and considering the growing need for new tailored fibril-forming peptides, herein we report a computational approach, based on coarse-grained molecular dynamics (CG-MD),18–23 enabling the identification of new promising peptide sequences at a relatively low cost. As a confirmation of the reliability of the proposed method, experimental studies – including single crystal X-ray diffraction – on a small set of pentapeptides among those computed, were performed.
Experimental validation of the used computational method revealed the importance of the nature of the N-term amino acid in the resulting supramolecular behavior, and led to the identification of theoretical conditions which may be related to the propensity of peptides in forming fibrils or crystalline self-assemblies.
Experimental section
Materials
All reagents and solvents for peptide synthesis and purification were purchased from Iris Biotech GmbH (Marktredwitz, Germany), Novabiochem (Darmstadt, Germany), Carlo Erba (Rodano) and Sigma-Aldrich (Steinheim, Germany). All solvents for solid-phase peptide synthesis (SPPS) were used without further purification. HPLC grade acetonitrile (ACN) and ultrapure 18.2 Ω water (Millipore-MilliQ) were used for the preparation of all buffers for liquid chromatography. The chromatographic columns were from Phenomenex (Torrance CA, USA). Analytical RP-HPLC was performed on a Shimadzu Prominence HPLC (Shimadzu) using a Shimadzu Shimpack GWS C18 column (5 micron, 4.6 mm i.d. × 150 mm). Preparative RP-HPLC was performed on a Shimadzu HPLC prominence system using a Gemini, Shimadzu, C18 column (10 micron, 21.2 mm i.d. × 250 mm).Single-crystal XRD analysis
XRD data of peptides GFEDF, GFTEF, HFEEF and SFVEF were collected at the XRD1 and XRD2 beamlines of the Elettra Synchrotron, Trieste (Italy).24 The crystals were dipped in NHV oil (Jena Bioscience, Jena, Germany) and mounted on the goniometer head with Kapton loops (MiTeGen, Ithaca, USA). Complete datasets were collected at 100 K (nitrogen stream supplied through an Oxford Cryostream 700) through the rotating crystal method. Data were acquired using monochromatic wavelengths of 0.700 Å or 0.620 Å on Pilatus hybrid-pixel area detectors (DECTRIS Ltd., Baden-Daettwil, Switzerland). The diffraction data were indexed, integrated and scaled using XDS.25 Fourier analysis and refinement were performed by the full-matrix least-squares methods based on F2 implemented in SHELXL (Version 2018/3).26 The Coot program was used for modeling.27 Anisotropic thermal motion refinement has been used for all atoms with occupancies higher than 50%. Pictures were prepared using Ortep-3,28 CCDC Mercury,29 Coot27 and Pymol software.30Transmission electron microscopy (TEM)
TEM bright field images were acquired using a Philips CM200 electron microscope operating at 200 kV equipped with a field emission gun filament. A Gatan US 1000 CCD camera was used and 2048 × 2048 pixel images with 256 grey levels were recorded. Some other images were recorded with a DeLong Instruments LVEM5, equipped with a field emission gun, and operating at 5 kV. Peptide samples were placed onto a 200-mesh carbon-coated copper grid and air dried for several hours before analysis. No negative staining was used.Fourier transform infrared spectroscopy (FTIR)
Infrared spectra were recorded at room temperature using a Nicolet iS50 FT-IR spectrometer equipped with a DTGS detector. Freeze-dried peptide samples obtained from 0.4 mM water solutions were analyzed in ATR mode. Spectra represent an average of 64 scans recorded in a single beam mode with a 2 cm−1 resolution and corrected for the background.Peptide synthesis and purification
Peptides were assembled using CTC resin (1.6 mmol g−1 loading) by stepwise microwave-assisted Fmoc-SPPS on a Biotage ALSTRA Initiator+ peptide synthesizer, operating in a 0.1 mmol scale. Activation of entering Fmoc-protected amino acids (0.3 M solution in DMF) was performed using 0.5 M oxyma in DMF/0.5 M DIC in DMF (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 molar ratio), with a 5-equivalent excess over the initial resin loading. Coupling steps were performed for 20 minutes at 50 °C. Fmoc-deprotection steps were performed by treatment with a 20% piperidine solution in DMF at room temperature (1 × 10 min). Following each coupling or deprotection step, peptidyl-resin was washed with DMF (4 × 3.5 mL). Upon complete chain assembly, resin was washed with DCM (5 × 3.5 mL) and gently dried under a nitrogen flow.
:1:1 molar ratio), with a 5-equivalent excess over the initial resin loading. Coupling steps were performed for 20 minutes at 50 °C. Fmoc-deprotection steps were performed by treatment with a 20% piperidine solution in DMF at room temperature (1 × 10 min). Following each coupling or deprotection step, peptidyl-resin was washed with DMF (4 × 3.5 mL). Upon complete chain assembly, resin was washed with DCM (5 × 3.5 mL) and gently dried under a nitrogen flow.
After cleavage from the resin, peptides were purified though RP-HPLC using a binary gradient of mobile phase A (A: 97.5% H2O, 2.5% ACN, 0.7% TFA) and mobile phase B (30% water, 70% acetonitrile, 0.1% trifluoroacetic acid). The following chromatographic method was used: 0% B to 90% B in 45 min; flow rate, 14 ml min−1. Pure RP-HPLC fractions (>95%) were combined and lyophilized.
More details on resin loading, cleavage from resin, work-up and analytical HPLC are available in the ESI.†
Rheometry
A KINEXUS Pro+ rheometer (MalvernPanalytical, UK) was used to measure the viscoelastic properties of the tested samples. Samples were pre-formed and directly transferred on the bottom rheometer plate. The upper geometry Cone 60 mm was lowered until it was in conformal contact with the top surface of the hydrogel, corresponding to gap distances of 1.0–1.5 mm. Temperature was controlled with a Peltier device and maintained at 25 °C. Each analysis was repeated at least 3 times, and representative measures are reported.Coarse grain molecular dynamics
Pentapeptide coordinate files were created using VMD scripting tools and converted to coarse grain (CG) representation in the MARTINI force field (version 2.2) using martinize.py.31 As the secondary structure needs to be defined in the force field, the flag –ss = EEE was used, (E = extended β-sheet) leading to Qa, Nda and Qd beads for the backbone particles. This choice was made as β-sheet-like conformations are often observed in peptide nanostructures.32 Using the GROMACS code version 4.5.3,33 a cubic box of 10 × 10 × 10 nm3 containing 90 zwitterionic pentapeptides was created giving a peptide concentration of 0.1 mol L−1 in explicit standard CG water, with side chains in their most prevalent charge state at pH 7. The box was energy minimized and equilibrated for 500000 steps of 25 fs (equivalent to 50 ns of simulated time, considering the 4× acceleration in CG dynamics), at a temperature of 303 K and a pressure of 1 bar. For the pentapeptides selected for further study, we also tested polarizable water (PW)34 and longer simulations of 400 ns effective time. The MD simulations were performed on local workstations equipped with 12 CPUs and 1 NVIDIA Quadro RTX5000 GPU. For our models (≈9000 particles) the system provided a performance of ≈3 μs per day. The overall simulated time is 8000 × 50 ns (400 μs), thus requiring ≈130 days of computation.
Results and discussion
Numerous studies have focused on molecular self-assembly in the context of supramolecular materials using computational approaches.35,36 Inspired by these theoretical models, the self-assembly propensity of different well-known peptide sequences such as NFGAIL,37,38 FF,39,40 FFF41 and KLVFFAE42 was studied.Human calcitonin (hCT) is a 32 amino acid polypeptide hormone produced by thyroid C lymphocytes that is involved in calcium homeostasis.43 This sequence, although much less studied than Aβ, displays a notable tendency to aggregate into fibrils; indeed, the formation of amyloids composed of hCT has long been associated with medullary thyroid carcinoma.44
Some of us recently reported the high-resolution single crystal X-ray structure of an iodinated variant of DFNKF (a small fragment derived from hCT), which revealed the crucial role of aromatic interactions involving phenylalanines in the resulting β-sheet architecture.45 Considering the intrinsic amyloid propensity of this sequence and the established importance of aromatic interactions, we selected DFNKF as a reference for our coarse-grained model.
Specifically, a wild-type peptide library was computed starting from the model sequence xFxxF, where x represents any of the 20 natural amino acids. Notably, the phenylalanine residues have been kept in the same position as in DFNKF, while all the possible peptide combinations (set of 203) were obtained through CG-MD upon varying the three x amino acids. For each computed pentapeptide, we estimated the aggregation propensity (AP). The AP value is defined as the ratio of solvent accessible surface areas (SASAs) at the beginning and at the end of a MD run according to the following equation:

 | ||
| Fig. 2 Graphical representation of solubility (ΔGwater–octanol kcal mol−1) vs. aggregation propensity (AP) values for the 8000 computed pentapeptides. Colored circles are the sequences that were selected for experimental studies. The reference peptide (DFNKF) is in red. | ||
A limited set of peptide sequences (colored circles in Fig. 2) were selected to validate experimentally the reliability of the CG-MD results, namely prove that the expected self-assembly behaviors are consistent with reality. These peptides were chosen with the intent to be as much as possible representative of the ΔGwater–octanolvs. AP distribution. A higher number of peptides laying in the central-left region of the graph were selected, since poor water solubility may be a major practical issue, especially in view of potential large-scale preparation and exploitation. We selected this set of peptides also to assess the role of the N-term amino acid in the self-assembly. Namely, we chose peptides roughly located in the same regions of the graph and differing in the nature (polar, nonpolar, aromatic) of the N-term residue (e.g., GFEDF and HFEEF, located in the same area of the graph and featuring a nonpolar –G– and an aromatic –H– N-term amino acid, respectively).
As the formation of good quality fibrils results from the subtle balance between hydration and aggregation phenomena, it would be oversimplified to think that the best fibril-forming sequences can be found in the upper right region of the plot in Fig. 2, thus experimental confirmation is needed.
The selected sequences were prepared through solid-phase peptide synthesis (SPPS) and characterized for their self-assembly propensity in MilliQ water at the common concentration of 0.4 mM. This value was selected to have a direct comparison among all the pentapeptides, including the ones showing a lower water solubility, namely FFVCF and YFMDF. This finding is coherent with the CG-MD study reported in Fig. 2, as these two sequences are positioned in the right region of the plot.
Of note, four out of twelve of the synthesized peptides (Scheme 1) formed crystals suitable for single crystal X-ray diffraction from the same 0.4 mM solutions within two weeks. Since obtaining good-quality peptide crystals is generally nontrivial, we may argue that the CG-MD applied on the model sequence xFxxF can be considered as a promising tool to statistically forecast not only the self-assembly behaviour, but also the theoretical conditions which may translate into the intrinsic crystallinity of peptide-based systems. In this regard, it may be no coincidence that the crystallized sequences are all located in the same area of the graph in Fig. 2 (lower-left region). This could be an interesting example of computations applied to biomimetic crystal engineering.
 | ||
| Scheme 1 Chemical structure of the peptide sequences whose crystal structures have been reported in this work. Phenylalanine residues, whose position was kept in the CG-MD study, are depicted in red. | ||
In all the reported crystal structures (Fig. 3 and 4) the peptide strands assemble in β-sheets, which is consistent with the fact that these pentapeptides have been derived from the reference sequence DFNKF, which is well-known for its amyloidogenic propensity. The strand–strand separation, fixed by multiple N–H⋯O hydrogen bonds (Table S4†) involving the amide moieties of stacked strands, is in the range of 4.7–4.9 Å, in line with the crystal structure of other small peptide fragments previously reported.46,47
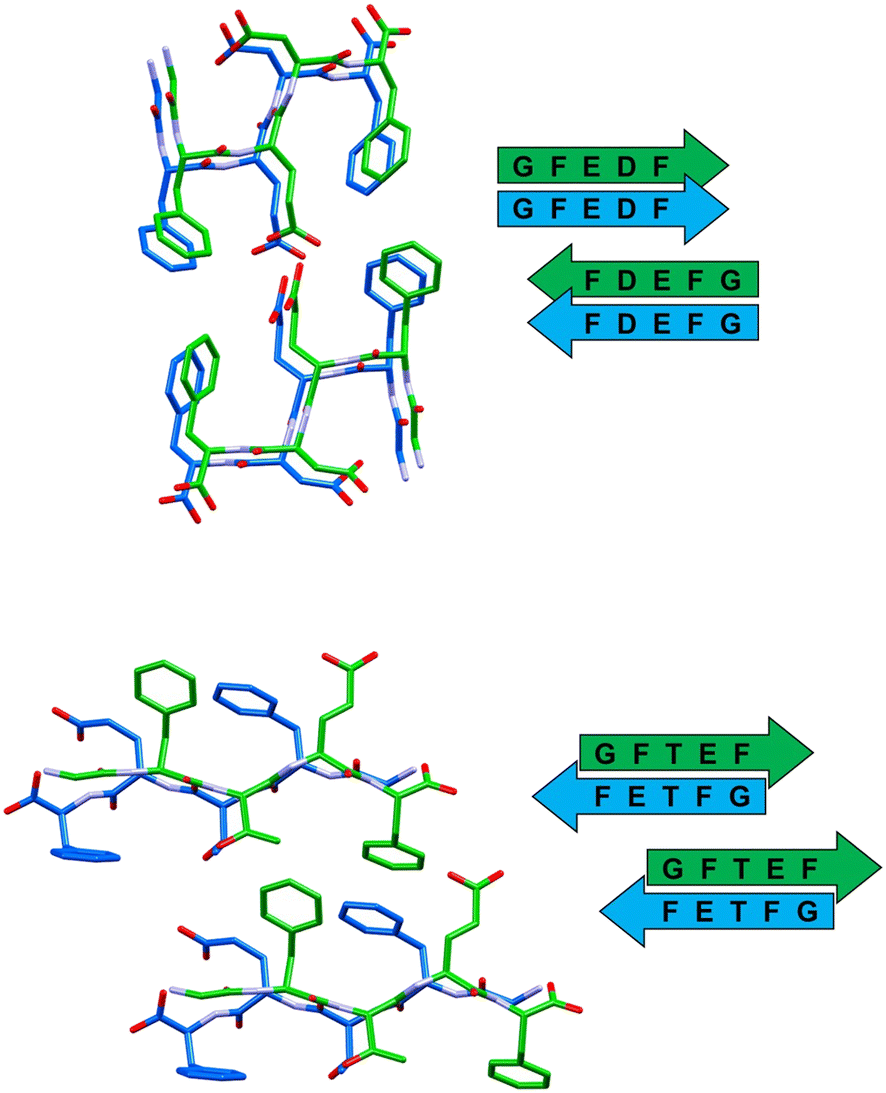 | ||
| Fig. 3 Stick representation (Mercury 2022.1.0) of crystal structures of peptide 1 (top) and peptide 2 (bottom), showing the essential packing features of the β-sheet arrangements. Hydrogen atoms have been omitted for clarity. Colour code: carbon, green and blue; nitrogen, violet; oxygen, red. | ||
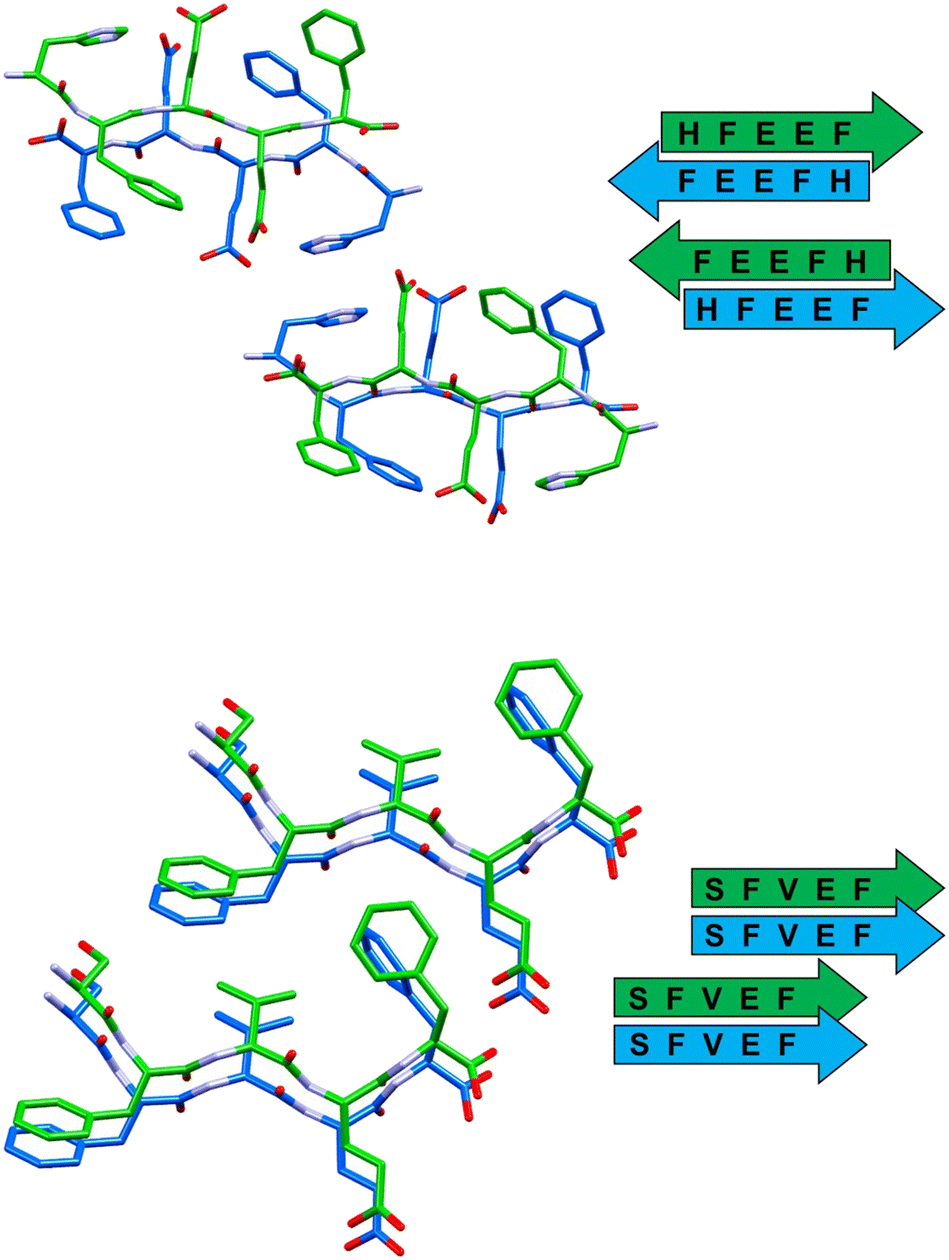 | ||
| Fig. 4 Stick representation (Mercury 2022.1.0) of crystal structures of peptide 3 (top) and peptide 4 (bottom), showing the essential packing features of the β-sheet arrangements. Hydrogen atoms have been omitted for clarity. Colour code as in Fig. 3. | ||
Each observed β-sheet and the resulting cross-β spines,48 namely the pairing of β-sheets running along the fibril axis, present distinct structural features (Table 1), suggesting that each residue, not only the fixed phenylalanines, has a non-negligible role in driving the self-assembly of the system.
| β-Sheet orientation | Paired β-sheets | Steric zipper | |
|---|---|---|---|
| GFEDF (1) | Parallel | Antiparallel | Class 1 |
| GFTEF (2) | Antiparallel | Parallel | Class 6 |
| HFEEF (3) | Antiparallel | Antiparallel | Class 5 |
| SFVEF (4) | Parallel | Parallel | Class 2 |
In detail, stacked strands of peptide 1 (GFEDF) give rise to parallel β-sheets, which form the cross-β motif as a result of reciprocal antiparallel pairing between facing β-sheet units (Fig. 3, top). This supramolecular arrangement, with the consequent interdigitation between the side-chains of paired β-sheets, generally dubbed as “steric zipper”,49 can be classified as a homosteric zipper of class 1 (parallel, up–up, face-to-face). Specifically, this self-complementarity is strengthened by the occurrence of a strong hydrogen bond involving facing glutamic acid side-chains (OE1A⋯HE24 distance is 2.15 Å).
Peptide 2 (GFTEF) forms antiparallel β-sheets, which pack in a steric zipper of class 6 (antiparallel, up![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) down, face-to-back). Here, additional stabilization of the zipper arrangement is given by π–π interactions involving the phenylalanine residues of facing β-sheets (Fig. 3, bottom).
down, face-to-back). Here, additional stabilization of the zipper arrangement is given by π–π interactions involving the phenylalanine residues of facing β-sheets (Fig. 3, bottom).
The self-assembly of sequence 3 (HFEEF) results in a homosteric zipper of class 5 (antiparallel, updown, face-to-face), where the most notable interaction is the π–π stacking between imidazole rings of histidine residues (Fig. 4, top).
β-Sheets of peptide 4 (SFVEF) display a parallel orientation (Fig. 4, bottom), forming a steric zipper of class 2 (parallel, up–up, face-to-back), where a strong hydrogen bond between the glutamic acid side chain and the C-terminal of facing sheets (OXT_B5⋯HE2_B4, 1.73 Å) aids in keeping the tight interdigitation of the zipper, in addition to other weaker interactions such as C–H⋯π (involving a –CH3 group of valine or a –CH2– of glutamic acid, i.e. C–H donors, and the aromatic rings of facing phenylalanines, i.e. acceptors).
Overall, these peptide strands adopting an extended conformation interact laterally by means of salt bridges connecting the C-terminal carboxylate and the protonated N-terminal of flanked molecules. This is not the case of 3 (HFEEF), where the C-terminal is not charged and the presence of trifluoroacetate ions functions as a bridge connecting the flanked peptide strands.
The same 0.4 mM water solutions, from which peptides 1–4 were crystallized, were analysed by TEM 48 hours after preparation, in order to visualize the morphology of the supramolecular structures formed by the synthesized peptide sequences. TEM images of the twelve studied peptides display a variety of nanostructures (Fig. 5), whose typology seems to be strongly related to the nature of the amino acid at the N-terminal. Specifically, in the case of charged side-chains (D, aspartic acid; E, glutamic acid; K, lysine) amorphous aggregates (Fig. S2†) or short and poorly defined fibrils (DFNKF, Fig. 5) are observed. Meanwhile, in the case of aromatic or non-polar amino acids (F, phenylalanine; H, histidine; Y, tyrosine; G, glycine) well-developed fibril networks or straight ribbons can be observed. Once the N-terminal residue is a polar, non-charged amino acid (C, cysteine; S, serine), the resulting morphology may be more connected to other factors, like the nature of the other residues. Indeed, CFFVF forms straight ribbons, while SFVEF (4) presents amorphous structures (Fig. S2†). Of note, the observed morphology for peptide 4 seems in contrast with the crystal structure previously described and showing amyloid features (Fig. 4, bottom). In this case the aging time is a key factor in the aggregation kinetics, since TEM images were recorded after 48 hours of aging, while crystals of the same peptide formed in a longer time.
 | ||
| Fig. 5 TEM images of the supramolecular structures formed in water (0.4 mM, aging 48 h) by some of the pentapeptides studied in this work. | ||
FTIR spectroscopy (Table S2†) of freeze-dried 48 h aged 0.4 mM solutions confirms the amyloid nature of the non-amorphous nanostructures detected through TEM. Here, the amide I region presents sharp bands located around 1630 cm−1 that are peculiar of β-sheet structures.50 Peptides forming amorphous aggregates show a huge prevalence of signals around 1660 cm−1, consistent with the unstructured material.51 Interestingly, FTIR reveals that a small β-sheet component, albeit not enough to result in a defined morphology visible by TEM, is even present in most of the ‘amorphous’ peptides. This may indicate that all the studied peptides, in suitable experimental conditions (e.g., higher concentration, varying pH or ionic strength), can potentially self-assemble into fibrils. Considering these findings and the ΔG vs. AP plot (Fig. 2), we may infer that solubility in most cases exerts a stronger impact than AP in the fibrillation propensity of the peptide. Indeed, none of the peptides having lysine at the N-terminal (KFDGF, KFEWF, KFKEF) showed fibril formation neither from 1.5 mM water solutions, that is almost four times the starting concentration that was used for our studies. Thus, peptide solubility should be a crucial factor to consider in view of selecting potential efficient fibril forming sequences from the computations.
In order to assess the formation of a stable supramolecular network, rheological analysis has been conducted to determine eventually the mechanical properties. Shear stress rheological measurements (Fig. S3†) were carried out on the same 0.4 mM solutions (48 h aged) analyzed through TEM. This analysis revealed that most of the peptide solutions behave as liquids, displaying a very scattered signal in the studied time lapse. The only exceptions are FFVCF and YFMDF (the least soluble peptides), which form soft viscoelastic fluid (G′ > G′′) with low elastic modules. Moreover, the strain sweep experiment suggests an improvement of the LVER region for the FFVCF pentapeptide. These results confirm that the hydrophobic nature of amino acids at the N-terminal strongly influences the stability of the fibril networks.
Conclusions
In this contribution we applied a computational method, based on CG-MD, to peptide systems featuring remarkable structural complexity (five amino acids). Notably, similar methods reported in the literature have been applied on simpler systems (tripeptides). Specifically, starting from a reference sequence (DFNKF) known for its amyloidogenic behavior, we selected a model peptide xFxxF (x = any natural amino acid) where the key residues for fibrillation (F) were kept, and all the possible pentapeptide combinations (203) were computed to identify new highly efficient fibril forming sequences. The applied computational model was validated by selecting a limited number of peptide sequences, which were then synthesized and experimentally studied.TEM and FTIR analyses of the studied peptides confirm the overall reliability of the “fibrillation forecast” resulting from computations (ΔG vs. AP plot), especially considering the many competing/synergic factors playing a role in the self-assembly process of such complex biomimetic systems. In detail, the nature of the residue at the N-terminal was found crucial in the resulting nanostructures formed by these pentapeptides, at least in the tested experimental conditions.
Rheological studies demonstrated that some of these sequences formed nanostructures that confer viscoelastic properties to the water solutions in which they are present. This is a good result in view of the potential employment of these peptides as structuring additive components in materials that need a strengthening of their mechanical performances.
Of note, one third of the synthesized pentapeptides formed good crystals suitable for single crystal X-ray diffraction analysis, which is a remarkable result considering that this kind of system is generally poorly prone to crystallize, especially without the presence of heavy atoms. Importantly, all the crystallized sequences are located in the same sector of the ΔG vs. AP plot (lower-left region, Fig. 6), characterized by medium-high solubility and medium-low AP, which may contain peptides with the tendency to form crystals, possibly thanks to the balance between solubility and low aggregation propensity that allows for slow and ordered (long-range periodicity) self-assembly.
 | ||
| Fig. 6 Representation of the same ΔG vs. AP plot of Fig. 2, where some macro regions associated with specific self-assembly behaviour have been identified based on experimental observations on the synthesized pentapeptides. | ||
This finding suggests that there should be a reasonable correlation between observed self-assembly behaviors and areas of the ΔG vs. AP graph where certain nanostructures are more likely to be found.
Indeed, we noticed reasonable correlations not only for crystalline assemblies, but also for other observed nanostructures. In detail (Fig. 6), a second area characterized by medium-high solubility and high AP may be populated by peptides that aggregate into amorphous structures, possibly due to the faster aggregation process during solvent evaporation. Finally, a third broad region may feature peptides with medium-to-low solubility and medium AP, which leads to fibril-forming peptides.
Once the calculated ΔG is around zero, subtle variations of the applied experimental conditions may play a major role in the resulting self-assembly (borderline region, Fig. 6). This is the case of EFGFF (amorphous structure) or SFVEF, with the latter forming crystals under slow evaporation conditions or amorphous aggregates under fast evaporation on the TEM grid.
Considering peptide crystallinity, our results may have a huge impact. Indeed, in recent years many efforts have been made in the development peptide-based active pharmaceutical ingredients (APIs), as they guarantee several advantages such as biocompatibility and low production costs.52 As crystallinity is usually an important factor in the industrial production and purification of an API, the choice of a reliable CG-model to forecast peptide crystallinity can drastically reduce computational times and costs in the development of peptide-based drugs.
Conflicts of interest
“There are no conflicts to declare”.Acknowledgements
The authors acknowledge Elettra-Sincrotrone Trieste for providing beamtime under proposal 20185487. P. M. acknowledge the European Research Council (ERC) for Proof-of-Concept grant no. 789815.Notes and references
- G. M. Whitesides and B. Grzybowski, Science, 2002, 295, 2418–2421 CrossRef CAS PubMed.
- S. Scheiner, Noncovalent Forces, Springer, 2010, vol. 19 Search PubMed.
- C. M. Dobson, Trends Biochem. Sci., 1999, 24, 329–332 CrossRef CAS PubMed.
- P. T. Lansbury and H. A. Lashuel, Nature, 2006, 443, 774–779 CrossRef CAS PubMed.
- F. Chiti and C. M. Dobson, Annu. Rev. Biochem., 2006, 75, 333–366 CrossRef CAS PubMed.
- A. Levin, T. A. Hakala, L. Schnaider, G. J. L. Bernardes, E. Gazit and T. P. J. Knowles, Nat. Rev. Chem., 2020, 4, 615–634 CrossRef CAS.
- A. Lampel, R. V. Ulijn and T. Tuttle, Chem. Soc. Rev., 2018, 47, 3737–3758 RSC.
- P. Besenius, G. Portale, P. H. H. Bomans, H. M. Janssen, A. R. A. Palmans and E. W. Meijer, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 17888–17893 CrossRef CAS PubMed.
- J. Wang, K. Liu, L. Yan, A. Wang, S. Bai and X. Yan, ACS Nano, 2016, 10, 2138–2143 CrossRef CAS PubMed.
- M. A. Sequeira, M. G. Herrera and V. I. Dodero, Phys. Chem. Chem. Phys., 2019, 21, 11916–11923 RSC.
- P. W. J. M. Frederix, G. G. Scott, Y. M. Abul-Haija, D. Kalafatovic, C. G. Pappas, N. Javid, N. T. Hunt, R. V. Ulijn and T. Tuttle, Nat. Chem., 2014, 7, 30–37 CrossRef PubMed.
- A. Pizzi, C. Pigliacelli, G. Bergamaschi, A. Gori and P. Metrangolo, Coord. Chem. Rev., 2020, 411, 213242 CrossRef CAS.
- A. Marchetti, A. Pizzi, G. Bergamaschi, N. Demitri, U. Stollberg, U. Diederichsen, C. Pigliacelli and P. Metrangolo, Chem. – Eur. J., 2022, 28, e202104089 CAS.
- A. Pizzi, C. Pigliacelli, A. Gori, N. Nonappa, O. Ikkala, N. Demitri, G. Terraneo, V. Castelletto, I. W. Hamley, F. Baldelli Bombelli and P. Metrangolo, Nanoscale, 2017, 9, 9805–9810 RSC.
- A. Bertolani, L. Pirrie, L. Stefan, N. Houbenov, J. S. Haataja, L. Catalano, G. Terraneo, G. Giancane, L. Valli, R. Milani, O. Ikkala, G. Resnati and P. Metrangolo, Nat. Commun., 2015, 6, 7574 CrossRef PubMed.
- L. Sori, A. Pizzi, N. Demitri, G. Terraneo, A. Frontera and P. Metrangolo, CrystEngComm, 2022, 24, 7255–7260 RSC.
- A. Pizzi, L. Sori, C. Pigliacelli, A. Gautieri, C. Andolina, G. Bergamaschi, A. Gori, P. Panine, A. M. Grande, M. B. Linder, F. Baldelli Bombelli, M. Soncini and P. Metrangolo, Small, 2022, 18, 2200807 CrossRef CAS PubMed.
- M. Fayaz-Torshizi and E. A. Müller, Mol. Syst. Des. Eng., 2021, 6, 594–608 RSC.
- A. Kantardjiev, Soft Matter, 2021, 17, 2753–2764 RSC.
- K. Shmilovich, R. A. Mansbach, H. Sidky, O. E. Dunne, S. S. Panda, J. D. Tovar and A. L. Ferguson, J. Phys. Chem. B, 2020, 124, 3873–3891 CrossRef CAS PubMed.
- A. Jain, C. Globisch, S. Verma and C. Peter, J. Chem. Theory Comput., 2019, 15, 1453–1462 CrossRef CAS PubMed.
- A. Banerjee, C. Y. Lu and M. Dutt, Phys. Chem. Chem. Phys., 2022, 24, 1553–1568 RSC.
- S. Chakraborty, C. M. Berac, M. Urschbach, D. Spitzer, M. Mezger, P. Besenius and T. Speck, ACS Appl. Polym. Mater., 2022, 4, 822–831 CrossRef CAS.
- A. Lausi, M. Polentarutti, S. Onesti, J. R. Plaisier, E. Busetto, G. Bais, L. Barba, A. Cassetta, G. Campi, D. Lamba, A. Pifferi, S. C. Mande, D. D. Sarma, S. M. Sharma and G. Paolucci, Eur. Phys. J. Plus, 2015, 130, 43 CrossRef.
- W. Kabsch, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 125–132 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- P. Emsley, B. Lohkamp, W. G. Scott and K. Cowtan, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 486–501 CrossRef CAS PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849–854 CrossRef CAS.
- C. F. Macrae, I. Sovago, S. J. Cottrell, P. T. A. Galek, P. McCabe, E. Pidcock, M. Platings, G. P. Shields, J. S. Stevens, M. Towler and P. A. Wood, J. Appl. Crystallogr., 2020, 53, 226–235 CrossRef CAS PubMed.
- L. Schrodinger and W. DeLano, The PyMOL Molecular Graphics System, Schrodinger, LLC, 2020 Search PubMed.
- martinize.py, can be found under http://md.chem.rug.nl/cgmartini/index.php/downloads/tools/204-martinize Search PubMed.
- S. Fleming, P. W. J. M. Frederix, I. Ramos Sasselli, N. T. Hunt, R. V. Ulijn and T. Tuttle, Langmuir, 2013, 29, 9510–9515 CrossRef CAS PubMed.
- B. Hess, C. Kutzner, D. van der Spoel and E. Lindahl, J. Chem. Theory Comput., 2008, 4, 435–447 CrossRef CAS PubMed.
- S. O. Yesylevskyy, L. V. Schäfer, D. Sengupta and S. J. Marrink, PLoS Comput. Biol., 2010, 6, 1–17 CrossRef PubMed.
- M. McCullagh, T. Prytkova, S. Tonzani, N. D. Winter and G. C. Schatz, J. Phys. Chem. B, 2008, 112, 10388–10398 CrossRef CAS PubMed.
- O.-S. Lee, V. Cho and G. C. Schatz, Nano Lett., 2012, 12, 4907–4913 CrossRef CAS PubMed.
- C. A. E. Hauser, R. Deng, A. Mishra, Y. Loo, U. Khoe, F. Zhuang, D. W. Cheong, A. Accardo, M. B. Sullivan, C. Riekel, J. Y. Ying and U. A. Hauser, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 1361–1366 CrossRef CAS PubMed.
- C. Wu, H. Lei and Y. Duan, Biophys. J., 2004, 87, 3000–3009 CrossRef CAS PubMed.
- P. W. J. M. Frederix, R. V. Ulijn, N. T. Hunt and T. Tuttle, J. Phys. Chem. Lett., 2011, 2, 2380–2384 CrossRef CAS PubMed.
- C. Guo, Y. Luo, R. Zhou and G. Wei, ACS Nano, 2012, 6, 3907–3918 CrossRef CAS PubMed.
- C. Guo, Y. Luo, R. Zhou and G. Wei, Nanoscale, 2014, 6, 2800–2811 RSC.
- D. Thirumalai, Curr. Opin. Struct. Biol., 2003, 13, 146–159 CrossRef CAS PubMed.
- L. A. Austin and H. Heath 3rd, N. Engl. J. Med., 1981, 304, 269–278 CrossRef CAS PubMed.
- I. Kedar, M. Ravid and E. Sohar, Isr. J. Med. Sci., 1976, 12, 1137–1140 CAS.
- A. Bertolani, A. Pizzi, L. Pirrie, L. Gazzera, G. Morra, M. Meli, G. Colombo, A. Genoni, G. Cavallo, G. Terraneo and P. Metrangolo, Chem. – Eur. J., 2017, 23, 2051–2058 CrossRef CAS PubMed.
- A. Pizzi, V. Dichiarante, G. Terraneo and P. Metrangolo, Pept. Sci., 2018, 110, e23088 CrossRef PubMed.
- A. Pizzi, L. Catalano, N. Demitri, V. Dichiarante, G. Terraneo and P. Metrangolo, Pept. Sci., 2020, 112, e24127 CAS.
- R. Nelson, M. R. Sawaya, M. Balbirnie, A. Ø. Madsen, C. Riekel, R. Grothe and D. Eisenberg, Nature, 2005, 435, 773–778 CrossRef CAS PubMed.
- M. R. Sawaya, S. Sambashivan, R. Nelson, M. I. Ivanova, S. A. Sievers, M. I. Apostol, M. J. Thompson, M. Balbirnie, J. J. W. Wiltzius, H. T. McFarlane, A. Ø. Madsen, C. Riekel and D. Eisenberg, Nature, 2007, 447, 453–457 CrossRef CAS PubMed.
- J. Seo, W. Hoffmann, S. Warnke, X. Huang, S. Gewinner, W. Schöllkopf, M. T. Bowers, G. von Helden and K. Pagel, Nat. Chem., 2017, 9, 39–44 CrossRef CAS PubMed.
- Y. Guo and J. Wang, ChemPhysChem, 2012, 13, 3901–3908 CrossRef CAS PubMed.
- S. S. Usmani, G. Bedi, J. S. Samuel, S. Singh, S. Kalra, P. Kumar, A. A. Ahuja, M. Sharma, A. Gautam and G. P. S. Raghava, PLoS One, 2017, 12, e0181748 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: CCDC 2174444–2174447. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ce00495c |
| ‡ These authors equally contributed to this work. |
| This journal is © The Royal Society of Chemistry 2023 |