
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Direct carbonate electrolysis into pure syngas†
Yurou Celine
Xiao
 a,
Christine M.
Gabardo
a,
Shijie
Liu
a,
Geonhui
Lee
b,
Yong
Zhao
a,
Colin P.
O’Brien
a,
Rui Kai
Miao
a,
Yi
Xu
a,
Jonathan P.
Edwards
a,
Mengyang
Fan
a,
Jianan Erick
Huang
b,
Jun
Li
a,
Panagiotis
Papangelakis
a,
Tartela
Alkayyali
a,
Armin
Sedighian Rasouli
b,
Jinqiang
Zhang
ab,
Edward H.
Sargent
b and
David
Sinton
*a
a,
Christine M.
Gabardo
a,
Shijie
Liu
a,
Geonhui
Lee
b,
Yong
Zhao
a,
Colin P.
O’Brien
a,
Rui Kai
Miao
a,
Yi
Xu
a,
Jonathan P.
Edwards
a,
Mengyang
Fan
a,
Jianan Erick
Huang
b,
Jun
Li
a,
Panagiotis
Papangelakis
a,
Tartela
Alkayyali
a,
Armin
Sedighian Rasouli
b,
Jinqiang
Zhang
ab,
Edward H.
Sargent
b and
David
Sinton
*a
aDepartment of Mechanical and Industrial Engineering, University of Toronto, 5 King's College Road, Toronto, ON M5S 3G8, Canada. E-mail: dave.sinton@utoronto.ca
bDepartment of Electrical and Computer Engineering, University of Toronto, 10 King's College Road, Toronto, ON M5S 3G4, Canada
First published on 14th November 2022
Abstract
Syngas, a mixture of carbon monoxide (CO) and hydrogen (H2), is a feedstock for a wide variety of chemical processes and is currently produced from fossil fuels. The need to reduce carbon dioxide (CO2) emissions motivates the production of syngas from atmospheric CO2, powered by renewable electricity. Current CO2 electrolyzers require costly separation processes to purify the CO2 reactant stream and to remove unreacted CO2 from the product stream. We demonstrate direct carbonate electrolysis (DCE) in a reactive capture system that avoids the initial CO2 purification process and produces pure syngas with sufficient CO content for direct industrial use (H2/CO ratios of 1–2). The DCE system incorporates a composite CO2 diffusion layer (CDL) that attains high CO selectivity by achieving high alkalinity and available CO2 concentration at the cathode. Applying this strategy, we produce pure syngas in the cathode outlet gas stream with a H2/CO ratio of 1.16 at 200 mA cm−2, corresponding to a CO faradaic efficiency (FE) of 46% and an energy intensity of 52 GJ tsyngas−1. By eliminating intensive upstream and downstream processes, DCE achieves syngas production with 13% less energy than CO2 electrolysis combined with water electrolysis, 39% less energy than past carbonate reduction work, and 75% fewer emissions than the conventional fossil fuel based route.
Broader contextThe electrochemical conversion of captured CO2 into CO could reduce CO2 emissions while producing the carbon content of a valuable feedstock, syngas, for upgrade into long-chain hydrocarbons. Most CO2 electrolyzers require pure gaseous CO2 streams, and, thus, incur substantial capital and operational costs for CO2 capture liquid regeneration and CO2 purification. Additionally, CO2 reactant can be lost to carbonates in the electrolyzer which crossover to the anode, regenerate, and mix with the O2 rich anode gas stream. Excess CO2 in the cathode gas product stream also demands separation. These sequential purification steps are costly and limit the viability of electroproduced chemicals and fuels. We demonstrate a reactive capture system that produces pure syngas with an industrial H2/CO ratio through direct electrolysis of a CO2 post-capture solution. We designed a CO2 diffusion layer that achieves high local CO2 reactant concentration and high alkalinity favourable for CO2 conversion. This report illustrates the potential for the renewable electroproduction of syngas in a net-zero emissions future. |
Introduction
Syngas is a commodity feedstock used in the production of hydrocarbons and oxygenates via methanol routes and Fischer–Tropsch synthesis.1,2 Syngas is currently produced from fossil-fuels via coal gasification and/or methane reforming.3,4 These pathways are energy intensive and have high CO2 emission intensities (1.5 tCO2e tsyngas−1).5Electrochemical syngas production methods use renewable electricity to produce syngas with a lower carbon footprint. These methods combine CO from CO2 electrolysis and H2 from water electrolysis.6–8 However, current CO2 electrolyzers require high-purity gaseous CO2 feeds,9–12 and sourcing this CO2 from air – via direct air capture – or from industrial sources, is costly.13–16 Within current electrolyzers, utilization of reactant CO2 is low, and CO2 is lost to carbonates and crossover to the anode.17–19 As a result, both the anodic and the cathodic outlet streams require CO2 separation.20,21
Reactive capture is an electrolysis approach that shortens this process (Fig. 1A). This pathway avoids the thermally-driven capture liquid regeneration from the post-capture solution (carbonate electrolyte in the case of hydroxide-based direct air capture) and the subsequent CO2 dehydration, compression, and transportation steps.22–26 In the electrolyzer, protons are generated by the anodic oxygen evolution reaction (OER) and transported to the cathode through a cation exchange membrane (CEM) (Fig. 1B). The protons then react with the carbonate ions to regenerate CO2in situ. Syngas is produced through co-synthesis of CO from the regenerated CO2 and H2 from the aqueous solution. Unreacted CO2 is recaptured by hydroxide ions (OH−), a by-product of CO and H2 evolution, to form carbonate.
 | ||
| Fig. 1 Direct carbonate electrolysis to produce syngas enabled by a CDL. The chemical balance of carbon capture from air, anodic OER, CO2 regeneration, CO evolution reaction, H2 evolution reaction, and in situ CO2 recapture by OH− are presented in eqn (1)–(6), respectively. (A) Schematic of DCE integrated with carbon capture in a reactive capture system. (B) Schematic of the conversion of carbonate into CO2 facilitated by the CDL and protons, and subsequent conversion of CO2 into CO via electroreduction on the Ag catalyst. Unreacted CO2 is recaptured by OH− generated as a by-product of CO and H2 evolution. The carbonate electrolyte is recirculated. | ||
Previous studies of reactive capture using bicarbonate electrolyte have demonstrated high CO selectivity (CO faradaic efficiency (FE) > 50%). However, the limited CO2 recapture capacity of bicarbonate electrolyte results in a product gas stream diluted with CO2.26–28 Direct electrolysis of carbonate (rather than bicarbonate) electrolyte allows for the collection of high-purity gaseous products, evidenced by the lack of CO2 (<400 ppm) detected in the gas stream.29 Carbonate electrocatalytic conversion into syngas has previously been achieved with a H2/CO ratio of 3 (CO FE of 25%) and energy intensity of 86 GJ tsyngas−1. However, this mixture does not have sufficient CO content to meet industrial syngas standards.30–34
Here, we present an adlayer strategy that modulates the cathode pH and maximizes CO2 conversion to produce syngas with a H2/CO ratio in the industrially relevant range (1–2, and corresponding to a CO FE between 33% to 50%). We develop a composite CO2 diffusion layer (CDL) that enables cathode alkalinity to favour CO2 electrolysis and increases CO selectivity by limiting the diffusion of protons to the cathode. We achieve a H2/CO ratio of 1.16 (CO FE of 46%) at a current density of 200 mA cm−2. An energy intensity of 52 GJ tsyngas−1 was achieved, resulting in a 39% energy saving compared to the previous carbonate electrolysis report.
Results & discussion
Increasing CO2 conversion
Previous (bi)carbonate electrolyzers have employed a bipolar membrane (BPM) to dissociate water and provide protons and OH− to the cathode and anode, respectively.26,29,35 To reduce the membrane overvoltage, thereby reducing the required energy input, we used a cation exchange membrane (CEM) to transport protons directly from the anolyte or anodic OER, resulting in a voltage reduction of ca. 0.5 V (Fig. S1, ESI†). A zero-gap configuration was first investigated by assembling the electrolyzer with an Ag electrocatalyst directly in contact with the CEM. We found that the maximum FE towards CO was 17% (H2/CO ratio of 4.88) at 100 mA cm−2 (Fig. 2A). The only other carbon-based product detected was methane with <0.2% FE. Hydrogen evolution reaction (HER) accounted for the remaining FE.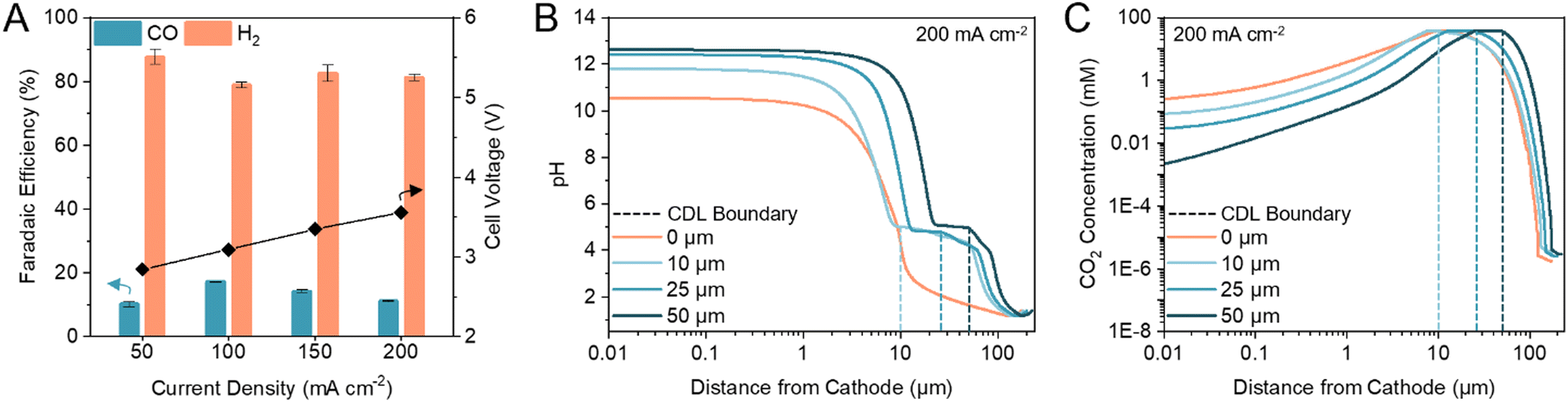 | ||
| Fig. 2 CDL thickness modulates cathode pH and CO2 concentration. (A) FE towards CO and H2 in a zero-gap configuration at current densities between 50 to 200 mA cm−2. Corresponding full cell voltages are noted on the secondary y-axis. Error bars represent the standard deviation of at least three samples measured under identical conditions. (B) One-dimensional multi-physics modelling of pH at distances from the cathode and current density of 200 mA cm−2 for CDL with thickness of 0 (zero-gap), 10, 25, and 50 μm. (C) One-dimensional multi-physics modelling of CO2 concentration at distances from the cathode and current density of 200 mA cm−2 for CDL with thickness of 0 (zero-gap), 10, 25, and 50 μm. Accompanying models of HCO3− and CO32− concentrations are provided in the ESI† (Fig. S4). | ||
We hypothesized that the low CO FE was caused by a CEM-induced acidic environment in which the kinetically more favourable HER outcompetes CO2 electrolysis,36–38 and, thus, modulating the cathode pH would improve CO2 conversion.39–41 We confirmed that CO2 was the electroreduction reactant, as opposed to the (bi)carbonate ions, by replacing the acidic anolyte with an alkaline electrolyte to suppress in situ CO2 regeneration. The only product detected was H2 (Fig. S2, ESI†). To increase CO2 conversion, we hypothesized that a CO2 diffusion adlayer between the CEM and the catalyst would limit proton diffusion to the cathode and separate the acidic CO2 regeneration region from the alkaline CO2 electrolysis region.
We developed multi-physics models of the carbonate electrolyzer with varying CDL thicknesses of 0 (zero-gap configuration), 10, 25, and 50 μm (Supplementary note 2, ESI†). We found that increasing the CDL thickness increased the pH at the cathode which favoured CO2 reduction over HER (Fig. 2B).42,43 However, increasing the CDL thickness also reduced the CO2 concentration at the cathode due to the recapturing of in situ CO2 within the extended alkaline region (Fig. 2C). The CDL must achieve high local cathode alkalinity and CO2 concentration to produce syngas with sufficient CO content for direct industrial application.
CDL design strategy
We first inserted commercially available hydrophilic microporous filters of different thicknesses between the catalyst and the CEM to separate the acidic CO2 regeneration region from the alkaline electrolysis region. We found that the filters increased the selectivity towards CO (Fig. S6, ESI†). However, the performance of the inserted filters was inconsistent due to trapped CO2 bubbles and material incompatibilities with the high and low pH extremes in this system (11 < pH < 2). We were motivated to design a robust and tuneable composite CDL. The engineered CDL needed to facilitate (bi)carbonate diffusion, hinder proton transport to the catalyst, sustain both high and low pH conditions, and have porous networks that allow mass transfer of in situ generated CO2. We selected TiO2 particles as the main substrate in view of their chemical stability and hydrophilic nature.44,45 For a substrate binder, we chose a hydrophilic ionomer that is anion-permeable to transport (bi)carbonate ions to the CEM.46,47 The wettability of the substrate and ionomer is important because hydrophobic elements will hinder ion transport, limiting the availability of carbonate ions in the CO2 regeneration region.48 When a hydrophobic substrate or ionomer was incorporated into the CDL, the CO FE was lower than the fully hydrophilic system (Fig. S7A and B, ESI†). The TiO2 and ionomer mixture was air-brushed evenly onto an Ag-catalyst layer (Fig. 3A and B) with corresponding energy dispersive X-ray (EDX) spectroscopy images confirming a distinct and uniform CDL (Fig. 3C).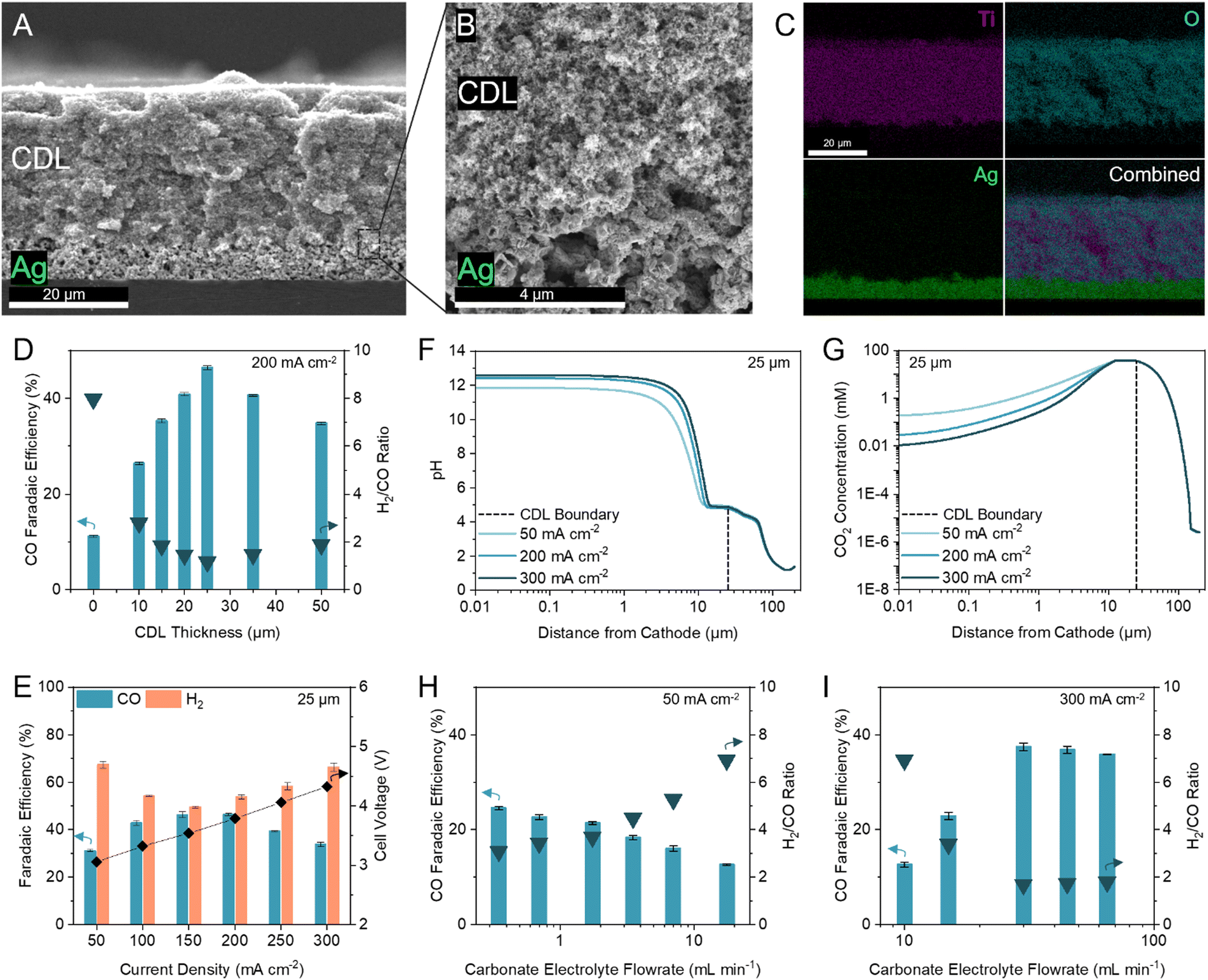 | ||
| Fig. 3 Optimization of the CDL for industrial H2/CO ratio. (A–C) Cross-sectional scanning electron microscopy (SEM) image of the CDL evenly air-brushed onto the Ag catalyst atop a silicon wafer (A), with seamless interfacial contact between the CDL and Ag catalyst (B), and corresponding energy dispersive X-ray (EDX) spectroscopy elemental mapping of Ti, O, and Ag (C). (D) FE towards CO and H2/CO ratio at 200 mA cm−2 with thicknesses of the CDL between 0 and 50 μm. (E) FE towards CO and H2 at current densities between 50 to 300 mA cm−2 with 25 μm CDL. Corresponding full cell voltages are noted on the secondary y-axis. (F) One-dimensional multi-physics modelling of pH at distances from the cathode and current densities of 50, 200, and 300 mA cm−2 for 25 μm CDL. (G) One-dimensional multi-physics modelling of CO2 concentration at distances from the cathode and current densities of 50, 200, and 300 mA cm−2 for 25 μm CDL. Accompanying models of HCO3− and CO32− concentrations are provided in the ESI† (Fig. S5). (H) FE towards CO and H2/CO ratio at 50 mA cm−2 with carbonate electrolyte flowrates between 0.35 to 17.5 mL min−1 showing an increase in CO FE with decreasing flowrate. (I) FE towards CO and H2/CO ratio at 300 mA cm−2 with carbonate electrolyte flowrates between 10 to 65 mL min−1 showing an increase in CO FE with increasing flowrate. Typical carbonate electrolyte flowrates are provided in the ESI† (Table S5). Error bars represent the standard deviation of at least three samples measured under identical conditions. | ||
To optimize the CDL for high CO FE, we screened TiO2 particle sizes (5, 25, 200, and 1500 nm) and TiO2/ionomer weight ratios between 5–25 (Fig. S7C and D, ESI†). We found that a combination of 25 nm TiO2 and a TiO2/ionomer ratio of 15 balanced the diffusion of (bi)carbonate ions and protons and enabled the local generation of CO2 to result in peak CO FE. The size of TiO2 nanoparticles and the ionomer volume fraction contribute to the permeability of the CDL. A high permeability failed to sufficiently hinder proton transport and resulted in hydrogen generation. A low permeability resulted in insufficient in situ regeneration of reactant CO2.49
We varied the CDL thickness between 10 to 50 μm and achieved a maximum CO FE of 46% (H2/CO ratio of 1.16) at 200 mA cm−2 with a 25 μm CDL (Fig. 3D). Achieving a high CO FE requires both a sufficiently alkaline local pH and adequate CO2 availability. Thinner CDLs have a smaller gap between the catalyst and the CEM which shortens the proton diffusion distance and results in a lower cathode pH. Despite having the highest CO2 concentrations in our simulations, the selectivity of the system with thinner CDLs was not optimal and approached the performance of the zero-gap configuration due to insufficient cathode alkalinity (Fig. 2B). As the CDL thickness is increased, the pH at the cathode increased but the local CO2 concentration decreased (Fig. 2C). With the cathode pH plateauing at thicknesses greater than 25 μm, this CDL thickness provided sufficient CO2 availability while maintaining an alkaline cathode environment to suppress HER.
We compared the difference in iR-compensated voltage of a zero-gap configuration to an otherwise identical system with a 25 μm CDL, with both experiments using an identical hydrogen-evolving catalyst (Supplementary note 3, ESI†). At 200 mA cm−2, a voltage increase of 117 mV was observed for the CDL system which corresponded to an increase in pH of 2, consistent with the multi-physics model which predicted a pH increase of 1.7 (Fig. 2B and Fig. S8, ESI†). These findings suggest that the CDL increases the pH of the cathode environment to favour CO2 conversion and thereby yielded a higher CO FE.
We screened the selectivity and full cell voltage of the optimized 25 μm CDL at current densities between 50 to 300 mA cm−2 and found that the CDL resulted in minimal voltage penalties while significantly increasing CO FE compared to the zero-gap configuration (+0.23 V and +35.3% CO FE at 200 mA cm−2) (Fig. 3E). Above 200 mA cm−2, the CO selectivity decreased, and the CO partial current plateaued, which indicated a CO2 mass transfer limit at the cathode (Fig. S9, ESI†). We hypothesized that the decrease in CO FE at the lower and higher current densities were due to an imbalance of cathode alkalinity and CO2 concentration at these extremes. The multi-physics model showed that compared to operating at 200 mA cm−2, the pH is lower at 50 mA cm−2, while the CO2 concentration is lower at 300 mA cm−2 (Fig. 3F and G). To investigate further, we decreased the carbonate flow rate while operating at 50 mA cm−2 and found that the CO FE increased (Fig. 3H). A slower flowrate increases the local pH due to the accumulation of OH−. At 300 mA cm−2, the CO FE increased as the carbonate electrolyte flowrate increased up to 30 mL min−1; however, further increases in flowrate resulted in similar, or slightly decreased, CO FE (Fig. 3I). This result suggests that at high current densities, the local environment is excessively alkaline, and CO2 availability is low. Increasing the carbonate electrolyte flowrate lowered the local pH and thereby increased the availability of CO2 for reaction.
To assess the long-term stability of the engineered CDL, we operated the DCE system with continuous CO2 capture and recycling of regenerated alkaline capture fluid (Fig. S10A, ESI†). At a constant current density of 100 mA cm−2, the full cell voltage, CO FE, H2/CO ratio, and capture solution pH were stable for over 23 hours of operation (Fig. 4). The pH of the anode electrolyte remained constant, (Fig. S10C, ESI†) and negligible CO2 was detected in the cathode and anode gas streams (<400 ppm), yielding a pure syngas (99.91 vol%, dry basis). Scanning electron microscopy (SEM) before and after prolonged operation showed minimal change in the CDL structure (Fig. S11, ESI†).
 | ||
| Fig. 4 Long-term operation of DCE over 23 hours with CO FE, H2/CO ratio, full cell voltage, and capture solution pH noted. Experiment conducted at a constant current density of 100 mA cm−2 with a 1 cm2 active area. Schematic and picture of the experimental set-up are provided in the ESI† (Fig. S10A and B). | ||
Comparison to alternative syngas production methods
We compared the energy intensity of syngas production via three CO2 electrolysis pathways: thermocatalytic reverse water gas shift (rWGS) (Fig. 5A); low-temperature CO2 electrolysis combined with water electrolysis (CE-WE) (Fig. 5B); and DCE (Fig. 5C) (Supplementary note 4, ESI†). In all cases, the feedstock CO2 is captured from the atmosphere using an alkaline capture liquid.13 The rWGS pathway produces syngas with H2/CO ratio of 1 and requires 57 GJ tsyngas−1 (Fig. 5D). The CE-WE and DCE pathways produce syngas with H2/CO of 1.16, requiring 60 and 52 GJ tsyngas−1, respectively. Syngas production via DCE circumvents intensive upstream and downstream processes and thereby results in a 13% energy saving compared to the CE-WE pathway and 8% energy savings compared to the rWGS method. The capture and dehydration steps in the DCE required only 0.85% of the energy demand for the DCE pathway, because the most energy intensive step of the capture process (regeneration) is avoided, and the output syngas stream is pure. | ||
| Fig. 5 Comparison of three syngas production methods. (A–C) Schematic showing process pathways, major chemical inputs and outputs, and energy source of rWGS (A), CE-WE (B), and DCE (C). (D) Energy intensity comparison to produce one tonne syngas using rWGS, CE-WE, and DCE. Syngas dehydration energies for CE-WE and DCE are too small to be seen in this figure. Detailed breakdown available in the ESI† (Table S3). (E) CO2e emissions to produce one tonne syngas using rWGS, CE-WE, and DCE. The CO2e associated with the energy input in each process is considered. Detailed breakdown available in the ESI† (Table S4). | ||
Of the three syngas production methods, DCE is the only pathway that is fully electrically driven, whereas CE-WE requires thermal energy input during CO2 capture and rWGS requires thermal energy in two major processes. Comparing the operational CO2e emissions of the three syngas production pathways, DCE was the only method that offered a low CO2 intensity (0.36 tCO2e tsyngas−1), while both CE-WE and rWGS exhibited high net CO2 emissions (1.39 and 2.48 tCO2e tsyngas−1, respectively) even when using renewable electricity (Fig. 5E). Compared to the fossil-based method (1.5 tCO2e tsyngas−1), DCE offers a 75% reduction in CO2e emissions.
Conclusions
We developed a strategy for the efficient electroproduction of syngas with sufficient CO-content and purity for direct industrial application. Through a one-dimensional multi-physics model, we found that the selectivity towards CO can be improved by separating the acidic CO2 regeneration region from the alkaline CO2 electrolysis region. We engineered a composite CDL positioned between the cathode and CEM of a DCE system to modulate the local pH and improve local CO2 conversion. The CDL was comprised of TiO2 nanoparticles bound by hydrophilic ionomer and was conformally coated onto the Ag catalyst. By determining the optimal CDL thickness that balanced the cathode alkalinity with CO2 concentration, a H2/CO ratio of 1.16 (CO FE 46%) was achieved with an energy intensity of 52 GJ tsyngas−1 and a CO2 intensity of 0.36 tCO2e tsyngas−1. Syngas production via DCE can provide a 13% energy saving compared to conventional CO2 electrolysis methods, and a 75% CO2e emissions reduction compared to fossil-fuel methods. These savings suggest that DCE is a promising pathway toward energy efficient syngas – a foundational feedstock for renewable chemicals and fuels in a net-zero emissions future.Experimental
Reagents
Potassium hydroxide (KOH) (>85%) and sulfuric acid (H2SO4) were purchased from Bioshop. All reagents were of analytical grade and used without further purification. All solutions were prepared using Milli-Q grade water (18.2 MΩ).Electrode preparation
The cathode was fabricated by air-brushing an Ag nanoparticle ink onto commercially available hydrophilic carbon paper (AvCarb MGL190, Fuel Cell Store) on a hot plate at 75 °C to achieve a loading of approximately 3 mg cm−2. The loading was measured by weighing the carbon paper before and after air-brushing. The catalyst ink was prepared with 150 mg of Ag nanoparticle (99.99%, 20 nm, metal basis, US Research Nanomaterials), 150 mg of Nafion dispersion (5 wt%, Fuel Cell Store), and 6 mL of methanol for a 25 cm2 substrate and sonicated for 1 hour prior to air-brushing. A commercially available titanium-based anode was used (Magneto Special Anodes, Evoqua Water Technologies).CDL preparation
Four sizes of TiO2 nanoparticle were used: 5 nm (anatase, 99.5%, US Research Nanomaterials), 25 nm (Aeroxide TiO2 P25, Evonik), 100 nm (anatase, 99.9%, US Research Nanomaterials), and 1500 nm (anatase, 99.9%, US Research Nanomaterials). Two ionomers were used: Nafion (Hydrophobic) and Aemion (Hydrophilic). Nafion ionomer can create strongly hydrophobic nanoporous networks due to its polytetrafluoroethylene (PTFE) (–CF2–CF2–) back-bone.50,51 Aemion ionomer contains poly-(benzimidazole) units which are more hydrophilic than the PTFE in Nafion.52 The Aemion dispersion was prepared by adding 388 mg of ionomer powder (AP1-CNN5-00-X, Ionomr) to 40 mL of ethanol and 10 mL of acetone and sonicated until fully dissolved.The CDL was fabricated by air-brushing a TiO2 nanoparticle ink onto the fabricated cathode to achieve the desired thickness. For the optimized CDL, the ink was prepared with 50 mg of 25 nm TiO2, 333 mg of the prepared Aemion dispersion, and 4 mL of ethanol for a 6.25 cm2 cathode and was sonicated for 1 hour prior to air-brushing. The CDL coated cathode was cut to a 1 cm2 size prior to electrolyzer assembly. CDLs were characterized using SEM at the Centre for Nanostructure Imaging at the University of Toronto using an FEI Quanta FEG 250 environmental SEM.
Two hydrophilic microporous membrane filters were used: 125 μm PVDF (Filter 1, 0.45 μm pore size) was purchased from Sigma Aldrich and 100 μm nylon (Filter 2, 5 μm pore size) was purchased from Sterlitech. Both filters were used as received.
Operation of the electrochemical cell
The carbonate electrolysis experiments were performed in a 1 cm2 electrolyzer with serpentine flow channels ingrained in both the stainless-steel cathode and the titanium anode current collectors. The electrolyzer was assembled by placing a CEM (Nafion 117) over the cathode, then placing the anode on the membrane. In all experiments, unless otherwise specified, the cathode feedstock was a carbonate electrolyte prepared by purging CO2 at 80 sccm into 85 mL of 2 M KOH for 40 minutes, similar to a previous report.29 The anode was fed with 0.05 M H2SO4. After starting the experiment, the carbonate electrolyte was continuously purged with Ar gas flowing at 20 mL min−1. The first gas sample is typically collected 20 minutes after the start of the experiment to ensure complete purging of excess CO2 from the electrolyte preparation process and even mixing of gaseous products. The electrochemical measurements were performed with a potentiostat (Autolab PGSTAT204) and the full cell voltages reported are not iR compensated unless otherwise specified.Product analysis
The cathode gas outlet stream was analyzed in 1 mL sample volumes by a gas chromatograph (PerkinElmer Clarus 590) coupled with a thermos conductivity detector (TCD) and flame ionization detector (FID). The gas chromatograph used Ar gas as the carrier (99.999%, Linde) and was equipped with a Molecular Sieve 5A Capillary Column and a packed Carboxen-1000 Column. Liquid product detection was performed using proton nuclear magnetic resonance spectroscopy (1H NMR) on an Agilent DD2 600 spectrometer in D2O using water suppression mode, with dimethyl sulfoxide (DMSO) as the internal standard. Calculation of FE and energy efficiency are included in Supplementary note 1 in the ESI.† pH measurements were conducted using Apera Instruments AI311 Premium Series PH60.Author contributions
D. S. and E. H. S. supervised the project. Y. C. X. and C. M. G. conceived the study. Y. C. X. designed and performed all the electrochemical experiments and generated the figures. G. L., C. P. O., R. K. M., and M. F. assisted with electrochemical experiments. C. M. G., G. L., C. P. O., and J. Z. assisted with CDL design. T. A. assisted with pH investigation experiment design. Y. C. X. and Y. X. performed gaseous product analysis. J. L., P. P., and J. E. H. performed liquid product analysis. J. P. E. and R. K. M. assisted with energy analysis. Y. Z. performed SEM imaging. S. L. performed system modelling. A. S. R. assisted with system modelling. Y. C. X. and D. S. wrote the manuscript. All authors discussed the results and assisted during manuscript preparation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Dr Paul J. Corbett, Dr Ben Rowley, and Dr Kai Han for helpful discussions. The authors acknowledge support from the Natural Sciences and Engineering Research Council (NSERC) of Canada and Natural Resources Canada – Clean Growth Program. Support from Canada Research Chairs Program is gratefully acknowledged, as is support from an NSERC E. W. R. Steacie Fellowship to D. S. Y. C. X. thanks Hatch for their support through graduate scholarships.References
- M. E. Dry, Catal. Today, 2002, 15 Search PubMed.
- D. J. Wilhelm, D. R. Simbeck, A. D. Karp and R. L. Dickenson, Fuel Process. Technol., 2001, 71, 139–148 CrossRef CAS.
- T. A. Adams and P. I. Barton, Fuel Process. Technol., 2011, 92, 639–655 CrossRef CAS.
- P. Gangadharan, A. Zanwar, K. Zheng, J. Gossage and H. H. Lou, Comput. Chem. Eng., 2012, 39, 105–117 CrossRef CAS.
- A. Schreiber, A. Peschel, B. Hentschel and P. Zapp, Front. Energy Res., 2020, 8, 533850 CrossRef.
- S. R. Foit, I. C. Vinke, L. G. J. de Haart and R.-A. Eichel, Angew. Chem., Int. Ed., 2017, 56, 5402–5411 CrossRef CAS PubMed.
- Y. Zheng, J. Wang, B. Yu, W. Zhang, J. Chen, J. Qiao and J. Zhang, Chem. Soc. Rev., 2017, 46, 1427–1463 RSC.
- V. Dieterich, A. Buttler, A. Hanel, H. Spliethoff and S. Fendt, Energy Environ. Sci., 2020, 13, 3207–3252 RSC.
- D. Higgins, C. Hahn, C. Xiang, T. F. Jaramillo and A. Z. Weber, ACS Energy Lett., 2019, 4, 317–324 CrossRef CAS.
- H. Xie, S. Chen, F. Ma, J. Liang, Z. Miao, T. Wang, H.-L. Wang, Y. Huang and Q. Li, ACS Appl. Mater. Interfaces, 2018, 10, 36996–37004 CrossRef CAS PubMed.
- A. Hauch, R. Küngas, P. Blennow, A. B. Hansen, J. B. Hansen, B. V. Mathiesen and M. B. Mogensen, Science, 2020, 370, eaba6118 CrossRef CAS PubMed.
- R. Küngas, J. Electrochem. Soc., 2020, 167, 044508 CrossRef.
- D. W. Keith, G. Holmes, D. St Angelo and K. Heidel, Joule, 2018, 2, 1573–1594 CrossRef CAS.
- K. Z. House, A. C. Baclig, M. Ranjan, E. A. van Nierop, J. Wilcox and H. J. Herzog, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 20428–20433 CrossRef CAS PubMed.
- M. Fasihi, O. Efimova and C. Breyer, J. Cleaner Prod., 2019, 224, 957–980 CrossRef CAS.
- P. Bains, P. Psarras and J. Wilcox, Prog. Energy Combust. Sci., 2017, 63, 146–172 CrossRef.
- C. M. Gabardo, C. P. O’Brien, J. P. Edwards, C. McCallum, Y. Xu, C.-T. Dinh, J. Li, E. H. Sargent and D. Sinton, Joule, 2019, 3, 2777–2791 CrossRef CAS.
- J. A. Rabinowitz and M. W. Kanan, Nat. Commun., 2020, 11, 5231 CrossRef CAS PubMed.
- E. Jeng and F. Jiao, React. Chem. Eng., 2020, 5, 1768–1775 RSC.
- T. Alerte, J. P. Edwards, C. M. Gabardo, C. P. O’Brien, A. Gaona, J. Wicks, A. Obradović, A. Sarkar, S. A. Jaffer, H. L. MacLean, D. Sinton and E. H. Sargent, ACS Energy Lett., 2021, 6, 4405–4412 CrossRef CAS.
- J. B. Greenblatt, D. J. Miller, J. W. Ager, F. A. Houle and I. D. Sharp, Joule, 2018, 2, 381–420 CrossRef CAS.
- G. Lee, Y. C. Li, J.-Y. Kim, T. Peng, D.-H. Nam, A. Sedighian Rasouli, F. Li, M. Luo, A. H. Ip, Y.-C. Joo and E. H. Sargent, Nat. Energy, 2021, 6, 46–53 CrossRef CAS.
- R. Sen, A. Goeppert, S. Kar and G. K. S. Prakash, J. Am. Chem. Soc., 2020, 142, 4544–4549 CrossRef CAS PubMed.
- J. Kothandaraman, J. Saavedra Lopez, Y. Jiang, E. D. Walter, S. D. Burton, R. A. Dagle and D. J. Heldebrant, ChemSusChem, 2021, 14, 4812–4819 CrossRef CAS PubMed.
- A. J. Welch, E. Dunn, J. S. DuChene and H. A. Atwater, ACS Energy Lett., 2020, 5, 940–945 CrossRef CAS.
- Z. Zhang, E. W. Lees, F. Habibzadeh, D. A. Salvatore, S. Ren, G. L. Simpson, D. G. Wheeler, A. Liu and C. P. Berlinguette, Energy Environ. Sci., 2022, 15, 705–713 RSC.
- Z. Zhang, E. W. Lees, S. Ren, B. A. W. Mowbray, A. Huang and C. P. Berlinguette, ACS Cent. Sci., 2022, 8, 749–755 CrossRef CAS.
- H. Li, J. Gao, Q. Du, J. Shan, Y. Zhang, S. Wu and Z. Wang, Energy, 2021, 216, 119250 CrossRef CAS.
- Y. C. Li, G. Lee, T. Yuan, Y. Wang, D.-H. Nam, Z. Wang, F. P. García de Arquer, Y. Lum, C.-T. Dinh, O. Voznyy and E. H. Sargent, ACS Energy Lett., 2019, 4, 1427–1431 CrossRef CAS.
- I. Wender, Fuel Process. Technol., 1996, 48, 189–297 CrossRef CAS.
- X. Song and Z. Guo, Energy Convers. Manage., 2006, 47, 560–569 CrossRef CAS.
- H. R. Shahhosseini, D. Iranshahi, S. Saeidi, E. Pourazadi and J. J. Klemeš, J. Cleaner Prod., 2018, 180, 655–665 CrossRef CAS.
- X. D. Peng, A. W. Wang, B. A. Toseland and P. J. A. Tijm, Ind. Eng. Chem. Res., 1999, 38, 4381–4388 CrossRef CAS.
- Y. Cao, Z. Gao, J. Jin, H. Zhou, M. Cohron, H. Zhao, H. Liu and W. Pan, Energy Fuels, 2008, 22, 1720–1730 CrossRef CAS.
- T. Li, E. W. Lees, M. Goldman, D. A. Salvatore, D. M. Weekes and C. P. Berlinguette, Joule, 2019, 3, 1487–1497 CrossRef CAS.
- K. D. Kreuer, J. Membr. Sci., 2001, 185, 29–39 CrossRef CAS.
- K. A. Mauritz and R. B. Moore, Chem. Rev., 2004, 104, 4535–4586 CrossRef CAS PubMed.
- J. E. Huang, F. Li, A. Ozden, A. Sedighian Rasouli, F. P. García de Arquer, S. Liu, S. Zhang, M. Luo, X. Wang, Y. Lum, Y. Xu, K. Bertens, R. K. Miao, C.-T. Dinh, D. Sinton and E. H. Sargent, Science, 2021, 372, 1074–1078 CrossRef CAS PubMed.
- K. Yang, M. Li, S. Subramanian, M. A. Blommaert, W. A. Smith and T. Burdyny, ACS Energy Lett., 2021, 6, 4291–4298 CrossRef CAS PubMed.
- K. Xie, R. K. Miao, A. Ozden, S. Liu, Z. Chen, C.-T. Dinh, J. E. Huang, Q. Xu, C. M. Gabardo, G. Lee, J. P. Edwards, C. P. O’Brien, S. W. Boettcher, D. Sinton and E. H. Sargent, Nat. Commun., 2022, 13, 3609 CrossRef CAS PubMed.
- C. M. Gabardo, A. Seifitokaldani, J. P. Edwards, C.-T. Dinh, T. Burdyny, M. G. Kibria, C. P. O’Brien, E. H. Sargent and D. Sinton, Energy Environ. Sci., 2018, 11, 2531–2539 RSC.
- C.-T. Dinh, T. Burdyny, M. G. Kibria, A. Seifitokaldani, C. M. Gabardo, F. P. García de Arquer, A. Kiani, J. P. Edwards, P. De Luna, O. S. Bushuyev, C. Zou, R. Quintero-Bermudez, Y. Pang, D. Sinton and E. H. Sargent, Science, 2018, 360, 783–787 CrossRef CAS PubMed.
- J. P. Edwards, Y. Xu, C. M. Gabardo, C.-T. Dinh, J. Li, Z. Qi, A. Ozden, E. H. Sargent and D. Sinton, Appl. Energy, 2020, 261, 114305 CrossRef CAS.
- C. Luo, X. Ren, Z. Dai, Y. Zhang, X. Qi and C. Pan, ACS Appl. Mater. Interfaces, 2017, 9, 23265–23286 CrossRef CAS PubMed.
- Y. Xu, J. P. Edwards, J. Zhong, C. P. O’Brien, C. M. Gabardo, C. McCallum, J. Li, C.-T. Dinh, E. H. Sargent and D. Sinton, Energy Environ. Sci., 2020, 13, 554–561 RSC.
- C. P. O’Brien, R. K. Miao, S. Liu, Y. Xu, G. Lee, A. Robb, J. E. Huang, K. Xie, K. Bertens, C. M. Gabardo, J. P. Edwards, C.-T. Dinh, E. H. Sargent and D. Sinton, ACS Energy Lett., 2021, 6, 2952–2959 CrossRef.
- Y. Xu, R. K. Miao, J. P. Edwards, S. Liu, C. P. O’Brien, C. M. Gabardo, M. Fan, J. E. Huang, A. Robb, E. H. Sargent and D. Sinton, Joule, 2022, 6, 1333–1343 CrossRef CAS.
- E. W. Lees, M. Goldman, A. G. Fink, D. J. Dvorak, D. A. Salvatore, Z. Zhang, N. W. X. Loo and C. P. Berlinguette, ACS Energy Lett., 2020, 5, 2165–2173 CrossRef CAS.
- Y. Kim, E. W. Lees and C. P. Berlinguette, ACS Energy Lett., 2022, 7, 2382–2387 CrossRef CAS.
- H.-L. Lin, T. L. Yu, C.-H. Huang and T.-L. Lin, J. Polym. Sci., Part B: Polym. Phys., 2005, 43, 3044–3057 CrossRef CAS.
- H.-G. Haubold, Th Vad, H. Jungbluth and P. Hiller, Electrochim. Acta, 2001, 46, 1559–1563 CrossRef CAS.
- O. D. Thomas, K. J. W. Y. Soo, T. J. Peckham, M. P. Kulkarni and S. Holdcroft, J. Am. Chem. Soc., 2012, 134, 10753–10756 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ey00046f |
| This journal is © The Royal Society of Chemistry 2023 |