
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chemical Chartographisis: a contemporary perspective in molecular design and synthesis†
George E.
Kostakis

Department of Chemistry, School of Life Sciences, University of Sussex, Brighton, BN1 9QJ, UK. E-mail: G.Kostakis@sussex.ac.uk
First published on 11th November 2023
Abstract
The use of flexible molecular systems in solution, without strictly controlling their behaviour, has frequently been productive. Their potential could increase by a more holistic view of the reaction(s) process(es) in which they are involved. In this perspective, we introduce a broader approach – “Chemical Chartographisis” – and discuss three projects in detail to illustrate its potential. The topics involve bimetallic 3d/4f species and coordination compounds built from benzotriazole-based and (a)symmetric salan ligands and focus on catalytic and, in less detail, biological-related examples.
Introduction
Coordination chemistry explores the synthesis of molecular or infinite entities. These are the products of Lewis acid–base reactions in which a ligand(s) (Lewis base) are covalently bound to a metal atom(s) (Lewis acid). Alfred Werner, the patriarch of Coordination Chemistry, set the barriers and limitations for the design, control and synthesis of such entities.1,2 These entities exhibit properties as solids or in solution. For the latter case, we use the term “dynamic system” to describe processes that require ligand exchange e.g., catalysis or sensing, or in situ formation of covalent bonds e.g., chelation therapy, for the function to emerge. Guiding a dynamic system into the correct structural and parameter space subset is challenging. It develops through extensive control experiments relying on chemical intuition. Synthetic strategies have been directed, driven by theory, towards robust and rigid compounds and limited to dynamic systems that are well understood, providing examples with interesting biological3 and catalytic4 properties. These strategies have hitherto excluded flexible, labile examples, and property investigations have excluded vital operational parameters, e.g., complex solubility and stability under different conditions, and unprecedented but significant transformations, subsequently confining a significant amount of knowledge. In other words, the potential of dynamic systems hasn't been realised because of the inability to control/understand their behaviour. For example, one-pot, tandem reactions dominate synthetic chemistry,5 and metal salts ease these organic transformations; however, using well-characterised flexible systems can lead to unprecedented but interesting transformations (see results below).6 Also, recent unprecedented findings question Irving–Williams's empirical rule regarding the relative stabilities of bivalent transition complexes, providing flexible molecules to treat metal intoxication.7We propose that the applications of coordination chemistry can be extended by the use of an evidence-based methodology that draws on two new sources:
(a) data collected by the latest analytical techniques
(b) archived crystallographic data and detailed measurements of physical properties on systems similar to those under study.
Both categories are made available by the rapid increase in computer power. Both correlations are examples of systematisation, i.e., they systematically set present data in the context of previous information.
Systematization in Coordination Chemistry has been inextricably linked with Graph Theory to explain or predict structures.8,9 This is based on vertices and links and is used in many areas of physics and mathematics. The implementation of Graph Theory in the early stages of systematic advances in infinite entities was the key to their description and, subsequently, their synthesis. In less than three decades, industrial zeolitic catalysts were replaced by metal–organic frameworks.10 At the beginning of our work, we envisaged that Graph Theory would be beneficial to understanding the properties of dynamic systems. However, at the molecular level, dynamic systems are more complicated. As our studies progressed, we found that systematisation is required not to explain or predict structures but to classify operational parameters for every individual dynamic system. To facilitate the discovery process, we borrow from Graph Theory the term topology, viz., the description and study of the specific spacial functionality of a molecular entity. We introduce an extended, complementary, more logical, evidence-based methodology called Chemical Chartographisis. The Greek “Xημική Xαρτoγράφηση” from which this word is derived implies that the ‘map writing’ for a particular dynamic system is systematically refreshed (Fig. 1). It draws on methodologies and insights from classical Coordination Chemistry, and applications are found in organic synthesis and biological chemistry.
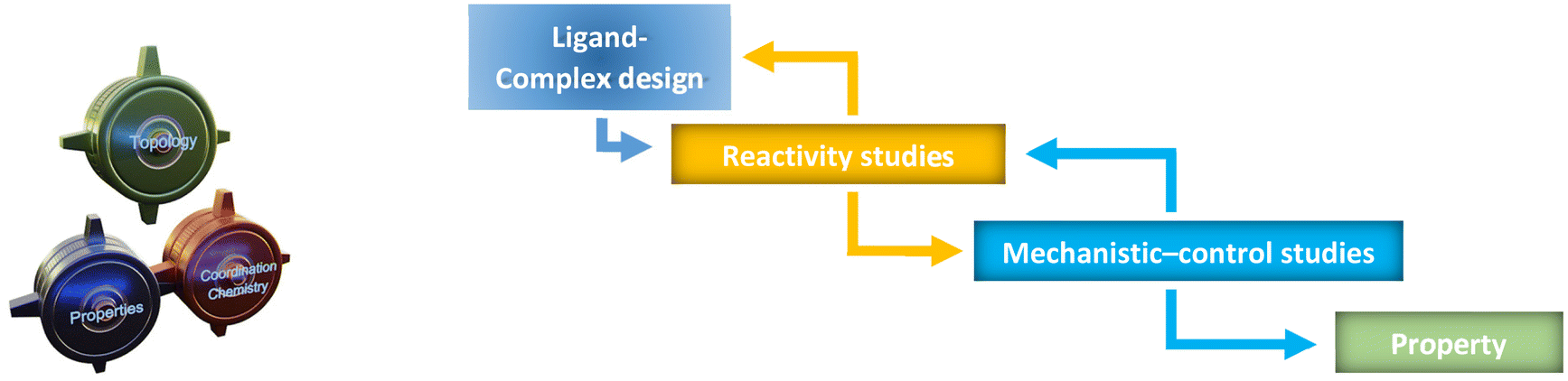 | ||
| Fig. 1 A three-interacting wheel explanation process of Chemical Chartographisis (left) and an arrow-based design, reactivity and control studies figure to rationalise the property of a dynamic system (right). | ||
Some of its essential features are shown in bold. It
(a) uses equivalent topological systems or motifs and describes them as isoskeletal,11
(b) considers every dynamic system individually. Information about unsuccessful or unexpected control experiments is not discarded; it is made available for related studies in the future.
(c) exploits compositional parameters and correlates structure and property (Structure–Activity Relationship; SAR).
(d) contains compounds with a range of similar-sized metals (metal independence). In some cases, it may be possible to vary the oxidation state of the metal throughout a reaction.
(e) requires ligands that can coordinate similarly, and their variation should not alter the motif (ligand independence),
(f) accounts for other species that lie outside the topologically defining unit; this includes solvents of solvation, substrates and anions (coordination geometry independence),
(g) can be explored through a variety of spectroscopic techniques in both the solid state and in solution (structure mapping and reaction monitoring).
A functionality comparison of a set of equivalent topological systems will then (a) rationalise experimental data without the need for the theory to drive the synthesis but to support the findings and (b) identify the reactivity and/or superiority of the studied system. Inactivity or extraordinary performance may be attributed to the components within a data series. Chemical Chartographisis lacks predictive character when applied in a new dynamic system. However, it can incrementally evolve, and prediction may become evident with increased data population. In this perspective, we detail the results obtained in our group in the last decade, with unavoidable self-citation, to certify the validity of the approach.
Dynamic systems in bimetallic 3d/4f chemistry, case 1
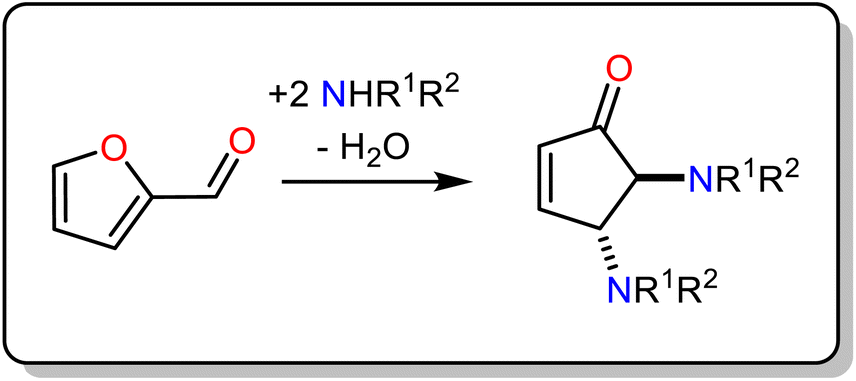
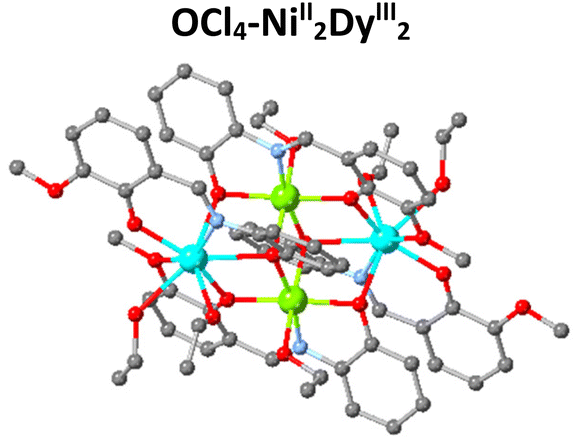
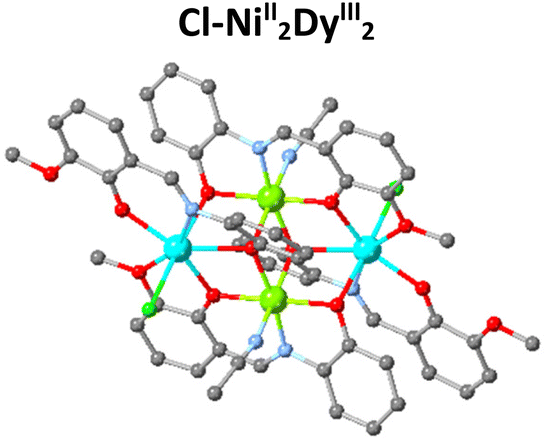
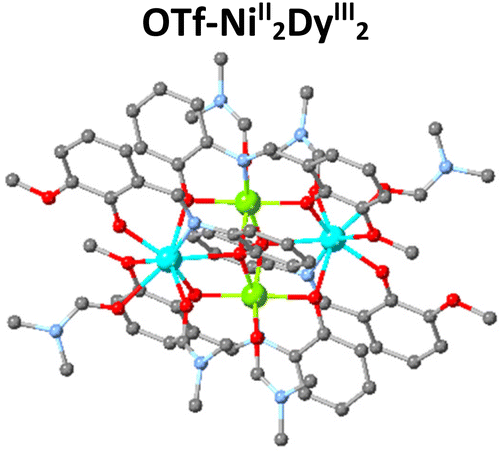
The magnetic properties of 3d/4f complexes have been extensively studied, underpinning the benefits and challenges of cooperativity between the metal centres.12–15 In 2014, inspired by the pioneer works in catalysis,16,17 we initiated a project to investigate the catalytic efficacy of well-characterised 3d/4f species for Lewis acid-promoted organic transformations.18 This process is challenging compared to the magnetic studies carried out in the solid state since imminently structural changes (ligand, solvent exchange or transformations) can occur upon desolvation. Our first trial, inspired by Batey's pioneer work,19 focused on developing heterometallic (3d/4f) moieties to convert furfural, a biomass derivative produced in million-ton quantities annually,20 into trans-4,5-diaminocyclopent-2-enones, a core which is the basis for several natural products (Table 1). This organic transformation proceeds via a domino path and generates water as a by-product.21 Our first effort incorporated weakly coordinating anions to facilitate, if any, ligand-substrate exchange and a tetranuclear heterometallic moiety built from an easy-to-make in gram scale ligand (E)-2-(2-hydroxy-3-methoxybenzylidene-amino)phenol (H2L) {[NiII2DyIII2L4(EtOH)6](ClO4)2·2(EtOH)}-OCl4/NiII2DyIII2.22,23,24 Post catalytic reactions, aiming to obtain a holistic understanding of the process and show whether OCl4/NiII2DyIII2 retains its structure, we noted an unexpected anion transformation (ClO4− to Cl−), which proved favourable for developing the catalytic protocol. The {[NiII2DyIII2L4Cl2(CH3CN)2]·2(CH3CN)}-Cl/NiII2DyIII2 derivative outperformed compared to Dy(OTf)3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 20 and OCl4/NiII2DyIII2. We then expanded our study to investigate the impact of the secondary coordination sphere and a Ln moiety but with insignificant improvement.25 In 2018, Afonso et al.26 reported a protocol that used Cu(OTf)2 as the catalyst, which proved inspirational for synthesising the {[Ni2Dy2L4(DMF)6] 2(OTf) 2(DMF)}-OTf/NiII2DyIII2 analogue.27 The use of the latter species significantly increases the reaction rate (three orders of magnitude less catalyst loading), and the new protocol also applies to primary amines and derivatives of furfural, reasoning our choice to develop this further. Aiming to increase our holistic understanding of this dynamic system behaviour, we (a) provided crystallographic evidence for the Stenhouse salt intermediate and (b) performed theoretical studies which suggested that Lewis acids affect the initial, that is, the ring-opening, path of this domino transformation followed by an energetically barrierless 4π-electrocyclization pathway.27 Control experiments prove the negligible contribution of the Ni moiety,25 therefore, based on the evidence we collected from these studies, we envisage the metal (Dy) and anion (OTf) cooperatively contribute to promoting this transformation and given the exciting recent advances in this field,28,29 more detailed studies are required to elucidate this mechanism fully.
20 and OCl4/NiII2DyIII2. We then expanded our study to investigate the impact of the secondary coordination sphere and a Ln moiety but with insignificant improvement.25 In 2018, Afonso et al.26 reported a protocol that used Cu(OTf)2 as the catalyst, which proved inspirational for synthesising the {[Ni2Dy2L4(DMF)6] 2(OTf) 2(DMF)}-OTf/NiII2DyIII2 analogue.27 The use of the latter species significantly increases the reaction rate (three orders of magnitude less catalyst loading), and the new protocol also applies to primary amines and derivatives of furfural, reasoning our choice to develop this further. Aiming to increase our holistic understanding of this dynamic system behaviour, we (a) provided crystallographic evidence for the Stenhouse salt intermediate and (b) performed theoretical studies which suggested that Lewis acids affect the initial, that is, the ring-opening, path of this domino transformation followed by an energetically barrierless 4π-electrocyclization pathway.27 Control experiments prove the negligible contribution of the Ni moiety,25 therefore, based on the evidence we collected from these studies, we envisage the metal (Dy) and anion (OTf) cooperatively contribute to promoting this transformation and given the exciting recent advances in this field,28,29 more detailed studies are required to elucidate this mechanism fully.
|
|
|||||
|---|---|---|---|---|---|
|
|
|
|
|||
| Compound | Dy(OTf)3 | OCl4-NiII2DyIII2 | Cl-NiII2DyIII2 | Cu(OTf)2 | OTf-NiII2DyIII2 |
| Catalyst loading (%) | 10 | 10 | 1 | 1 | 0.01 |
| Temperature | 80 °C | 80 °C | r.t. | r.t. | r.t. |
| Solvent | CH3CN | CH3CN | CH3CN | H2O | Solventless |
| Secondary amines (Y/N) | Y | Y | Y | Y | Y |
| Primary amines (Y/N) | N | Not applicable | Y | N | Y |
| Time | 16 h | 16 h | 2 h | 1 min | 40 min |
| Ref. | 19 | 22 | 22 | 26 | 27 |
Dynamic systems in bimetallic 3d/4f chemistry, case 2
We synthesised the isostructural compounds formulated as [ZnII2LnIII2L4(NO3)2(DMF)2] where Ln is Y (ZnY), Sm (ZnSm), Eu (ZnEu) Gd (ZnGd), Dy (ZnDy), Tb (ZnTb) and Yb (ZnYb) and tested their efficacy for the reaction between indole and benzaldehyde to provide the corresponding bis-indole.30 All compounds behave as Lewis Acids and catalyse the reaction at the same rate. However, aiming to obtain a holistic understanding of the process, we used the ZnY, ZnDy and ZnGd species to map the solid and solution structures and underpin mechanistic evidence for the organic transformation. ESI-MS data for the three compounds provided mono- and di-cationic fragments with perfect isotropic distributions. EPR studies of ZnGd in both solid and solution phases indicate that GdIII ion retains its coordination environment in solution. 1H NMR studies of the diamagnetic ZnY in solution show all characteristic peaks consistent with the solid-state structure, whereas variable temperature studies exclude complex decomposition. All these notions suggested that the tetranuclear entities retain their structure in the solution upon desolvation. Due to solubility issues, efforts to monitor the organic transformation and structural changes on the core of the tetranuclear entity via NMR proved insufficient. In contrast, UV-Vis studies of the bimetallic entities and the substrates identified that the aldehyde binds to Ln over Zn. Considering all these data and based on control experiments, we proposed a mechanistic pathway involving hemiketal formation. We then substituted the ligands and altered the transition element, retaining the tetranuclear core unaltered to expand our work on investigating the use of similar species as cooperative catalysts for highly diastereoselective Michael addition reactions (Fig. 2, left).31 Ligand modification does not impact the catalytic efficacy; however, tuning the metal centres has provided evidence that two metal centres act cooperatively to orchestrate these rapid and highly diastereoselective reactions. NMR, UV-Vis and EPR (Fig. 2, right) studies confirm structural integrity in the solution. Given the proximity of the two Lewis acidic metal centres in these structures, we proposed a cooperative mechanistic path.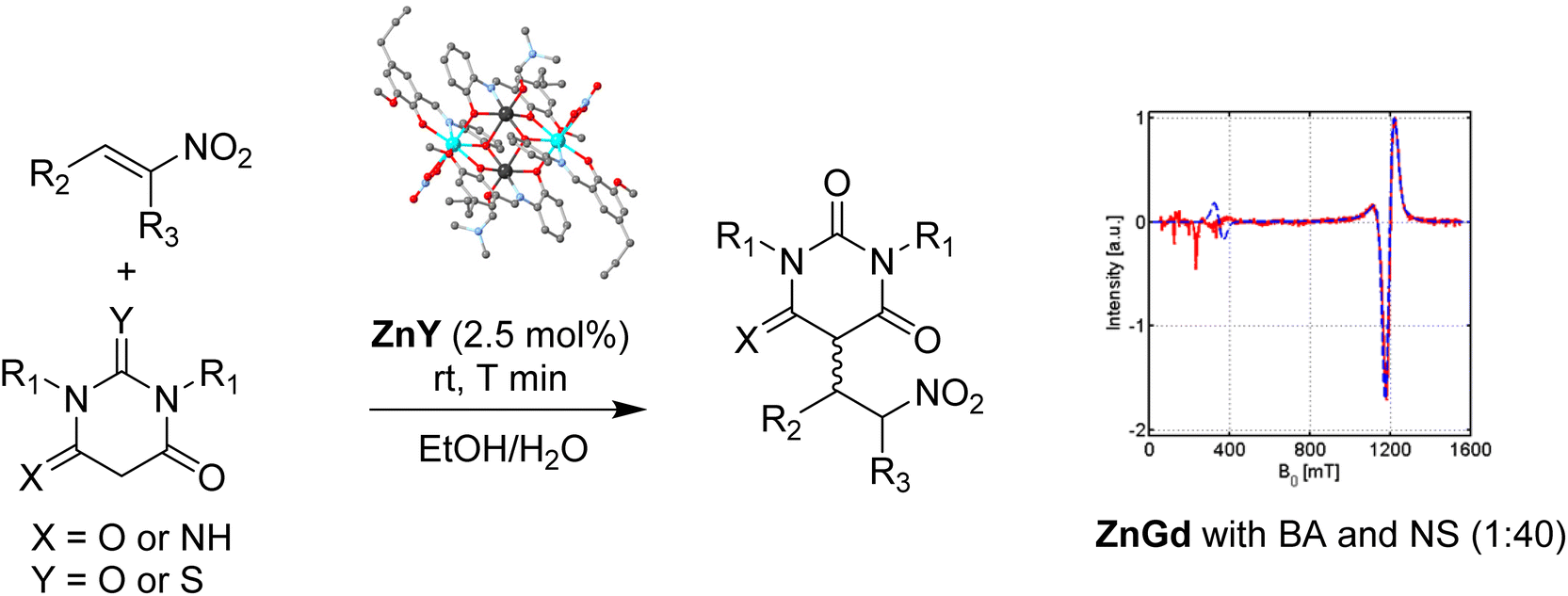 | ||
| Fig. 2 (Left) The Michael addition reaction between barbituric acid and nitrostyrene (right) the EPR data, in solution, of the ZnGd analogue in a catalytic amount 2.5:100:100 and fitting (red line) indicating the structural integrity. Adapted with permission from ref. 31. Copyright 2017 American Chemical Society. | ||
Dynamic systems in one-dimensional coordination polymers, case 1
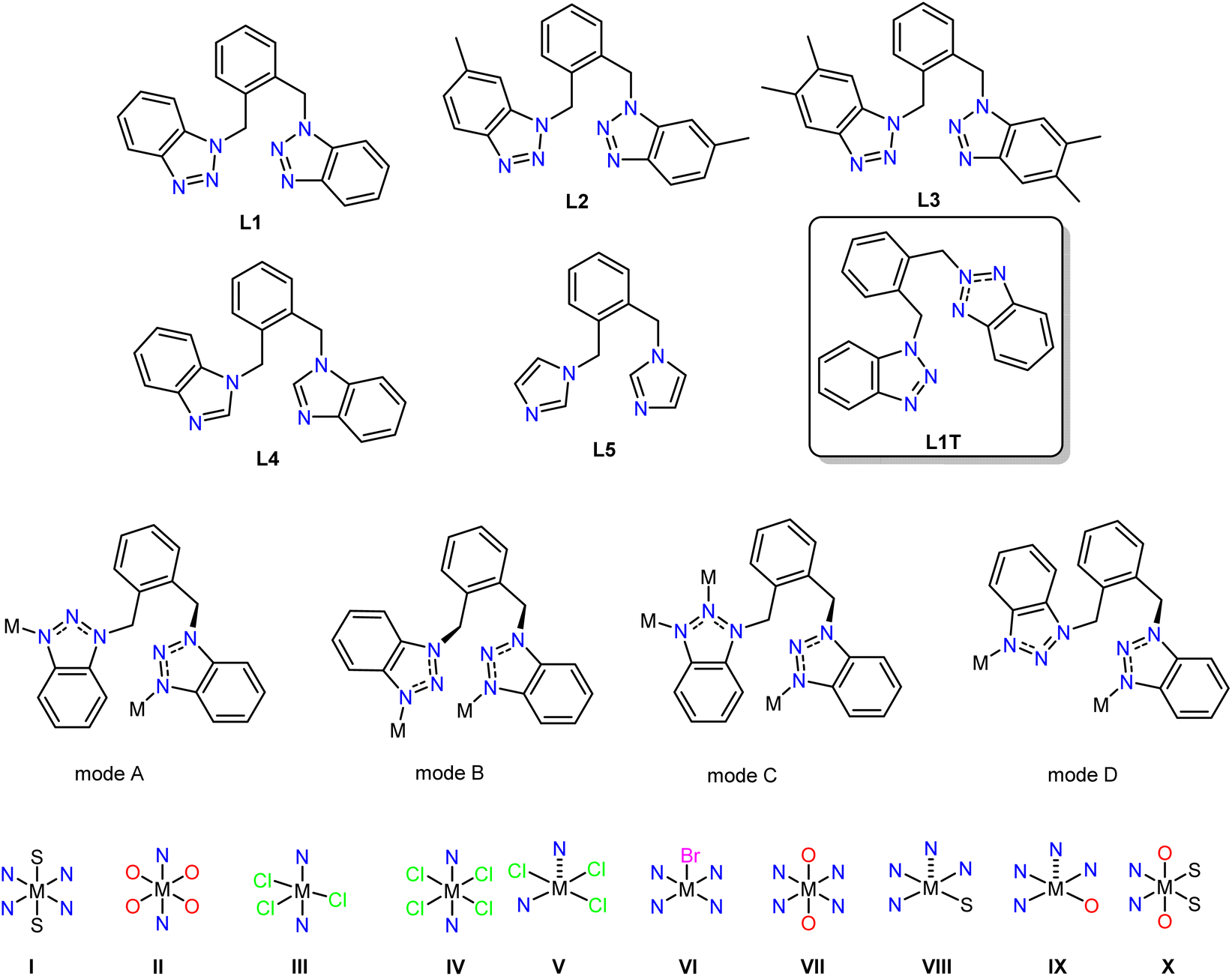
In 2011, Leong and Vittal reviewed the complexity and diversity in structure properties and application of one-dimensional coordination polymers, which proved inspirational for our work.32 The outcome of a complexation reaction of a previously unknown ligand and a metal salt is unpredictable, and the formation of several products is favourable. Several studies are required to set rules, limitations and barriers to their coordination chemistry. We investigated the coordination chemistry of the previously unknown neutral ligands shown in Table 2. And we studied the applications of the corresponding coordination compounds for the following reasons: (a) benzotriazole is an inexpensive redox heterocycle widely popular in organic and coordination chemistry,33,34 thus, the ligands can be produced on a multigram scale, and several control experiments can be performed to enhance our understanding, (b) introducing the xylyl–CH2 moieties imposes limited flexibility, thus restricting the numerous configurations of the formed coordination compounds and (c) the set of the chosen ligands imposes electronic effects that may affect the studied property. All the ligands can be synthesised in one high-yielding step, and we investigated their coordination behaviour with CuII salts.6,35,36 As anticipated, some ligands and coordination compounds exhibit tautomerism (for example, L1TTable 2) in the solid and solution phases, resulting from differentiating the synthetic protocols.37,38 However, we aimed to isolate ligands L1–L5, not the tautomer, and perform complexation reactions under the same conditions to isolate the topological equivalent components. These ligands can coordinate similarly, therefore, we screened the impact of several parameters on the final motif, i.e. metal salt, metal–ligand ratio, temperature and solvent. Table 2 summarises data for twelve compounds, which crystallise between two to seven days. Weakly binding anions result, in principle, in an octahedral geometry, in which the axial positions are occupied by solvent or anion moieties and the equatorial positions by four different ligands. Different morphologies are obtained when strongly or medium coordinating anions are used. The plurality in coordination modes and the coordination geometries signify the level of difficulty in controlling the synthesis and behaviour of these systems. We attempted to map the solid and solution structures with EPR35 and UV-Vis. However, solvation immediately changes the dynamic structure; therefore, we abandoned further attempts as time and resource-consuming. Given the challenging task of obtaining only one product from these very complicated systems, we decided to use only the crystalised forms, not the bulk powder formed from these reactions, for the catalytic studies.6,36,39,40|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Compound | Ligand | Coordination mode | Metal salt | Geometry of M(II) | Dimension |
| 1 | [CuII(L1)2(MeCN)2]·2(ClO4)·MeCN | L1 | A | Cu(OCl4)2 6H2O | I | 1D |
| 2 | [CuII(L1)(NO3)2]·MeCN | L1 | A | Cu(NO3)2 | II | 2D |
| 3 | [ZnII(L1)2(H2O)2]·2(ClO4)·2MeCN | L1 | A | Zn(ClO4)2·6H2O | I | 1D |
| 4 | [CuII(L1)2Cl2]2 | L1 | B | CuCl2 | III | 0D |
| 5 | [CuII5(L1)2Cl10] | L1 | C | CuCl2 | IV & V | 2D |
| 6 | [CuII2(L1)4Br2]·4MeCN·(CuII2Br6) | L1 | D | CuBr2 | VI | 0D |
| 7 | [CuII(L1)2(MeCN)2]·2(BF4) | L1 | A | Cu(BF4)2 | I | 1D |
| 8 | [CuII(L1)2(OTf)2] | L1 | A | Cu(OTf)2·H2O | VII | 1D |
| 9 | [CuII2(L2)4(H2O)2]·4(OTf)·4Me2CO | L2 | A | Cu(OTf)2·H2O | VIII | 0D (dimer) |
| 10 | [CuII2(L3)4(OTf)2]·2(OTf)·Me2CO | L3 | A | Cu(OTf)2·H2O | IX | 0D (dimer) |
| 11 | [CuII(L4)(MeCN)2(OTf)2] | L4 | A | Cu(OTf)2·H2O | X | 1D |
| 12 | [CuII(L5)2(OTf)2] | L5 | A | Cu(OTf)2·H2O | VII | 1D |
Our first, not chronologically communicated, diagnostic effort focused on exploring the unconventional use of compounds (Table 2, entries 1–8) in promoting the multicomponent amine, aldehyde, alkyne (A3) coupling reaction (Scheme 1, upper).35 This organic transformation involves an in situ imine formation with concomitant activation of the alkyne to yield the propargylamine, and it is well established to be promoted by CuI species.41 Our protocol required elevated temperatures, thus prohibiting monitoring with EPR, and the triflate component (entry 8) surpasses perchlorate (entry 1). Cyclic voltammetry of [CuII(L1)2(OTf)2] identified a reversible one-electron redox process. Given the required high loadings (5%) to promote the reaction, we proposed a possible mechanism with an in situ generated CuI species. To validate the in situ CuI generation notion, we performed a diagnostic study against the benchmark click reaction between alkyne and azide, and [CuII(L1)2(OTf)2] proved more efficient (Scheme 1, lower).36 Ligands L2 and L3 can coordinate similarly to L1, but their complexation reactions with the same metal salt yielded compounds of different dimensions, and coordination geometry, which performed poorly. This result signifies that the substitution impacts the performance due to structural differentiation and/or electronic effects. The performance of compounds formed by ligands L4 and L5 (the coordination compounds can be considered isostructural) was moderate, indicating possible electronic effects as the source of the different behaviour. Several scenarios (structural configurations) can be in place to promote this click reaction, but the in situ genesis of a {CuILx} species at elevated temperatures became evident.
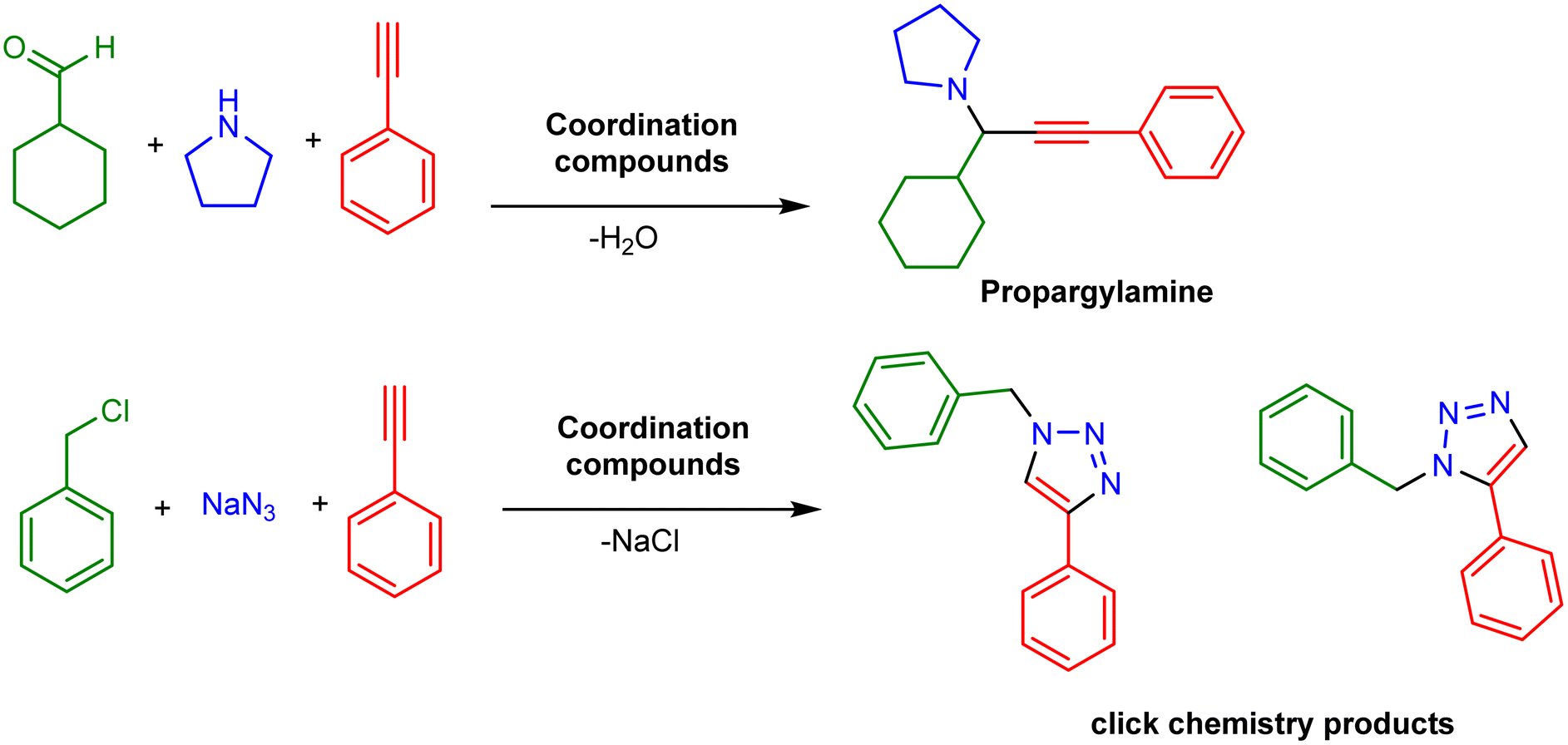 | ||
| Scheme 1 The two organic transformations that we tested the efficacy of compounds listed in Table 2. | ||
Dynamic systems in one-dimensional coordination polymers, case 2
The use of the compounds mentioned above found a good application in the following two examples. We used this family of compounds to catalyse a multicomponent reaction that involves aldehyde, amine, and nitrostyrene to synthesise pyrroles (Fig. 3).39,40 From the organic perspective, the protocols apply in an extended scope; however, from the Chemical Chartographisis point of view, that is, the holistic view mentioned earlier, the control experiments suggest that compound [CuII(L1)2(OTf)2] outperformed. It minimises by-product(s) formation and provides the anticipated products in yields over 85%. It is worth noting that (a) the control experiments with the metal salts and our compounds used a 20% and 1% loading, respectively, and (b) these reactions can be self-catalysed; however, using our compounds, efficacy significantly improves. The reactions are performed at elevated temperatures; thus, monitoring is inconvenient. However, we aim to develop a protocol that works at room temperature to underpin the input of every component in this reaction.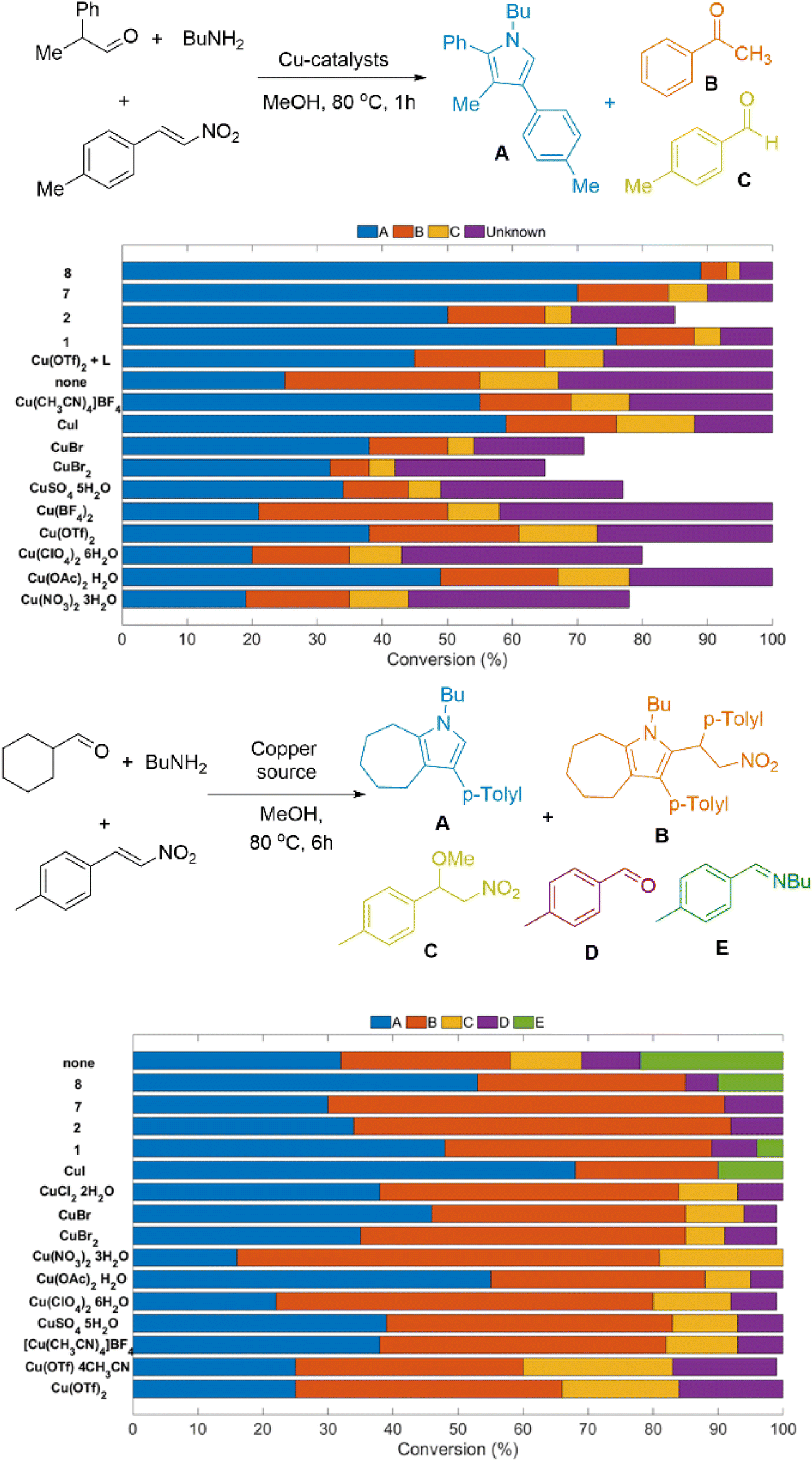 | ||
| Fig. 3 Control experiments for synthesising poly-substituted (upper) and seven-membered ring fused (lower) pyrroles. 1, 2, 7 and 8 correspond to entry numbers of Table 2. | ||
The second example relates to the unprecedented multicomponent synthesis of dihydropyridines, a product of the reaction 1,2-bis ((E)-4-methylbenzylidene) hydrazine (azine) and (m)ethyl propiolate (Fig. 4).6 The reaction of these two substrates provides two products: product A is the result of the presence of Cu source, which we call the catalytic product (Fig. 4) and product B, the non-catalytic, which is the result of the organic transformation that can occur at elevated temperatures (Fig. 4). Control experiments suggest that copper salts yield unknown products or decompose the starting material. We tested three compounds and various metal salts. Compounds [CuII(L1)2(MeCN)2]·2(ClO4)·MeCN and [ZnII(L1)2(H2O)2]·2(ClO4)·2MeCN are isoskeletal, altering only the metal centre (CuII to ZnII), whereas, compound [CuII(L1)(NO3)2]·MeCN, exhibits different coordination geometry and dimensionality. To our surprise, the reaction is promoted only by [CuII(L1)2(MeCN)2]·2(ClO4)·MeCN, in moderate yields. In contrast, the reaction with the ZnII analogue yields the non-catalytic product. The substrates remain intact when [CuII(L1)(NO3)2]·MeCN is used. The reactivity ceases when substituted, -at the ortho-position azines are incorporated-, imposing strong covalent (6-membered chelated ring, see Fig. 4 bottom right) Cu-azine bonding. The use of asymmetric azines, gave the expected products, suggesting they do not dissociate during the catalytic reaction. In contrast, the use of electron-donating additives retard the reaction. The functionality comparison of this set of equivalent topological systems rationalises the experimental data and suggests that covalent bonding of the substrate(s) to the Cu centre is vital. Notably, post-catalysis, we isolated and crystallographically characterised a yellow solid formulated as [CuIL1Cl], which is catalytically inactive. This finding indicates that during the organic transformation, ClO4− converts to Cl−; this anion transformation was observed in 3d/4f case 1 study. We envisage that Cl− binds to the in situ generated CuI species, thus terminating the catalytic process and explaining the moderate yields.42 More evidence and, if possible, reaction monitoring are required to validate the mechanism, and these studies are currently investigated.
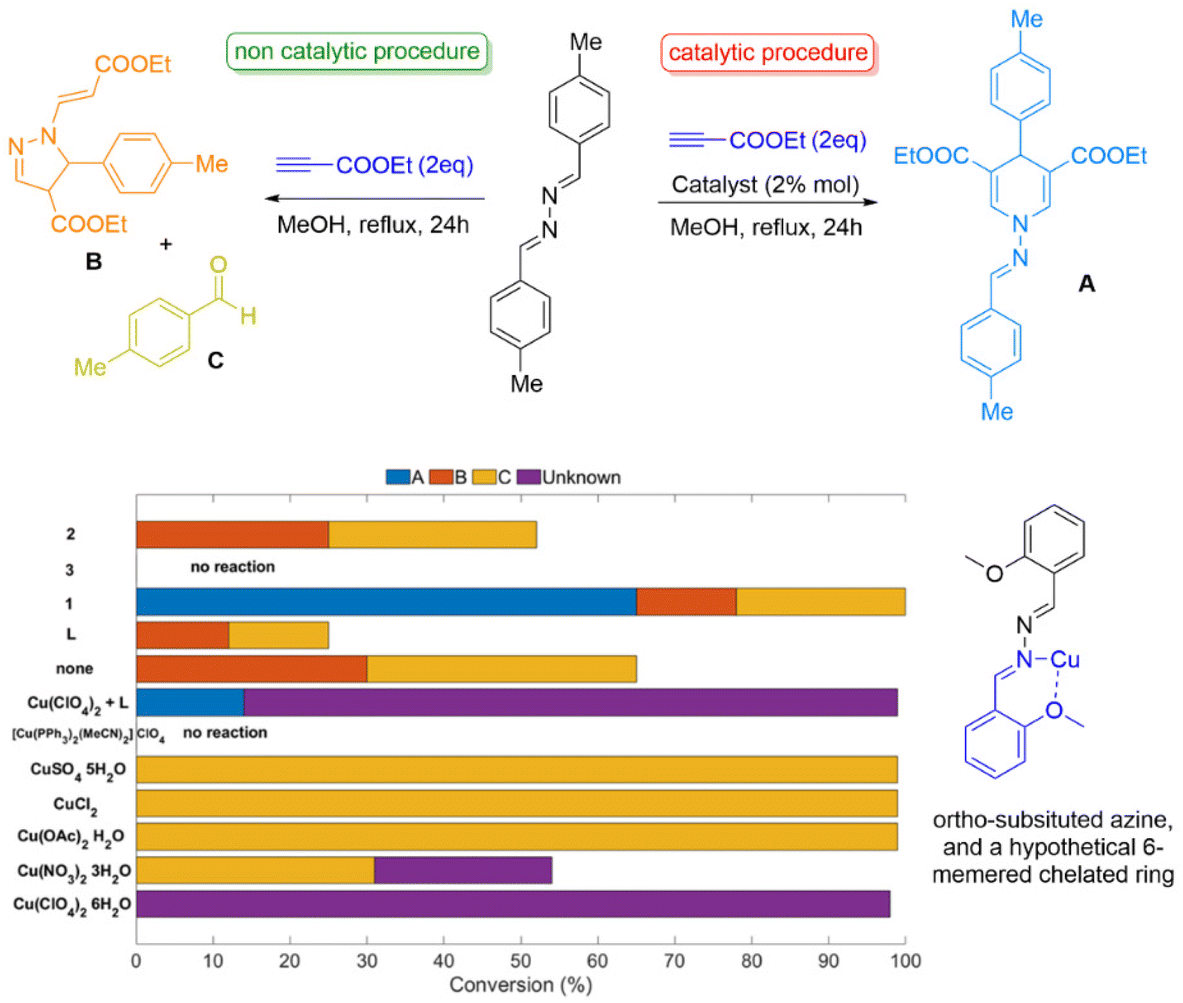 | ||
| Fig. 4 Control experiments for the synthesis of dihydropyridines. 1 corresponds to [CuII(L1)2(MeCN)2]·2(ClO4)·MeCN, 2 to [ZnII(L1)2(H2O)2]·2(ClO4)·2MeCN and 3 to [CuII(L1)(NO3)2]·MeCN (see Table 2 for more info). | ||
Dynamic systems in hybrid Cu-benzotriazole-pyridine systems
Inspired by the results mentioned above and identifying the numerous problems imposed by the flexible character of these dynamic systems, we embarked on investigating the use of other benzotriazole ligands. We focused on the coordination chemistry of the less investigated hybrid bidentate 1-(2-pyridyl)benzotriazole (pyb, Fig. 5, left),43 which was proved to be more electron-deficient when compared with that of 2,2′-bipyridine (bpy).44 We synthesised trans-[CuII(OTf)2(pyb)2]2(CH3CN) (Fig. 5 left), in which CuII sits in the centre of an almost ideal octahedron and adopts a trans geometry, whereas the bpy analogue gives compound cis-[CuII(OTf)2(bpy)2] (Fig. 5, left) in which the CuII centre adopts a cis geometry. The compound trans-[CuII(OTf)2(pyb)2]2(CH3CN) enables the A3 coupling in 1% loading, at room temperature and under N2 atmosphere; however, this is not feasible when the cis-[CuII(OTf)2(bpy)2] species is used. From the Chemical Chartographisis point of view, the holistic approach in investigating this dynamic system indicates that the stereochemistry of the pre-catalyst and the nature of the N,N′-bidentate ligand are essential parameters to consider when designing such molecules. An extensive set of control experiments suggested the in situ generation of a catalytic active [CuI(OTf)(pyb)] species (Fig. 5, right).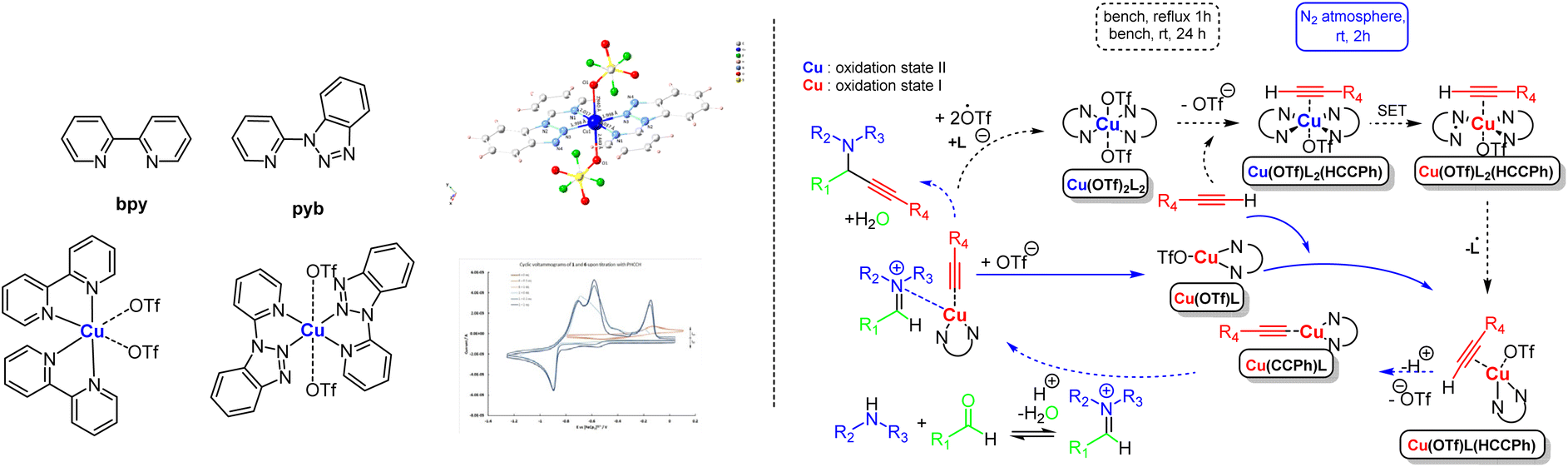 | ||
| Fig. 5 The complex and the proposed interlinked mechanism. Adapted with permission from ref. 43. Copyright 2021 Wiley Materials. | ||
Dynamic systems in Cu–salen systems
Inspired by well-characterised CuII–salen complexes that efficiently catalysed the A3 coupling reaction at elevated temperatures,45,46 we investigated this dynamic system and obtained mechanistic evidence to ease the process. We chose a well-known framework47 and repurposed its use for the following reasons: (a) this framework is known to generate radicals, a process that would be helpful for the reduction of the metallic centre from CuII to CuI, (b) the complex is neutral so we can discard anion transformation, (c) the use of the tert-butyl groups has a twofold character; to prevent self-poylmerisation48 and to increase solubility for organic solvents, aiming to use a non-coordinating solvent for the catalytic reaction. We followed already-known protocols to synthesise the chiral CuII complex (CuL6) (Fig. 6) where H2L6 is ligand formed from the condensation reaction of (1S,2S)-(−)-1,2-diaminocyclohexane with two equivalents of 3,5-di-tert-butyl-2-hydroxybenzaldehyde, in bulk and perform extensive control experiments and screenings.49 We developed two protocols, one under microwave conditions and one in the open air at room temperature, providing twenty propargylamines in excellent yields. In the latter protocol, the reactions are complete in 72 hours and depend on concentration. Reactions under N2 or Ar atmosphere yield the targeted propargylamines in the same yield as the reaction in the open air, indicating that atmospheric oxygen is not involved in the reaction. The recovered complex retained its structure and activity after ten runs (shown by single crystal X-ray diffraction). The reaction in the presence of a radical additive trap, TEMPO, failed to give the final product; this reaction was repeated four times to justify the notion, all giving the same response. Then, we synthesised the salan derivative (shown in grey in Fig. 6), which also promoted the reaction in excellent yields. All these experiments with the use of topological equivalent compounds identified that vital to the success of this protocol is the use of the phenoxido salen-based ligand, which orchestrates topological control permitting alkyne binding with concomitant activation of the C–H bond, the rate-determining step, and simultaneously acting as template temporarily accommodating the abstracted acetylenic proton, and continuous generating, via in situ formed radicals and Single Electron Transfer (SET) mechanism, of a transient CuI active site to facilitate this transformation. Theoretical calculations further supported our mechanistic proposition.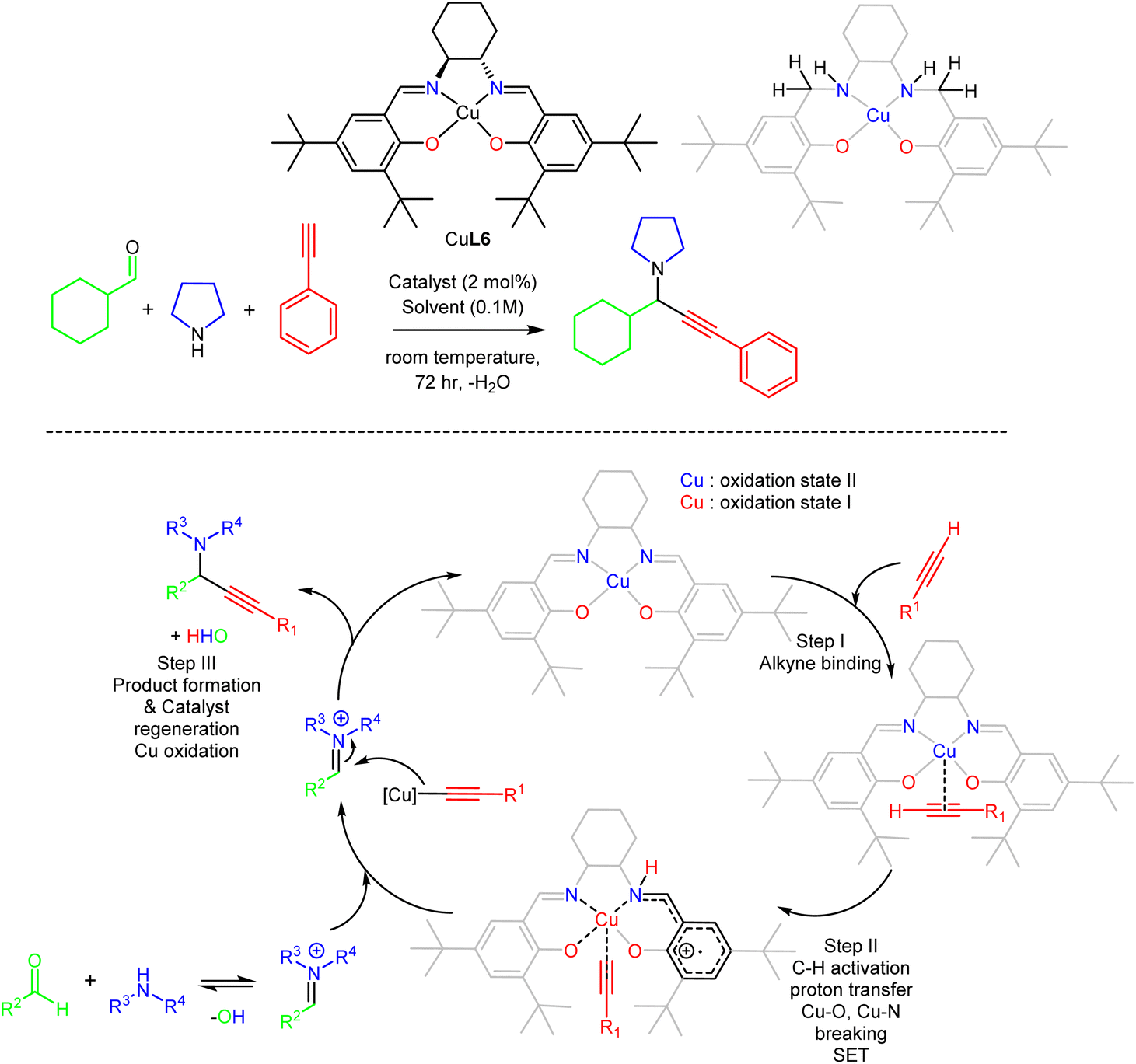 | ||
| Fig. 6 The proposed A3 coupling mechanism with the tetradentate-based CuII complex. Adapted from ref. 49 with permission from The Royal Society of Chemistry. | ||
From the coordination chemistry perspective, we established a convenient route to access asymmetric salan and salen frameworks in bulk.50 This route yielded several tetradentate asymmetric CuII salen/salan complexes, which were found to promote the A3 coupling reaction at the same rate and suggested mere electronic and/or steric effect(s) accounted for the rate. We concluded that using tetradentate salen frameworks is the limiting factor to improving catalytic efficacy, possibly because the ligand saturates the coordination sphere of the metal centre, and we hypothesised that a CuII complex built from a tridentate ligand and weakly coordinating anion would be an ideal component to overcome this obstacle and increase the reaction rate. With this in mind, we synthesised compound [CuII2(L7)2(μ2-Cl)Cl] in gram scale, where L7 is the anion of trans-2-(Aminocyclohexyl(imino)methyl)-4,6-di-tert-butylphenol, to permit extensive control experiments (Fig. 7).51 Single crystal structure determination suggests the presence of a dimeric species, with the chlorides acting as bridges in the solid state. However, upon dissolution in DCM, the compound exists in a monomeric [CuIIL7Cl] (85%), – dimeric (15%) equilibrium and treatment with phenylacetylene populates the monomeric species. This compound promotes the A3 reaction in 16 hours; however, this dynamic system is now sensitive to atmospheric oxygen; so, reactions should be carried out under N2 or Ar balloons. Monitoring, with EPR, of the complex/phenylacetylene interaction identifies the in situ transient radical generation, whereas monitoring with IR, identifies a weakening of the C–H alkyne bond. Finally, post-catalysis, crystallographic studies confirmed the framework's stability and showed that the metal centre preserves its oxidation state. From the organic perspective, this unusual C–H activation method is applicable for late stage functionalization of important bioactive molecules. We envisage that the radical initiates an alkyne C–H bond activation process via a four-membered ring (CuII–O⋯H–Calkyne) intermediate. In all, the functionality comparison of these non equivalent topological systems (Fig. 6 & 7) suggest the design of the ligand (number of heteroatoms) was the key to minimizing the time of the reaction).
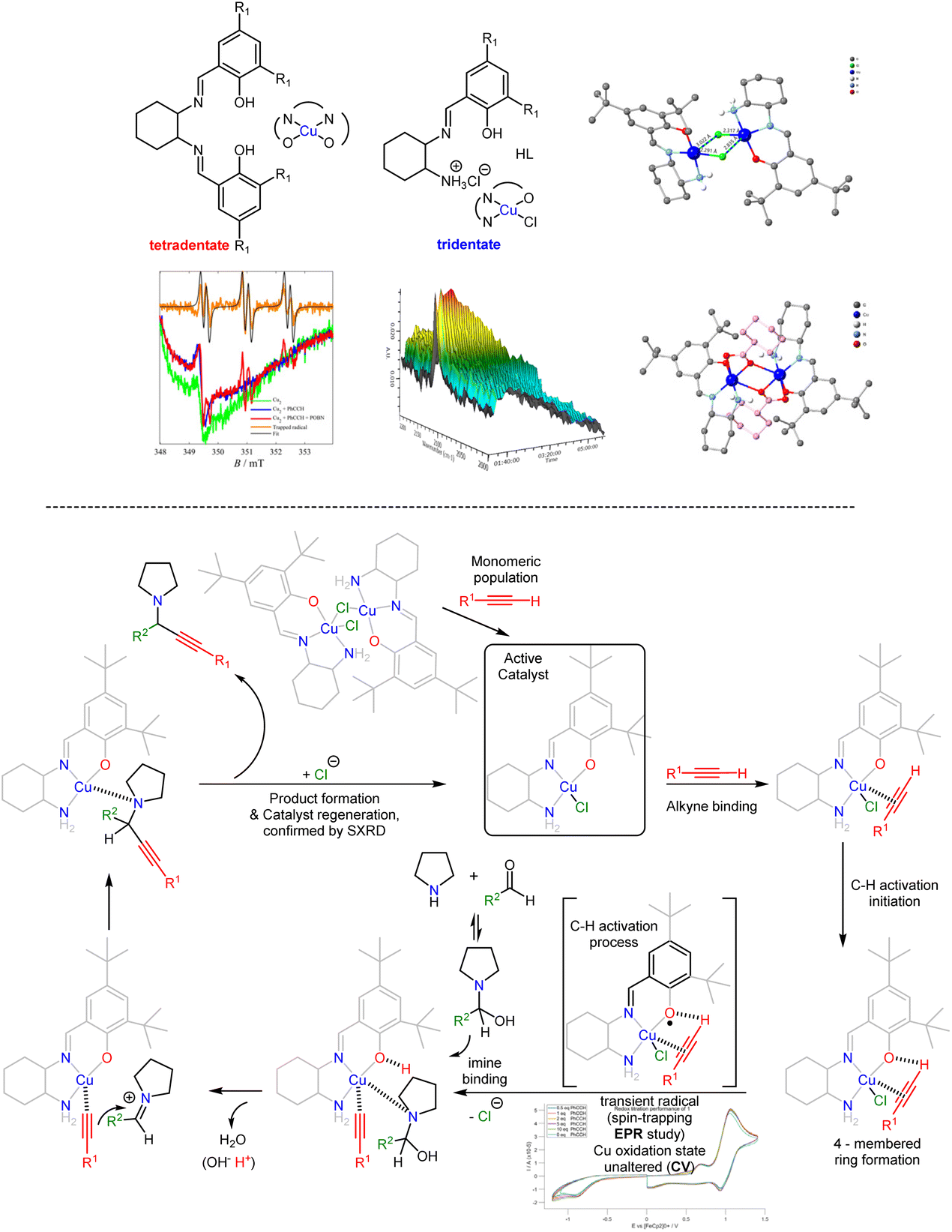 | ||
| Fig. 7 The proposed A3 coupling mechanism with the tridentate-based CuII complex. Adapted from ref. 51 with permission from Copyright 2021 American Chemical Society. | ||
The second part of this perspective focuses on the biological aspects of Chemical Chartographisis.
Dynamic systems in 3d/4f species and peptidic systems
Building on our results on bimetallic 3d/4f systems that retain their structure in solution, we investigated their ability to form covalent complexes with biomolecules.52 Hexapeptides have been particularly useful in forming highly ordered amyloid cores, and a cross-β structural model for the self-assembling peptide sequence HIS-TYR-PHE-ASN-ILE-PHE (HYFNIF) has been proposed.53 This amyloid-forming peptide contains groups ideal for functionalisation, including histidine residues, the most widely studied in coordination chemistry, due to their presence in vital metalloproteins.54 We considered the centrosymmetric Zn2Dy2 entity an ideal multimetal group to bind with histidine or tyrosine-containing biomolecules. It comprises organic ligands which partially saturate the coordination environment of all metal centres, and the metal centres in the Zn2Dy2 unit are quite close (3.3 Å). Dy is oxophilic, and Zn can coordinate to N-atoms, thus permitting either the Zn (A), or Dy (B) or both (A & B) centres to bind to one or two positions in the biomolecule (Fig. 8, left). A range of techniques suggests that despite the significant flexibility the Zn2Dy2 moiety possesses, and the use of protic solvent for the assembly, successful binding to the HYFNIF fibril is achieved. The architecture of the HYFNIF fibril remains unchanged as a core template. The success of the proposed methodology is attributed to the limited coordination sites in the Zn2Dy2 entity available for coordination with HYFNIF and the presence of the N-donor histidine groups. Lastly, we showed that the functionalized HYFNIF-Zn2Dy2 entity behaves as a Lewis acid.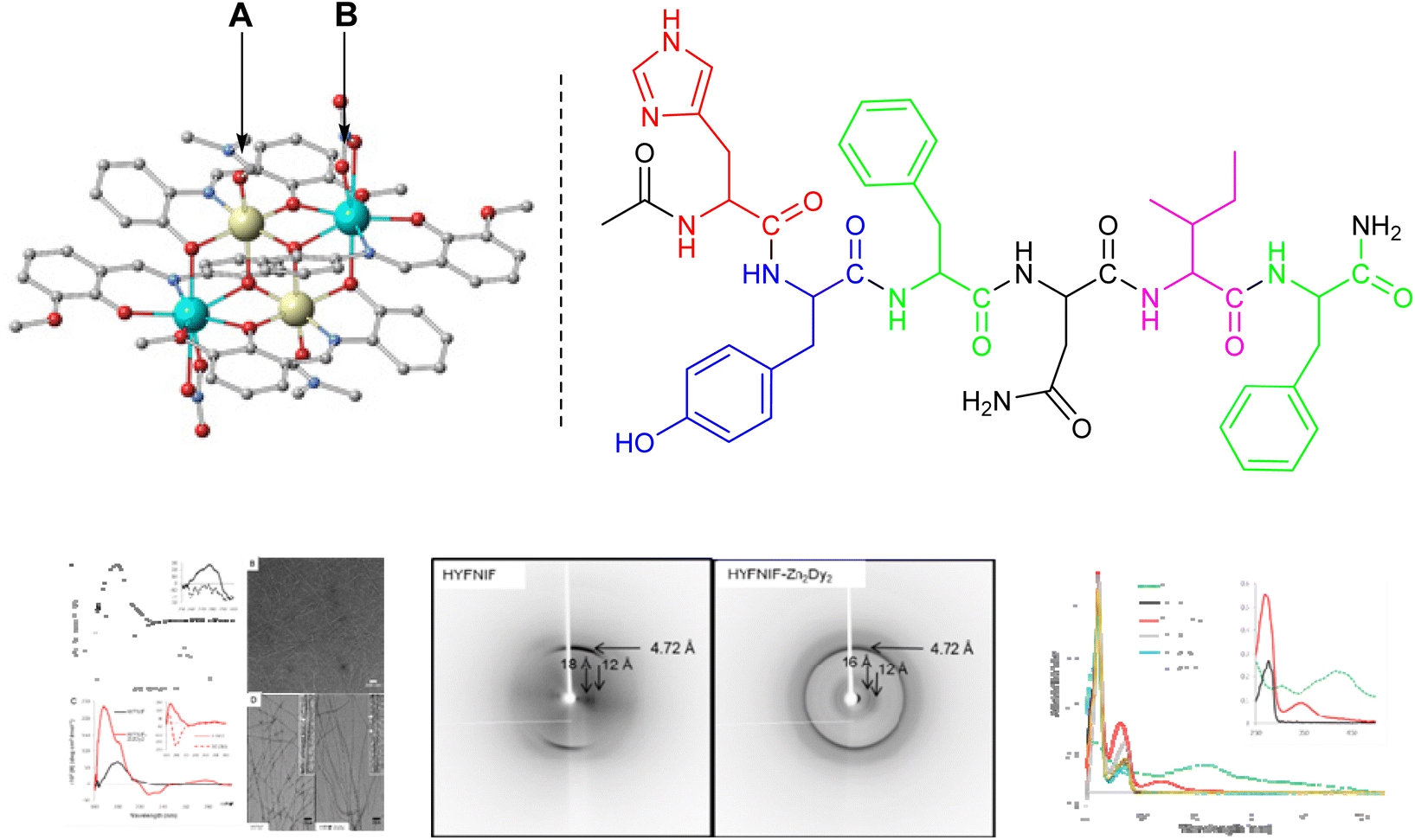 | ||
| Fig. 8 The Zn2Dy2 molecule and HYFNIF and characterisation of the resulting entity. Adapted with permission from ref. 52 with permission from The Royal Society of Chemistry. | ||
Dynamic systems in 3d/4f species for amine sensing
Swager55 on a square Pd complex and Song56 on an octahedral Rh complex (Fig. 9) described a 19F chemosensory method to detect multiple chiral amines. Both methods depend on a host–guest complex through interaction between a chiral substrate sample and a chiral detector molecule that transfers chiral information and induces a change in the chiral environment, which is observed as split signals of precise chemical shifts in the corresponding NMR spectrum. The broad detection window of 19F NMR gives easily distinguishable split signals and narrow overlapping resonances, increasing spectral resolution and providing simple deconvolution of complicated spectra. Following these results and given our excellent understanding of 3d/4f dynamic systems, we thought that the introduction of several F-antenna into a propeller-shaped bimetallic molecule,57–59 and synthesis of the F–CoIII3YIII analogue, would represent an ideal platform for amine sensing.60 Our hypothesis was based on the following points: (a) the high ionic radii of 4f ions make it difficult to control ligand exchange with solvent-substrates. (b) Amines are known to coordinate to lanthanide metal centres in organic media;61,62 (c) the usual coordination geometry of the Co centres is realised, and the 4f ion is captured in a trigonal antiprism, which limits access to solvent–substrate molecules via only axial sites, thus replicating the coordination sphere of the non-labile Pd and Ru analogues.55,56 The F–CoIII3YIII complexes are air-stable and easy to make, built from non-toxic and inexpensive metals and retain their structure in the solution for a prolonged period. Extensive control experiments, further supported by theoretical calculations, suggest that the complex can sense a specific type of analyte bearing an NH2CX–CHX–OH sequence via a dynamic ligand exchange mechanism (Fig. 9, bottom right). The method has a limited analyte scope but imposes an extreme sensor:analyte ratio (1:20 or 1:50) and broad sensing windows (8 ppm over 0.8 ppm, as seen in other studies), which are better than those for other techniques. In all, the functionality comparison of these equivalent topological systems suggests that these systems should be possible to detect and recognize organic or biological molecules bearing this specific motif at millimolar concentrations.
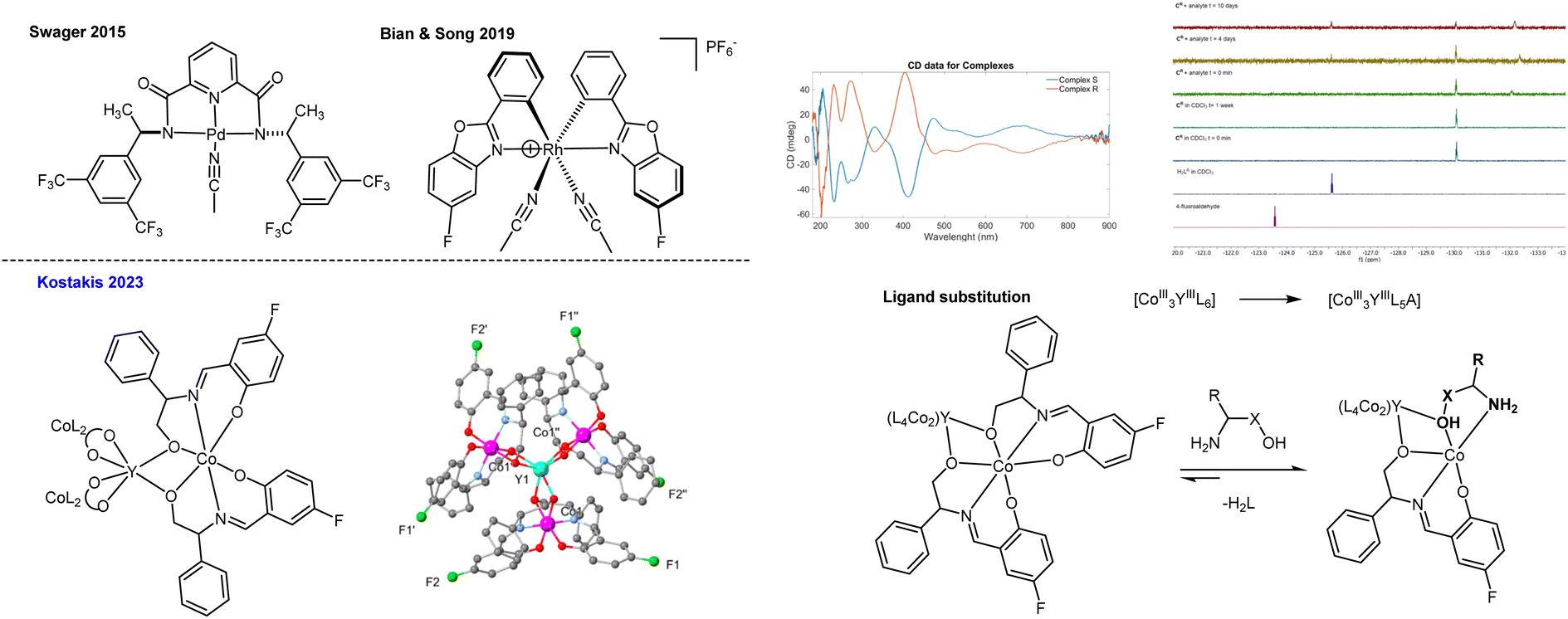 | ||
| Fig. 9 Previously known complexes built from non-labile complexes and the F–CoIII3YIII propeller-shaped complex that senses some amines via a very slow ligand substitution process. Adapted from ref. 60 with permission from Copyright 2023 American Chemical Society. | ||
Dynamic systems in developing Cu selective chelators
The last project relates to developing efficient and selective copper chelators. This challenging task finds applications in several biological (dys)functions. Hureau et al. reviewed the pros and cons of several organic scaffolds to selectively bind to Cu ions.63 The majority of these scaffolds build on hydroxyquinoline frameworks; however we noted that the symmetric tetradentate N4 Bis(pyridine), (ENDIP),64 and N2O2 tetrahydrosalen (Salan) ligands65 have very good selectivity for CuII. When considering a biological application, the organic framework should bear both hydrophilic and hydrophobic groups to permit studies in aqueous media and cell penetration. The salan frame has been decorated with carbohydrate66 or sulfonate groups,67 but these frameworks are expected to result in poor Blood Brain Barrier permeability. From the functionality comparison of the previous known dynamic systems and our expertise in accessing asymmetric salan frameworks in bulk,50 we rationally designed a highly modifiable tetradentate N3O copper chelating scaffold, Salpyran. This exhibits selectivity comparable to the state-of-the-art components with CuII selectivity over ZnII (Fig. 10).68 Solid and solution studies corroborate variation in coordination behaviour at different pH values but confirm the existence of only one dominant species at physiological pH values in aqueous solutions. Under physiological pH values and anaerobic conditions, the [CuIIsalpyran]+ complex remains intact for at least two days, but an oxidation procedure occurs in the presence of H2O2. Antioxidant studies, including ascorbate, tau, and human prion protein assays, reveal that Salpyran prevents the formation of reactive oxygen species from the binary CuII/H2O2 system. Notably, the species distribution of the binary Cu–salpyran system significantly changes when the investigation takes place in a mixture of DMSO–H2O(Fig. 10). This notion should be considered when applying any chelator for medical treatment(s). Due to its drug-likeness, desirable coordination behaviour, high hydrophilicity, antioxidant properties, and tunability, Salpyran is an alternative scaffold to 8-hydroxy/aminoquinolines for further pharmaceutical development of CuII targeting drugs.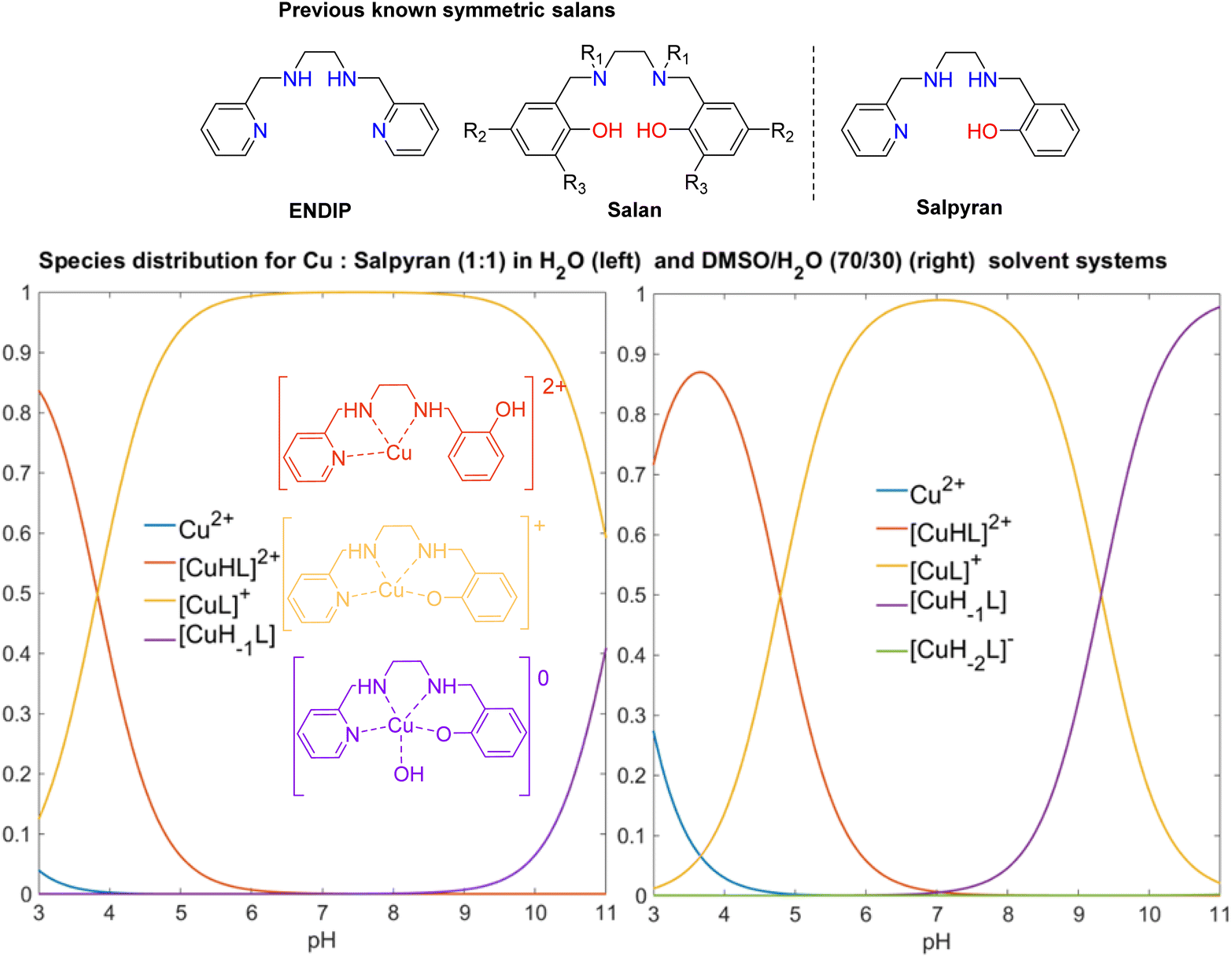 | ||
| Fig. 10 A comparison of the known tetradentate ligands and salpyran and species distribution of the Cu:salpyran dynamic system in 1:1 ratio at a different solvent system, emphasising the solvent impact on species formation. Adapted from ref. 68 with permission from Copyright 2021 American Chemical Society. | ||
Features
In the initial steps of our work, use of easy-to-make ligands and complexes allowed us to test the hypothesis and give enough evidence to support and develop the concept of Chemical Chartographisis. As the idea matured, it became evident that we could minimise the synthetic time by expanding the number of control experiments using equivalent topological systems. The examples above draw attention to vital points not accounted for when designing a chelator or a complex or catalytic experiment. The lessons, not possible to be digitalised at the moment, learnt by systematising and classifying the operational parameters for every individual system are as follows:(a) We noted twice, at different paradigms (Dynamic systems in bimetallic 3d/4f chemistry, case 1 & Dynamic systems in one-dimensional coordination polymers, case 2), the conversion of perchlorate to chloride during a catalytic reaction at elevated temperatures; this notion can be vital for the reaction(s) but neglected when drawing organic reactions that occur at elevated temperatures,
(b) The substitution of the benzotriazole moiety (Dynamic systems in one-dimensional coordination polymers, case 1) may induce electronic effects affecting the rate of the catalytic reaction, but our evidence suggests the formation of a completely different species compared to the parent-targeted molecules,
(c) The use of metal salts for organic transformations, especially multicomponent reactions, may not always be a panacea; the formation of dihydropyridines is only promoted by the well-characterized 1D polymers (Dynamic systems in one-dimensional coordination polymers, case 2),
(d) We noted that the stereochemistry (trans–cis) of CuII complexes built by bidentate ligands plays a vital role in the C–H activation (Dynamic systems in hybrid Cu–benzotriazole–pyridine systems) of a specific system; this notion may be discovered in other paradigms.
(e) An elegant ligand design may allow the use of CuII complexes for the activation of alkynes at room temperature, in contrast to already known protocols that account CuI species; the recovered complexes identify that the metal centre retains its oxidation state (Dynamic systems in Cu–salen systems),
(f) Polynuclear species can be used to sense amines with 19F NMR, but the mechanism involves a slow ligand exchange process rather than via an expected covalent bond formation (Dynamic systems in 3d/4f species for amine sensing),
(g) Solvent variation differentiates the species distribution (Fig. 10) when investigating the potentiometric behaviour of a chelator; this notion should be strongly accounted for when aiming to design chelators for treatment/biological purposes (Dynamic systems in developing Cu selective chelators).
Conclusions
Chemical Chartographisis complements and extends established approaches to molecular design and synthesis. As it systematically draws on data from previous projects – whether used or not in the original context – to inform and shape new areas of interest, it is especially likely to find applications in blue sky projects. It can potentially repurpose information, thus giving coordination chemistry new pnoea. As the three examples given in this perspective article have demonstrated, current technical advances in characterisation and monitoring both transient species and product composition have made exploration of further similar projects particularly timely. We suggest that Chemical Chartographisis has the potential to become an evidence-based strategy for synthesis development, not only for systems where isolable intermediates can be characterised, but also for one-pot cascade reactions in solution and reactions on transient biological surfaces.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work summarised in this perspective is mainly based on the evidence collected by five talented scientists, the real heroes: Kieran Griffiths, Edward Loukopoulos, Stavroula Sampani and Jack Devonport (PhD) and Gabrielle Audsley (MChem). Ultimate thanks to all collaborators, especially in-house John Spencer, Alfredo Vargas, Louise Serpell, Cristina Pubill-Ulldemolins, and overseas Ioannis Lykakis (Thessaloniki, Greece) and Csilla Kállay (Debrecen, Hungary), who have contributed to receiving evidence and developing this discovery method. Special thanks to John F. C. Turner and David Smith for valuable intellectual discussions that helped the idea mature and develop. We thank the EPSRC (UK, grant number EP/M023834/1) and the University of Sussex for financial support (K. G., E. L., J. D PhD).References
- E. C. Constable and C. E. Housecroft, Chem. Soc. Rev., 2013, 42, 1429–1439 RSC.
- G. B. Kauffman, in Encyclopedia of Inorganic and Bioinorganic Chemistry, John Wiley & Sons, Ltd, 2011 Search PubMed.
- P. Cieslik, P. Comba, B. Dittmar, D. Ndiaye, É. Tóth, G. Velmurugan and H. Wadepohl, Angew. Chem., Int. Ed., 2022, 61, e202115580 CrossRef CAS PubMed.
- M. Okamura, M. Kondo, R. Kuga, Y. Kurashige, T. Yanai, S. Hayami, V. K. K. Praneeth, M. Yoshida, K. Yoneda, S. Kawata and S. Masaoka, Nature, 2016, 530, 465–468 CrossRef CAS PubMed.
- E. V. Van Der Eycken and U. K. Sharma, Multicomponent Reactions towards Heterocycles, Wiley, 2021 Search PubMed.
- M. Kallitsakis, E. Loukopoulos, A. Abdul-Sada, G. J. Tizzard, S. J. Coles, G. E. Kostakis and I. N. Lykakis, Adv. Synth. Catal., 2017, 359, 138–145 CrossRef CAS.
- D. Ndiaye, P. Cieslik, H. Wadepohl, A. Pallier, S. Même, P. Comba and É. Tóth, J. Am. Chem. Soc., 2022, 144, 22212–22220 CrossRef CAS PubMed.
- I. Langmuir, Science, 1921, 54, 59–67 CrossRef CAS PubMed.
- K. Wade, J. Chem. Soc. D, 1971, 792–793 RSC.
- H. Kim, S. Yang, S. R. Rao, S. Narayanan, E. A. Kapustin, H. Furukawa, A. S. Umans, O. M. Yaghi and E. N. Wang, Science, 2017, 356, 430–434 CrossRef CAS PubMed.
- K. Griffiths, V. N. Dokorou, J. Spencer, A. Abdul-Sada, A. Vargas and G. E. Kostakis, CrystEngComm, 2016, 18, 704–713 RSC.
- Y. Peng and A. K. Powell, Coord. Chem. Rev., 2021, 426, 213490 CrossRef CAS.
- Y. Peng, H. Kaemmerer and A. K. Powell, Chem. – Eur. J., 2021, 27, 15044–15066 CrossRef PubMed.
- K. Liu, W. Shi and P. Cheng, Coord. Chem. Rev., 2015, 289290, 74–122 CrossRef.
- H. Li, X. Meng, M. Wang, Y. X. Wang, W. Shi and P. Cheng, Chin. J. Chem., 2019, 37, 373–377 CrossRef CAS.
- S. Handa, V. Gnanadesikan, S. Matsunaga and M. Shibasaki, J. Am. Chem. Soc., 2007, 129, 4900–4901 CrossRef CAS PubMed.
- G. Maayan and G. Christou, Inorg. Chem., 2011, 50, 7015–7021 CrossRef CAS PubMed.
- K. Griffiths and G. E. Kostakis, Dalton Trans., 2018, 47, 12011–12034 RSC.
- S.-W. Li and R. A. Batey, Chem. Commun., 2007, 3759–3761 RSC.
- J. P. Lange, E. Van Der Heide, J. Van Buijtenen and R. Price, ChemSusChem, 2012, 5, 150–166 CrossRef CAS PubMed.
- C. Verrier, S. Moebs-Sanchez, Y. Queneau and F. Popowycz, Org. Biomol. Chem., 2018, 16, 676–687 RSC.
- K. Griffiths, C. W. D. Gallop, A. Abdul-Sada, A. Vargas, O. Navarro and G. E. Kostakis, Chem. – Eur. J., 2015, 21, 6358–6361 CrossRef CAS PubMed.
- K. C. Mondal, G. E. Kostakis, Y. Lan, W. Wernsdorfer, C. E. Anson and A. K. Powell, Inorg. Chem., 2011, 50, 11604–11611 CrossRef CAS PubMed.
- K. C. Mondal, A. Sundt, Y. Lan, G. E. Kostakis, O. Waldmann, L. Ungur, L. F. Chibotaru, C. E. Anson and A. K. Powell, Angew. Chem., Int. Ed., 2012, 51, 7550–7554 CrossRef CAS PubMed.
- K. Griffiths, P. Kumar, J. D. Mattock, A. Abdul-Sada, M. B. Pitak, S. J. Coles, O. Navarro, A. Vargas and G. E. Kostakis, Inorg. Chem., 2016, 55, 6988–6994 CrossRef CAS PubMed.
- R. F. A. Gomes, N. R. Esteves, J. A. S. Coelho and C. A. M. Afonso, J. Org. Chem., 2018, 83, 7509–7513 CrossRef CAS PubMed.
- S. I. Sampani, A. McGown, A. Vargas, A. Abdul-Sada, G. J. Tizzard, S. J. Coles, J. Spencer and G. E. Kostakis, J. Org. Chem., 2019, 84, 6858–6867 CrossRef CAS PubMed.
- R. F. A. Gomes, B. M. F. Gonçalves, K. H. S. Andrade, B. B. Sousa, N. Maulide, G. J. L. Bernardes and C. A. M. Afonso, Angew. Chem., Int. Ed., 2023, e202304449 CAS.
- R. F. A. Gomes, J. R. Vale and C. A. M. Afonso, Org. Lett., 2023, 25, 4188–4192 CrossRef CAS PubMed.
- K. Griffiths, P. Kumar, G. R. Akien, N. F. Chilton, A. Abdul-Sada, G. J. Tizzard, S. J. Coles and G. E. Kostakis, Chem. Commun., 2016, 52, 7866–7869 RSC.
- K. Griffiths, A. C. Tsipis, P. Kumar, O. P. E. Townrow, A. Abdul-Sada, G. R. Akien, A. Baldansuren, A. C. Spivey and G. E. Kostakis, Inorg. Chem., 2017, 56, 9563–9573 CrossRef CAS PubMed.
- W. L. Leong and J. J. Vittal, Chem. Rev., 2011, 111, 688–764 CrossRef CAS PubMed.
- E. F. V. Scriven and C. A. Ramsden, Heterocyclic chemistry in the 21st Century : a tribute to Alan Katritzky, Elsevier, 1st edn, 2017 Search PubMed.
- E. Loukopoulos and G. E. Kostakis, Coord. Chem. Rev., 2019, 395, 193–229 CrossRef CAS.
- E. Loukopoulos, M. Kallitsakis, N. Tsoureas, A. Abdul-Sada, N. F. Chilton, I. N. Lykakis and G. E. Kostakis, Inorg. Chem., 2017, 56, 4898–4910 CrossRef CAS PubMed.
- E. Loukopoulos, A. Abdul-Sada, G. Csire, C. Kállay, A. Brookfield, G. J. Tizzard, S. Coles, I. Lykakis and G. E. Kostakis, Dalton Trans., 2018, 47, 10491–10508 RSC.
- F. Tomás, J. L. M. Abboud, J. Laynez, R. Notario, L. Santos, S. O. Nilsson, J. Catalán, R. M. Claramunt and J. Elguero, J. Am. Chem. Soc., 1989, 111, 7348–7353 CrossRef.
- G. E. Kostakis, P. Xydias, E. Nordlander and J. C. Plakatouras, Inorg. Chim. Acta, 2012, 383, 327–331 CrossRef CAS.
- D. Andreou, M. G. Kallitsakis, E. Loukopoulos, C. Gabriel, G. E. Kostakis and I. N. Lykakis, J. Org. Chem., 2018, 83, 2104–2113 CrossRef CAS PubMed.
- D. Andreou, N. B. Essien, C. Pubill-Ulldemolins, M. A. Terzidis, A. N. Papadopoulos, G. E. Kostakis and I. N. Lykakis, Org. Lett., 2021, 23, 6685–6690 CrossRef CAS PubMed.
- J. Farhi, I. N. Lykakis and G. E. Kostakis, Catalysts, 2022, 12, 660 CrossRef CAS.
- The redox potential of CuII to CuI is +0.15eV, and for ClO4 to Cl is +0.65eV.
- S. I. Sampani, V. Zdorichenko, J. Devonport, G. Rossini, M. C. Leech, K. Lam, B. Cox, A. Abdul-Sada, A. Vargas and G. E. Kostakis, Chem. – Eur. J., 2021, 27, 4394–4400 CrossRef CAS PubMed.
- C. Richardson and P. J. Steel, Dalton Trans., 2003, 34, 992–1000 RSC.
- B. Agrahari, S. Layek, R. Ganguly and D. D. Pathak, New J. Chem., 2018, 42, 13754–13762 RSC.
- M. Tajbaksh, M. Farhang, H. R. Mardani, R. Hosseinzadeh and Y. Sarrafi, Chin. J. Catal., 2013, 34, 2217–2222 CrossRef CAS.
- W. Zhang, J. L. Loebach, S. R. Wilson and E. N. Jacobsen, J. Am. Chem. Soc., 1990, 112, 2801–2803 CrossRef CAS.
- D. S. Peters, F. E. Romesberg and P. S. Baran, J. Am. Chem. Soc., 2018, 140, 2072–2075 CrossRef CAS PubMed.
- S. I. Sampani, V. Zdorichenko, M. Danopoulou, M. C. Leech, K. Lam, A. Abdul-Sada, B. Cox, G. J. Tizzard, S. J. Coles, A. Tsipis and G. E. Kostakis, Dalton Trans., 2020, 49, 289–299 RSC.
- J. Devonport, J. Spencer and G. E. Kostakis, Dalton Trans., 2021, 50, 12069–12073 RSC.
- J. Devonport, L. Sully, A. K. Boudalis, S. Hassell-Hart, M. C. Leech, K. Lam, A. Abdul-Sada, G. J. Tizzard, S. J. Coles, J. Spencer, A. Vargas and G. E. Kostakis, JACS Au, 2021, 1, 1937–1948 CrossRef CAS PubMed.
- S. I. Sampani, Y. K. Al-Hilaly, S. Malik, L. C. Serpell and G. E. Kostakis, Dalton Trans., 2019, 48, 15371–15375 RSC.
- K. L. Morris, A. Rodger, M. R. Hicks, M. Debulpaep, J. Schymkowitz, F. Rousseau and L. C. Serpell, Biochem. J., 2013, 450, 275–283 CrossRef CAS PubMed.
- M. Laitaoja, J. Valjakka and J. Jänis, Inorg. Chem., 2013, 52, 10983–10991 CrossRef CAS PubMed.
- Y. Zhao and T. M. Swager, J. Am. Chem. Soc., 2015, 137, 3221–3224 CrossRef CAS PubMed.
- W. Wang, X. Xia, G. Bian and L. Song, Chem. Commun., 2019, 55, 6098–6101 RSC.
- Y. Zhang, J. P. Calupitan, T. Rojas, R. Tumbleson, G. Erbland, C. Kammerer, T. M. Ajayi, S. Wang, L. A. Curtiss, A. T. Ngo, S. E. Ulloa, G. Rapenne and S. W. Hla, Nat. Commun., 2019, 10, 1–9 CrossRef PubMed.
- Y. Shimizu, Y. Shoji, D. Hashizume, Y. Nagata and T. Fukushima, Chem. Commun., 2018, 54, 12314–12317 RSC.
- A. Cornia, M. Mannini, R. Sessoli and D. Gatteschi, Eur. J. Inorg. Chem., 2019, 552–568 CrossRef CAS.
- G. Audsley, H. Carpenter, N. B. Essien, J. Lai-Morrice, Y. Al-Hilaly, L. C. Serpell, G. R. Akien, G. J. Tizzard, S. J. Coles, C. P. Ulldemolins and G. E. Kostakis, Inorg. Chem., 2023, 62, 2680–2693 CrossRef CAS PubMed.
- G. K. Veits and J. Read de Alaniz, Tetrahedron, 2012, 68, 2015–2026 CrossRef CAS.
- D. Yu, V. T. Thai, L. I. Palmer, G. K. Veits, J. E. Cook, J. Read De Alaniz and J. E. Hein, J. Org. Chem., 2013, 78, 12784–12789 CrossRef CAS PubMed.
- C. Esmieu, D. Guettas, A. Conte-Daban, L. Sabater, P. Faller and C. Hureau, Inorg. Chem., 2019, 58, 13509–13527 CrossRef CAS PubMed.
- A. Lakatos, É. Zsigó, D. Hollender, N. V. Nagy, L. Fülöp, D. Simon, Z. Bozsó and T. Kiss, Dalton Trans., 2010, 39, 1302–1315 RSC.
- J. C. Pessoa and I. Correia, Coord. Chem. Rev., 2019, 388, 227–247 CrossRef CAS.
- T. Storr, M. Merkel, G. X. Song-Zhao, L. E. Scott, D. E. Green, M. L. Bowen, K. H. Thompson, B. O. Patrick, H. J. Schugar and C. Orvig, J. Am. Chem. Soc., 2007, 129, 7453–7463 CrossRef CAS PubMed.
- S. Noël, F. Perez, J. T. Pedersen, B. Alies, S. Ladeira, S. Sayen, E. Guillon, E. Gras and C. Hureau, J. Inorg. Biochem., 2012, 117, 322–325 CrossRef PubMed.
- J. Devonport, N. Bodnár, A. McGown, M. B. Maina, L. C. Serpell, C. Kállay, J. Spencer and G. E. Kostakis, Inorg. Chem., 2021, 60, 15310–15320 CrossRef CAS PubMed.
Footnote |
| † In memory of Prof Nick Hadjiliadis. |
| This journal is © The Royal Society of Chemistry 2023 |