
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
2D noble metals: growth peculiarities and prospects for hydrogen evolution reaction catalysis
Ivan
Shtepliuk

Semiconductor Materials Division, Department of Physics, Chemistry and Biology-IFM, Linköping University, S-58183 Linköping, Sweden. E-mail: ivan.shtepliuk@liu.se
First published on 23rd February 2023
Abstract
High-performance electrocatalysts for the hydrogen evolution reaction are of interest in the development of next-generation sustainable hydrogen production systems. Although expensive platinum-group metals have been recognized as the most effective HER catalysts, there is an ongoing requirement for the discovery of cost-effective electrode materials. This paper reveals the prospects of two-dimensional (2D) noble metals, possessing a large surface area and a high density of active sites available for hydrogen proton adsorption, as promising catalytic materials for water splitting. An overview of the synthesis techniques is given. The advantages of wet chemistry approaches for the growth of 2D metals over deposition techniques show the potential for kinetic control that is required as a precondition to prevent isotropic growth. An uncontrolled presence of surfactant-related chemicals on a 2D metal surface is however the main disadvantage of kinetically controlled growth methods, which stimulates the development of surfactant-free synthesis approaches, especially template-assisted 2D metal growth on non-metallic substrates. Recent advances in the growth of 2D metals using a graphenized SiC platform are discussed. The existing works in the field of practical application of 2D noble metals for hydrogen evolution reaction are analyzed. This paper shows the technological viability of the “2D noble metals” concept for designing electrochemical electrodes and their implementation into future hydrogen production systems, thereby providing an inspirational background for further experimental and theoretical studies.
1. Advantages of 2D noble metals
In the long-term, hydrogen technologies are foreseen to be the cornerstones of sustainable and renewable energy sources,1,2 and will replace traditional fossil fuels.3 With this target in mind, it is imperative to pay special attention to the implementation of new strategies of green hydrogen production and the improvement of existing technologies towards new hydrogen storage materials4,5 and highly efficient catalysts for the hydrogen evolution reaction (HER).6–10 The HER is a fundamentally important process in electrochemical water splitting for clean hydrogen production.11–13 Even though platinum group metals (PGMs) are considered to be superior catalysts for the HER,14,15 they are referred to as critical raw materials due to their limited abundance.16 Indeed, the natural scarcity of noble metals (NMs) largely determines their high cost. Efficient catalysts should contain a high NM content to ensure a good performance, which impedes their large-scale production and implementation. More efficient approaches should therefore be applied to ensure a scalable cost-efficient production of HER catalysts beyond the conventional bulk PGM-based ones. One of the possible solutions to address this challenge involves replacing PGM.17–19 In this context, advanced non-PGM catalysts with a large surface area, high electrical conductivity, low toxicity, and appropriate catalytic activity (i.e., near-to-zero Gibbs free energy of hydrogen adsorption (ΔGH*), as low as possible water decomposition barrier, wide potential window, fast charge transfer, and good stability, are highly sought. Despite huge efforts dedicated to developing advanced non-PGM catalytic materials,20–22 parameters describing their catalytic activity in the HER are, in most cases, noticeably lagging behind the indicators of NM-based catalysts. PGMs are still the gold standard for the hydrogen evolution reaction. For this reason, even the lower cost of non-PGM materials does not play a decisive role. With an appropriate strategy, catalysts based on precious metals can be, however, re-designed and re-engineered towards less NM consumption and more cost-efficient technologies. This implies designing HER catalysts with as low a NM content as possible. To some extent, this can be achieved via a 3D-to-2D phase transformation. The main goal is to create planar (2D) arrays of metal atoms with low coordination numbers that will eventually lead to the discovery of atomically thin structures with a high density of coordinatively unsaturated metal sites (CUMS) and a large electrochemically active surface area. This will enable not only the production of low-cost and mass-efficient large-area metal-based catalysts that is important in terms of rational PGM use, but will also improve the overall catalytic performance towards HERs by utilizing CUMS as catalytically active centers. Indeed, surficial atoms of 2D metal nanosheets have low coordination numbers and, thus, can facilitate hydrogen proton adsorption for the efficient HER. This is because almost all the metal atoms in the 2D catalyst will be involved in the catalytic reaction. Such highly exposed CUMS can eventually increase the utilization efficiency of NMs while maintaining their outstanding catalytic properties. Furthermore, as demonstrated by the specific example of subnanometric Ru clusters,23 low-coordinated Rh clusters exhibit upshifted d band centres and hence are more catalytically active in the HER compared to highly coordinated Rh structures. A similar effect of the coordination number on the d band centre was also reported for Pt24 and Au25 nanoclusters. The above highlights the exceptional importance of 2D metals with an abundance of low-coordinated atoms as promising novel HER catalysts.This work provides a brief overview of the currently existing approaches and challenges in the growth of 2D noble metals and summarizes the use of selected 2D metals as catalysts for the hydrogen evolution reaction.
2. Advances in the synthesis of 2D noble metals
Modern HER electrocatalyst design concepts require a more rational utilization of NMs especially PGMs to achieve their catalytic activity, comparable to that of the state-of-the-art commercial Pt/C catalysts. A possible way toward the sustainable use of these precious resources is coordination engineering of NMs that can be efficiently realized through the creation of atomically thin metal catalysts. In this regard, a profound theoretical and experimental comprehension of the fundamental mechanisms behind the formation of low-coordinated 2D NMs is of particular importance. From a theoretical point of view, most two-dimensional metals are predicted to be stable, at least when they have hexagonal and honeycomb structures.26 This prediction was mainly reached based on an estimation of the cohesive energy and in-plane elastic constants at 0 K. However, the room-temperature stability of 2D metal nanosheets is still an unaddressed issue. Intuitively, one can expect a 2D-to-3D transition for metals possessing a relatively low 0 K-cohesive energy for their 2D structure and a comparatively large 0 K-cohesive energy for their 3D counterpart. According to Fig. 1, close-packed hexagonal 2D metal lattices are energetically less stable than their bulk counterparts. Taking the above into account and considering the connection between cohesive energy and surface energy (the stronger the cohesion, the higher the surface energy), it can be concluded that the growth of 2D noble metals is quite a challenging task due to their strong tendency to agglomerate to form 3D close-packed structures.27 In other words, there are inherent thermodynamic restrictions to prevent anisotropic growth of metal nanosheets, which can be, however, eliminated or avoided through realization of the kinetically controlled synthesis. Nevertheless, due to the adaptability and flexibility of conventional physical and chemical methods (like atomic layer deposition, magnetron sputtering, chemical vapor deposition, thermal evaporation, electrodeposition) to meet strict conditions for thin-film deposition at an extreme thickness limit, these techniques are also broadly used for the formation of 2D metals. However, in this case, the need to make an appropriate choice of substrate comes to the forefront. Indeed, the wetting nature of the metal film on the substrate, and hence the film growth regime (2D or 3D), is strongly dependent on the metal-substrate interaction.28 The possible routes for the synthesis of 2D noble metals are introduced in the following subsections.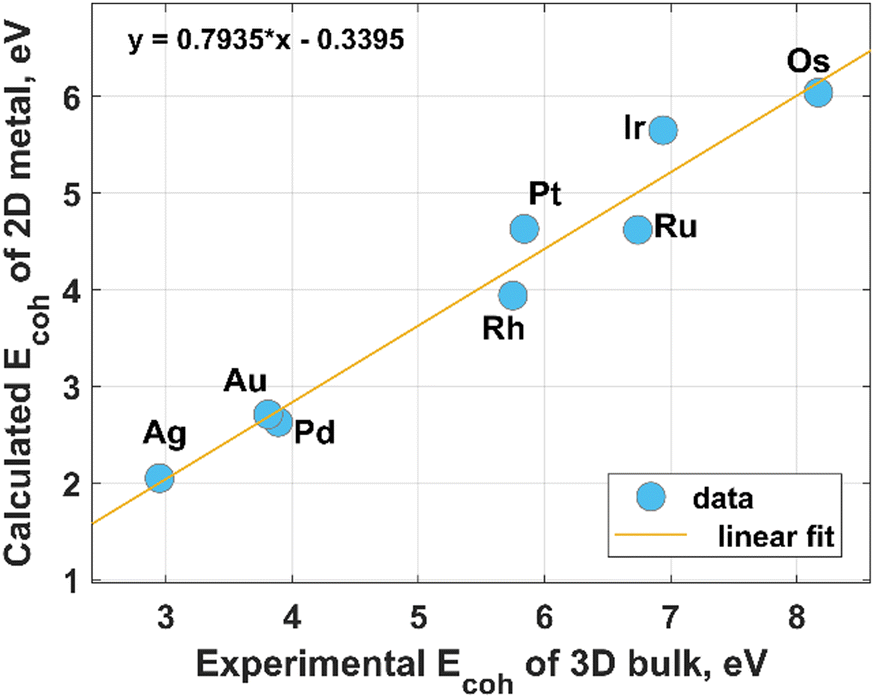 | ||
| Fig. 1 Relationship between the experimental values of the cohesive energy of bulk noble metals and the theoretically predicted cohesive energies for their 2D counterparts. The experimental data were extracted from the literature,29 while the theoretical data for hexagonal structures were reported by Nevalaita and Koskinen.26 | ||
2.1. Ruthenium
In their recent study, Chen et al.30 reported the synthesis of two-dimensional ruthenium nanocrystals using an O2-mediated solution-based colloidal method. More specifically, 2D Ru is formed in three stages, including: (i) the initial reduction of the RuCl3 precursor (at 180 °C) followed by the appearance of small irregularly shaped Ru nanosheets, (ii) a gradual in-plane dendritization and (iii) a final filling of the empty channels between the separated Ru branches by the Ru atoms, respectively (Fig. 2). From a kinetics point of view, the growth process can be understood as an aggregation of freshly reduced zero-valent Ru atoms (Ru0) towards the formation of early-stage nuclei and enlarged Ru0n+1 clusters:| Ru3+ + 3e → Ru0 | (1) |
| n(Ru0) → Ru0n | (2) |
| Ru0n + Ru3+ + 3e− → Ru0n+1 | (3) |
 | ||
| Fig. 2 Time evolution of the morphology of Ru nanosheets. Transmission electron microscopy (TEM) images were made at different reaction times: (a) 180 min, (b) 190 min, (c) 200 min, (d) 220 min, and (e) 420 min. Reproduced with permission from ref. 31. Copyright 2020 Wiley-VCH GmbH. | ||
A similar solvothermal approach was applied to form Ru nanosheets of ∼1.0–1.2 nm thickness (approximately 5–7 atomic layers) through the reduction of Ru acetylacetonate at 180 °C over 18 h in the presence of urea and isopropanol.32 It was stated that the isopropanol is responsible for the directional (anisotropic) growth of Ru nanosheets, while the urea can stave off the aggregation of the initially separated Ru nanosheets, thereby promoting the in-plane growth mode.
Several efforts were also made to grow ultrathin Ru films on glass,33 Au (111)34 and Si(100)35 substrates by atomic layer deposition (ALD), electrochemical deposition and dc-magnetron sputtering, respectively. In most cases, the supporting substrate may significantly affect the growth mode and physicochemical properties of the deposited films. However, the mentioned examples concern not only different substrates, but also different growth methods. This brings additional method-related influencing factors on Ru growth. Therefore, a direct comparison between the corresponding growth modes would be incorrect. At the same time, it can be noted that the Ru film on gold is relatively homogeneous.34 This is due to a small lattice misfit between Au(111) and Ru(0001) that promotes Ru heteroepitaxy on gold surfaces. Meanwhile, Ru film growth on more lattice-mismatched substrates like silicon35 and glass33 resulted in the formation of the polycrystalline phase (grained films). The presence of the grain boundaries in polycrystalline film leads to the appearance of unique catalytically active sites that are missing in the continuous layers. This can eventually affect the HER performance of Ru catalysts. Furthermore, as was recently demonstrated,36 the substrate properties may influence the catalytic performance of 2D metal. It was revealed that modulation of the charge transfer at the 2DAu/SiC interface affected the d band center of the gold layer and the Gibbs free energy of hydrogen adsorption (generally accepted descriptors of the HER). The same phenomena can be expected in the case of Ru film. Nevertheless, since in almost all cases the thickness of the resulting Ru films exceeds 2 nm, they cannot be classified as true atomically thin Ru layers. The catalytic properties of such Ru layers may be interesting in their own right.
One more interesting approach for the synthesis of 2D Ru was originally proposed by Fukuda et al.37 This method called topotactic metallization involves the reduction of pre-synthesized exfoliated RuO2 nanosheets with subsequent formation of the ruthenium monolayer (an average thickness of ∼0.6 nm) with hexagonal symmetry (Fig. 3). The successful reduction was achieved through treatment in a gas mixture atmosphere (5% H2 + 95% N2) at 200 °C. Since Ru is believed to be one of the best catalysts for ammonia synthesis,38 then the question arises as to whether the use of a (5% H2 + 95% N2) gas mixture in the presence of Ru catalyst can cause a NH3 synthesis reaction. Since the authors in ref. 37 have not commented on the possibility of ammonia production by the conventional Haber–Bosch process, one can assume that the proposed synthesis conditions are far different from those that initiate the reaction using a Ru/C catalyst (T = 460 °C, P = 50–100 bar, H2/N2 ratio of 1.5).39
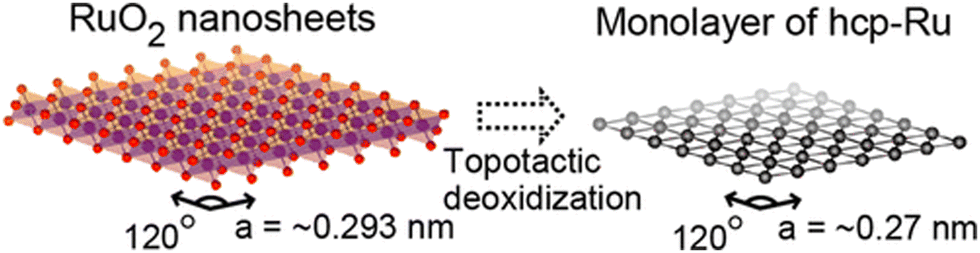 | ||
| Fig. 3 An illustration of the transformation of exfoliated RuO2 nanosheets into a Ru monolayer. Reproduced with permission from ref. 37. Copyright 2013 American Chemical Society. | ||
2.2. Iridium
From Fig. 1 it is clear that iridium has quite a high value of cohesive energy for the 3D phase that makes the synthesis of atomically thin Ir layers extremely challenging. For this reason, only several studies on the growth of ultrathin Ir layers have been published to date.40,41 The performance of the wet-chemical method for the synthesis of freestanding ultrathin Ir nanosheets (with a thickness of 5–6 atomic layers) has been explored in ref. 40. This method is based on the thermal decomposition of formic acid into H2 and CO components at 100 °C for 5 h that is accompanied with a reduction of IrCl3 precursor using released reducing agents (Fig. 4). It was also emphasized that CO plays a dual role of the reducing agent and a surface-confining agent. The latter is a necessary condition for the lateral growth of Ir over the (111) plane. Moreover, partial hydroxylation of the surface was suggested as a possible reason for the minimization of the surface energy and the formation of the stable 2D structure of iridium. These results provide a good scientific background for further experimental work towards obtaining the iridium monolayer.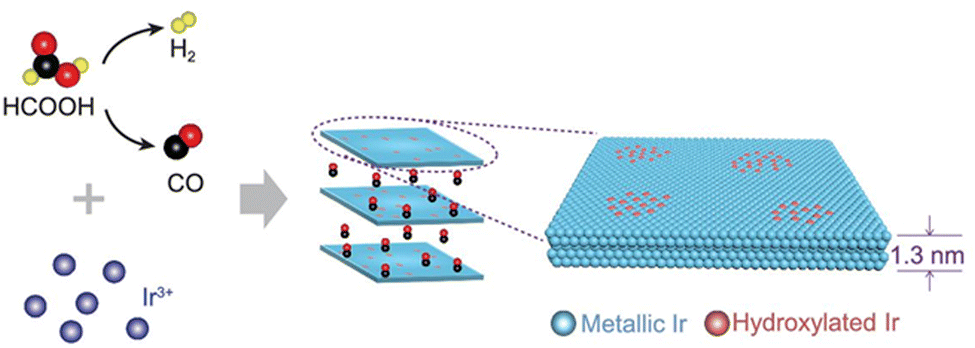 | ||
| Fig. 4 The proposed mechanism underlying the formation of the Ir nanosheets. Reproduced with permission from ref. 40. Copyright The Author(s) 2020. Published by Oxford University Press on behalf of China Science Publishing & Media Ltd. | ||
Attempts to grow the nanometre iridium films onto a porous anodic alumina (PAA) template by using an atomic layer deposition technique have also been reported.41 However, the measured thickness of 3 nm is pretty far from the true 2D limit. It remains to be seen whether the atomic layer deposition can be used to synthesize a large-area iridium monolayer. Probably, the key to solving this problem lies in the optimization of the reaction chamber and the iridium source temperatures during ALD cycles.
2.3. Rhodium
Belonging to platinum-group metals and possessing superior catalytic activity, Rh in 2D form is of special interest for catalysis due to its internal physical and chemical properties.42 Like other 2D noble metals, Rh nanosheets are synthesized mainly in the presence of surface-capping agents.43–45 Polyallylamine hydrochloride (PAH) was used as a capping and shape directing agent to form carbon nanotube (CNT)-supported 2D rhodium nanosheets with a thickness of 0.9 nm43 and free-standing ultrathin rhodium nanosheet layers with a thickness of 0.8 nm that correspond to 6 atomic layers.44 In both cases, the mechanism underlying the formation of 2D Rh nanosheets lies in inhibiting the isotropic growth by the attached polyallylamine hydrochloride molecules (Fig. 5A). The strong interaction between PAH and RhCl3 precursor molecules lowers the reduction rate of RhCl3 precursor, enabling kinetically controlled synthesis. The process starts with reducing Rh3+ to Rh0 under heating conditions (at 120 °C) followed by the formation of small Rh nanoparticles with a size of 3 nm after 30 min and their gradual transformation to dendritic Rh nanosheet nanoassemblies over 1.5 h. The points of attachment of capping molecules provide necessary conditions for the appearance of 2D Rh nuclei and eventual formation of atomically thick ultrathin Rh nanosheets. The synthesis time of 6 h was identified as the time frame sufficient enough to form a final product – Rh nanosheet nanoassemblies with a mean size of 95 nm.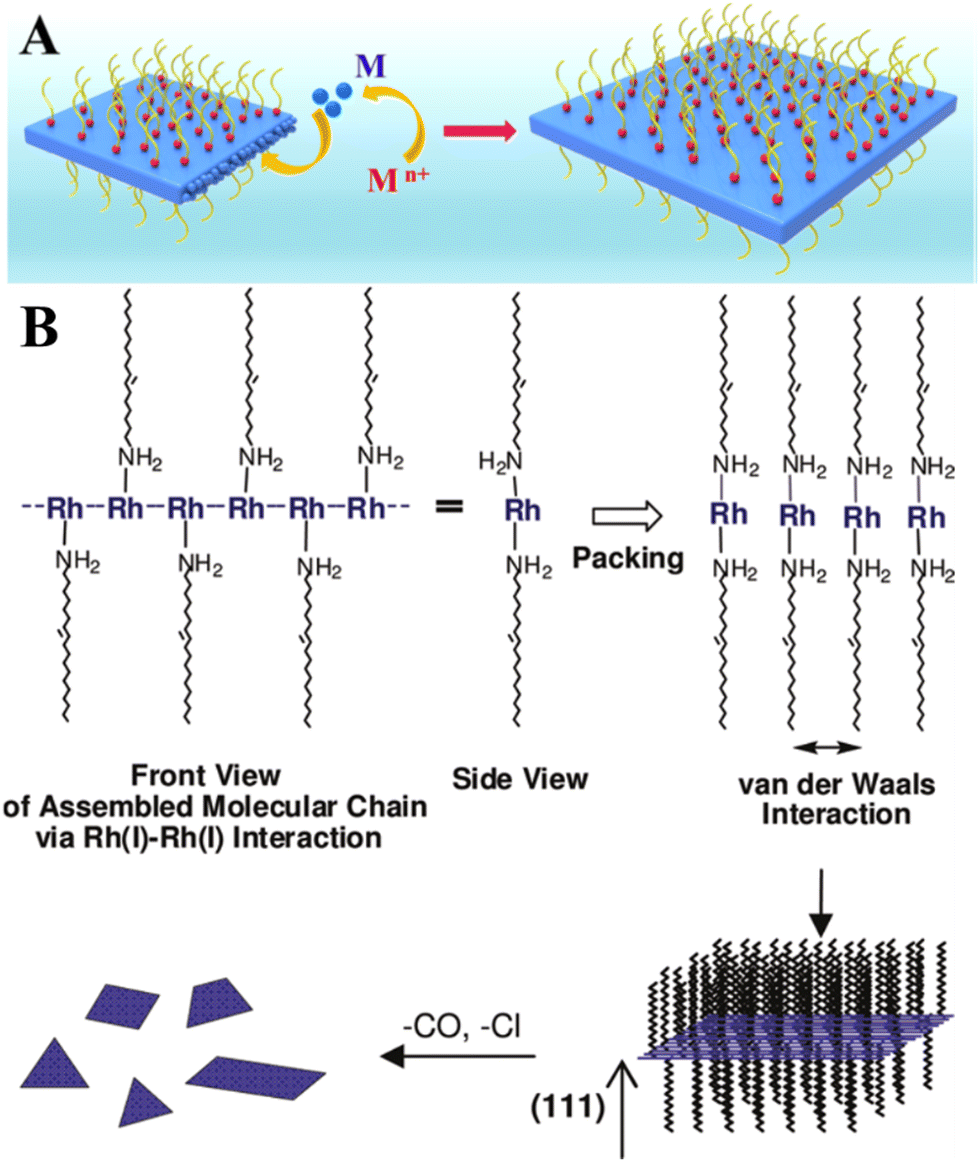 | ||
| Fig. 5 (A) The illustration of the confinement growth of 2D Rh nanosheets at 120 °C for 6 h. Reproduced with permission from ref. 44. Copyright © 2016 American Chemical Society. (B) A possible growth mechanism of 2D Rh nanoplates proposed by Jang et al.36 Reproduced with permission from ref. 47. Copyright © 2010 American Chemical Society. | ||
Another research study45 described the growth recipe of single-layered rhodium nanosheets (with a thickness of ∼4 Å) that is based on the reduction of Rh(acac)3 at 180 °C for 8 h in the presence of poly(vinylpyrrolidone) (PVP) surfactant. This method enables Rh nanosheets to be obtained with an edge length of approximately 500–600 nm. Despite the efficiency of these methods, it is felt that using of surface-capping agents is in principle undesirable and should be avoided.46 This is due to the fact that the capping molecules may hinder the chemical reaction between the metal surface and targeted substances (for example, hydrogen protons, water molecules, hydronium ions), thereby adversely affecting the catalytic performance of 2D metal layers especially towards the HER. In this regard, the cyanogel-reduction method proposed by Liu et al.46 implies the capping agent-free synthesis of ultrathin rhodium nanosheets (3–4 atomic layers) through the reduction of a jelly-like RhCl3–K3Co(CN)6 cyanogel with formaldehyde at 180 °C for 12 h. It was argued that the 2D structural units of the 3D cyanogel skeleton serve as a self-platform for the formation of 2D Rh nanosheets. In line with this, earlier work by Jang and others47 showed that extremely thin Rh nanoplates (average thickness of 1.3 ± 0.2 nm) can be synthesized through the reaction between [Rh(CO)2Cl]2 precursor and oleylamine. It was hypothesized that the formation mechanism is based on the strong metal-metal interaction between the dissolved Rh precursor molecules. Since the resulting Rh-based complex has a chain structure, the van der Waals interaction between the neighboring chains (via alkyl groups) may result in the formation of a lamellar structure and a gradual self-assembling of reduced Rh0 species into 2D plates (Fig. 5B).
Another example of the surfactant-free synthesis of the ultrathin Rh layers with thicknesses from 1 to a few monolayers is a direct layer-by-layer growth of Rh onto single-crystal Ru(0001) by means of an e-beam evaporator.48 Rh films were grown in an ultrahigh vacuum chamber at a growth temperature of 650 K and a base pressure of 10−10 mbar. These conditions enable us to avoid atom intermixing at the Rh/Ru interface and undesired adsorption of residual gases. It was revealed that the first Rh layer grows pseudomorphically, completely reproducing the hcp structure of the Ru substrate. However, the second one and next layers stack in an fcc sequence.
2.4. Osmium
There is practically no information on the existence of osmium in 2D form (having only one monolayer). This is partly due to its highest cohesive energy (for both 2D and 3D cases), as was shown in Fig. 1, and hence a high melting point that makes it impossible to easily grow the Os layers. Other drawbacks limiting the Os-involved experimental studies include its natural scarcity, and its tendency to form highly toxic and volatile OsO4 tetroxide by reacting with atmospheric oxygen under ambient conditions.49 Wet chemical synthesis approaches enable the growth of only Os nanoparticles of very small size (<2 nm).50 This specificity of osmium complicates the formation of large area 2D Os layers by using the kinetically controlled process. Meanwhile, growth by deposition techniques is an appealing alternative to form the continuous Os monolayer via using, for example, strongly-interacting substrates. This eventually might lead to good wetting characteristics of the Os film. A systematic review of the existing literature, including reports on the growth of thin osmium films by using a differential potential pulse deposition method,51 atomic layer deposition,52 chemical vapor deposition,53–55 and magnetron sputtering,56 has highlighted a potential pathway for the synthesis of true 2D Os layers. In particular, a more beneficial combination of the molecular precursors for ALD/CVD growth and supporting substrates could facilitate a template-assisted 2D growth regime of osmium. In this context, a recent report on the modification of graphene by osmium adatoms (up to reaching 18% coverage)57 indicates that graphene can be potentially used as a substrate for the formation of 2D Os nanopatches (due to the strong tendency of the metal adatoms to form the clusters). Adjusting the osmium-graphene interaction is a promising tool to control the Os cluster planarity and a transition between 2D and 3D cluster shapes. In general, there are different approaches to controlling a metal–graphene interaction. This control can be achieved through defect,58 strain,59 and substrate36 engineering. Moreover, appropriate surface modification of graphene by functional chemical groups also enables fine tuning of the metal–graphene interaction.60 Although these techniques were initially exploited to alter the strength of the interaction between graphene and other metals than osmium, their scope can be also extended to the Os case.2.5. Palladium
Probably due to a comparatively low cohesive energy (Fig. 1), palladium is more prone than the previously considered metals to forming extremely thin 2D nanosheets by means of top-down methods. According to earlier reports, successful deposition of a Pd monolayer onto Al (111),61 Pt(111),62 Ni(111),63 Mo (100),64,65 Ta (110),66 Re{0001},67 and HOPG68 can be achieved through a thermal evaporation approach. Among the most common methods for the synthesis of 2D Pd layers is CO-mediated growth,69–73 electrochemical deposition,74–76 and the reduction of a PdCl2-graphite intercalation compound precursor.77–80 An unusual route to grow two-dimensional palladium dendritic nanostructures was proposed by Zhu et al.81The method is based on in situ liquid-cell transmission electron microscopy (TEM). Within this approach, electron beam irradiation of the liquid tetrachloropalladate precursor surrounded by two SiN membranes resulted in the reduction of the palladium ions to Pd0 followed by the formation of the initial Pd nuclei on SiN and a gradual growth of branched nanostructures by a diffusion-limited aggregation mechanism (Fig. 6).
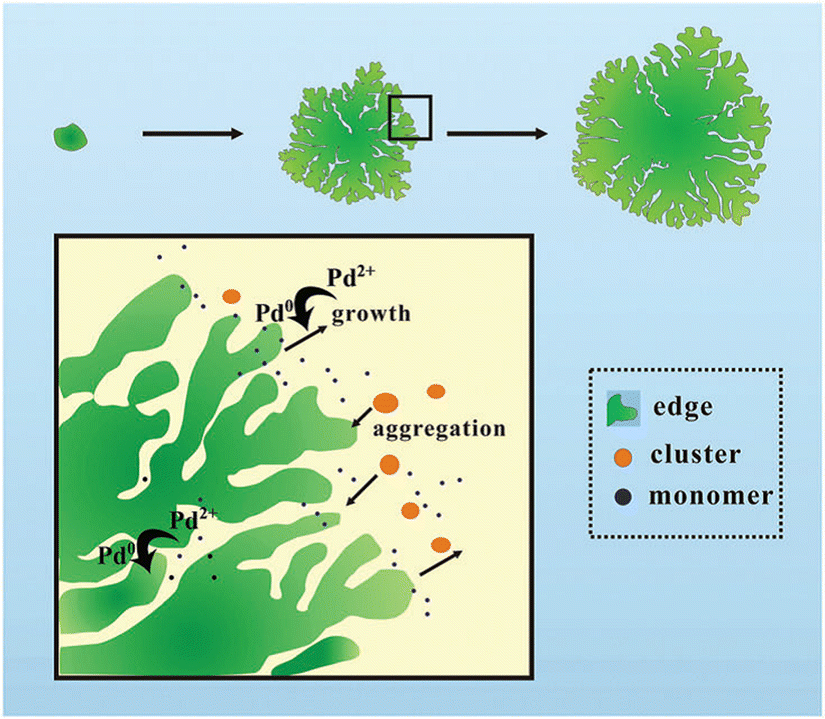 | ||
| Fig. 6 A sketch of the mechanism of the formation of the 2D Pd dendritic nanostructures. Reproduced with permission from ref. 81. Copyright © 2014 The Royal Society of Chemistry. | ||
2.6. Platinum
Due to its outstanding catalytic performance, platinum has received great attention from the research community for a long time. However, the high cost of bulk platinum hinders the large-scale production of Pt-based catalysts and is a trigger for the development of new technologies assuming reduction of the platinum consumption with a simultaneous increase in active surface area. In this context, the implementation of 2D Pt-based technologies is a reliable strategy to meet the above-mentioned challenge.In earlier works, Madey et al.82 and Schröder et al.83 reported the deposition of atomically thin Pt layers on metallic substrates like W and Cu surfaces by the evaporation technique. It was shown that 2D Pt film growth at room temperature proceeds through layer-by-layer growth rather than by island growth mode (at least for the first two monolayers). The detrimental role of the oxygen atmosphere on the stability of the platinum monolayer was underlined.82
As was shown later, the growth of continuous 2D Pt layers on non-metallic substrates (Si(111),84 TiO2-terminated SrTiO3 (001),85 SiO2/Si86) by different deposition techniques (magnetron sputtering and electron beam evaporation) is quite a challenging task due to their unsuitable wettability. Instead, the formation of Pt nanocluster/nanoisland arrays is usually observed. However, since the height of the Pt islands/clusters is controllable (until reaching 0.13 nm84) they are of interest to catalytic applications in their own way. Concomitantly, an ALD technique was found to be the more effective method to forming continuous ultrathin Pt layers on graphene oxide87 and titanium carbide (TiC),88 respectively. For example, the use of MeCpPtMe3 as a Pt precursor and ozone as a co-reactant enabled the formation of stable Pt films with thicknesses ranging from 1 to 6 monolayers.88
Surface-limited redox replacement (SLRR) is another promising approach to form a Pt bilayer or monolayer on graphene89 or gold.90 The core of this method is based on the galvanic displacement on a underpotentially deposited (UPD) Cu layer (sacrificial metal) by the platinum. In contrast, there are a few reports on more economically beneficial galvanic-replacement-free electrochemical deposition of Pt overlayers on Ag nanocrystals91 and 111 textured Au thin films.92
Surfactant-directed solution-phase synthesis was proposed as a facile route to grow two-dimensional ultrathin single-crystalline platinum nanodendrites in aqueous solution under ambient conditions.93 The formation mechanism implies the initial chemical reaction between C22N–COOH (Br−) surfactant that acts as a structure-directing template and facet-capping agent and H2PtCl6 in the presence of ascorbic acid (reducing agent). This reaction is followed by stabilization of the C22N–COOH/PtCl62− lamellar mesophase with subsequent reduction of PtCl62− into 2D Pt nanocrystals (Fig. 7). Although the thickness of these metal nanodendrites is 2.3 nm, there is ample room for their thinning via adjusting the growth conditions.
 | ||
| Fig. 7 The mechanism of the step-by-step in-the-plane epitaxial growth of platinum nanodendrites. Reproduced with permission from ref. 93. Copyright © 2019 American Chemical Society. | ||
A carbon-rich (6√3 × 6√3) R30° surface reconstruction of SiC also called a buffer layer and zero-layer graphene was recently identified as a promising template for growth of 2D Pt (with a thickness of 3–4 Å) by a physical vapor deposition technique.94 Due to the fact that approximately 30% of carbon atoms belonging to the buffer layer are covalently bonded to the silicon atoms of the SiC substrate, the buffer layer is corrugated, acting as a unique sp2–sp3 hybrid platform that is capable of preventing the formation of 3D Pt islands at early growth stages. This arises from the nonuniform potential energy landscape caused by the presence of sp3-bonded carbon atoms.
Masayuki Shirai and their colleagues95,96 proposed that the hydrogen reduction of platinum tetrachloride intercalated between graphite layers can also be used as a possible method for the synthesis of two-dimensional platinum nanosheets (1–3 nm in thickness and 100–500 nm in width) between the graphite layers.
2.7. Gold
According to Fig. 1 the cohesive energy of gold is comparable to that of the Pd. This creates good preconditions for the synthesis of 2D gold nanosheets by using top-down approaches. In particular, two research groups reported the synthesis of free-standing two-dimensional gold membranes using in situ transmission electron microscopy.97,98 One of the approaches suggests the initial welding of two Au nanocrystals followed by the induction of in-plane tensile strain near the grain boundary region.97 This manipulation resulted in the extreme mechanical thinning of the local Au bicrystal region and a final formation of the free-standing 2D Au membrane (Fig. 8). The resulting atomically thick membrane had a simple-hexagonal lattice. A similar approach was based on in situ dealloying of the Au–Ag alloy during the TEM.98 E-beam exposure of the alloy causes much faster sublimation of the Ag atoms from the exposed region compared to Au atoms. As a result, the remaining gold atoms diffuse to vacancy sites and gradually form a hexagonally close-packed Au monolayer.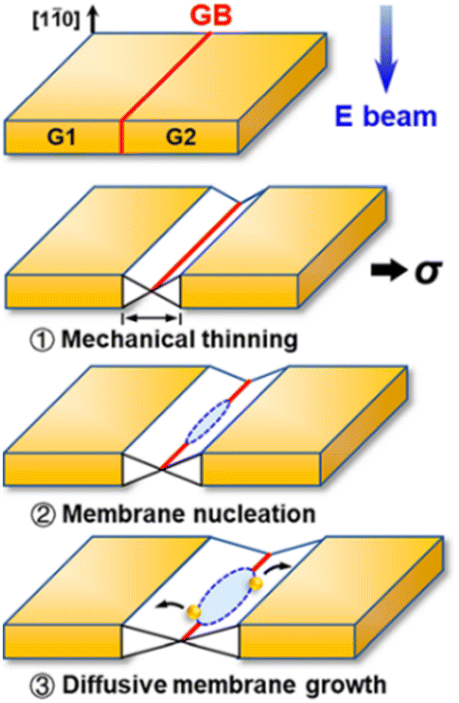 | ||
| Fig. 8 A sketch of the mechanical thinning mechanism of the formation of a 2D gold membrane using in situ TEM. Reproduced with permission from ref. 97. Copyright © 2020 American Chemical Society. | ||
Another type of 2D Au synthesis method involves directly depositing extremely thin gold layers on foreign substates, which makes it possible to facilitate the using of 2D Au for catalytic applications through avoiding process steps related to 2D Au transfer to the preferred substrate. In this regard, some efforts have been dedicated to forming 2D Au on C60 covered HOPG,99 defect-free boron nitride nanotubes (BNNTs),100 Au(111)/HOPG101 and MgO/Ag(001).102 However, despite that in all mentioned cases the obtained 2D Au product is atomically thin, it can be referred to the planar islands or even small 2D clusters rather than to large-area monolayers. It is interesting to note that recent progress in the sublimation growth of high-quality and large-area buffer layers on SiC enabled this platform to be exploited to host the gold atoms.103 More specifically, the annealing of the vapor-deposited gold nanostructures on the buffer layer led to the intercalation of the gold species below the buffer layer, with subsequent conversion of the buffer layer to quasi-free-standing graphene. This enables confining the gold atoms and their self-assembly into a 2D Au layer with semiconducting properties.
Wet chemistry synthesis strategies have also been applied to grow 2D Au nanosheets.104–107 Jeong et al.104 used L-arginine as the reductant and the capping agent to reduce hydrogen tetrachloroaurate trihydrate (HAuCl4·3H2O) at 95 °C for 2 h in order to form single-layered Au nanosheets with a mean lateral size of 11.65 μm. Another reported method dealt with the array of uniform 2D gold clusters formed on Si using colloidal solutions in the presence of liquid CO2.105 A promising wet chemistry method for the facile synthesis of 2D Au nanosheets (only 1–2 atomic layers) using a layered double hydroxide (LDH) template was proposed by Wang et al.106 This method involves the reaction between Au precursor (AuCl4) and Mg/Al-LDH that accompanied the insertion of AuCl4 species between layers of LDH and their reduction by using NaBH4 (Fig. 9).
 | ||
| Fig. 9 A possible growth mechanism of ultra-thin 2D Au nanosheets proposed by Wang et al.106 Reproduced with permission from ref. 106. Copyright © 2015 Macmillan Publishers Limited. | ||
A further example of an aqueous approach is represented by a methyl orange (MO)-mediated synthesis method that exploits aqueous solutions of HAuCl4, Na3C6H5O7 and MO to form highly catalytically active 2D Au nanosheets with a thickness of only 0.47 nm under ambient conditions.107 In this case, methyl orange acts as a confining agent.
2.8. Silver
Historically, the growth of extremely thin silver layers including 2D Ag has been studied over the years. Like other noble metals, vapor-deposited Ag monolayers on different substrates (Ge(111),108 Ru(001),109 Si(111),110 Ni(111),111 W(110),112 Ni(001),113 Cu(001),114 graphite115) were initially in focus. In parallel with this, a set of experiments related to the electrochemical deposition of 2D Ag layers under Langmuir films (negatively charged organic monolayers116–118 or even neutral amphiphilic Schiff bases119) was carried out in the 1990s. In most cases, a 2D growth regime manifests as the formation and evolution of the dendritic silver nanostructures below organic monolayers. It was stressed that the morphology of such nanostructures is defined by a balance between the surface pressure of the Langmuir monolayer, an applied voltage, and a silver ion concentration.More recent approaches utilize organic or non-organic layered templates (redox-active tyrosine-mediated Peptide120 and layered double hydroxide (LDH),121 for example) to synthesize 2D Ag in the interlayer space by using wet chemistry. In the former case, the key step is the binding of silver ions to the peptide at the cysteine sites. As a result, Ag ion-coordinated peptide monomers interact with each other so that the multilayer stacking in the peptide template is formed at the air/water interface through π–π interactions between aromatic side chains (Fig. 10A). At the final stage, the reduced Ag species located between the peptide layers tend to self-assemble into 2D layers. Although further practical application of synthesized 2D Ag seems to be challenging due to the possibly detrimental effects of the organic template on the electronic properties of the metal nanosheets, the authors in ref. 120 argued that their structures possess excellent electrical properties even without the removal of the organic surfactant.
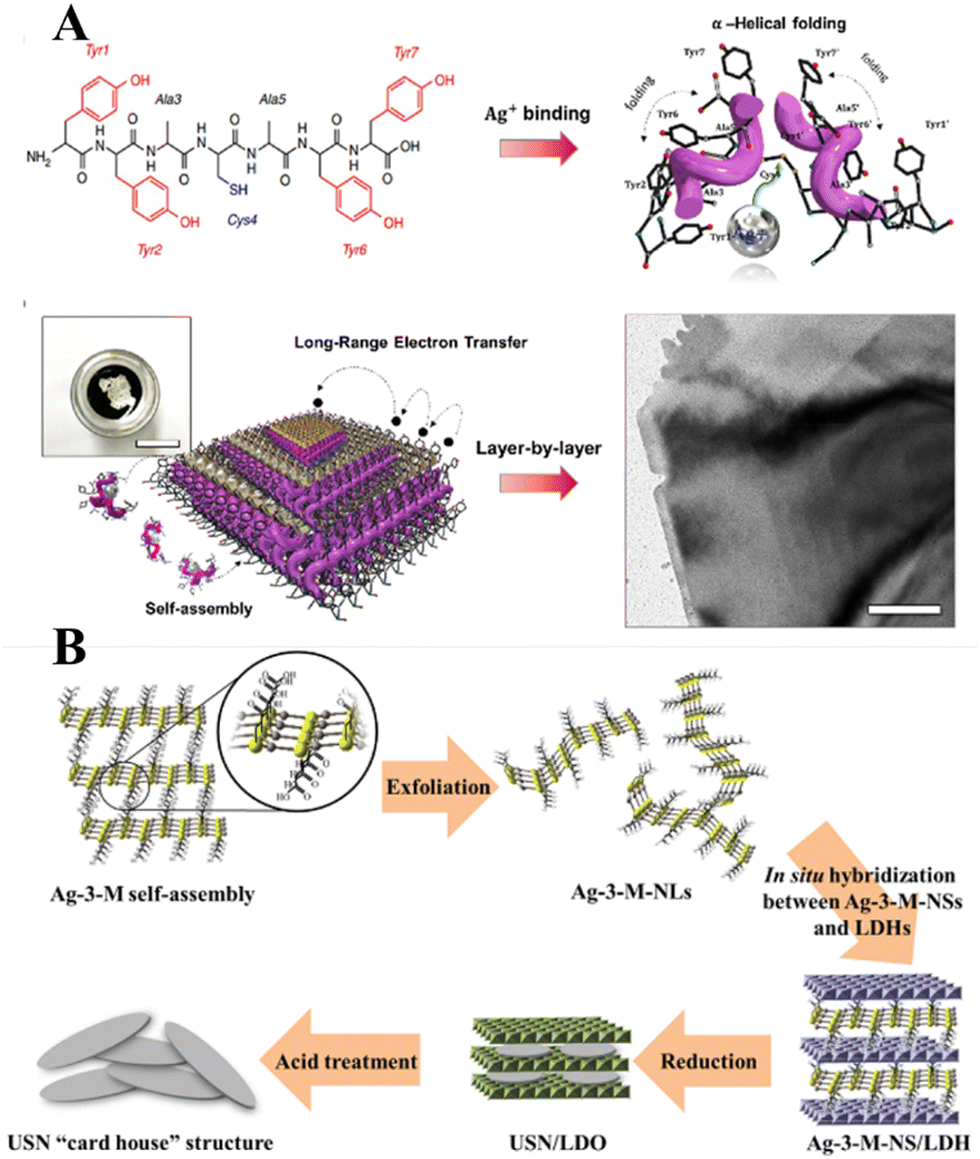 | ||
| Fig. 10 (A) Schematics of the peptide-template-mediated formation of 2D Ag layers.120 Reproduced with permission from ref. 120. Copyright © 2020 American Chemical Society. (B) Illustration of the formation process of single Ag layers within the interlayer space of LDH. Reproduced with permission from ref. 121. Copyright © 2019 American Chemical Society. | ||
The use of a hard inorganic LDH template to provide the conditions for anisotropic growth of 2D Ag is another promising approach. This method suggests exploitation of a 2-dimensional silver carboxylthiolate (Ag-3-M-NS) as the precursor (Fig. 10B). It was shown that the layered structure of LDH acts as an effective platform for interlayer confining of the silver carboxylthiolate and its subsequent reduction to Ag0. It was emphasized that a worthwhile hard inorganic LDH template can be removed via acid treatment while keeping the 2D nature of the silver nanosheets intact.
As was already mentioned, graphenized SiC can be utilized as a host template to accommodate 2D metal layers. In particular, two different cases were considered: (i) the growth of 2D Pt on buffer layer/SiC and (ii) Au intercalation beneath the buffer layer followed by the formation of 2D Au in the space between the decoupled buffer layer and SiC. In contrast, Briggs and colleagues122 have gone further by utilizing defective monolayer epitaxial graphene on SiC to grow the confined 2D Ag layer. In this regard, the defects provide extra intercalation channels, thereby facilitating the penetration of the evaporated Ag species beneath both the topmost graphene layer and the buffer layer, respectively (Fig. 11).
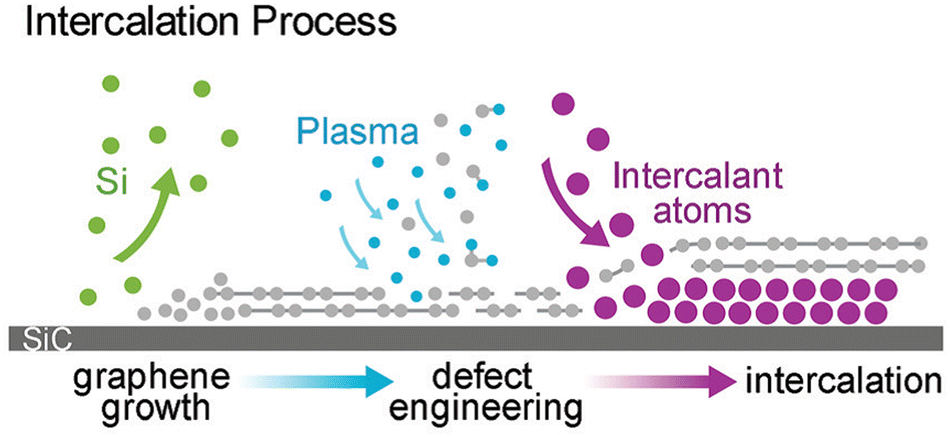 | ||
| Fig. 11 A sketch of the formation of 2D Ag film using epitaxial graphene on SiC as a confining template. Reproduced with permission from ref. 122. Copyright © 2019 The Royal Society of Chemistry. | ||
3. 2D noble metal-based catalysts for the hydrogen evolution reaction
Despite the huge potential of 2D noble metal-based catalysts in the hydrogen evolution reaction, literature analysis has revealed that the HER performance of 2D metals is still relatively understudied. There are only a few experimental studies discussing the benefits of 2D Ru,30,32 2D Ir,40 2D Rh43 and 2D Pt93 as HER electrocatalysts. Chen et al.30 revealed that among different electrodes including Ru nanowires, Ru nanoparticles and commercial Ru electrodes (Fig. 12A and B), 2D Ru nanosheets synthesized by the O2-mediated solution-based colloidal method exhibit the most superior HER catalytic activity (with a Tafel slope of 58.6 mV dec−1) under acidic conditions (H2-saturated 0.1 M HClO4 aqueous solution). This is achieved through favourable combination of electrochemically active specific surface area and Gibbs free energy of the hydrogen adsorption. Improved reaction kinetics arising from the 2D nature of ruthenium was also identified as the main reason of the excellent catalytic properties of 2D Ru formed by a solvothermal approach.32 Possessing a Tafel slope and overpotential to achieve a current density of 10 mA mg−1 that are comparable to those of commercial Pt/C electrodes (46 mV dec−1 and 20 mV vs. 31 mV dec−1 and 5 mV in in 0.5 M H2SO4, respectively), 2D Ru-based electrodes have significant advantages in terms of the metal price, which makes them a nice alternative to platinum (Fig. 12C and D). Based on the analysis of the Tafel slope, it was suggested the Volmer–Heyrovsky mechanism dominates the HER mechanism for 2D Ru. This is in contrast to the Pt electrocatalyst, for which the HER follows the Volmer–Tafel mechanism. For 2D Ru, the rate of the HER was limited by the desorption process.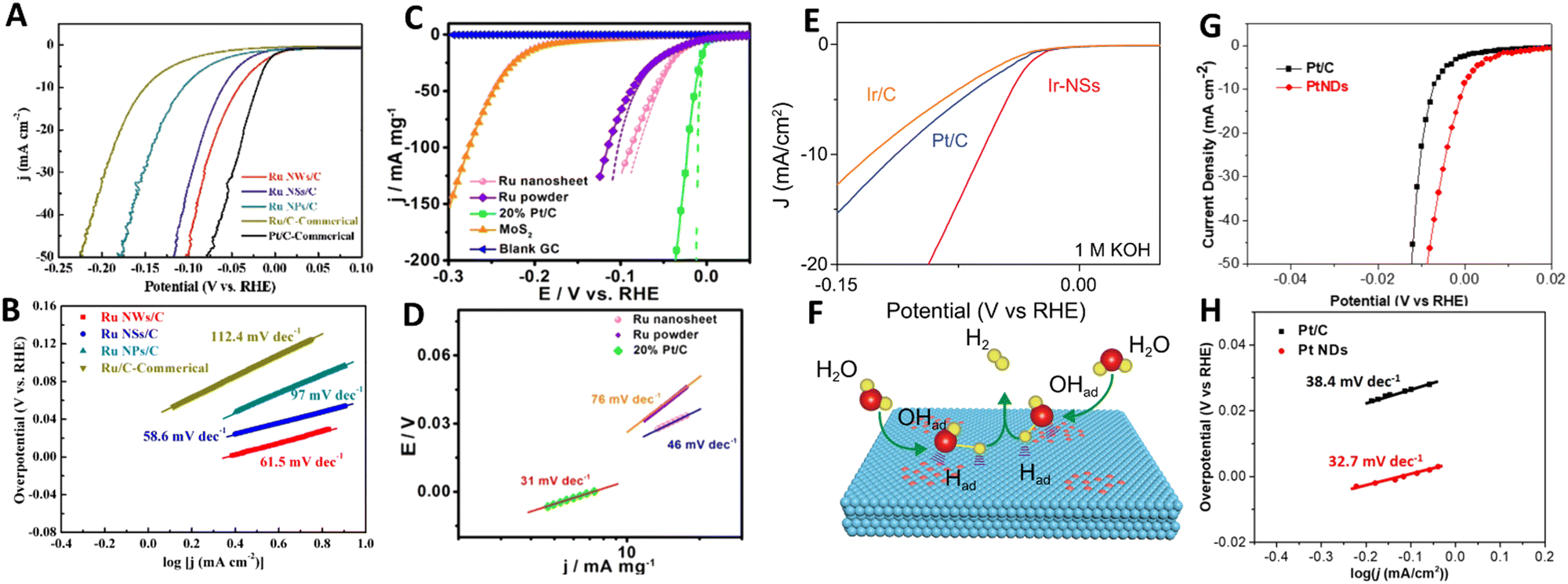 | ||
| Fig. 12 (A) Linear sweep voltammetry (LSV) curves and (B) Tafel plots of Ru nanosheets for the HER in 0.1 m HClO4 solution. Reproduced with permission from ref. 30. Copyright © 2020 Wiley-VCH GmbH. (C) LSV curves and (D) Tafel plots of Ru nanosheets for the HER in 0.5 M H2SO4 electrolyte with pH = 0. Reproduced with permission from ref. 32. Copyright © 2016 American Chemical Society. (E) Polarization curves of Ir nanosheets for the HER in 1 M KOH and (F) an illustration of the HER mechanism for Ir nanosheets. Reproduced with permission from ref. 40. Copyright © Copyright The Author(s) 2020. (G) LSV curves and (H) Tafel Plots of ultrathin Pt nanodendrites. Reproduced with permission from ref. 93. Copyright © 2019 American Chemical Society. | ||
Switching to other noble metals, it is worth noting that the electrochemical electrodes based on partially hydroxylated 2D iridium nanosheets show an outstanding HER catalytic performance under both acidic (0.5 M H2SO4) and alkaline conditions (in 1 M KOH) manifested as the 50 mV-improvement in overpotential (at 10 mA cm−2) compared to commercial Pt/C and Ir/C electrodes (Fig. 12E).40 Those authors underlined the critically important role of the partial hydroxylation in the reaction kinetics at the 2D Ir surface. Under acidic conditions H* adsorption mainly occurs at the pristine 2D Ir surface, however, hydroxyl groups on an Ir surface favour H2O adsorption under alkaline conditions (Fig. 12F). Rhodium is another prominent representative of the catalytically active noble metal groups. Specifically, a unique 2D structure of rhodium nanosheets functionalized by polyallylamine and supported by CNT offers excellent conditions (large electrochemically active specific surface area, good stability, fast electron transport, etc) for the hydrogen evolution reaction in acidic medium (N2-saturated 0.5 M H2SO4).43 A comparison of the catalytic characteristics of commercial Pt nanocrystals and the synthesized nanosheets is in favor of the latter. In this case, 2D Rh exhibited a lower onset potential compared to that of Pt nanocrystals (−1 mV vs. 10.4 mV, respectively) and a similar Tafel slope (30 mV dec−1vs. 31 mV dec−1). The latter also highlights that the HER at 2D Rh proceeds in a manner similar to that of Pt, with a dominating Tafel reaction mechanism. Two main reasons for the improved HER catalytic activity were singled out: (i) the 2D nature of rhodium nanosheets and (ii) the proton enrichment at the electrode/electrolyte interface due to functionalization of the Rh nanosheets with polyallylamine.
A final example of the 2D-metal-based hydrogen electrocatalyst concerns the platinum that is regarded as a “gold standard” metal for the HER. A transition from bulk Pt to 2D Pt is accompanied by changes in the electrochemical activity towards the HER under acidic conditions.93 This is mainly caused by an increase in the electrocatalytic active surface area of Pt from 46.4 m2 g−1 for commercial Pt/C to 51.4 m2 g−1 for 2D Pt nanosheets. As a direct consequence of that, 2D Pt demonstrates a smaller overpotential of <0.01 V at a current density of 50 mA cm−2 and lower Tafel slope (32.7 mV dec−1) compared to Pt/C, indicating its better electrochemical activity and faster reaction kinetics (Fig. 12G and H). This is however possible not only due to the extraordinary internal properties of 2D Pt, but also due to the presence of the remaining surfactant-related chemical groups at the 2D Pt surface which may alter the hydrogen proton adsorption.
4. Concluding remarks
The current status of research investigations regarding 2D noble metals has been highlighted. Special focus was placed on the synthesis methods and applications of 2D noble metals for the hydrogen evolution reaction. Although the anisotropic growth of 2D metals is a technologically intricate process, which is mainly due to the large surface energy of most metals, a kinetic control approach offers a promising route to synthesize extremely thin 2D metal nanosheets. However, since in most cases the kinetically controlled growth methods require the presence of the surfactant or capping agents the quality of the resulting 2D metal nanosheets suffers from the remaining functional groups on their surfaces that can affect the catalytic performance of 2D metal in unexpected ways. This explains the research efforts to develop surfactant-free and capping agent-free synthesis methods. In line with this, one-step template-assisted growth of 2D metals by using deposition techniques is of special interest. An appropriate choice of substrate and a fine control of the synthesis conditions (precursor type, temperature, time, pressure etc.) are keys to ensure a 2D growth regime. Although a traditional way to produce metal monolayers implies a pseudomorphic epitaxy on other metal surfaces, more recent approaches include the formation of 2D metals on non-metallic substrates like graphene, graphite, graphenized SiC, etc. The latter one is considered to be a rich platform to host vapor-deposited 2D metal layers atop the surface and to confine them by employing the intercalation process.2D noble metals are characterized by a high density of coordinatively unsaturated metal sites and large surface area, which make them excellent electrocatalysts for the hydrogen evolution reaction. Even though there are not much experimental data on the catalytic activity of 2D metals towards HERs, the existing results suggest that with appropriate design of selected 2D metals the HER performance can be even better than that of commercial Pt electrodes. This provides both experimentalists and theoreticians with an inspirational framework for further application-focused investigations of 2D metal-based electrocatalysis and more sophisticated design of 2D noble metals.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The author acknowledges the support from Ångpanneföreningens Forskningsstiftelse (Grant 21-112).References
- Hydrogen's growing role in sustainable energy systems, Research*eu, European Commission, 2020, 94, 1-12. https://cordis.europa.eu/article/id/421533-hydrogens-growing-role-in-sustainable-energy-systems.
- S. van Renssen, Nat. Clim. Chang., 2020, 10, 799–801 CrossRef.
- Towards Fossil-Free Energy In 2050, European Climate Foundation, 2019 Search PubMed.
- R. Moradi and K. M. Groth, Int. J. Hydrogen Energy, 2019, 44(23), 12254–12269 CrossRef CAS.
- Y. Huang, Y. Cheng and J. Zhang, Ind. Eng. Chem. Res., 2021, 60(7), 2737–2771 CrossRef CAS.
- J. Zhu, L. Hu, P. Zhao, L. Yoon Suk Lee and K.-Y. Wong, Chem. Rev., 2020, 120(2), 851–918 CrossRef CAS PubMed.
- M. Ďurovič, J. Hnát and K. Bouzek, J. Power Sources, 2021, 493, 229708 CrossRef.
- Z. Li, R. Ge, J. Su and L. Chen, Adv. Mater. Interfaces, 2020, 7, 2000396 CrossRef CAS.
- S. Bai, M. Yang, J. Jiang, X. He, J. Zou, Z. Xiong, G. Liao and S. Liu, npj 2D Mater Appl., 2021, 5, 78 CrossRef CAS.
- J. Zheng, X. Sun, C. Qiu, Y. Yan, Z. Yao, S. Deng, X. Zhong, G. Zhuang, Z. Wei and J. Wang, J. Phys. Chem. C, 2020, 124(25), 13695–13705 CrossRef CAS.
- S. Wang, A. Lu and C. J. Zhong, Nano Convergence, 2021, 8, 4 CrossRef CAS PubMed.
- C. C. L. McCrory, S. Jung, I. M. Ferrer, S. M. Chatman, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2015, 137(13), 4347–4357 CrossRef CAS PubMed.
- T. Mallouk, Nat. Chem., 2013, 5, 362–363 CrossRef CAS PubMed.
- C. Li and J.-B. Baek, ACS Omega, 2020, 5(1), 31–40 CrossRef CAS PubMed.
- J. K. Nørskov, T. Bligaard, A. Logadottir, J. R. Kitchin, J. G. Chen, S. Pandelov and U. Stimming, J. Electrochem. Soc., 2005, 152(2), J23–J26 CrossRef.
- Communication from the Commission to the European Parliament, The Council, the European Economic and Social Committee and the Committee of the Regions – Critical Raw Materials Resilience: Charting a Path towards Greater Security and Sustainability, 2020.
- L. M. Salonen, D. Y. Petrovykh and Yu. V. Kolen'ko, Mater. Today Sustainability, 2021, 11–12, 100060 CrossRef.
- S. Zhang, X. Zhang, Y. Rui, R. Wang and X. Li, Green Energy Environ., 2021, 6(4), 458–478 CrossRef CAS.
- M. Datt Bhatt and J. Yong Lee, Energy Fuels, 2020, 34(6), 6634–6695 CrossRef.
- Y. Chen, K. Yang, B. Jiang, J. Li, M. Zeng and L. Fu, J. Mater. Chem. A, 2017, 5, 8187–8208 RSC.
- W. Moschkowitsch, O. Lori and L. Elbaz, ACS Catal., 2022, 12(2), 1082–1089 CrossRef CAS.
- S. Zhang, X. Zhang, Y. Rui, R. Wang and X. Li, Green Energy Environ., 2021, 6(4), 458–478 CrossRef CAS.
- Q. Hu, K. Gao, X. Wang, H. Zheng, J. Cao, L. Mi, Q. Huo, H. Yang, J. Liu and C. He, Nat. Commun., 2022, 13, 3958 CrossRef CAS PubMed.
- E. Toyoda, R. Jinnouchi, T. Hatanaka, Y. Morimoto, K. Mitsuhara, A. Visikovskiy and Y. Kido, J. Phys. Chem. C, 2011, 115(43), 21236–21240 CrossRef CAS.
- A. Visikovskiy, H. Matsumoto, K. Mitsuhara, T. Nakada, T. Akita and Y. Kido, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 165428 CrossRef.
- J. Nevalaita and P. Koskinen, Phys. Rev. B: Condens. Matter Mater. Phys., 2018, 97, 035411 CrossRef CAS.
- Y. Chen, Z. Fan, Z. Zhang, W. Niu, C. Li, N. Yang, B. Chen and H. Zhang, Chem. Rev., 2018, 118, 6409–6455 CrossRef CAS PubMed.
- D. Bonn, J. Eggers, J. Indekeu, J. Meunier and E. Rolley, Rev. Mod. Phys., 2009, 81, 739 CrossRef CAS.
- C. Kittel, Introduction to Solid State Physics, John Wiley & Sons, Inc., Hoboken, NJ, 8th edn, 2005, p. 50 Search PubMed.
- Y. B. He, M. Knapp, E. Lundgren and H. Over, J. Phys. Chem. B, 2005, 109, 21825 CrossRef CAS PubMed.
- L. Chen, Y. Li and X. Liang, Adv. Funct. Mater., 2021, 31, 2007344 CrossRef CAS.
- X. Kong, K. Xu, C. Zhang, J. Dai, S. N. Oliaee, L. Li, X. Zeng, C. Wu and Z. Peng, ACS Catal., 2016, 6, 1487–1492 CrossRef CAS.
- K. Gregorczyk, P. Banerjee and G. W. Rubloff, Mater. Lett., 2012, 73, 43–46 CrossRef CAS.
- O. Mann, W. Freyland, O. Raz and Y. Ein-Eli, Chem. Phys. Lett., 2008, 460, 178–181 CrossRef CAS.
- Y. B. He, A. Goriachko, C. Korte, A. Farkas, G. Mellau, P. Dudin, L. Gregoratti, A. Barinov, M. Kiskinova, A. Stierle, N. Kasper, S. Bajt and H. Over, J. Phys. Chem. C, 2007, 111, 10988–10992 CrossRef CAS.
- I. Shtepliuk and R. Yakimova, RSC Adv., 2023, 13, 1125–1136 RSC.
- K. Fukuda, J. Sato, T. Saida, W. Sugimoto, Y. Ebina, T. Shibata, M. Osada and T. Sasaki, Inorg. Chem., 2013, 52, 2280–2282 CrossRef CAS PubMed.
- N. Saadatjou, A. Jafari and S. Sahebdelfar, Chem. Eng. Commun., 2015, 202(4), 420–448 CrossRef CAS.
- I. Rossetti, N. Pernicone, F. Ferrero and L. Forni, Ind. Eng. Chem. Res., 2006, 45(12), 4150–4155 CrossRef CAS.
- Z. Cheng, B. Huang, Y. Pi, L. Li, Q. Shao and X. Huang, Natl. Sci. Rev., 2020, 7, 1340–1348 CrossRef CAS PubMed.
- B. Fang, C. Yang, C. Pang, W. Shen, X. Zhang, Y. Zhang, W. Yuan and X. Liu, Appl. Phys. Lett., 2017, 110, 141103 CrossRef.
- J. Yin, J. Wang, Y. Ma, J. Yu, J. Zhou and Z. Fan, ACS Materials Lett., 2021, 3, 121–133 CrossRef CAS.
- J. Bai, S.-H. Xing, Y.-Y. Zhu, J.-X. Jiang, J.-H. Zeng, Y. Chen and J. Power, Sources, 2018, 385, 32–38 CrossRef CAS.
- J. Bai, G.-R. Xu, S.-H. Xing, J.-H. Zeng, J.-X. Jiang and Y. Chen, ACS Appl. Mater. Interfaces, 2016, 8, 33635–33641 CrossRef CAS PubMed.
- H. Duan, N. Yan, R. Yu, C.-R. Chang, G. Zhou, H.-S. Hu, H. Rong, Z. Niu, J. Mao, H. Asakura, T. Tanaka, P. J. Dyson, J. Li and Y. Li, Nat. Commun., 2014, 5, 3093 CrossRef PubMed.
- H.-M. Liu, S.-H. Han, Y. Zhao, Y.-Y. Zhu, X.-L. Tian, J.-H. Zeng, J.-X. Jiang, B. Y. Xia and Y. Chen, J. Mater. Chem. A, 2018, 6, 3211 RSC.
- K. Jang, H. J. Kim and S. Uk Son, Chem. Mater., 2010, 22(4), 1273–1275 CrossRef CAS.
- Y. He, A. P. Seitsonen and H. Over, J. Chem. Phys., 2006, 124, 034706 CrossRef PubMed.
- C. E. Housecroft and A. G. Sharpe, Inorganic Chemistry, Prentice Hall, 2nd edn, 2004,710, 671–673 Search PubMed.
- J. Quinson, Open Research Europe, 2022, 2, 39 Search PubMed.
- A. Ignaszak and E. Gyenge, Electrochim. Acta, 2013, 95, 268–274 CrossRef CAS.
- J. Hämäläinen, T. Sajavaara, E. Puukilainen, M. Ritala and M. Leskelä, Chem. Mater., 2012, 24(1), 55–60 CrossRef.
- Y. Chi, H.-L. Yu, W.-L. Ching, C.-S. Liu, Y.-L. Chen, T.-Y. Chou, S.-M. Peng and G.-H. Lee, J. Mater. Chem., 2002, 12, 1363–1369 RSC.
- Y. Senzaki, W. L. Gladfelter and F. B. McCormick, Chem. Mater., 1993, 5(12), 1715–1721 CrossRef CAS.
- C. Li, W. K. Leong and K. P. Loh, Appl. Organometal. Chem., 2009, 23(5), 196–199 CrossRef CAS.
- S. L. Li, C. Y. Ma, Q. Y. Zhang, X. P. Liu, C. Zhang and Z. Yi, Surf. Coat. Technol., 2015, 282, 1–5 CrossRef CAS.
- J. A. Elias and E. A. Henriksen, Ann. Phys., 2020, 532, 1900294 CrossRef CAS.
- I. Shtepliuk and R. Yakimova, Phys. Chem. Chem. Phys., 2018, 20, 21528–21543 RSC.
- W. Gong, W. Zhang, C. Ren, X. Ke, S. Wang, P. Huai, W. Zhang and Z. Zhu, Appl. Phys. Lett., 2013, 103, 143107 CrossRef.
- S. M. Elgengehi, S. El-Taher, M. A. A. Ibrahim, J. K. Desmarais and K. E. El-Kelany, Appl. Surf. Sci., 2020, 507, 145038 CrossRef CAS.
- B. Prick and K. Jacobi, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 4408 CrossRef PubMed.
- M. Han, P. Mrozek and A. Wieckowski, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 48, 8329 CrossRef CAS PubMed.
- S. Terada, T. Yokoyama, N. Saito, Y. Okamoto and T. Ohta, Surf. Sci., 1999, 433–435, 657–660 CrossRef CAS.
- J. M. Heitzinger, S. C. Gebhard and B. E. Koel, Surf. Sci., 1992, 275, 209–222 CrossRef CAS.
- J. M. Heitzinger, S. C. Gebhard, D. H. Parker and B. E. Koel, Surf. Sci., 1992, 260, 151–162 CrossRef CAS.
- A. Sellidj and B. E. Koel, Surf. Sci., 1993, 284, 139–153 CrossRef CAS.
- H. A. Etman, Z. V. Zheleva, G. Held and R. A. Bennett, J. Phys. Chem. C, 2011, 115, 4191–4199 CrossRef CAS.
- E. De Biasi, A. de Siervo, F. Garcia, F. Vicentin, R. Landers and M. Knobel, J. Electron Spectrosc. Relat. Phenom., 2007, 156–158, 332–335 CrossRef CAS.
- M. Z. Yazdan-Abad, M. Noroozifar and N. Alfi, J. Colloid Interface Sci., 2018, 532, 485–490 CrossRef CAS PubMed.
- C. He, J. Tao and P. Kang Shen, ACS Catal., 2018, 8, 910–919 CrossRef CAS.
- X. Yin, X. Liu, Y.-T. Pan, K. A. Walsh and H. Yang, Nano Lett., 2014, 14, 7188–7194 CrossRef CAS PubMed.
- H. Li, G. Chen, H. Yang, X. Wang, J. Liang, P. Liu, M. Chen and N. Zheng, Angew. Chem., Int. Ed., 2013, 52, 8368–8372 CrossRef CAS PubMed.
- P. F. Siril, L. Ramos, P. Beaunier, P. Archirel, A. Etcheberry and H. Remita, Chem. Mater., 2009, 21, 5170–5175 CrossRef CAS.
- A. Gil, A. Clotet, J. M. Ricart, F. Illas, B. Álvarez, A. Rodes and J. Miguel Feliu, J. Phys. Chem. B, 2001, 105(30), 7263–7271 CrossRef CAS.
- Y. Su, E. Prestat, C. Hu, V. K. Puthiyapura, M. Neek-Amal, H. Xiao, K. Huang, V. G. Kravets, S. J. Haigh, C. Hardacre, F. M. Peeters and R. R. Nair, Nano Lett., 2019, 19, 4678–4683 CrossRef CAS PubMed.
- C. Lebouin, Y. S. Olivier, E. Sibert, P. Millet, M. Maret and R. Faure, J. Electroanal. Chem., 2009, 626, 59–65 CrossRef CAS.
- J. Walter and H. Shioyama, Phys. Lett. A, 1999, 254(1–2), 65–71 CrossRef CAS.
- J. Walter, Philos. Mag. Lett., 2000, 80(4), 257–262 CrossRef CAS.
- J. Walter, J. Heiermann, G. Dyker, S. Hara and H. Shioyama, J. Catal., 2000, 189(2), 449–455 CrossRef CAS.
- D. Mendoza, F. Morales, R. Escudero and J. Walter, J. Phys.: Condens. Matter, 1999, 11, L317 CrossRef CAS.
- G. Zhu, Y. Jiang, F. Lin, H. Zhang, C. Jin, J. Yuan, D. Yang and Z. Zhang, Chem. Commun., 2014, 50, 9447 RSC.
- T. E. Madey, K.-J. Song, C.-Z. Dong and R. A. Demmin, Surf. Sci., 1991, 247(2–3), 175–187 CrossRef CAS.
- U. Schröder, R. Linke, J.-H. Boo and K. Wandelt, Surf. Sci., 1996, 357–358, 873–878 CrossRef.
- H. Yasumatsu, T. Hayakawa, S. Koizumi and T. Kondowa, J. Chem. Phys., 2005, 123, 124709 CrossRef PubMed.
- W. Guo, A. B. Posadas and A. A. Demkov, J. Vac. Sci. Technol., B, 2017, 35, 061203 CrossRef.
- R. N. Esfahani, G. Jordan Maclay and G. W. Zajac, Thin Solid Films, 1992, 219(257–265), 257 CrossRef CAS.
- B. Karasulu, R. H. J. Vervuurt, W. M. M. Kessels and A. A. Bol, Nanoscale, 2016, 8, 19829–19845 RSC.
- D. Göhl, H. Rueß, S. Schlicht, A. Vogel, M. Rohwerder, K. J. J. Mayrhofer, J. Bachmann, Y. Román-Leshkov, J. M. Schneider and M. Ledendecker, ChemElectroChem, 2020, 7, 2404 CrossRef.
- A. W. Robertson, G.-D. Lee, S. Lee, P. Buntin, M. Drexler, A. A. Abdelhafiz, E. Yoon, J. H. Warner and F. M. Alamgir, ACS Nano, 2019, 13, 12162–12170 CrossRef CAS PubMed.
- M. Hakamada, S. Sakakibara, N. Miyazawa, S. Deguchi and M. Mabuchi, Sci. Rep., 2020, 10, 9594 CrossRef PubMed.
- H. Liu, P. Zhong, K. Liu, L. Han, H. Zheng, Y. Yin and C. Gao, Chem. Sci., 2018, 9, 398 RSC.
- S. H. Ahn, Y. Liu and T. P. Moffat, ACS Catal., 2015, 5, 2124–2136 CrossRef CAS.
- D. Xu, H. Lv, H. Jin, Y. Liu, Y. Ma, M. Han, J. Bao and B. Liu, J. Phys. Chem. Lett., 2019, 10, 663–671 CrossRef PubMed.
- K. Ho Kim, H. He, M. Rodner, R. Yakimova, K. Larsson, M. Piantek, D. Serrate, A. Zakharov, S. Kubatkin, J. Eriksson and S. Lara-Avila, Adv. Mater. Interfaces, 2020, 7, 1902104 CrossRef.
- M. Shirai, Chem. Rec., 2019, 19, 1263–1271 CrossRef CAS PubMed.
- M. Shirai, K. Kubo, M. Sodeno and H. Nanao, Chem. – Asian J., 2021, 16, 2035–2040 CrossRef CAS PubMed.
- Q. Zhu, Y. Hong, G. Cao, Y. Zhang, X. Zhang, K. Du, Z. Zhang, T. Zhu and J. Wang, ACS Nano, 2020, 14, 17091–17099 CrossRef CAS PubMed.
- X. Wang, C. Wang, C. Chen, H. Duan and K. Du, Nano Lett., 2019, 19, 4560–4566 CrossRef CAS PubMed.
- L. Guo, Y. Wang, D.-L. Bao, H.-H. Jia, Z. Wang, S. Du and Q. Guo, Nanoscale, 2020, 12, 21657 RSC.
- S. Bhandari, B. Hao, K. Waters, C. H. Lee, J.-C. Idrobo, D. Zhang, R. Pandey and Y. K. Yap, ACS Nano, 2019, 13, 4347–4353 CrossRef CAS PubMed.
- M. R. Fard and Q. Guo, J. Phys. Chem. C, 2018, 122, 7801–7805 CrossRef.
- C. Stiehler, Y. Pan, W.-D. Schneider, P. Koskinen, H. Häkkinen, N. Nilius and H.-J. Freund, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 88, 115415 CrossRef.
- S. Forti, S. Link, A. Stöhr, Y. Niu, A. A. Zakharov, C. Coletti and U. Starke, Nat. Commun., 2020, 11, 2236 CrossRef CAS PubMed.
- E. J. Jeong, E. Im, D. C. Hyun, J. W. Leed and G. D. Moon, J. Ind. Eng. Chem., 2020, 89, 204–211 CrossRef CAS.
- J. S. Wang, C. M. Wai, G. J. Brown and S. D. Apt, Langmuir, 2016, 32, 4635–4642 CrossRef CAS PubMed.
- L. Wang, Y. Zhu, J.-Q. Wang, F. Liu, J. Huang, X. Meng, J.-M. Basset, Y. Han and F.-S. Xiao, Nat. Commun., 2015, 6, 6957 CrossRef CAS PubMed.
- S. Ye, A. P. Brown, A. C. Stammers, N. H. Thomson, J. Wen, L. Roach, R. J. Bushby, P. L. Coletta, K. Critchley, S. D. Connell, A. F. Markham, R. Brydson and S. D. Evans, Adv. Sci., 2019, 6, 1900911 CrossRef CAS PubMed.
- G. Le Lay, R. L. Johnson, R. Seemann, F. Grey, R. Feidenhans'l and M. Nielsen, Surf. Sci., 1993, 287–288(1), 539–544 CrossRef CAS.
- K. Wandelt, K. Markert, P. Dolle, A. JabŁonski and J. J. Niemantsverdriet, Surf. Sci. Lett., 1987, 189–190, A396 CrossRef.
- K. Spiegel, Surf. Sci., 1967, 7(2), 125–142 CrossRef CAS.
- S. Mróz, Z. Jankowski and M. Nowicki, Surf. Sci., 2000, 454–456, 702–706 CrossRef.
- A. Elbe, G. Meister and A. Goldmann, Surf. Sci., 1998, 397, 346–353 CrossRef CAS.
- S. Mróz and Z. Jankowski, Surf. Sci., 1996, 349, 111–118 CrossRef.
- M. Nowicki and S. Mróz, Vacuum, 1996, 47(5), 445–449 CrossRef CAS.
- F. Patthey and W.-D. Schneider, Surf. Sci., 1995, 334, L715–L718 CrossRef CAS.
- S. Ravaine, C. Breton, C. Mingotaud and F. Argoul, Mater. Sci. Eng., C, 1999, 8–9, 437–444 CrossRef.
- G. P. Luo, Z. M. Ai, Z. H. Lu and Y. Wei, Phys. Rev. E, 1994, 50, 409 CrossRef CAS PubMed.
- G. Luo, Z. Al and Y. We, Chin. Phys. Lett., 1994, 11, 6341 Search PubMed.
- Z. Tai, G. Zhang and X. Qian, Langmuir, 1993, 9, 1601–1603 CrossRef CAS.
- H. Lee, H.-S. Jang, D.-H. Cho, J. Lee, B. Seong, G. Kang, Y.-S. Park, K. T. Nam, Y.-S. Lee and D. Byun, ACS Nano, 2020, 14, 1738–1744 CrossRef CAS PubMed.
- G.-H. Gwak, M.-K. Kim, W.-J. Lee, D.-G. Jeung, J. Kuen Park, S.-M. Paek and J.-M. Oh, Inorg. Chem., 2020, 59, 2163–2170 CrossRef CAS PubMed.
- N. Briggs, Z. M. Gebeyehu, A. Vera, T. Zhao, K. Wang, A. De La Fuente Duran, B. Bersch, T. Bowen, K. L. Knappenberger, Jr. and J. A. Robinson, Nanoscale, 2019, 11, 15440 RSC.
| This journal is © the Owner Societies 2023 |