
Electrochemical nitration for organic C–N bond formation: a current view on possible N-sources, mechanisms, and technological feasibility
Nils
Kurig
 * and
Regina
Palkovits
* and
Regina
Palkovits
Institute for Technical und Macromolecular Chemistry, RWTH Aachen University, Worringerweg 2, 52074 Aachen, Germany. E-mail: Kurig@itmc.rwth-aachen.de
First published on 25th August 2023
Abstract
Growing awareness of the environmental impact caused by the chemical industry drives considerations towards a circular economy and the use of renewable electricity. A key role will be played by ammonia (NH3) currently produced in the Haber Bosch process at elevated temperature and pressure from fossil hydrogen leaving an enormous CO2 footprint. A more sustainable production is to be realized by electrochemically generated hydrogen or all-electrochemical synthesis. Aside from the crucial role in fertilizer production, NH3 is necessary for the synthesis of organic intermediates and monomers via C–N bond formation reactions. This perspective highlights different strategies for electrochemical C–N functionalisation focusing on electrophilic nitration. Based on literature from one century, syntheses involving different inorganic nitrogen sources are discussed and their potential is evaluated. Finally, a perspective for more sustainable electrochemical nitration using NH3 and atmospheric nitrogen (N2) is presented. This way, a possibility to bypass the Haber Bosch process shall be demonstrated especially for decentralized small-scale productions.
 Nils Kurig | Nils Kurig obtained his Ph.D. in Chemistry from RWTH Aachen (2019–2023), working with Prof. Regina Palkovits in the cluster of excellence “The Fuel Science Center” supported by a scholarship from “Studienstiftung des deutschen Volkes e. V.” His research focused on efficient biomass conversion into fuels and commodities, whilst advancing electrochemical flow methods, C–N bond formation, coupled processes, and spectroelectrochemistry. Recently promoted to project leader, he now leads research on electrocatalytic C–N bond formation and complex redox mediators for biomass transformation. |
 Regina Palkovits | Regina Palkovits is a full professor for Heterogeneous Catalysis & Chemical Technology at RWTH Aachen University. She graduated in Chemical Engineering from Technical University Dortmund and finished her Ph.D. with Prof. Ferdi Schüth at the Max-Planck-Institut für Kohlenforschung. Since 2010 she has been Professor at RWTH Aachen University. She has received numerous awards, including the 2019 EFCATS Young Researcher Award and the 2016 DECHEMA Award. She is a Max Planck Fellow at the Max Planck Institute for Chemical Energy Conversion and as of 2020, a member of the North Rhine-Westphalian Academy of Sciences, Humanities and the Arts. |
Introduction
Rising global average temperature and scarcity of fossil resources show that a transition towards circular energy economy is imperative. A key player in this regard is the electrification of industrial processes utilizing ecologically friendly harvested energy from wind, water, or solar power.That is why sustainability nowadays extends to the largest chemical productions such as Haber–Bosch.1 Conventional ammonia (NH3) synthesis via the Haber–Bosch process for its application in fertilizer production is worth 1–2% of global energy consumption and a similar share of CO2 emissions.2–4 This is because, fossil hydrogen (H2)4 is pressurised to 150–250 bar at 400–450 °C to react with nitrogen (N2).5 Although, H2 may in the future be taken from water electrolysis6 to reduce the carbon footprint, the energy demand for the process itself remains. Therefore, a direct N2 reduction is highly desired. However, in the renaissance of electrochemistry as a green tool for the chemical industry,7–11 N2 activation remains a great challenge.12–15 A detailed overview of the scientific approaches and crucial experimental considerations was provided by Stephens, Chorkendorff, Shao-Horn, and co-workers in 2021.16 Even if an atom-, redox-, and energy-economic synthesis of NH3 or also nitrate (NO3−) is realised, bulk chemical production is currently limited by the economies of scale.17,18
Meanwhile, a second branch of electrochemical research focuses on products providing a higher margin19–21 or downstream processing of biogenic materials.22–24 These approaches of organic electrosynthesis usually name inherent advantages of electrochemistry for a decentralized production matching the upcoming energy infrastructure.25 For example, the elimination of harsh reaction temperatures and high pressures provides easier access to larger-scale production equipment. Also, high flexibility towards fluctuating substrate streams and energy supply can be achieved.8,18
Since multiple classes of target molecules such as monomers, pesticides, and pharmaceuticals possess C–N bonds,26 the formation of these bonds by electrochemical methods is receiving increasing attention.27–33 In 2018, Kärkäs presented a comprehensive review article describing electrochemical C–H activation for, among other things, C–N functionalization.34 Most of the included studies consider organic nitrogen sources (N-sources), i.e. the reaction of two organic molecules.
On the other hand, the inorganic electrochemistry of nitrogen compounds has been intensively studied, for example, by Koper's group in 2009.35 Here, the authors described the nitrogen cycle across eight oxidation states comparing natural and synthetic conversions. This article was later supplemented with an important review article on NH3 oxidation by Bunce and Bejan.36 They focused on electrocatalytic conversion using different electrocatalysts. The number of review articles on electrochemical N2 reduction is vast14,37–40 and the respective conversions are not in the scope of this perspective.
Currently, the literature lacks a linkage between the organic and inorganic scientific approaches. The activation of N2 has hitherto encountered difficulties on technical scales due to energy or cost efficiency. However, for organic target compounds, the margin is much higher.41,42 Activation of inorganic nitrogen compounds for introduction into organic structures would therefore have the potential to exploit the higher value increase of organic products. An envisioned way to realise this is the electrochemical nitration reaction, which is the subject of this review article.
Accordingly, the first section comprises existing approaches to electrochemical nitration. Starting from early studies to understand the selectivity of electrophilic aromatic substitution (SEAr) reactions,43–45 over different N-sources employed.46–48 A connection will be made to the state of the art findings in sustainable electrochemical nitration recently presented by the Waldvogel group.49 Then, further strategies of electrochemical C–N bond formation will be discussed in both organic and inorganic reactions.50,51 Alongside, the available insights in activation mechanisms of nitrogen species are reviewed including spectroscopic and computational studies.52–55 At the end, a perspective that evaluates the opportunities of electrochemical N2 activation for organic C–N bond formation and scientific starting points for further research is provided.
Electrochemical nitration
In organic synthesis, ammonia represents one way to introduce nitrogen into large molecules like pharmaceuticals, pesticides, or generally advanced intermediates. Due to the higher margin of these fine chemicals,41,42 the hourly production rate is of less importance than for the bulk synthesis of ammonia. Therefore, electrochemical nitrogen activation should be reassessed in this context targeting organic nitro compounds instead of ammonia or nitric acid. Direct incorporation of N2 into organic molecules would additionally reduce the number of reaction steps leading to an overall more elegant and thus more sustainable synthesis. To realize this, first, active nitrogen species must be identified.For functionalizing organic molecules, especially oxidative pathways yielding electrophilic nitrating agents are of interest. Typically, this is realised by the activation of nitric acid using and acidic catalyst like sulfuric acid.56,57 Industrial processes are conducted on a scale of several million tons per year58 and recently, effort is made to explore more sustainable alternatives.57 Despite that, the organic nitration reaction is not a particularly active research field, which provides room for innovation for groups wishing to enter the field. Furthermore, already in 1971, the electrochemical nitration of aromatic compounds in the presence of nitrate was reported.43 Recently, an environmentally benign and hazard reduced pathway involving a soluble nitrite salt was developed.49 The field of electrochemical nitration hence comprises a period of 50 years in which conceptual knowledge was collected. This provides the basis for a fresh discussion of electrochemical nitration in the context of new issues such as the efficient use of electricity and decentralised production. The revision of literature is started with the question about the active species in electrophilic nitration using the mixture of sulfuric acid and nitric acid, sometimes called nitrating acid. Here, it is suggested that the textbook mechanism involving the nitronium ion (NO2+)59 gives no conclusive explanation of the observed intramolecular selectivity without intermolecular selectivity. While from a mixture of aromatics all react at a similar rate,60 the position of the nitro group in the target compounds represents the selectivity of an electrophilic attack.61 Olah explained this by a complex formed from aromatic and NO2+.45
Perrin later states that a single electron transfer from the π-system to NO2+ leaves a radical pair ArH˙+/NO2˙ that collapses to form the organic nitro compound (Fig. 1). Evidence is given by electrochemical formation of the aryl cation and subsequent reaction with NO2˙ yielding the same selectivity.44 This is later on affirmed in studies on naphthalene which is repeatedly used in electrochemical nitration.62,63 The most common electrode material in these studies is platinum.63–65 In fact, yields of up to 75% of nitronaphthalene were reported for the electrochemical nitration using N2O4 which is oxidized to NO2+ in anhydrous sulfolane.65 Generally, reactions at potentials suitable for the oxidation of the aromatic compound63,64 are discriminated from reactions at potentials above 1.75 vs. Ag/Ag+ at which both aryl and nitrogen dioxide can be oxidized.66 This already indicates the possibility of using nitrite and nitrate as nitrating agent precursors by the recombination of ArH+ and the negatively charged nitrogen species. Exploring the opportunities of electrochemical nitration to avoid toxic nitrating agents such as NO2˙/N2O4, this approach highlights two potentially attractive active species to be generated in situ. As mentioned above, the abundant and highly soluble nitrate (NO3−) was investigated alongside this discussion. Fig. 2 shows the reaction mechanisms using NO2˙ or NO3− as nitrating agents. Nyberg reported the nitration of mesitylene with tetrabutylammonium nitrate in nitromethane. The highest selectivity of 39% was obtained using a carbon anode at 1.5 V (presumably vs. SHE) and a mesitylene![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :nitrate ratio of 5:1. Accordingly, mesitol was the main product with 56% selectivity.43
:nitrate ratio of 5:1. Accordingly, mesitol was the main product with 56% selectivity.43
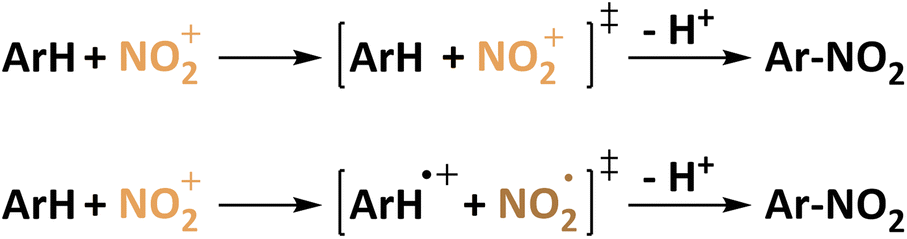 | ||
| Fig. 1 Proposed mechanism for the nitration of aromatics according to Olah (top) and Perrin (bottom). | ||
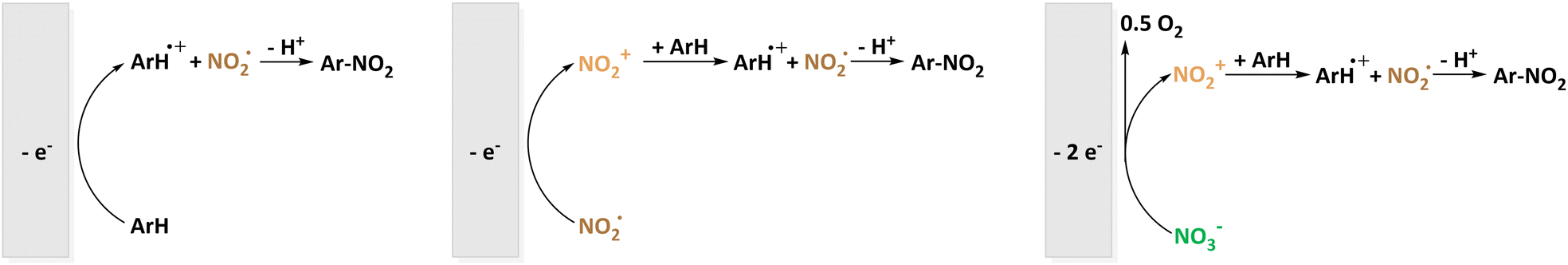 | ||
| Fig. 2 Electrochemical nitration using NO2˙ as nitrating agent below (left) and above (middle) its potential of oxidation to NO2+ and similarity to the reaction pathway using NO3− as source of NO2˙ (right). | ||
The more common electrochemical nitrating agent compared to nitrate is nitrite (NO2−). Very recently, the Waldvogel group published an environmentally benign method for the nitration of 20 aromatic compounds using NBu4NO2 as soluble nitrite source in MeCN. The employed graphite electrodes represent a metal-free, low-cost material underlining the demonstrated scalability of the reaction to gram-scale maintaining satisfactory yields of 85%.49 Previously, electrochemical nitration with NO2− was shown for the prominent model substance naphthalene in MeCN by the Sereno group as well.48 In the discussion of the reaction mechanism, NO2˙/N2O4 is referred to as the active species. The different pathways are depicted in Fig. 3.
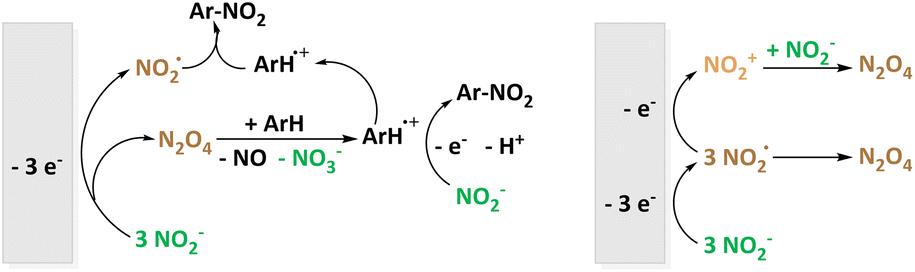 | ||
| Fig. 3 Electrochemical nitration using soluble NO2− as source of the nitrating agent according to the groups of Waldvogel (left) and Sereno (right). | ||
Interestingly, NO2˙/N2O4 formation is suggested to be not only due to NO2− oxidation but also via recombination of NO2− and NO2+ to N2O4 if there is an excess of NO2−.48 In follow-up investigations, the effect of the non-ionic surfactant polyoxyethylen(23)-laurylether on the same nitration system was examined. The pre-concentration of both compartmentalized substrate and nitrating agent reactants enhances the desired reaction pathway.46,47 Laurent et al. also studied nitration starting from a nitrite source reacting with ArH˙+.67 Additionally, they described the interconversion of NOx species under oxidative conditions including NO2−, N2O4−, NO3−, N2O3, N2O5, and NO2+ underlining the versatility of these reaction systems.
Aside from electrochemical nitration, NO2− and NO3− can be used to electrochemically form N–N bonds in an N-nitrosation or N-nitration of secondary amines.68,69 In this process, a carbon anode facilitates the oxidation of organic substrates, whereas the active NOx species are generated thermally from Fe(NO3)3. This mechanism was verified by radical capture tests. Here, NO˙ recombined with 2,2,6,6-tetramethyl-1-piperidi-nyloxy (TEMPO) and 2,4-di-tert-butyl-4-methylphenol (BHT) with the pyrazole radical indicating a biradical mechanism. Follow-up DFT calculations confirmed opposite selectivity of pyrazole and morpholine towards NO˙ and NO2˙ leading to either the nitrosamine or the nitramine.69 This emphasises the versatility of the NOx radical chemistry and possible reaction pathways for the targeted C–N functionalization.
Further strategies for C–N bond formation and nitrogen activation
In 2018, Kärkäs revised organic C–N bond formation reactions.34 Examples are the C–N coupling of pyrrolidones to anilines in a Shono oxidation,70 intramolecular amine formation from olefines,71,72 synthesis of primary amines of aromatics,73,74 and α-amination of ketones.75 Additionally, recent publications describe reductive amination of biogenic substrates for C–N bond formation in the context of amino acids31,33 or pyrrolidone synthesis.27 Most of these conversions are realized by coupling two organic molecules whereas there is a limited number of examples of inorganic N-sources such as NH3 or N3−.34Further strategies for C–N bond formation from small, inorganic N-sources are recently discussed in the field of CO2 utilization.51,76–79 For the strategies discussed here, a comparison of reaction pathways from urea synthesis displayed in Fig. 4 is of particular interest.
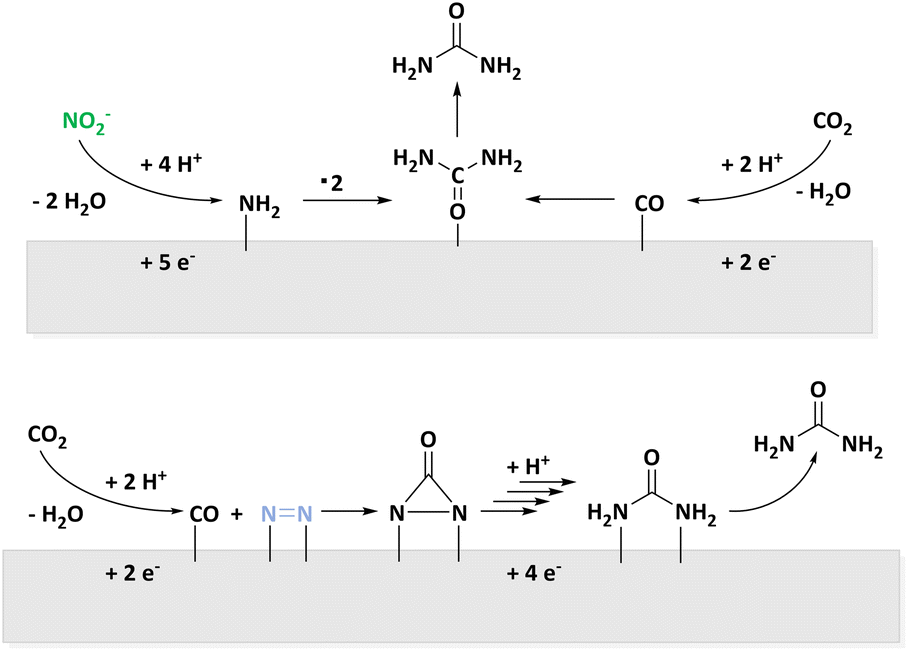 | ||
| Fig. 4 Reaction pathways of electrochemical urea synthesis via NO2− (top) or N2 (bottom) activation and parallel CO2 reduction to CO. | ||
On one hand, the C–N coupling is achieved by reduction of NO3−,80–82 NO2−,50,83,84 or NO85 to a *NH2 surface species which attacks surface bound *CO. Not only the number of electrons transferred in the process but also the activation energy indicate that excess CO is formed. In this regard, bifunctional catalysts are applied as adsorption and activation of different molecules has to be facilitated.86 On the other hand, activation of N2 for an attack of CO originating from CO2RR is discussed. According to DFT calculations, this leads to a *NCON* species described as tower-like which is selectively hydrogenated to form *NCONH*. A combined process of hydrogenation and CO release to build intermediates of NH3 formation is unfavoured according to DFT calculation because *N2H is energetically disadvantageous. Once again, bifunctional materials are necessary. The PdCu alloy provides comparably negatively charged Cu and facilitates the addition of H atoms to Cu-bound nitrogen.51 In both reaction systems, urea formation competes with CO2RR and NRR and outlines the variety of active carbon and nitrogen species accessible with electrochemical methods. For a detailed discussion of reactions pathways of electrocatalytic urea synthesis from CO2 and NOx species, we refer the interested reader to a recent review by the Jin group.87
In this perspective, different nitrogen species have been elaborated, which can potentially be used for electrochemical nitration. One opportunity to access them is the ammonia oxidation reaction (AOR). Research in this field is mostly driven by the hydrogen-fuel properties of ammonia88 and its removal from wastewater.89 In 2011, Bunce and Bejan described the electrocatalytic AOR mechanisms in a review article.36 These reactions target dinitrogen as product and consider nitrogen oxides unwanted side products. Therefore, faradaic efficiencies are often not discussed for the latter. Below the potential range of the oxygen evolution reaction (OER) in aqueous medium, ammonia is electrolyzed at platinum in alkaline solution to form N2 and NO3−.90 Under these conditions, NO2− was only formed in traces, unless the solution additionally contained Cu(OH)2.91 The key intermediate in the reaction sequence leading to NO2− and NO3− is NH2OH,52 whereas adsorbed NHy species are attributed to N2 formation.53 To enhance the NH2OH and thus NO2− and NO3− production, potentials in the range of both AOR and OER must be applied to couple N–O. Therefore, the pathways were divided into above and below OER potential as displayed in Fig. 5.
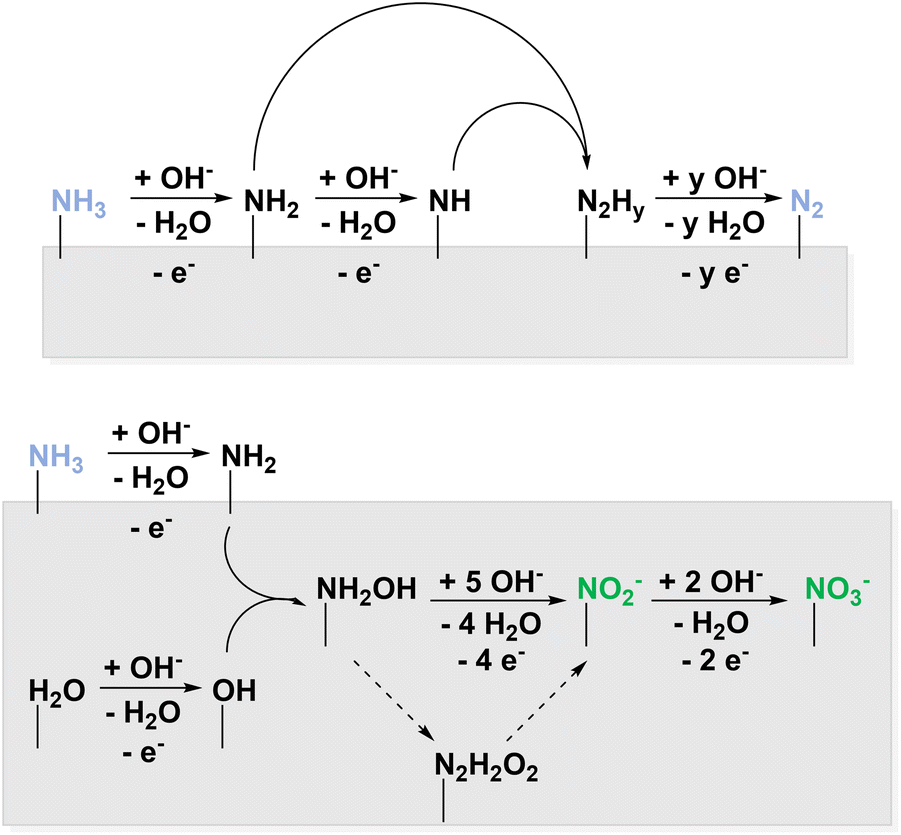 | ||
| Fig. 5 Electrochemical NH3 oxidation below (top) and above (bottom) the potential of OER. | ||
The possibility of oxidizing NH2OH to NO2− and NO3− at potentials around 0.6 V vs. Ag/AgCl was confirmed on a rotating ring disk electrode (RRDE) providing a Ni ring.92 These elevated potentials yielded traces of the same products also when Pt-based materials were used.93,94 This verifies that in a potential range of both AOR and OER, mixed oxidized products NzOx can be observed.94 In acidic solution, also NO˙ is detected at elevated oxidation potentials on Pt.95 Following the RRDE investigation described above, Ni/Ni(OH)2 was employed yielding NO3− and a non-specified mixture of N2 and NOx. Additionally, a strong pH dependence of the reaction occurred indicating the effective oxidation of NH3 and not soluble NH4+ species at elevated pH.96 Furthermore, NH3 oxidation on boron-doped diamond (BDD) was studied with the unexpected result that, unlike most organics, the oxidation does not proceed via hydroxyl radicals giving access to NO3− and NO2˙ in different concentrations with respect to the reaction conditions.97 Recently, the Klinkova group reported Ni(OH)2 to catalyse the AOR with current efficiencies of either 72% to NO3− or 60% to NO2− through slight changes in reaction conditions.98 Adaptive catalytic systems of this kind are particularly interesting for decentralised, flexible production. To summarize, AOR yields NOx, NO2−, and NO3− at potentials where parallel OER is observed. Here, NH2OH is involved instead of NHy species which are intermediates in N2 formation.36 AOR is therefore a possible starting point for electrochemical nitration of organic compounds. For full electrification of the process, ammonia might be used that was produced electrochemically.
As the electrochemical ammonia production remains challenging,13–15,99 this step might be circumvented, by using N2 gas directly as the source of nitrating agents. Also, oxidative pathways to activate molecular nitrogen have been reported. Like in AOR, the competition to OER is the main restriction. The direct electrochemical oxidation of N2 to NO3− bypasses not only energy-intensive Haber–Bosch but also the subsequent Ostwald process and can therefore be considered a dream reaction.89 In an experimental study from 2019 by Wang et al., the electrochemical synthesis of NO3− from N2 on Pt was achieved with a faradaic efficiency of around 1% and a maximum productivity of 0.06 μmol h−1 cm−2.55 The proposed mechanism proceeds via surface bound *NO which desorbs from the surface to form HNO3 and HNO2 in aqueous, oxygen containing environment (Fig. 6).
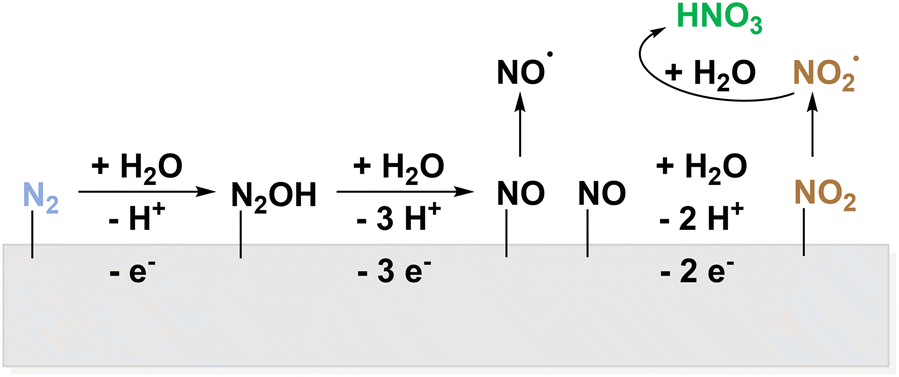 | ||
| Fig. 6 Electrochemical activation of N2via surface bound *N2OH and *NO to yield NO2˙. | ||
Also, consecutive oxidation to *NO2 before desorption and formation of HNO3 as depicted in Fig. 7 is discussed.55
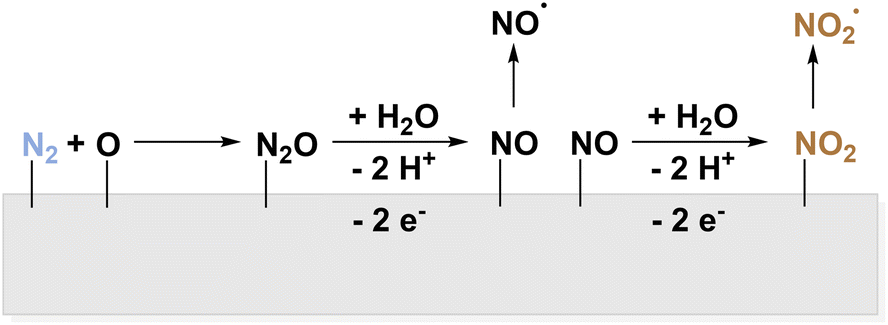 | ||
| Fig. 7 Electrochemical activation of N2via surface bound *N2O and *NO to yield NO2˙. | ||
The role of *NO is supported by a computational study identifying the reaction of N2 to *N2O as the rate-determining step before further oxidation and dissociation.54 This is most likely to occur by the activation with *O and therefore preferably on metal oxides with weak *O adsorption. Additionally, strong N2 binding is necessary which is in line with experimental results for Ru-doped TiO2 showing a maximum faradaic efficiency of 26%.100 Other experimental studies using mixed metal oxides supplement this trend.101,102
The second reaction pathway in the computational study involves *N2OH as intermediate species while adsorption of OH on metal oxides impedes the activation of N2.54 However, under the addition of sulphate, this pathway is promoted on Rh nanoparticles. The study also confirmed the general presence of *NO and *NO2 in the NOR by in situ IR spectroscopy as well as NO˙ formation via gas analysis.103 The third possibility of direct N2 activation and bond cleavage proved to be unlikely for the investigated metal oxides.54 Another computational study compared pathways for OER and NOR for both a good (IrO2) and a poor (TiO2) OER catalyst in detail. In either case, OH is most likely to adsorb to the surface and N2 is not directly activated. Consecutively, *O is formed and reacts to N2O over TiO2 because of its decreased stability. Still, the reaction is energetically difficult, and the article cannot account for a complete description of possible oxidation pathways.104
Evaluation of electrochemical nitration
Different examples of electrochemical nitration of organic molecules were presented. The first study was published in 197143 when organic electrochemistry was a niche technique driven by scientific curiosity.19,20,105 In contrast, the most recent investigation from 2021 follows the principles of green chemistry and directly aims at technical applicability.49 Aside few similarities like aromatic substrates and using standard anode materials like Pt and graphite, the overall reaction conditions differ a lot as does the focus of the collected publications. Because of this very heterogenous literature base, we decided to select chemical yield as a common performance indicator to compare existing technologies as shown in Table 1. Depending on the reaction conditions employed (N-source, solvent, electrode material and electrolyte), maximum yields between 26–96% were obtained in the electrochemical nitration reaction. The performance of NH3 or N2 oxidation is evaluated for selected examples of the demonstrated literature.| N-Source | Y m (%) | Product | Solvent | pH | Electrode | Electrolyte | ref. | |
|---|---|---|---|---|---|---|---|---|
| a Very similar conditions and results were obtained from ref. 47 and 46. b Maximum current efficiency from two separate experiments under different conditions. c NO3− yield is given as production per catalyst and time. d The stoichiometric composition was not defined in 100 but rather Ru-doping of TiO2 in wt%. | ||||||||
| NO2˙/N2O4 | 26 ± 5 | Nitronaphthalene | MeCN | — | Pt | LiBF4 | 63 | |
| 59 | Nitronaphthalene | DCM | — | Pt | Bu4NPF6 | 64 | ||
| 75 | Nitronaphthalene | Sulfolane | — | Pt | — | 65 | ||
| NO2− | 92 | Aromatics/olefins | MeCN | — | Pt | AgNO2 | 67 | |
| 85 | Nitronaphthalene | MeCN | — | Pt | NaNO2 | 48 | ||
| 32 | Nitronaphthalenea | Water + surfactant | Neutral | Pt | NaNO2 + NaClO4 | 47 | ||
| 32 | Low | Pt | 46 | |||||
| 96 | Aromatics | MeCN + HFIP | Low | C | Bu4NNO2 | 49 | ||
| NO3− | 39 | Nitromesitylene | MeNO2 | — | C | Bu4NNO3 | 43 | |
| NH3 | >99 | N2 | Water | High | Pt | KOH | 53 | |
| 63b | N2 | Water | High | Pt | NaOH | 90 | ||
| 37b | NO3− | |||||||
| 11 | NO3− | Water | High | Ni/Ni(OH)2 | NaOH | 96 | ||
| 72b | NO3− | Water | High | Ni(OH)2 | K2SO4 | 98 | ||
| 60b | NO2− | NaOH | ||||||
| N2 | 0.06 | NO3−c | μmol cm−2 h−1 | Water | Neutral | Pt-foil | K2SO4 | 55 |
| 77.7 | μmol gcat−1 h−1 | High | Pd0.9Ru0.1O2 | KOH | 101 | |||
| 161.9 | μmol gcat−1 h−1 | Neutral | RurTitO2d |
Na2SO4 | 100 | |||
| 130 | μmol gcat−1 h−1 | High | ZnFe0.4Co1.6O4 | KOH | 102 | |||
With this comparison at hand, we evaluate the potential of the presented technologies based on particular advantages and disadvantages of the N-sources. As the two soluble sources, NO2− and NO3−, require very similar process design, they will be considered together in the following. The same applies to the novel nitrating agents NH3 and N2. To provide an easy approach to evaluating the different N-sources, a simple list of advantages and disadvantages is provided in Table 2.
| NO2˙/N2O4 | NO2−, NO3− | NH3, N2 | |
|---|---|---|---|
| + | Active species reported | Soluble N-source | Abundant (if NH3 is decentralised) |
| Reactions proceed via NO2˙ for every N-source | Active species reported | Design of sustainable production route | |
| − | Toxic, pressurized gas | Previous production of N-precursor | Active species not described yet |
| Solubility and multiphase system | Solubility and multiphase system | ||
Overall, we think that already existing pathways using NO2˙/N2O4 or soluble N-sources are promising and their rejuvenation should be pushed as demonstrated by the Waldvogel group. This will lead to a broader library of electrocatalysts and methods as well as mechanistically improved understanding of the electrochemical nitration using different N-sources. Thereby, more challenging activation mechanisms as for NH3 and N2 can be addressed by research to potentially find even more elegant and benign synthesis of organic nitro compounds.
Perspective
In recent years, the electrochemical oxidation of N2 was achieved to a certain extent and efforts were made to understand the reaction mechanism using in situ spectroscopy, online analytics, and DFT calculations. Nonetheless, the active species and rate-determining steps have not conclusively been identified and are, as is often the case, material dependent. The efficient conversion of N2 to (inorganic) bulk chemicals therefore remains challenging. An alternative is the activation of N2 to produce (surface) intermediates that can potentially react with organic molecules exposed to them in a nitration reaction to fine chemicals. A key step is the simultaneous production of reactive N- and C-species for C–N-bond formation. This was already demonstrated in numerous studies of electrochemical nitration during the last century and very recently in the context of sustainable electrocatalysis, i.e. urea synthesis. To activate NH3 or N2 for electrochemical nitration, three main challenges must be overcome: first, suitable electrode materials and geometries are essential to catalyse the reaction. Furthermore, to work at the gas–liquid interface, the design of an appropriate reactor is required. Finally, the reaction conditions should be optimized regarding yield and faradaic efficiency as well as a sufficiently broad substrate scope.Catalyst design
The most atom-efficient and environmentally benign C–N bond formation strategy is to use atmospheric nitrogen in an electrochemical nitration reaction although the mechanisms to activate nitrogen are challenging. Currently, an overview that summarizes the different N-sources and oxidation mechanisms, e.g. nitrogen and ammonia, is absent. For the AOR, platinum was discussed and provides access to NH2OH, NO2− and NO3− as well as different NzOx species. Other electrodes of major interest are Ni/Ni(OH)2 and BDD.96,97 The investigation of BDD also presented the opportunity to deliver ammonia in the form of NH4ClO4 matching the supporting electrolyte.97A comparable situation can be expected for N2 activation. Here, also noble metal catalysts or alloys were employed to access different NzOx intermediates.55,100,101 Additionally, a noble metal free spinel oxide was reported to be active.102 Based on this, mechanisms are proposed to capture the active nitrogen oxide species with organic aryls displayed in Fig. 8. The active nitrating agents are NO2˙, NO2−, and NO2+. The species detected in experimental studies and predicted with computational methods suggest that basically all conceivable radical or ionic nitrogen oxides are accessible from NH3 or N2. However, it should be investigated whether a species active for nitration must desorb from the catalyst surface before reacting with the organic molecules. Considering the reaction of NO2˙ and NO2−, parallel oxidation of the organic compound is necessary. To deepen the mechanistic understanding and rationalising the catalyst design, in situ spectroscopic insights can be very useful. Therefore, the already introduced ATR-SEIRAS103 should be considered alongside advanced electrochemical techniques like RRDE measurements92 for the development of new materials.
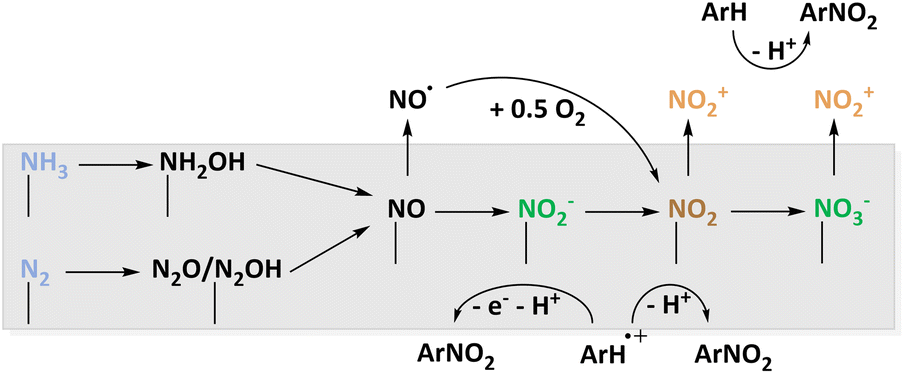 | ||
| Fig. 8 Proposed reaction sequence leading to different surface intermediates suitable for electrochemical nitration which are caught by organic molecules. | ||
Regarding the gaseous substrates NH3 and N2, the mass transport will be of major importance like in CO2 electrolysis.107,108 Also, suitable reaction conditions are sought to access nitrogen species active for nitration of aromatic substrates. An expected challenge is that AOR and NOR take place in aqueous media, while electrochemical nitration is usually performed in organic solvents. Here, surface modification of the electrode to tailor hydrophobicity109,110 may be an important tool in combination with the choice of solvent and electrolyte. As an interplay of hydroxide and nitrogen oxidation is mentioned in AOR and NOR literature, an aqueous reaction system may be inevitable.36,54,104 To deliver the organic substrate, also a surfactant could be utilized as demonstrated for nitration using NO2˙.47
Based on this, the research with a bulk Pt catalyst and the perspective of dispersed noble metal oxides with a final goal of (noble) metal free materials can be considered as potential direction to develop efficient electrode materials. Besides, after a proof of enhanced catalytic activity, the development of a gas diffusion electrode (GDE) geometry and surface modification to tailor the solid–gas–liquid three-phase interface will be necessary to improve the overall performance.
Reactor design
While development and testing of electrocatalysts is mostly performed in H-cells or even undivided 3-electrode cells, plant-scale setups demand for a different level of sophistication. As the interest in modular electrolysers for decentralised application is increasing, the development of cells gains importance. The envisioned process of electrochemical nitration using NH3 or N2 N-sources works at a gas–liquid interphase. While ammonia dissolves relatively easily in many aqueous electrolytes,111 the solubility of nitrogen in them is very low.112 Thus, mass transport is of utmost importance and is currently discussed for the NRR.113 Further inspiration may once again be found looking at developments for CO2RR. Generally, (micro)flow electrolysers and membrane electrode assemblies (MEA) are used in CO2RR.114,115 Flow electrolysers enhance the mass transport using gas-diffusion electrodes trough which a gaseous substrate is brought in contact with the liquid electrolyte. Material properties like pore dimension and wetting behaviour allow for a tailored mass transport and high current densities. MEAs are derived from fuel cell technology in which the electrocatalyst is in direct contact with the membrane. Also here, GDEs are employed but the electrolyte may be humidified CO2 gas in case of the CO2RR.115 For the rapid-prototyping of electrochemical reactors, additive manufacturing techniques recently gain importance.116–120 Such 3D-printed reactors are also tested in CO2RR, although different challenges like chemical tolerance of the materials or tightness occur.118,121Therefore, to start, reliable catalytic activity should be demonstrated in an analytical H-cell reactor configuration. The electrosynthesis can then directly be transferred to a flow electrolyser allowing the use of GDEs.
Optimisation
As for any electrochemical system, a rigorous screening study of reaction conditions such as solvent, electrolyte, pH, current density, and potential is necessary. In the vision of a sustainable process, the solvent of choice is water which is common to electrochemistry and already discussed for the oxidation of NH3 or N2. Nonetheless, literature on electrochemical nitration is built around organic solvents because of the solubility of the organic substrates. In electrochemistry, often also mixed solvents improve the reaction. A suitable electrolyte enables electric conductivity without interfering with the actual reaction, which is why strong acids or bases are often desired and N-containing molecules should be circumvented in this process to control the actual N-source. Furthermore, it can mediate the solubility of aromatic substrates. The pH must be adapted to the electrode material and allows certain control over OER as major side reaction. While noble metals are stable over a broad range of pH, the use of other materials may require rigorous pH control. Finally, current density and potential are crucial to control selectivity, mass transport, and productivity.Hence, we expect that an aqueous system and emulsifying electrolyte at elevated pH can enable electrochemical nitration using NH 3 or N 2 . Still, both gases show descent solubility in organic solvents122–124so that conditions closer to the original literature on electrochemical nitration can be chosen. Based on industrially established electrochemical processes for organic fine chemicals,125a current density of 10–40 mA cm−2is desirable to be productive and should be targeted when tailoring the catalyst, cell, and conditions.
Additionally, to understand the selectivity of the envisioned electrochemical nitration process, a broad substrate scope should be considered. This includes typical examples of nitration substrates such as naphthalene or mesitylene as well as functionalized aromatics to understand electronic effects. The tolerance of halogens, methoxy groups, and already multi-substituted aromatics forms a typical scope. Finally, for certain medicinal molecules it would be interesting to evaluate which impurities may result from an electrochemical process. For example, the antibiotic nitrofurantoin can be synthesised from 5-nitrofurfural126 which is an example for nitrofuranes127 accessible from the biogenic platform molecule furfural128,129via electrophilic nitration.
Author contributions
Nils Kurig: conceptualisation, formal analysis, visualisation, writing – original draft. Regina Palkovits: conceptualisation, resources, funding acquisition, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Part of this work was supported by the Cluster of Excellence Fuel Science Center (EXC 2186, ID: 390919832) funded by the Excellence Initiative by the German Federal and State Governments to promote science and research at German Universities. We also acknowledge funding by the Federal Ministry of Food and Agriculture granted by the Agency for Renewable Resources (FNR, 2220NR101X).References
- A. Hermann, Physikalische Blätter, 1965, 21, 168–171 CrossRef.
- P. J. Fischer and F. E. Kühn, Chem. Unserer Zeit, 2019, 53, 112–124 CrossRef CAS.
- V. Kyriakou, I. Garagounis, A. Vourros, E. Vasileiou and M. Stoukides, Joule, 2020, 4, 142–158 CrossRef CAS.
- J. Brightling, Johnson Matthey Technol. Rev., 2018, 62, 32–47 CrossRef CAS.
- C. Smith, A. K. Hill and L. Torrente-Murciano, Energy Environ. Sci., 2020, 13, 331–344 RSC.
- T. Grundt and K. Christiansen, Int. J. Hydrogen Energy, 1982, 7, 247–257 CrossRef CAS.
- B. A. Frontana-Uribe, R. D. Little, J. G. Ibanez, A. Palma and R. Vasquez-Medrano, Green Chem., 2010, 12, 2099–2119 RSC.
- F. J. Holzhäuser, J. B. Mensah and R. Palkovits, Green Chem., 2020, 22, 286–301 RSC.
- M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230–13319 CrossRef CAS PubMed.
- S. R. Waldvogel, S. Möhle, M. Zirbes, E. Rodrigo, T. Gieshoff and A. Wiebe, Angew. Chem., 2018, 130, 2–28 CrossRef.
- A. Wiebe, T. Gieshoff, S. Möhle, E. Rodrigo, M. Zirbes and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 5594–5619 CrossRef CAS PubMed.
- M. Capdevila-Cortada, Nat. Catal., 2019, 2, 1055–1055 CrossRef.
- J. Kibsgaard, J. K. Nørskov and I. Chorkendorff, ACS Energy Lett., 2019, 4, 2986–2988 CrossRef CAS.
- B. Yang, W. Ding, H. Zhang and S. Zhang, Energy Environ. Sci., 2021, 14, 672–687 RSC.
- J. Choi, B. H. R. Suryanto, D. Wang, H.-L. Du, R. Y. Hodgetts, F. M. Ferrero Vallana, D. R. MacFarlane and A. N. Simonov, Nat. Commun., 2020, 11, 5546 CrossRef CAS PubMed.
- H. Iriawan, S. Z. Andersen, X. Zhang, B. M. Comer, J. Barrio, P. Chen, A. J. Medford, I. E. L. Stephens, I. Chorkendorff and Y. Shao-Horn, Nat. Rev. Methods Primers, 2021, 1, 56 CrossRef CAS.
- M. J. Orella, Y. Román-Leshkov and F. R. Brushett, Curr. Opin. Chem. Eng., 2018, 20, 159–167 CrossRef.
- S. Palkovits and R. Palkovits, Chem. Ing. Tech., 2019, 91, 699–706 CrossRef CAS.
- Y. Kawamata and P. S. Baran, Joule, 2020, 4, 701–704 CrossRef.
- M. Yan, Y. Kawamata and P. S. Baran, Angew. Chem., Int. Ed., 2018, 57, 4149–4155 CrossRef CAS PubMed.
- B. Zhang, Y. Gao, Y. Hioki, M. S. Oderinde, J. X. Qiao, K. X. Rodriguez, H.-J. Zhang, Y. Kawamata and P. S. Baran, Nature, 2022, 606, 313–318 CrossRef CAS PubMed.
- F. Harnisch and U. Schröder, ChemElectroChem, 2019, 6, 4126–4133 CrossRef CAS.
- H. G. Cha and K.-S. Choi, Nat. Chem., 2015, 7, 328–333 CrossRef CAS PubMed.
- Y. Kwon, S. E. F. Kleijn, K. J. P. Schouten and M. T. M. Koper, ChemSusChem, 2012, 5, 1935–1943 CrossRef CAS PubMed.
- S. Yang, A. Verdaguer-Casadevall, L. Arnarson, L. Silvioli, V. Čolić, R. Frydendal, J. Rossmeisl, I. Chorkendorff and I. E. L. Stephens, ACS Catal., 2018, 8, 4064–4081 CrossRef CAS.
- K.-S. Ju and R. E. Parales, Microbiol. Mol. Biol. Rev., 2010, 74, 250–272 CrossRef CAS PubMed.
- S. D. Mürtz, N. Kurig, F. J. Holzhäuser and R. Palkovits, Green Chem., 2021, 23, 8428–8433 RSC.
- S. Kasemthaveechok and N. von Wolff, DOI:10.26434/chemrxiv-2022-n306b-v2.
- M. Quertenmont, I. Goodall, K. Lam, I. Markó and O. Riant, Org. Lett., 2020, 22, 1771–1775 CrossRef CAS PubMed.
- J. He, L. Chen, S. Liu, K. Song, S. Yang and A. Riisager, Green Chem., 2020, 22, 6714–6747 RSC.
- T. Fukushima and M. Yamauchi, Chem. Commun., 2019, 55, 14721–14724 RSC.
- X. Shao, Y. Zheng, L. Tian, I. Martín-Torres, A. M. Echavarren and Y. Wang, Org. Lett., 2019, 21, 9262–9267 CrossRef CAS PubMed.
- Y. Xiao, C. W. Lim, J. Chang, Q. Yuan, L. Wang and N. Yan, Green Chem., 2023, 25, 3117–3126 RSC.
- M. D. Kärkäs, Chem. Soc. Rev., 2018, 47, 5786–5865 RSC.
- V. Rosca, M. Duca, M. T. DeGroot and M. T. M. Koper, Chem. Rev., 2009, 109, 2209–2244 CrossRef CAS PubMed.
- N. J. Bunce and D. Bejan, Electrochim. Acta, 2011, 56, 8085–8093 CrossRef CAS.
- C. Tang and S. Z. Qiao, Chem. Soc. Rev., 2019, 48, 3166–3180 RSC.
- S. L. Foster, S. I. P. Bakovic, R. D. Duda, S. Maheshwari, R. D. Milton, S. D. Minteer, M. J. Janik, J. N. Renner and L. F. Greenlee, Nat. Catal., 2018, 1, 490–500 CrossRef.
- X. Guo, H. Du, F. Qu and J. Li, J. Mater. Chem. A, 2019, 7, 3531–3543 RSC.
- X. Chen, Y. Guo, X. Du, Y. Zeng, J. Chu, C. Gong, J. Huang, C. Fan, X. Wang and J. Xiong, Adv. Energy Mater., 2020, 10, 1903172 CrossRef CAS.
- P. Pollak, Fine Chemicals The Industry and the Business, 1st edn, 2007 Search PubMed.
- J. Seidler, J. Strugatchi, T. Gärtner and S. R. Waldvogel, MRS Energy Sustain., 2020, 7, 42 CrossRef.
- K. Nyberg, A.-B. Hörnfeldt, J. J. Lindberg, H. Okinaka, K. Kosuge and S. Kachi, Acta Chem. Scand., 1971, 25, 3246–3254 CrossRef CAS.
- C. L. Perrin, J. Am. Chem. Soc., 1977, 99, 5516–5518 CrossRef CAS.
- G. A. Olah, Acc. Chem. Res., 1971, 4, 240–248 CrossRef CAS.
- M. N. Cortona, N. R. Vettorazzi, J. J. Silber and L. E. Sereno, J. Electroanal. Chem., 1999, 470, 157–165 CrossRef CAS.
- M. N. Cortona, N. R. Vettorazzi, J. J. Silber and L. E. Sereno, J. Braz. Chem. Soc., 1997, 8, 377–382 CrossRef CAS.
- M. N. Cortona, N. Vettorazzi, J. J. Silber and L. Sereno, J. Electroanal. Chem., 1995, 394, 245–251 CrossRef.
- S. P. Blum, C. Nickel, L. Schäffer, T. Karakaya and S. R. Waldvogel, ChemSusChem, 2021, 14, 4936–4940 CrossRef CAS PubMed.
- Y. Feng, H. Yang, Y. Zhang, X. Huang, L. Li, T. Cheng and Q. Shao, Nano Lett., 2020, 20, 8282–8289 CrossRef CAS PubMed.
- C. Chen, X. Zhu, X. Wen, Y. Zhou, L. Zhou, H. Li, L. Tao, Q. Li, S. Du, T. Liu, D. Yan, C. Xie, Y. Zou, Y. Wang, R. Chen, J. Huo, Y. Li, J. Cheng, H. Su, X. Zhao, W. Cheng, Q. Liu, H. Lin, J. Luo, J. Chen, M. Dong, K. Cheng, C. Li and S. Wang, Nat. Chem., 2020, 12, 717–724 CrossRef CAS PubMed.
- F. Fichter, Z. Elektrochem. Angew. Phys. Chem., 1912, 18, 647–654 Search PubMed.
- H. Gerischer and A. Mauerer, J. Electroanal. Chem. Interfacial Electrochem., 1970, 25, 421–433 CrossRef CAS.
- H. Wan, A. Bagger and J. Rossmeisl, J. Phys. Chem. Lett., 2022, 13, 8928–8934 CrossRef CAS PubMed.
- Y. Wang, Y. Yu, R. Jia, C. Zhang and B. Zhang, Natl. Sci. Rev., 2019, 6, 730–738 CrossRef CAS PubMed.
- A. A. Kulkarni, Beilstein J. Org. Chem., 2014, 10, 405–424 CrossRef PubMed.
- G. Yan and M. Yang, Org. Biomol. Chem., 2013, 11, 2554–2566 RSC.
- G. R. Maxwell, Synthetic Nitrogen Products, Kluwer Academic Publishers, Boston, 2005 Search PubMed.
- C. K. Ingold, Structure and mechanism in organic chemistry, Cornell University Press, Ithaca, 2nd edn, 1969, vol. 2 Search PubMed.
- S. R. Hartshorn, R. B. Moodie, K. Schofield and M. J. Thompson, J. Chem. Soc. B, 1971, 2447 RSC.
- L. M. Stock and H. C. Brown, in Advances in Physical Organic Chemistry, 1963, pp. 35–154 Search PubMed.
- G. A. Olah, S. C. Narang and J. A. Olah, Proc. Natl. Acad. Sci. U. S. A., 1981, 78, 3298–3300 CrossRef CAS PubMed.
- L. Eberson, L. Jönsson, F. Radner, M. Sponholtz and T. Matsuno, Acta Chem. Scand., 1978, 32b, 749–753 CrossRef.
- L. Eberson, F. Radner, V. Sundström, S.-K. Kan and M. Lounasmaa, Acta Chem. Scand., 1980, 34b, 739–745 CrossRef.
- A. Boughriet, C. Bremard and M. Wartel, J. Electroanal. Chem. Interfacial Electrochem., 1987, 225, 125–137 CrossRef CAS.
- J. M. Achord and C. L. Hussey, J. Electrochem. Soc., 1981, 128, 2556–2561 CrossRef CAS.
- A. Laurent, E. Laurent and P. Locher, Electrochim. Acta, 1975, 20, 857–862 CrossRef CAS.
- Y. Wang, S. You, M. Ruan, F. Wang, C. Ma, C. Lu, G. Yang, Z. Chen and M. Gao, Eur. J. Org. Chem., 2021, 3289–3293 CrossRef CAS.
- J. P. Zhao, L. Ding, P. C. Wang, Y. Liu, M. J. Huang, X. L. Zhou and M. Lu, Adv. Synth. Catal., 2020, 362, 5036–5043 CrossRef CAS.
- M. Gong and J. M. Huang, Chem. – Eur. J., 2016, 22, 14293–14296 CrossRef CAS PubMed.
- H. C. Xu and K. D. Moeller, J. Am. Chem. Soc., 2008, 130, 13542–13543 CrossRef CAS PubMed.
- H. C. Xu and K. D. Moeller, Org. Lett., 2010, 12, 5174–5177 CrossRef CAS PubMed.
- T. Morofuji, A. Shimizu and J. I. Yoshida, J. Am. Chem. Soc., 2013, 135, 5000–5003 CrossRef CAS PubMed.
- S. R. Waldvogel and S. Möhle, Angew. Chem., Int. Ed., 2015, 54, 6398–6399 CrossRef CAS PubMed.
- S. Liang, C. C. Zeng, H. Y. Tian, B. G. Sun, X. G. Luo and F. Z. Ren, J. Org. Chem., 2016, 81, 11565–11573 CrossRef CAS PubMed.
- Y. Zhong, H. Xiong, J. Low, R. Long and Y. Xiong, eScience, 2023, 3, 100086 CrossRef.
- J. E. Kim, S. Choi, M. Balamurugan, J. H. Jang and K. T. Nam, Trends Chem., 2020, 2, 1004–1019 CrossRef CAS.
- J. Li, Y. Zhang, K. Kuruvinashetti and N. Kornienko, Nat. Rev. Chem., 2022, 6, 303–319 CrossRef CAS PubMed.
- K. Kuruvinashetti, J. Li, Y. Zhang, H. Bemana, M. McKee and N. Kornienko, Chem. Phys. Rev., 2022, 3, 021306 CrossRef CAS.
- J. Qin, N. Liu, L. Chen, K. Wu, Q. Zhao, B. Liu and Z. Ye, ACS Sustainable Chem. Eng., 2022, 10, 15869–15875 CrossRef CAS.
- J. Geng, S. Ji, M. Jin, C. Zhang, M. Xu, G. Wang, C. Liang and H. Zhang, Angew. Chem., Int. Ed., 2023, 62, e202210958 CrossRef CAS PubMed.
- N. Meng, X. Ma, C. Wang, Y. Wang, R. Yang, J. Shao, Y. Huang, Y. Xu, B. Zhang and Y. Yu, ACS Nano, 2022, 16, 9095–9104 CrossRef CAS PubMed.
- M. Shibata and N. Furuya, J. Electroanal. Chem., 2001, 507, 177–184 CrossRef CAS.
- N. Meng, Y. Huang, Y. Liu, Y. Yu and B. Zhang, Cell Rep. Phys. Sci., 2021, 2, 100378 CrossRef CAS.
- Y. Huang, R. Yang, C. Wang, N. Meng, Y. Shi, Y. Yu and B. Zhang, ACS Energy Lett., 2022, 7, 284–291 CrossRef CAS.
- M. B. Ross, P. De Luna, Y. Li, C.-T. Dinh, D. Kim, P. Yang and E. H. Sargent, Nat. Catal., 2019, 2, 648–658 CrossRef CAS.
- M. Jiang, M. Zhu, M. Wang, Y. He, X. Luo, C. Wu, L. Zhang and Z. Jin, ACS Nano, 2023, 17, 3209–3224 CrossRef CAS PubMed.
- M. Muthuvel and G. G. Botte, Trends in Ammonia Electrolysis, in Modern Aspects of Electrochemistry 45, ed. R. E. White, Springer, New York, NY, 2009, pp. 207–245 Search PubMed.
- V. Rosca, M. Duca, M. T. DeGroot and M. T. M. Koper, Chem. Rev., 2009, 109, 2209–2244 CrossRef CAS PubMed.
- E. Müller and F. Spitzer, Z. Elektrotech. Elektrochem., 1905, 11, 917–931 CrossRef.
- W. Traube and A. Biltz, Ber. Dtsch. Chem. Ges., 1904, 37, 3130–3138 CrossRef CAS.
- K. Endo, Y. Katayama and T. Miura, Electrochim. Acta, 2005, 50, 2181–2185 CrossRef CAS.
- J. F. E. Gootzen, A. H. Wonders, W. Visscher, R. A. van Santen and J. A. R. van Veen, Electrochim. Acta, 1998, 43, 1851–1861 CrossRef CAS.
- S. Wasmus, E. J. Vasini, M. Krausa, H. T. Mishima and W. Vielstich, Electrochim. Acta, 1994, 39, 23–31 CrossRef CAS.
- R. Halseid, J. S. Wainright, R. F. Savinell and R. Tunold, J. Electrochem. Soc., 2007, 154, B263 CrossRef CAS.
- A. Kapałka, A. Cally, S. Neodo, C. Comninellis, M. Wächter and K. M. Udert, Electrochem. Commun., 2010, 12, 18–21 CrossRef.
- N.-L. Michels, A. Kapałka, A. A. Abd-El-Latif, H. Baltruschat and C. Comninellis, Electrochem. Commun., 2010, 12, 1199–1202 CrossRef CAS.
- J. J. Medvedev, Y. Tobolovskaya, X. V. Medvedeva, S. W. Tatarchuk, F. Li and A. Klinkova, Green Chem., 2022, 24, 1578–1589 RSC.
- I. McPherson and J. Zhang, Joule, 2020, 4, 12–14 CrossRef.
- M. Kuang, Y. Wang, W. Fang, H. Tan, M. Chen, J. Yao, C. Liu, J. Xu, K. Zhou, Q. Yan, M. Kuang, W. Fang, H. Tan, M. Chen, J. Yao, Q. Yan, Y. Wang, K. Zhou, C. Liu and J. Xu, Adv. Mater., 2020, 32, 2002189 CrossRef CAS PubMed.
- T. Li, S. Han, C. Wang, Y. Huang, Y. Wang, Y. Yu and B. Zhang, ACS Catal., 2021, 11, 14032–14037 CrossRef CAS.
- C. Dai, Y. Sun, G. Chen, A. C. Fisher and Z. J. Xu, Angew. Chem., Int. Ed., 2020, 59, 9418–9422 CrossRef CAS PubMed.
- T. Li, S. Han, C. Cheng, Y. Wang, X. Du, Y. Yu and B. Zhang, Angew. Chem., 2022, 134, e202204541 CrossRef.
- M. Anand, C. S. Abraham and J. K. Nørskov, Chem. Sci., 2021, 12, 6442–6448 RSC.
- C. Kingston, M. D. Palkowitz, Y. Takahira, J. C. Vantourout, B. K. Peters, Y. Kawamata and P. S. Baran, Acc. Chem. Res., 2020, 53, 72–83 CrossRef CAS PubMed.
- L. R. Snyder, J. Chromatogr. Sci., 1978, 16, 223–234 CAS.
- Z. Xing, K. Shi, X. Hu and X. Feng, J. Energy Chem., 2022, 66, 45–51 CrossRef CAS.
- B. J. M. Etzold, U. Krewer, S. Thiele, A. Dreizler, E. Klemm and T. Turek, Chem. Eng. J., 2021, 424, 130501 CrossRef CAS.
- Y. Peng, Y. Ning, X. Ma, Y. Zhu, S. Yang, B. Su, K. Liu, L. Jiang, Y. Peng, Y. Ning, S. Yang, K. Liu, L. Jiang, X. Ma, Y. Zhu, B. Su and L. Jiang Beijing, Adv. Funct. Mater., 2018, 28, 1800712 CrossRef.
- M. Röhe, D. Franzen, F. Kubannek, B. Ellendorff, T. Turek and U. Krewer, Electrochim. Acta, 2021, 389, 138693 CrossRef.
- S. L. Clegg and P. Brimblecombe, J. Phys. Chem., 1989, 93, 7237–7248 CrossRef CAS.
- H. Shen, C. Choi, J. Masa, X. Li, J. Qiu, Y. Jung and Z. Sun, Chem, 2021, 7, 1708–1754 CAS.
- W. Bi, N. Shaigan, A. Malek, K. Fatih, E. Gyenge and D. P. Wilkinson, Energy Environ. Sci., 2022, 15, 2259–2287 RSC.
- D. M. Weekes, D. A. Salvatore, A. Reyes, A. Huang and C. P. Berlinguette, Acc. Chem. Res., 2018, 51, 910–918 CrossRef CAS PubMed.
- U. O. Nwabara, E. R. Cofell, S. Verma, E. Negro and P. J. A. Kenis, ChemSusChem, 2020, 13, 855–875 CrossRef CAS PubMed.
- M. P. Browne, E. Redondo and M. Pumera, Chem. Rev., 2020, 120, 2783–2810 CrossRef CAS PubMed.
- M. Cheng, R. Deivanayagam and R. Shahbazian-Yassar, Batteries Supercaps, 2020, 3, 130–146 CrossRef CAS.
- N. Kurig, J. Meyers, E. Richter, S. Palkovits and R. Palkovits, Chem. Ing. Tech., 2022, 94, 786–790 CrossRef CAS.
- A. Ambrosi and M. Pumera, Chem. Soc. Rev., 2016, 45, 2740–2755 RSC.
- A. Ambrosi, R. R. S. Shi and R. D. Webster, J. Mater. Chem. A, 2020, 8, 21902–21929 RSC.
- A. B. Navarro, A. Nogalska and R. Garcia-Valls, Membranes, 2023, 13, 90 CrossRef CAS PubMed.
- R. Battino, T. R. Rettich and T. Tominaga, J. Phys. Chem. Ref. Data, 1984, 13, 563–600 CrossRef CAS.
- M. C. Rehbein, C. Meier, P. Eilts and S. Scholl, Energy Fuels, 2019, 33, 10331–10342 CrossRef CAS.
- I. Short, A. Sahgal and W. Hayduk, J. Chem. Eng. Data, 1983, 28, 63–66 CrossRef CAS.
- D. S. P. Cardoso, B. Šljukić, D. M. F. Santos and C. A. C. Sequeira, Org. Process Res. Dev., 2017, 21, 1213–1226 CrossRef CAS.
- E. Colacino, A. Porcheddu, I. Halasz, C. Charnay, F. Delogu, R. Guerra and J. Fullenwarth, Green Chem., 2018, 20, 2973–2977 RSC.
- D. R. Guay, Drugs, 2001, 61, 353–364 CrossRef CAS PubMed.
- Shin Nippon Biomedical Lab Ltd, EP 3018125A1, 2016.
- Nanjing Ruosai Pharmaceutical Tech Co Ltd, CN 108003031A, 2018.
| This journal is © The Royal Society of Chemistry 2023 |