
Unlocking the performance of ternary metal (hydro)oxide amorphous catalysts via data-driven active-site engineering†
Doudou
Zhang
 a,
Haobo
Li
*bd,
Haijiao
Lu
c,
Zongyou
Yin
c,
Zelio
Fusco
a,
Asim
Riaz
a,
Karsten
Reuter
d,
Kylie
Catchpole
a and
Siva
Karuturi
*a
a,
Haobo
Li
*bd,
Haijiao
Lu
c,
Zongyou
Yin
c,
Zelio
Fusco
a,
Asim
Riaz
a,
Karsten
Reuter
d,
Kylie
Catchpole
a and
Siva
Karuturi
*a
aSchool of Engineering, College of Engineering, Computing and Cybernetics, The Australian National University Canberra, ACT 2601, Australia. E-mail: siva.karuturi@anu.edu.au
bSchool of Chemical Engineering, The University of Adelaide, Adelaide, South Australia 5005, Australia. E-mail: haobo.li@adelaide.edu.au
cResearch School of Chemistry, The Australian National University, Canberra, Australian Capital Territory 2601, Australia
dFritz-Haber-Institut der Max-Planck-Gesellschaft, 14195 Berlin, Germany
First published on 4th September 2023
Abstract
Ternary metal (hydro)oxide amorphous catalysts are attractive oxygen evolution reaction (OER) catalysts due to their high performance and cost-effectiveness, but a fundamental understanding of their structure–property relationships remains elusive. Herein, we fabricate a highly active ternary metal (hydro)oxide (NiFeCo) OER catalyst, showing an overpotential of 146 mV at 10 mA cm−2 and ∼300 hours of durability in 1 M KOH. Inspired by this finding, a dataset with first-principles adsorption energies of reaction intermediates at over 300 active-site structures for both oxides and hydroxides is computed and used to train a machine-learning model for screening the dominant factors and unveiling their intrinsic contributions. The computational work confirms that adding Fe and Co makes the original Ni (hydro)oxide reach ultra-low overpotentials below 200 mV through the modulation from hydroxide towards oxide and the formation of active-sites of ternary metallic components. A series of physical properties of the Fe, Co and Ni atoms in the active-sites are further included in the analysis, and it is found that the magnetic moment (mag) plays an important role in the OER activity. This work demonstrates the application of machine-learning methods in atomic-scale active-site engineering to understand the activity mechanism of ternary metal (hydro)oxide amorphous catalysts for water oxidation, and it has the potential to be extended to wider applications.
Broader contextThe cost-effective production of hydrogen from renewable sources via electrochemical water splitting is crucial for climate goals and emission reduction. Transition metal oxides have shown potential as catalysts for the oxygen evolution reaction (OER) due to their affordability and high performance. However, understanding the complex relationship between atomic compositions, magnetic properties, adsorption properties, and electronic structures of multi-metallic catalysts remains a challenge. In this study, a theoretical workflow combining machine learning (ML) and density functional theory (DFT) is introduced to engineer active sites in complex (hydro)oxide catalysts. The qualitative role of Fe and Co in enhancing the OER activity of Ni-based catalysts is demonstrated using ML. The study involves a DFT-calculated dataset of over 300 NiFeCo hydroxide combinations, achieving exceptional OER performance with low overpotential and high durability. The analysis emphasizes the significant influence of the magnetic moment on OER activity. This pioneering research integrates ML, DFT, and experimental verification to understand the OER mechanism in multi-metallic NiFeCo-based oxides/hydroxides and provides valuable composition guidance for achieving superior OER performance. The developed ML model, electronic database, and fabrication method can be extended to other multi-metallic (hydro)oxides, showcasing the application of Artificial Intelligence in electrolysis. The cost-effective and scalable corrosion synthesis processes presented in this study facilitate industrial implementation. |
Introduction
The production of hydrogen as a high-energy-density and carbon-free fuel via water electrocatalysis has been considered as a critical step in energy transition.1,2 However, the oxygen evolution reaction (OER), as one of the half-reactions of water splitting, is an energy-intensive process involving multiple proton/electron-coupled steps and suffers from a more sluggish mechanism than the hydrogen evolution reaction (HER).3 Therefore, numerous investigations have been performed to explore efficient OER catalysts/electrodes with sufficient catalytic activity, stability and ease of fabrication.3,4 Recently, besides noble-metal-based catalysts such as IrO25 and RuO2,6 several nonprecious OER catalysts such as multiple-metallic oxides,7,8 layered double hydroxides,9,10 spinel type oxides11 or perovskite oxides12 have been developed. Although the studies have discussed the active species, the nanostructure and the components, their intrinsic structure–property relationships are missing.13,14 Moreover, the components or the crystal phases are often influenced in a complicated way by the synthesis process, which makes it challenging to achieve reproducibility in large-scale synthesis and practical applications.Multiple-metallic catalysts based on transition-metal (oxy)hydroxides attract intensive attention due to the modification of the electronic and structural properties from the participation of multiple elements.8,15,16 For example, it has been demonstrated that iron cations are active sites in NiFe layered double hydroxides (LDHs), while other evidence supports nickel ions with high valence as active sites.17–19 Furthermore, it has been shown that distinct primary components within Ni–Fe–Co electrocatalysts synergistically accelerate the kinetics of water oxidation across varying ranges of overpotentials.20 The commonly highlighted mechanisms based on as-fabricated catalysts include observation of a unique crystalline phase, synergistic effects, defect engineering, heterogeneous structures, etc. Moreover, the optimal ranges of the ingredients are too broad to repeat, and the resultant structure–property relationships of the catalysts are not accurate. Thus, the synthesis of multi-metallic catalysts with complex ingredients based on an accurate relationship between the atomic compositions, metal magnetic properties, adsorption properties and electronic structure and catalytic behavior is still a major challenge.
Recent advances in machine-learning (ML) methods have demonstrated their efficacy and practicality in predicting the performance of energy materials, particularly in the design of electrocatalysts.21–23 These ML techniques are particularly useful when the geometric structure and elemental composition of catalytically active sites on material surfaces are complex, making it difficult to confirm and differentiate structures using conventional experimental characterization methods. The acceleration achieved by ML methods in comparison to traditional first-principles calculations therefore enables a more extensive and systematic sampling and analysis of geometric and chemical species. Examples include spinel,11 high-entropy alloy24 or doped oxides25 with multi-metal components, and covalent organic framework (COF) materials with various M–NxOy active-site structures.26 Zhao et al. studied the trends in oxygen electrocatalysis of 3d-layered (hydro)oxides and used linear regression models to identify descriptors to relate adsorption energy with material properties.27 Furthermore, interpretable ML methods can provide insights into the physical relationship between the adsorption energy of intermediates and the composition and structure of active sites in electrocatalytic processes. The use of the sure independence screening and sparsifying operator (SISSO) for alloys28 and subgroup discovery (SGD) for single-atom catalysts29 has for instance already helped to deepen our understanding of these relationships.
Here, we employ such an ML-driven approach to explore complex structure–activity relationships of binary or ternary multi-metallic catalysts. This reveals the inner covalency or competition among the various metals, oxides, hydroxide species, or different-strength metal–oxygen bonds that lead to the exposure of different active sites. Specifically for the case of metal constituents with similar properties, such as Fe, Co and Ni, we then aim to exploit the gained understanding of atomic-scale active-site engineering, i.e. the deliberate generation of multi-metallic active-site structures. As a starting point, we fabricated a ternary NiFeCo catalyst via a corrosion-engineering strategy that exhibits an overpotential below 146 mV (@10 mA cm−2) and ∼300 hours of durability in 1 M KOH, thus surpassing the performance of hitherto reported Ni, Fe, Co-based monometallic, binary and ternary metallic based catalysts.5,30 To rationalize the dominant features of amorphous tri-metallic Ni, Fe, and Co (hydro)oxides that enable excellent OER activity, we analyze a density-functional theory (DFT) calculated dataset of more than 300 possible active-site structures of Ni, Fe, and Co (hydro)oxides via an ML approach. This demonstrates that the tri-metallic catalyst composition is optimal for achieving higher OER activity than that of mono- and bi-metallic catalysts and identifies the critical role of the magnetic moment in the relationship between the active-site structure and activity. This work methodologically establishes a theoretical workflow for active-site engineering on the atomic scale of ternary metal components for structurally complex (hydro)oxide catalysts, where ML is applied to qualitatively illustrate the role of Fe and Co in increasing the oxidation state and participating in the composition of the active-site of Ni-based catalysts to enhance the OER activity. Such rational design of tri-metallic active sites for electrocatalysts that closely combines quantum chemical computation, ML and experimental verification is expected to be extended to a wider range of electrocatalytic systems.
Results and discussion
Experimental investigation of the NiFeCo electrocatalyst
The NiFeCo ternary metallic catalyst was experimentally synthesized to investigate the synergistic effect of multiple metallic components in the OER. Inspired by their strong penetrating ability and passivation performance, nitrate ions (NO3−)31 were introduced by adsorption on nickel foam to corrode the nickel foam (NF) surface for fabrication of metallic hydroxyl electrodes, and both binary and ternary transition metallic hydroxyl electrodes were fabricated for comparison in this work. As shown in Fig. 1a, the reaction mechanism of monometallic, binary metallic and ternary metallic nitrate involves the corrosion of NF and reaction with hydroxyl in a single container with a low concentration of a mixture of different nitrates (e.g. nickel nitrate (Ni(NO3)2), ferrous nitrate (Fe(NO3)2·6H2O) and cobalt nitrate(Co(NO3)2). | ||
| Fig. 1 Illustrations and structural characterization of NixFeyCoz(OH)m. (a) Schematic illustrations of the formation and microstructure of the electrodes. The specific reactions that occur with the corrosion of nickel substrates in an uncovered Petri dish; Mx+ cations and hydroxyl cause the spontaneous electrochemical reaction on the nickel foam (NF). The nitrate ions present in the solution etch the surface of NF. Ternary NiFeCo hydroxyl oxide nanosheets are formed uniformly distributed along the skeleton structure of the NF. (b) X-ray diffraction patterns of the ternary NiFeCo hydroxyl layers. (c) AFM image of ternary NiFeCo hydroxyl layers and their corresponding thicknesses along the marked lines (d) 1 and (e) 2. (f)–(h) The transmission electron microscopy (TEM) image, the corresponding high-resolution transmission electron microscopy (HRTEM) image and the selected-area electron diffraction (SAED) pattern, respectively, collected from the square area marked in (f). (i)–(m) TEM images from the selected area and the corresponding elemental mappings of ternary NiFeCo alloy hydroxyl layers. | ||
It was necessary for oxygen ions, nitrate ions, and ferrous ions to all be present for forming NiFe or NiFeCo hydroxyl on NF. We confirmed that catalysts were not generated on NF substrates if nitrate ions were replaced by sulfate ions (SO42−). The concentration of the nitrate ions in our experiments was kept at the mM level; otherwise, they damaged the mechanical strength of the substrates after several hours of the reaction. From the linear sweep voltammetry (LSV) curves (Fig. S1, ESI†), the optimum concentration of NO3− is 2 mM at room temperature for a reaction time of 48 hours, which favored NO3− ions adsorbing on the surface of the NF and expedited the local corrosion process. With the NO3− concentration increases from 2 mM to 10 mM, the morphology of nanosheets on the surface of the NF has disappeared (Fig. S2, ESI†). It is noted that a high concentration of nitrate ions or a longer corrosion time quickly caused the substrate to have poor mechanical strength or even to dissolve in the solution.
Additionally, we found the corrosion reaction did not occur if the solution did not contain Fe2+, which suggested the transition of a spontaneous electrochemical potential from Fe2+ to Fe3+.32 Thus, the mechanism for fabricating multi-metallic hydroxide catalysts supported on NF can be proposed as follows: firstly, the nitrate ions continuously migrate to and get adsorbed on the NF surface, followed by corrosion of the NF substrate to Ni2+ ions. Meanwhile, Fe2+ ions quickly oxidise to Fe3+ with a low concentration of NO3−, accompanied by Ni2+ in the precursor generally enriched in the anode region. Then, O2 dissolved in the solution or from the air reacts with water to form hydroxyl (OH−) in the cathode region. The inherent driving force for multi-metallic hydroxyl formation on NF is the electric potential difference between Fe2+/Fe3+ and O2/OH−, as shown in the half and overall electrochemical reaction equations in Fig. 1a. Finally, the M+x cations (including Ni2+, Fe3+ and Co2+) synchronously react with OH− to precipitate and form NiFeCo ternary hydroxide nanosheets on the NF. The experimental details are described in the ESI.† Consistent with previous reports, when comparing the as-prepared electrodes’ performances, the ternary metallic hydroxide electrodes (Ni2+, Fe2+ and Co2+) exhibited superior OER activity compared with the binary metallic NiFe, FeCo, and NiCo hydroxide catalyst, while the latter showed superior performance compared to the monometallic Ni hydroxide or Fe hydroxide (Fig. S3, ESI†). However, the growth process of the catalysts differs from previously reported multimetallic NiFeCo precursor electrodeposition7 or ion-exchange processes.33 This highlights the significance of the slow and spontaneous reaction sequence, and our aim is to uncover the intrinsic factors that govern the OER activity of these high-performance catalysts.
Structural characterization of NixFeyCoz(OH)m
Due to the intentional introduction of three divalent cations (Ni2+, Fe2+ and Co2+) in the corrosive environment, ternary NiFeCo hydroxyl (NixFeyCoz(OH)m) is spontaneously generated on the NF instead of the commonly-formed iron rusts. Additionally, NixFeyCoz(OH)m generated exists in an amorphous form, which is distinct from the reported NiFe-LDHs that exhibit a crystalline structure.32 This is evident from the absence of distinct peaks in the XRD patterns, except for those attributed to the NF substrate in Fig. S4 (ESI†) within the range of 10–80°. However, in contrast, it is evident that there are poor crystalline peaks of both Ni hydroxide and Fe hydroxide synthesized using the sole cation precursors in a similar procedure. Fig. 1b presents amplified XRD patterns from 10 to 40° to distinguish the set of peaks. There is a peak at 17.5° attributed to β-Ni(OH)2 (JCPDS#14-0117) and another at 28.7° matching NiOOH (JCPDS#06-0075) in the nickel nitrate precursor, while for sole ferrous nitrate, the XRD result reveals two peaks of β-FeOOH (JCPDS#75-1594) at 18.7° and 28.6°. However, the peaks of β-Ni(OH)2 and NiOOH and the major peak of β-FeOOH disappeared when Ni and Fe divalent cations coexisted in the precursor solution, and only the minor peak of β-FeOOH at 28.6° remained. Furthermore, with Co2+ added into the solution, the XRD results showed that the NixFeyCoz(OH)m catalyst completely transformed into the amorphous structure instead of the LDHs.As mentioned in Fig. 1a, the NixFeyCoz(OH)m catalyst was well-oriented, with grain boundary-enriched thin nanosheet arrays, of which advantageous microstructural features (i.e., nanosheet array architecture and abundant grain boundaries) are believed to be beneficial for electrochemical reactions.32 The ultrathin nanosheets exhibited a lateral size of approximately 40 nm without detectable stacking via AFM (Fig. 1c). In addition, the thickness of the nanosheets was calculated by scanning along the lines marked “line 1” and “line 2,” which indicates that the thicknesses of a single layer and five layers are 1 nm (Fig. 1d) and 4.7 nm (Fig. 1e), respectively.
Transmission electron microscopy (TEM) and high-resolution transmission electron microscopy (HRTEM) images were obtained to investigate the morphology, distribution of each element, and composition of NixFeyCoz(OH)m nanosheet arrays on NF (Fig. 1f–m). It is clearly seen that the as-fabricated catalysts consist of compact nanosheet arrays assembled like flower petals with lateral sizes of 40–50 nm without detectable stacking, which agrees with the AFM results. The magnified HRTEM image from the marked square in Fig. 1f shows that the nanosheets are composed of several layers but with no clear lattice fringes, suggested by the clear edges of regions with different brightnesses (Fig. 1g). The corresponding selected-area electron diffraction (SAED) pattern (Fig. 1h) confirms the NixFeyCoz(OH)m structure was amorphous by the fuzzy and broad halos. The corresponding energy-dispersive X-ray spectroscopy (EDS) elemental-mapping images of NixFeyCoz(OH)m provide further evidence confirming the coexistence and uniform distribution of elemental Ni, Fe and Co in the nanosheets (Fig. 1i–m).
Additionally, from the combined elemental mappings (Fig. 1m), it is evident that all Fe and Co elements are present inside the nanosheets, while Ni is distributed at the surface of the NF. We conclude that Fe2+ and Co2+ first become enriched on the NF after NO3− etching and react with OH−. When the concentrations of the divalent cations decrease, freshly dissolved Ni2+ surrounds the surface of the nanosheets, revealing the formation of homogeneously distributed multi-metallic hydroxides.
To reveal the chemical valence and bonding states in NixFeyCoz(OH)m hydroxides, typical metal–OH hydroxides were characterized by X-ray photoelectron spectroscopy (XPS) (Fig. S5, ESI†). For the initial Ni 2p spectra in Fig. S5a (ESI†), 2p3/2 and 2p1/2 spin–orbit peaks are situated at 855.8 and 873.5 eV, along with two satellite peaks identified as “Sat.” at 861.8 and 879.8 eV,34,35 which suggests that most Ni elements in the nanosheets are in the Ni2+ oxidation state. In Fig. S5b (ESI†), the Fe 2p XPS spectrum displays two peaks, one at 713.1 eV for Fe 2p3/2 and another at 725.2 eV for Fe 2p1/2, indicating the presence of the Fe3+ oxidation state,34 which matches well with the mechanism of Fe2+ transitioning to Fe3+ in the fabrication process. Co 2p in Fig. S5c (ESI†) exhibits two different doublets situated at Co 2p3/2 (a lower-energy band 781.5 eV) and Co 2p1/2 (a higher-energy band 797.0 eV) with a gap of over 15 eV, suggesting the existence of Co2+ and Co3+ species in coordination with −OH.36 Meanwhile, the binding energy position at 782.3 eV is influenced by the Auger effect with Ni.37 For the O 1s spectra in Fig. S5d (ESI†), the peak at 531 eV corresponds to typical metal–OH bonds, and the peak at 532.8 eV is assigned to surface-adsorbed water molecules in ternary NixFeyCoz(OH)m.38 Furthermore, we confirm from the XPS results that the surface atomic ratio of Ni![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Fe:Co is 94.36%:2.04%:3.6% and the surface composition is a combination of oxides and hydroxides.
:Fe:Co is 94.36%:2.04%:3.6% and the surface composition is a combination of oxides and hydroxides.
Electrocatalytic performance of NixFeyCoz(OH)m
To evaluate the electrocatalytic performance, we first assessed the OER activity of the as-prepared NixFeyCoz(OH)m and NiFe(OH)x catalysts and plain NF in a 1 M KOH electrolyte for comparison. From the OER polarization curves displayed in Fig. 2a, the NixFeyCoz(OH)m electrode showed a significant enhancement for OER compared with the NF and NiFe(OH)x. To deliver the current densities of 10 and 100 mA cm−2, the required overpotentials of the NixFeyCoz(OH)m electrode are 146 and 220 mV, respectively, which are lower than those of NiFe(OH)x by approximately 51 mV and 56 mV, not to mention the plain NF substrates. In order to assess the specific intrinsic activities of NixFeyCoz(OH)m, their electrocatalytic activities were normalized by the electrochemically active surface area (ECSA) measured by the double layer capacitance method on the basis of cyclic voltammetry in a 1 M KOH electrolyte (Fig. S6, ESI†). The higher double layer capacitance (Cdl) of NixFeyCoz(OH)m (4.88 mF cm−2) compared to that of NiFe(OH)x (3.15 mF cm−2) indicates that NixFeyCoz(OH)m possesses a higher ECSA.8,39 This further confirms that the nanosheets array morphology of NixFeyCoz(OH)m electrode plays an important role in boosting the catalytic activity. The ultralow overpotential shown by ternary NixFeyCoz(OH)m further proves that ternary Fe, Co, Ni (hydro)oxide catalyst achieves higher activity than single or binary components. Moreover, the NixFeyCoz(OH)m electrode initially does not exhibit any pre-oxidation peak compared to NiFe(OH)x, likely due to the presence of fully covered Ni2+ hydroxide, which is proved by the Fig. 1m and Fig. S5 (ESI†). In addition, the NixFeyCoz(OH)m electrode exhibited the smallest Tafel slope of 50 mV dec−1 (Fig. 2b) compared with those of NiFe(OH)x (104 mV dec−1) and NF (122 mV dec−1), suggesting faster OER catalytic kinetics. Impressively, the NixFeyCoz(OH)m catalyst remained stable for almost 300 hours (Fig. 2c). Comparing the LSV curves of the initial NixFeyCoz(OH)m and that after 300 hours of operation in Fig. S7 (ESI†), the activity increased substantially with a prominent pre-oxidation peak, confirming that the composition of the catalyst on the surface of the electrodes has changed. The changed surface ratio of Ni:Fe:Co was confirmed by the XPS result to be 18.03%:0.84%:0.91%, which indicates that the Fe content remained almost the same but that of Co was slightly increased on the surface. TEM and HRTEM images showed that the edges of the nanosheets have become blurry and sloppy (Fig. S8a and b, ESI†), and the layers have become thicker than in Fig. 1g. Moreover, the corresponding SAED pattern (Fig. S8c, ESI†) further confirmed the NixFeyCoz(OH)m was amorphous after 300 hours of stability testing, as manifested by the fuzzy and broad halos similar to those in Fig. 1h. By contrast, a comparison of the HRTEM and corresponding elemental mapping in Fig. S8(d)–(h) (ESI†) with the results in Fig. 1(i)–(m) shows that the distribution of the metals has changed evidently with Ni aggregated as nanoparticles surrounded by Fe and Co. Fe and Co dominated most of the skeleton of the nanosheet from the elemental mapping results. The good match of the measured faradaic efficiency of the NixFeyCoz(OH)m catalyst with the calculated amounts of the evolved O2 gas (Fig. 2d) proves that there were no undesirable side reactions. Overall, the corrosion-derived NixFeyCoz(OH)m from NF in this work demonstrated superior OER activity and stability in alkaline electrolytes compared with the other binary Ni, Fe-based catalysts, e.g. LDHs, or those having delicately fabricated nanostructures (Fig. 2e). Additionally, it outperformed the recently reported NiFe-based (oxy)-hydroxide catalysts in alkaline media, as well as many non-noble metal catalysts (Table S1, ESI†). More importantly, the synthesis process of the NixFeyCoz(OH)m catalyst via this corrosion strategy is much more efficient in terms of saving energy and resources than the other reported OER catalysts. It is highly likely that large-size electrodes can be manufactured with low energy consumption by a roll-to-roll solution approach. Furthermore, this Ni-derived NixFeyCoz(OH)m catalyst can be directly utilized as an OER electrode after a simple fabrication procedure that avoids the use of an expensive polymer binder. However, the superficial morphology, the amorphous structure and the broad composition range of ternary Ni, Fe, Co (hydro)oxide catalysts make it challenging to elicit their complicated structure–property relationships, which are important for further development of these high-performance catalysts. Thus, we introduce a DFT-assisted ML methodology to set up a dataset and evaluate the various features dominating the OER activity.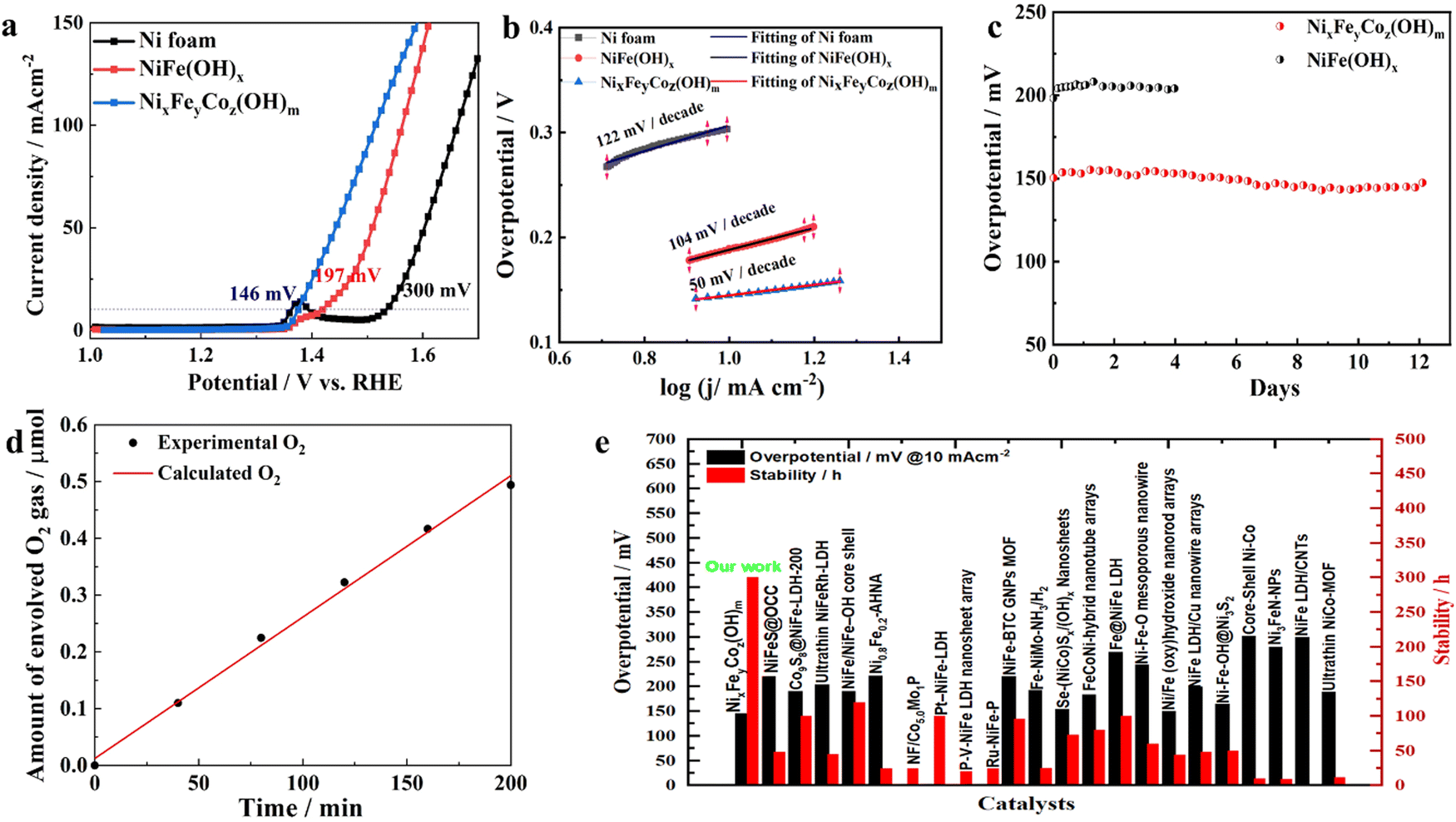 | ||
| Fig. 2 OER performance. (a) Polarisation curves after IR-compensation and (b) corresponding Tafel plots of the NF, NiFe(OH)x, and NixFeyCoz(OH)m electrodes in 1 M KOH. (c) Long-term stability tests at a constant current density of 10 mA cm−2 for the NiFe(OH)x, and NixFeyCoz(OH)m electrodes in 1 M KOH. (d) O2 amounts detected by gas chromatography (dark dots) and the calculated amounts of O2 (red line) by NixFeyCoz(OH)m catalyst. The measurement was performed at jOER = 10 mA cm−2 in 1 M KOH. (e) Comparison of this work and reported overpotential required to achieve current densities of 10 mA cm−2 and the related stability for various catalysts in 1 M KOH. RHE stands for reversible hydrogen electrode. | ||
Active-site structure engineering
Extensive theoretical works have revealed that the OER active sites on the surface of metal (hydro)oxides are metal sites formed by the vacancy of surface oxygen atoms.40 We have built a two-dimensional model of oxides (MO2) and hydroxides (M(OH)2) based on LDHs40 nanosheets to simulate such metal active-site structures with 3 first-nearest neighbor metal atoms, 3 second-nearest metal atoms and the O or OH groups between the metals (Fig. S9, ESI†). Assuming that chemical adsorption depends mostly on the short range, only the first and second nearest neighbor metal atoms are included in active-site engineering. The adsorption active site is in the center of atoms 1–6 (marked by the yellow circle shown in Fig. S9c, ESI†). Atoms 1–6 can be Fe, Co or Ni atoms, which creates 36 = 729 possible combinations. Considering both the triple and axial symmetry of the structure results in 161 inequivalent active-site configurations each for oxides and hydroxides. All 322 configurations constitute a dataset for active-site engineering of ternary OER catalysts.Another key reason for constructing this dataset for oxides and hydroxides is the uncertainty of the degree of hydrogenation of the oxides under alkaline working conditions. The surface structure of the catalyst under electrochemical conditions can be determined by conventional ab initio thermodynamics40,41 calculations and comparison of surface free energies to obtain the Pourbaix diagram.40 The computed surface Pourbaix diagrams of pure Fe, Co and Ni (hydro)oxide (Fig. S10, ESI†) show that under experimental conditions (E = 1.1–1.7 V vs. RHE, pH ∼13.8), Ni has both oxide and hydroxide structures, while Fe and Co are more stable as oxides. This matches well with the XPS results that showed that Ni maintained a hybrid of oxides and hydroxides and was aggregated as nanoparticles surrounded by Fe and Co oxides. The pre-oxidation peak's appearance after the long stability test also confirms that the surface of the catalyst is rich in Fe and Co oxides that transform into higher oxidation states. We suspect, they are the dominant reason that the catalyst is stable in alkaline electrolytes and has even higher activity than the initial state, as Co2+ and Co3+ species were shown to exhibit a self-healing property.42,43 Although the theoretical phase diagram is only an estimation, it still illustrates the high structural complexity of the amorphous ternary (hydro)oxides. As shown in the experimental results and discussion, when Fe, Co and Ni are randomly distributed, the surface will be a combination of oxide and hydroxide, and the hydrogenation structure cannot be determined due to the interaction among Fe, Co, and Ni. Therefore, a separate database of oxides and hydroxides must be built and further analyzed via advanced methods such as ML to find reasonable descriptors and influencing factors.
To test the computational parameters, the overpotential of pure Ni hydroxide was calculated to compare with the experimental value as a benchmark. The theoretical overpotential of 0.59 V (Fig. S11, ESI†) is very close to the experimental value of a planar Ni electrode at ∼0.55 V (Fig. S12, ESI†). This provides further evidence that the Ni-based catalyst is mainly hydroxylated under experimental conditions. Previous studies have demonstrated a strong agreement between the thermodynamic overpotential obtained from DFT calculations for the OER and experimental values, including the (hydro)oxide system studied in this work.44,45 This correspondence validates the reliability and accuracy of our theoretical method in predicting experimental outcomes, and in particular the meaningful relative trends. Then, extensive DFT calculations were performed to investigate the adsorption strength of all the intermediates and through this the reactivity of each active-site structure was derived.
Recent experimental and theoretical works,44,46,47 have indicated that the OER at metal (hydro)oxide surfaces under alkaline conditions follows the Mars–Van Krevelen (MvK) mechanism, i.e., the elementary steps and corresponding Gibbs free energy change are as follows:
| *OH + OH− → *O + H2O + e− ΔG1 = ΔGad(*O) − ΔGad(*OH) | (1) |
| *O + OH− → *OOH + e− ΔG2 = ΔGad(*OOH) − ΔGad(*O) | (2) |
| *OOH + OH− → *O2 + H2O + e− ΔG3 = ΔGad(*O2) − ΔGad(*OOH) | (3) |
| *O2 + OH− → O2 + *OH + e− ΔG4 = 4.92 eV − ΔG1 − ΔG2 − ΔG3 | (4) |
In our work, an estimate of the overpotential (η) of the overall reaction is then obtained as the difference between the highest thermodynamic energy barrier and the equilibrium potential:
| η = max(ΔG1, ΔG2, ΔG3, ΔG4)/e − 1.23 V |
Density functional theory calculations for the OER activity
The DFT-calculated adsorption free energies (Gad) of each intermediate (*OH, *O, *OOH, *O2) and the deduced overpotentials (η) of the reaction are shown in Fig. 3. Unlike the well-known scaling relation between Gad of the OER intermediates observed across the metal oxides,48Gad at the ternary active-sites still has a certain range distribution of over 1.5 eV but with low linear correlations (Fig. 3a, d and Fig. S13, ESI†). This directly leads to the result that the η calculated from Gad no longer has a good correlation with Gad of certain intermediates such as *O (Fig. 3b and e). Therefore, utilizing the Gad of individual adsorbates as descriptors for catalytic reactivity poses challenges in this system. It becomes imperative to devise alternative descriptors that can comprehensively encapsulate Gad of multiple adsorbates simultaneously.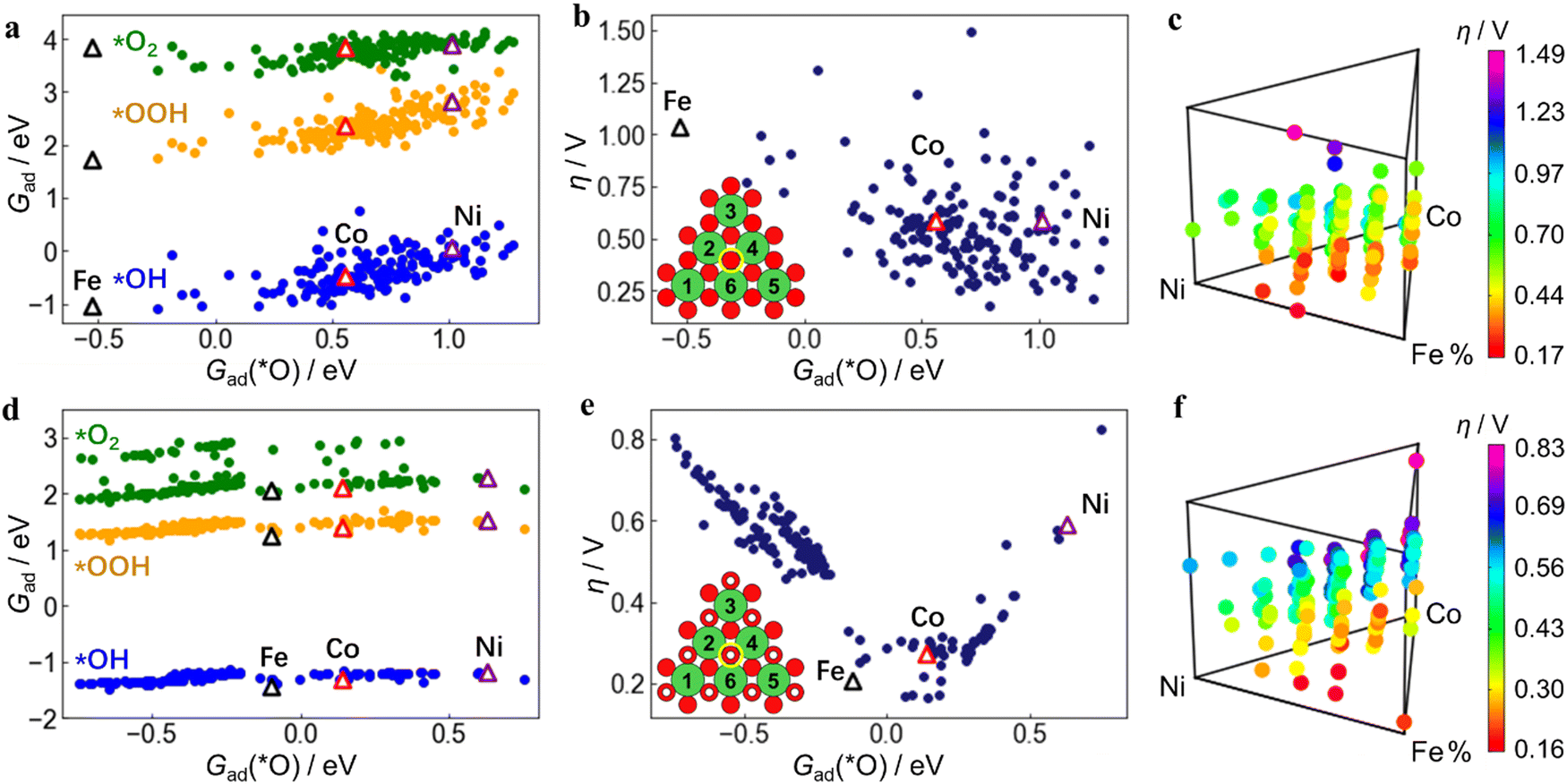 | ||
| Fig. 3 DFT simulation across active-sites through structure engineering. DFT calculation results on Fe, Co, Ni-ternary MO2 (a)–(c) and M(OH)2 (d)–(f) active-sites. (a) and (d) Relationship between the adsorption free energies (Gad) of the 3 intermediate adsorbates (*OH, *OOH, *O2) with the adsorbate *O. The positions of pure Fe, Co, or Ni (hydro)oxide are marked by triangles. (b) and (e) OER overpotential (η) derived from the MvK mechanism vs. Gad(*O). The positions of pure Fe, Co, or Ni (hydro)oxide are marked by triangles. The atomic structures of the active-sites are shown as insets. Each active center is marked by a yellow circle, and the metal, O, and H atoms are green, red and white spheres, respectively. (c) and (f) η shown in height above the Fe, Co, Ni ternary compositional map. Each dot represents an active-site structure of the computation. Different structures can have the same composition ratio, so some dots overlap vertically. | ||
From the composition of the 6 nearest neighboring metal atoms (insets of Fig. 3b and e), the relationship between the elemental composition of the active site and η was further obtained. Each dot represents an active site structure with a discrete composition separated by 18.33% (1/6). The composition of different active site structures can be the same, so some dots overlap vertically, which shows that even with the same elemental composition, the active-site structure and reactivity can vary significantly. The relationship between η and Fe, Co, Ni content is further shown in Fig. 3c and f, which are 4D mappings including the content of each metal component as the 3D triangle diagram and the η value as the 4th dimension. In order to enhance the figure readability, each of Fig. 3c and f is plotted separately in three 2D diagrams according to Fe, Co, and Ni contents to display the trend (Fig. 4). It can be seen that η also presents a wide distribution due to the influence from the adsorption free energy of the intermediates and has a complicated relationship with Fe, Co, and Ni content. A lower value of η corresponds to a higher catalytic activity, and the ideal OER catalyst occurs when η is close to 0 V. When only considering the oxide MO2 structures (Fig. 4(a)–(c)), Fe, Co and Ni monometallic oxides can not reach an overpotential (η) below 200 mV, which is consistent with literature49–51 and our experimental results (Fig. S3, ESI†).
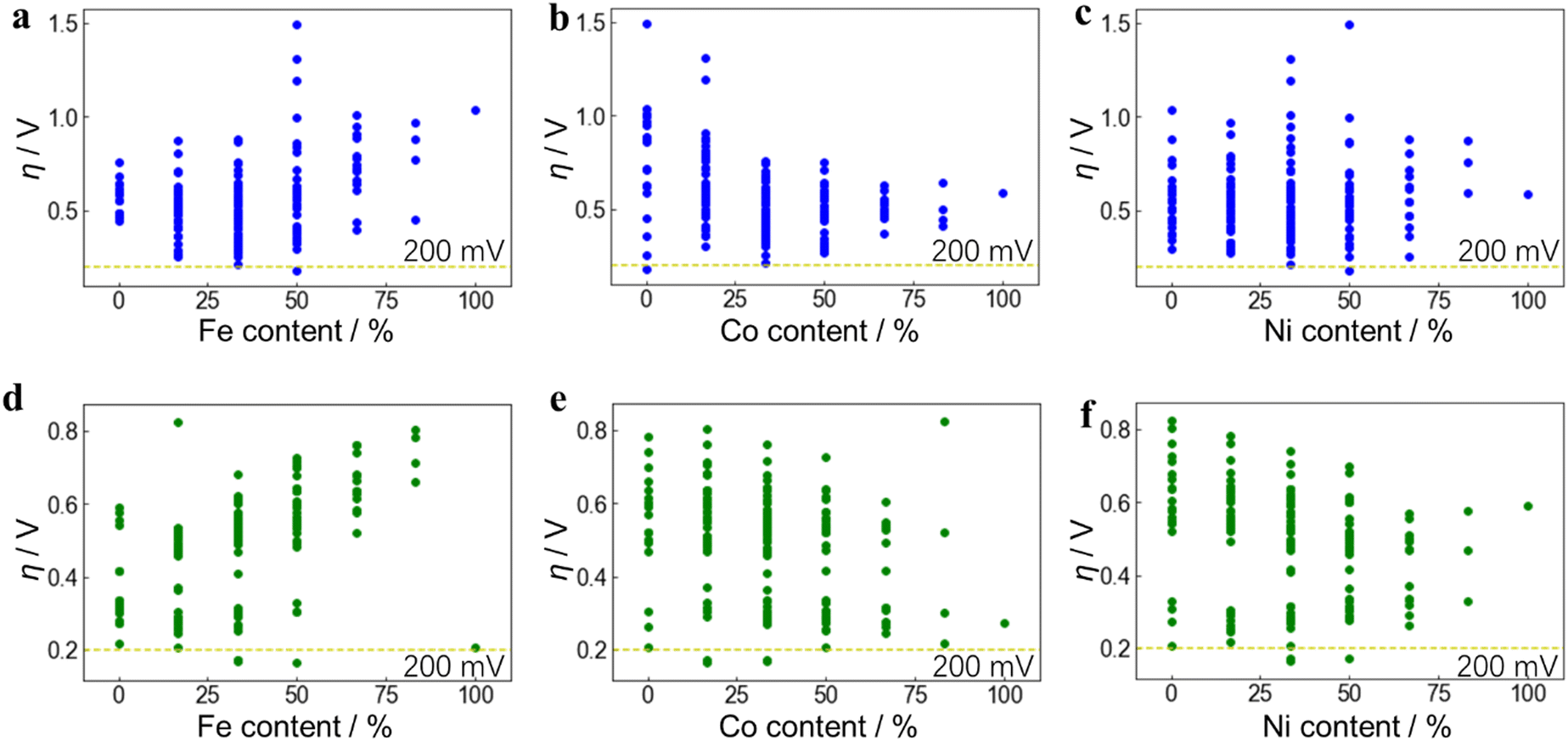 | ||
| Fig. 4 Relationship between OER activity and Fe, Co, Ni contents. DFT-calculated OER overpotential (η) on Fe, Co, Ni-ternary MO2 ((a)–(c)) and M(OH)2 ((d)–(f)) active sites vs. Fe, Co, Ni content from the site structure, respectively, as two-dimensional expansions of Fig. 3c and f. Under the same Fe, Co, or Ni content, the other two components can have different proportions, resulting in different overpotentials, so a series of points with a wide distribution is formed. The position where η is 200 mV is indicated by the yellow dotted lines, and the dots below them are regarded as active-site structures with high OER activity. | ||
Meanwhile, considering the hydroxide M(OH)2 in Fig. 4(d)–(f), only Fe (OH)2 or its compounds (FeOOH) have the possibility to reach low overpotentials below 200 mV. Again, this is consistent with previous reports52–54 and matches with the result in this work that NiFe(OH)x demonstrates an overpotential of only 197 mV (Fig. 2a). Interestingly, multi-metallic MO2 and M(OH)2 structures can both achieve an ultra-low η value well below 200 mV. However, it is still difficult to observe the role of different metal components and the in-depth physical and chemical influencing factors from this 2D diagram. It is thus necessary to further analyze the data with the assistance of machine learning methods.
A machine-learning approach to search for intrinsic descriptors
Aiming to unveil meaningful trends and draw definitive conclusions from the extensive dataset generated by DFT computational, ML analysis methods were further applied. A compressed-sensing method was used to identify the descriptors for the catalyst activity. The SISSO46 method identifies the best features and predicts the target quantity, i.e. the adsorption free energy (Gad), through a formula for mutual operation between features. The 16 primary features used are listed in Table 1. Features related to the Fe, Co, and Ni metal atoms, metal bulk and metal (hydro)oxide surface were included to better describe the active-site properties and achieve more predictive performance via ML. As it is often considered to be related to adsorption performance, the d-band center and width were calculated to describe the Fe, Co, and Ni sites. Especially for oxide catalysts, additional potential descriptors frequently mentioned in previous reviews55 such as the p-band center of the O atom (εp), charge transfer energy (CTE) and eg-orbital filling of the metal atom (eg), were included. Moreover, to take the unique magnetic properties of catalysts containing Fe, Co, and Ni into account, the magnetic moment (mag) was also specially calculated as a feature.| Class | Name | Abbrev. | Unit |
|---|---|---|---|
| Metal atom | Pauling electronegativity | PE | None |
| Ionization potential | IP | eV | |
| Electron affinity | EA | eV | |
| Number of electrons in the outermost d-shell | n d | None | |
| Metal bulk | Radius of d-orbitals | r d | Å |
| Coupling matrix element squared | V ad 2 | None | |
| Cohesive energy | E coh | eV | |
| Surface | Work function | WF | eV |
| Fermi energy level | E Fermi | eV | |
| Site | d-band center | ε d | eV |
| d-bandwidth | W d | eV | |
| p-band center | ε p | eV | |
| Charge transfer energy | CTE | eV | |
| e g-Orbital filling | e g | eV | |
| Bader charge | q | e | |
| Magnetic moment | mag | Bohr |
To describe the atomic structure of an active site, the 6 surrounding metal atoms (Fig. S14, ESI†) are divided into first-shell (metal atoms 2,4,6) and second-shell (metal atoms 1,3,5) to the reactivity center. Taking the interaction and relative position between the first-shell and second-shell atoms into consideration, the features for first-shell (F1) and second-shell (F2) atoms are processed by:
| F1 = (f2 + f4 + f6)/3 | (5) |
| F2 = ((f2 × f6/f1) + (f2 × f4/f3) + (f4 × f6)/f5)/3 | (6) |
Multi-task learning was used to identify common descriptors to predict the Gad values of all the intermediates (*OH, *O, *OOH, *O2) simultaneously so that the overpotential can then be calculated through the features. In this way, it enables the use of the same set of physical attributes to describe multiple Gad values of different adsorbates, providing deeper insights into the underlying structure–property relationships. The ten most-correlated features in the training are shown in Fig. 5a and d. For oxides, the top 4 correlated features are ionization potential (IP), magnetic moment (mag), Bader charge (q) and eg-orbital filling (eg), all from the first neighboring shell. The next three (5th, 6th, and 7th) correlated features are from the second neighboring shell, and include the CTE, p-band center (εp) and radius of d-orbitals (rd). For hydroxides, the similarity is that the top 4 correlated features are also from the first neighboring shell, including the work function (WF), CTE, rd and cohesive energy (Ecoh). The difference is that features from the second neighboring shell such as eg only have small correlations. The influence on adsorption energy is not only attributed to features of first-neighbor metal atoms, but also extends to second-neighbor metal atoms, exemplified by CTE of the second-neighbor in oxides. This underscores the significance of engineering the composition of secondary neighbors in the active site.
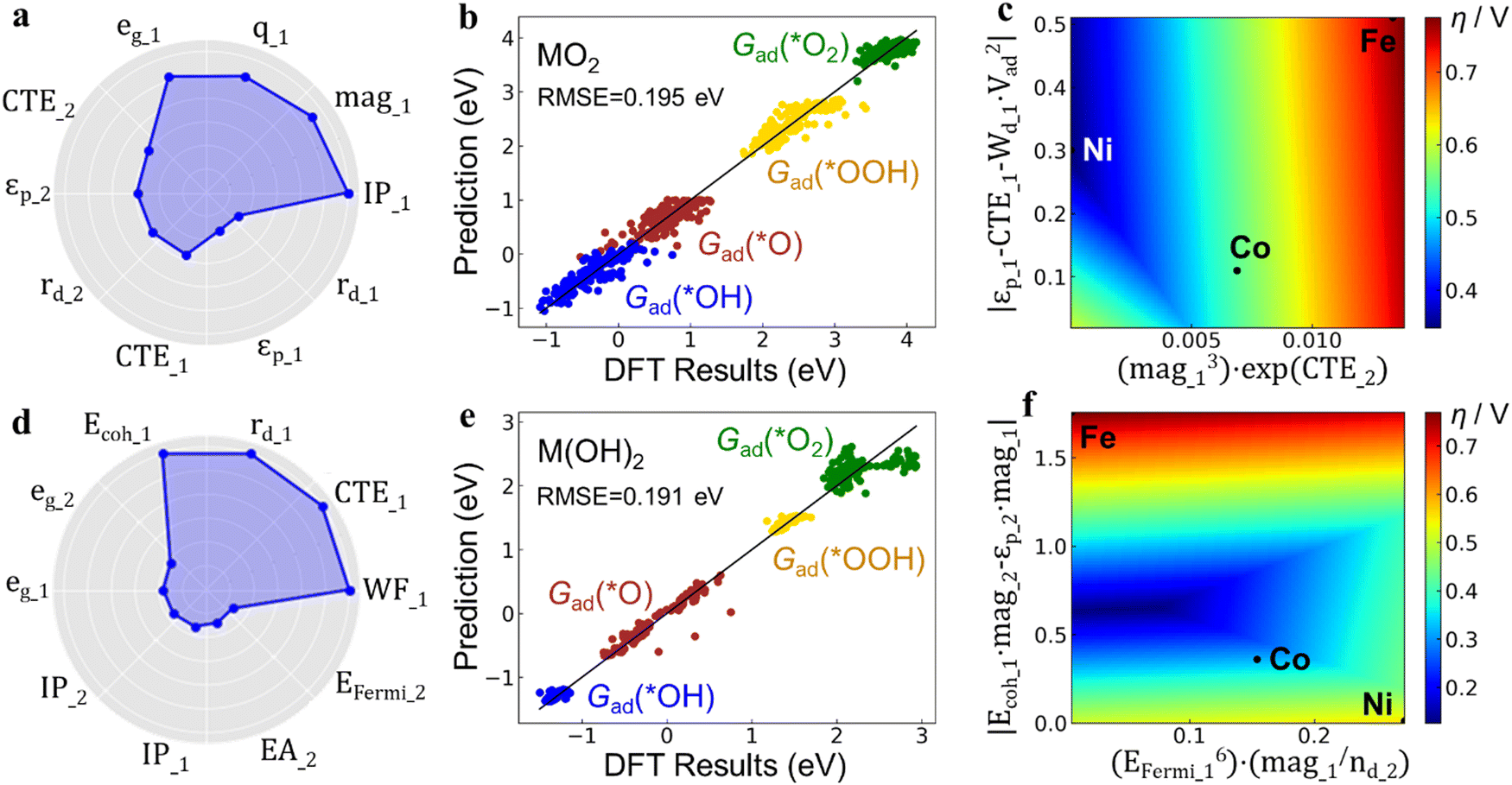 | ||
| Fig. 5 Data analysis and prediction through machine-learning. SISSO predictions on Fe, Co, Ni-ternary MO2 (a)–(c) and M(OH)2 (d)–(f) catalysts. (a) and (d) Top 10 most-correlated features. The distance of each dot from the center is proportional to its correlation coefficient. The features corresponding to the first- and second-neighbor metal atoms are denoted by subscripts 1 and 2, respectively. (b) and (e) Predicted adsorption free energies (Gad) vs. DFT calculations of the 4 reaction intermediates. The root mean square error (RMSE) is calculated as a 5-fold cross-validation prediction error. (c) and (f) OER overpotentials (η) calculated from the predicted Gad values. The x- and y-axis of the 2D-plot for overpotentials are the two operations of features from the 2D-training. The positions of pure Fe, Co, or Ni (hydro)oxide are marked by dots on the colormap. | ||
The binding strength of the reaction intermediates predicted by the ML method through the primary features reproduces the DFT calculation results quite well, which is shown by the dots straddling the perfect correlation line with a root mean square error (RMSE) below 0.2 eV (Fig. 5b, e and Fig. S15, ESI†). Conventional OER activity theory research is based on the linear scaling relationship between the adsorption free energy of different intermediates (*OOH, *O, *OH, etc.) found on the surface of metals, alloys and oxides, so that the reaction activity (overpotential) can be calculated from the descriptors in the form of adsorption energies. However, in the active-site engineering case in this work, this basic assumption of the linear scaling relation is weak, and it presents a challenge to the investigation of the structure–activity relationship of the catalyst. Through the ML approach adopted herein, for different adsorption intermediates, the feature types and operations formula used are the same, only with different coefficients. In this way, several Gad values with low linear correlation can be linked with physical quantities to generate physically inspired descriptors, which will be further used to realize the screening and prediction of unconventional catalyst structures generated by active-site engineering that do not satisfy the scaling relations.
Through the operation formula derived from the ML model, a relationship was built between the physical properties of the active sites and their OER activity (Fig. 5c and f). Fig. 5c shows that the Ni sites in MO2 exhibits the lowest η, indicating the optimal OER activity. The calculations and experiments suggest that Ni-based catalysts primarily consist of M(OH)2, while Fe and Co are mainly present as MO2. Incorporating Fe and Co into the catalyst increases the oxidation state of Ni active-sites, transforming them from M(OH)2 to MO2, thereby enhancing their activity. Furthermore, Fig. 5f demonstrates that for the M(OH)2 structure, the Fe, Co and Ni sites all exhibit high η values. However, there exists an intermediate region with physical properties in between that have even lower η than MO2. This finding indicates that ternary active-sites composed of Fe, Co, and Ni can achieve higher OER activity. Interestingly, the descriptor formula used to construct the two axes of the 2D mapping of Fig. 5c and f frequently includes magnetic moment features (mag1, mag2). This suggests that the magnetic moment of the constituent atoms in the active-site significantly influences activity optimization. This importance of the magnetic moment for Fe, Co, and Ni-containing catalysts complements the understanding of activity descriptors for (hydro)oxides. Recent works by Cao et al.56 and Wang et al.57 have also emphasized the role of magnetism in catalytic activity. Subsequent electronic structure calculations have corroborated the discernible distinctions in magnetic moments among the Fe, Co, and Ni active site atoms within the Fe, Co, and Ni (hydro)oxide (Fig. S16, ESI†). Hydroxide materials manifest stronger magnetic moments than oxides, with Fe demonstrating the highest, followed by Co, and Ni exhibiting the weakest. These variations exert an impact on adsorption properties, thereby influencing reactivity. Here we theoretically demonstrate that the Fe, Co, and Ni ternary (hydro)oxide catalysts hold promise for achieving higher activity compared to single or binary component catalysts. The theoretical predictions align well with experimental results, where the tri-metallic NiCoFe catalyst shows higher activity than the bimetallic NiFe component (Fig. 2a and Fig. S3, ESI†). This research strategy can be expected to extend to atomic engineering of active sites in tri-metallic component catalysts, particularly when the properties of the three metals are similar and conventional theoretical models are not applicable.
Conclusions
In summary, we synthesized a highly active ternary metal (hydro)oxide (NiFeCo) OER catalyst via a corrosion-engineering strategy consisting of both metal oxides and hydro oxides with a Ni:Fe:Co atomic ratio of 94.36%:2.04%:3.60%. The activity reached an ultra-low overpotential of 146 mV at 10 mA cm−2 and over 300 hours of stability in a 1 M KOH electrolyte due to its unique ternary component formation and amorphous structure. The DFT-calculated dataset composed of over 300 Fe, Co, Ni (hydro)oxide structures obtained through atomic-scale active-site engineering shows a wide distribution of reaction intermediates' adsorption energies and OER overpotentials. The relationship between the Fe, Co, and Ni components and the OER overpotential is especially complicated. Through the analysis of a data-driven machine-learning method based on the physical properties of the compound metals in active-site structures, the ternary component is expected to obtain higher OER activity than monometallic or binary catalysts. Among various physical properties, the magnetic moment shows an important influence on the OER activity for this type of catalyst.
Local structures within amorphous catalysts can exhibit considerable complexity, while the active site structures remain a pivotal factor influencing their activity. Active-site engineering involves the comprehensive construction and calculation of all possible hundreds of active site structures to encompass the entire spectrum of possible motifs, which introduces fresh perspectives for investigating the intricate interplays among the composition elements within amorphous catalysts. These findings open an avenue for the rational prediction and design of multi-metallic (hydro)oxide materials with improved performance.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The financial support from the Australian government through the Australian Research Council (ARC) and the Australian Renewable Energy Agency (ARENA) is gratefully acknowledged. Access to fabrication and characterization facilities of the Australian National Fabrication Facility (ANFF) and Centre for Advanced Microscopy is also gratefully acknowledged. We thank Dr Olivier Lee Cheong Lem, Dr Felipe Kremer and Dr Li Li for their support. The authors gratefully acknowledge Dr Christoph Scheurer for the technical support and assistance. H. Li acknowledges Dr Martin Deimel for technical support. The authors gratefully acknowledge the Gauss Centre for Supercomputing e.V. (GCS) (https://www.gauss-centre.eu) for funding this project by providing computing time through the John von Neumann Institute for Computing (NIC) on the GCS Supercomputer JUWELS at Juelich Supercomputing Centre (JSC). Open Access funding provided by the Max Planck Society.References
- R. C. Armstrong, C. Wolfram, K. P. De Jong, R. Gross, N. S. Lewis, B. Boardman, A. J. Ragauskas, K. Ehrhardt-Martinez, G. Crabtree and M. Ramana, Nat. Energy, 2016, 1, 1–8 CrossRef.
- I. Staffell, D. Scamman, A. V. Abad, P. Balcombe, P. E. Dodds, P. Ekins, N. Shah and K. R. Ward, Energy Environ. Sci., 2019, 12, 463–491 RSC.
- N.-T. Suen, S.-F. Hung, Q. Quan, N. Zhang, Y.-J. Xu and H. M. Chen, Chem. Soc. Rev., 2017, 46, 337–365 RSC.
- T. Reier, M. Oezaslan and P. Strasser, ACS Catal., 2012, 2, 1765–1772 CrossRef CAS.
- L. Li, B. Wang, G. Zhang, G. Yang, T. Yang, S. Yang and S. Yang, Adv. Energy Mater., 2020, 10, 2001600 CrossRef CAS.
- H. Over, ACS Catal., 2021, 11, 8848–8871 CrossRef CAS.
- X. Wang, W. Ma, C. Ding, Z. Xu, H. Wang, X. Zong and C. Li, ACS Catal., 2018, 8, 9926–9935 CrossRef CAS.
- D. Zhang, J. Z. Soo, H. H. Tan, C. Jagadish, K. Catchpole and S. K. Karuturi, Adv. Energy Sustainability Res., 2021, 2, 2000071 CrossRef CAS.
- P. F. Liu, S. Yang, B. Zhang and H. G. Yang, ACS Appl. Mater. Interfaces, 2016, 8, 34474–34481 CrossRef CAS PubMed.
- B. W. Xue, C. H. Zhang, Y. Z. Wang, W. W. Xie, N.-W. Li and L. Yu, Nanoscale Adv., 2020, 2, 5555–5566 RSC.
- Y. Sun, H. Liao, J. Wang, B. Chen, S. Sun, S. J. H. Ong, S. Xi, C. Diao, Y. Du and J.-O. Wang, Nat. Catal., 2020, 3, 554–563 CrossRef CAS.
- C. Sun, J. A. Alonso and J. Bian, Adv. Energy Mater., 2021, 11, 2000459 CrossRef CAS.
- B. Zhang, X. Zheng, O. Voznyy, R. Comin, M. Bajdich, M. García-Melchor, L. Han, J. Xu, M. Liu and L. Zheng, Science, 2016, 352, 333–337 CrossRef CAS PubMed.
- H. Wang, H.-W. Lee, Y. Deng, Z. Lu, P.-C. Hsu, Y. Liu, D. Lin and Y. Cui, Nat. Commun., 2015, 6, 1–8 Search PubMed.
- K. Liang, L. Guo, K. Marcus, S. Zhang, Z. Yang, D. E. Perea, L. Zhou, Y. Du and Y. Yang, ACS Catal., 2017, 7, 8406–8412 CrossRef CAS.
- J. T. Mefford, A. R. Akbashev, M. Kang, C. L. Bentley, W. E. Gent, H. D. Deng, D. H. Alsem, Y.-S. Yu, N. J. Salmon and D. A. Shapiro, Nature, 2021, 593, 67–73 CrossRef CAS PubMed.
- S. Zou, M. S. Burke, M. G. Kast, J. Fan, N. Danilovic and S. W. Boettcher, Chem. Mater., 2015, 27, 8011–8020 CrossRef CAS.
- M. Görlin, P. Chernev, J. Ferreira de Araujo, T. Reier, S. R. Dresp, B. Paul, R. Krähnert, H. Dau and P. Strasser, J. Am. Chem. Soc., 2016, 138, 5603–5614 CrossRef PubMed.
- D. K. Bediako, B. Lassalle-Kaiser, Y. Surendranath, J. Yano, V. K. Yachandra and D. G. Nocera, J. Am. Chem. Soc., 2012, 134, 6801–6809 CrossRef CAS PubMed.
- M. L. Lindstrom, R. Gakhar, K. Raja and D. Chidambaram, J. Electrochem. Soc., 2020, 167, 046507 CrossRef CAS.
- M. Umer, S. Umer, M. Zafari, M. Ha, R. Anand, A. Hajibabaei, A. Abbas, G. Lee and K. S. Kim, J. Mater. Chem. A, 2022, 10, 6679–6689 RSC.
- R. Palkovits and S. Palkovits, ACS Catal., 2019, 9, 8383–8387 CrossRef CAS.
- H. Li, Y. Jiao, K. Davey and S. Z. Qiao, Angew. Chem., 2022, e202216383 Search PubMed.
- T. A. Batchelor, J. K. Pedersen, S. H. Winther, I. E. Castelli, K. W. Jacobsen and J. Rossmeisl, Joule, 2019, 3, 834–845 CrossRef CAS.
- Z. Wang, Y. Gu, L. Zheng, J. Hou, H. Zheng, S. Sun and L. Wang, Adv. Mater., 2021, 2106776 Search PubMed.
- W. Zhou, L. Yang, X. Wang, W. Zhao, J. Yang, D. Zhai, L. Sun and W. Deng, JACS Au, 2021, 1, 1497–1505 CrossRef CAS PubMed.
- Z. Zhao, P. Schlexer Lamoureux, A. Kulkarni and M. Bajdich, ChemCatChem, 2019, 11, 3423–3431 CrossRef CAS.
- C. Ren, S. Lu, Y. Wu, Y. Ouyang, Y. Zhang, Q. Li, C. Ling and J. Wang, J. Am. Chem. Soc., 2022, 144, 12874–12883 CrossRef CAS PubMed.
- H. Li, Y. Liu, K. Chen, J. T. Margraf, Y. Li and K. Reuter, ACS Catal., 2021, 11, 7906–7914 CrossRef CAS.
- J. Lim, D. Park, S. S. Jeon, C. W. Roh, J. Choi, D. Yoon, M. Park, H. Jung and H. Lee, Adv. Funct. Mater., 2018, 28, 1704796 CrossRef.
- W. Xu, B. Zhang, Y. Deng, L. Yang and J. Zhang, Electrochim. Acta, 2020, 137660 Search PubMed.
- Y. Liu, X. Liang, L. Gu, Y. Zhang, G.-D. Li, X. Zou and J.-S. Chen, Nat. Commun., 2018, 9, 1–10 CrossRef PubMed.
- K. Yue, J. Liu, Y. Zhu, C. Xia, P. Wang, J. Zhang, Y. Kong, X. Wang, Y. Yan and B. Y. Xia, Energy Environ. Sci., 2021, 14, 6546–6553 RSC.
- L. Yu, L. Wu, B. McElhenny, S. Song, D. Luo, F. Zhang, Y. Yu, S. Chen and Z. Ren, Energy Environ. Sci., 2020, 13, 3439–3446 RSC.
- D. Zhang, J. Shi, Y. Qi, X. Wang, H. Wang, M. Li, S. Liu and C. Li, Adv. Sci., 2018, 5, 1801216 CrossRef PubMed.
- Z. Li, W. Niu, L. Zhou and Y. Yang, ACS Energy Lett., 2018, 3, 892–898 CrossRef CAS.
- O. Keski-Rahkonen and J. Ahopelto, J. Phys. C-Solid State Phys., 1980, 13, 471 CrossRef CAS.
- S. Hao, L. Chen, C. Yu, B. Yang, Z. Li, Y. Hou, L. Lei and X. Zhang, ACS Energy Lett., 2019, 4, 952–959 CrossRef CAS.
- D. Zhang, H. Li, A. Riaz, A. Sharma, W. Liang, Y. Wang, H. Chen, K. Vora, D. Yan and Z. Su, Energy Environ. Sci., 2022, 15, 185–195 RSC.
- J. Rossmeisl, Z.-W. Qu, H. Zhu, G.-J. Kroes and J. K. Nørskov, J. Electroanal. Chem., 2007, 607, 83–89 CrossRef CAS.
- K. Reuter, Catal. Lett., 2016, 146, 541–563 CrossRef CAS.
- D. A. Lutterman, Y. Surendranath and D. G. Nocera, J. Am. Chem. Soc., 2009, 131, 3838–3839 CrossRef CAS PubMed.
- A. E. Thorarinsdottir, S. S. Veroneau and D. G. Nocera, Nat. Commun., 2022, 13, 1243 CrossRef CAS PubMed.
- I. C. Man, H. Y. Su, F. Calle-Vallejo, H. A. Hansen, J. I. Martínez, N. G. Inoglu, J. Kitchin, T. F. Jaramillo, J. K. Nørskov and J. Rossmeisl, ChemCatChem, 2011, 3, 1159–1165 CrossRef CAS.
- D. Friebel, M. W. Louie, M. Bajdich, K. E. Sanwald, Y. Cai, A. M. Wise, M.-J. Cheng, D. Sokaras, T.-C. Weng and R. Alonso-Mori, J. Am. Chem. Soc., 2015, 137, 1305–1313 CrossRef CAS PubMed.
- F. Dionigi, Z. Zeng, I. Sinev, T. Merzdorf, S. Deshpande, M. B. Lopez, S. Kunze, I. Zegkinoglou, H. Sarodnik and D. Fan, Nat. Commun., 2020, 11, 1–10 CrossRef PubMed.
- J. Ferreira de Araújo, F. Dionigi, T. Merzdorf, H. S. Oh and P. Strasser, Angew. Chem., Int. Ed., 2021, 60, 14981–14988 CrossRef PubMed.
- E. M. Fernández, P. G. Moses, A. Toftelund, H. A. Hansen, J. I. Martínez, F. Abild-Pedersen, J. Kleis, B. Hinnemann, J. Rossmeisl and T. Bligaard, Angew. Chem., Int. Ed., 2008, 47, 4683–4686 CrossRef PubMed.
- M. Yu, E. Budiyanto and H. Tüysüz, Angew. Chem., Int. Ed., 2022, 61, e202103824 CrossRef CAS PubMed.
- K. N. D. Van Chinh Hoanga and V. G. Gomesa, 2019.
- A. A. Sh, R. Guseynova, U. Gurbanova, D. Babanly, V. Fateev, I. Pushkareva and D. Tagiyev, Kimya Problemleri, 2018, 283–306 Search PubMed.
- C. Liang, P. Zou, A. Nairan, Y. Zhang, J. Liu, K. Liu, S. Hu, F. Kang, H. J. Fan and C. Yang, Energy Environ. Sci., 2020, 13, 86–95 RSC.
- L. Yang, Z. Liu, S. Zhu, L. Feng and W. Xing, Mater. Today Phys., 2021, 16, 100292 CrossRef CAS.
- Y. Wang, D. Yan, S. El Hankari, Y. Zou and S. Wang, Adv. Sci., 2018, 5, 1800064 CrossRef PubMed.
- B. Wang and F. Zhang, Angew. Chem., 2022, 134, e202111026 CrossRef.
- A. Cao, V. J. Bukas, V. Shadravan, Z. Wang, H. Li, J. Kibsgaard, I. Chorkendorff and J. K. Nørskov, Nat. Commun., 2022, 13, 2382 CrossRef CAS PubMed.
- Z. Wang, W. A. Goddard III and H. Xiao, Nat. Commun., 2023, 14, 4228 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ee01981k |
| This journal is © The Royal Society of Chemistry 2023 |