
Role of polyoxometalate precursors and supports in the selective oxidation of methane into formaldehyde using supported metal oxide subnanocluster catalysts†
Keiju
Wachi
,
Tomohiro
Yabe
 *,
Takaaki
Suzuki
,
Kentaro
Yonesato
,
Kosuke
Suzuki
and
Kazuya
Yamaguchi
*
*,
Takaaki
Suzuki
,
Kentaro
Yonesato
,
Kosuke
Suzuki
and
Kazuya
Yamaguchi
*
Department of Applied Chemistry, School of Engineering, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-8656, Japan. E-mail: tyabe@g.ecc.u-tokyo.ac.jp; kyama@appchem.t.u-tokyo.ac.jp
First published on 10th July 2023
Abstract
The direct synthesis of useful chemicals from methane (CH4) is desirable; however, the products are prone to nonselective overoxidation, leading to the formation of CO2. A previous study developed a supported iron oxide subnanocluster catalyst with high thermal stability using iron-containing polyoxometalates (POMs) as precursors to selectively produce formaldehyde (HCHO) and CO. Herein, we investigated various supported POM-based catalysts to further improve the selectivity to HCHO via CH4 oxidation, specifically by suppressing the pyrolysis and overoxidation of HCHO. After examining various metal-containing POM precursors and supports, we found that catalysts prepared using mononuclear- and dinuclear-iron-containing POM precursors supported on SiO2 with a high specific surface area were effective and yielded relatively high quantities of HCHO. In situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) measurements under HCHO flow demonstrated that the pyrolysis and oxidation of HCHO were suppressed on SiO2, while the pyrolysis of HCHO was promoted on Al2O3. Furthermore, in situ DRIFTS measurements conducted at different temperatures revealed that HCHO was not decomposed or oxidized at 500 °C in the absence of catalysts.
Introduction
Methane (CH4), the main component of natural gas, is abundantly available worldwide, including in unconventional resources, such as biogas (including bio-methanation gas)1–3 and CH4 hydrates.4 Furthermore, the production of carbon-neutral CH4, such as in power-to-gas technology, is expected to increase in the future. This production involves synthesizing CH4via methanation processes that utilize green hydrogen produced from renewable energy sources, such as water electrolysis and CO2 emitted from power plants and factories.5–7 Thus, CH4 is gaining attention as a major energy resource and a promising chemical raw material.8–11 However, CH4, with its tetrahedral shape and four identical C–H bonds, has the highest C–H bond energy (439 kJ mol−1) among sp3-hybridized hydrocarbons and is known as the least reactive alkane, making it very challenging to chemically convert into useful compounds.12There are two methods for CH4 conversion: indirect conversion via synthesis gas (syngas) and direct conversion to useful compounds, such as formaldehyde (HCHO), in one step.13 Indirect conversion involves a syngas process, which is a high-temperature and high-pressure process that requires complicated and expensive equipment and significant energy consumption. Therefore, an economically viable direct conversion process of CH4 into high-value-added chemicals is desired. One approach to economically achieve direct CH4 conversion is to selectively oxidize CH4 to oxygenates using O2. However, the target products are susceptible to thermal decomposition or sequential oxidation under the harsh oxidation conditions required for CH4 activation. Therefore, developing a catalytic process that selectively synthesizes the desired products while suppressing sequential reactions is an urgent challenge for ensuring the stable supply of CH4 oxidation-derived chemicals.
Catalysts for synthesizing HCHO from CH4 have been developed since the 1980s, and various types of catalyst systems have been proposed, such as oxide catalysts,14,15 supported catalysts,16–19 and zeolite catalysts.20,21 Commonly investigated oxide catalysts include molybdenum,16,17,19,22 vanadium,16,23 iron,24 copper,22,25 cobalt,21 and tungsten.14,26 However, bulk oxide and large nanoparticle catalysts have demonstrated relatively low selectivity to HCHO. For example, large nanoparticles of VOx on the VOx/Al2O3 catalyst reduced both CH4 conversion and HCHO selectivity in CH4 oxidation.23 Additionally, Zhang et al. reported that in molybdenum oxide catalysts, CH4 conversion and HCHO selectivity were related to the density of Mo![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds; namely, MoO bonds on Zr(MoO4)2 were responsible for HCHO production, whereas excess lattice oxygen and bulk MoO3 caused overoxidation of HCHO.27 In the case of iron oxide catalysts, isolated active lattice oxygen atoms on the surface of iron oxides helped to suppress overoxidation during CH4 oxidation.28 Thus, supported metal oxide nanocluster catalysts have attracted attention, with reported examples including supported CuOx,29 FeOx,30,31 MoOx,32 and VOx nanocluster catalysts.33
O bonds; namely, MoO bonds on Zr(MoO4)2 were responsible for HCHO production, whereas excess lattice oxygen and bulk MoO3 caused overoxidation of HCHO.27 In the case of iron oxide catalysts, isolated active lattice oxygen atoms on the surface of iron oxides helped to suppress overoxidation during CH4 oxidation.28 Thus, supported metal oxide nanocluster catalysts have attracted attention, with reported examples including supported CuOx,29 FeOx,30,31 MoOx,32 and VOx nanocluster catalysts.33
The choice of catalyst supports is also significant in selective CH4 oxidation, and various oxide supports have been investigated, with SiO2 being recognized as the most suitable support. Kobayashi et al. discovered that highly dispersed Fe3+ species on SiO2 with isolated tetrahedrally coordinated Fe–O species significantly promoted selective HCHO production.34 Although detailed mechanistic studies are still in progress, it is evident that highly dispersed active sites in FeOx nanoclusters on SiO2 are essential for achieving selective HCHO production. Similarly, there is ongoing debate regarding the reaction mechanism of pyrolysis and overoxidation on SiO2 and other supports.
A polyoxometalate (POM) is an anionic metal oxide cluster consisting of metal–oxygen polyhedral units, such as {WO6}.35,36 The structure formed by eliminating some of the polyhedral units is called a lacunary POM, which functions as an inorganic multidentate ligand and allows the incorporation of different metals.37–39 Recent studies have reported a new synthesis method for introducing various multinuclear metal oxide clusters into lacunary POMs in organic solvents.40–44 Furthermore, a previous study used a diiron-containing POM (Fe2) as a precursor to in situ form FeOx subnanoclusters on SiO2, which served as the active species to selectively converted CH4 into HCHO and CO.45 This catalyst maintained the catalytic activity at 600 °C for 72 h, because the FeOx subnanoclusters were dispersed in tungsten oxide species and inhibited from aggregation and deactivation. Another study successfully developed a method to achieve highly dispersed POM tetra-n-butylammonium (TBA) salts on various supports.46 Therefore, using POMs with different multinuclear metal oxide cores as catalyst precursors allows the investigation of various highly stable metal oxide subnanoclusters for the selective conversion of CH4 to HCHO.
In this study, to further enhance the HCHO yield, we investigated CH4 oxidation by using various POMs supported on SiO2 as catalyst precursors (Fig. 1). Catalysts prepared using mononuclear (Fe1)- and dinuclear (Fe2)-iron-containing POM precursors supported on SiO2 produced relatively high yields of HCHO. Furthermore, we investigated CH4 oxidation using various supports. The results demonstrated that although CH4 conversion increased when using Fe2/Al2O3 and Fe2/CeO2, overoxidation to CO2 was promoted. We observed that catalysts prepared using SiO2 (i.e., Fe2/SiO2) exhibited significantly higher HCHO yields than catalysts prepared using other supports. In addition, to elucidate the mechanism underlying the suppression of overoxidation of HCHO on Fe2/SiO2, we investigated HCHO oxidation using Al2O3 and SiO2 with different specific surface areas. We found that SiO2 suppressed HCHO pyrolysis, and the suppression was enhanced as the specific surface area of SiO2 increased. Moreover, we conducted temperature variation tests on HCHO oxidation and observed that HCHO was minimally decomposed or oxidized at 500 °C in the absence of catalysts.
 | ||
| Fig. 1 Schematic diagram of the catalytic performance for CH4 oxidation using supported metal oxide subnanocluster catalyst prepared from polyoxometalates. | ||
Experimental
Catalyst characterization
Cold-spray ionization (CSI)-mass spectra were recorded on a JEOL JMS-T100CS spectrometer. Infrared (IR) spectra were measured on a JASCO FT/IR-4100 spectrophotometer using KBr disks. Inductively coupled plasma atomic emission spectroscopy (ICP-AES) analysis was performed on a Shimadzu ICPS-8100 instrument. Brunauer–Emmett–Teller (BET) surface areas were measured by N2 adsorption at −196 °C using a Micromeritics TriStar II Plus instrument. Raman spectra were recorded on a JASCO NRS-5100 spectrometer. The measurement conditions included an irradiation laser wavelength of 532 nm and a laser power of 10.2 mW, and the data were collected twice with a measurement time of 3 min. X-ray absorption spectroscopy (XAS) of the Fe K-edge was performed using the transmission and fluorescence method at the BL14B2 beamline of SPring-8 (proposal numbers 2022B1656 and 2023A1512). The X-ray beam was monochromatized using a Si (111) monochromator, and the energy was calibrated using an Fe metal foil for the Fe K-edge XAS. X-ray absorption near-edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) data were analyzed using Athena and Artemis software (Demeter, version 0.9.26; Bruce Ravel). The k3-weighted EXAFS spectra were Fourier-transformed into R-space in the range of 3–12 Å−1 for Fe. The XAS measurement methods and EXAFS analyses are explained in detail in the ESI.†Reagents
Dichloromethane, diethyl ether, and acetonitrile were purchased from Kanto Chemicals. SiO2 (CARiACT Q-6, Q-10, Q-30, or Q-50, Fuji Silysia Chemical Ltd.), Al2O3 (KHS-46, Sumitomo Chemical Co. Ltd.), ZrO2 (JRC-ZRO-6, Daiichi Kigenso Kagaku Kogyo Co., Ltd), CeO2 (JRC-CEO-5, Daiichi Kigenso Kagaku Kogyo Co., Ltd.), TiO2 (ST-01, ISHIHARA SANGYO KAISHA, Ltd), Nb2O5 (JRC-NBO-1, Companhia Brasileira de Metalurgia e Mineração), hydroxyapatite (HAP, Cat. No. 012-14882 FUJIFILM Wako Pure Chemical Corporation), and boron nitride (BN, Cat. No. 028-02281, FUJIFILM Wako Pure Chemical Corporation) were acquired from commercial sources. To represent SiO2 with different specific surface areas, CARiACT Q-6, CARiACT Q-30, and CARiACT Q-50 are denoted as SiO2-Q6, SiO2-Q30, and SiO2-Q50, respectively. SiO2 without any specific indication refers to CARiACT Q-10. Fe3O(CH3CO2)7(H2O)3 (ref. 47) and TBA4[SiW9O28(OCH3)6] (SiW9–OMe)48,49 were synthesized according to the reported procedures.Synthesis of various POMs
TBA8H4Fe2O(SiW10O36)2 (Fe2),45 TBA6Ti4O2(OH)4(PW10O36)2 (Ti4),50 TBA4V2O2(OH)2SiW10O36 (V2),51 TBA4VOPMo11O39 (V1Mo),52 TBA7HMn5(OH)2(SiW9O34)2 (Mn5),42 TBA8H4Co4(OH)4(SiW10O36)2 (Co4),53 TBA8H4Cu2(SiW10O36)2 (Cu2),40 TBA6Ce4(H2O)2(CH3CN)2O(SiW10O36)2 (Ce4),41 and TBA4HFe(H2O)SiW11O39 (Fe1)54,55 were synthesized according to the reported procedures. In addition, TBA4Fe4(OH)3(CH3CO2)3SiW9O34 (Fe4) was synthesized according to the following procedure. SiW9–OMe (500 mg, 152 μmol) was added to a dichloromethane solution (40 mL) of Fe3O(CH3CO2)7(H2O)3 (134 mg, 205 μmol), and the resulting solution was stirred for 24 h at room temperature. Then, the remaining precipitate was filtered off, and the filtrate was dropped into diethyl ether (40 mL). Finally, the generated precipitate was collected by filtration through a membrane filter. After vacuum drying, a powder sample of Fe4 was obtained. The anionic structure of Fe4 was estimated from the IR spectra (Fig. S1†), ICP-AES elemental analysis, and CSI-mass spectrum (Fig. S2†), as described below. The IR spectra illustrated acetate peaks slightly shifted from those of Fe3O(CH3CO2)7(H2O)3. The elemental analysis confirmed that four Fe3+ ions were introduced into an [SiW9O34]10− (SiW9) unit. The CSI-mass spectrum indicated the presence of a molecule containing four iron atoms, an SiW9 unit, three hydroxide ligands, and three acetate ligands (Fig. S2†). These results suggested that Fe4 had a cubane-type structure, as illustrated in Fig. 1, since a similar structure was reported for a manganese analogue according to the IR results.56 IR (KBr pellet, cm−1): 3441, 2962, 2874, 1631, 1581, 1544, 1485, 1460, 1382, 1275, 1152, 1106, 1000, 958, 909, 804, 672, 524, 376, 359, 324, 319, 303, 297, 290, 282, 278, 271, 257, 252. Elemental analysis: calcd (%) for TBA4Fe4(OH)3(CH3CO2)3SiW9O34·2CH2Cl2, Si 0.74, Fe 5.85, W 43.33; found Si 0.74, Fe, 5.87, W 43.38. Positive-ion CSI-mass (acetonitrile): m/z = 3891.36, [TBA5Fe4(OH)3(CH3CO2)3SiW9O34]+ (theoretical m/z = 3890.48).Preparation of POM-supported catalysts
Various types of POMs were generally dispersed on supports with the loading amount of 10 wt% using the incipient wetness method.46 The preparation method of Fe2/SiO2 is described as a typical example. A TBA salt of Fe2 (100 mg) was dissolved in acetonitrile (2 mL). Then, the resulting solution was dropped onto a thin layer of SiO2 (900 mg) spread on an evaporation tray. The resulting powder was dried at 100 °C for 5 h and then calcined at 600 °C for 5 h under air atmosphere, giving the Fe2/SiO2 catalyst. Various kinds of supported catalysts were prepared by the same method using POM precursors shown in Fig. 1. These supported catalysts were denoted using the abbreviation of the POM precursors and supports, such as Fe1/SiO2, Fe2/SiO2, and Fe4/SiO2. As for Fe2/SiO2, the catalyst with 35 wt% Fe2 (35Fe2/SiO2) was also prepared to align the loading amount of iron with Fe4/SiO2. Additionally, Cs–Fe2/SiO2 was prepared following our previous report.46 Briefly, TBA–Fe2 was supported on SiO2 in acetonitrile using the incipient wetness method. Then, Cs–Fe2/SiO2 was obtained by cation exchange from TBA–Fe2/SiO2 using cesium trifluoromethanesulfonate as the Cs source in ethanol.Evaluation of CH4 and HCHO oxidation performance
Catalytic performance tests for CH4 oxidation were conducted using a fixed-bed flow-type reactor schematically illustrated in Fig. S3a.† After 100 mg of catalyst was loaded into a quartz tube reactor (6.0 mm i.d.), the reactant gas (CH4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :O2:Ar = 2:1:7, total flow rate: 50 mL min−1) was introduced into the reactor. Then, the input temperature of the tube furnace containing the catalyst was increased to 600 °C at a rate of 10 °C min−1. The inlet and outlet gases were heated to 100 °C to reduce the temperature variation in the catalyst bed and to suppress the condensation of products. When the furnace temperature reached 600 °C, it was maintained for 1 h to allow the activity to stabilize. Then, HCHO was trapped in 20 mL of an aqueous solution containing 2 g of Na2SO3 and 50 μL of H2SO4, and the amount of trapped HCHO was determined by titration with an aqueous solution of NaOH (0.1 M). CO and CO2 were analyzed immediately before and after HCHO collection using a Nexis GC-2030 gas chromatograph equipped with a barrier discharge ionization detector (Shimadzu Corporation) and a Shincarbon-ST packed column. The respective calculation formulas for CH4 conversion, product selectivity, and product yield in this study are as follows:
:O2:Ar = 2:1:7, total flow rate: 50 mL min−1) was introduced into the reactor. Then, the input temperature of the tube furnace containing the catalyst was increased to 600 °C at a rate of 10 °C min−1. The inlet and outlet gases were heated to 100 °C to reduce the temperature variation in the catalyst bed and to suppress the condensation of products. When the furnace temperature reached 600 °C, it was maintained for 1 h to allow the activity to stabilize. Then, HCHO was trapped in 20 mL of an aqueous solution containing 2 g of Na2SO3 and 50 μL of H2SO4, and the amount of trapped HCHO was determined by titration with an aqueous solution of NaOH (0.1 M). CO and CO2 were analyzed immediately before and after HCHO collection using a Nexis GC-2030 gas chromatograph equipped with a barrier discharge ionization detector (Shimadzu Corporation) and a Shincarbon-ST packed column. The respective calculation formulas for CH4 conversion, product selectivity, and product yield in this study are as follows: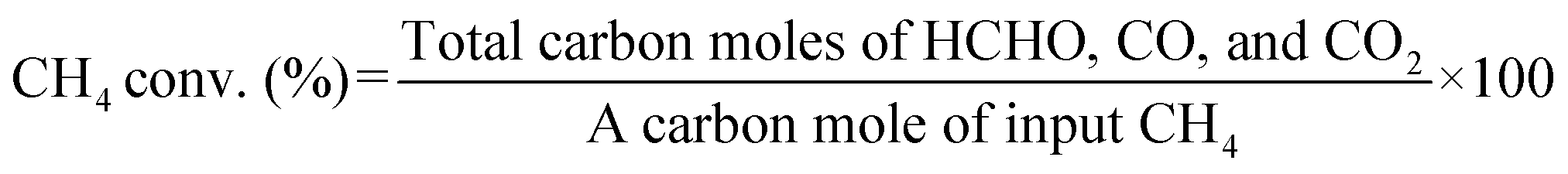
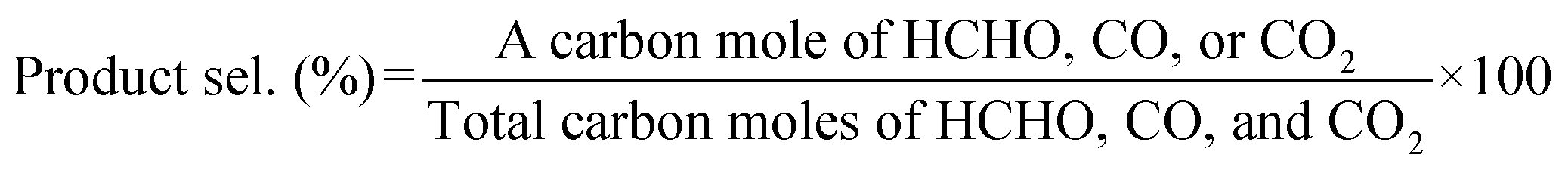
| Product yield (%) = CH4 conv. (%) × Product sel. (%) ÷ 100 |
In situ DRIFTS measurement
Diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) was performed using a JASCO FT/IR-6700 spectrometer equipped with an in situ sample cell in the range of 1500–4000 cm−1 at a resolution of 4 cm−1 (number of scans: 64). The samples were loaded onto a sample plate (6 mm diameter) and placed on the heater in the IR cell. The sample was pretreated by heating in a vacuum at 600 °C for 1 h, and the background spectrum was measured. After the pretreatment, the sample was exposed to approximately 1% HCHO in N2 (20 mL min−1) for 1 h. Then, the cell was evacuated into a vacuum at 600 °C, and the DRIFTS spectra were measured both in the reactant gas and in a vacuum. HCHO was generated in the same manner as described above.Results and discussion
CH4 oxidation using various catalysts
First, the catalytic performance for CH4 oxidation was investigated for various POM-based catalysts at 600 °C under atmospheric pressure, and the results are presented in Table 1. Ti4/SiO2, V2/SiO2, V1Mo/SiO2, Cu2/SiO2, and Ce4/SiO2 exhibited relatively low CH4 conversion (Table 1, entries 2–4, 7, and 8). Although there have been several reports16,17,19,22,23,25 on the oxidation of CH4 to HCHO using vanadium, molybdenum, and copper oxides, the CH4 oxidation performance using these POM precursors supported on SiO2 was much lower than that using Fe2/SiO2 (Table 1, entry 1). Particularly, the use of Ce4/SiO2 resulted in minimal HCHO formation, with significant CO2 formation from complete CH4 oxidation (Table 1, entry 8). In contrast, although Fe2/SiO2, Mn5/SiO2, and Co4/SiO2 exhibited relatively high CH4 conversion, the overoxidation to COx (CO and CO2) was more pronounced for Mn5/SiO2 and Co4/SiO2 than for Fe2/SiO2 (Table 1, entries 1, 5, and 6). In the case of iron, reducing the cluster size promoted the partial oxidation of CH4 to HCHO, while the catalytic performance of manganese and cobalt nanoclusters remained similar to that of bulk oxides57 or nanoparticles.58 Thus, the catalytic performance for CH4 oxidation on SiO2-supported metal oxide subnanocluster catalysts prepared from various POM precursors indicated that Fe2/SiO2 catalysts exhibited the highest HCHO yield among the metal oxide subnanocluster catalysts with 3d metal or Ce metal multinuclear structures.| Entry | Catalyst | Conv. [%] | Sel. [%] | Yield [%] | ||
|---|---|---|---|---|---|---|
| CH4 | HCHO | CO | CO2 | HCHO | ||
|
a Reaction conditions: 10 wt% supported catalyst (100 mg), CH4:O2:Ar = 2:1:7, 50 mL min−1, 1 atm, 600 °C.
|
||||||
| 1 | Fe2/SiO2 | 1.4 | 41 | 45 | 14 | 0.57 |
| 2 | Ti4/SiO2 | 0.40 | 82 | 5 | 13 | 0.33 |
| 3 | V2/SiO2 | 0.44 | 55 | 27 | 18 | 0.24 |
| 4 | V1 Mo /SiO2 | 0.52 | 63 | 22 | 15 | 0.33 |
| 5 | Mn5/SiO2 | 0.97 | 25 | 45 | 30 | 0.24 |
| 6 | Co4/SiO2 | 1.4 | 22 | 52 | 26 | 0.30 |
| 7 | Cu2/SiO2 | 0.69 | 47 | 27 | 26 | 0.33 |
| 8 | Ce4/SiO2 | 0.56 | — | 66 | 34 | — |
| 9 | Fe1/SiO2 | 1.4 | 54 | 32 | 13 | 0.73 |
| 10 | Fe4/SiO2 | 3.0 | 11 | 62 | 28 | 0.33 |
| 11 | 35Fe2/SiO2 | 1.9 | 29 | 58 | 13 | 0.55 |
| 12 | Cs–Fe2/SiO2 | 0.67 | 73 | 20 | 7 | 0.49 |
Next, the catalytic performance for CH4 oxidation was investigated for various Fe–POM-based catalysts prepared using iron-containing POM precursors with various multinuclear oxide cores at 600 °C under atmospheric pressure. The results are summarized in Table 1. The iron content of the supported iron catalysts is also summarized in Table S1.† Among the catalysts prepared from POMs with different numbers of iron nuclei, Fe1/SiO2 and Fe2/SiO2 exhibited relatively high HCHO yields of 0.73% and 0.57%, respectively (Table 1, entries 1, 9, and 10). Fe4/SiO2 exhibited a relatively high CH4 conversion of 3% but a low HCHO selectivity of 11%, resulting in a low HCHO yield of 0.33%. 35 wt% Fe2/SiO2 (35Fe2/SiO2) was prepared using Fe2 with the same iron loading amount as that of Fe4/SiO2, and the reaction was performed using 35Fe2/SiO2. As a result, 35Fe2/SiO2 exhibited lower CO2 selectivity than Fe4/SiO2 (Table 1, entries 10 and 11). In addition, Cs–Fe2/SiO2 was prepared to improve the thermal stability of the anionic structure of Fe2,46 and the CH4 oxidation performance of Cs–Fe2/SiO2 was investigated under the same reaction conditions. However, despite its high selectivity toward HCHO (73%), Cs–Fe2/SiO2 exhibited relatively low CH4 conversion (0.67%) (Table 1, entry 12).
To investigate the structural differences between Fe2/SiO2, 35Fe2/SiO2 and Fe4/SiO2 after the reaction, Fe K-edge XAFS measurements were performed, with the XANES and EXAFS spectra displayed in Fig. S4 and S5,† respectively. The Fe K-edge XANES results indicated that the valence of Fe in each catalyst prepared from Fe2 and Fe4 was not different. In a previous study, Fe2/SiO2 was directly used for the reaction without any pretreatment.45 In contrast, Fe2/SiO2 in this study was used after calcination. The results of Fe K-edge XANES and EXAFS suggested that Fe2/SiO2 prepared by calcination under air atmosphere formed a structure similar to that of Fe2/SiO2 treated by the reaction gas (Fig. S4a, S5a and b†).45 In our previous report on Fe2/SiO2 (used for CH4 oxidation without precalcination),45 it was revealed that Fe2 was decomposed into FeOx subnanoclusters (<1 nm) and WOx nanoclusters (approximately 3 nm) on SiO2 under CH4 oxidation conditions at 600 °C. Various control experiments and characterizations also revealed that the FeOx subnanoclusters were the active species. Furthermore, the FeOx subnanoclusters were dispersed on the WOx nanoclusters, and as a result, excessive aggregation and deactivation of the effective FeOx subnanoclusters were suppressed. Thus, a similar structure was expected to form over Fe4/SiO2. The Fe K-edge XANES spectra of 35Fe2/SiO2 and Fe4/SiO2 suggested that they also had similar structures to the previously reported Fe2/SiO2 (Fig. S4b†).45 On the other hand, we found that the peak intensity corresponding to the Fe–O bond in the first coordination sphere for Fe2/SiO2 and 35Fe2/SiO2 was higher than that for Fe4/SiO2 in the Fe K-edge EXAFS spectra (Fig. S5c and d†). This may be due to the larger number of WOx nanoclusters near the FeOx subnanoclusters in Fe2/SiO2 and 35Fe2/SiO2 than that in Fe4/SiO2 since the amount of tungsten per iron in Fe2 was larger than that in Fe4. Furthermore, in the Raman spectrum of Fe2/SiO2, three main vibrational peaks were observed, corresponding to ν(WO) at 980–960 cm−1, ν(W–Oa–W) at 840–780 cm−1, and ν(W–Ob–W) at 720–680 cm−1 (Fig. S6†).59,60 The terminal oxo species (WO) corresponds to an isolated tungsten species. In contrast, in the spectrum of Fe4/SiO2, two peaks corresponding to ν(W–Oa–W) and ν(W–Ob–W) were observed and no ν(WO) peak was observed. Considering that Fe2/SiO2 and Fe4/SiO2 contained almost the same amount of tungsten, 5.3 wt% and 4.5 wt%, respectively, there was a sufficient amount of WOx species near the FeOx subnanoclusters. The remaining WOx species on Fe2/SiO2 formed the isolated tungsten species, possibly because the amount of tungsten per iron of Fe2 was larger than that of Fe4. Thus, these results were consistent with the EXAFS spectra (Fig. S5†). Therefore, the difference in the iron oxide subnanocluster structures formed on SiO2 was reflected in the catalytic performance. In addition, 35Fe2/SiO2 exhibited higher HCHO selectivity than Fe4/SiO2, possibly due to the higher tungsten content and the presence of a larger number of tungsten oxide clusters, which inhibited the aggregation of FeOx subnanoclusters. In summary, the iron oxide subnanocluster catalysts prepared from Fe1 and Fe2 exhibited the highest HCHO yield in CH4 oxidation.
Support effect
The catalytic performance for CH4 oxidation was investigated for Fe2-based catalysts prepared using various supports at 600 °C under atmospheric pressure. The results and specific surface areas of supported are summarized in Table 2. The results indicated that higher specific surface areas of SiO2 supports resulted in higher CH4 conversion (Table 2, entries 1–4). This suggested that a higher dispersion state of the Fe2 precursor on SiO2 led to the generation of more dispersed iron oxide nanoclusters, resulting in increased CH4 conversion and HCHO yield. The supported Fe2 catalyst prepared using SiO2-Q6, which had a very high specific surface area of 386 m2 g−1, exhibited the highest CH4 conversion and HCHO yield of 2.0% and 0.73%, respectively. In contrast, despite having lower specific surface areas than SiO2, the supported Fe2 catalysts prepared using Al2O3, ZrO2, and CeO2 exhibited higher CH4 conversion, resulting in the production of only COx (Table 2, entries 5–7). In addition, the reaction was performed at 500 °C with Fe2/CeO2, but the selectivity to HCHO did not increase, although the CH4 conversion decreased (Table 2, entry 8). Similar catalytic performance has been reported for FePO4 catalysts supported on Al2O3, SiO2, TiO2, and ZrO2, suggesting that the choice of support affects the overoxidation of HCHO.18| Entry | Support | BET surface area [m2 g−1] | Conv. [%] | Sel. [%] | Yield [%] | ||
|---|---|---|---|---|---|---|---|
| CH4 | HCHO | CO | CO2 | HCHO | |||
|
a Reaction conditions: supported Fe2 catalyst (100 mg), CH4:O2:Ar = 2:1:7, 50 mL min−1, 1 atm, 600 °C.
b 500 °C.
|
|||||||
| 1 | SiO2-Q50 | 67 | 0.71 | 57 | 35 | 8 | 0.41 |
| 2 | SiO2-Q30 | 100 | 0.96 | 43 | 44 | 13 | 0.42 |
| 3 | SiO2 | 278 | 1.4 | 41 | 45 | 14 | 0.57 |
| 4 | SiO2-Q6 | 386 | 2.0 | 36 | 46 | 18 | 0.73 |
| 5 | Al2O3 | 158 | 6.1 | — | 82 | 18 | — |
| 6 | ZrO2 | 40 | 3.3 | — | 78 | 22 | — |
| 7 | CeO2 | 43 | 6.8 | — | 65 | 35 | — |
| 8 | CeO2b | 43 | 0.79 | — | 69 | 31 | — |
| 9 | Nb2O5 | 145 | 0.84 | 39 | 51 | 10 | 0.33 |
| 10 | TiO2 | 72 | 1.2 | 7 | 64 | 29 | 0.08 |
| 11 | BN | 6 | 0.28 | 57 | 24 | 19 | 0.16 |
| 12 | HAP | 9 | 0.12 | 67 | n.d. | 33 | 0.08 |
The surface acidity of these supports may promote the easy activation of CH4, resulting in overoxidation of the products.61 In addition, HAP, BN, Nb2O5, and TiO2 had smaller specific surface areas than SiO2, resulting in lower CH4 conversion when used as supports for Fe2 catalysts (Table 2, entries 9–12). Therefore, the effect of the support on CH4 oxidation was found to depend on the type of support and its specific surface area. Moreover, it was found that the Fe2 catalyst supported on SiO2, which was inert and had a high specific surface area, exhibited the highest HCHO yield.
Pyrolysis and oxidation tests of HCHO
As the support significantly affected the overoxidation of HCHO, pyrolysis and oxidation tests of HCHO on various supports and Fe2/SiO2 were investigated (Fig. 2). The results indicated that HCHO was decomposed by 80% or more even in the absence of catalysts at 600 °C (Fig. 2, entry 1). We found that HCHO pyrolysis was significantly suppressed in the presence of SiO2 (Fig. 2, entry 2), and this suppression increased with the specific surface area of SiO2 (Fig. 2, entry 3). Additionally, HCHO pyrolysis was suppressed by using Fe2/SiO2 (Fig. 2, entry 4). In contrast, HCHO pyrolysis on Al2O3 was promoted (Fig. 2, entry 5). HCHO overoxidation on SiO2 was more suppressed even in the presence of O2 than in the absence of catalysts (Fig. 2, entries 6 and 7). Furthermore, HCHO conversion using Fe2/SiO2 was lower than that in the absence of catalysts (Fig. 2, entry 8). These findings suggested that the reason for the high HCHO yield when using Fe2/SiO2 was the suppression of HCHO pyrolysis by SiO2 and the use of Fe2 as a catalyst precursor. Meanwhile, even with an inert support such as SiO2, HCHO was sequentially oxidized in the presence of O2 at 600 °C. Since a significant amount of HCHO was thermally decomposed even at 600 °C, pyrolysis and oxidation tests without catalysts were conducted at various temperatures, as illustrated in Fig. 3. The results indicated that both pyrolysis and oxidation were suppressed at lower temperatures and that almost no pyrolysis or oxidation occurred at 500 °C. In our previous study, however, the CH4 conversion over Fe2/SiO2 at 550 °C was lower than 1%, and the CH4 conversion at 500 °C predicted based on the Arrhenius plots was below 0.4%.45 Therefore, FeOx subnanocluster active site should be modified and improved to archive higher HCHO yield at 500 °C.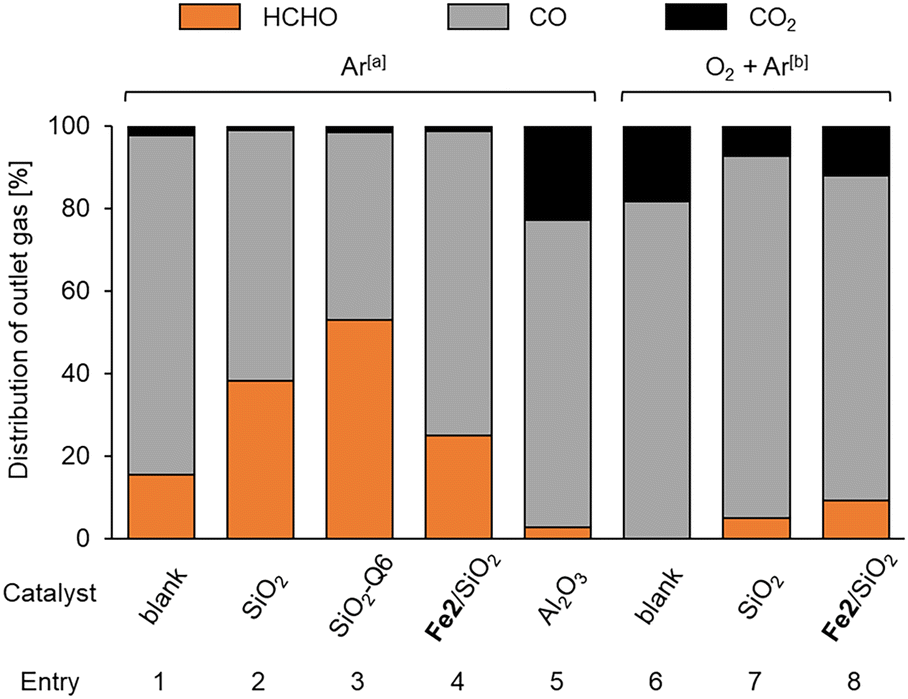 | ||
| Fig. 2 HCHO pyrolysis and oxidation tests on various supports and Fe2/SiO2. Reaction conditions: catalyst (100 mg), HCHO (1%), 1 atm, 600 °C. [a] Ar (50 mL min−1), [b] O2 (5 mL min−1), Ar (45 mL min−1). | ||
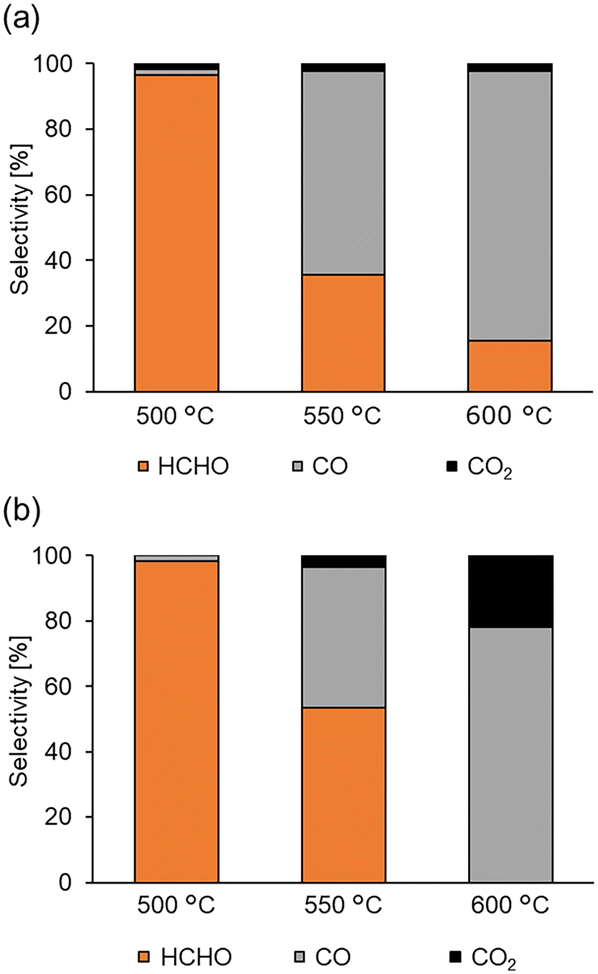 | ||
| Fig. 3 Pyrolysis and oxidation tests without catalysts at various temperatures. Reaction conditions: no catalyst, 1 atm, HCHO (ca. 0.6%), (a) Ar (50 mL min−1), (b) O2 (5 mL min−1), Ar (45 mL min−1). | ||
Proposed mechanism of the suppression of HCHO pyrolysis
To elucidate the mechanism by which pyrolysis of HCHO was promoted on Al2O3 and suppressed on SiO2, in situ DRIFTS measurements were conducted at 600 °C. The DRIFTS spectra measured 60 min after evacuation following HCHO flow were presented in Fig. 4. The observed peaks of DRIFTS spectra and their assignments are summarized in Table S2.†62–64 Furthermore, time variation of in situ DRIFTS spectra of Al2O3 and SiO2 at 600 °C is illustrated in Fig. S7.† In the spectrum of Al2O3, three peaks observed at 1572, 2906, and 2997 cm−1 were attributed to formate species (Fig. 4a). In addition, the peaks at 1630 and 3751 cm−1 were attributed to H2O and an aluminol group, respectively, indicating that these species possibly involved in the mechanism.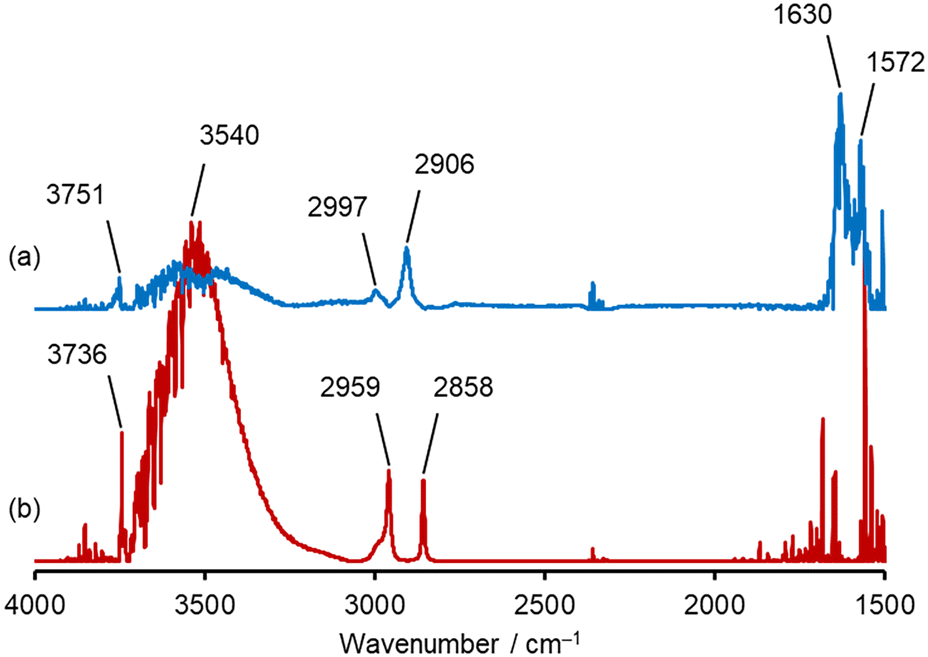 | ||
| Fig. 4 In situ DRIFTS spectra on (a) Al2O3 and (b) SiO2. Analysis conditions: HCHO/N2 mixture gas (20 mL min−1) flowed at 600 °C for 60 min. Then, the cell was evacuated, and DRIFTS spectra were measured 60 min later. The absorbance intensity on the vertical axis was Kubelka–Munk-transformed. | ||
On the other hand, in the spectrum of SiO2, two sharp peaks at 2959 and 2858 cm−1 were observed (Fig. 4b). From the previously reported data summarized in Table. S2,† these peaks can be attributed to dioxymethylene species. As well as Al2O3, the peaks derived from adsorbed H2O (ref. 65) at 3540 cm−1 and a silanol group66 at 3736 cm−1 were observed. These findings suggested that the adsorbed HCHO was decomposed into formate on Al2O3, while not on SiO2.
We proposed a surface reaction mechanism of HCHO on Al2O3 and SiO2 based on the results of in situ DRIFTS, as displayed in Fig. 5. In the gas phase, CO is generated by HCHO pyrolysis via the formyl radical.67 In the presence of Al2O3, HCHO reacts with aluminol groups on the surface to form dioxymethylene species. Then, the dioxymethylene species reacts with another HCHO to afford formate and methoxide species via the Cannizzaro reaction.62 Then, these species further react with H2O to form formic acid and methanol; however, due to the high temperature of 600 °C, they are possibly sequentially oxidized to COx. Therefore, Al2O3 undesirably promotes the overoxidation of HCHO to COx.
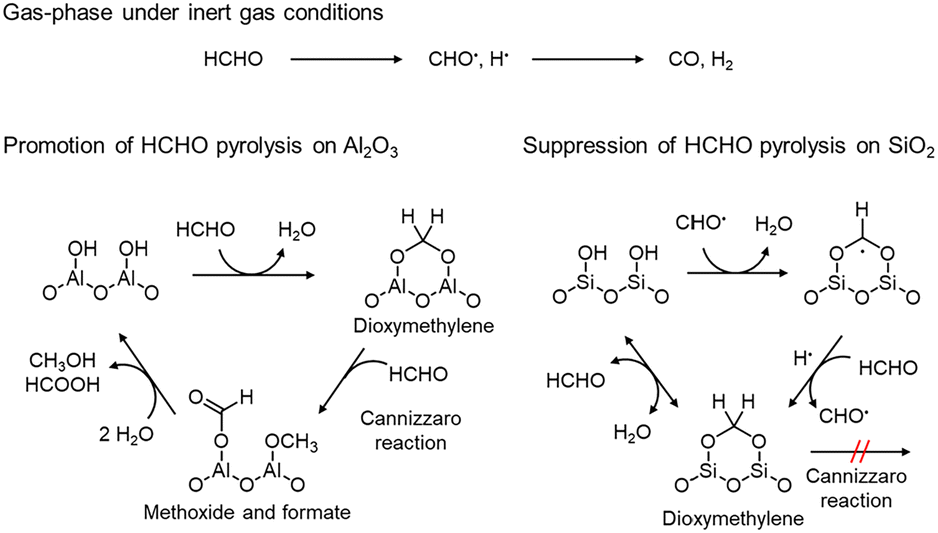 | ||
| Fig. 5 Proposed mechanism of the suppression of HCHO pyrolysis on SiO2 and the promotion of HCHO pyrolysis on Al2O3. | ||
In contrast, the dioxymethylene species adsorbed on SiO2 do not cause the Cannizzaro reaction because SiO2 possesses no strong acidic or basic sites. Moreover, the formyl radical reacts with silanol species, and adsorbed on SiO2. Then, the adsorbed radical species reacts with hydrogen atom or accepts hydrogen atom from HCHO to form dioxymethylene. Since the adsorbed dioxymethylene species reacts with H2O to generate HCHO, we consider that HCHO pyrolysis can be suppressed on SiO2. SiO2-Q6 suppressed HCHO pyrolysis more than SiO2 possibly because SiO2-Q6 possessed more silanol groups to react with formyl radicals due to higher specific surface area.
Conclusions
The catalytic performance for CH4 oxidation using SiO2-supported metal oxide subnanocluster catalysts prepared from various POM precursors indicated that Fe1/SiO2 and Fe2/SiO2 exhibited the highest HCHO yields among oxide subnanocluster catalysts with 3d metal or Ce metal multinuclear oxide cores. In addition, we found that Fe2-based catalysts prepared using SiO2 with a high specific surface area improved both the CH4 conversion and HCHO yield. Pyrolysis and oxidation tests of HCHO on various supports revealed that HCHO was almost completely decomposed even in the absence of catalysts at 600 °C. Moreover, the pyrolysis and overoxidation of HCHO were suppressed when SiO2 was used as the support. In situ DRIFTS measurement under HCHO flow demonstrated that the pyrolysis and oxidation of HCHO were suppressed on SiO2, while the pyrolysis of HCHO was promoted on Al2O3. Thus, to achieve even higher HCHO yields in the future, it is necessary to suppress the pyrolysis and oxidation of HCHO in the gas phase. As pyrolysis and oxidation did not occur below 500 °C in the absence of catalysts, it is necessary to develop more effective catalysts that can lower the temperature of CH4 activation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by JSPS KAKENHI Grant Number 22K14539, 22H04971 and JST CREST Grant Number JPMJCR17P4. We greatly appreciate Dr. Hironori Ofuchi (Japan Synchrotron Radiation Research Institute, SPring-8) for the support of XAFS measurements at BL14B2, Proposal Number 2022B1656 and 2023A1512.Notes and references
- D. Sutton, B. Kelleher and J. R. H. Ross, Fuel Process. Technol., 2001, 73, 155–173 CrossRef CAS.
- J. Kopyscinski, T. J. Schildhauer and S. M. A. Biollaz, Fuel, 2010, 89, 1763–1783 CrossRef CAS.
- G. Strobel, B. Hagemann, T. M. Huppertz and L. Ganzer, Renewable Sustainable Energy Rev., 2020, 123, 109747 CrossRef CAS.
- J. Kondori, S. Zendehboudi and L. James, Fuel, 2019, 249, 264–276 CrossRef CAS.
- F. M. Mota and D. H. Kim, Chem. Soc. Rev., 2019, 48, 205–259 RSC.
- M. Held, D. Schollenberger, S. Sauerschell, S. Bajohr and T. Kolb, Chem. Ing. Tech., 2020, 92, 595–602 CrossRef CAS.
- A. Giocoli, V. Motola, N. Scarlat, N. Pierro and S. Dipinto, Renewable Sustainable Energy Transition, 2023, 3, 100051 CrossRef.
- M. C. Alvarez-Galvan, N. Mota, M. Ojeda, S. Rojas, R. M. Navarro and J. L. G. Fierro, Catal. Today, 2011, 171, 15–23 CrossRef CAS.
- C. Hammond, S. Conrad and I. Hermans, ChemSusChem, 2012, 5, 1668–1686 CrossRef CAS PubMed.
- P. Schwach, X. Pan and X. Bao, Chem. Rev., 2017, 117, 8497–8520 CrossRef CAS PubMed.
- N. F. Dummer, D. J. Willock, Q. He, M. J. Howard, R. J. Lewis, G. Qi, S. H. Taylor, J. Xu, D. Bethell, C. J. Kiely and G. J. Hutchings, Chem. Rev., 2023, 123, 6359–6411 CrossRef CAS PubMed.
- T. J. Hall, J. S. J. Hargreaves, G. J. Hutchings, R. W. Joyner and S. H. Taylor, Fuel Process. Technol., 1995, 42, 151–178 CrossRef CAS.
- J. H. Lunsford, Catal. Today, 2000, 63, 165–174 CrossRef CAS.
- K. Otsuka and M. Hatano, J. Catal., 1987, 108, 252–255 CrossRef CAS.
- E. M. Coda, E. Mulhall, R. van Hoek and B. K. Hodnett, Catal. Today, 1989, 4, 383–387 CrossRef.
- A. Parmaliana and F. Arena, J. Catal., 1997, 167, 57–65 CrossRef CAS.
- K. Aoki, M. Ohmae, T. Nanba, K. Takeishi, N. Azuma, A. Ueno, H. Ohfune, H. Hayashi and Y. Udagawa, Catal. Today, 1998, 45, 29–33 CrossRef CAS.
- R. L. McCormick and G. O. Alptekin, Catal. Today, 2000, 55, 269–280 CrossRef CAS.
- N. Ohler and A. T. Bell, J. Phys. Chem. B, 2006, 110, 2700–2709 CrossRef CAS PubMed.
- A. de Lucas, J. L. Valverde, L. Rodriguez, P. Sanchez and M. T. Garcia, Appl. Catal., A, 2000, 203, 81–90 CrossRef CAS.
- N. V. Beznis, A. N. C. van Laak, B. M. Weckhuysen and J. H. Bitter, Microporous Mesoporous Mater., 2011, 138, 176–183 CrossRef CAS.
- T. Akiyama, R. Sei and S. Takenaka, Catal. Sci. Technol., 2021, 11, 5273–5281 RSC.
- S. Pei, B. Yue, L. Qian, S. Yan, J. Cheng, Y. Zhou, S. Xie and H. He, Appl. Catal., A, 2007, 329, 148–155 CrossRef CAS.
- A. Matsuda, H. Tateno, K. Kamata and M. Hara, Catal. Sci. Technol., 2021, 11, 6987–6998 RSC.
- T. Akiyama, M. Shimakawa and S. Takenaka, Chem. Lett., 2022, 51, 511–514 CrossRef CAS.
- A. de Lucas, J. L. Valverde, P. Cañizares and L. Rodriguez, Appl. Catal., A, 1999, 184, 143–152 CrossRef CAS.
- Q. Zhang, W. Yang, X. Wang, Y. Wang, T. Shishido and K. Takehira, Microporous Mesoporous Mater., 2005, 77, 223–234 CrossRef CAS.
- F. Arena, G. Gatti, G. Martra, S. Coluccia, L. Stievano, L. Spadaro, P. Famulari and A. Parmaliana, J. Catal., 2005, 231, 365–380 CrossRef CAS.
- Y. Li, D. An, Q. Zhang and Y. Wang, J. Phys. Chem. C, 2008, 112, 13700–13708 CrossRef CAS.
- Q. Zhang, Y. Li, D. An and Y. Wang, Appl. Catal., A, 2009, 356, 103–111 CrossRef CAS.
- J. He, Y. Li, D. An, Q. Zhang and Y. Wang, J. Nat. Gas Chem., 2009, 18, 288–294 CrossRef CAS.
- Y. Kim, T. Y. Kim, C. K. Song, K. R. Lee, S. Bae, H. Park, D. Yun, Y. S. Yun, I. Nam, J. Park, H. Lee and J. Yi, Nano Energy, 2021, 82, 105704 CrossRef CAS.
- H. Berndt, A. Martin, A. Brückner, E. Schreier, D. Müller, H. Kosslick, G.-U. Wolf and B. Lücke, J. Catal., 2000, 191, 384–400 CrossRef CAS.
- T. Kobayashi, N. Guilhaume, J. Miki, N. Kitamura and M. Haruta, Catal. Today, 1996, 32, 171–175 CrossRef CAS.
- M. Sun, J. Zhang, P. Putaj, V. Caps, F. Lefebvre, J. Pelletier and J.-M. Basset, Chem. Rev., 2014, 114, 981–1019 CrossRef CAS PubMed.
- S.-S. Wang and G.-Y. Yang, Chem. Rev., 2015, 115, 4893–4962 CrossRef CAS PubMed.
- P. Putaj and F. Lefebvre, Coord. Chem. Rev., 2011, 255, 1642–1685 CrossRef CAS.
- D.-L. Long, R. Tsunashima and L. Cronin, Angew. Chem., Int. Ed., 2010, 49, 1736–1758 CrossRef CAS PubMed.
- A. Tézé, G. Hervé, R. G. Finke and D. K. Lyon, Inorg. Synth., 1990, 27, 85–96 Search PubMed.
- K. Suzuki, M. Shinoe and N. Mizuno, Inorg. Chem., 2012, 51, 11574–11581 CrossRef CAS PubMed.
- K. Suzuki, F. Tang, Y. Kikukawa, K. Yamaguchi and N. Mizuno, Angew. Chem., Int. Ed., 2014, 53, 5356–5360 CrossRef CAS PubMed.
- K. Suzuki, R. Sato, T. Minato, M. Shinoe, K. Yamaguchi and N. Mizuno, Dalton Trans., 2015, 44, 14220–14226 RSC.
- Y. Sunada, K. Yamaguchi and K. Suzuki, Coord. Chem. Rev., 2022, 469, 214673 CrossRef CAS.
- Y. Koizumi, K. Yonesato, K. Yamaguchi and K. Suzuki, Inorg. Chem., 2022, 61, 9841–9848 CrossRef CAS PubMed.
- K. Wachi, T. Yabe, T. Suzuki, K. Yonesato, K. Suzuki and K. Yamaguchi, Appl. Catal., B, 2022, 314, 121420 CrossRef CAS.
- T. Suzuki, T. Yabe, K. Wachi, K. Yonesato, K. Suzuki and K. Yamaguchi, ChemNanoMat, 2023, 9, e202200428 CAS.
- A. K. Pandey, T. Gupta and B. P. Baranwal, Transition Met. Chem., 2004, 29, 370–375 CrossRef CAS.
- T. Minato, K. Suzuki, K. Kamata and N. Mizuno, Chem. – Eur. J., 2014, 20, 5946–5952 CrossRef CAS PubMed.
- T. Minato, K. Suzuki, K. Yamaguchi and N. Mizuno, Chem. – Eur. J., 2017, 23, 14213–14220 CrossRef CAS PubMed.
- E. Takahashi, K. Kamata, Y. Kikukawa, S. Sato, K. Suzuki, K. Yamaguchi and N. Mizuno, Catal. Sci. Technol., 2015, 5, 4778–4789 RSC.
- N. Nakagawa, K. Uehara and N. Mizuno, Inorg. Chem., 2005, 44, 9068–9075 CrossRef PubMed.
- K. Nomiya, K. Yagishita, Y. Nemoto and T. Kamataki, J. Mol. Catal. A: Chem., 1997, 126, 43–53 CrossRef CAS.
- Y. Kuriyama, Y. Kikukawa, K. Suzuki, K. Yamaguchi and N. Mizuno, Chem. – Eur. J., 2016, 22, 3962–3966 CrossRef CAS PubMed.
- M. S. Balula, J. A. Gamelas, H. M. Carapuça, A. M. V. Cavaleiro and W. Schlindwein, Eur. J. Inorg. Chem., 2004, 2004, 619–628 CrossRef.
- F. Zonnevijlle, C. M. Tourne and G. F. Tourne, Inorg. Chem., 1982, 21, 2751–2757 CrossRef CAS.
- R. Al-Oweini, B. S. Bassil, J. Friedl, V. Kottisch, M. Ibrahim, M. Asano, B. Keita, G. Novitchi, Y. Lan, A. Powell, U. Stimming and U. Kortz, Inorg. Chem., 2014, 53, 5663–5673 CrossRef CAS PubMed.
- Y.-F. Han, L. Chen, K. Ramesh, Z. Zhong, F. Chen, J. Chin and H. Mook, Catal. Today, 2008, 131, 35–41 CrossRef CAS.
- H. Wang, C. Chen, Y. Zhang, L. Peng, S. Ma, T. Yang, H. Guo, Z. Zhang, D. S. Su and J. Zhang, Nat. Commun., 2015, 6, 7181 CrossRef CAS PubMed.
- E. I. Ross-Medgaarden and I. E. Wachs, J. Phys. Chem. C, 2007, 11, 15089–15099 CrossRef.
- S. Chen, L. Zeng, H. Tian, X. Li and J. Gong, ACS Catal., 2017, 7, 3548–3559 CrossRef CAS.
- W. H. Cheng, J. Catal., 1996, 158, 477–485 CrossRef CAS.
- G. Busca, J. Lamotte, J.-C. Lavalley and V. Lorenzelli, J. Am. Chem. Soc., 1987, 109, 5197–5202 CrossRef CAS.
- D. B. Clarke, D. K. Lee, M. J. Sandoval and A. T. Bell, J. Catal., 1994, 150, 81–93 CrossRef CAS.
- S. Kattel, B. Yan, Y. Yang, J. G. Chen and P. Liu, J. Am. Chem. Soc., 2016, 138, 12440–12450 CrossRef CAS PubMed.
- D. B. Clarke and A. T. Bell, J. Catal., 1995, 154, 314–328 CrossRef CAS.
- M. A. Bañares, L. J. Alemany, M. L. Granados, M. Faraldos and J. L. G. Fierro, Catal. Today, 1997, 33, 73–83 CrossRef.
- R. Klein, M. D. Scheer and L. J. Schoen, J. Am. Chem. Soc., 1956, 78, 50–52 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cy00750b |
| This journal is © The Royal Society of Chemistry 2023 |