
3D acceptors with multiple A–D–A architectures for highly efficient organic solar cells†
Hongbin
Chen‡
a,
Zhe
Zhang‡
a,
Peiran
Wang
a,
Yunxin
Zhang
b,
Kangqiao
Ma
a,
Yi
Lin
c,
Tainan
Duan
 d,
Tengfei
He
b,
Zaifei
Ma
c,
Guankui
Long
b,
Chenxi
Li
a,
Bin
Kan
*b,
Zhaoyang
Yao
*a,
Xiangjian
Wan
a and
Yongsheng
Chen
*a
d,
Tengfei
He
b,
Zaifei
Ma
c,
Guankui
Long
b,
Chenxi
Li
a,
Bin
Kan
*b,
Zhaoyang
Yao
*a,
Xiangjian
Wan
a and
Yongsheng
Chen
*a
aState Key Laboratory and Institute of Elemento-Organic Chemistry, The Centre of Nanoscale Science and Technology and Key Laboratory of Functional Polymer Materials, Renewable Energy Conversion and Storage Center (RECAST), College of Chemistry, Nankai University, Tianjin 300071, China. E-mail: yschen99@nankai.edu.cn; zyao@nankai.edu.c
bSchool of Materials Science and Engineering, National Institute for Advanced Materials, Renewable Energy Conversion and Storage Center (RECAST), Nankai University, 300350, Tianjin, China. E-mail: kanbin04@nankai.edu.cn
cState Key Laboratory for Modification of Chemical Fibers and Polymer Materials, Center for Advanced Low-dimension Materials, College of Materials Science and Engineering, Donghua University, Shanghai 201620, China
dChongqing Institute of Green and Intelligent Technology, Chongqing School, University of Chinese Academy of Sciences (UCAS Chongqing), Chinese Academy of Sciences, Chongqing, China
First published on 9th March 2023
Abstract
Compared to the most-studied non-fullerene acceptors (NFAs) with linear skeletons, multi-dimensional NFAs with largely conjugated extensions in multiple directions may contribute to more efficient organic solar cells (OSCs) due to the potentially improved absorption, molecular packing and charge transport dynamics. Herein, a conjugated-skeleton connection mode assembled from central units of NFAs is developed, extending conventionally linear molecular skeletons towards three-dimensions (3D). The afforded 3D NFAs with specific A–D–A architectures in two directions demonstrate extremely low reorganization energy, fibrillar network film morphology, improved charge transport behavior and enhanced stability. After molecular geometry control by elaborate fluorine-induced noncovalently conformational locks, an exciting efficiency of 17.05% is achieved by CH8-1-based binary bulk-heterojunction OSCs, which is the highest value afforded by multi-dimensional acceptors thus far. Our success in constructing efficient 3D NFAs through easy central unit connection blazes a new trail in further molecular structural optimization of state-of-the-art NFAs.
Broader contextPlaying a dominant role in the active layers of organic solar cells (OSCs), recently developed non-fullerene acceptors (NFAs) with a distinctive A–D–A structural feature (such as Y-series NFAs) have contributed to their rapidly surging power conversion efficiencies (PCEs). Our recent success in A–D–A-featuring CH-series NFAs with sufficient modification sites on molecular skeletons inspires us to explore their potential for constructing three-dimensional (3D) acceptors in light of the merits of the improved absorption, molecular packing and charge transport dynamics in multiple directions of multi-dimensional molecules. Herein, three 3D NFAs (CH8-0, CH8-1 and CH8-2) with the same skeleton but controlled fluorination are constructed through an exotic central unit connection mode, providing a feasible/effective strategy to construct novel 3D NFA platforms with sufficient structural modification sites on molecular backbones. These 3D NFAs with specific A–D–A architectures in two directions demonstrate extremely low reorganization energy, fibrillar network film morphology and improved charge transport behavior. An exciting efficiency of 17.05% is achieved by OSCs based on CH8-1 with moderate fluorination, which is the highest value for binary OSCs based on multi-dimensional acceptors thus far. Furthermore, the excellent device stability and environmentally friendly features of device processing using non-halogenated solvents demonstrate the great potential of these 3D NFAs for industrial applications in the future. |
Introduction
Due to their remarkable properties of light weight, low cost, mechanical flexibility, tunable transparency and solution processing, organic solar cells (OSCs) have been regarded as one of the most promising solar energy conversion technologies and we already stand on the eve of commercialization on a large scale.1–4 Notably, the most attractive advantage of OSCs, which endows them with great potential for achieving efficiencies comparable to or even beyond those of state-of-the-art silicon solar cells,5,6 is the tremendous adjustability of organic semiconductor materials via precise molecular design and modification.7 Playing a dominant role in active layers, recently developed non-fullerene acceptors (NFAs) with a distinctive A–D–A structural feature have contributed to the rapidly surging power conversion efficiencies (PCEs) of OSCs due to their superior charge separation/transport and reduced energy loss (Eloss).8 Thus far, an exciting PCE of over 19%9–11 has been achieved by state-of-the-art Y-series NFAs after extensive structural modification on well-known Y612 acceptors, including side chain engineering, alternation of end groups, elemental replacement and so on.13–15 However, the potential for further optimizing chemical structures and achieving record-breaking OSCs based on Y-series NFAs has been significantly limited due to the lack of modification sites on their conjugated skeletons.16Recently, a series of efficient NFAs (CH-series)16–18 featuring conjugation-extended central units have been developed by our group, providing sufficient modification sites on molecular skeletons and making further great structural optimization possible. More importantly, a unique and more favorable molecular packing mode of dual “end unit to central unit” can be also established by CH-series NFAs when extending the conventionally linear backbones of NFAs towards two dimensions through central unit extension.16–18 As a consequence, excellent OSCs have been achieved on the basis of this highly promising platform of CH-series NFAs due to their enhanced charge transport characteristics and reduced energy losses.16 Our success in CH-series NFAs makes us wonder whether the OSCs could work much better if the linear NFAs skeletons were further extended towards three dimensions,19e.g. directly coupling with another NFA through the conjugated-skeleton connection mode of central units to dramatically extend the conjugated backbones of NFAs. Theoretically, such a 3D molecular in its single-molecule state should generally possess enhanced absorption coefficients and reduced reorganization energy due to its greatly enlarged conjugation plane, which will thus give rise to strong light-harvesting capability and superior charge recombination dynamics.19,20 Moreover, given the extremely crucial role of end units in molecular packing,16,21–25 greatly enhanced π–π stacking of the surrounding molecules could be expected because more end units can be assembled in such a 3D molecule.23 Last but not least, 3D acceptors with a suitably twisted molecular plane can usually form a more robust packing network not only through strong π–π stacking but also through sufficient noncovalent interactions, which should be beneficial for the morphological stability of active layers.26,27 When taking all the above advantages into consideration, such a 3D NFA is expected to generate highly efficient and stable OSCs, thus making multi-dimensional NFAs very promising for the coming large-scale commercialization of OSCs. Despite the great potential of 3D NFAs, their exploration still confronts huge challenges in terms of both molecular design and chemical construction.20,28 Therefore, it is no wonder that very few 3D NFAs have been reported19,23 and the PCEs of the resulting OSCs are mostly below 12%,19 lagging far behind OSCs based on linear NFAs.9
Bearing these thoughts in mind, a feasible study to construct a 3D molecular platform is proposed in this contribution by linking two CH-series NFAs with the same type of A–D–A16–18 directly through a conjugated-skeleton connection mode of central units (Fig. 1a and Fig. S1, ESI†). All three newly formed 3D NFAs (CH8-0, CH8-1 and CH8-2) possess the same backbones with four fluorine-substituted end groups. However, extra fluorination on both phenazine and bridging units can be observed for CH8-2, compared to fluorination only on the phenazine unit for CH8-1 and without fluorination for CH8-0 (Fig. 1b). A comprehensive investigation reveals that the greatly extended conjugation enables all three molecules to possess extremely low electron recombination energy, which is favorable for better charge transport in OSCs. Moreover, the different types of fluorination on either central or bridged units established noncovalently conformational locks with different strengths through F–S/F–H secondary interactions,29 leading to gradually decreased dihedral angles30,31 between the two monomers from CH8-0 to CH8-2. The gradually planar geometries of the NFAs in combination with the increasing fluorine density on the molecular skeletons result in increased crystallinity of the molecules and also enlarged fiber sizes in blended films, which should account for the improved charge transport behavior in CH8-1 and CH8-2 based OSCs compared to that of CH8-0.32 After making a subtle tradeoff between open-circuit voltage (VOC) and short current density (JSC), an excellent PCE of 17.05% can be achieved by CH8-1 based binary OSCs along with a VOC of 0.923 V, JSC of 24.89 mA cm−2 and fill factor (FF) of 74.2%. Note that a PCE of over 16% for OSCs based on multi-dimensional molecules has not been achieved in binary BHJ OSCs before.23 Our work has not only afforded a feasible strategy to construct 3D acceptors but also demonstrated that multi-dimensional molecules are very promising for highly efficient OSCs.
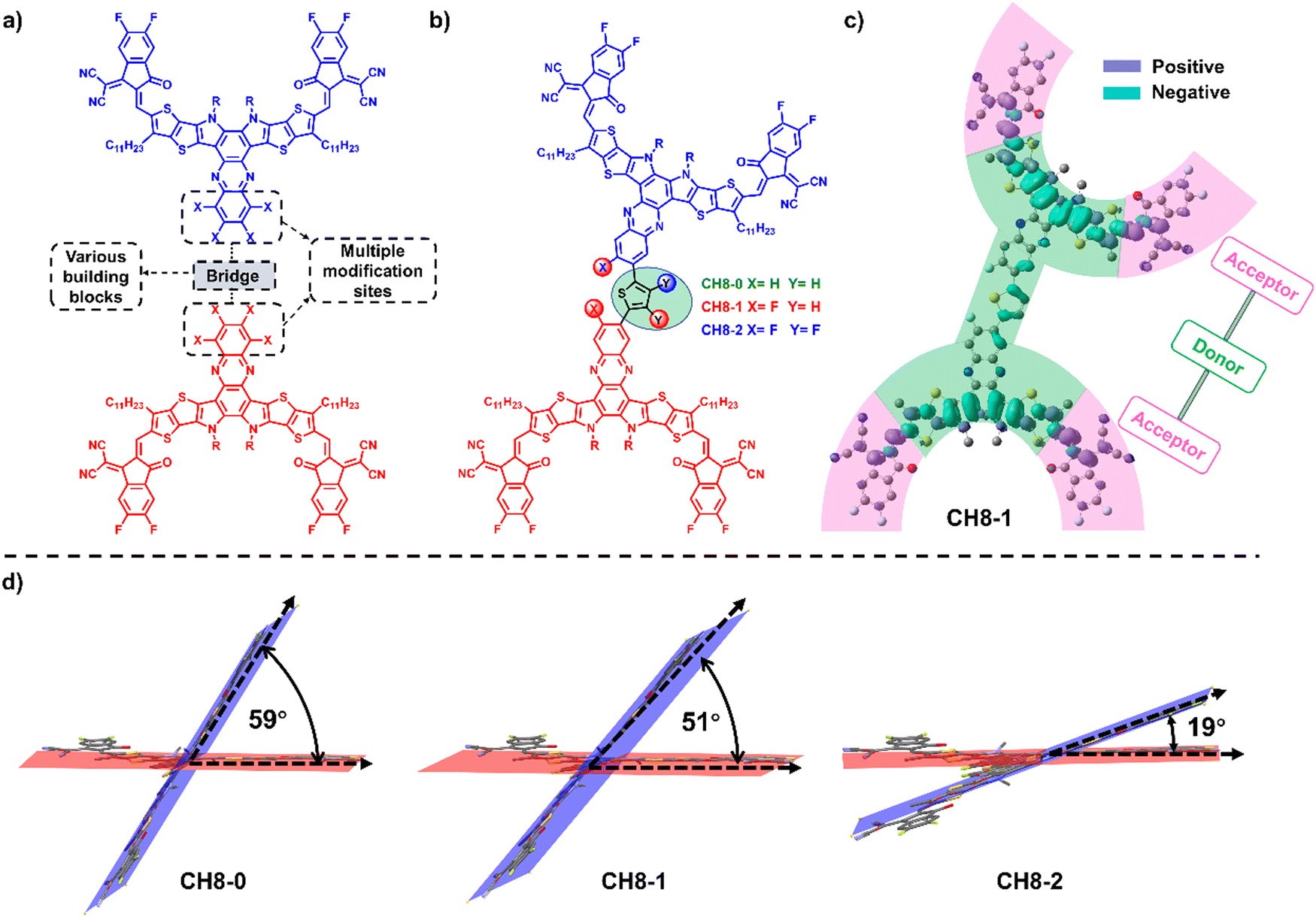 | ||
| Fig. 1 (a) Molecular design of 3D molecules with sufficient chemical modification sites on the molecular skeleton. The two monomers are marked with red and blue colors, respectively. (b) Chemical structures of CH8-0, CH8-1 and CH8-2. The two monomers are marked with red and blue colors, respectively. (c) Theoretical density distribution ΔQ (ΔQ = Ψ2LUMO − Ψ2HOMO) along the longest axis (backbone) of CH8-1. (d) Ground-state geometries of CH8-0, CH8-1 and CH8-2 calculated by the DFT method. The degrees of dihedral angles between the two monomers are also marked. Red and blue indicate the planes where the two monomers are located. | ||
Results and discussions
Synthesis and physical properties
The successful syntheses of CH8-0, CH8-1 and CH8-2 are described in Scheme S1 (ESI†). Given their similar structures, herein we just take the synthetic route for CH8-0 as an example. First, compound 2–1 with four formyl groups was generated through a Stille coupling reaction between 2,5-bis-trimethylstannanyl-thiophene and compound 1–1 in a moderate yield of 67%. Then a four-fold Knoevenagel condensation reaction between 2–1 and an electron-withdrawing terminal of 2-(5,6-difluoro-3-oxo-2,3-dihydro-1H-inden-1-ylidene) malononitrile (INCN-2F) was further performed to afford CH8-0 with a good yield of 79%. The synthetic details and characterization parameters of CH8-0, CH8-1 and CH8-2 are illustrated in ESI.† In order to investigate the energy levels, distribution of frontier molecular orbitals and molecular geometries for these three NFAs, density functional theory (DFT) calculations were first resorted to.33 As shown in Fig. S2 (ESI†), both the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) energy levels downshift gradually as fluorination increases from CH8-0 to CH8-2, which should be attributed to the large electronegativity of fluorine atoms. Note that a relatively larger variation on HOMO energy levels (downshifting by ∼40 meV) than those of LUMOs (downshifting by ∼20 meV) from CH8-0 to CH8-2 can be observed. This tendency is very consistent with the clear A–D–A feature in two directions not only along the conventionally longest x axis but also the vertical y axis, which is indicated by the characteristic peak–valley–peak curves for their frontier orbital charge density differences (ΔQ) (Fig. 1c and Fig. S3, ESI†).8 It has been proposed before that a distinctive A–D–A feature of NFAs will usually contribute to enhanced molecular packing, better exciton separation, facilitated charge transport, smaller energy losses and thus improved photovoltaic performance.8 As regards the molecular geometries, all three NFAs possess a relatively large dihedral angle between the conjugated planes of the two monomers, moving the linear molecular skeletons towards multi-dimensions. It is worth noting that the molecular geometries are inclined to become more and more planar from CH8-0 to CH8-2 with the increased fluorination on the central and bridge units. This should be ascribed to the gradually strengthened noncovalently conformational locks caused by the F–S/F–H secondary interactions.30,31,33 Generally, the unique 3D structures with great conjugation extension in multiple directions and the elaborately tuned molecular geometries are expected to give rise to very different and favorable morphological features in blended films, which will be discussed in detail below.As presented in Fig. S4 (ESI†) and Fig. 2a, the absorption spectra of CH8-0, CH8-1 and CH8-2 in both solutions and neat films were measured in order to evaluate their light-harvesting capacities. The maximum absorption peaks (λmax) of CH8-0, CH8-1 and CH8-2 are located at 740, 736 and 732 nm in dilute chloroform solution, respectively. As expected, a much larger molar extinction coefficient of ∼4 × 105 M−1 cm−1 is afforded by CH8-0, CH8-1 and CH8-2 compared to that of ∼2 × 105 M−1 cm−1 for their corresponding mono-like molecules.16–18 At shown in Fig. 2a, the λmax of CH8-0, CH8-1 and CH8-2 in the solid state are located at 801, 788 and 788 nm, respectively, retaining a similar variation tendency to those in solution. The maximum coefficients for CH8-0, CH8-1 and CH8-2 films are 8.19 × 104 cm−1, 9.42 × 104 cm−1 and 1.00 × 105 cm−1, respectively. Furthermore, the optical bandgaps (Eoptg) can be determined from the thin-film absorption edge (λonset) of 881, 871 and 863 nm for CH8-0, CH8-1 and CH8-2, respectively, corresponding to a gradually enlarged Eoptg of 1.41, 1.42 and 1.44 eV (Table S1, ESI†). Note that efficient light utilization in the near-infrared region for all three NFAs well meets the fundamental requirements for high-performance OSCs based on our previously proposed semi-empirical model.34 Both the blue-shifted λmax in solutions/neat films and the enlarged Eoptg are very consistent with the downshifted HOMO energy levels caused by the increased fluorination on the central and bridged skeletons from CH8-0 to CH8-2. The experimental HOMO and LUMO energy levels of CH8-0, CH8-1 and CH8-2 can be evaluated by cyclic voltammetry (CV) measurements. Among them, the experimental HOMO energy levels can be determined from the following equation: EHOMO = –(4.80 + Eonsetox) eV, where Eonsetox represents the onset potential of the oxidized peak shown in Fig. S5 (ESI†). As illustrated in Fig. 2b, the HOMOs of CH8-0, CH8-1 and CH8-2 are −5.61, −5.69 and −5.71 eV, respectively. Similarly, the LUMO energy levels of CH8-0, CH8-1 and CH8-2 can also be estimated as −3.78, −3.80 and −3.81 eV, respectively, according to the onset potential of the reduced peak of the CVs displayed in Fig. S6 (ESI†). The relative alignments of both HOMO and LUMO energy levels are in good agreement with those predicted by DFT calculation. Note that the gradually downshifted HOMO energy levels caused by increased fluorination could in theory give rise to an enlarged driving force for exciton dissociation and thus contribute to a more efficient charge generation process.35 In addition, the decomposition temperatures of CH8-0, CH8-1 and CH8-2 are all over 330 °C as measured by the thermal gravimetric analysis (TGA), demonstrating the outstanding thermal stability of all three molecules (Fig. S7, ESI†). The corresponding detailed physicochemical data have been summarized in Table S1 (ESI†).
 | ||
| Fig. 2 (a) Normalized absorption spectra in the solid state. (b) Energy level diagram of PM6, CH8-0, CH8-1 and CH8-2 derived from CVs. (c) J–V curves of OSCs based on PM6:CH8-0, PM6:CH8-1 and PM6:CH8-2 blends; the inset shows a histogram of the PCEs of OSCs, which have been fitted with Gaussian distributions (solid lines). (d) EQE plots of OSCs based on PM6:CH8-0, PM6:CH8-1 and PM6:CH8-2 blends. (e) A summary of the JSC and PCE of binary OSCs based on multi-dimensional acceptors (detailed data are displayed in Table S8, ESI†). Herein, the “3D acceptor” refers to molecules with a largely conjugated extension in multiple directions, thus forming a 3D molecular conjugated skeleton/configuration rather than a planar one (Fig. S8, ESI†). Compared to the most-studied NFAs with linear skeletons (such as Y6 or CH17), the “3D acceptor” generally possesses more end units and a relatively large dihedral angle between its two planes (a detailed comparison of the configurations of CH17 and CH8-1 is presented in Fig. S8, ESI†). (f) PCE variation versus operating time in a glovebox filled with nitrogen at room temperature. | ||
Photovoltaic performance
As we have discussed above, such a 3D NFA, like CH8-0, CH8-1 and CH8-2, is expect to render highly efficient and stable OSCs; therefore, the photovoltaic properties based on these NFAs were evaluated by fabricating OSCs with a conventional architecture of ITO Glass/PEDOT:PSS/Active layer/PNDIT-F3N/Ag. The widely used polymeric donor PM636 (Fig. S9, ESI†) was chosen to blend with CH8-0, CH8-1 and CH8-2 to compose active layers. The corresponding J–V curves and optimized photovoltaic parameters of the OSCs are presented in Fig. 2c and Table 1, respectively. After device optimization, a champion PCE of 17.05% was achieved by the CH8-1 based OSC along with a VOC of 0.923 V, JSC of 24.89 mA cm−2 and FF of 74.2%, which is much better than that of 15.26% for the CH8-0 based OSC with a slightly larger VOC of 0.936 V but an inferior JSC of 22.61 mA cm−2 and FF of 72.1%. Obviously, the improvement in PCE for the CH8-1 based OSC compared to that with CH8-0 should be attributed to its significantly improved JSC and FF, which should be induced by the facilitated charge generation/transport dynamics after fluorination on the central units (see the detailed discussions below). Moreover, the CH8-2 based OSC afforded a comparable but slightly lower PCE of 16.84% with respect to that of CH8-1. The narrower absorption range of CH8-2 with respect to that of CH8-1 should account for its inferior PCE, especially for the smaller JSC of the CH8-2 based OSC. As shown in Fig. 2d, both CH8-1 and CH8-2 based OSCs show much higher external quantum efficiencies (EQEs) than that of the CH8-0 based one, suggesting improved charge generation/transport dynamics after fluorination. In spite of the comparable but slightly higher EQEs for CH8-2 compared to that of CH8-1, a smaller integrated JSC of 23.60 mA cm−2 for CH8-2 than that of 23.97 mA cm−2 for CH8-1 is obtained due to the inefficient light harvesting of CH8-2 in the range of 850–900 nm, which also remains in good agreement with the tendency of JSC derived from J–V curves and is consistent with absorption range of the blended films in Fig. S10 (ESI†). Fig. 2c presents the PCE distribution histogram of 15 independently measured OSCs, indicating the good reproducibility of 3D NFA based devices (detailed device parameters are listed in Tables S2–S7, ESI†). A brief summary of the JSC and PCE of binary BHJ OSCs based on multi-dimensional acceptors is displayed in Fig. 2e and Table S8 (ESI†), where CH8-1 and CH8-2 based OSCs show the largest values of both JSCs and PCEs thus far. It is also worth noting that a PCE of over 16% has not been achieved by binary OSCs23 using multi-dimensional molecules as primary NFAs except for CH8-1 and CH8-2 based ones, demonstrating our successful molecular design in constructing 3D NFAs directly through central unit connection. Furthermore, an excellent PCE of 15.67% was also achieved by the CH8-1 based OSC along with a VOC of 0.911 V, JSC of 24.11 mA cm−2 and FF of 71.3% by using an inverted device structure of ITO/ZnO/PFN-Br/Active layer/MoOx/Ag (Fig. S11, ESI†). A moderate PCE of 15.12% was achieved along with a VOC of 0.935 V, JSC of 24.07 mA cm−2 and FF of 67.1% by an OSC based on D18:CH8-1 with a conventional architecture of ITO Glass/PEDOT:PSS/Active layer/PNDIT-F3N/Ag (Fig. S12, ESI†). In addition, a decent PCE of 16.42% with a VOC of 0.921 V, JSC of 23.73 mA cm−2 and FF of 75.2% is afforded by an CH8-1 based OSC by processing with a green solvent of xylene (Fig. S13, ESI†). Using non-halogenated solvents for device processing has been regarded as an effective strategy to increase the environmentally friendly features of OSCs.37| Active layer | V OC (V) | J SC (mA cm−2) | Calc. JSCb (mA cm−2) | FF (%) | PCE (%) |
|---|---|---|---|---|---|
| a Statistical and optimal results are listed inside parentheses and outside parentheses, respectively. The average parameters were calculated from 15 independent devices. b Current densities calculated from EQE curves. | |||||
| PM6:CH8-0 | 0.936 (0.932 ± 0.002) | 22.61 (22.46 ± 0.22) | 21.77 | 72.1 (71.4 ± 0.6) | 15.26 (15.05 ± 0.13) |
| PM6:CH8-1 | 0.923 (0.926 ± 0.005) | 24.89 (24.54 ± 0.45) | 23.97 | 74.2 (74.2 ± 0.8) | 17.05 (16.87 ± 0.12) |
| PM6:CH8-2 | 0.928 (0.925 ± 0.004) | 24.24 (24.27 ± 0.28) | 23.60 | 74.9 (74.1 ± 1.1) | 16.84 (16.63 ± 0.13) |
More importantly, the PCEs of all the CH8-0, CH8-1 and CH8-2 based OSCs could be maintained above 90% or even around 95% compared to their original PCEs after 2700 h at room temperature (Fig. 2f). Meanwhile, the PCEs of CH8-0, CH8-1 and CH8-2 based OSCs could be maintained above 78%, 80% and 85% compared to their original PCEs after 360 h with heat treatment at 65 °C, respectively (Fig. S14, ESI†). Besides, as shown in Fig. S15 (ESI†), the PCE of the CH8-1 based OSC could also remain near 85% with respect to its original PCE after 200 h under continuous 1 sun illumination simulated by LED arrays under maximum power point (MPP) tracking in a nitrogen-filled glovebox, demonstrating satisfactory stability in spite of the conventional device structure. Please note that the good storage, thermal and photo stabilities of the OSCs make these three 3D acceptors among the best systems in OSCs and indicate their great potential for industrial application.38,39 Considering the crucial role of morphological stability in the stability of the final device, the excellent stability of multi-dimensional NFAs based OSCs may partially originate from their potentially robust 3D packing network formed not only by the strong intermolecular π–π stacking but also by the weak noncovalent interactions.26,27,40
Charge transport, charge recombination, exciton dissociation and energy loss analysis
To further reveal the charge generation and extraction behaviors of CH8-0, CH8-1 and CH8-2 based OSCs, the dependence of photocurrent density (Jph) on the effective voltage (Veff) was first characterized.41 As shown in Fig. 3a, CH8-0, CH8-1 and CH8-2 based OSCs afforded exciton dissociation efficiencies (ηdiss) of 95.3%, 97.0% and 97.2%, respectively. Meanwhile, the charge collection efficiencies (ηcoll) can also be estimated as 83.2%, 84.9% and 87.2% for CH8-0, CH8-1 and CH8-2 based OSCs, respectively. Generally, ηdiss is closely related to the energy offset at the D/A interfaces of blended films, which usually determines the driving force for charge generation. Due to the gradually downshifted energy levels and thus increasing driving forces for charge generation from CH8-0 to CH8-2, it is no wonder that a stepwise improvement in ηdiss can be observed.35 Meanwhile, the photoluminescence quenching yields (ηPLQ) of PM6:CH8-0, PM6:CH8-1 and PM6:CH8-2 blends were estimated to be 72%, 78% and 85% (Fig. S16, ESI†), respectively, agreeing well with the trend of ηdiss variation discussed above. Whereas the slightly improved ηcoll from CH8-0 to CH8-2 should be attributed to the more favorable molecular packing and optimized film morphology, which will be discussed in detail below. Note that the gradual improvement in ηdiss and ηcoll from CH8-0 to CH8-2 based OSCs is consistent with the stepwise enlarged EQE values of the resulting OSCs.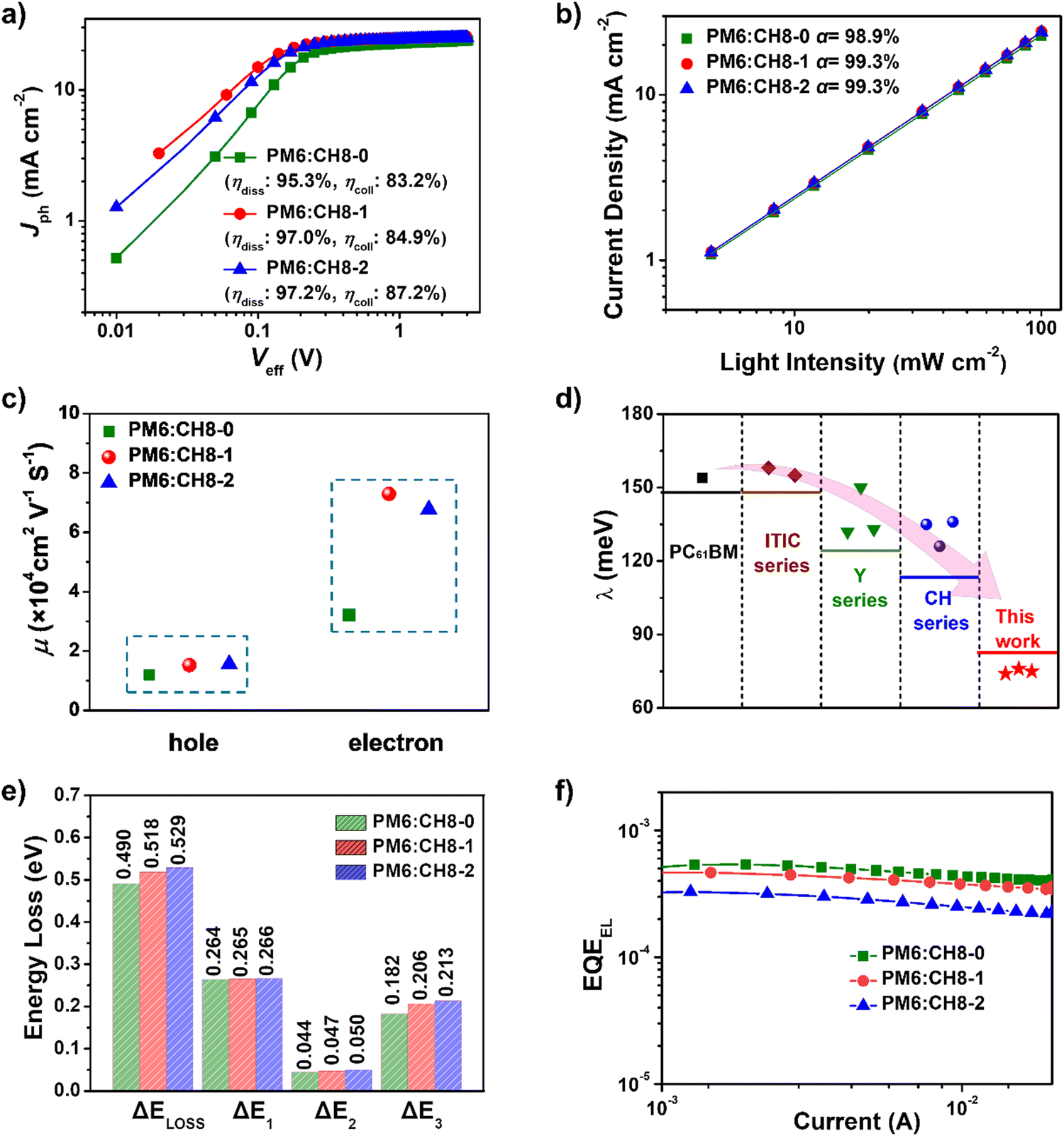 | ||
| Fig. 3 (a) Plots of Jphversus Veff. (b) Dependence of current density (Jsc) on Plight of optimized OSCs. (c) Hole and electron mobilities of PM6:CH8-0, PM6:CH8-1 and PM6:CH8-2 based devices. (d) A summary of the electron reorganization energy of several representative series of electron acceptors. (e) Detailed Eloss of CH8-0, CH8-1 and CH8-2 based devices. (f) EQEEL spectra of PM6:CH8-0, PM6:CH8-1 and PM6:CH8-2 based OSCs; EQEEL values for CH8-0, CH8-1 and CH8-2 based devices are 5.15 × 10−4, 4.55 × 10−4 and 3.24 × 10−4, respectively. | ||
Moreover, the dependence of JSC on light intensity (Plight) is plotted in Fig. 3b to reveal the bimolecular charge recombination behavior in blended films. Note that bimolecular recombination has been significantly suppressed, as indicated by the similar α values close to unity (98.9% for CH8-0, 99.3% for CH8-1 and 99.3% for CH8-2, respectively).42 Then, the charge transport abilities in blended films were further evaluated through the method of space-charge-limited current (SCLC) by using hole-only and electron-only devices. As displayed in Fig. 3c and Fig. S17, Table S9 (ESI†), the electron and hole mobilities (μe and μh) are determined to be 3.21 × 10−4 and 1.20 × 10−4 cm2 V−1 s−1 for CH8-0 based devices, 7.29 × 10−4 and 1.52 × 10−4 cm2 V−1 s−1 for CH8-1 based devices, and 6.77 × 10−4 and 1.56 × 10−4 cm2 V−1 s−1 for CH8-2 based devices, respectively. The enlarged μe and μh of CH8-1 and CH8-2 based devices compared to those of their CH8-0 counterparts indicate the more efficient charge transport in CH8-1 and CH8-2 blends, which agrees well with their higher ηcoll and should partially account for the improved JSC and FF.43 It is also worth noting that all three 3D NFAs demonstrate an extremely low electron reorganization energy of only 75 ± 1 meV, which is dramatically lower than those of more than 120 meV for several representative acceptors, including PCBM,19 ITIC series,44 Y6 series12,45 and CH series16–19 (Fig. 3d). The extremely low electron reorganization energy will favor the excellent electron transport property based on Marcus charge-transfer theory.46 The detailed computational methods of electron reorganization energy are presented in ESI.† The detailed photodynamic parameters and electron reorganization energies have been summarized in Tables S9 and S10 (ESI†).
Given that the relatively high VOC for these 3D NFA-based OSCs may be caused by reduced energy losses (Eloss),45 a detailed Eloss analysis was then carried out based on detailed balance theory.47,48 A detailed decomposition of energy loss is displayed in Fig. 3e and Table S11 (ESI†). As shown in Fig. S18 (ESI†), the optical bandgaps (Eg) of blended films could be estimated from the derivatives of the EQE curves45 (the detailed method is described in ESI†), which are 1.426, 1.441 and 1.457 eV for CH8-0, CH8-1 and CH8-2, respectively. Therefore, the corresponding Eloss for CH8-0, CH8-1 and CH8-2 based OSCs are 0.490, 0.518 and 0.529 eV, respectively, employing the following equation: Eloss = Eg – qVOC (where q is the elementary charge). Among the three parts of energy losses, the radiative recombination energy losses above the bandgap (ΔE1) for the three OSCs are all about 0.265 eV, and the radiative recombination energy losses below the bandgap (ΔE2) are in a small range of 0.044–0.050 eV. Note that the most-concerning non-radiative recombination energy losses (ΔE3) in OSCs for CH8-0, CH8-1 and CH8-2 based systems are as low as 0.182, 0.206 and 0.213 eV, respectively, ranking among the smallest values in high-performance OSCs.16,49–52 Such a low non-radiative recombination energy loss has also been verified by the relatively large EQEEL values in Fig. 3f. According to the following equation, 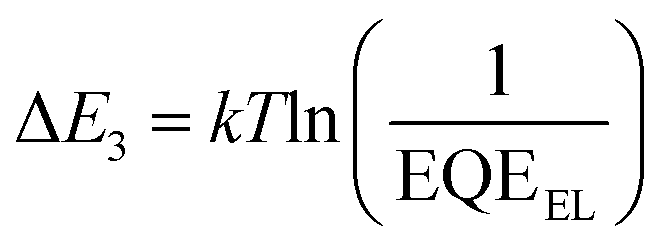 , where k represents the Boltzmann constant and T is the Kelvin temperature, the non-radiative recombination energy losses can be calculated as 0.195, 0.199 and 0.207 eV for CH8-0, CH8-1 and CH8-2 based OSCs, respectively. The variation tendency in non-radiative recombination energy losses calculated by the two different methods remained in good agreement. Then, the energetic differences (ΔECT) between the local exciton (LE) and charge-transfer (CT) states were measured by fitting the corresponding highly sensitive EQEs (sEQE) and electroluminescence (EL) spectra (Fig. S19, ESI†), being 0.03, 0.04 and 0.04 eV for CH8-0, CH8-1 and CH8-2 based OSCs, respectively.53,54 As has been proposed, the smaller ΔECT of the CH8-0 based OSC may induce a more effective hybridization between LE and CT states, which could result in the lower non-radiative recombination rate through an intensity borrowing mechanism.53,55 Furthermore, the Urbach energies (EU) were measured through an exponential fitting of corresponding sEQE tails, being 22.17, 21.98 and 21.84 meV for CH8-0, CH8-1 and CH8-2 based OSCs, respectively (Fig. S20, ESI†). The comparable but low EU is indicative of the highly ordered molecular packing for these 3D NFAs and should account for the largely suppressed ΔE2 and ΔE3.48,56
, where k represents the Boltzmann constant and T is the Kelvin temperature, the non-radiative recombination energy losses can be calculated as 0.195, 0.199 and 0.207 eV for CH8-0, CH8-1 and CH8-2 based OSCs, respectively. The variation tendency in non-radiative recombination energy losses calculated by the two different methods remained in good agreement. Then, the energetic differences (ΔECT) between the local exciton (LE) and charge-transfer (CT) states were measured by fitting the corresponding highly sensitive EQEs (sEQE) and electroluminescence (EL) spectra (Fig. S19, ESI†), being 0.03, 0.04 and 0.04 eV for CH8-0, CH8-1 and CH8-2 based OSCs, respectively.53,54 As has been proposed, the smaller ΔECT of the CH8-0 based OSC may induce a more effective hybridization between LE and CT states, which could result in the lower non-radiative recombination rate through an intensity borrowing mechanism.53,55 Furthermore, the Urbach energies (EU) were measured through an exponential fitting of corresponding sEQE tails, being 22.17, 21.98 and 21.84 meV for CH8-0, CH8-1 and CH8-2 based OSCs, respectively (Fig. S20, ESI†). The comparable but low EU is indicative of the highly ordered molecular packing for these 3D NFAs and should account for the largely suppressed ΔE2 and ΔE3.48,56
Morphological analysis
As we have revealed above, facilitated charge transport, suppressed energy loss and encouraging performance have been achieved by OSCs based on such a distinctive 3D NFA system. Given the dominant role of film morphology in device performance, a systematic morphological analysis of active layers was further carried out. Firstly, in order to shed light on the molecular crystallinity, packing behavior and orientations of CH8-0, CH8-1 and CH8-2, we resorted to grazing incident wide-angle X-ray scattering (GIWAXS) measurements.57 As shown in Fig. 4a and Fig. S21, Table S12 (ESI†), all the neat and blended films for these three NFAs displayed a clear (100) diffraction peak in the in-plane (IP) direction and a strong (010) diffraction peak in the out-of-plane (OOP) direction, demonstrating the good molecular crystallinity and favorable face-on orientation of the molecular packing. A clear (010) diffraction peak at 1.65–1.66 Å−1 in the OOP direction for both neat and blended films of the three 3D NFAs can be observed, corresponding to a similar π–π stacking distance of 3.79–3.80 Å. It is worth noting that similar crystal coherence lengths (CCLs) of over 20 Å can be afforded by both neat and blended films, demonstrating the ordered molecular packing for all these 3D NFAs. To sum up, compact and ordered molecular packing has been constructed by all these 3D NFA based films, as indicated by the relatively small intermolecular π–π stacking distances and also large CCLs. In combination with the good PCE and stability of the resulting OSCs, there is a logical conclusion that multi-dimensional NFAs could also establish highly efficient OSCs after an elaborate molecular design, in spite of their relatively large steric hindrance which has traditionally been regarded as an obstruction to efficient molecular packing. More importantly, a bundle-like nanofiber network has been formed in the blended films of CH8-0, CH8-1 and CH8-2 (Fig. 4b and Fig. S22, ESI†).58 The suitable size of nanofibers has been proven to play an important role in facilitating charge separation/transport and reducing carrier recombination, thus leading to greatly improved photovoltaic parameters for OSCs.9,32 Therefore, a statistical size analysis of the bundle-like nanofibers was conducted and is displayed in Fig. 4c and Fig. S23 (ESI†), being 8.5, 10.5 and 14.2 nm for CH8-0, CH8-1 and CH8-2, respectively.9 The enlarged nanofibers from CH8-0 to CH8-2 should be caused by their gradually increased crystallinity, and also the reduced miscibility with the PM6 donor, as indicated by the incremental Flory–Huggins interaction parameter χ (0.01 for CH8-0, 0.07 for CH8-1 and 0.37 for CH8-2; Fig. S24 and Table S13, ESI†).59 The variation in crystallinity and miscibility should be attributed to the decreased dihedral angles with the growing fluorination on the central and bridged skeletons.60 Note that the relatively larger nanofibers for CH8-1 and CH8-2 than those of CH8-0 should partially account for the gradually improved JSC and FF for CH8-1 and CH8-2 based OSCs.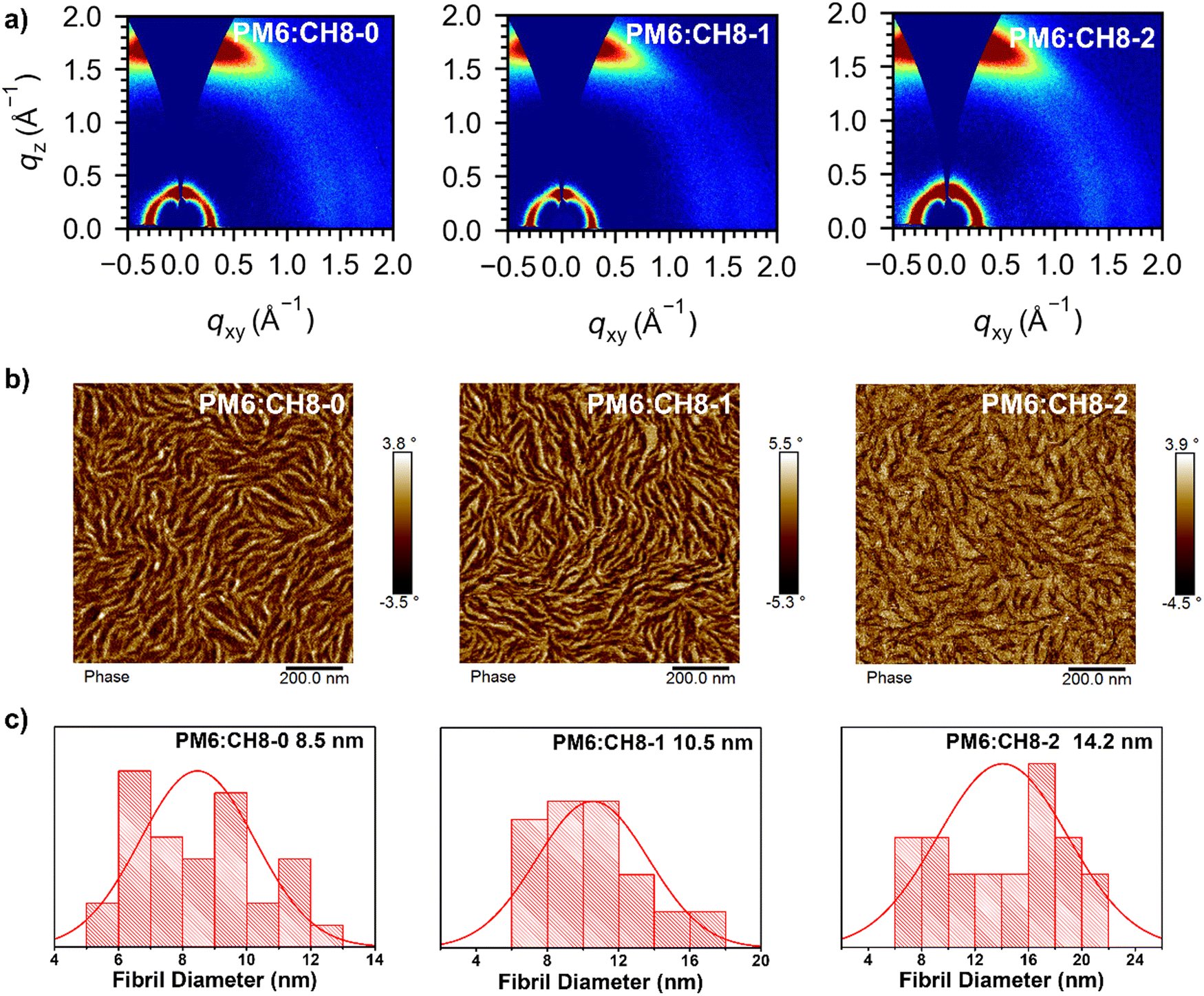 | ||
| Fig. 4 (a) 2D GIWAXS patterns of optimized PM6:CH8-0, PM6:CH8-1 and PM6:CH8-2 based films. (b) AFM phase images of PM6:CH8-0, PM6:CH8-1 and PM6:CH8-2 based films. (c) The statistical distribution of the fibril width for PM6:CH8-0, PM6:CH8-1 and PM6:CH8-2 based blended films (see Fig. S22 and S23 for the details, ESI†). | ||
Conclusions
A series of 3D electron acceptors (CH8-0, CH8-1 and CH8-2) with extended conjugation in multiple directions have been constructed through a conjugated-skeleton connection mode of central units. A comprehensive investigation reveals that all three 3D NFAs possess an extremely low electron recombination energy and two-fold A–D–A architectures, which favors better charge transport in OSCs. Moreover, fluorination on either central or bridged units has established noncovalently conformational locks with different strengths through intramolecular secondary interactions, leading to gradually decreased steric hindrance and increased crystallinity from CH8-0 to CH8-2. Finally, a preferable bundle-like nanofiber network can be afforded by all these 3D NFAs and the greatly enlarged size of the nanofibers in CH8-1 and CH8-2 blended films with respect to that of CH8-0 should result in superior charge separation/transport and reduced charge recombination. An attractive PCE of 17.05% can be achieved by CH8-1 based binary BHJ OSCs, which is the best result for binary BHJ OSCs based on multi-dimensional acceptors. Furthermore, OSCs based on this 3D material class show excellent device stability. Our work has not only afforded a novel and feasible strategy to construct multi-dimensional acceptors through central unit connections but also demonstrated the great potential of multi-dimensional acceptors for highly efficient and stable OSCs.Author contributions
The synthetic works were carried out by H. C. and Z. Z., and three acceptors were characterized by H. C., P. W. and Y. Z., and B. K. carried out most of the device fabrication and measurements. T. H., H. C. and G. L. carried out the theoretical computation of three acceptors. Y. L. and Z. M. performed the EL, EQEEL and sEQE experiments and analyzed the data. K. M. and T. D. performed the morphology characterization and analyzed the data. C. L. helped to analyze the data and revise the manuscript. Y. C., Z. Y., B. K. and X. W. supervised and directed this project. Y. C., H. C., Z. Y. and B. K. wrote the manuscript. All authors discussed the results and commented on the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors gratefully acknowledge the financial support from MoST (2022YFB4200400, 2019YFA0705900) of China and NSFC (21935007, 52025033, 51873089), Tianjin city (20JCZDJC00740, 22JCQNJC00530), 111 Project (B12015), the Fundamental Research Funds for the Central Universities, Nankai University (023-ZB22000105, 020-ZB22000110 and 020-92220002) and Haihe Laboratory of Sustainable Chemical Transformations.References
- A. J. Heeger, Adv. Mater., 2014, 26, 10–28 CrossRef CAS PubMed.
- R. F. Service, Science, 2011, 332, 293 CrossRef CAS PubMed.
- L. X. Chen, ACS Energy Lett., 2019, 4, 2537–2539 CrossRef CAS.
- H. Fu, J. Yao, M. Zhang, L. Xue, Q. Zhou, S. Li, M. Lei, L. Meng, Z.-G. Zhang and Y. Li, Nat. Commun., 2022, 13, 3687 CrossRef CAS PubMed.
- K. Yoshikawa, H. Kawasaki, W. Yoshida, T. Irie, K. Konishi, K. Nakano, T. Uto, D. Adachi, M. Kanematsu, H. Uzu and K. Yamamoto, Nat. Energy, 2017, 2, 17032 CrossRef CAS.
- Z. Zheng, J. Wang, P. Bi, J. Ren, Y. Wang, Y. Yang, X. Liu, S. Zhang and J. Hou, Joule, 2022, 6, 171–184 CrossRef CAS.
- G. Zhang, F. R. Lin, F. Qi, T. Heumüller, A. Distler, H.-J. Egelhaaf, N. Li, P. C. Y. Chow, C. J. Brabec, A. K. Y. Jen and H.-L. Yip, Chem. Rev., 2022, 122, 14180–14274 CrossRef CAS PubMed.
- X. Wan, C. Li, M. Zhang and Y. Chen, Chem. Soc. Rev., 2020, 49, 2828–2842 RSC.
- L. Zhu, M. Zhang, J. Xu, C. Li, J. Yan, G. Zhou, W. Zhong, T. Hao, J. Song, X. Xue, Z. Zhou, R. Zeng, H. Zhu, C.-C. Chen, R. C. I. MacKenzie, Y. Zou, J. Nelson, Y. Zhang, Y. Sun and F. Liu, Nat. Mater., 2022, 21, 656–663 CrossRef CAS PubMed.
- C. He, Y. Pan, Y. Ouyang, Q. Shen, Y. Gao, K. Yan, J. Fang, Y. Chen, C.-Q. Ma, J. Min, C. Zhang, L. Zuo and H. Chen, Energy Environ. Sci., 2022, 15, 2537–2544 RSC.
- J. Gao, N. Yu, Z. Chen, Y. Wei, C. Li, T. Liu, X. Gu, J. Zhang, Z. Wei, Z. Tang, X. Hao, F. Zhang, X. Zhang and H. Huang, Adv. Sci., 2022, 9, 2203606 CrossRef CAS PubMed.
- J. Yuan, Y. Zhang, L. Zhou, G. Zhang, H.-L. Yip, T.-K. Lau, X. Lu, C. Zhu, H. Peng, P. A. Johnson, M. Leclerc, Y. Cao, J. Ulanski, Y. Li and Y. Zou, Joule, 2019, 3, 1140–1151 CrossRef CAS.
- Y. Liu, B. Liu, C.-Q. Ma, F. Huang, G. Feng, H. Chen, J. Hou, L. Yan, Q. Wei, Q. Luo, Q. Bao, W. Ma, W. Liu, W. Li, X. Wan, X. Hu, Y. Han, Y. Li, Y. Zhou, Y. Zou, Y. Chen, Y. Li, Y. Chen, Z. Tang, Z. Hu, Z.-G. Zhang and Z. Bo, Sci. China: Chem., 2022, 65, 224–268 CrossRef CAS.
- Y. Liu, B. Liu, C.-Q. Ma, F. Huang, G. Feng, H. Chen, J. Hou, L. Yan, Q. Wei, Q. Luo, Q. Bao, W. Ma, W. Liu, W. Li, X. Wan, X. Hu, Y. Han, Y. Li, Y. Zhou, Y. Zou, Y. Chen, Y. Liu, L. Meng, Y. Li, Y. Chen, Z. Tang, Z. Hu, Z.-G. Zhang and Z. Bo, Sci. China: Chem., 2022, 65, 1457–1497 CrossRef CAS.
- W. Gao, B. Fan, F. Qi, F. Lin, R. Sun, X. Xia, J. Gao, C. Zhong, X. Lu, J. Min, F. Zhang, Z. Zhu, J. Luo and A. K. Y. Jen, Adv. Funct. Mater., 2021, 31, 2104369 CrossRef CAS.
- H. Chen, Y. Zou, H. Liang, T. He, X. Xu, Y. Zhang, Z. Ma, J. Wang, M. Zhang, Q. Li, C. Li, G. Long, X. Wan, Z. Yao and Y. Chen, Sci. China: Chem., 2022, 65, 1362–1373 CrossRef CAS.
- H. Chen, H. Liang, Z. Guo, Y. Zhu, Z. Zhang, Z. Li, X. Cao, H. Wang, W. Feng, Y. Zou, L. Meng, X. Xu, B. Kan, C. Li, Z. Yao, X. Wan, Z. Ma and Y. Chen, Angew. Chem., Int. Ed., 2022, 61, e202209580 CAS.
- Y. Zou, H. Chen, X. Bi, X. Xu, H. Wang, M. Lin, Z. Ma, M. Zhang, C. Li, X. Wan, G. Long, Y. Zhaoyang and Y. Chen, Energy Environ. Sci., 2022, 15, 3519–3533 RSC.
- H. Chen, X. Cao, X. Xu, C. Li, X. Wan, Z. Yao and Y. Chen, Chin. J. Polym. Sci., 2022, 40, 921–927 CrossRef CAS.
- S. Li, W. Liu, M. Shi, J. Mai, T.-K. Lau, J. Wan, X. Lu, C.-Z. Li and H. Chen, Energy Environ. Sci., 2016, 9, 604–610 RSC.
- T. J. Aldrich, M. Matta, W. Zhu, S. M. Swick, C. L. Stern, G. C. Schatz, A. Facchetti, F. S. Melkonyan and T. J. Marks, J. Am. Chem. Soc., 2019, 141, 3274–3287 CrossRef CAS PubMed.
- X. Shi, L. Zuo, S. B. Jo, K. Gao, F. Lin, F. Liu and A. K. Y. Jen, Chem. Mater., 2017, 29, 8369–8376 CrossRef CAS.
- X. Meng, M. Li, K. Jin, L. Zhang, J. Sun, W. Zhang, C. Yi, J. Yang, F. Hao, G.-W. Wang, Z. Xiao and L. Ding, Angew. Chem., Int. Ed., 2022, 61, e202207762 CAS.
- G. Zhang, X.-K. Chen, J. Xiao, P. C. Y. Chow, M. Ren, G. Kupgan, X. Jiao, C. C. S. Chan, X. Du, R. Xia, Z. Chen, J. Yuan, Y. Zhang, S. Zhang, Y. Liu, Y. Zou, H. Yan, K. S. Wong, V. Coropceanu, N. Li, C. J. Brabec, J.-L. Bredas, H.-L. Yip and Y. Cao, Nat. Commun., 2020, 11, 3943 CrossRef CAS PubMed.
- T.-J. Wen, Z.-X. Liu, Z. Chen, J. Zhou, Z. Shen, Y. Xiao, X. Lu, Z. Xie, H. Zhu, C.-Z. Li and H. Chen, Angew. Chem., Int. Ed., 2021, 60, 12964–12970 CrossRef CAS PubMed.
- D. Meng, R. Wang, J. B. Lin, J. L. Yang, S. Nuryyeva, Y.-C. Lin, S. Yuan, Z.-K. Wang, E. Zhang, C. Xiao, D. Zhu, L. Jiang, Y. Zhao, Z. Li, C. Zhu, K. N. Houk and Y. Yang, Adv. Mater., 2021, 33, 2006120 CrossRef CAS PubMed.
- D. Meng, J. L. Yang, C. Xiao, R. Wang, X. Xing, O. Kocak, G. Aydin, I. Yavuz, S. Nuryyeva, L. Zhang, G. Liu, Z. Li, S. Yuan, Z.-K. Wang, W. Wei, Z. Wang, K. N. Houk and Y. Yang, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 20397–20403 CrossRef CAS PubMed.
- D. Liu, T. Wang, Z. Chang, N. Zheng, Z. Xie and Y. Liu, J. Mater. Chem. A, 2021, 9, 2319–2324 RSC.
- Q. Shen, C. He, S. Li, L. Zuo, M. Shi and H. Chen, Acc. Mater. Res., 2022, 3, 644–657 CrossRef CAS.
- T. Duan, Q. Chen, Q. Yang, D. Hu, G. Cai, X. Lu, J. Lv, H. Song, C. Zhong, F. Liu, D. Yu and S. Lu, J. Mater. Chem. A, 2022, 10, 3009–3017 RSC.
- T. Duan, J. Gao, T. Xu, Z. Kan, W. Chen, R. Singh, G. P. Kini, C. Zhong, D. Yu, Z. Xiao, Z. Xiao and S. Lu, J. Mater. Chem. A, 2020, 8, 5843–5847 RSC.
- Q. Liu, Y. Jiang, K. Jin, J. Qin, J. Xu, W. Li, J. Xiong, J. Liu, Z. Xiao, K. Sun, S. Yang, X. Zhang and L. Ding, Sci. Bull., 2020, 65, 272–275 CrossRef CAS PubMed.
- Y. Cui, P. Zhu, X. Liao and Y. Chen, J. Mater. Chem. C, 2020, 8, 15920–15939 RSC.
- L. Meng, Y. Zhang, X. Wan, C. Li, X. Zhang, Y. Wang, X. Ke, Z. Xiao, L. Ding, R. Xia, H.-L. Yip, Y. Cao and Y. Chen, Science, 2018, 361, 1094–1098 CrossRef CAS PubMed.
- B. Kan, J. Zhang, F. Liu, X. Wan, C. Li, X. Ke, Y. Wang, H. Feng, Y. Zhang, G. Long, R. H. Friend, A. A. Bakulin and Y. Chen, Adv. Mater., 2018, 30, 1704904 CrossRef PubMed.
- M. Zhang, X. Guo, W. Ma, H. Ade and J. Hou, Adv. Mater., 2015, 27, 4655–4660 CrossRef CAS PubMed.
- B. Liu, H. Sun, J.-W. Lee, J. Yang, J. Wang, Y. Li, B. Li, M. Xu, Q. Liao, W. Zhang, D. Han, L. Niu, H. Meng, B. J. Kim and X. Guo, Energy Environ. Sci., 2021, 14, 4499–4507 RSC.
- W. Yang, Z. Luo, R. Sun, J. Guo, T. Wang, Y. Wu, W. Wang, J. Guo, Q. Wu, M. Shi, H. Li, C. Yang and J. Min, Nat. Commun., 2020, 11, 1218 CrossRef CAS PubMed.
- Y. Liang, D. Zhang, Z. Wu, T. Jia, L. Lüer, H. Tang, L. Hong, J. Zhang, K. Zhang, C. J. Brabec, N. Li and F. Huang, Nat. Energy, 2022, 7, 1180–1190 CrossRef CAS.
- H. Gao, C. Han, X. Wan and Y. Chen, Ind. Chem. Mater., 2023, 1, 60–78 RSC.
- A. K. K. Kyaw, D. H. Wang, V. Gupta, W. L. Leong, L. Ke, G. C. Bazan and A. J. Heeger, ACS Nano, 2013, 7, 4569–4577 CrossRef CAS PubMed.
- L. J. A. Koster, V. D. Mihailetchi, R. Ramaker and P. W. M. Blom, Appl. Phys. Lett., 2005, 86, 123509 CrossRef.
- X. Zhang, C. Li, J. Xu, R. Wang, J. Song, H. Zhang, Y. Li, Y.-N. Jing, S. Li, G. Wu, J. Zhou, X. Li, Y. Zhang, X. Li, J. Zhang, C. Zhang, H. Zhou, Y. Sun and Y. Zhang, Joule, 2022, 6, 444–457 CrossRef CAS.
- S. M. Swick, J. M. Alzola, V. K. Sangwan, S. H. Amsterdam, W. Zhu, L. O. Jones, N. Powers-Riggs, A. Facchetti, K. L. Kohlstedt, G. C. Schatz, M. C. Hersam, M. R. Wasielewski and T. J. Marks, Adv. Energy Mater., 2020, 10, 2000635 CrossRef CAS.
- G. Li, X. Zhang, L. O. Jones, J. M. Alzola, S. Mukherjee, L.-W. Feng, W. Zhu, C. L. Stern, W. Huang, J. Yu, V. K. Sangwan, D. M. DeLongchamp, K. L. Kohlstedt, M. R. Wasielewski, M. C. Hersam, G. C. Schatz, A. Facchetti and T. J. Marks, J. Am. Chem. Soc., 2021, 143, 6123–6139 CrossRef CAS PubMed.
- V. Stehr, R. F. Fink, M. Tafipolski, C. Deibel and B. Engels, WIREs Comput. Mol. Sci., 2016, 6, 694–720 CrossRef CAS.
- Y. Wang, D. Qian, Y. Cui, H. Zhang, J. Hou, K. Vandewal, T. Kirchartz and F. Gao, Adv. Energy Mater., 2018, 8, 1801352 CrossRef.
- J. Liu, S. Chen, D. Qian, B. Gautam, G. Yang, J. Zhao, J. Bergqvist, F. Zhang, W. Ma, H. Ade, O. Inganäs, K. Gundogdu, F. Gao and H. Yan, Nat. Energy, 2016, 1, 16089 CrossRef CAS.
- C. Li, J. Zhou, J. Song, J. Xu, H. Zhang, X. Zhang, J. Guo, L. Zhu, D. Wei, G. Han, J. Min, Y. Zhang, Z. Xie, Y. Yi, H. Yan, F. Gao, F. Liu and Y. Sun, Nat. Energy, 2021, 6, 605–613 CrossRef CAS.
- S. Liu, J. Yuan, W. Deng, M. Luo, Y. Xie, Q. Liang, Y. Zou, Z. He, H. Wu and Y. Cao, Nat. Photonics, 2020, 14, 300–305 CrossRef CAS.
- Y. Shi, Y. Chang, K. Lu, Z. Chen, J. Zhang, Y. Yan, D. Qiu, Y. Liu, M. A. Adil, W. Ma, X. Hao, L. Zhu and Z. Wei, Nat. Commun., 2022, 13, 3256 CrossRef CAS PubMed.
- X. Gu, Y. Wei, N. Yu, J. Qiao, Z. Han, Q. Lin, X. Han, J. Gao, C. Li, J. Zhang, X. Hao, Z. Wei, Z. Tang, Y. Cai, X. Zhang and H. Huang, CCS Chemistry, 2023 DOI:10.31635/ccschem.023.202202575.
- H. Liu, M. Li, H. Wu, J. Wang, Z. Ma and Z. Tang, J. Mater. Chem. A, 2021, 9, 19770–19777 RSC.
- K. Vandewal, K. Tvingstedt, A. Gadisa, O. Inganäs and J. V. Manca, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 125204 CrossRef.
- X.-K. Chen, D. Qian, Y. Wang, T. Kirchartz, W. Tress, H. Yao, J. Yuan, M. Hülsbeck, M. Zhang, Y. Zou, Y. Sun, Y. Li, J. Hou, O. Inganäs, V. Coropceanu, J.-L. Bredas and F. Gao, Nat. Energy, 2021, 6, 799–806 CrossRef CAS.
- J. Yuan, C. Zhang, B. Qiu, W. Liu, S. K. So, M. Mainville, M. Leclerc, S. Shoaee, D. Neher and Y. Zou, Energy Environ. Sci., 2022, 15, 2806–2818 RSC.
- P. Müller-Buschbaum, Adv. Mater., 2014, 26, 7692–7709 CrossRef PubMed.
- Y. Huang, E. J. Kramer, A. J. Heeger and G. C. Bazan, Chem. Rev., 2014, 114, 7006–7043 CrossRef CAS PubMed.
- S. Nilsson, A. Bernasik, A. Budkowski and E. Moons, Macromolecules, 2007, 40, 8291–8301 CrossRef CAS.
- L. Ye, B. A. Collins, X. Jiao, J. Zhao, H. Yan and H. Ade, Adv. Energy Mater., 2018, 8, 1703058 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ee03902h |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |