
Integrating electrocatalytic seawater splitting and biomass upgrading via bifunctional nickel cobalt phosphide nanorods†
Yunyi
Yang
a,
Ren
Zou
a,
Jianyun
Gan
a,
Yujia
Wei
a,
Zhongxin
Chen
c,
Xuehui
Li
 a,
Shimelis
Admassie
d,
Yunpeng
Liu
*b and
Xinwen
Peng
*a
a,
Shimelis
Admassie
d,
Yunpeng
Liu
*b and
Xinwen
Peng
*a
aState Key Laboratory of Pulp and Paper Engineering, South China University of Technology, Guangzhou 510641, Guangdong, China. E-mail: fexwpeng@scut.edu.cn
bSchool of Materials Science and Hydrogen Energy, Foshan University, Foshan 528231, Guangdong, China. E-mail: l.yunpeng@fosu.edu.cn
cSchool of Science and Engineering, The Chinese University of Hong Kong, Shenzhen 518172, Guangdong, China
dSchool of Materials Science and Engineering, South China University of Technology, Guangzhou 510640, Guangdong, China
First published on 19th April 2023
Abstract
The high overpotential of the oxygen evolution reaction (OER) and the undesirable chlorine evolution reaction (ClER) severely limit the hydrogen production efficiency of seawater electrolysis. The organic electrooxidation reactions, involving C–H bond oxidation with low bond dissociation energy in the rate-limiting step, can possess lower oxidation potential than the OER as well as prevent the ClER. Herein, we demonstrate a strategy for replacing the sluggish OER by thermodynamically favorable biomass oxidation for scaling up the electrolysis of alkaline seawater. A xylose electrooxidation reaction (XOR) holds great promise for superseding the OER in seawater electrolysis, which not only simultaneously produces valuable formic acid and hydrogen, but also improves energy utilization. By utilizing NiCoP metal phosphide as the bifunctional electrode, the XOR shows an apparent overpotential reduction of 290 mV at 100 mA cm−2 as compared with the OER in the alkaline seawater electrolyte. Furthermore, the xylose-assisted seawater electrolysis exhibits a high formic acid selectivity of at least 94.6% and a high hydrogen production rate of 150 mL cm−2. This work provides a universal and an effective pathway for hydrogen production from seawater and biomass upgrading.
Introduction
Solar and wind energy are considered as alternative sources for reducing the dependence on fossil fuels.1 However, the high cost of electrical transmission and inherent intermittence hinder the development of sustainable energy.2 Seawater electrolysis for hydrogen (H2) production powered by solar and wind is a carbon-neutral and promising solution for growing energy demand.3 To date, numerous efforts have been devoted to high-efficiency electrocatalysts for the oxygen evolution reaction (OER) and the hydrogen evolution reaction (HER), including non-metals,4,5 noble metals6–8 and transition metals.9–11 However, some critical issues of the present electrolysis technology remain unresolved, such as its high cost,12 low efficiency13,14 and poor durability,15 which hinder the scalable development of seawater electrolysis. Theoretically, the energy threshold for the water-splitting reaction is 1.23 V, but the voltage required by loads is always larger than 1.8 V because of the sluggish kinetics of the OER.16 The substantial overpotential results in high electricity input,17 which is the major bottleneck for seawater splitting. Furthermore, the low value of oxygen and the additional energy consumption to avoid explosion of mixed gases sharply reduce the economic benefits of seawater electrolysis.18,19 In addition, the abundant chloride anions in seawater participate in the chlorine evolution reaction (ClER) involving only two-electron transfer steps, which may compete with other anodic reactions such as the OER.20 Another critical challenge of seawater electrolysis is the short-term stability of the electrocatalyst, because chloride corrosion and the formation of insoluble magnesium/calcium hydroxides will cover the electrode surface.21,22An alternative strategy to the OER is the biomass electrooxidation reaction,23,24 which can significantly reduce costs and prevent the ClER process during the seawater electrolysis. Biomass electrooxidation, which requires a much lower overpotential, can inhibit the ClER, protecting the electrocatalyst from chloride corrosion.25 As a renewable carbon neutral resource, biomass also offers the opportunity to obtain high-value-added products at the anode. Marketable organic acids can be obtained, in principle, from biomass electrooxidation by controlling the potential applied to the cell.26 Xylose, the major structural unit of xylan, is the second most abundant saccharide in plants. Currently, an efficient xylose oxidation reaction (XOR) for organic acids production is reported,27 exhibiting many advantages such as mild reaction conditions, high-value liquid products and low overpotential. Recently, Jiang et al. have reported a PtRh catalyst for decoupled hydrogen evolution by integrating ethylene glycol oxidation in alkaline seawater electrolyte,28 creating new opportunities for seawater electrolysis. Biomass electrooxidation can suppress the undesired byproducts in seawater electrolysis and provide valuable chemicals at a low potential. However, H2 evolution assisted by biomass electrooxidation from seawater remains a great challenge, and non-noble metals as electrocatalysts for hybrid electrolysis have been rarely reported.
Herein, we proposed a hybrid electrolyser, where green hydrogen and value-added formic acid (FA) were simultaneously produced in alkaline water or seawater. The self-supported transition metal phosphide of NiCoP on nickel foam (NF) was prepared as the bifunctional electrocatalyst. The NiCoP electrode exhibits an excellent XOR performance, which requires only 1.35 V versus a reversible hydrogen electrode (vs. RHE) to reach a current density of 100 mA cm−2 in alkaline seawater with 100 mM xylose. In addition, an optimal yield of FA is nearly 50% and the selectivity is not less than 95%. Furthermore, due to the acceptable HER activity, a two-electrode cell can deliver a current density of 100 mA cm−2 at 1.57 V in seawater, and the lowest selectivity of FA remains 94.6% at 1.9 V with no oxygen production. Selective biomass electrooxidation coupled with hydrogen evolution will provide a sustainable approach for seawater electrolysis and high-value utilization of biomass.
Experimental section
Materials
Chemicals including hydrochloric acid (HCl, 36.0%), cobalt nitrate hexahydrate (Co(NO3)2·6H2O, 98%), urea (CO(NH2)2, 99%), ammonium fluoride (NH4F, 97%), sodium hypophosphite monohydrate (NaH2PO2·H2O, 98%), xylose (C5H10O5, 98%) and potassium hydroxide (KOH, 95%) were purchased from Aladdin and used as received.Synthesis of the NiCo hydroxide precursor on Ni foam
NiCo hydroxide was prepared according to a reported method.29 A piece of 3 × 3.5 cm2 nickel foam (NF) was carefully cleaned with 3 M HCl, deionized (DI) water and ethyl alcohol in sequence for 20 min. The pre-treated NF was immersed in 30 mL of water solution containing 873 mg of Co(NO3)2·6H2O, 901 mg of urea and 296 mg of NH4F in a 50 mL Teflon-lined autoclave. The autoclave was then heated at 120 °C for 6 h to obtain NiCo hydroxide as the precursor, which was labeled as NiCoOH.Synthesis of NiCoP bimetallic phosphide
The obtained NiCoOH precursor was placed at the end of a porcelain boat and 1 g of NaH2PO2·H2O was placed at the upstream side near NiCoOH. The porcelain boat was placed in a furnace and heated at 300 °C under an N2 atmosphere for 2 h, with a heating rate of 2 °C min−1. Then the sample was rinsed with DI water thoroughly and then dried to obtain NiCoP bimetallic phosphide (denoted as NiCoP). For comparison, NF was also annealed under an N2 atmosphere with the same conditions to obtain an NiP electrocatalyst.Characterization studies
Scanning electron microscopy (SEM) was performed on a HITACHI SU5000 at 3.0 kV. Transmission electron microscopy (TEM) was carried out on a Philips-FEI Tecnai G2S-Twin microscope at an acceleration voltage of 200.0 kV, and the powder used for TEM characterization was obtained from NF that had been treated by ultrasonication rather than directly using the NF samples. X-ray powder diffraction (XRD) patterns were recorded using a Bruker D8 Focus diffractometer with Cu-Kα radiation. X-ray photoelectron spectroscopy (XPS) was carried out on a Thermo Scientific K-Alpha+ system, and the energy correction was calibrated by referencing the C 1s peak of adventitious carbon to 284.8 eV. Raman spectra were recorded on a Horiba LabRAM HR Evolution instrument with an excitation wavelength of 532 nm. The content of Co and P was measured by inductively coupled plasma-optical emission spectrometry (ICP-OES, Agilent 720ES).Electrocatalytic measurements
The electrochemical measurements were evaluated using a CHI 760E electrochemical analyzer (Shanghai, China) with a standard three-electrode cell system at room temperature. The as-prepared electrocatalysts (0.5 × 0.5 cm2), an Ag/AgCl (standard KCl) electrode and a platinum sheet electrode served as the working electrode, the reference and the counter electrodes, respectively. The measured potential was calibrated to the reversible hydrogen electrode (RHE) using the following equation: V(vs. RHE) = V(vs. Ag/AgCl) + 0.197 + 0.0591 × pH. The OER and HER were tested in 1.0 M KOH solution, and the electrolyte for the XOR was 1.0 M KOH with 0.1 M xylose. The linear sweep voltammetry (LSV) tests were applied to measure the activities of the electrocatalysts at a scan rate of 2 mV s−1 with 85% iR compensation. All electrocatalysts were activated by the repetitive LSV experiments until the data were consistent. LSV was scanned at a potential range of −0.8 to −1.6 V (vs. Ag/AgCl) for the HER and 0 to 0.7 V (vs. Ag/AgCl) for the OER and XOR.Product detection
H2 was analyzed using a gas chromatograph (GC-9790II, Fuli, China) equipped with a thermal conductivity detector (TCD) and a 5A molecular sieve column (3 mm × 1 m). 0.1 mL volumes of liquid samples were immediately extracted, diluted in distilled water (0.9 mL) and analyzed by high-performance liquid chromatography (HPLC, LC-5090, Fuli, China) equipped with an ultraviolet detector at 210 nm and a differential refractive index detector. The liquid samples were eluted by 5 mM H2SO4 with a flow rate of 0.6 mL min−1 through a Bio-Rad Aminex HPX-87H column (300 mm × 7.8 mm × 9 μm) at 35 °C. The Faraday efficiency is calculated using the equation: faradaic efficiency (%) = (ne × n × F)/Q × 100%, where ne is the number of electrons exchanged, n is the mole of products, F is Faraday's constant, and Q is the total charge. The yields and selectivity of the products were calculated using eqn (1) and (2), respectively: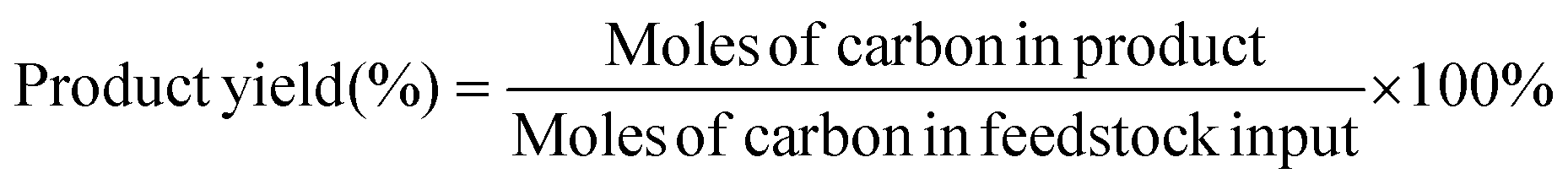 | (1) |
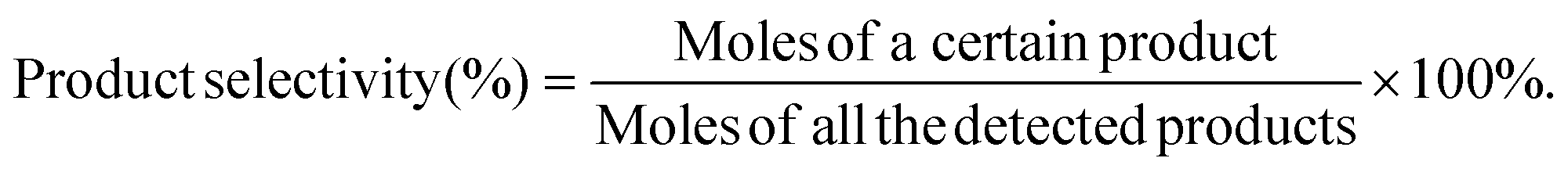 | (2) |
Results and discussion
Structural analysis of materials
As depicted in Fig. 1a, the NiCoOH precursor was first grown on NF through a hydrothermal process, and subsequently converted to NiCoP by phosphorization treatment. Digital photographs in Fig. S1† show that the colors of Ni foam after hydrothermal and phosphorization treatments had turned to purple for the NiCoOH precursor and black for NiCoP, respectively. The surface topography changes in the preparation process of electrocatalysts were investigated by SEM and TEM. Differing from the smooth surface of NF (Fig. S2†) and the rough surface of NiP (Fig. S3†), the NiCoOH precursor shows a smooth nanorod structure with a diameter of around 1.5 nm, whose nanorods are uniformly distributed across the NF (Fig. S4a†). Such a structure is beneficial for the mass transfer of the electrolyte and offers a large active surface area with abundant active sites for the XOR.30 HRTEM images of the NiCoOH in Fig. S4b† further reveal the lattice fringe spacing of 0.228 nm and 0.255 nm, which accurately index the (015) plane of CoNi LDH and the (111) plane of Ni(OH)2,31,32 respectively. The energy-dispersive X-ray spectroscopy (EDS) elemental mapping images confirm that only Co, Ni and O exist inside each NiCoOH nanorod (Fig. S4c†). The SEM (Fig. S5†) and TEM (Fig. 1b and c) images of NiCoP demonstrate that the nanorod array remains uniformly distributed on NF after phosphorization treatment. The HRTEM results (Fig. 1d) exhibit the lattice spacings of 0.279, 0.166 and 0.283 nm, which are consistent with the (101) and (211) planes of NiCoP33,34 and the (011) planes of CoP,33,35,36 respectively. The EDS mapping images (Fig. 1e and Fig. S5c†) verify the uniform distributions of Ni, Co, P and O elements in the nanorod, further demonstrating that the bimetallic phosphide NiCoP was fabricated successfully. | ||
| Fig. 1 (a) Schematic of the preparation of the NiCoP electrocatalyst and the corresponding SEM images of Ni foam, NiCoOH and NiCoP samples. (b and c) TEM, (d) HRTEM images and (e) elemental mappings of NiCoP nanorods. | ||
The crystal structures of NiCoOH and NiCoP are shown in Fig. 2a. The peaks of NiCoOH at 46.9 and 53.1° demonstrate the NiCo-LDH phase (PDF#33-0429)37,38 and the peaks at 33.7, 39.0 and 59.6° relate to the Ni(OH)2 (PDF#22-0444) phase,39 confirming the formation of metal hydroxides in NiCoOH. After phosphorization, the diffraction peaks centering at 35.5, 41.0, 47.6 and 55.3° are ascribed to the (200), (111), (210) and (211) planes of CoNiP (JCPDS 71-2336), respectively.40 The diffraction peaks at 2θ = 31.6, 48.1 and 56.0° corresponding to the CoP phase (PDF#29-0497) are also observed.41 Except for the strong peaks of Ni (PDF#04-0850),42 there are no diffraction peaks of NiCo-LDH or Ni(OH)2 phases in the spectra of NiCoP, demonstrating that the NiCoOH precursor is entirely converted to the bimetallic phosphide NiCoP. The detailed surface chemical composition and elemental state of the electrocatalysts were analyzed by XPS spectra. As shown in Fig. 2b, the survey spectrum of NiCoP reveals the presence of P, Co, Ni and O. The peaks of Co, Ni and O are also observed in the survey spectrum of NiCoOH, but without the signal for P. These results are consistent with the EDS analysis. The Ni 2p high-resolution spectrum of NiCoP can be deconvoluted into three spin orbit doublets, accompanied by satellite peaks (Sat.). The peaks at binding energies of 856.2 and 873.9 eV can be assigned to Ni2+,43 and the other two peaks at 858.9 and 876.3 eV correspond to Ni3+ (Fig. 2c).44 Furthermore, the peaks with binding energies at 853.1 and 870.7 eV are attributed to Ni0, indicating the presence of Ni–P.34,36 Similarly, these spin orbit doublets caused by Co2+, Co3+ and Co occur in Co 2p high-resolution spectra of NiCoP (Fig. 2d).45,46Fig. 2e shows the P 2p region, where the peaks centering at 130.8 and 129.6 eV are associated with P 2p1/2 and P 2p3/2, relating to the metal–phosphide (M–P) bond.47 The predominant peak at 133.9 eV corresponds to the oxidized PO43− species on the surface of NiCoP, originating from the inevitable surface oxidation of NiCoP.48,49 The O 1s XPS spectra (Fig. 2f) indicate that both NiCoOH and NiCoP show the peaks of a metal–hydroxy group (M–OH) and metal–oxo bonding (M–O).27 Moreover, the O 1 s peak of NiCoP is lower than that of NiCoOH, further implying the realization of conversion from NiCoOH to NiCoP.
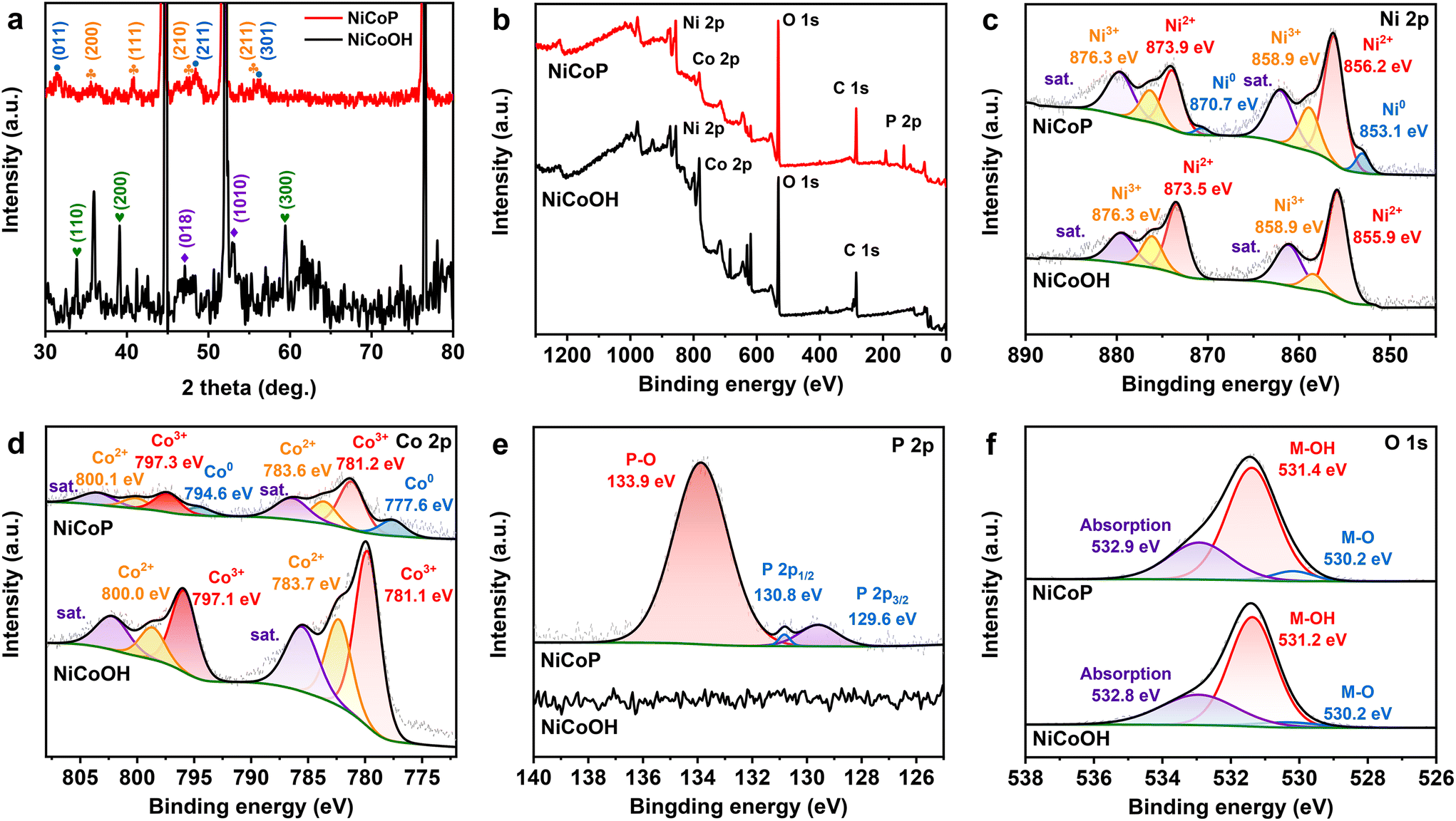 | ||
| Fig. 2 (a) XRD patterns and XPS spectra of (b) broad scans, (c) Ni 2p, (d) Co 2p, (e) P 2p and (f) O 1s of NiCoP and NiCoOH electrocatalysts. | ||
Electrocatalytic performances for xylose oxidation
The electrocatalytic performances of NiCoP were investigated in a typical three-electrode configuration at a scan rate of 2 mV s−1 with an Ag/AgCl reference electrode and Pt foil as a counter electrode. Before investigating the XOR performance, the NiCoP catalyst was activated by LSV from 1.0 to 1.7 V (vs. RHE) until the OER performance stabilized. In alkaline water (1.0 M KOH), the LSV curves with 100 mM xylose plotted in Fig. 3a show that the current density for the XOR is significantly larger than that of the OER. NiCoP exhibits an anodic potential of 1.57 V (vs. RHE) at a current density of 100 mA cm−2 for the OER, which strikingly reduces to 1.29 V (vs. RHE) for the XOR, owing to the thermodynamically and kinetically more favorable XOR over the OER.27 In comparison with other electrocatalysts applied in organic chemicals (Table S1†), the NiCoP catalyst exhibits a lower potential at the same current density, demonstrating the advantage of the NiCoP electrode. Additionally, NiCoOH, NiP and Ni electrocatalysts (Fig. 3a and Fig. S6†) require higher overpotentials to deliver a current density of 100 mA cm−2 for the OER, and lower overpotentials in the XOR, as compared with those for NiCoP. The enhancement of catalytic performance should be attributed to the richer oxidation reaction sites of the NiCoP bimetallic phosphides.50 The XOR performance of NiCoP was also examined at different concentrations of xylose (Fig. S7†). When xylose concentrations increases from 50 mM to 100 mM, both the onset potential and the overpotential at a current density of 100 mA cm−2 decrease. The xylose oxidation reaction was controlled by the surface process. At low xylose concentrations, the reaction rate relates to the diffusion transport of xylose. However, under the condition of high concentrations of xylose, the excess xylose may hamper the formation of active intermediates CoOOH and NiOOH.51,52 Hence, the current density changes little with further increases of xylose concentration from 100 mM to 150 mM. Compared with OER, the current densities of the XOR at 1.2, 1.3, 1.4, 1.5 and 1.6 V (vs. RHE) remarkably increase by 10.9, 102.8, 361.6, 649.8 and 734.5 mA cm−2, respectively (Fig. 3b), strongly confirming the better energy conversion efficiency of the XOR.18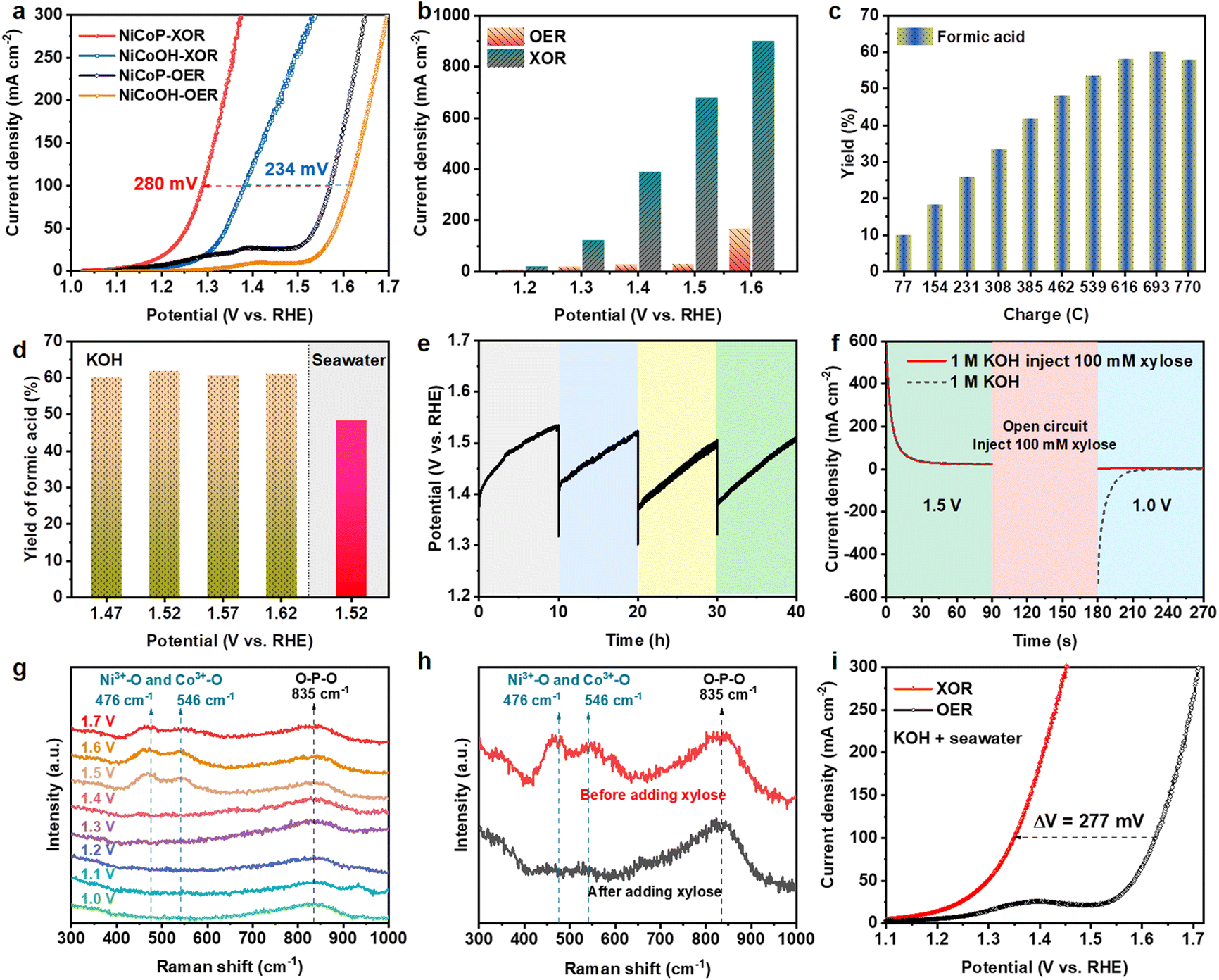 | ||
| Fig. 3 (a) LSV curves of the NiCoP electrocatalyst in 1.0 M KOH with and without 100 mM xylose. (b) The corresponding current densities at various potentials with and without 100 mM xylose. (c) The yield of FA after passing different charges at 1.52 V (vs. RHE). (d) The yield of FA in 8 mL of 100 mM xylose at different potentials. (e) Four consecutive runs of the XOR at a constant current of 100 mA cm−2 with the intermittent addition of 100 mM xylose. (f) Multi-potential chronoamperometry of NiCoP in 1.0 M KOH and 1.0 M KOH when injecting 100 mM xylose during open circuit time. The Raman spectra of NiCoP for (g) the OER and (h) the XOR. (i) LSV curves of the NiCoP electrocatalyst in 1 M KOH seawater electrolytes with and without 100 mM xylose. | ||
The performance of xylose oxidation was further evaluated by chronoamperometric measurements conducted at a constant potential and the products were detected by high-performance liquid chromatography (HPLC) via the calibration curve (Fig. S8†). The HPLC results indicate that the products mainly contain formic acid (FA), lactic acid (LA) and acetic acid (AA) (Fig. S9†). As shown in Fig. 3c, the optimal yield of FA after passing a charge of 693 C at 1.52 V (vs. RHE) is 61.75%, with an FA selectivity of 97.35%. The faradaic efficiency of FA is 68.60% (Fig. S10†), when the conversion of xylose is 89.90% (Fig. S11†). Moreover, the yields and faradaic efficiencies of FA are all over 60% (Fig. 3d and S12†) at different potentials. The possible mechanism of xylose electrooxidation to formic acid is proposed in Fig. S13.† Then the stability of the NiCoP electrode for xylose oxidation was evaluated by chronopotentiometry measurements at 100 mA cm−2 with 50 mM and 100 mM xylose (Fig. 3e and S14†). The electrocatalytic activity is well maintained after four successive cycles, implying the excellent durability of the NiCoP electrode for the XOR. The increasing potential during the reaction is attributed to the decrease of xylose substrate and pH of the solution due to the production of organic acid (Fig. S15†).53 The SEM images (Fig. S16†) and XPS spectra (Fig. S17†) of NiCoP after consecutive runs indicate that the structure of NiCoP remains unchanged during the electrooxidation reaction.
Different potentials were applied on NiCoP to separate the proton deintercalation of the catalyst and oxidation of xylose (Fig. 3f). At a high potential of 1.5 V (vs. RHE), the oxidation current is attributed to the conversion of Co2+/Ni2+ (M2+) to Co3+/Ni3+ (M3+), and a reduction current of M3+ to M2+ was observed after changing the potential to 1.0 V (vs. RHE) in 1.0 M KOH. When 100 mM xylose was added during the open-circuit state, the reduction current disappeared, indicating the spontaneous reduction from Co3+/Ni3+ to Co2+/Ni2+ by accepting protons from xylose.53 Raman spectra were used to reveal this process, as shown in Fig. 3g. In the electrolyte of 1.0 M KOH, the peak at 835 cm−1 corresponded to O–P–O stretching vibrations associated with metal phosphide.54 When the applied potential was raised to 1.5 V (vs. RHE), two bands located at 476 and 546 cm−1 are attributed to the eg bending and the A1g stretching vibration mode of M3+–O, respectively,53,55 demonstrating that the metal oxyhydroxides are generated from metal phosphide. However, the peaks related to M3+–O disappear in the presence of xylose (Fig. 3h), further indicating the reduction of M3+ by xylose.56 The XRD pattern after the XOR shows almost no peak (Fig. S18†), suggesting that the electrogenerated metal oxyhydroxides are amorphous. The contents of Co and P were then tested by ICP-OES (Table S2†). The contents of Co in fresh and used NiCoP electrocatalysts changed little, and were 12.81 and 11.14 wt%, respectively. But the contents of P in fresh and used NiCoP decrease from 6.86 to 2.78 wt% owing to the conversion from metal phosphide to metal oxyhydroxides. These results demonstrate that M3+ ions in oxyhydroxides generated by electrochemical activation are the active sites of xylose electrooxidation. A schematic of the reversible redox cycles of Ni2+/Ni3+ and Co2+/Co3+ during the XOR with the HER is displayed in Fig. S19.†
Then the XOR activity was also measured in alkaline natural seawater (1.0 M KOH seawater). The LSV scans of NiCoP in alkaline seawater with or without the addition of 100 mM xylose are shown in Fig. 3i. The overpotential to achieve a current density of 100 mA cm−2 is significantly reduced by 277 mV compared with the OER. At 1.2, 1.3, 1.4, 1.5 and 1.6 V (vs. RHE), the current densities of the XOR are significantly raised to 14.188, 51.84, 189.16, 408.4 and 695.6 mA cm−2 compared with the OER (Fig. S20†). These results suggest that the XOR is thermodynamically and kinetically more favorable than the OER in alkaline seawater electrolyte. The organic products of the XOR in alkaline seawater were also tested by chronoamperometric measurements at 1.52 V (vs. RHE). The optimal FA yield still reaches 48.20% with a high selectivity of 95.12% after passing a charge of 540 C (Fig. S21†). Furthermore, the faradaic efficiency of FA is 68.84% (Fig. S12†) and the xylose conversion is 69.13% (Fig. S22†). Additionally, in 1.0 L of alkaline seawater with the addition of 100 mM xylose, the potential changes negligibly at a constant current density of 50 mA cm−2 during 25 h of chronopotentiometry testing (Fig. S23†), demonstrating the acceptable stability of NiCoP in seawater. In summary, biomass electrooxidation shows great promise for energy-saving and cost-effective seawater electrolysis, which is of great significance to large-scale H2 production from seawater.
Electrocatalytic performances for the HER
The NiCoP electrode also presents prominent performance for the HER under alkaline conditions. The LSV curves in Fig. 4a and Fig. S24† prove that NiCoP shows a remarkable advantage over the NiCoOH precursor, NiP and bare NF, indicating that the NiCoP bimetallic phosphide possesses abundant active sites for the HER. The NiCoP electrocatalyst requires an overpotential of only 119 mV to drive a current density of 100 mA cm−2, which is much smaller than that of NiCoOH (201.6 mV). As compared with NiP (312.6 mV), the lower overpotential of bimetallic phosphide further demonstrates the more efficient HER catalytic activity. Meanwhile, the Tafel slope of the NiCoP electrode for the HER is calculated as 87.52 mV dec−1, less than that of NiCoOH (112.11 mV dec−1) and NiP (219.97 mV dec−1), which demonstrates the higher transfer coefficient and the favorable reaction kinetics of the HER.57 Such performance is competitive among the noble metal electrocatalysts reported to date (Fig. 4c and Table S3†). Additionally, the NiCoP catalyst shows a comparable HER activity with slight degradation after introducing 50 and 100 mM xylose, illustrating the xylose tolerance of the HER (Fig. S25†). The hydrogen production agrees well with theoretical amounts, demonstrating an approximately 100% faradaic efficiency for the HER (Fig. S26†). Furthermore, after continuous chronopotentiometry measurement for 45 h conducted at 100 and 200 mA cm−2, the required potential barely increases by 7.1 and 0.5 mV, respectively (Fig. 4d), suggesting excellent stability of the NiCoP catalyst for the HER. Then the HER performances of NiCoOH and NiCoP were assessed in alkaline seawater. As depicted in Fig. 4e, NiCoP still displays excellent HER activity with an overpotential of 152.6 mV at a current density of 100 mA cm−2. The HER performance of NiCoP has limited degradation in alkaline seawater, which is caused by a decrease of conductivity in complicated natural seawater.58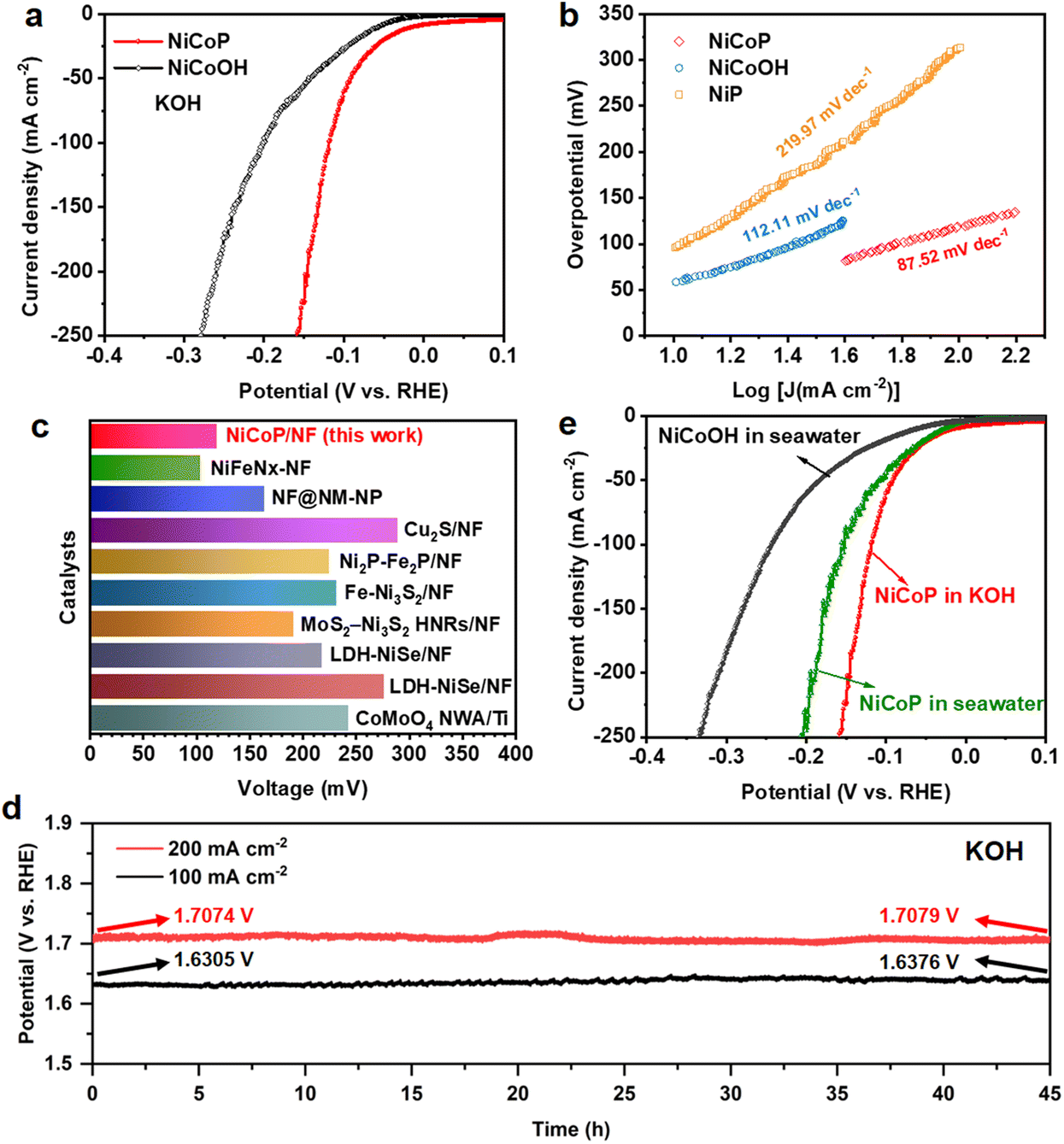 | ||
| Fig. 4 (a) LSV curves and (b) the corresponding Tafel plots of NiCoOH and NiCoP electrocatalysts in 1.0 M KOH. (c) A comparison of the HER in 1.0 M KOH at a current density of 100 mA cm−2 between NiCoP and other noble-metal catalysts reported to date. (d) 45 h stability test of NiCoP for a cathodic HER at 100 and 200 mA cm−2 and (e) LSV curves of the NiCoOH and NiCoP electrocatalysts in 1.0 M KOH and alkaline seawater. | ||
Electrochemical property of a two-electrode system
Considering that the NiCoP electrode simultaneously possessed optimal electrocatalytic activity in the XOR and HER in alkaline natural seawater, a two-electrode electrolytic cell employing NiCoP as a bifunctional electrocatalyst was constructed with 1.0 M KOH seawater as the electrolyte (Fig. 5a). As presented in Fig. 5b, the overpotential of NiCoP to afford a current density of 100 mA cm−2 is 1.86 V in the absence of xylose, while the cell voltage reduces to 1.57 V to deliver a current density of 100 mA cm−2 in the presence of xylose, which is nearly 290 mV lower than that for overall water splitting. Besides, after adding 100 mM xylose, the current densities to obtain the voltages of 1.6, 1.7, 1.8, 1.9 and 2.0 V are reduced by 115.64, 206.47, 275.24, 302.00 and 296.92 mA cm−2, respectively (Fig. 5c). The gas products in the hybrid system were investigated at 1.9 V (Fig. 5d). After chronoamperometric measurement for 60 min, the H2 production rate in the presence of xylose reaches 150 mL cm−2, which is much faster than the production rate in the absence of xylose (36.67 mL cm−2). No bubbles were observed at the anode, implying that no oxygen is produced, owing to the dominance of the XOR. The reaction products after passing 693 C were also investigated by HPLC. The FA selectivity was retained as high as 94.6%, while the selectivity values of LA and AA were 4.6% and 0.7%, respectively (Fig. 5e). These results indicate that our strategy for biomass-assisted seawater electrolysis is low-cost and highly efficient in producing hydrogen and chemicals with high selectivity.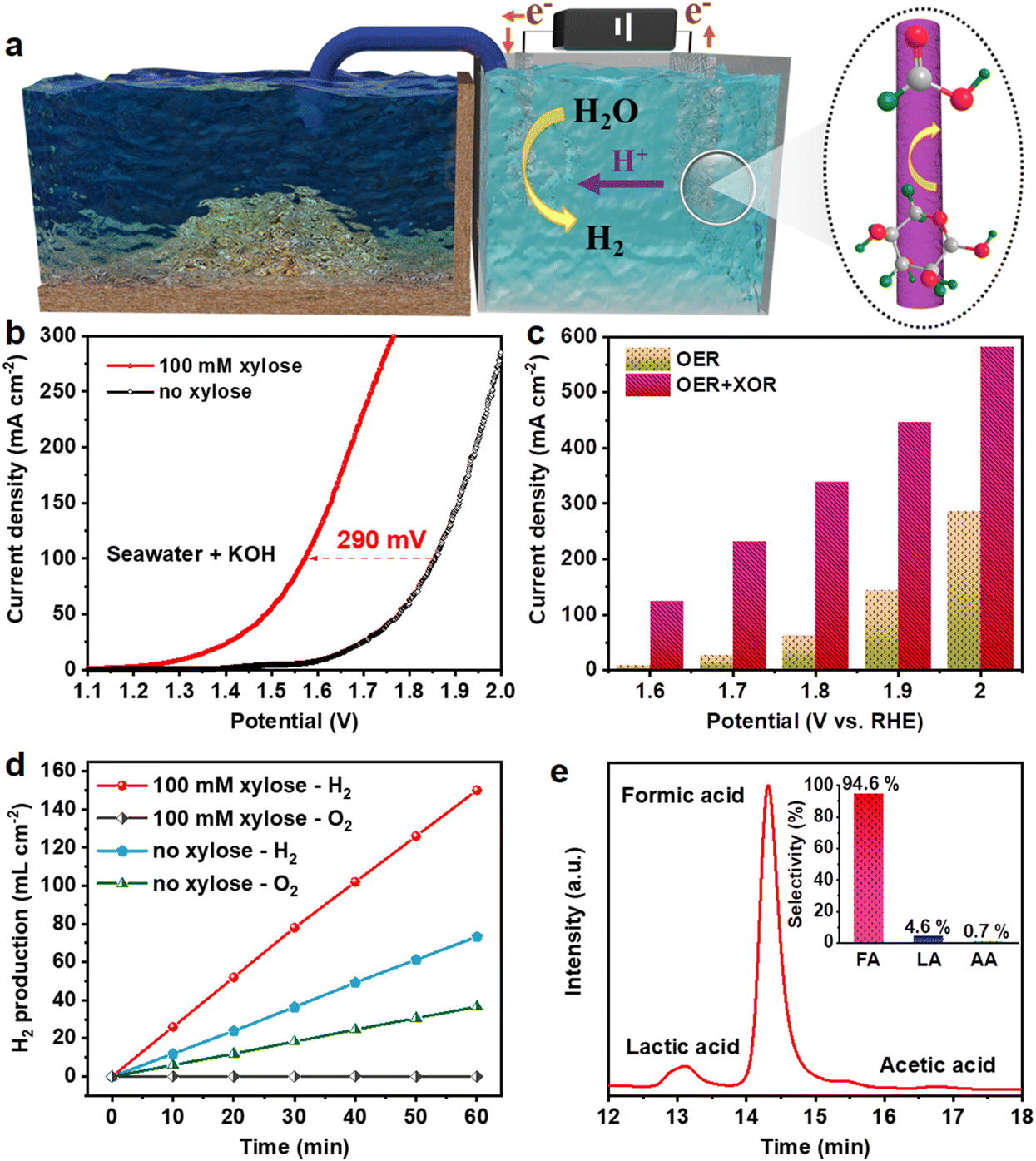 | ||
| Fig. 5 Electrochemical performances of NiCoP || NiCoP. (a) A schematic illustration of the hybrid seawater electrolysis system. (b) LSV curves of the NiCoP electrocatalyst in 1.0 M KOH seawater with and without 100 mM xylose. (c) The corresponding current densities at various potentials with (green bars) and without (orange bars) 100 mM xylose. (d) H2 and O2 production rates with and without 100 mM xylose at 1.9 V. (e) The HPLC traces of products; the inset exhibits the selectivities of FA, LA and AA. | ||
Conclusions
In summary, we designed a hybrid electrolyser that coupled xylose electrooxidation and the HER to coproduce value-added organic acid and H2 using NiCoP as a bifunctional electrode, aiming to boost the economic benefit of seawater electrolysis. Thermodynamically favorable xylose-assisted seawater electrolysis required less energy than traditional overall water splitting. In a three-electrode cell system with alkaline seawater as the electrolyte, the overpotential of the XOR to achieve a current density of 100 mA cm−2 was reduced by 277 mV as compared with that of the OER, while the two-electrode electrolytic cell only required 1.57 V to deliver a current density of 100 mA cm−2 in the presence of xylose. Furthermore, formic acid was obtained with high selectivity of 94.6% and the H2 production rate was 150 mL cm−2 without oxygen. Therefore, our work offers an energy-saving xylose-assisted seawater electrolysis system to produce pure H2 and valuable chemicals, which is promising for seawater electrolysis and biomass valorization.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (31971614) and the State Key Laboratory of Pulp and Paper Engineering (2022PY02). We acknowledge the financial support from the National Program for Support of Top-notch Young Professionals (Grant no. x2qsA4210090) and the Guangdong Basic and Applied Basic Research Foundation (Grant no. 2021A1515110622, 2023A1515012589).References
- Q. Liu, Y. Sun and M. Wu, J. Cleaner Prod., 2021, 295, 126459 CrossRef
.
- D. Jang, K. Kim, K.-H. Kim and S. Kang, Energy Convers. Manage., 2022, 263, 115695 CrossRef CAS
- O. S. Ibrahim, A. Singlitico, R. Proskovics, S. McDonagh, C. Desmond and J. D. Murphy, Renewable Sustainable Energy Rev., 2022, 160, 112310 CrossRef CAS
- Z. Zhang, Y. Lei and W. Huang, Chin. Chem. Lett., 2021, 33, 3623–3631 CrossRef
- X. Zhang, T. Liu, T. Guo, X. Han, Z. Mu, Q. Chen, J. Jiang, J. Yan, J. Yuan, D. Wang, Z. Wu and Z. Kou, Chin. J. Catal., 2021, 42, 1798–1807 CrossRef CAS
- N. Q. Tran, B. T. N. Le, T. N.-M. Le, L. T. Duy, T. B. Phan, Y. Hong, T. K. Truong, T. L. H. Doan, J. Yu and H. Lee, J. Phys. Chem. Lett., 2022, 13, 8192–8199 CrossRef CAS PubMed
- N. Wen, Y. Xia, H. Wang, D. Zhang, H. Wang, X. Wang, X. Jiao and D. Chen, Adv. Sci., 2022, 9, 2200529 CrossRef CAS PubMed
- S. Liu, S. Ren, R. T. Gao, X. Liu and L. Wang, Nano Energy, 2022, 98, 107212 CrossRef CAS
- J. Chang, G. Wang, Z. Yang, B. Li, Q. Wang, R. Kuliiev, N. Orlovskaya, M. Gu, Y. Du and G. Wang, Adv. Mater., 2021, 33, 2101425 CrossRef CAS PubMed
- S. Seenivasan and D. H. Kim, J. Mater. Chem. A, 2022, 10, 9547–9564 RSC
- Q. Liu, E. Wang and G. Sun, Chin. J. Catal., 2020, 41, 574–591 CrossRef CAS
- M. A. Khan, T. Al-Attas, S. Roy, M. M. Rahman, N. Ghaffour, V. Thangadurai, S. Larter, J. Hu, P. M. Ajayan and M. G. Kibria, Energy Environ. Sci., 2021, 14, 4831–4839 RSC
- J. Cheng and D. Wang, Chin. J. Catal., 2022, 43, 1380–1398 CrossRef CAS
- C. Wang, H. Shang, L. Jin, H. Xu and Y. Du, Nanoscale, 2021, 13, 7897–7912 RSC
- S. R. Dresp, F. Dionigi, M. Klingenhof and P. Strasser, ACS Energy Lett., 2019, 4, 933–942 CrossRef CAS
- H. Luo, J. Barrio, N. Sunny, A. Li, L. Steier, N. Shah, I. E. L. Stephens and M. M. Titirici, Adv. Energy Mater., 2021, 11, 2101180 CrossRef CAS
- Y. Zhang, B. Zhou, Z. Wei, W. Zhou, D. Wang, J. Tian, T. Wang, S. Zhao, J. Liu, L. Tao and S. Wang, Adv. Mater., 2021, 33, 2104791 CrossRef CAS PubMed
- Y. Li, X. Wei, L. Chen, J. Shi and M. He, Nat. Commun., 2019, 10, 1–12 CrossRef PubMed
- X. Wei, S. Wang, Z. Hua, L. Chen and J. Shi, ACS Appl. Mater. Interfaces, 2018, 10, 25422–25428 CrossRef CAS PubMed
- C. Wang, M. Zhu, Z. Cao, P. Zhu, Y. Cao, X. Xu, C. Xu and Z. Yin, Appl. Catal., B, 2021, 291, 120071 CrossRef CAS
- L. Wu, L. Yu, F. Zhang, B. McElhenny, D. Luo, A. Karim, S. Chen and Z. Ren, Adv. Funct. Mater., 2021, 31, 2006484 CrossRef CAS
- H. Wang, L. Chen, L. Tan, X. Liu, Y. Wen, W. Hou and T. Zhan, J. Colloid Interface Sci., 2022, 613, 349–358 CrossRef CAS PubMed
- Z. Zhou, C. Chen, M. Gao, B. Xia and J. Zhang, Green Chem., 2019, 21, 6699–6706 RSC
- C. Chen, Z. Zhou, J. Liu, B. Zhu, H. Hu, Y. Yang, G. Chen, M. Gao and J. Zhang, Appl. Catal., B, 2022, 307, 121209 CrossRef CAS
- G. Yang, Y. Jiao, H. Yan, Y. Xie, A. Wu, X. Dong, D. Guo, C. Tian and H. Fu, Adv. Mater., 2020, 32, 2000455 CrossRef CAS PubMed
- T. Wu, B. H. Nguyen, M. C. Daugherty and K. D. Moeller, Angew. Chem., 2019, 131, 3600–3603 CrossRef
- Y. Liu, R. Zou, B. Qin, J. Gan and X. Peng, Chem. Eng. J., 2022, 136950 CrossRef CAS
- X. Jiang, Z. Dong, Q. Zhang, G. Xu, J. Lai, Z. Li and L. Wang, J. Mater. Chem. A, 2022, 10, 20571–20579 RSC
- Q. Lv, J. Han, X. Tan, W. Wang, L. Cao and B. Dong, ACS Appl. Energy Mater., 2019, 2, 3910–3917 CrossRef CAS
- C. Huang, Y. Zhong, J. Chen, J. Li, W. Zhang, J. Zhou, Y. Zhang, L. Yu and Y. Yu, Chem. Eng. J., 2021, 403, 126304 CrossRef CAS
- H. Zhang, Y. Tong, J. Xu, Q. Lu and F. Gao, Chem. – Eur. J., 2018, 24, 400–408 CrossRef CAS PubMed
- W. Wei, W. Ye, J. Wang, C. Huang, J. B. Xiong, H. Qiao, S. Cui, W. Chen, L. Mi and P. Yan, ACS Appl. Mater. Interfaces, 2019, 11, 32269–32281 CrossRef CAS PubMed
- W. Du, Q. Zong, J. Zhan, H. Yang and Q. Zhang, ACS Appl. Energy Mater., 2021, 4, 6667–6677 CrossRef CAS
- K. Dai, X. Gao, L. Yin, Y. Feng, X. Zhou, Y. Zhao and B. Zhang, Appl. Surf. Sci., 2019, 494, 22–28 CrossRef CAS
- C. Zhang, Y. Huang, Y. Yu, J. Zhang, S. Zhuo and B. Zhang, Chem. Sci., 2017, 8, 2769–2775 RSC
- J. Li, G. Wei, Y. Zhu, Y. Xi, X. Pan, Y. Ji, I. V. Zatovsky and W. Han, J. Mater. Chem. A, 2017, 5, 14828–14837 RSC
- Y. Wang, C. Shi, Y. Chen, D. Li, G. Wu, C. Wang and L. Guo, Electrochim. Acta, 2021, 376, 138040 CrossRef CAS
- F. Zhu, W. Liu, Y. Liu and W. Shi, Chem. Eng. J., 2020, 383, 123150 CrossRef CAS
- X. Zhang, A. Wu, X. Wang, C. Tian, R. An and H. Fu, J. Mater. Chem. A, 2018, 6, 17905–17914 RSC
- F. Meng, Y. Yu, D. Sun, S. Lin, X. Zhang, T. Xi, C. Xu, H. OuYang, W. Chu and L. Shang, ACS Appl. Energy Mater., 2021, 4, 2471–2480 CrossRef CAS
- H. Liu, X. Ma, H. Hu, Y. Pan, W. Zhao, J. Liu, X. Zhao, J. Wang, Z. Yang and Q. Zhao, ACS Appl. Mater. Interfaces, 2019, 11, 15528–15536 CrossRef CAS PubMed
- C. Liao, P. Hung, Y. Cheng, S. Chang and P. Wu, Mater. Lett., 2018, 215, 152–156 CrossRef CAS
- S. Chen, J. Duan, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2013, 52, 13567–13570 CrossRef CAS PubMed
- Q. Yang, J. Dai, K. Li and J. Chen, Chin. J. Catal., 2013, 34, 1201–1207 CrossRef CAS
- J. Liu, Y. Gao, X. Tang, K. Zhan, B. Zhao, B. Y. Xia and Y. Yan, J. Mater. Chem. A, 2020, 8, 19254–19261 RSC
- X. Ding, H. Huang, Q. Wan, X. Guan, Y. Fang, S. Lin, D. Chen and Z. Xie, J. Energy Chem., 2021, 62, 415–422 CrossRef CAS
- R. Boppella, J. Tan, W. Yang and J. Moon, Adv. Funct. Mater., 2019, 29, 1807976 CrossRef
- Q. Zong, H. Yang, Q. Wang, Q. Zhang, J. Xu, Y. Zhu, H. Wang, H. Wang, F. Zhang and Q. Shen, Dalton Trans., 2018, 47, 16320–16328 RSC
- Z. Shao, J. Sun, N. Guo, F. He, K. Huang, F. Tian and Q. Wang, J. Power Sources, 2019, 422, 33–41 CrossRef CAS
- H. Wang, C. Li, J. An, Y. Zhuang and S. Tao, J. Mater. Chem. A, 2021, 9, 18421–18430 RSC
- C. Borrás, C. Berzoy, J. Mostany, J. C. Herrera and B. R. Scharifker, Appl. Catal., B, 2007, 72, 98–104 CrossRef
- O. Sahin and H. Kivrak, Int. J. Hydrogen Energy, 2013, 38, 901–909 CrossRef CAS
- Z. He, J. Hwang, Z. Gong, M. Zhou, N. Zhang, X. Kang, J. W. Han and Y. Chen, Nat. Commun., 2022, 13, 1–10 Search PubMed
- J. M. Benevides, S. A. Overman and G. J. Thomas Jr., J. Raman Spectrosc., 2005, 36, 279–299 CrossRef CAS
- Y. J. Wu, J. Yang, T. X. Tu, W. Q. Li, P. F. Zhang, Y. Zhou, J. F. Li, J. T. Li and S. G. Sun, Angew. Chem., Int. Ed., 2021, 60, 26829–26836 CrossRef CAS PubMed
- P. Zhang, X. Sheng, X. Chen, Z. Fang, J. Jiang, M. Wang, F. Li, L. Fan, Y. Ren and B. Zhang, Angew. Chem., 2019, 131, 9253–9257 CrossRef
- X. Sun, N. Habibul and H. Du, Chin. J. Catal., 2021, 42, 235–243 CrossRef CAS
- L. Wu, L. Yu, B. McElhenny, X. Xing, D. Luo, F. Zhang, J. Bao, S. Chen and Z. Ren, Appl. Catal., B, 2021, 294, 120256 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3gc00684k |
| This journal is © The Royal Society of Chemistry 2023 |