
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIn situ solvothermal reduction engineering enables delicate control over surface-rich oxygen vacancies on Bi2WO6 for highly efficient photocatalytic CO2 reduction†
Huanhuan
Liu‡
a,
Yanxu
Chen‡
a,
Wentao
Wang‡
 b,
Xiaoyue
He
a,
ZiXu
He
a,
Lei
Li
c,
Suyuan
Zeng
d,
Ruiguo
Cao
a and
Genqiang
Zhang
*a
b,
Xiaoyue
He
a,
ZiXu
He
a,
Lei
Li
c,
Suyuan
Zeng
d,
Ruiguo
Cao
a and
Genqiang
Zhang
*a
aHefei National Research Center for Physical Sciences at the Microscale, CAS Key Laboratory of Materials for Energy Conversion, Department of Materials Science and Engineering, University of Science and Technology of China, Hefei, Anhui 230026, China. E-mail: gqzhangmse@ustc.edu.cn
bGuizhou Provincial Key Laboratory of Computational Nano-Material Science, Guizhou Education University, Guiyang, Guizhou 550018, China
cSchool of Chemistry and Chemical Engineering, Hefei University of Technology, Hefei, Anhui 230009, China
dDepartment of Chemistry and Chemical Engineering, Liaocheng University, Liaocheng, Shandong 252059, China
First published on 13th March 2023
Abstract
The high binding energy of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds in CO2 severely inhibits the photocatalytic CO2 reduction (PCR) activity. Constructing oxygen vacancies (Vo) is considered as a fascinating strategy to promote the capture and activation of CO2 molecules. The main challenge, however, lies in the delicate control of the Vo distribution, since the bulk Vo can act as a recombination centre for photogenerated electron–hole pairs. Here, an in situ solvothermal reduction strategy is presented by programming temperature to accurately control the Vo distribution on the catalytic surface. Taking Bi2WO6 as a model, surface-rich Vo on Bi2WO6 (Sur-Vo-BWO) were generated. The governable Vo distribution strategy remarkably increases the PCR dynamics with a high CO evolution rate for Sur-Vo-BWO (18.73 μmol g−1 h−1), which is 8.32 times more than that of the Vo-free control sample. This work paves a new pathway to implement delicate control of the Vo distribution on the catalysts for precisely tuned catalytic activity.
O bonds in CO2 severely inhibits the photocatalytic CO2 reduction (PCR) activity. Constructing oxygen vacancies (Vo) is considered as a fascinating strategy to promote the capture and activation of CO2 molecules. The main challenge, however, lies in the delicate control of the Vo distribution, since the bulk Vo can act as a recombination centre for photogenerated electron–hole pairs. Here, an in situ solvothermal reduction strategy is presented by programming temperature to accurately control the Vo distribution on the catalytic surface. Taking Bi2WO6 as a model, surface-rich Vo on Bi2WO6 (Sur-Vo-BWO) were generated. The governable Vo distribution strategy remarkably increases the PCR dynamics with a high CO evolution rate for Sur-Vo-BWO (18.73 μmol g−1 h−1), which is 8.32 times more than that of the Vo-free control sample. This work paves a new pathway to implement delicate control of the Vo distribution on the catalysts for precisely tuned catalytic activity.
Broader contextThe presence of oxygen vacancies on the surface of the photocatalysts is believed to play a significant role in the absorption and activation of CO2 molecules, which in turn contribute to the conversion of CO2 molecules into activated intermediates. However, controllable regulation of the oxygen vacancy distribution is a great challenge. Herein, we present an in situ solvothermal reduction strategy to generate surface-rich oxygen vacancy (Vo) on Bi2WO6 (Sur-Vo-BWO) in a controlled manner by the programmable temperature. The steerable Vo distribution strategy can significantly increase the CO2 reduction performance with high CO evolution rates, which can immensely mitigate the greenhouse effect. This strategy can be applied to assemble many other materials where the distribution of Vo is finely controlled. |
Increasing concentrations of CO2 in the atmosphere have been considered as one of the fatal factors in global warming and environmental degradation.1–3 The conversion of CO2 to chemical feedstocks holds great expectation to decrease the concentration of CO2 around the globe.4–6 Just as in plant photosynthesis, CO2 and H2O can be converted into chemical feedstocks over photocatalysts with a power input from solar energy.7–9 Presently, a great variety of photocatalytic materials (such as TiO2,10 In2O3,11 C3N412 and BiOX13) have been investigated for photocatalytic CO2 reduction (PCR); however, the main thermodynamic bottleneck of CO2 conversion has not been thoroughly conquered because of the high binding energy of the C
O bond (∼750 kJ mol−1).8
Recently, some studies instituted that an oxygen vacancy (Vo) could enhance the adsorption and activation of CO2 by regulating the charge density distribution of CO2 on the surface of the catalysts14 and can also extend the light absorption region by introducing some defect levels.15–17 Generally, catalysts with Vo are obtained by annealing the prepared material at high temperature with a gaseous reductant (such as H2, CH4 and CO), ion plasma etching, light etching or charge compensation by heteroatom doping.8,18 Nevertheless, these reductive gases are usually dangerous and harmful. In addition, several studies have manifested that these reductive gases can access the bulk, leading to abundant Vo in the bulk.19 Besides, the Vo caused by charge compensation also lead to the Vo in the bulk, which is the recombination centre of electron–hole (e−–h+) pairs.20,21 High energy input, expensive equipment, and harsh conditions are generally inevitable during ion plasma etching, and are undesirable for cost-effective mass production. The content of Vo is usually limited by light etching. For example, the Vo concentration of the BiOBr atomic layer does not increase after UV irradiation for more than eight hours.13 Although solvothermal reduction has been used to prepare Vo, the traditional one-step strategy inevitably generates Vo in the bulk. Therefore, it is necessary and difficult to develop a mild yet effective strategy that can delicately control the Vo distribution on the surface and thus precisely tune the photocatalytic activity.
Bi2WO6 (BWO) is a promising candidate for PCR due to the nontoxic and inexpensive properties, high chemical and thermal stability, and suitable electronic band structures.22 In addition, as a typical Aurivillius-phase oxide, an internal static electric field exists in the BWO, which is beneficial for the transfer of e−–h+ pairs.23 However, several issues have impeded the further application of BWO in PCR, such as the low visible light utilization induced by the wide bandgap, and the low surface active site-derived insufficient CO2 adsorption capacity.24 Therefore, scholars have paid close attention to improving the separation capacity of photoinduced e−–h+ pairs through elemental impurities, morphological regulation, structural heterojunctions, and defect engineering.25–27 Among them, the construction of Vo is considered as a significant tactic to effectively prevent the recombination of photogenerated e−–h+ pairs.28,29 However, refined control of the Vo distribution is the main challenge. Herein, we propose an in situ solvothermal reduction strategy to delicately control the generation of BWO with rich Vo on the surface (Sur-Vo-BWO) in a refined control manner over a programmable temperature during the reaction time. The reduction strategy can precisely regulate the Vo distribution over Sur-Vo-BWO with the uppermost CO evolution rate (18.73 μmol g−1 h−1), which is 8.32 times more than that of the Vo-free control sample (BWO-C, 2.25 μmol g−1 h−1). Theoretical calculations disclose that the energy barrier for the rate-limiting step is drastically decreased in the attendance of Vo, which extremely facilitates CO evolution. This work presents a new pathway for the construction of surface-Vo-rich photocatalysts to enhance PCR activity.
The Sur-Vo-BWO was assembled by in situ solvothermal reduction engineering, as depicted in Fig. 1A. Firstly, the precursors were synthesized at low solvothermal temperature (120 °C), where EG acts as the reaction solvent. Subsequently, the Sur-Vo-BWO was obtained by in situ increasing the temperature to a higher value (180 °C), where EG is a mild reducing agent for despoiling surface lattice oxygen atoms from the catalysts surface at high temperatures.29,30 The morphology of the Sur-Vo-BWO and control samples was observed by scanning electron microscope (SEM), as shown in Fig. 1B and Fig. S1 (ESI†) and transmission electron microscope (TEM) images (Fig. 1C and Fig. S2, ESI†). As revealed in Fig. S3 (ESI†), the thickness of the as-prepared BWO photocatalysts (approximately 8 nm) does not change significantly, presumably because the morphology has already been formed at low temperatures. Increasing the temperature to higher values only forms oxygen vacancies and increases the crystallinity of the BWO. The interplanar spacings of 0.274 and 0.309 nm in the high-resolution TEM (HRTEM) image (Fig. 1D) are regarded as the (060) and (131) planes of Sur-Vo-BWO, respectively, which are the two lattice planes most exposed in Sur-Vo-BWO based on the XRD result. As shown in Fig. S4 (ESI†), the elemental mapping images for Bi, W and O are uniformly distributed, indicating that Sur-Vo-BWO is manufactured without impurities. The XRD patterns of the as-obtained samples at 2θ = 28.3, 32.7, 47.1, 55.8, 58.5 and 68.7° (Fig. 1E and Fig. S5, ESI†) were indexed to the (131), (060), (202), (133), (262) and (004) planes of the orthorhombic BWO materials (JCPDS 79-2381). The absence of impurities in the XRD patterns manifests that the BWO is a pure phase.
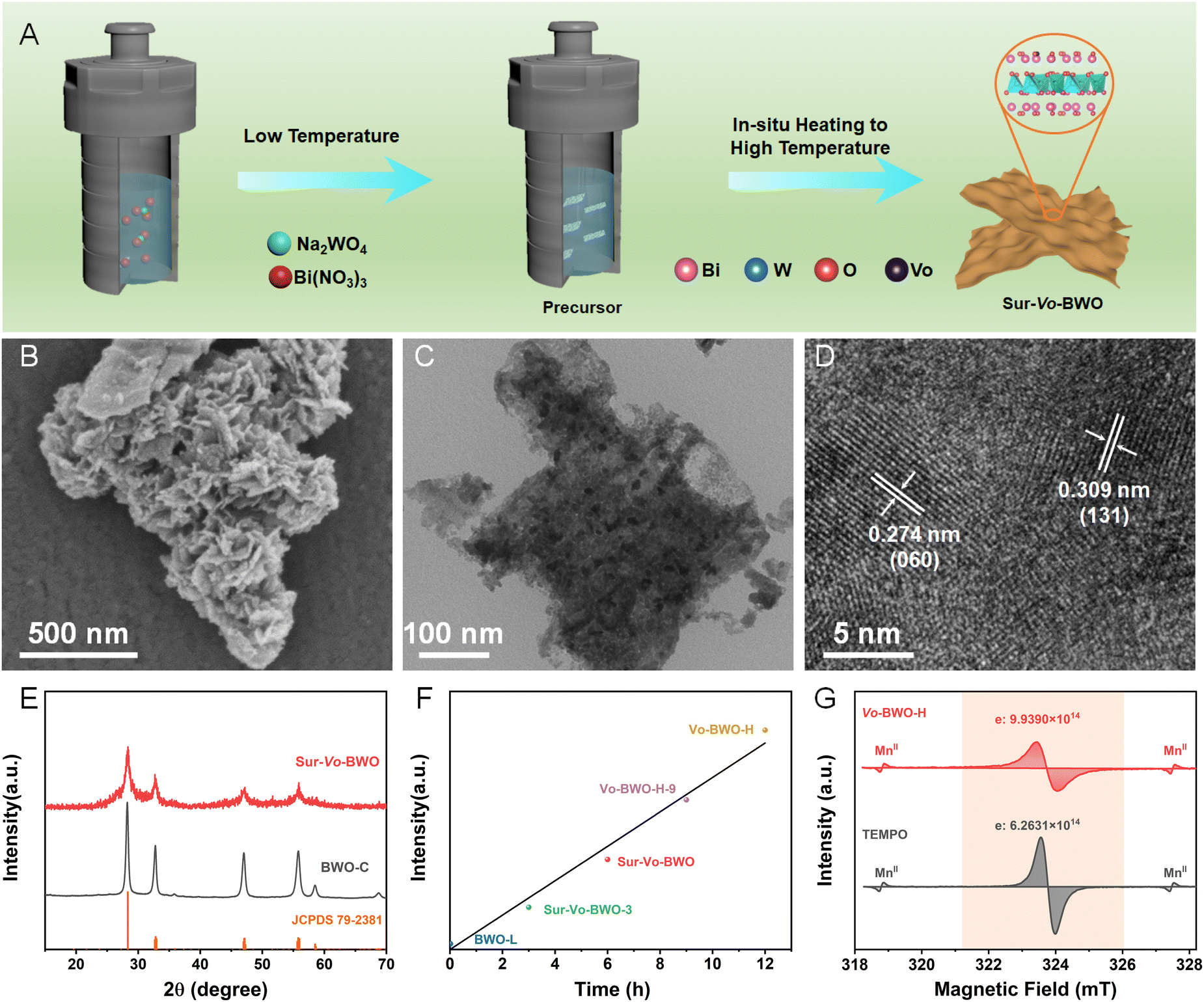 | ||
| Fig. 1 (A) Schematic illustration of the formation process of Sur-Vo-BWO photocatalysts by programming temperature to accurately control the Vo distribution on the catalytic surface; (B) SEM image, (C) TEM image and (D) HRTEM image of Sur-Vo-BWO, respectively. (E) XRD pattern; (F) the relation between EPR intensity and solvothermal time at high temperature (180 °C); (G) quantitative study of the Vo density for Vo-BWO-H (the weight of Vo-BWO-H in the test is 0.4991 g). | ||
The electron paramagnetic resonance (EPR) was utilized to verify the existence of Vo as it can obtain information about the defect states in materials.31 As exhibited in Fig. S6 (ESI†), the Sur-Vo-BWO displays an obvious EPR signal at g = 2.001, which is identified as the unpaired electrons at the oxygen defect sites.21 However, the BWO synthesized at the low solvothermal temperature (named BWO-L) and Vo free BWO-C do not present an apparent EPR signal, indicating that Vo is not formed at low temperatures and can be removed by calcining Sur-Vo-BWO in the air atmosphere. The EPR signal increases when the precursors stay for a longer time (12 h) at high temperature (denoted as Vo-BWO-H). Fascinatingly, the EPR intensity displays a linear relation with the solvothermal temperature at high values (Fig. 1F), indicating that the Vo is acquired due to the extended time at high temperatures. For the quantitative analysis of Vo, 2,2,6,6-tetramethylpiperidinyloxyl (TEMPO) and MnII were employed as the standard sample and reference samples, respectively, using the EPR methods.32 As demonstrated in Fig. 1G, the number of unpaired electrons (one unpaired electron denotes one Vo) in Vo-BWO-H is about 1.99 × 1015 e− g−1Catalyst. Therefore, the Vo concentrations of BWO-C, BWO-L, Sur-Vo-BWO-3, Sur-Vo-BWO and Vo-BWO-H-9 were estimated as 0, 5.13 × 1013, 3.83 × 1014, 8.16 × 1014 and 1.36 × 1015 e− g−1Catalyst, respectively. These above results indicate that the Vo-rich state is controllably obtained by increasing the reaction time at higher temperatures.
A schematic illustration of the Ar+ etching is shown in the inset of Fig. 2A. The Ar+ etching technique can scrape away the surface information of the catalyst, so that the inner information of the catalyst can be detected.33,34 Therefore, the depth information of Vo distribution was investigated by X-ray photoelectron spectroscopy (XPS) through time coursed Ar+ etching. The O 1s XPS spectra of Sur-Vo-BWO at Fig. 2A present two peaks located at 529.2 and 530.6 eV, which are considered as lattice oxygen and Vo, respectively.24 Notably, the binding energy of the lattice oxygen for Sur-Vo-BWO moves to the BWO-C site with increasing Ar+ etching time. In addition, the content of Vo in Sur-Vo-BWO determined by the O 1s XPS spectra (Table S1, ESI†) decreases from 22.66% to 16.26% after 60 s Ar+ etching, which is close to the Vo content of BWO-C (15.02%). The O 1s XPS spectra results indicate that Vo is mainly generated on the surface of Sur-Vo-BWO. The peaks at 164.5 and 159.2 eV for Sur-Vo-BWO in Fig. 2B are ascribed to the Bi 4f5/2 and Bi 4f7/2 of the Bi3+ chemical state, respectively.35 Moreover, two prominent peaks for Sur-Vo-BWO at 162.4 and 157.0 eV are identified as the Bi(3−x)+ chemical state due to the partial reduction of Bi atoms by localized electrons trapped in the Vo.36 The peaks of Bi(3−x)+ for Sur-Vo-BWO weaken after Ar+ etching, indicating that rich Vo exists over Sur-Vo-BWO due to the despoilment of the lattice oxygen linked with Bi species in the reductive solvent of EG at high temperature. The W 4f XPS spectra of Sur-Vo-BWO (Fig. 2C) consisted of two peaks at 35.3 and 37.4 eV, representing the W 4f7/2 and W 4f5/2 in the W6+ chemical state, respectively.23 The W 4f XPS sites of binding energy for BWO-C and Sur-Vo-BWO are immobile, manifesting that the W–O bond might not be cracked during the solvothermal process.37 According to the above-mentioned results, the BWO with rich Vo on the surface by cracking the Bi–O bond has been manufactured by an in situ solvothermal reduction strategy.
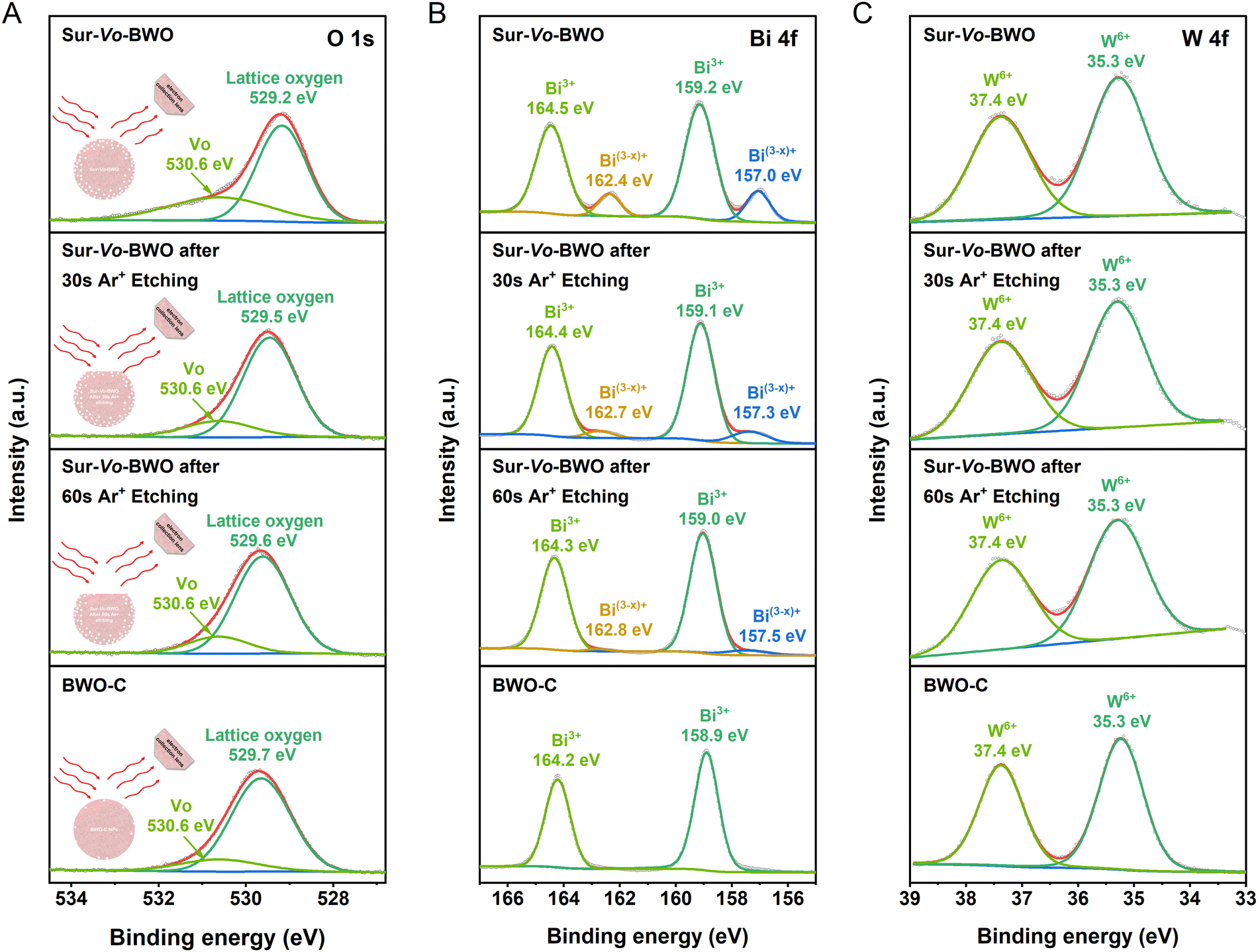 | ||
| Fig. 2 XPS spectra for Sur-Vo-BWO and BWO-C before and after Ar+ etching: (A) O 1s XPS spectra, (B) Bi 4f XPS spectra and (C) W 4f XPS spectra, respectively. A schematic illustration of Ar+ etching is shown in the inset of (A). | ||
The photocatalytic activity and selectivity of the Sur-Vo-BWO and control samples were evaluated under simulated sunlight without any co-catalysts or sacrificial agents. As given in Fig. 3A, B and Fig. S7, S8 in the ESI,† the evolution of CO and CH4 demonstrates a linear process with increasing illumination time. Notably, the Sur-Vo-BWO exposes the highest CO evolution rate (18.73 μmol g−1 h−1), selectivity (99.0%), and total electron transfer rate (RTET) (38.99 μmole g−1 h−1) as shown in Table S2 (ESI†), because of the appropriate concentration of surface Vo over Sur-Vo-BWO. The Vo-induced defect state may act as an initial charge carrier's acceptor to inhibit the recombination of e−–h+ pairs and promote the interfacial charge transfer from the excited Sur-Vo-BWO to preactivated CO2 on the Vo sites. The high selectivity also avoids complex and expensive purification processes, which is advantageous for subsequent industrial utilization.8 The highest CO evolution rate for Sur-Vo-BWO also outperforms commercial P25 and most of the reported BWO-based catalysts (Table S3, ESI†), demonstrating that Sur-Vo-BWO is a promising candidate for PCR. The CO evolution rate decreases to 6.18 μmol g−1 h−1 with RTET of 13.60 μmole g−1 h−1 for Vo-BWO-H, separately, which may be the redundant Vo in the bulk as an electron trapping centre to facilitate surface recombination.38 Besides, the BWO-C reveals the lowest CO evolution rate (2.25 μmol g−1 h−1) because of the poor concentration of Vo over BWO-C. The emergence of trace CH4 could be a stronger interaction between Vo and intermediates of the CO2 photoreduction process, which is verified by the CO temperature-programmed desorption in Fig. S9 (ESI†). The apparent quantum yield (AQY) was also measured to illustrate the photon conversion efficiency. The AQY (Fig. S10 and S11, ESI†) achieved 0.0224%, 0.00394% and 0.00328% for Sur-Vo-BWO at 420, 500 and 600 nm band-pass filter irradiation, respectively, comparable to the reported BWO photocatalyst.35 By comparison, the AQY for BWO-C was only 0.0118%, 0.000509% and 0.000330% at 420, 500 and 600 nm irradiation, respectively. The result indicates that Vo plays an important role in light absorption and CO evolution. No products were detected in the absence of CO2, light source or photocatalyst. As exposed in Fig. S12, ESI,† although no water was added to the reactor, a trace of CO was spotted, which may be the adsorption of water vapor from the air due to the abundant oxygen vacancies in Sur-Vo-BWO as an oxidant to consume the photogenerated holes. 13CO2 isotope labelling experiments were performed to further shed light on the origin of the photocatalytic products. As presented in Fig. 3C, a clear signal at m/z = 29 is attributed to 13CO formed by 13CO2 reduction, implying that the CO is generated by CO2 reduction. This presents that the products are generated by PCR and that H2O plays a crucial role in the reaction system. The PCR activity (Fig. 3D) remains 97.2% after 4 cycles with the unbroken morphology (Fig. S13, ESI†) and crystal structure (Fig. S14, ESI†), demonstrating the excellent durability of Sur-Vo-BWO. Interestingly, the higher CO evolution rate in the second times than in the first could be due to the strong interaction of the Sur-Vo-BWO with CO2 and C1 intermediates, which avoids the activated time from CO2 to C1 intermediate in the second times than in the first. Hence, a slight increase in the CO evolution rate is detected in Fig. 3D. The Sur-Vo-BWO and BWO-C after photocatalytic testing were measured by XPS. The XPS peaks at 164.4 and 159.1 eV for Sur-Vo-BWO in Fig. S15A (ESI†) are ascribed to the Bi 4f5/2 and Bi 4f7/2 of the Bi3+ chemical state, respectively.39 Noteworthily, the Bi(3−x)+ of Sur-Vo-BWO is indistinct after the cycling test, which is deemed to be the adsorption of CO2 and water on the Vo sites and bond with Bi species. The W 4f XPS spectra (Fig. S15B, ESI†) at 35.2 and 37.3 eV for Sur-Vo-BWO represent the W 4f7/2 and W 4f5/2 in the W6+ chemical state, correspondingly.23 The O 1s XPS peak (Fig. S15C, ESI†) located at 529.7 eV is deemed as the lattice oxygen of Sur-Vo-BWO. In addition, the surface hydroxyl group and CO of the carboxylate were detected at 530.9 and 531.8 eV, separately.40 The increased relative ratio of surface hydroxyl groups and carboxylate for Sur-Vo-BWO over BWO-C (Table S4, ESI†) manifests that the Vo enormously accelerates the adsorption and activation of CO2 and water over Sur-Vo-BWO. The C–OH and CO peaks also emerged at 286.1 and 288.3 eV in the C 1s XPS spectra (Fig. S15D, ESI†).41 The increased relative ratio of C–OH and CO peaks for Sur-Vo-BWO than BWO-C (Table S5, ESI†) is in line with the increased relative ratio of surface hydroxyl and carboxylate groups in the O 1s XPS spectra. The XPS results indicate that surface Vo can accelerate the adsorption and activation of water and CO2 molecules. As emerged in Fig. S16 (ESI†), the vague EPR response of Sur-Vo-BWO after photocatalytic testing declares that the Vo is occupied by adsorbed water and carboxylate species. Surprisingly, the Vo was recurrent when the sample was illuminated under an argon atmosphere, which clarifies that the Vo is the active site and it can be released during PCR.
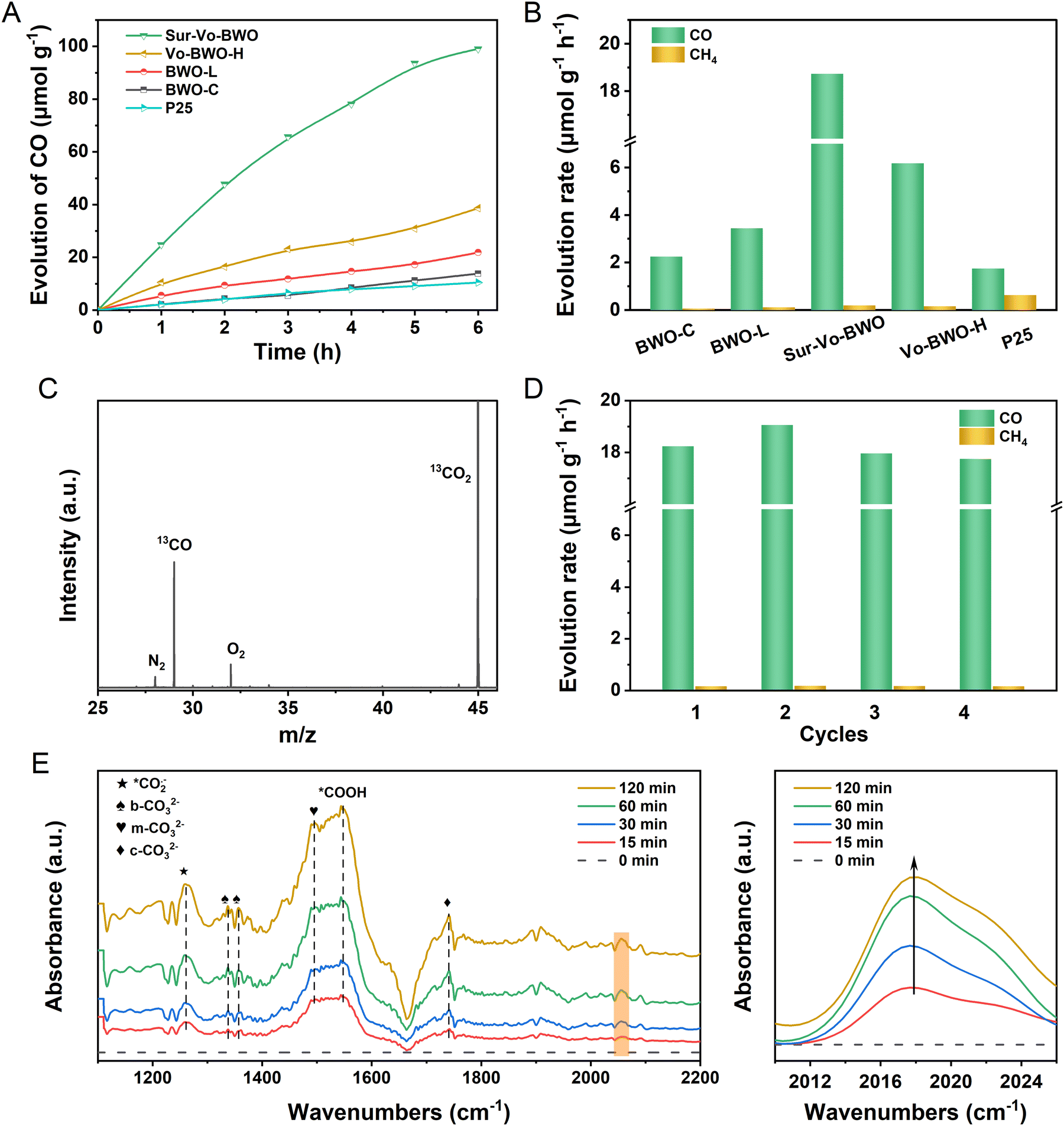 | ||
| Fig. 3 (A) Time courses of photocatalytic CO evolution curves and (B) CO and CH4 evolution rate of Sur-Vo-BWO and control samples; (C) mass spectrum of 13CO produced over Sur-Vo-BWO by a 13CO2 isotope labelling experiment; (D) CO and CH4 evolution rate of Sur-Vo-BWO under stability test; (E) the in situ FTIR study of Sur-Vo-BWO. | ||
To understand the PCR mechanism over Sur-Vo-BWO, in situ FTIR measurements were implemented, as shown in Fig. 3E. The FTIR peak at 1261 cm−1 is indexed to the carbonate species during the PCR.13,42 The peak at 1548 cm−1 is attributed to the typical intermediate of *COOH for CO2 reduction.43 In the meantime, the monodentate carbonate (m-CO32−) at 1496 cm−1, the bidentate carbonate (b-CO32−) at 1340 and 1356 cm−1 and the chelating bridged carbonate (c-CO32−) at 1740 cm−1 are detected.44,45 In addition, the *CO absorption band at 2056 cm−1 is also detected in the photocatalytic process, and it is enhanced with the increase of the irradiation time, indicating that abundant CO intermediates (*CO) are generated over Sur-Vo-BWO.8,16 According to the in situ FTIR analysis, the pathway of photoreduction from CO2 to CO over Sur-Vo-BWO may be recommended as the formate (*COOH) pathway.
Considering the influence of specific surface areas in different samples, N2 adsorption–desorption tests were carried out. As shown in Fig. S17 (ESI†), the Brunauer–Emmett–Teller (BET) surface areas of BWO-C, BWO-L, Sur-Vo-BWO and Vo-BWO-H are 11.934, 13.261, 25.421 and 30.523 m2 g−1, respectively. The trapping of N2 can be facilitated by localized excess electrons due to the coordinated unsaturated nature of metal atoms caused by Vo.8,46,47 Therefore, Vo-BWO-H reveals the highest specific surface areas due to the existence of exorbitant Vo. However, the specific area normalized CO evolution rate for Sur-Vo-BWO is 0.737 μmol m−2 h−1 (Fig. S18 and Table S6, ESI†), about 3.92, 2.83 and 3.65 times higher than that of BWO-C (0.188 μmol m−2 h−1), BWO-L (0.260 μmol m−2 h−1) and Vo-BWO-H (0.202 μmol m−2 h−1), respectively. This result indicates that the intrinsic photocatalytic activity of Sur-Vo-BWO is enhanced by the surface Vo, and that the specific surface area is not the most important factor to enhance the PCR activity. The presence of Vo also reinforces CO2 adsorption (Fig. 4A and Fig. S19, ESI†), which is thanks to the coordinatively unsaturated nature of the Bi atoms near the Vo site. CO2 is likely to aggregate at the site of localized excess electrons, which is conducive for the CO2 adsorption and activation.8 Time-resolved photoluminescence (TRPL) decay spectroscopy (Fig. 4B) was employed to investigate the charge transfer dynamics. The fitting parameters are summarized in Table S7 (ESI†). The average lifetime (τav) for Sur-Vo-BWO is 2.69 ns, which is about double the lifetime of BWO-C (1.57 ns). These results indicate that surface Vo plays a great role in improving the lifetime of photocarriers.48 Steady-state PL spectroscopy was employed to understand the carrier recombination process caused by Vo. As exposed in Fig. S20 (ESI†), BWO-C exhibits the highest PL intensity due to the strong radiative recombination behaviour. The PL is gradually quenched as Vo is generated over BWO, confirming that the quenching of the PL intensity is directly triggered by Vo. The Sur-Vo-BWO emerges the lowest PL intensity, manifesting an obvious suppression of the charge recombination probability for the photogenerated carriers.49 However, the redundant Vo is pernicious for the charge separation due to the excessive existence of electron trapping in the bulk of Vo-BWO-H, which acts as a recombination centre to aggrandize the charge recombination. The result can be further verified by the stronger PL emission peak, the larger arc radius of the Nyquist plots (Fig. S21, ESI†) and the depressed transient photocurrent response curves (Fig. S22, ESI†) of Vo-BWO-H compared to Sur-Vo-BWO.50 These results clearly indicate that Sur-Vo-BWO exhibits stronger electron transfer properties than that of the control samples.
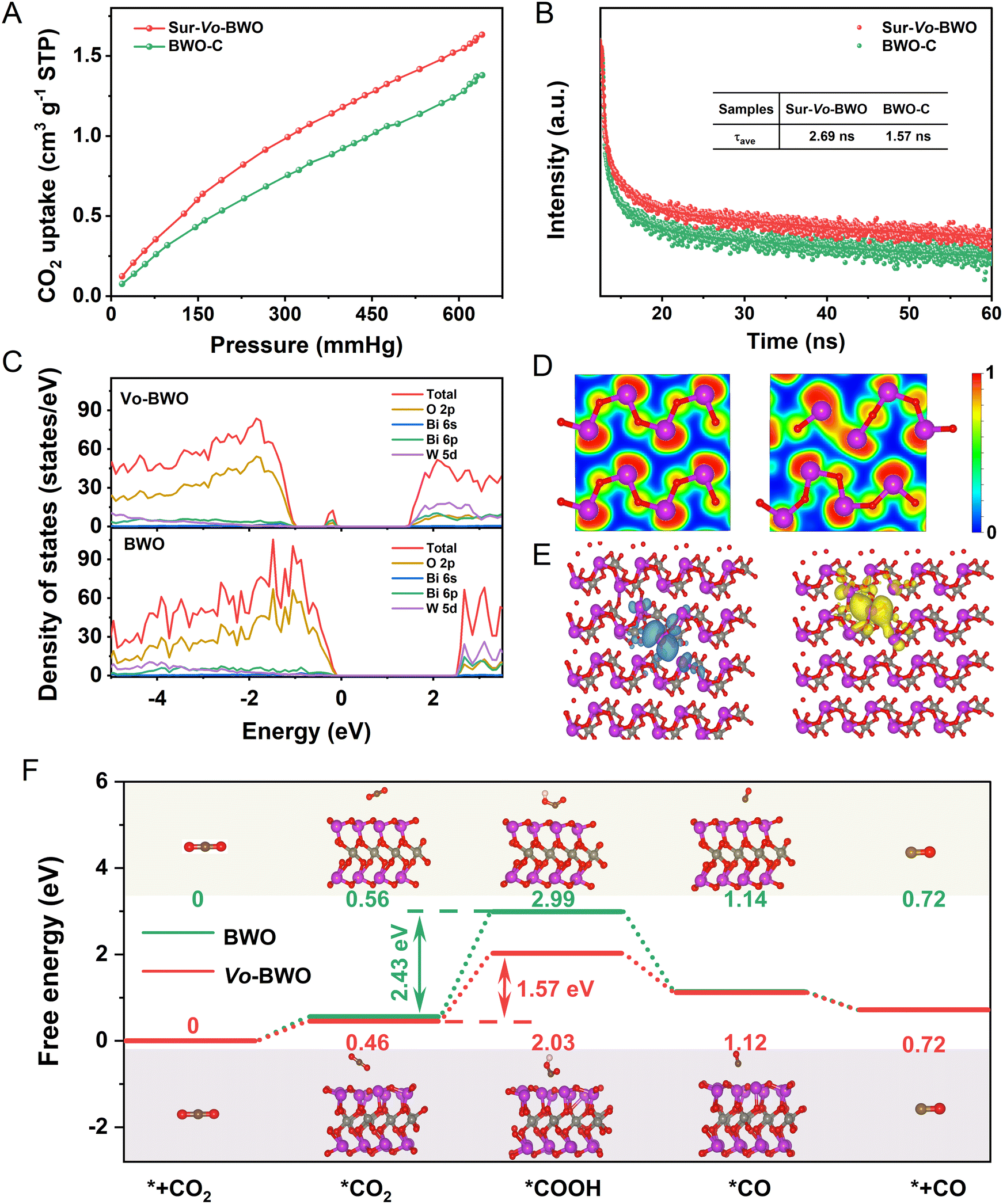 | ||
| Fig. 4 (A) CO2 adsorption isotherms and (B) TRPL spectra of Sur-Vo-BWO and BWO-C; (C) total and projected DOS of Vo-BWO and BWO; (D) the ELF of BWO (left) and Vo-BWO (right). The value of function closer to 1 means that the electrons are more localized; (E) distribution of photogenerated electrons (left) and photogenerated holes (right) for the Vo-BWO in the excited state; (F) free energy diagrams of CO2 photoreduction to CO for the Vo-BWO and BWO. | ||
DFT simulation was performed to understand the electronic structure and the reaction energy barrier. According to the density of states (Fig. 4C), the VBM for BWO is mainly formed by O 2p orbitals, while the CBM is comprised by W 5d orbitals. When the Vo is produced in the BWO (named Vo-BWO), the band gap reduces from 2.50 eV for BWO to 1.30 eV for Vo-BWO. Simultaneously, a mid-gap state near the VBM of Vo-BWO appears and consists of Bi 6p and O 2p orbitals. The adventitious energy levels can induce electronic transitions from the VBM to the intermediate state and then to the CBM, which is identical with extended light absorption in the longer wavelength region by the ultraviolet-visible diffuse reflection spectra result in Fig. S23 (ESI†). The intermediate state can serve as the reactive sites and trap the separated photocarriers for the subsequent surface reaction. Consequently, there is less chance for e−–h+ pairs to meet and recombine. The narrower band gap and extended absorption in the visible-light region improve the utilization of low energy light, the major component of sunlight on the earth.49,51 The electron localization function (ELF) maps reveal strong covalent interactions of Bi with neighbouring O atoms in BWO (Fig. 4D). However, the electron is localized surrounding Bi sites for Vo-BWO when the O atom is removed. This result demonstrates that the Vo in the ground state can trap electrons. Excited state calculations provide a deeper explanation of the role of Vo on charge separation in photocatalytic reactions. The distance of Bi–vacancy–Bi for Vo-BWO is 3.16 Å in the ground state (Fig. S24A, ESI†). When an electron is excited from the valence band to the conduction band, that is, the electron is transferred from a Bi atom (HOMO) to neighbouring Bi atom (LUMO), charge repulsion between the two Bi atoms will exist. Accordingly, the distance of Bi–vacancy–Bi extended to 4.42 Å in the excited state (Fig. S24B, ESI†). This result also leads to the spatial separation of the HOMO and LUMO orbitals, as revealed in Fig. 4E. Hence, the extended carrier lifetime and enhanced charge separation become a reality. On the contrary, the electrons and holes disperse on the whole region for BWO in the excited state (Fig. S25, ESI†). These results indicate that Vo as an active site can preferably promote adsorption and activation of CO2 molecules, and encourage diffusion of e−–h+ pairs. The photoreduction pathways from CO2 to CO (the main product) were calculated to reveal the adsorption and activation energy over BWO and Vo-BWO. Usually, the CO2 photoreduction mainly contains five steps: (i) the adsorption and (ii) activation of CO2 molecules; (iii) hydrogenation of activated CO2 molecules; (iv) decomposition of the C–O bond with removal of water and (v) desorption of reductive products from the active sites.16,52 The hydrogenation step of CO2 molecules is regarded as a potential-determining step for PCR. As displayed in Fig. 4F, the Gibbs free energies of Vo-BWO with *CO2−, *COOH and *CO are 0.46, 1.57 and 1.12 eV, while the Gibbs free energy is 0.56, 2.99 and 1.14 eV for BWO, respectively. What needs to be emphasized is that the energy barriers of the potential-determining step are reduced obviously from 2.43 eV for BWO to 1.57 eV for Vo-BWO by the hydrogenation of adsorbed *CO2− to *COOH, which immensely facilitates CO evolution.
Conclusions
In conclusion, the Sur-Vo-BWO has been synthesized by a delicate in situ solvothermal reduction strategy in a controlled manner over programmable temperature during the reaction time, which can avoid the superfluous Vo in the bulk. The Sur-Vo-BWO shows the highest CO evolution rate and selectivity. The surface Vo extends the photoresponse in the visible-spectrum region, increases the adsorption and activation capacity of CO2, suppresses the recombination of photogenerated charges and decreases the energy barrier of the potential-determining step, which are demonstrated by experimental and theoretical results. This work opens a novel avenue to manage the Vo distribution of the photocatalysts to enhance the CO2 photoreduction capability.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the financial support from the National Natural Science Foundation of China (Grant No. 52072359) and the Recruitment Program of Global Experts and the Fundamental Research Funds for the Central Universities (WK2060000016). The numerical calculations in this paper have been done in the Supercomputing Center of University of Science and Technology of China and TianHe-2 at LvLiang Cloud Computing Center of China.Notes and references
- D. U. Nielsen, X.-M. Hu, K. Daasbjerg and T. Skrydstrup, Nat. Catal., 2018, 1, 244–254 CrossRef CAS.
- S. Deutz and A. Bardow, Nat. Energy, 2021, 6, 203–213 CrossRef CAS.
- X. Zhi, A. Vasileff, Y. Zheng, Y. Jiao and S.-Z. Qiao, Energy Environ. Sci., 2021, 14, 3912–3930 RSC.
- S. Mehla, A. E. Kandjani, R. Babarao, A. F. Lee, S. Periasamy, K. Wilson, S. Ramakrishna and S. K. Bhargava, Energy Environ. Sci., 2021, 14, 320–352 RSC.
- Y. Zhao, G. I. N. Waterhouse, G. Chen, X. Xiong, L. Z. Wu, C. H. Tung and T. Zhang, Chem. Soc. Rev., 2019, 48, 1972–2010 RSC.
- W. Lai, Y. Qiao, J. Zhang, Z. Lin and H. Huang, Energy Environ. Sci., 2022, 15, 3603–3629 RSC.
- S. Gao, B. Gu, X. Jiao, Y. Sun, X. Zu, F. Yang, W. Zhu, C. Wang, Z. Feng, B. Ye and Y. Xie, J. Am. Chem. Soc., 2017, 139, 3438–3445 CrossRef CAS PubMed.
- H. Liu, F. Zhang, H. Wang, J. Xue, Y. Guo, Q. Qian and G. Zhang, Energy Environ. Sci., 2021, 14, 5339–5346 RSC.
- S. Karmakar, S. Barman, F. A. Rahimi and T. K. Maji, Energy Environ. Sci., 2021, 14, 2429–2440 RSC.
- B.-H. Lee, E. Gong, M. Kim, S. Park, H. R. Kim, J. Lee, E. Jung, C. W. Lee, J. Bok, Y. Jung, Y. S. Kim, K.-S. Lee, S.-P. Cho, J.-W. Jung, C.-H. Cho, S. Lebègue, K. T. Nam, H. Kim, S.-I. In and T. Hyeon, Energy Environ. Sci., 2022, 15, 601–609 RSC.
- Y. X. Pan, Y. You, S. Xin, Y. Li, G. Fu, Z. Cui, Y. L. Men, F. F. Cao, S. H. Yu and J. B. Goodenough, J. Am. Chem. Soc., 2017, 139, 4123–4129 CrossRef CAS PubMed.
- R. Kuriki, M. Yamamoto, K. Higuchi, Y. Yamamoto, M. Akatsuka, D. Lu, S. Yagi, T. Yoshida, O. Ishitani and K. Maeda, Angew. Chem., Int. Ed., 2017, 56, 4867–4871 CrossRef CAS PubMed.
- J. Wu, X. Li, W. Shi, P. Ling, Y. Sun, X. Jiao, S. Gao, L. Liang, J. Xu, W. Yan, C. Wang and Y. Xie, Angew. Chem., Int. Ed., 2018, 57, 8719–8723 CrossRef CAS PubMed.
- S. Sun, M. Watanabe, J. Wu, Q. An and T. Ishihara, J. Am. Chem. Soc., 2018, 140, 6474–6482 CrossRef CAS PubMed.
- S. Yin, X. Zhao, E. Jiang, Y. Yan, P. Zhou and P. Huo, Energy Environ. Sci., 2022, 15, 1556–1562 RSC.
- X. D. Li, Y. F. Sun, J. Q. Xu, Y. J. Shao, J. Wu, X. L. Xu, Y. Pan, H. X. Ju, J. F. Zhu and Y. Xie, Nat. Energy, 2019, 4, 690–699 CrossRef CAS.
- W. Gao, S. Li, H. He, X. Li, Z. Cheng, Y. Yang, J. Wang, Q. Shen, X. Wang, Y. Xiong, Y. Zhou and Z. Zou, Nat. Commun., 2021, 12, 4747 CrossRef CAS PubMed.
- W. Wang, F. Xiong, S. Zhu, J. Chen, J. Xie and Q. An, eScience, 2022, 2, 278–294 CrossRef.
- Y. Liu, Y. Li, S. Yang, Y. Lin, J. Zuo, H. Liang and F. Peng, ChemSusChem, 2018, 11, 2766–2775 CrossRef CAS PubMed.
- K. Wang, D. Liu, L. Liu, J. Liu, X. Hu, P. Li, M. Li, A. S. Vasenko, C. Xiao and S. Ding, eScience, 2022, 2, 518–528 CrossRef.
- N. Zhang, A. Jalil, D. Wu, S. Chen, Y. Liu, C. Gao, W. Ye, Z. Qi, H. Ju, C. Wang, X. Wu, L. Song, J. Zhu and Y. Xiong, J. Am. Chem. Soc., 2018, 140, 9434–9443 CrossRef CAS PubMed.
- L. Liang, F. Lei, S. Gao, Y. Sun, X. Jiao, J. Wu, S. Qamar and Y. Xie, Angew. Chem., Int. Ed., 2015, 54, 13971–13974 CrossRef CAS PubMed.
- Y. Liu, D. Shen, Q. Zhang, Y. Lin and F. Peng, Appl. Catal., B, 2021, 283, 119630 CrossRef CAS.
- J. G. Hou, S. Y. Cao, Y. Z. Wu, F. Liang, Y. F. Sun, Z. S. Lin and L. C. Sun, Nano Energy, 2017, 32, 359–366 CrossRef CAS.
- D. Tan, X. Cheng, J. Zhang, Y. Sha, X. Shen, Z. Su, B. Han and L. Zheng, Green Chem., 2021, 23, 2913–2917 RSC.
- Y. Zhou, Y. Zhang, M. Lin, J. Long, Z. Zhang, H. Lin, J. C. Wu and X. Wang, Nat. Commun., 2015, 6, 8340 CrossRef PubMed.
- N. Zhang, R. Ciriminna, M. Pagliaro and Y. J. Xu, Chem. Soc. Rev., 2014, 43, 5276–5287 RSC.
- Y. Bo, H. Wang, Y. Lin, T. Yang, R. Ye, Y. Li, C. Hu, P. Du, Y. Hu, Z. Liu, R. Long, C. Gao, B. Ye, L. Song, X. Wu and Y. Xiong, Angew. Chem., Int. Ed., 2021, 60, 16085–16092 CrossRef CAS PubMed.
- X. Yang, S. Wang, N. Yang, W. Zhou, P. Wang, K. Jiang, S. Li, H. Song, X. Ding, H. Chen and J. Ye, Appl. Catal., B, 2019, 259, 118088 CrossRef CAS.
- Z. Cai, Y. Bi, E. Hu, W. Liu, N. Dwarica, Y. Tian, X. Li, Y. Kuang, Y. Li, X.-Q. Yang, H. Wang and X. Sun, Adv. Energy Mater., 2018, 8, 1701694 CrossRef.
- K. Xie, N. Umezawa, N. Zhang, P. Reunchan, Y. Zhang and J. Ye, Energy Environ. Sci., 2011, 4, 4211–4219 RSC.
- C. Mao, H. Cheng, H. Tian, H. Li, W.-J. Xiao, H. Xu, J. Zhao and L. Zhang, Appl. Catal., B, 2018, 228, 87–96 CrossRef CAS.
- Y. Sakai, S. Ninomiya and K. Hiraoka, Surf. Interface Anal., 2012, 44, 938–941 CrossRef CAS.
- D.-P. Kim, K.-T. Kim, C.-I. Kim and A. M. Efremov, Thin Solid Films, 2004, 447–448, 343–348 CrossRef CAS.
- C. Lu, X. Li, Q. Wu, J. Li, L. Wen, Y. Dai, B. Huang, B. Li and Z. Lou, ACS Nano, 2021, 15, 3529–3539 CrossRef CAS PubMed.
- X. Y. Kong, Y. Y. Choo, S. P. Chai, A. K. Soh and A. R. Mohamed, Chem. Commun., 2016, 52, 14242–14245 RSC.
- X. Cao, Z. Chen, R. Lin, W.-C. Cheong, S. Liu, J. Zhang, Q. Peng, C. Chen, T. Han, X. Tong, Y. Wang, R. Shen, W. Zhu, D. Wang and Y. Li, Nat. Catal., 2018, 1, 704–710 CrossRef CAS.
- B. Wang, X. Wang, L. Lu, C. Zhou, Z. Xin, J. Wang, X.-K. Ke, G. Sheng, S. Yan and Z. Zou, ACS Catal., 2017, 8, 516–525 CrossRef.
- B. He, H. Liu, Z. Lin, L. Yan, J. Ning, Y. Zhong, C. Zheng, Z. Zhang and Y. Hu, Chem. Eng. J., 2019, 359, 924–932 CrossRef CAS.
- B. Yang, K. Niu, F. Haag, N. Cao, J. Zhang, H. Zhang, Q. Li, F. Allegretti, J. Björk, J. V. Barth and L. Chi, Angew. Chem., Int. Ed., 2022, 61, e202113590 CAS.
- D. Sun, B. Luo, H. Wang, Y. Tang, X. Ji and L. Wang, Nano Energy, 2019, 64, 103937 CrossRef CAS.
- P. Galhotra, J. G. Navea, S. C. Larsen and V. H. Grassian, Energy Environ. Sci., 2009, 2, 401–409 RSC.
- J. Di, C. Chen, S. Z. Yang, S. Chen, M. Duan, J. Xiong, C. Zhu, R. Long, W. Hao, Z. Chi, H. Chen, Y. X. Weng, J. Xia, L. Song, S. Li, H. Li and Z. Liu, Nat. Commun., 2019, 10, 2840 CrossRef PubMed.
- Y. He, H. Rao, K. Song, J. Li, Y. Yu, Y. Lou, C. Li, Y. Han, Z. Shi and S. Feng, Adv. Funct. Mater., 2019, 29, 1905153 CrossRef CAS.
- Q. Liu, H. Cheng, T. Chen, T. W. B. Lo, Z. Xiang and F. Wang, Energy Environ. Sci., 2022, 15, 225–233 RSC.
- J. Li, H. Li, G. Zhan and L. Zhang, Acc. Chem. Res., 2017, 50, 112–121 CrossRef CAS PubMed.
- P. Li, Z. Zhou, Q. Wang, M. Guo, S. Chen, J. Low, R. Long, W. Liu, P. Ding, Y. Wu and Y. Xiong, J. Am. Chem. Soc., 2020, 142, 12430–12439 CrossRef CAS PubMed.
- Z. Luo, Y. Fang, M. Zhou and X. Wang, Angew. Chem., Int. Ed., 2019, 58, 6033–6037 CrossRef CAS PubMed.
- H. Liu, L. Li, C. F. Guo, J. Q. Ning, Y. J. Zhong and Y. Hu, Chem. Eng. J., 2020, 385, 123929 CrossRef CAS.
- Z. Dai, F. Qin, H. P. Zhao, J. Ding, Y. L. Liu and R. Chen, ACS Catal., 2016, 6, 3180–3192 CrossRef CAS.
- H. Liu, K. Tian, J. Ning, Y. Zhong, Z. Zhang and Y. Hu, ACS Catal., 2019, 9, 1211–1219 CrossRef CAS.
- L. Wang, W. Chen, D. Zhang, Y. Du, R. Amal, S. Qiao, J. Wu and Z. Yin, Chem. Soc. Rev., 2019, 48, 5310–5349 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ey00019b |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |