
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Selective demethylation reactions of biomass-derived aromatic ether polymers for bio-based lignin chemicals
Florian M.
Harth
 a,
Brigita
Hočevar
a,
Tina Ročnik
Kozmelj
a,
Edita
Jasiukaitytė-Grojzdek
a,
Jana
Blüm
b,
Michael
Fiedel
b,
Blaž
Likozar
*a and
Miha
Grilc
*a
a,
Brigita
Hočevar
a,
Tina Ročnik
Kozmelj
a,
Edita
Jasiukaitytė-Grojzdek
a,
Jana
Blüm
b,
Michael
Fiedel
b,
Blaž
Likozar
*a and
Miha
Grilc
*a
aDepartment of Catalysis and Chemical Reaction Engineering, National Institute of Chemistry, Hajdrihova 19, 1000 Ljubljana, Slovenia. E-mail: blaz.likozar@ki.si; miha.grilc@ki.si; Tel: +386 1 4760 281 Tel: +386 1 4760 540
bEvonik Operations GmbH, Goldschmidtstraße 100, 45127 Essen, Germany
First published on 25th October 2023
Abstract
The selective demethylation of aryl methyl ethers is a reaction of high relevance both in the context of deprotection in organic syntheses as well as in the modification of lignin and lignin-derived chemicals. The resulting vicinal diol or triol configurations originating from guaiacyl or synapyl lignin moieties are particularly relevant for adhesive properties, while other positions are applicable for condensation polymerization. Throughout the years, a plethora of methods have been developed to efficiently cleave the particularly stable aryl methyl bond. This review provides, on the one hand, a comprehensive overview of conventional approaches associated with the field of organic chemistry, with a focus on recently developed methods with a focus on aspects of green chemistry. On the other hand, acid- and base-catalyzed as well as (hydro)thermal methods are discussed as well as the occurrence of demethylation reactions under metal-catalyzed hydrotreatment conditions, which, however, proved to be the most environmentally friendly principle. While the different chemical approaches are first presented for the conversion of selected model compounds, a subsequent section presents the application of these methods to lignin. The selective cleavage of aryl methyl ether moieties of lignin to increase the amount of phenolic hydroxyl groups has recently gained considerable attention, and research can benefit from the emerging knowledge gained from the new approaches on model compound demethylation. In sum, we provide a multi-faceted and critical overview of demethylation strategies to obtain phenol and catechol moieties from the renewable lignin and related compounds.
1. Introduction and scope
Catechol (1,2-dihydroxybenzene) is an important chemical intermediate for the synthesis of pesticides, drugs and perfumes and serves as a polymerization inhibitor for polymer production. Catechol itself is used as a photographic developer, an analytical reagent and as an antioxidant.1 Furthermore, catechol and its derivatives are well suited as adhesion promoters due to their strong and versatile interaction with surfaces.2Despite being present as a common motif in nature, catechol and its derivatives are commercially produced together with hydroquinone by the direct hydroxylation of phenol with hydrogen peroxide in the presence of catalytic amounts of either acids or ferrocene-based catalysts.1 Since phenol is most commonly produced by the oxidation of benzene-derived cumene via the Hock process,3 the overall production of catechol and its derivatives heavily relies on the utilization of fossil resources.
A renewable alternative as a feedstock for phenolics as well as catechol and its derivatives is lignin. Being the underutilized fraction (between 1/6 and 1/3) of lignocellulosic biomass, lignin is a heterogeneous macromolecular compound consisting mainly of three monomer units. These are the guaiacyl, the syringyl and the p-hydroxyphenyl units. The most common one among them is the guaiacyl unit, making up 50 to 95% of lignin depending on its source.4–7 This structural unit consists of an aromatic moiety with one hydroxyl group and one methoxy group in the neighboring ortho position. Selectively demethylating the methoxy group results in the desired catechol motif. In the simplest case, catechol can in this way be derived from guaiacol. Similarly, 4-allyl(pyro)catechol can be obtained via the demethylation of eugenol, which is commercially available as the main component (≥50%) of clove oils.8,9 Therefore, O-demethylation is a crucial reaction to obtain catechol-based structures from renewable plant-based feedstock.10 Overall, demethylation of lignin is a means to increase the amount of phenolic hydroxyl groups, which is desirable for adhesion11,12 or polymer applications.12,13
Several strategies and methods have been developed to selectively cleave aryl ethers, which are the subject of comprehensive review articles from the field of organic chemistry.14–16 In this context, the methylation of phenolic hydroxyl groups is a common strategy to protect this functional group, which then requires efficient demethylation procedures for subsequent deprotection. A common finding is that in particular aryl methyl ether bonds, which are present e.g. in guaiacol, anisole or eugenol, are highly stable.17,18 This makes their cleavage inherently challenging, especially when other functional groups need to be preserved.
This literature review aims at providing a comprehensive overview over a broad range of demethylation methods for aryl methyl ethers. It is divided into three main sections representing different classes of demethylation approach, which will be assessed regarding their previous usage for different demethylation applications, their strengths and weaknesses as well as aspects of green chemistry. The first section comprises different methods originating mostly from the field of organic chemistry that are commonly used for the production of pharmaceuticals and fine chemicals as well as in the total syntheses of complex molecules. The use of stoichiometric amounts of reactants is characteristic, as is typically high selectivity. These methods are mostly harmful to the environment and use chemicals that are harmful to health, so there is an urgent need to find alternative, more environmentally friendly methods. In this context, new developments regarding AlI3-based demethylation methods were reported in recent years. The second section includes comparatively simple, aqueous-phase methods based on either hydrolysis or acid-catalyzed demethylation. This includes studies under supercritical conditions as well as the utilization of different mineral acids and Lewis acids. The third section discusses demethylation under hydrotreatment conditions. Both non-catalytic as well as transition metal-catalyzed approaches are presented. Metal-catalyzed reactions are the most environmentally friendly of all, as they usually occur under milder reaction conditions and the catalyst can be easily recycled and reused. In terms of substrates, the first three sections of this review article are limited to the common lignin model compounds guaiacol, eugenol and similar functionalized guaiacols. Furthermore, biotechnological demethylation approaches19–24 are not considered. Eventually, in the final section the application of different methods from all the previous categories to lignin samples is discussed. Here, the focus moves away from the model compound-based discussion and the issue of product selectivity to the broader aspect of lignin defunctionalization and modification by demethylation.
2. Stoichiometric approaches in organic solvents
In this section different, demethylation methods that have emerged from the field of organic chemistry are presented, with a focus on guaiacol and substituted 1-hydroxy-2-methoxybenzene compounds. Approaches discussed here have in common that they are conducted in organic solvents and require stoichiometric or excess amounts of reactants. The most common methods are based on Lewis acids (BBr3, AlX3), oxidizing agents (often iodine-containing) or nucleophiles. Almost all approaches presented show good to excellent product yield and high tolerance towards other functional groups. Among them, however, AlI3-based approaches with acid scavengers (section 2.2.2.) and a method using 2-iodoxybenzoic acid as oxidizing agent followed by hydrolysis and mild reduction (section 2.5.1.) appear especially promising. Moreover, safety, health and environmental aspects are discussed qualitatively, primarily based on the classification of the chemicals used in each method using safety data sheet information.2.1. Boron-based Lewis acids
One recent example for a BBr3 demethylation of a guaiacol derivative is that reported in ref. 27. 2-Bromo-6-methoxyphenol was dissolved in anhydrous dichloromethane and 1 equivalent of BBr3 was added slowly at 0 °C. The mixture was stirred overnight at room temperature before 10% aqueous NaHCO3 was added. After extraction and purification, a 98% yield of 3-bromobenzen-1,2-diol was obtained.27
In a study by Bovicelli et al.,28 BBr3 was used for the demethylation of homovanillyl alcohol (4-(2-hydroxyethyl)-2-methoxyphenol; (1)), which has a hydroxyethyl substituent (Fig. 1). The primary alcohol (as well as the phenol-OH) had to be acetylated prior to the demethylation to avoid undesired bromination. During the reaction with BBr3, which was conducted in dichloromethane at −20 °C, both demethylation and deacetylation of the hydroxybenzene groups take place. With 1.5 eq. BBr3, hydroxytyrosyl acetate (3) was obtained in yields of 80% after 2.5 hours of reaction.
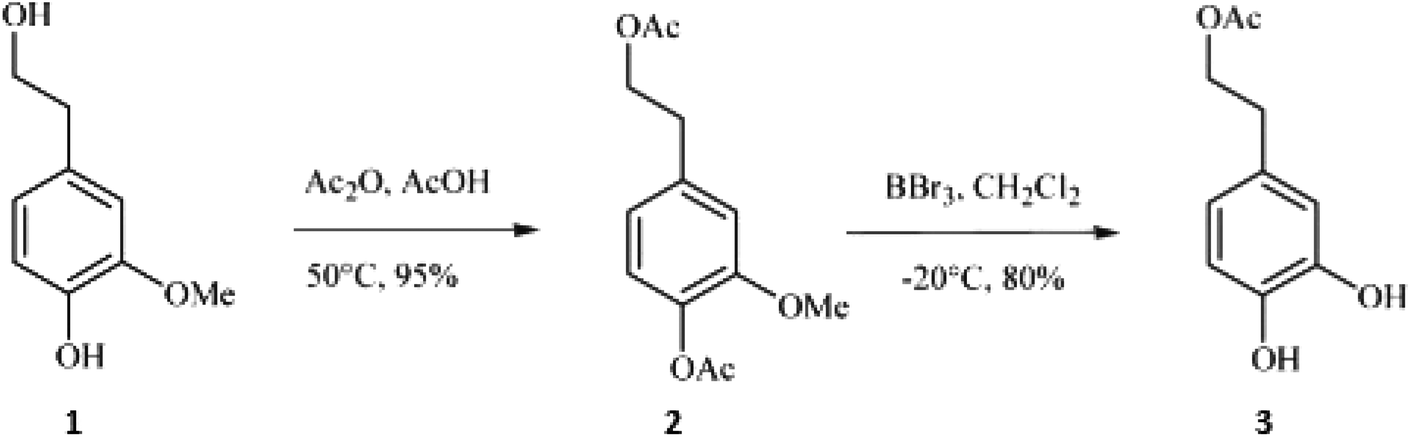 | ||
| Fig. 1 Reaction sequence consisting of protection of the primary aliphatic alcohol by acetylation followed by aryl methyl cleavage with Lewis acid BBr3.28 | ||
In recent years, the mechanism of the ether cleavage reaction with BBr3 has attracted research attention. Both a uni- and a bimolecular mechanism were discussed,29,30 which is beyond the scope of this review article. DFT calculations showed that one 1 eq. of BBr3 can demethylate 3 eq. of methyl aryl ethers.30 In typical experimental procedures, however, excess amounts are used.
The BBr3 method can be modified e.g. by using the BBr3·S(CH3)2 complex or by using other boron trihalides as has been discussed in previous review articles.14,15 While the use of BBr3 enables the successful demethylation of guaiacol-type compounds under mild conditions it has to be noted that BBr3 is highly hazardous, it is toxic and it reacts violently with water and alcohols forming HBr, which also makes handling difficult.31 In addition, the reaction is currently tied to the use of DCM as solvent, which is considered a hazardous solvent.32 In sum, however, the BBr3-based approach, while highly efficient from a conventional perspective, is highly problematic from a green chemistry standpoint and should thus be avoided if suitable alternatives exist for the application in question.
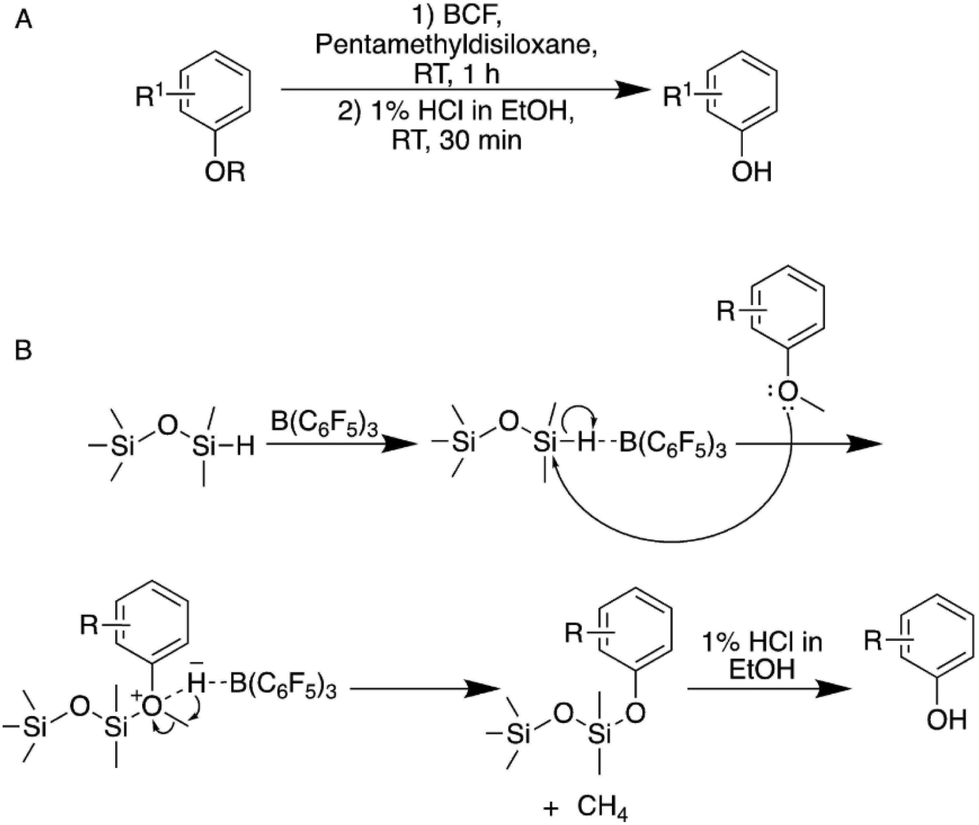 | ||
| Fig. 2 (A) Two-step demethylation approach via the Piers–Rubinsztajn reaction using tris(pentafluorophenyl)boran (BCF). (B) Mechanism of the Piers–Rubinsztajn demethylation reaction.33 | ||
Overall, the approach allows for the selective demethylation of differently substituted anisole derivatives in yields between 85 and 95% and it was shown that also complex guaiacol-type molecules could be demethylated selectively. It should be noted that both an allyl as well as a vinyl group could be tolerated without a significant decrease in selectivity when a polymerization inhibitor is added during the acidic work-up. Most importantly, BCF is stable in air and moisture and the reaction proceeds under ambient conditions in hexane, which makes it advantageous to e.g. the aforementioned BBr3-based methods.33 While using BCF rather than BBr3 presents a considerable advantage in terms of the health and safety aspects of the reaction, the use of hexane as solvent is highly problematic as the alkane is classified among the most harmful nonpolar solvents using green chemistry metrics.32
2.2. Aluminium halides
One recent example where the AlCl3 approach was applied to a guaiacol derivative is a study by Arifin et al.37 They selectively demethylated clove oil-derived eugenol to hydroxychavicol using 2.5 eq. AlCl3 and excess dimethyl sulfide in dichloromethane at 0 °C. After 24 hours reaction time the formed reaction intermediate was hydrolyzed by aqueous HCl to yield the demethylated product in yields around 30%. However, the authors argue that the reaction conditions chosen (0 °C, nitrogen atmosphere) prevent side reactions like e.g. double bond isomerization.37
In general, the use of AlCl3 bears the hazard of working with a corrosive compound that reacts violently with water, as applies to AlBr3 and AlI3. Moreover, the use of thiols is particularly problematic since, e.g. in the case of ethanethiol, the flammability as well as ecotoxicity, causing risks of severe environmental impacts, must be highlighted. Other thiols such as dimethyl sulfide are less harmful but strongly odorous, and thus should be avoided as well if possible.
Deffieux et al.39 tried to apply the reaction to eugenol but found that the allyl group was hydrogenated during the demethylation reaction with Al3. Instead of hydroxychavicol, an 81% yield of hydrogenated 1,2-dihydroxy-4-propylbenzene was obtained from eugenol after 0.5 hours with 2 eq. of AlI3 (freshly prepared) under reflux in cyclohexane. Besides, 2 eq. of tetrabutylammonium iodide were added following a procedure by Andersson et al.40
This study serves as a starting point for a series of studies published Sang and co-workers investigating the selective demethylation of eugenol and other guaiacol derivatives with AlI3.41–45 In their first article they investigated the mechanisms of allyl group hydrogenation.41 As shown in Fig. 3, they proposed that HI formed during the initial reaction of AlI3 with the phenolic OH group added to the allyl group via hydroiodination. Subsequent hydrodeiodination via AlI3-catalyzed hydrogen–halogen exchange was regarded as the origin of the hydrogenated products.
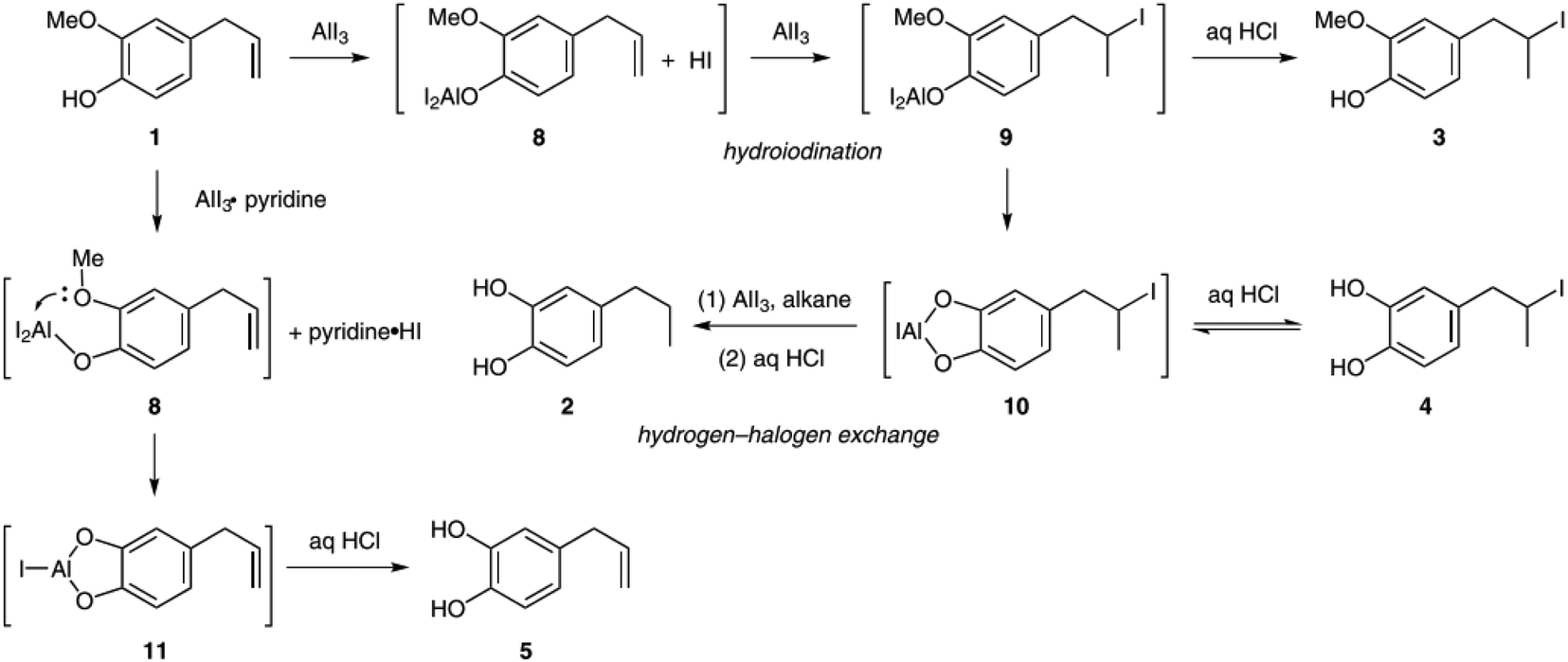 | ||
| Fig. 3 Mechanistic explanation of hydrogenation observed during the demethylation of eugenol with AlI3. The presence of the acid scavenger pyridine prevents the initial hydroiodination step, which results in the preservation of the allyl group.41 | ||
Having identified HI as the origin of the side product formation, different acid scavengers were used to suppress the Hydroiodination side reaction during the demethylation of eugenol with 1.1 eq. AlI3 in acetonitrile. While the initial investigation was based on pyridine (4.5 eq.) as acid scavenger, resulting in a 99% hydroxychavicol yield,41 similar results were obtained with 0.6 eq. carbodiimide.43 However, carbodiimide also allows for the complete demethylation of 1,2-dimethoxybenzene-type compounds whereas in the presence of pyridine no demethylation of such compounds is possible. In further articles inorganic bases and metal oxides (e.g. Na2CO3, CaO, CuO and ZnO; 1.5 eq.) were successfully used as acid scavengers,42 as well as dimethyl sulfoxide (1.1 eq.), which generally gave slightly lower hydroxychavicol yields (ca. 94%). Eventually, an up-scalable one-pot approach was developed, including the in situ formation of AlI3 from the elements, which yielded 75% of hydroxychchavicol (starting from 6 g of eugenol) after 18 hours at 80 °C in dichloromethane with dimethyl sulfoxide.45 Besides allyl groups, aldehyde, ketone or carboxylic acid functionalities can be tolerated, while other alkyl ether bonds can be cleaved as well.43
Among all approaches reported in this review the AlI3-based procedure using scavengers like pyridine or carbodiimide is the only one that gives an almost quantitative yield while tolerating the allyl group of eugenol as well as other functional groups. In combination with the comparatively accessible reactants, mild reaction conditions and its proven up-scaling it is probably the best-suited method for the demethylation of functionalized derivatives of guaiacol.
Considering additional factors, it needs to be highlighted that for the preparation of AlI3 required for this reaction, typically Al powder is used, which is a flammable solid, as well as elemental iodine, which poses considerable threats to human health as well as environmental hazards. While the solvent acetonitrile is less harmful than many of the aforementioned organic solvents used in other approaches, it is still to be considered problematic.32 In terms of acid scavengers, especially pyridine (toxic, flammable, environmental hazards) and carbodiimides such as N,N’-dicyclohexylcarbodiimide (toxic, known allergen and sensitizer) are very harmful and, from safety and environmental standpoints, the solid alternatives are by far preferable despite their lower efficiency. Dimethyl sulfoxide is also a less harmful alternative; however it was also listed as a problematic compound based on green chemistry metrics.32 In general, the recently developed AlI3-based method can be considered an improvement over other Lewis acid-based approaches also from a green chemistry standpoint. However, it is by no means deserving of the label “green” and effort should be invested into adjusting the method taking into consideration health, safety and environmental factors as well.
2.3. Lithium chloride in dimethyl formamide
The use of LiCl in dimethyl formamide for aryl ether dealkylation was reported by Bernard et al.46 The reaction was conducted with 3 eq. of LiCl in boiling dimethyl formamide for ca. 18 hours, after which aqueous work-up with NaOH solution and extraction was conducted. It was found that the demethylation reaction is benefited by electron-withdrawing substituents. On the other hand, the demethylation efficiency was limited for anisole as well as for dimethoxybenzenes. Moreover, when developing a more rapid microwave method, Fang et al.47 showed that the reaction of 3,4-dimethoxybenzaldehyde with LiCl results in the cleavage of only one aryl methyl group. This indicates that the cleavage of the second methoxy group, i.e. the demethylation of the guaiacol motif, is considerably more demanding. Fang et al.47 also suggested a reaction mechanism (Fig. 4), which is based on a nucleophilic attack by Li followed by the release of methyl chloride. Eventually, the phenolic hydroxyl group is formed upon hydrolysis.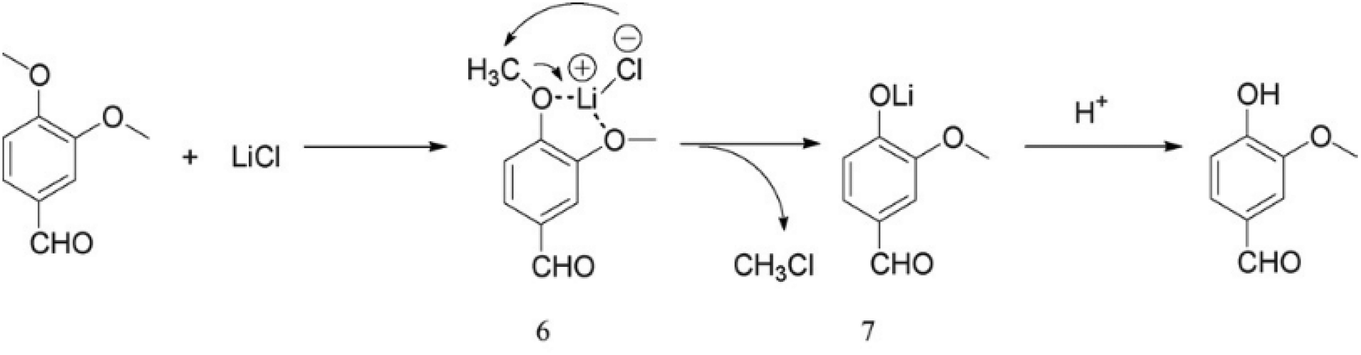 | ||
| Fig. 4 Cleavage of aryl methyl ethers with LiCl; the reaction mechanism proposed by Fang et al.47 | ||
Encouraged by recent results by Kraft and Eichenberger,48 Alni et al.49 attempted the demethylation of eugenol via the LiCl-dimethyl formamide method. They used 2 eq. of LiCl and conducted the reaction for 48 hours under reflux and in a flow of N2 gas before aqueous work-up and extraction. After purification only an 18% yield of hydroxychavicol was obtained, which was explained by the sensitivity of the product towards oxygen.49
It must be pointed out that the due to the use of dimethyl formamide this method cannot be recommended from a work safety standpoint unless its replacement by other solvents is feasible.32 Dimethyl formamide is particularly problematic due to its inherent health risks, especially for being suspected of harming the unborn child. LiCl, on the other hand, poses the hazard of affecting the human central nervous system.
2.4. Iodocyclohexane in dimethyl formamide
Zuo et al.50 discovered a demethylation method for aryl methyl ethers using iodocyclohexane in dimethyl formamide. When the reaction was e.g. conducted with guaiacol and 5 eq. of iodocyclohexane under reflux a 91% yield of catechol was obtained after 4 hours. Iodocyclohexane was considered to be a source for HI formed continuously during the reaction.Compared with conventional HI-based methods14,51 the new approach is characterized by milder reaction conditions and a slow in situ generation of HI. Due to a lack of substrates subjected to the iodocyclohexane demethylation method there is no information available on how the reagent behaves towards other functional groups like double bonds, or hydroxyl or carboxyl groups. As discussed above, the use of dimethyl formamide is highly problematic in terms of health-related hazards and if possible other methods should be used or dimethyl formamide should be replaced by alternative solvents.
2.5. Iodine-based oxidizing agents
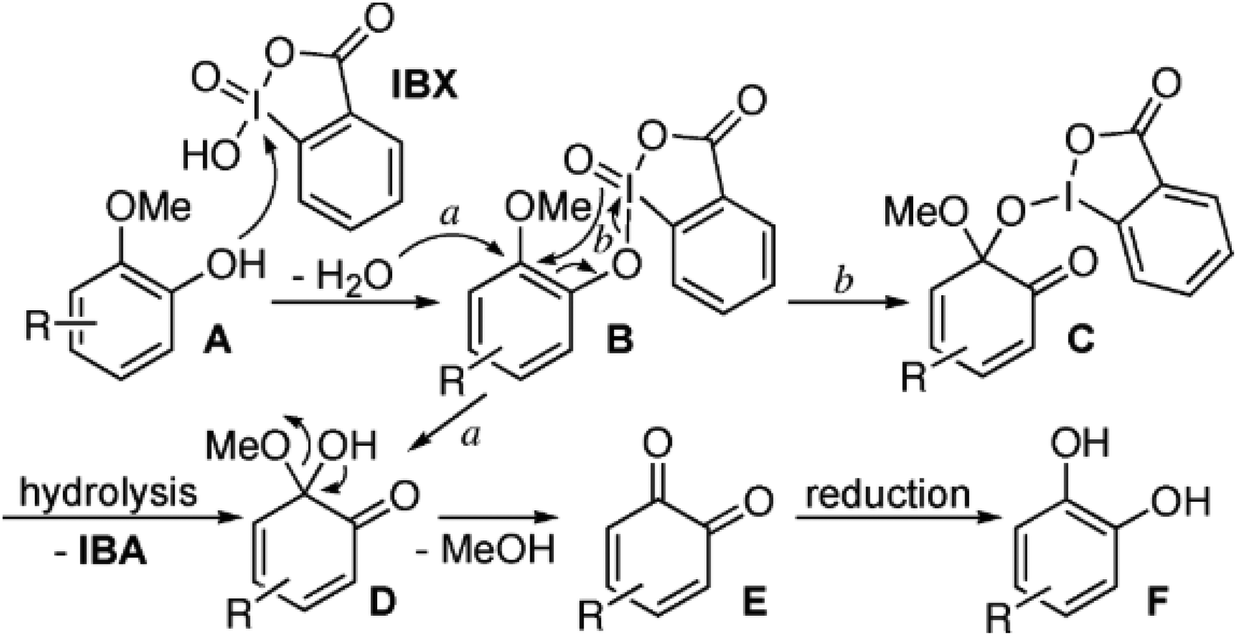 | ||
| Fig. 5 Mechanism proposed by Ozanne et al.52 explaining the oxidative demethylation with 2-iodoxybenzoic acid (IBX), including subsequent hydrolysis and reductive work-up. | ||
The demethylation of eugenol was performed with 2.1 eq. of stabilized 2-iodoxybenzoic acid in tetrahydrofuran. After 16 hours of reaction in the dark at room temperature the product was extracted, hydrolyzed with aqueous NaHCO3 and reduced with 6 eq. of Na2SO4. A 77% yield of hydroxychavicol was obtained.52
In a later study53 it was shown that besides an allyl group, substituents with hydroxyl groups may also be tolerated, e.g. in the case of demethylating homovanillyl alcohol into hydroxytyrosol. In the case of iso-vanillyl alcohol, however, the primary alcohol was reduced to an aldehyde. Moreover, when vanillyl alcohol was used as the substrate, no demethylation could be observed while the primary alcohol was converted into an aldehyde, thus yielding vanillin. The authors argue that the electron-withdrawing aldehyde function in the para position is interfering with the reaction of the oxidizing agent with the phenolic OH group.53
While stabilized 2-iodoxybenzoic acid has significant improvements compared with its unstabilized counterpart, which has explosive properties, it is still a strong oxidizing agent that needs to be handled cautiously and has to be deactivated before disposal.54 Moreover, IBX poses considerably hazards to human health and is used typically in combination with tetrahydrofuran, which is also considered a problematic solvent primarily due to health concerns.32 Thus, while the demethylation reaction with IBX is highly selective and good yields can be obtained, this approach is also found particularly wanting in terms of health, safety and environmental aspects and required further improvements in that regard.
2.6. Thiols in combination with bases
Feutrill and Mirrington57,58 developed a demethylation method based on the nucleophile thioethoxide (alkali ethanethiolate) in dimethyl formamide. Focusing on the selective monodemethylation of 1,2-dimethoxybenzene to guaiacol, they also reported that about 25% of the identified product was catechol. This indicates that guaiacol was further demethylated under the following reaction conditions: 5 eq. thioethoxide (from ethanethiol and NaOH), reflux in dimethylformamide for 3 hours under a N2 atmosphere, and aqueous work-up.58More recently, odorless thiols have been used for this reaction and different bases were investigated, e.g. tert-butoxides.59,60 However, no examples of guaiacol-type substrates were reported. As previously mentioned, thiols pose considerable health and environmental risks and, even more so does the solvent dimethyl formamide. Thus this type of demethylation cannot be recommended.
2.7. Other approaches
There are a variety of other methods for ether cleavage which are, however, not as commonly applied or as viable as the methods reviewed here. Many of these are included in previous review articles.14–16,51 These include demethylation using tetramethylsilyl iodide18 or pyridinium-based compounds.61 Furthermore, ionic liquids have been successfully used.62–64 Recently, Liu et al.65 reported a Cu2O-catalyzed demethylation procedure, which was conducted at 185 °C in methanol in the presence of sodium methanoate. A different catalytic approach was described by Duprat et al.,66 which is an oxidative method based on an Fe0/acetic acid/O2 system. Moreover, ether cleavage is known to be feasible with alkali organometallic compounds or alkali metals.67 Another classical approach, aqueous hydrolysis with Brønsted acids, will be discussed in the next section.3. Aqueous hydrolysis, acids and bases
In this section different aqueous-phase approaches to selectively demethylate guaiacol and its derivatives into catechols are reviewed. Like in the previous section, literature for guaiacol and its derivatives like e.g. eugenol was selected with a side focus on the fate of other functional groups during demethylation. The approaches discussed here include methods relying on either acids or bases as well as sub- and supercritical hydrothermal conditions. On the one hand, this means that the reagents required are comparatively simple and cheap. On the other hand, the reaction conditions are harsh and result in both enhanced corrosion and low selectivity for functionalized molecules. Moreover, this is associated with safety, health and environmental hazards.One aspect that is rarely mentioned is the low solubility of guaiacol and especially its alkylated derivatives. This explains why the use of phase-transfer catalysts can strongly improve this reaction, as will be shown for aqueous HBr, and should certainly play a role for the processes conducted in sub- or supercritical water, where the solubility of nonpolar compounds is drastically increased.68 On the other hand, in other cases a two-phase system may be present even at the reaction temperature unless the substrate is strongly diluted.
3.1. Mineral Brønsted acids
In other studies, acetic acid was added as a co-solvent. In a recent attempt, Alni et al.49 exposed eugenol to a mixture (50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 vol%) of acetic acid and aqueous HBr. Instead of the target compound hydroxychavicol only small amounts (ca. 1%) of what was identified as a dimer species could be obtained after 0.5 hours at 110 °C. The authors hypothesize that an allylic radical could be formed in the presence of oxygen that leads to dimerization.49 In general, the allyl group appears not to be stable under these conditions and it might be speculated whether besides dimerization also the addition of HBr to the double bond takes place.
:50 vol%) of acetic acid and aqueous HBr. Instead of the target compound hydroxychavicol only small amounts (ca. 1%) of what was identified as a dimer species could be obtained after 0.5 hours at 110 °C. The authors hypothesize that an allylic radical could be formed in the presence of oxygen that leads to dimerization.49 In general, the allyl group appears not to be stable under these conditions and it might be speculated whether besides dimerization also the addition of HBr to the double bond takes place.
One significant improvement of the conventional HBr system is the addition of phase-transfer catalysts, which was reported by Landini et al.70 Waghmode et al.71 used the quaternary ammonium salt methyltrioctylammonium chloride, known as Aliquat 336, as the phase-transfer catalyst when studying the demethylation of aryl methyl ethers with HBr. The reaction was conducted with 48% aqueous HBr (4.5 eq.) and 10 wt% (based on substrate) of the phase-transfer catalyst at 105 °C. With guaiacol as substrate this approach yielded 78% of catechol after 6 hours.71 It is noteworthy that the reaction is considerably faster compared with approaches without phase-transfer catalysts.
Both HI and HBr are strong acids and their concentrated solutions are strongly corrosive and have severe detrimental health effects when in contact with skin, inhaled or ingested. Moreover, HBr is known to be toxic to aquatic life, and thus it cannot be considered a green reagent. Of particularly high concern is especially the aforementioned phase-transfer catalyst Aliquat 366, which is toxic to reproduction and an environmental hazard. The use of this modified demethylation approach is particularly advised against.
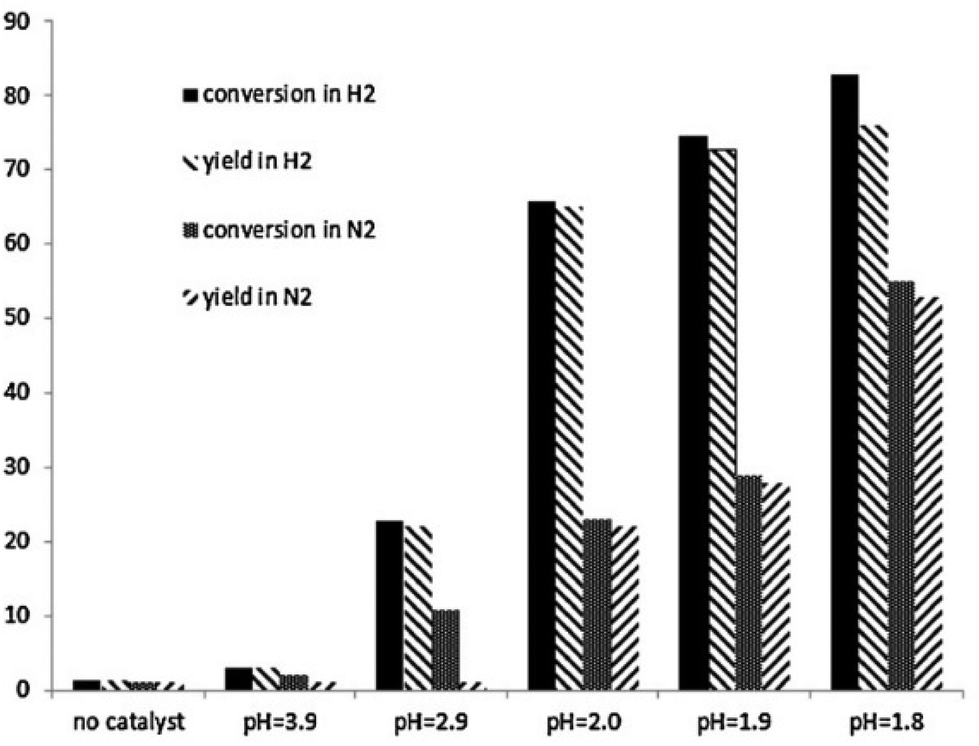 | ||
| Fig. 6 Demethylation of guaiacol at 280 °C in aqueous HCl at different pH values. Both conversion of guaiacol and catechol yield after 3 hours are shown. Reactions were conducted either under a N2 or H2 atmosphere. Reproduced from ref. 72 with permission from Elsevier, copyright 2013. | ||
Both lower pH, i.e. higher acid concentration, and H2 atmosphere are beneficial. At optimum conditions (10 bar H2, pH = 1.8, 280 °C) almost complete guaiacol conversion and an 89% yield of catechol were reported. The presence of H2 was suggested to open an additional reaction pathway resulting in the formation of methane besides the conventional hydrolysis product methanol. Furthermore, based on kinetic analysis the authors suggested a pH-dependent change in the reaction mechanism, shown in Fig. 7. It was proposed that below a certain pH threshold (ca. 3.9) a proton-catalyzed mechanism dominates while at higher pH water plays the role of a catalyst.72
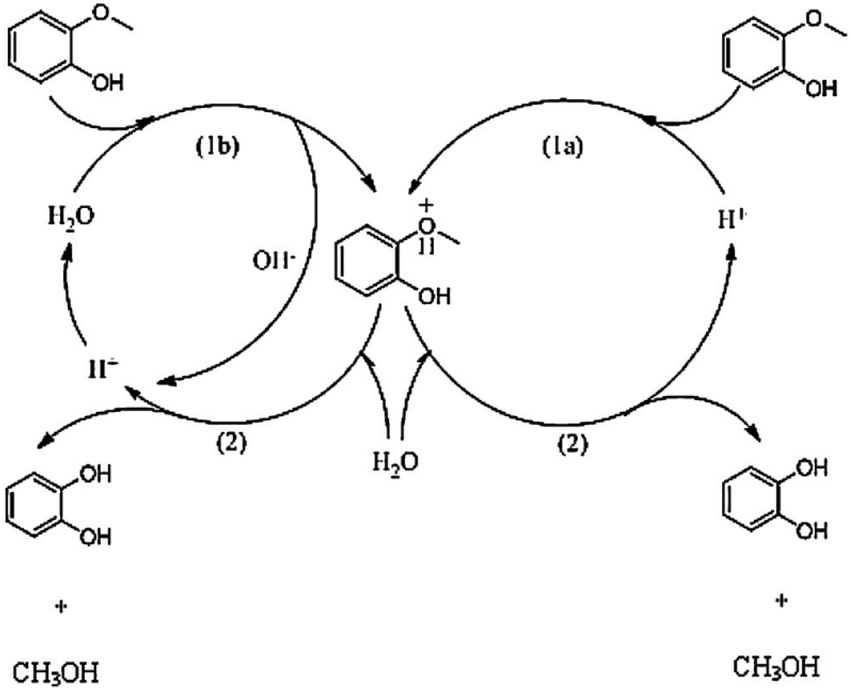 | ||
| Fig. 7 Competing water-catalyzed (left) and acid-catalyzed (right) mechanism of guaiacol demethylation in aqueous HCl proposed by Yang et al.72 The acid-catalyzed mechanism was suggested to dominate at a sufficiently low pH (<3.9). | ||
Very recently, Bomon et al.73 investigated the demethylation of differently functionalized guaiacol derivatives under similar conditions. Experiments were conducted with aqueous HCl at 250 °C under a N2 atmosphere for 3 hours. Their key finding was that, besides demethylation to yield a catechol derivative simultaneously, dealkylation was observed for certain substrates. This was the case for guaiacol derivatives with unsaturated alkyl chain substituents like e.g. 4-vinylguaiacol, eugenol and isoeugenol, where the main product was unsubstituted catechol (yields of 40%, 71% and 62%, respectively). For the saturated analogue 4-propylguaiacol, however, only demethylation took place, yielding 4-propylcatechol in excellent yield (97%).73
A mechanism was proposed (Fig. 8) to explain both acid-catalyzed reactions. The dealkylation was suggested to take place via a retro vinylogical aldol reaction after addition of water to the double bond. The cleaved alkyl fragment was confirmed to give an aldehyde as a byproduct. The acid-catalyzed demethylation, which eventually produces methanol as a byproduct, was proposed to be initialized by arene protonation and proceed vial a hemiacetal intermediate (E).73 This is in contrast to the mechanism by Yang et al.72
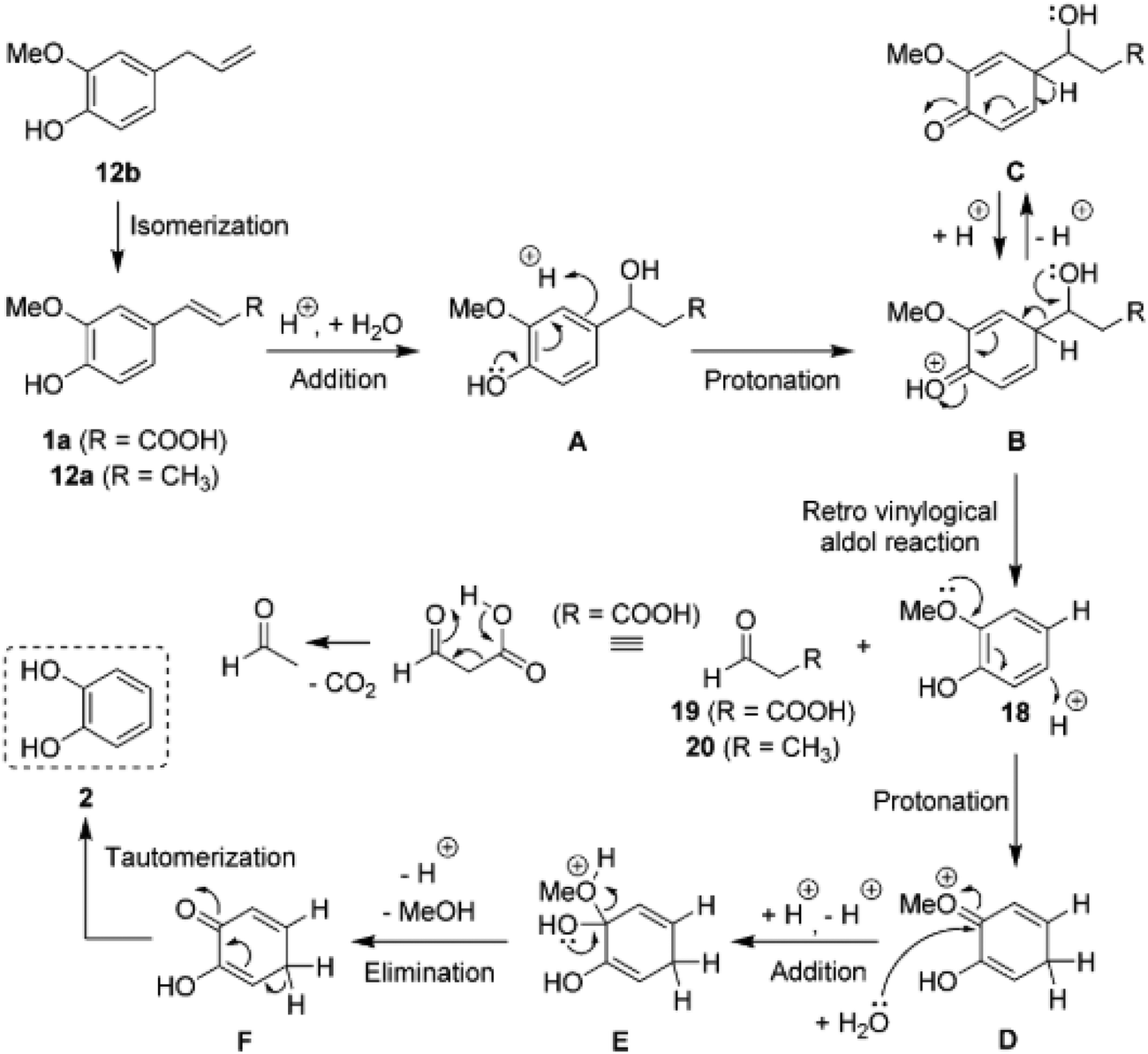 | ||
| Fig. 8 Mechanistic view of a sequence of acid-catalyzed reactions proposed by Bomon et al.73 for allyl- and vinyl-substituted guaiacol derivatives. For example, eugenol (12b) undergoes isomerization, dealkylation and demethylation resulting in the formation of catechol. | ||
Besides HCl, Bomon et al.73 tested several other acids and obtained similar results for H2SO4, however requiring a higher concentration. H3PO4 and triflic acid were significantly less active and acetic acid did not produce any catechol from ferulic acid. Overall, these results show that under acidic conditions double bonds are labile and prone to hydration and even dealkylation.
Compared with the aforementioned aqueous solutions of the acids HBr and HI used for demethylation, it is apparent that HCl and H2SO4, while highly corrosive, are due to their elemental composition less toxic and less environmentally harmful. The discussed variation of the process under H2 pressure of up to 10 bar is not only less desirable from an economic but also from a safety-centered perspective due to the explosion hazard and should be omitted if the process is feasible under inert gas.
3.2. Solid Brønsted acid catalysts
Using solid Brønsted acid catalysts rather than mineral acids offers solutions to the corrosiveness and limited recyclability of the previously presented, homogeneously catalyzed approaches. Wu et al.75 showed that in the heterogeneously catalyzed hydrolysis of 4-propylguaiacol over solid acids selective demethylation to 4-propylcatechol occurs (Fig. 9A) and selectivity comparable to mineral acids was observed. Probably due to its high density of acid sites (Fig. 9B), zeolite beta was found to be best suited among the different solid acids and 4-propylcatechol yields up to 90% were obtained at 275 °C. Moreover, the reusability of the zeolite catalyst was demonstrated but the catalyst lost ca. 60% of its initial activity over four recycling runs. Interestingly, silylation was shown to increase the stability of the catalyst, however at the cost of reduced catalytic activity. In addition, experiments showed lignin oil rich in 4-propylguaiacol could also be converted into 4-propylcatechol, achieving yields as high as 74%.75 Overall, the solid acid catalyst in an aqueous reaction environment is the best choice in terms of the environmental aspects of the process. The proven recyclability (even though it should be improved by developing more stable catalysts) in combination with the reasonably good product yields, the use of water as the solvent, the absence of any other additives and the low harm potential of the zeolite materials makes the process highly desirable. Nevertheless, the process will have to be shown to work on a significantly larger scale.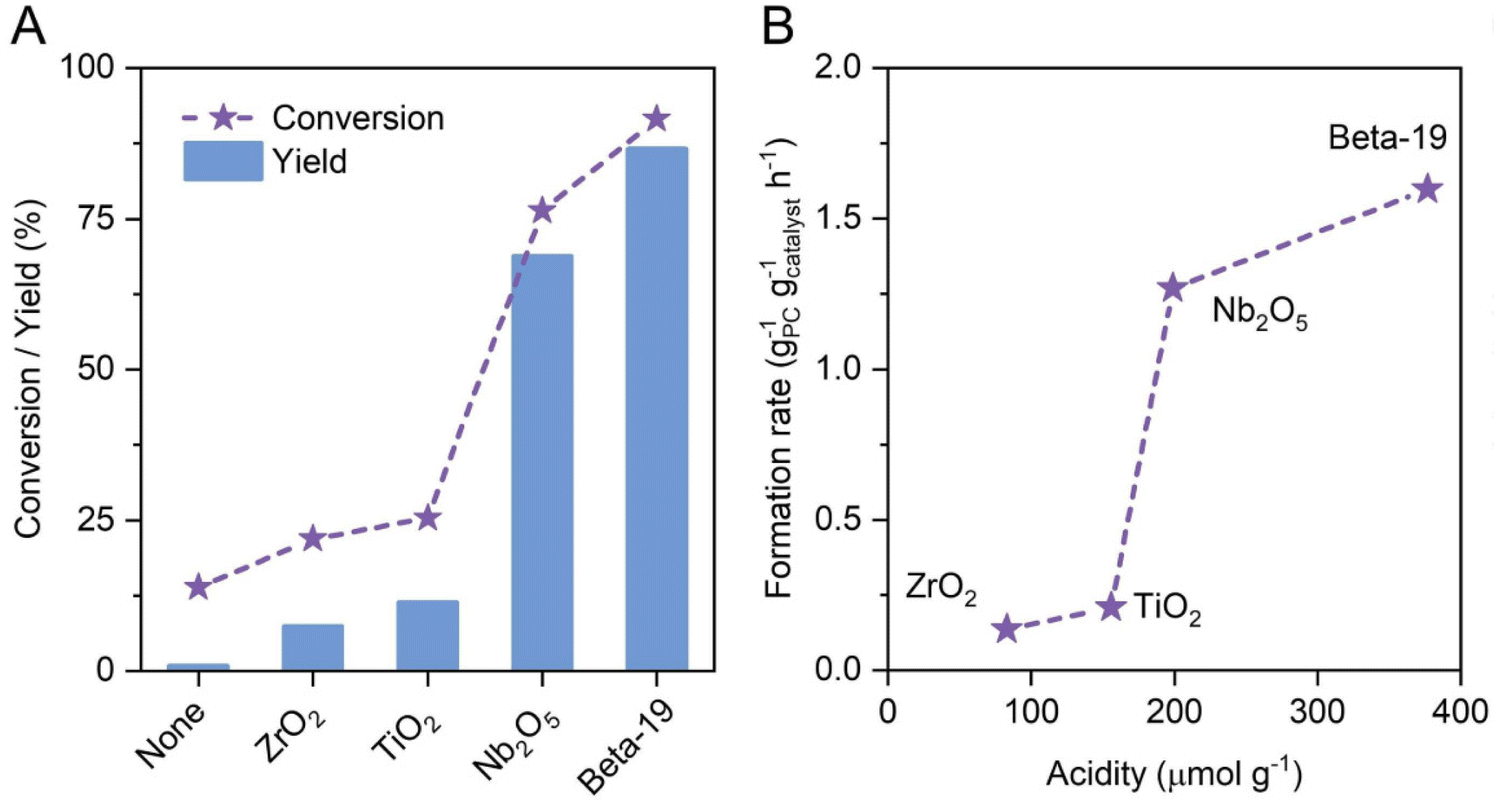 | ||
| Fig. 9 Conversion of 4-propylguaiacol and yield of 4-propylcatechol over different solid acid catalysts after 2 h at 260 °C in water (A). Catalytic activity (given as the product formation rate) plotted against catalyst acidity (B). Reproduced from ref. 75 with permission from John Wiley and Sons, copyright 2022. | ||
3.3. Aqueous bases
Aryl methyl ether hydrolysis is also feasible via a base-catalyzed pathway and can in certain cases be beneficial.76 Yang et al.72 also reported a single demethylation experiment under basic conditions. Guaiacol was demethylated in aqueous NaOH (pH = 10.4) at 280 °C under a H2 atmosphere, similar to their acid-catalyzed experiments discussed above. After 3 hours, it was confirmed that catechol was formed; however, no quantitative information was reported. Most importantly, significant amounts of “char” were found, indicating a strong contribution of undesired side reactions. Therefore, this approach was not pursued further.723.4. Hydrolysis in sub- and supercritical water
At elevated temperature and pressure, guaiacol can be demethylated in water alone. The underlying effects that play a significant role are the higher solubility of nonpolar compounds as well as the drastically increased ion concentration due to the auto-dissociation of water under sub- and especially supercritical conditions.77 In the 1980s the hydrolysis of guaiacol was investigated under supercritical conditions at 383 °C. Klein and co-workers78–80 investigated the decomposition of guaiacol without and with different amounts of water. Increasing contents of water were suggested to shift the dominating reaction mechanism from pyrolysis to hydrolysis, which results in the observed increasing selectivity for the hydrolysis products catechol and methanol with increasing density of water.78–80 Interestingly, at higher temperatures (400–500 °C) DiLeo et al.81 could not identify catechol or methanol.More recently, Wahyudiono et al.82 revisited the reaction and compared the hydrolysis under super- and subcritical conditions (Fig. 10). In the subcritical regime at 250 °C and 80 bar, guaiacol conversion barely reached 6% after 2 hours while selectivity for catechol was ca. 70%. On the other hand, under supercritical conditions (400 °C, 400 bar) guaiacol conversion reached 55% and catechol yield 40% within 0.5 hours. After 2 hours the guaiacol was almost completely decomposed, however without a clear increase in product yield.82 In the super-critical aqueous system the addition of salts (NaCl, CaCl2, FeCl3) was found to promote the hydrolysis of guaiacol to catechol, probably by increasing the polarity of the solvent.80
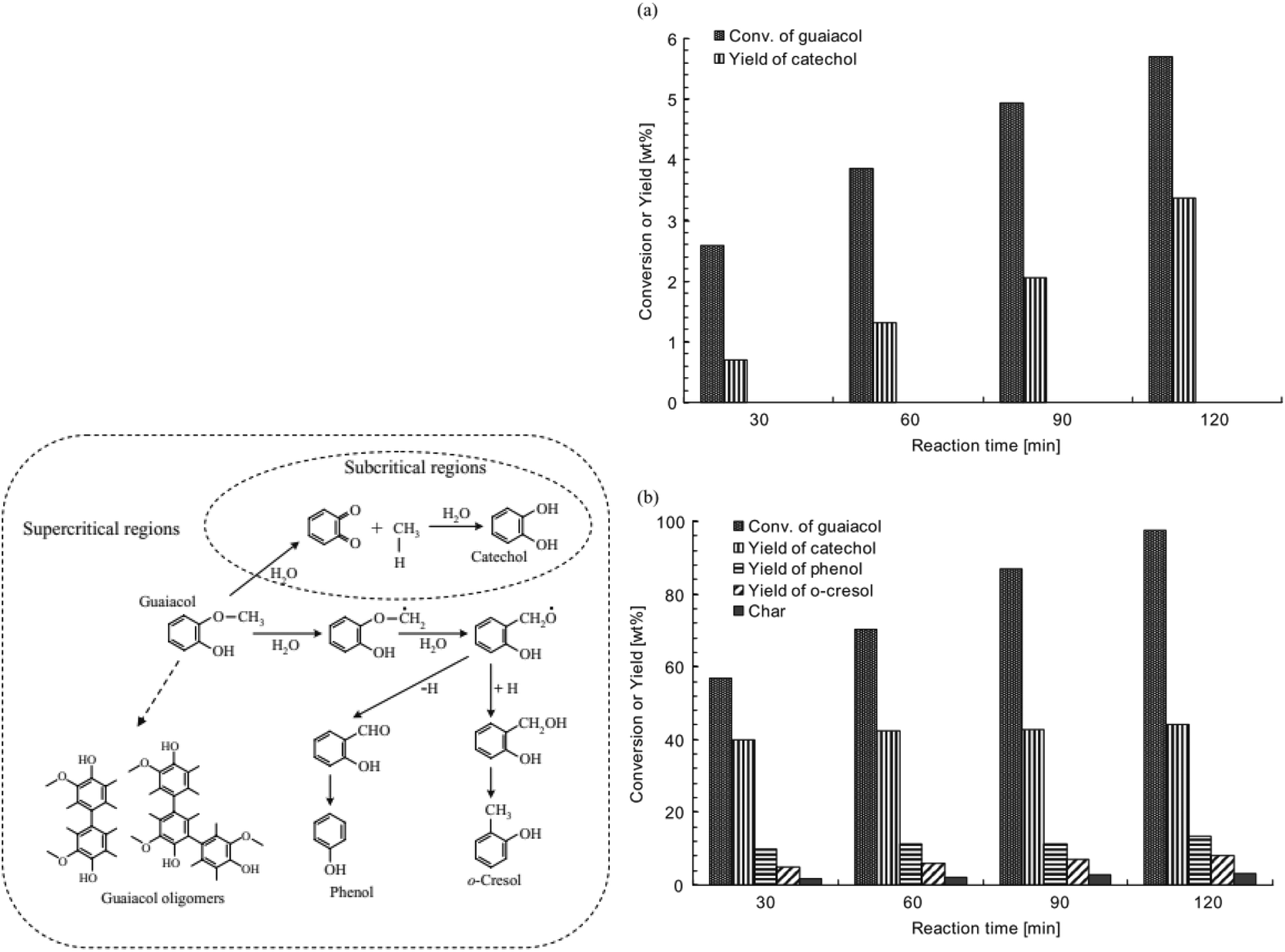 | ||
| Fig. 10 Left: probable reaction pathways for guaiacol decomposition to achieve the desired products; Right: yields of guaiacol decomposition products in sub- and supercritical water as a function of reaction time, 400 °C and 400 bar.82 | ||
From a green chemistry perspective this approach appears very attractive based on the fact that only water, considered a highly recommended solvent, and no other compounds in catalytic or even stoichiometric reactions are required. Comparing the reaction conditions, however, with the solid Brønsted acid-catalyzed approach (section 3.2) it becomes apparent that the considerably milder reaction conditions and higher product yield indicate that the heterogeneously catalyzed reaction is preferable, as it enables a sufficiently high reusability of the catalyst.
3.5. Thermolysis
Thermochemical conversion of biomass includes fast pyrolysis, gasification, gasification, combustion and vapor phase cracking. The later was used for eugenol thermolysis in the temperature range 300–900 °C and residence times of 1–3 s in a non-isothermal laminar flow-reactor operated at atmospheric pressure. Temperatures above 650 °C led to aromatic hydrocarbons, while at lower temperature oxygenates were observed with a retained aromatic ring. Under pyrolytic conditions, free-radical reactions are the major pathways responsible for the decomposition and formation of species. In eugenol, it is expected that the methoxy C–O bond in eugenol is the weakest and the source of the initiation reaction, as presented in Fig. 11. Even though the diol was formed according to the below presented mechanism, the propyl chain was cracked into a methyl group. The yield of 3-methyl-1,2-benzenediol reached 4% at 750 °C and residence time 1 s. The yield of catechol at the same reaction conditions reached 2.5%.83,84 | ||
| Fig. 11 Left: general reaction scheme for the vapor-phase cracking of eugenol; right: fractional yields of 2,3-dihydrobenzofuran, 2-coumaranone, 3-methyl-1,2-benzenediol, and 4-(2-propenyl)-phenol (clockwise from top left) as functions of the temperature at (●) τ = 1 s and (○) τ = 3 s.83 | ||
The first and major step in the thermolysis of guaiacol in tetralin in the presence of hydrogen has been shown to be a monomolecular dissociation into phenoxy and methyl radicals with subsequent stabilization of the radicals by hydrogen abstraction from tetralin. The results show that hydrogen does not participate in the reaction because no systematic difference is noted between nitrogen and hydrogen in the same solvent system.85,86 Afifi et al.87 investigated the thermal treatment of guaiacol in tetralin in the absence of catalysts. The major reaction products identified were catechol, o-cresol and phenol and an oligomeric/polymeric material. At 375 °C the conversion of guaiacol reached 15% with around a 5% yield of catechol. With increasing conversion, the yield of catechol decreased. An increased initial concentration of guaiacol in tetralin yielded a smaller amount of catechol.87
Supercritical water gasification (SCWG) of guaiacol was performed between 400 °C and 700 °C in the presence or absence of Ni wire as a heterogeneous catalyst. At 400–500 °C, guaiacol was completely converted in SCWG, but not all of it was gasified. Phenol and o-cresol were decomposition products and larger amounts of char also formed. The SCWG of phenol at 600–700 °C did not produce any char. The major liquid-phase decomposition product was benzene, which was in yields up to about 5% at 600 °C and 12% at 700 °C. Up to 99% of the carbon was gasified when using three Ni wires, and the major products were hydrogen and carbon dioxide in yields close to equilibrium.81 Overall, the non-catalytic thermolysis processes are far too unselective to be considered viable alternatives for the demethylation of methyl aryl ethers.
4. Hydrotreatment
The scope of the published literature is mostly focused on the total (hydro)deoxygenation (HDO) of the oxygenated lignin-based aromatics (eugenol, vanillin, guaiacol) rather than selective conversion into targeting value-added compounds. Especially 1,2-dihydroxy compounds are rarely mentioned in the existing literature. However, general conclusions can be outlined from the screened reaction conditions, used supported metals and mechanistic studies.Among the metals, noble (Pt, Pd, Ru) and transition metals (Ni, Mo, Co, Ir, Ce, Cu) as well as their bimetallic combinations were screened in numerous studies using eugenol or guaiacol as a reactant. Almost in all research studies high H2 pressure was used (10–60 bar), resulting in deoxygenated aromatic and aliphatic (due to the ring saturation) compounds. The selected metals were usually supported on neutral or acidic supports (C, SiO2, TiO2, MgO, Al2O3, ZrO2, CaCO3, HZSM, etc.). The choice of the metal and support is important for the selective conversion of compounds in particular chemical reactions, such as hydrogenation, deoxygenation, demethylation, decarboxylation, ring opening, etc.
It cannot be overstated that the selective demethylation of methyl aryl ether groups during metal-catalyzed hydrotreatment appears to be very challenging and, in particular, hydrogenation reactions are occurring in general more facilely. However, the reaction does occur and due to the lack of studies investigating this particular pathway it is difficult to estimate the true potential of this reaction. Nevertheless, it is certain that the demethylation pathway will probably be more prevalent under conditions less favorable to hydrogenation and with purposefully designed catalysts. As it stands, the low yields of demethylated products are currently the main impediment for this type of process and also problematic from a sustainability point of view.
While the use of solid and potentially reusable catalysts is promising regarding the green chemistry metrics of this process, the use of H2 gas at elevated pressure presents a severe harm potential that also may limit the applicability in industry. Significantly lowering the required H2 pressure would therefore be highly desirable. An additional hazard arises from the use of organic solvents, typically, which are considered hazardous solvents,32 which are used in some cases rather than the more desirable solvent water. Lastly, for the development of suitable solid catalysts the focus should also be placed on the health, safety and environmental impact of the components of the catalyst. The use of Ni should e.g. be avoided due to its carcinogenicity, while elements such as Ru, Mo or Pt are considered most “green” by Bystrzanowska et al.88
4.1. Noble metals (supported Pt, Pd, Ru)
Noble metals showed high activity for HDO and especially for hydrogenation (ring saturation) under high hydrogen pressure and at high reaction temperatures (>300 °C). In the presence of Pt catalyst significant hydrogenation of the propenyl chain took place besides HDO and deallylation when eugenol was used as a reactant. In the absence of H2, Al2O3 catalyzed similar reactions (isomerization and deallylation) to zeolites.74 Pd/C in a combination with HZSM-5 zeolite was used for aqueous phase HDO of eugenol under 20 bar of H2. The introduction of acidic active sites increased the dehydration of 4-propyl-1,2-cyclohexanediol, while Pd was responsible for the hydrogenation of the aromatic ring; the proposed reaction pathway is presented in Fig. 12. However, the hydrogenation is more pronounced.89 Carbon- and HZSM-5-supported Pd catalysts were tested for aqueous-phase HDO of methoxy-substituted lignin monomers (such as guaiacol and 2,6-dimethoxy-phenol) under 20 bar H2. Again, hydrogenation was the dominant reaction, resulting in aromatic ring saturation, which proceeded even before the HDO of oxygen-containing functional groups.90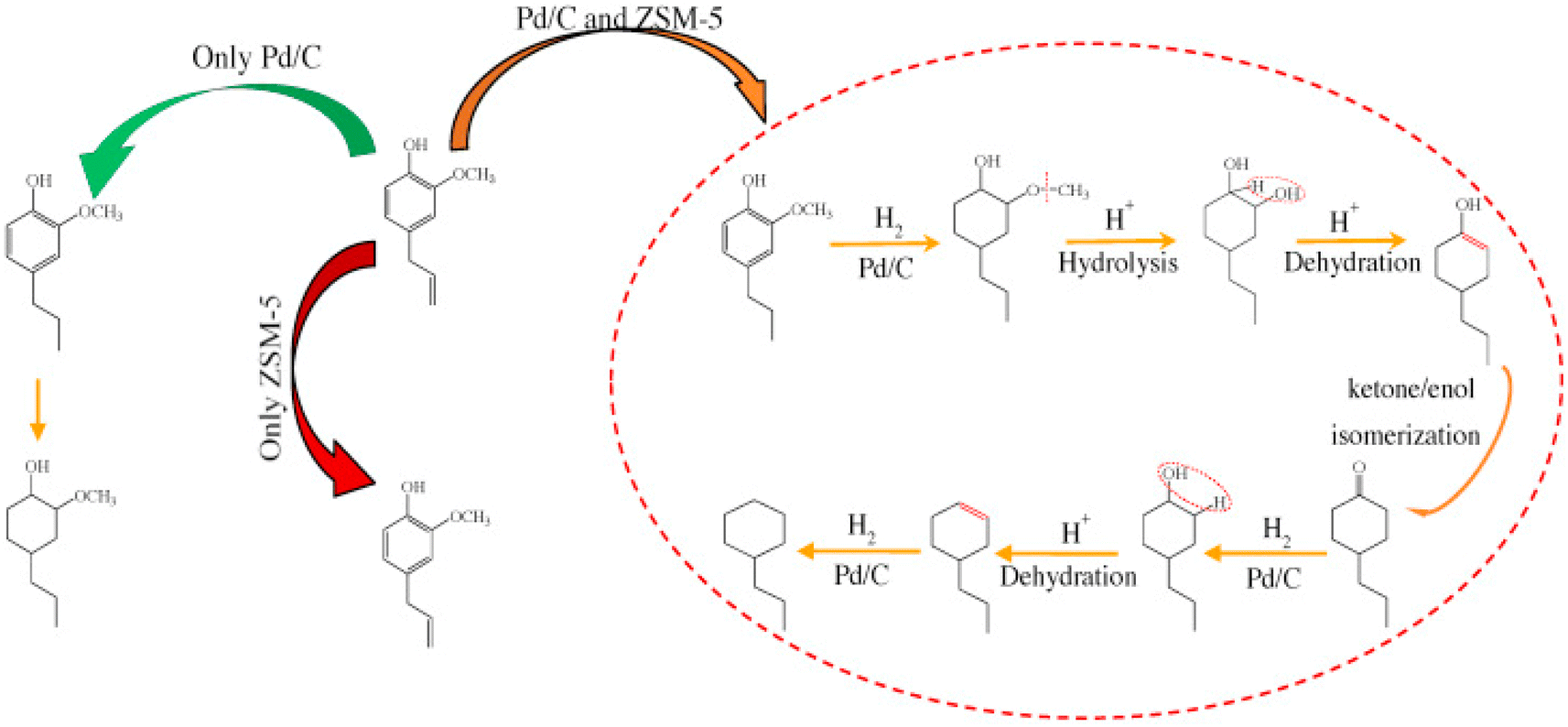 | ||
| Fig. 12 Reaction pathways of eugenol to cyclohexanes over Pd/C and HZSM-5 in the aqueous phase.89 | ||
The hydrothermal conversion of eugenol over a Pt/C catalyst at temperatures between 250 °C and 300 °C in a water/ethanol mixture in the absence of gaseous H2 was investigated using various catalysts. Under these conditions, the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond of the propenyl chain is hydrogenated, but the hydroxyl and methoxy groups of eugenol do not react.91
C double bond of the propenyl chain is hydrogenated, but the hydroxyl and methoxy groups of eugenol do not react.91
Pt was found to catalyze the demethylation of guaiacol to catechol, while acid sites on the support (HBeta and SiO2) were responsible for transalkylation and dehydroxylation reactions into monohydroxyl phenolic compounds.92 Besides silica-supported Pt, Rh was also tested for the HDO of p-methyl guaiacol in a fixed bed reactor at a temperature of 300 °C with a hydrogen pressure of 4 bar. Silica-supported Rh improved the selectivity towards 4-methyl-catechol in comparison with the respective Pt catalyst.93 Zhang et al.94 studied the aqueous-phase HDO of eugenol over Pd/C combined with HZSM-5 zeolite catalysts in the presence of 50 bar H2 at the reaction temperature of 240 °C. The results showed that hydrogenation is the dominant reaction; firstly the alkyl chain was saturated and next the aromatic ring.94
An interesting approach was used by González-Borja et al.,95 where inconel monoliths were impregnated with Pt and Sn and tested for the HDO of anisole and guaiacol under atmospheric pressure of H2/N2 (17:83 vol%) at 400 °C in a quartz tube. Guaiacol was vaporized in the gas mixture before entering the reactor. The results showed that guaiacol first produces catechol, followed by deoxygenation to phenol and finally to benzene. The decomposition of the methoxy group results in catechol or phenol from guaiacol and anisole, respectively. Thermodynamically, this CO bond is weaker than the CO bond connecting the methoxyl to the aromatic ring and weaker than the CO bond of the hydroxyl group. Therefore, producing catechol from guaiacol is more favorable than directly producing anisole or phenol. However, only traces of catechol were observed in the product mixture from guaiacol, which suggests that catechol is rapidly converted to phenol, perhaps even before it desorbs from the metal surface.95
MgO-supported Pt catalyzed the conversion of guaiacol into phenol, catechol and cyclopentanone. However, the dominant pathway was through a complete cleavage of the methoxy group, while a slightly higher yield of catechol was observed when Pt/Al2O3 was used as a catalyst. In the absence of acidic sites, catechol is expected to be formed over Pt/MgO, primarily by hydrogenolysis on the Pt, with methane as a co-product.96 Guaiacol was hydrotreated in the presence of Pt/HZSM-5 catalysts with different crystallite sizes. The reactions were performed in dodecane at 250 °C with 40 bar of hydrogen; within 120 min the guaiacol conversion reached near 100% in all cases, but no catechol was formed, except in minor concentrations (0.25 wt%) in the presence of Pt/NS-2 catalyst.97 When using a flow reactor with Pt/γ-Al2O3 catalyst, and HDO performed at 300 °C in the presence of H2 (1.4 bar), the major products from guaiacol conversion were phenol, catechol and 3-methylcatechol. The study by Runnebaum et al. showed that the higher reaction temperature did not influence the formation of catechol, while the higher H2 pressure decreased the catechol selectivity.98
When alumina-supported Pt catalyst was doped with different loadings of Nb2O5 the HDO activity increased and yielded more deoxygenated products. However, at a low hydrogen concentration (H2:guaiacol = 1 mol/mol) the selectivity towards catechol was very high (80%), but the conversion remained low (15%). These experiments were performed in a continuous flow reactor.99
Ru also showed very high activity towards the complete HDO of guaiacol even at low temperatures, while selective demethoxylation from aqueous guaiacol proceeded over Ru catalysts.100 The addition of MgO to the reaction media suppressed the unselective C–O dissociation.101 Similar behavior was observed with magnetically separable Ru/C catalyst, where 70% of the oxygen was removed from eugenol.102 CaCO3-supported Ru even surpassed the HDO activity compared with ZrO2- or MgO-supported Ru catalysts. The aromatic ring was saturated and complete deoxygenation was reached.103 Ru/HZSM-5 was used for the HDO of guaiacol in an aqueous environment at moderate reaction conditions (240 °C with 2 bar H2). The reaction pathway with guaiacol is presented in Fig. 13. In the presence of H2 Ru-catalyzed guaiacol conversion in aqueous phase proceeds along 4 different pathways: (1) hydrogenation to 2-methoxy-1-cyclohexanol, (2) hydrogenolysis of the C–O bond at position b to phenol and methanol with further deoxygenation to benzene, (3) hydrogenolysis of C–O bond at position c to catechol and (4) hydrogenolysis of C–O bond at position a to produce anisole. The results showed that the C–O bond cleavage at position b (to form phenol) was the primary and major step, with a selectivity of 85% at 120 min. The highest yield of catechol (40%) was reached in a solvent-free system at 240 °C with 2 bar of H2 and 6 bar of N2 in a batch system. A slightly lower yield of catechol (35%) was obtained in a fixed bed reactor under similar reaction conditions.104 When carbon black-supported Ru–MnOx/C catalyst was used, guaiacol was demethoxylated and then hydrogenated, forming cyclohexanol and methanol under relatively mild reaction conditions (160 °C and 15 bar H2). A lower H2 pressure and higher reaction temperature were advantageous to demethoxylation. The addition of MnOx species slowed down the reaction rate of total hydrogenation of the aromatic ring, which increased the relative rate of the elimination of methoxy groups to that of total hydrogenation before the elimination.105
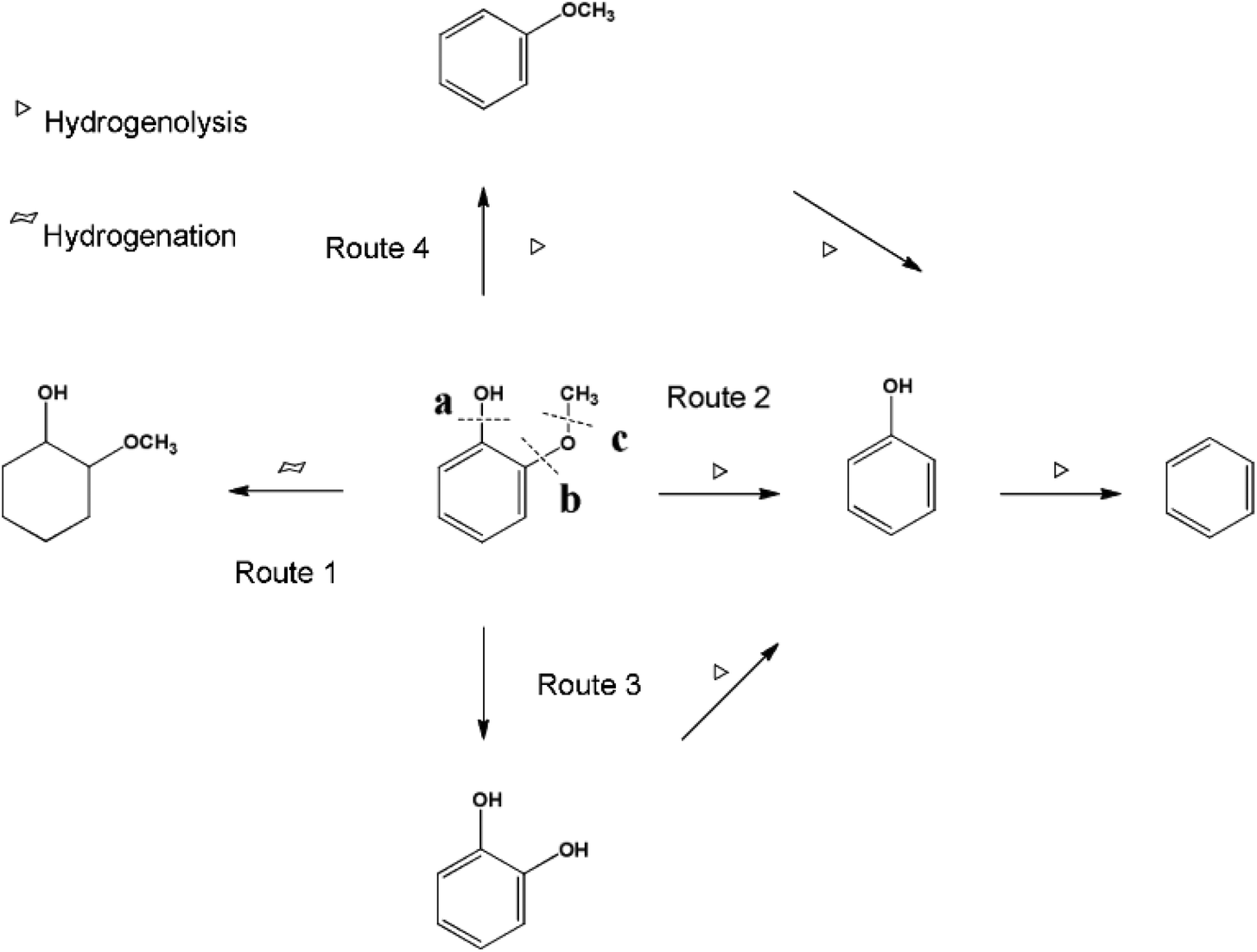 | ||
| Fig. 13 The plausible reaction pathways for guaiacol conversion with metal/acid bifunctional catalysts in the aqueous phase. Reproduced from ref. 104 with permission from the Royal Society of Chemistry, copyright 2016. | ||
Alumina-supported Ru was tested in a H2-free reaction system for the HDO of guaiacol, using a hydrogen donor solvent (iso-propyl alcohol). In the catalytic transfer hydrogenation firstly 2-metoxycyclohexanol was formed from guaiacol, which was further hydrodeoxygenated to cyclohexanol. Without the external source of hydrogen, there was a sufficient amount of in situ-formed hydrogen to saturate the aromatic ring first, even before the further hydrodeoxygenation of oxygen-containing functional groups (–OH, –O–CH3).106
Lee et al.107 tested different supported noble metals (Pt, Rh, Pd, Ru on Al2O3, SiO2–Al2O3 and NAC) for the HDO of guaiacol in n-decane under 40 bar of H2 at 250 °C. All the tested metals were highly active for HDO to the final completely or partially deoxygenated products (cyclohexane, cyclohexanol, cyclohexanone and 2-metoxycyclohexanol). Among the products catechol was not detected, and thus rather the demethylation demethoxylation proceeded under the selected reaction conditions.107
A combination of supported noble and transition metals was screened for guaiacol, eugenol and isoeugenol HDO in hexadecane or dodecane as solvents under high hydrogen pressure (30 to 50 bar) at elevated temperatures (200–350 °C). As expected, hydrogenation of the aromatic ring and propenyl chain was the dominant reaction, and no diol was found among the formed products.108–115 The comparison between transition metal catalysts (sulfided CoMo and NiMo) and noble metals (Rh, PtRh and PdRh) supported on ZrO2 showed that the latter (Rh) has higher HDO activity than CoMo or NiMo. However, the addition of Pt and Pd had no positive effect. The temperature and H2 pressure were supposed to be too high to allow the formation of catechol.116
Afifi et al.87 investigated the effect of homogeneous catalysis, Fe and Ru (FeCl3, RuCl3), on the conversion of guaiacol in tetralin as the hydrogen donor solvent to catechol. In the absence of catalyst and at low ratios of guaiacol to tetralin, the primary product is catechol; however, the yield of a secondary product phenol is increased with both Fe and Ru. The yield of catechol reached up to 55% with FeCl3 at 400 °C in the presence of hydrogen in addition to the hydrogen donor solvent (tetralin).87
4.2. Transition metals (supported Ni, Mo, Co, Re, Cu)
Transition metals offer a larger potential for the selective demethylation of eugenol or guaiacol, since they have lower hydrogenation activity. Moreover, supported Ni, Mo and Co are widely used in the chemical industry for hydrodeoxygenation and hydrodesulphurization reactions where oxygen and sulphur species are removed, respectively.A series of cobalt-based catalysts with different supports were prepared and used to catalyze lignin-derived phenols to cyclohexanols. Among the catalysts, TiO2-supported Co showed the highest HDO activity under 10 bar of hydrogen at 200 °C.In the first step, a complete –O-CH3 group was removed, followed by saturation of the aromatic ring. All the Co-based catalysts selected in the study by Liu et al.117 showed high activity to cleave the Caryl–OCH3 bond before the hydrogenation of the aromatic ring. Similar deoxygenation activity was obtained with an activated carbon-supported α-molybdenum carbide catalyst (α-MoC1−x/AC). The reaction was performed in supercritical ethanol at 340 °C. 87% conversion of guaiacol was reached with 85% selectivity for phenol and alkylphenols. The solvent selection (methanol, isopropanol, tetralin, n-hexane, water, ethanol) showed the best performance between 280 and 340 °C, where the reaction took place via demethylation followed by deoxygenation and transalkylation. Catechol, which was not detected in the products with organic solvents, was formed with a selectivity of 94% in water.118
ZrO2-supported Ni and Ir both showed a high activity towards the hydrodeoxygenation of isoeugenol, guaiacol and vanillin under high H2 pressure (30 bar). Ni metal was active in hydrogenation of the phenyl ring, but at the same time exhibited lower HDO activity; however, no diol products were detected.119 Supported Ni catalysts showed high activity for the hydrogenation and hydrogenolysis of eugenol, but again no diol products were detected.120–123 Surprisingly the catechol was obtained in a catalytic hydrotreating over nickel catalyst supported on red mud with a selectivity of 5.3% at 300 °C and with 50% conversion. The selectivity decreases with increased temperature and hydrogen pressure. With commercial Ni/SiO2–Al2O3 catalyst the obtained catechol selectivity reached 8.1% (Fig. 14).124
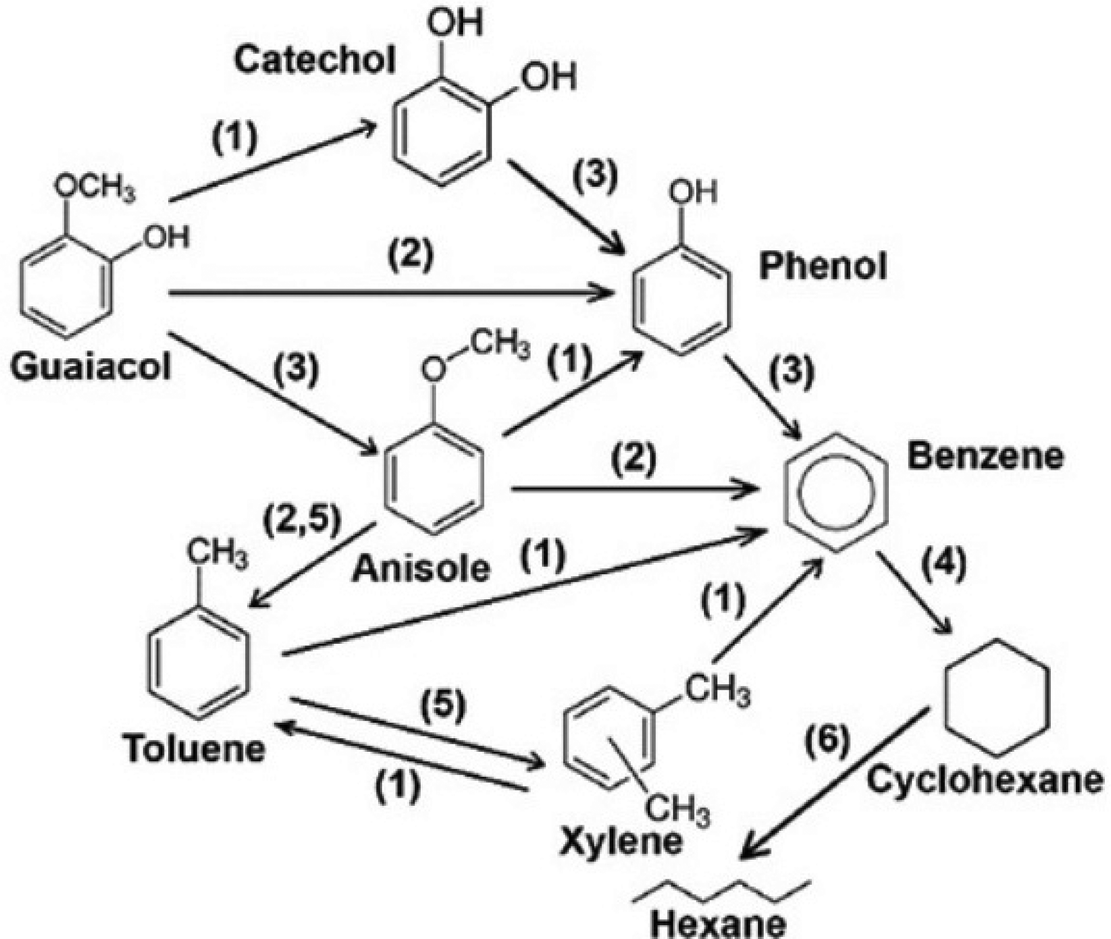 | ||
| Fig. 14 Reaction network of guaiacol HDO over Ni-based catalysts. 1: demethylation, 2: demethoxylation, 3: dehydroxylation, 4: hydrogenation, 5: transalkylation, 6: ring opening. Reproduced from ref. 124 with permission from Elsevier, copyright 2018. | ||
When performing the HDO of guaiacol over nickel phosphide (Ni2P) supported on ZSM-5 zeolite in acetic acid as solvent, surprisingly the obtained selectivity of catechol reached 30%. While the H2 pressure did not have an influence in the range of 10–30 bar, the increased reaction temperature (from 260 to 300 °C) significantly improved the catechol yield. Interestingly, the possibility of using carboxylic acids as a source of hydrogen, for the deoxygenation of guaiacol, is a potentially attractive route for the upgrading of bio-oils.125 Ni2P supported on SiO2 revealed that direct demethoxylation and dehydroxylation are major reaction pathways in guaiacol HDO, producing phenol and anisole as intermediates. Demethylation of guaiacol is of minor importance because only small amounts of catechol form even at very low contact times.126 Wu et al.127 tested different silica-supported Ni/P ratios (1–3) for the HDO of guaiacol, performed in a fixed-bed reactor at 300 °C with a continuous flow composed of an 80% H2/N2 stream at atmospheric pressure. The most favorable ratio of Ni/P for the formation of catechol was 2. The major influence was shown to be the weight hourly space velocity (WHSV); the highest WHSV gave the highest catechol yield. The highest yield of catechol (27.1%) was reached with Ni/P = 2 at 10.68 h−1 WHSV.127 Moreover, alumina and zirconia were tested as supports of Ni2P catalysts under the same reaction conditions. The catechol yield significantly increased. At 5.34 h−1 WHSV reached 67.7% over a ZrO2-supported catalyst, while a SiO2-supported catalyst yielded 46.7% of catechol at 2.67 h−1 WHSV. However, the high selectivity towards catechol was connected to the low conversion of guaiacol.128 HDO activity of the transition metal phosphides was tested on guaiacol as a model compound. At a lower contact time using Co2P/SiO2 and WP/SiO2 catalysts there was 99% and 88% of catechol formed (at 35 and 12% conversion, respectively), respectively. With a commercial Pd/Al2O3 the only formed product was catechol at 70% conversion; reaction conditions: contact time 0.339 min and space velocity 59 h−1 at 300 °C with a hydrogen:guaiacol ratio of 33.129
Peters et al.130 studied guaiacol HDO over Ni-based and Fe-based catalysts. Separately zeolites were also used as catalysts. The reaction was performed in gaseous phase in a fixed-bed reactor at 300–500 °C in the presence of hydrogen. Catechol was proposed as an intermediate even though it was not detected in the liquid phase; however, a significant concentration of methane (1–8%) was detected in the gaseous phase, which proved the formation of catechol from guaiacol.130
Molybdenum nitrides on commercial activated carbon were used for the HDO of guaiacol in decalin as solvent at 300 °C under 50 bar of H2, which was filled into the reactor at the plateau temperature. The highest yield of catechol was obtained over unsupported Mo2N catalyst. After 4 h of reaction and 4% conversion, the yield of catechol and phenol was 3% and 1%, respectively. HDO was more prominent over supported catalyst, resulting in higher yields of phenol.131 Moreover pre-sulphided MoS2 supported on activated carbon converted guaiacol into catechol with a yield of 10% at 300 °C under 50 bar of H2 in a batch regime in decalin as solvent.132
The influence of the presence of H2S in the reaction feed was studied for the HDO of guaiacol, catalyzed with pre-sulphided CoMo catalyst at 270 °C. The increased H2S concentration significantly improved the catechol formation but at the same time did not affect the guaiacol conversion.133
Jimenez et al.134 tested carbon-supported molybdenum catalysts for the HDO of guaiacol in aqueous and organic (dodecane) environments in the presence of hydrogen. The results showed relatively high selectivity towards catechol when reaction was performed in dodecane at 300 °C under an initial 40 bar of H2 and even better results in a water environment. However, guaiacol conversion was significantly better in the organic environment, reaching up to 91% with 24% selectivity towards catechol (with 20% Ac/CNT catalyst).134
Copper has a low tendency to hydrogenation, preventing over-reduction and thus improving the selectivity. Copper-doped PMO (Cu-PMO) was used for the HDO of eugenol in the temperature range between 22 °C and 180 °C under 40 bar of hydrogen, performed from 3–18 hours. Even though the most thermodynamically favorable product is predicted to be catechol (based on DFT calculations) in the experiments it was not detected.135
Rhenium oxide (Re2O7) converted guaiacol into phenol and alkyl phenols at 280–320 °C in ethanol as a (hydrogen donor) solvent under an inert gaseous atmosphere.136 Bimetallic RuRe catalyst supported on carbon showed considerably higher activity for HDO; after 4 hours of treatment at 240 °C under 20 bar of H2 almost all the guaiacol was converted into cyclohexane.137 When water was added to the reaction mixture using a ReS2/SiO2 catalyst the conversion of guaiacol was significantly lower. Moreover, the preferential adsorption of water on the acidic active sites inhibited the demethylation reaction pathway, resulting in less catechol.138
Many studies used supported bimetallic NiMo, CoMo or NiCo catalyst for the HDO of lignin-based monomers.139,140 Blanco et al.141 studied the HDO of guaiacol in the liquid phase at 300 °C under 50 bar of hydrogen, where nickel–cobalt bimetallic catalysts supported on high surface area graphite with different Ni/Co ratios were tested. The highest activity was reached with Ni/Co = 2:1, but the results showed that Ni was more active in the hydrogenation, favoring cyclohexanol production from phenol, while this was inhibited on the Co-containing catalysts. However, the major products were phenol and cyclohexanol.141 The demethylation of guaiacol was studied in the presence of alumina-supported NiMo and CoMo catalysts. It was found that selective demethylation occurs for a large part on the Lewis acid–base sites of the alumina support. The addition of hydrogen sulphide or water does not inhibit the demethylation, while the reaction is inhibited by the addition of a strong base as ammonia. The alumina support alone had a non-negligible activity for the conversion of guaiacol but only catechol was produced in low quantity. At 50% of guaiacol conversion, the ratio between phenol and catechol was 0.4 and 0.15 when no H2S is added using CoMo and NiMo catalysts, respectively. The addition of H2S decreased the obtained phenol/catechol ratio.142 NiMo/Al2O3 was found to be highly active and selective for the HDO of guaiacol in a fixed bed reactor at 300 °C and 71 bar of H2, leading to the hydrogenation of the aromatic ring, followed by demethylation and dihydroxylation to the final cyclohexane. Pt/TiO2 was more stable than alumina-supported catalysts (Ni and Pt). However, no catechol formation was detected, probably due to the excessive amount of hydrogen, used at high pressure.143 Doping the NiMo catalyst with Cu enhanced the reducibility of the NiMo catalyst, while the addition of Ce at an appropriate quantity induced a higher dispersion of active metals and made it difficult to reduce the catalyst. The addition of Ce could effectively inhibit coke formation during the HDO. 1,2-Benzenediol (catechol) was one of the major products formed in the HDO of guaiacol over all the tested catalysts (NiMo, NiMo4Cu, NiMo4Ce, NiMo8Ce); the product distribution can be seen in Fig. 15.144 However, Na-doped CoMo catalysts supported on alumina (γ-Al2O3) showed decreased activity for the HDO of guaiacol compared with undoped catalyst on supports with different Al/Si ratios.145
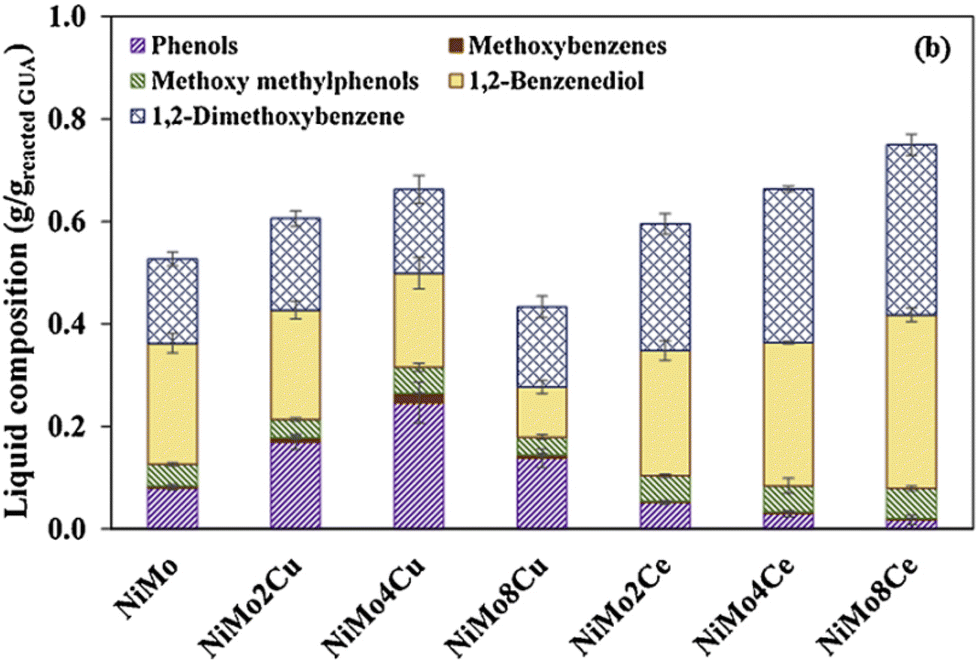 | ||
| Fig. 15 Effect of the promoter type and content on the liquid compositions obtained from the HDO of GUA (condition: catalyst concentration = 15 wt% based on the GUA content at 10 bar H2 pressure and 300 °C for 1 h.)144 | ||
Jongerius et al.146 focused in their study on the HDO of different aromatic monomers (o-cresol, p-cresol, anisole, 4-methylanisole, catechol, guaiacol, 4-methylguaiacol, 1,3-dimethoxybenzene, syringol, and vanillin) and dimers (mimicking the β-O-4 bond) using CoMo/Al2O3 catalyst under 50 bar H2 pressure, performing the reaction in dodecane at 300 °C. The proposed reaction network showed that HDO, demethylation and hydrogenation took place simultaneously. When guaiacol was used as a reactant, 11% of catechol was formed, but the major product was phenol, proposed to be formed in a demethoxylation reaction. Similarly, 4-methylcatechol was formed in 13% yield from 4-methylguaiachol, with the highest yield of methylphenol (42%) as a demethoxylation product. However, converting a dimer with a β-O-4 bond resulted in many monomeric products, but only 1% of catechol was formed among the diol products.146
Pre-reduced NiCu/MCM-41 and NiCu/Ti-MCM-41 catalyst were synthesized and tested for the HDO of guaiacol at 260 °C under 40–100 bar of H2. As expected, high hydrogen pressure led the reaction towards partially or totally deoxygenated products, among which no catechol was detected, but it was supposed to be one of the possible intermediates.116
5. Demethylation of lignin
Natural lignin does not consist of any structural unit that contains the catechol motif with both free hydroxyl groups. However, guaiacyl units are abundant and selective demethylation of these aryl methyl groups is a pathway that leads to the formation of 1,2-dihydroxybenzene structures. In a more general context, demethylation can be viewed as a method to increase the content of phenolic hydroxyl groups, which is desirable for adhesion11 as well as polymer applications.147 Some of the demethylation approaches reviewed above for guaiacol and its derivatives have been successfully applied to lignin. These specific methods will be discussed after a brief overview of catechol formation during more general lignin depolymerization approaches.5.1. Lignin functional groups analysis by NMR spectroscopy
Significant progress has been made in evaluating lignin's structural features by nuclear magnetic resonance (NMR) methodologies. A variety of lignin functional groups and structural insights have been identified by quantitative 31P NMR and semiquantitative two-dimensional heteronuclear (2D 1H–13C HSQC) NMR spectroscopy (Fig. 16).148–150 The technique of phosphitylation followed by 31P NMR spectroscopy extends straightforward advantages in measuring lignin hydroxyl (OH) groups at high single-spectrum resolution. Meng et al. published a well-defined protocol to ensure the scientific accuracy and uniform application of this methodology.149 With an appropriate phosphorus reagent, different lignin OH groups, which also depend on the biomass source, can be quantified by 31P NMR. Among the aliphatic, phenolic and carboxylic OH groups, the content of phenolic OH is the key lignin structural feature that affects its chemical properties and reactivity.151,152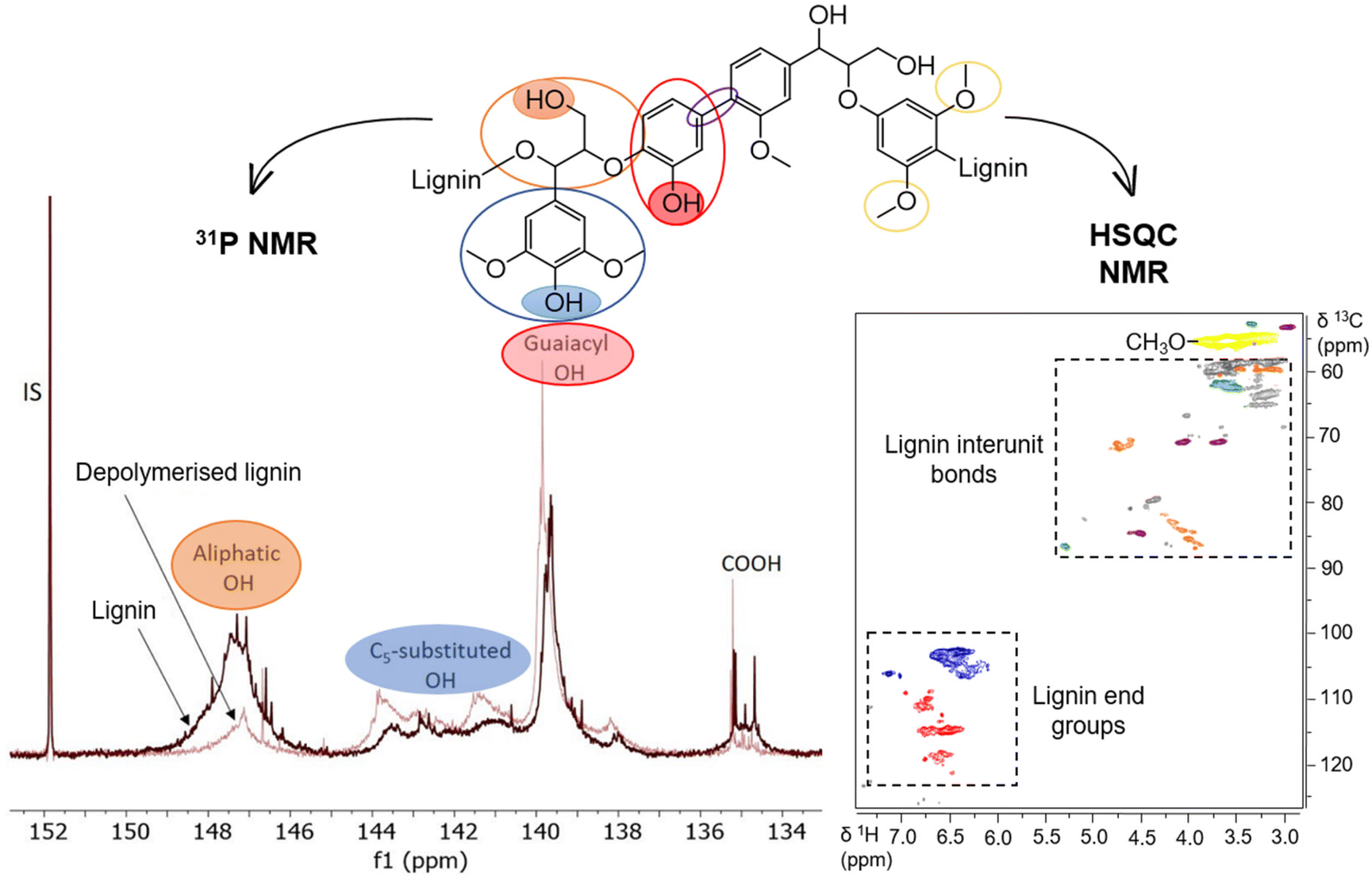 | ||
| Fig. 16 Representative quantitative 31P NMR spectrum (colored filled ellipses: hydroxyl groups) and semi-quantitative 2D HSQC NMR spectrum (colored ellipses: lignin structural units) of lignin samples. | ||
Critically, one-dimensional NMR spectroscopy (i.e., 1H and 13C NMR) can be employed to quantify the phenolic hydroxyl and methoxyl groups in lignin, but is usually associated with severe signal overlap and an inability to measure the aliphatic OH group content. On the other hand, the use of 2D HSQC NMR has brought advances in the determination of lignin subunits (aliphatic and aromatic regions, methoxyl groups, etc.) and lignin carbohydrate complexes. The principles, applications and comparisons of the extremely useful NMR methods for lignin characterization have been summarized in critical reviews.153,154 However, the use of only one NMR technique provides limited opportunities for the structural prediction of the efficient demethylation of lignin.
Lignin undergoes various structural modifications and rearrangements with each type of treatment (i.e., depolymerization, hydrogenation, demethylation, etc.).155 This means that the characterization of demethylated lignin cannot be limited to a single NMR method. 31P NMR provides the content of hydroxyl groups, while C5-substituted and guaiacyl phenolic OH groups overlap with either hydroxyl or methoxyl groups in the C3 or C5 position. Crestini et al. studied different lignin and tannin samples by the 31P NMR method; these have methoxyl and hydroxyl substituted functional groups, respectively.156 The results showed that the signals of guaiacyl and syringyl phenolic OH still overlapped in lignin and tannin even with differently substituted functional groups.149 Therefore, the higher content of phenolic OH groups does not necessarily reflect the demethylation of lignin after treatment, as previously reported.157–159 To detect the demethylation of lignin, HSQC or the molecular weight of lignin had to be performed and determined, respectively. The change in molecular weight of lignin with altered phenolic OH groups content indicates the cleavage of lignin into smaller lignin-based molecules and rearrangements rather than demethylation. On the other hand, the constant molecular weight of lignin and demethylated lignin samples suggests that efficient demethylation occurred. The HSQC method was used to determine the number of methoxyl groups per 100 C9 units by integrating the signals in the aromatic and methoxyl regions. Efficient demethylation of lignin is therefore evidenced by the reduced number of methoxyl groups per 100 C9 units or the reduced ratio of methoxyl groups to total aromatic units.150,160
5.2. Demethylation in the context of lignin depolymerization
In general, lignin depolymerization strategies, in particular ones that target phenolic compounds, are conducted under reaction conditions which also allow for aryl methyl ether cleavage. A review article by Schutyser et al.161 gives a good summary of the literature on acid- and base-catalyzed depolymerization and clearly highlights the formation of catechol and its derivatives. They conclude that catechol mainly appears at high reaction temperatures and typically towards the end of the reaction time;161 a similar behavior is observed during lignin pyrolysis.162 This is indicative of the previously mentioned comparatively high stability of the aryl methyl ether bond. Hydrothermal treatment (sub- or supercritical conditions) of lignin often gives catechol as the main product.163–168 A general problem of these methods relying on harsh reaction conditions is that myriad products are formed and yields for selected molecules are typically low. On the other hand, if these issues could be resolved by substantially improved reaction efficiencies as well as milder reaction conditions, the combination of lignin depolymerization and demethylation would be highly promising. From the viewpoint of green chemistry, the aqueous-phase demethylation approaches appear especially preferable when compared with the organic chemistry-based methods presented in section 5.3. Similarly, catalytic depolymerization and demethylation via hydrotreatment offers the advantage of combining several reaction steps in one pot, thus avoiding additional intermediate separation and purification steps. As highlighted in section 3 and 4, however, neither approach represents an ideal case of a green process; in particular the use hydrogen gas, which presents an explosion hazard, is problematic. It should also be noted that this limits the chances of implementing these processes in industry. Nevertheless, improvements have been reported using catalytic processes, and further developing more suitable catalysts could be a pathway to not only increase the sustainability but also the applicability of these approaches. Some relevant examples are shown in the following paragraph.It is noteworthy that in certain studies it was still possible to obtained unusually high catechol yields. They report reductive catalytic depolymerization approaches performed under conditions similar to the hydrotreatment methods reviewed in section 4. Onwudili and Williams169 found that the presence of formic acid during the catalytic depolymerization of lignin over Pd/C strongly enhanced the demethylation pathway under hydrothermal conditions. The highest catechol yield, ca. 18 wt% based on lignin, was obtained without the Pd catalyst but with formic acid after 6 hours at 265 °C and 65 bar.169 In other studies, not catechol but substituted C9 catechol derivatives could be obtained as primary products. Probably the highest yield of any catechol derivative from lignin was reported by Barta et al.,170 who conducted the demethylation of candlenut shell-derived organosolv lignin over a Cu-based catalyst. After 20 hours at 140 °C a total yield of 64% (four different C9 catechol species) was obtained. After purification, a 43% yield of 4-(3-hydroxypropyl)-catechol could be obtained.170 Another very interesting study was published recently that reported propylcatechol and propenylcatechol as the main products. The approach is based on the reductive catalytic fractionation of C-lignin over a Ni catalyst, which was conducted in a flow setup.171
5.3. Selective lignin demethylation
Besides the examples in the previous subsection that more or less deliberately make use of demethylation as one of the key reactions during depolymerization, a number of studies are reported that focus on demethylation reaction specifically. Furthermore, overviews of different lignin demethylation and defunctionalization methods were included in several recent review articles.11,172,173 It should be noted that in most cases reactions were carried out under inert gas to prevent side reactions.Several studies were published using aqueous HBr or HI for aryl methyl ether cleavage of lignin samples. Wang et al.147 used aqueous HBr and HI for the demethylation of alkali lignin at 130 °C. They obtained increases in the content of phenolic hydroxyl groups of 20–30%. The demethylation efficiency was slightly higher with HI compared with HBr.147 Aqueous HBr was also used in a study by Gao et al.174 in combination with microwave irradiation. After 2 hours at 90 °C the phenolic hydroxyl content of the lignin sample doubled, which made it suitable for polymer applications.174 Variations of this method were reported recently relying on acidic concentrated lithium bromide, i.e. HBr/LiBr, with a reported increase in phenolic OH of up to nearly 100%.175,176
Chai et al.177 applied different acid-based approaches discussed earlier to demethylate lignin. In particular, they used either HBr, BBr3 or AlCl3 as the demethylating agent at 115 °C in dimethyl formamide. BBr3 and AlCl3 proved to be considerably more active for demethylation than HBr, which was found to primarily result in depolymerization of the alkaline lignin. The phenol hydroxyl content could be increased by 80 (BBr3) and 172% (AlCl3) after 4 h. While using BBr3 also led to significant depolymerization, selectivity for demethylation was much higher with AlCl3, so that 75% of β-O-4 linkages could be retained. Moreover, it was shown that especially the reaction time is a crucial parameter of the demethylation reaction.
Song et al.159 applied the approach using iodocyclopropane in dimethyl formamide, which was described in section 2.4, to lignin. After 3 hours at 145 °C the phenolic hydroxyl group content more than tripled while at the same time the amount of methoxy groups decreased by 80%.159 Moreover, the thiolate-based demethylation method (section 2.6) can be used for lignin as well. The hydroxyl group content increased and methoxy group content decreased by ca. 30–40% when Kraft lignin was subjected to dodecanethiol and sodium methoxide in dimethyl formamide at 130 °C for 1 hour.178
A highly interesting study was conducted by Sawamura et al.,179 who compared three different methods, namely thiol-based, iodocyclohexane-based and HI-based demethylation, each under typical reaction conditions. In all cases not only demethylation but also cleavage of other linkages was observed. The iodine-containing reagents showed higher degrees of demethylation than the thiol, and in the case of iodocyclohexane recondensation of fragments was observed, which led to the conclusion that demethylation with iodocyclohexane is most promising.179
In a later study, Kim et al.180 applied Sawamura's approach to milled wood lignin and used iodocyclohexane in dimethyl formamide as demethylation agent at 100 °C. After 24 h the content of phenolic OH increased from 1.02 to 2.98 mmol g−1, indicating a high degree of demethylation, which was confirmed by 2D NMR, even though also a contribution from other aryl–ether bond cleavages is likely. Interestingly, in the study a subsequent depolymerization step by catalytic hydrogenolysis over Ru/C at 300 °C was conducted with the demethylated lignin. This allowed for a high selectivity of catechol-type compounds in the oil fraction (up to 80%). However, the higher content of phenolic OH resulted in a ca. 30% increase in char formation, indicating a tendency towards undesired condensation reactions.
Relying on the acid- and base-catalyzed aryl methyl ether cleavage reactions, Zhao et al.181 investigated the use of protic ionic liquids formed from organic amine bases and organic or mineral acids as reactive solvents for lignin demethylation. In particular, the combination of monoethanolamine and acetic acid resulted in a highly active demethylation agent, which selectively removed 73% of the aromatic methoxy groups after treatment at 90 °C for 2 h. The authors also showed the reusability of the ionic liquid and emphasize the benign reaction conditions.181
Finally, Podschun et al.157 used indium triflate as a Lewis acid for the aqueous-phase demethylation of organosolv lignin. The content of phenolic hydroxyl groups was found to more than double within 3 hours of reaction at 275 °C.157
In view of the discussion of safety, health and environmental factors presented in the previous sections, it may suffice to summarize here that many of the presented lignin demethylation reactions, while highly efficient, cannot be recommended and should be either considerably improved following green chemistry guidelines or avoided in favor of other suitable methods. Among the successfully applied methods for lignin demethylation, the approach using catalytic amounts of non-harmful Lewis acids in combination with water as the solvents reported by Podschun et al.157 stands out as a reaction coming closest to the case of a “green” reaction.
6. Conclusions
Cleavage of aryl methyl ethers is a reaction not only relevant for syntheses of organic molecules used as pharmaceuticals and pesticides, where phenolic hydroxyl groups often have to be initially protected by methylation. Demethylation has also gained attention in the context of lignin utilization where it is a crucial method to convert guaiacyl units into catechol motifs. Increasing the amount of phenolic hydroxyl groups in this way is a valuable means to tune the lignin properties for adhesion and polymer applications, which will be a crucial step towards the utilization of lignin-based resources and replacing conventional fossil feedstocks. With a focus on the lignin model compounds guaiacol and eugenol, different types of demethylation strategy were reviewed.The toolbox of organic chemistry offers a variety of different methods for selective aryl methyl ether cleavage. Here, in particular two comparatively recently modified approaches offer high selectivity while requiring comparatively mild reaction conditions. Stabilized 2-iodoxybenzoic acid can be used in an oxidative demethylation approach followed by a reductive work up. This method tolerates e.g. substituents with primary hydroxyl groups to a high degree. The second strategy is a variation of the long-known aluminium halide-based method. By applying acid scavengers like carbodiimide, pyridine or dimethyl sulfoxide the AlI3-mediated demethylation tolerates e.g. allyl substituents, which are labile e.g. under acidic demethylation conditions. Interestingly, these highly selective approaches have not been applied to lignin samples. Other organic chemistry-derived reagents like e.g. thiol in combination with base or iodocyclohexane, on the other hand, have recently been successfully applied to lignin as well. It should be pointed out, however, that none of these demethylation methods can be considered “green”. On the contrary, in some cases highly hazardous reactants, e.g. BBr3 or IBX, or solvents, e.g. dimethyl formamide or linear alkanes, are required. Thus, none of the reactions can be recommended without reservation.
Acid-catalyzed demethylation in an aqueous medium is a very popular demethylation approach during organic syntheses as well as for lignin modification since no elaborate reagents are needed. Unfortunately, it requires high temperatures and strong mineral acids, which can lead to severe corrosion and the environmental, health and safety hazards associated with it. Furthermore, demethylation under these conditions often suffers from the occurrence of side reactions. On the other hand, in certain cases this means that demethylation can be combined with dealkylation (as one example with eugenol showed) or accompany general lignin depolymerization. Similar selectivity challenges, often resulting in the formation of tar-like side products, also accompany base-catalyzed as well as sub- and supercritical water-based demethylation strategies when applied to pure chemicals like guaiacol. On the other hand, examples of lignin depolymerization show that these latter methods often yield catechol and its derivatives in high yields. For further development, the arguably most promising studies report the use of recyclable solid catalysts in aqueous solution. Considering safety, health and environmental aspects, these reactions are closest to a “green” demethylation procedure among the presented methods.
Finally, the heterogeneously catalyzed hydrotreatment of guaiacol and eugenol typically includes demethylation as one of the earliest reaction steps. In the case of eugenol, over most catalysts the allyl functionality is almost immediately hydrogenated followed by demethylation or demethoxylation. Here, the difficulty is to achieve sufficient selectivity for the demethylated product since competing demethoxylation as well as subsequent dehydroxylation can occur. While most hydrotreatment studies target highly deoxygenated and hydrogenated products up to cyclohexanes, the recent research focus has shifted more towards phenolic products. Catechol and its derivatives, however, have so far not been directly targeted by this approach, which is also evident from the reaction conditions in most studies, which are too harsh to effectively suppress the aforementioned side and follow-up reactions. Hydrotreatment is also relevant from the viewpoint of reductive catalytic lignin depolymerization, where in certain cases surprisingly large amounts of catechol could be obtained. While the true potential of the hydrotreatment approach is currently difficult to assess and remains to be unlocked by purposeful catalyst and process development, it must be pointed out that the industrial applicability will strongly depend on ways to mitigate the explosion hazard associated with hydrogen gas. Nevertheless, in terms of waste production and material balance the catalytic approaches are preferable from an environmental point of view and should not be disregarded per se.
In the overall picture, aryl methyl ether cleavage is a highly important reaction that draws attention from two opposite poles: on the one hand, fine chemical synthesis relies on highly selective demethylation methods while, on the other hand, lignin depolymerization and (de)functionalization more or less deliberately makes use of demethylation. Therefore, it is probably not too far-fetched to expect that selective lignin demethylation could greatly benefit from applying promising demethylation methods from a more organic chemistry background also to lignin. As described in section 5.2., a limited number of studies already pioneered this approach. In turn, the efficient conversion of lignin biomass into catechol and its derivatives has the potential to offer this renewable feedstock for a wide range of applications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Slovenian Research and Innovation Agency (research core funding P2–0152 and research project J2–2492).References
- H. Fiege, H.-W. Voges, T. Hamamoto, S. Umemura, T. Iwata, H. Miki, Y. Fujita, H.-J. Buysch, D. Garbe and W. Paulus, Phenol Derivates, in Ullman's Encycl. Ind. Chem, 2012, pp. 503–519. DOI:10.1002/14356007.a19.
- J. Saiz-Poseu, J. Mancebo-Aracil, F. Nador, F. Busqué and D. Ruiz-Molina, The Chemistry behind Catechol-Based Adhesion, Angew. Chem., Int. Ed., 2019, 58, 696–714, DOI:10.1002/anie.201801063.
- M. Weber, M. Weber and V. Weber, Phenol, in Ullmann's Encycl. Ind. Chem, Wiley, 2020, pp. 1–20. DOI:10.1002/14356007.a19_299.pub3.
- R. Rinaldi, R. Jastrzebski, M. T. Clough, J. Ralph, M. Kennema, P. C. A. Bruijnincx and B. M. Weckhuysen, Paving the Way for Lignin Valorisation: Recent Advances in Bioengineering, Biorefining and Catalysis, Angew. Chem., Int. Ed., 2016, 55, 8164–8215, DOI:10.1002/anie.201510351.
- T. Renders, G. Van den Bossche, T. Vangeel, K. Van Aelst and B. Sels, Reductive catalytic fractionation: state of the art of the lignin-first biorefinery, Curr. Opin. Biotechnol., 2019, 56, 193–201, DOI:10.1016/j.copbio.2018.12.005.
- P. Azadi, O. R. Inderwildi, R. Farnood and D. A. King, Liquid fuels, hydrogen and chemicals from lignin: A critical review, Renewable Sustainable Energy Rev., 2013, 21, 506–523, DOI:10.1016/j.rser.2012.12.022.
- M. V. Galkin and J. S. M. Samec, Lignin Valorization through Catalytic Lignocellulose Fractionation: A Fundamental Platform for the Future Biorefinery, ChemSusChem, 2016, 9, 1544–1558, DOI:10.1002/cssc.201600237.
- N. I. Bhuiyan, J. Begum, N. C. Nandi and F. Akter, Constituents of the essential oil from leaves and buds of clove (Syzigium caryophyllatum (L.) Alston), Afr. J. Plant Sci., 2010, 4, 451–454 Search PubMed https://www.researchgate.net/publication/228673419_Constituents_of_the_essential_oil_from_leaves_and_buds_of_clove_(Syzigium_caryophyllatum_(L.)_Alston) .
- L. Jirovetz, G. Buchbauer, I. Stoilova, A. Stoyanova, A. Krastanov and E. Schmidt, Chemical composition and antioxidant properties of clove leaf essential oil, J. Agric. Food Chem., 2006, 54, 6303–6307, DOI:10.1021/jf060608c.
- F. Coupé, L. Petitjean, P. T. Anastas, F. Caijo, V. Escande and C. Darcel, Sustainable oxidative cleavage of catechols for the synthesis of muconic acid and muconolactones including lignin upgrading, Green Chem., 2020, 22, 6204–6211, 10.1039/d0gc02157a.
- X. Luo and L. Shuai, Lignin–Based Adhesives, Green Adhes., 2020, 25–56, DOI:10.1002/9781119655053.ch2.
- X. Gong, Y. Meng, J. Lu, Y. Tao, Y. Cheng and H. Wang, A Review on Lignin–Based Phenolic Resin Adhesive, Macromol. Chem. Phys., 2022, 223, 2100434, DOI:10.1002/macp.202100434.
- E. Faure, C. Falentin-Daudré, C. Jérôme, J. Lyskawa, D. Fournier, P. Woisel and C. Detrembleur, Catechols as versatile platforms in polymer chemistry, Prog. Polym. Sci., 2013, 38, 236–270, DOI:10.1016/j.progpolymsci.2012.06.004.
- M. V. Bhatt and S. U. Kulkarni, Cleavage of Ethers, Synthesis, 1983, 1983, 249–282, DOI:10.1055/s-1983-30301.
- B. C. Ranu and S. Bhar, Dealkylation of ethers. A review, Org. Prep. Proced. Int., 1996, 28, 371–409, DOI:10.1080/00304949609356549.
- S. A. Weissman and D. Zewge, Recent advances in ether dealkylation, Tetrahedron, 2005, 61, 7833–7863, DOI:10.1016/j.tet.2005.05.041.
- C. D. Snyder and H. Rapoport, Oxidative Cleavage of Hydroquinone Ethers with Argentic Oxide, J. Am. Chem. Soc., 1972, 94, 227–231, DOI:10.1021/ja00756a040.
- M. E. Jung and M. A. Lyster, Quantitative Dealkylation of Alkyl Ethers via Treatment with Trimethylsilyl Iodide. A New Method for Ether Hydrolysis, J. Org. Chem., 1977, 42, 3761–3764, DOI:10.1021/jo00443a033.
- A. Dardas, D. Gal, M. Barrelle, G. Sauret-Ignazi, R. Sterjiades and J. Pelmont, The demethylation of guaiacol by a new bacterial cytochrome P-450, Arch. Biochem. Biophys., 1985, 236, 585–592, DOI:10.1016/0003-9861(85)90662-9.
- E. Lanfranchi, M. Trajković, K. Barta, J. G. de Vries and D. B. Janssen, Exploring the Selective Demethylation of Aryl Methyl Ethers with a Pseudomonas Rieske Monooxygenase, ChemBioChem, 2019, 20, 118–125, DOI:10.1002/cbic.201800594.
- C. Grimm, M. Lazzarotto, S. Pompei, J. Schichler, N. Richter, J. E. Farnberger, M. Fuchs and W. Kroutil, Oxygen-Free Regioselective Biocatalytic Demethylation of Methyl-phenyl Ethers via Methyltransfer Employing Veratrol- O-demethylase, ACS Catal., 2020, 10, 10375–10380, DOI:10.1021/acscatal.0c02790.
- S. González-Rodríguez, T. A. Lu-Chau, X. Chen, G. Eibes, A. Pizzi, G. Feijoo and M. T. Moreira, Functionalisation of organosolv lignin by enzymatic demethylation for bioadhesive formulation, Ind. Crops Prod., 2022, 186, 115253, DOI:10.1016/j.indcrop.2022.115253.
- A. C. Harlington, K. E. Shearwin, S. G. Bell and F. Whelan, Efficient O -demethylation of lignin monoaromatics using the peroxygenase activity of cytochrome P450 enzymes, Chem. Commun., 2022, 58, 13321–13324, 10.1039/D2CC04698A.
- A. Bleem, E. Kuatsjah, G. N. Presley, D. J. Hinchen, M. Zahn, D. C. Garcia, W. E. Michener, G. König, K. Tornesakis, M. N. Allemann, R. J. Giannone, J. E. McGeehan, G. T. Beckham and J. K. Michener, Discovery, characterization, and metabolic engineering of Rieske non-heme iron monooxygenases for guaiacol O-demethylation, Chem. Catal., 2022, 2, 1989–2011, DOI:10.1016/j.checat.2022.04.019.
- F. L. Benton and T. E. Dillon, The Cleavage of Ethers with Boron Bromide. I. Some Common Ethers, J. Am. Chem. Soc., 1942, 64, 1128–1129, DOI:10.1021/ja01257a035.
- J. F. W. McOmie, M. L. Watts and D. E. West, Demethylation of aryl methyl ethers by boron tribromide, Tetrahedron, 1968, 24, 2289–2292, DOI:10.1016/0040-4020(68)88130-X.
- L. Li, D. Qiu, J. Shi and Y. Li, Vicinal Diamination of Arenes with Domino Aryne Precursors, Org. Lett., 2016, 18, 3726–3729, DOI:10.1021/acs.orglett.6b01747.
- P. Bovicelli, R. Antonioletti, S. Mancini, S. Causio, G. Borioni, S. Ammendola and M. Barontini, Expedient synthesis of hydroxytyrosol and its esters, Synth. Commun., 2007, 37, 4245–4252, DOI:10.1080/00397910701575509.
- C. Sousa and P. J. Silva, BBr3-assisted cleavage of most ethers does not follow the commonly assumed mechanism, Eur. J. Org. Chem., 2013, 5195–5199, DOI:10.1002/ejoc.201300337.
- T. M. Kosak, H. A. Conrad, A. L. Korich and R. L. Lord, Ether Cleavage Re-Investigated: Elucidating the Mechanism of BBr3-Facilitated Demethylation of Aryl Methyl Ethers, Eur. J. Org. Chem., 2015, 7460–7467, DOI:10.1002/ejoc.201501042.
- A. Suzuki, S. Hara and X. Huang, Boron Tribromide, in Encycl. Reagents Org. Synth, John Wiley & Sons, Ltd, Chichester, UK, 2006. DOI:10.1002/047084289X.rb244.pub2.
- D. Prat, A. Wells, J. Hayler, H. Sneddon, C. R. McElroy, S. Abou-Shehada and P. J. Dunn, CHEM21 selection guide of classical- and less classical-solvents, Green Chem., 2015, 18, 288–296, 10.1039/C5GC01008J.
- A. A. Torrens, A. L. Ly, D. Fong and A. Adronov, Rapid and Mild Cleavage of Aryl–Alkyl Ethers to Liberate Phenols, Eur. J. Org. Chem., 2022, 2022(27), 65–70, DOI:10.1002/ejoc.202200570.
- E. G. Paul and P. S. C. Wang, Reaction of 2, 3, 4, 6-Tetramethoxybenzaldehyde with Aluminum Chloride. Selective Cleavage at Position 2 and Selective Ether Exchange at Position 3, J. Org. Chem., 1979, 44, 2307–2308, DOI:10.1021/jo01327a066.
- M. Node, K. Nishide, K. Fuji and E. Fujita, Hard Acid and Soft Nucleophile System. 2.1 Demethylation of Methyl Ethers of Alcohol and Phenol with an Aluminum Halide-Thiol System, J. Org. Chem., 1980, 45, 4275–4277, DOI:10.1021/jo01310a003.
- K. Nishide, S. I. Ohsugi, T. Miyamoto, K. Kumar and M. Node, Development of Odorless Thiols and Sulfides and Their Applications to Organic Synthesis, Monatsh. Chem., 2004, 135, 189–200, DOI:10.1007/s00706-003-0122-1.
- B. Arifin, D. F. Tang and S. S. Achmadi, Transformation of eugenol and safrole into hydroxychavicol, Indones. J. Chem., 2015, 15, 77–85, DOI:10.22146/ijc.21227.
- M. Vivekananda Bhatt and J. Ramesh Babu, New reagents 3: aluminium iodide - a highly regioselective ether-cleaving reagent with novel cleavage pattern, Tetrahedron Lett., 1984, 25, 3497–3500, DOI:10.1016/S0040-4039(01)91058-5.
- D. Deffieux, P. Gossart and S. Quideau, Facile and sustainable synthesis of the natural antioxidant hydroxytyrosol, Tetrahedron Lett., 2014, 55, 2455–2458, DOI:10.1016/j.tetlet.2014.02.134.
- S. Andersson, An Improvement of the Aluminium Iodide Method for Ether Cleavage: Catalysis by Quaternary Ammonium Iodides, Synthesis, 1985, 1985, 437–439, DOI:10.1055/s-1985-31235.
- D. Sang, M. Yao, J. Tian, X. Chen, L. Li, H. Zhan and L. You, Pyridine Improves Aluminum Triiodide Induced Selective Cleavage of Alkyl o -Hydroxyphenyl Ethers: A Practical and Efficient Procedure for the Preparation of Hydroxychavicol by Demethylation of Eugenol, Synlett, 2017, 28, 138–142, DOI:10.1055/s-0035-1588889.
- J. Tian, C. Yi, Z. He, M. Yao and D. Sang, Aluminum Triiodide-Mediated Cleavage of o-Hydroxyphenyl Alkyl Ethers Using Inorganic Bases and Metal Oxides as Acid Scavengers, ChemistrySelect, 2017, 2, 9211–9214, DOI:10.1002/slct.201701685.
- D. Sang, J. Wang, Y. Zheng, J. He, C. Yuan, Q. An and J. Tian, Carbodiimides as Acid Scavengers in Aluminum Triiodide Induced Cleavage of Alkyl Aryl Ethers, Synthesis, 2017, 49, 2721–2726, DOI:10.1055/s-0036-1588755.
- D. Sang, J. Tian, X. Tu, Z. He and M. Yao, Cleavage of Catechol Monoalkyl Ethers by Aluminum Triiodide-Dimethyl Sulfoxide, Synthesis, 2019, 51, 704–712, DOI:10.1055/s-0037-1610996.
- J. Tian, H. Yue, P. Yang and D. Sang, One-Pot Cleavage of Aryl Alkyl Ethers by Aluminum and Iodine in Acetonitrile, ChemistrySelect, 2019, 4, 38–41, DOI:10.1002/slct.201803469.
- A. M. Bernard, M. R. Ghiani, P. P. Piras and A. Rivoldini, Dealkylation of activated alkyl aryl ethers using lithium chloride in dimethylformamide, Synthesis, 1989, 1989, 287–289, DOI:10.1055/s-1989-27225.
- Z. Fang, G. C. Zhou, S. L. Zheng, G. L. He, J. L. Li, L. He and D. Bei, Lithium chloride-catalyzed selective demethylation of aryl methyl ethers under microwave irradiation, J. Mol. Catal. A: Chem., 2007, 274, 16–23, DOI:10.1016/j.molcata.2007.04.013.
- P. Kraft and W. Eichenberger, Conception, characterization and correlation of new marine odorants, Eur. J. Org. Chem., 2003, 3735–3743, DOI:10.1002/ejoc.200300174.
- A. Alni, M. D. Puspita and D. Mujahidin, Unexpected dimer from demethylization reaction of eugenol under acidic conditions, J. Math. Fundam. Sci., 2019, 51, 60–67, DOI:10.5614/j.math.fund.sci.2019.51.1.5.
- L. Zuo, S. Yao, W. Wang and W. Duan, An efficient method for demethylation of aryl methyl ethers, Tetrahedron Lett., 2008, 49, 4054–4056, DOI:10.1016/j.tetlet.2008.04.070.
- R. L. Burwell, The cleavage of ethers, Chem. Rev., 1954, 54, 615–685, DOI:10.1021/cr60170a003.
- A. Ozanne, L. Pouységu, D. Depernet, B. François and S. Quideau, A stabilized formulation of IBX (SIBX) for safe oxidation reactions including a new oxidative demethylation of phenolic methyl aryl ethers, Org. Lett., 2003, 5, 2903–2906, DOI:10.1021/ol0349965.
- L. Pouységu, T. Sylla, T. Garnier, L. B. Rojas, J. Charris, D. Deffieux and S. Quideau, Hypervalent iodine-mediated oxygenative phenol dearomatization reactions, Tetrahedron, 2010, 66, 5908–5917, DOI:10.1016/j.tet.2010.05.078.
- S. Quideau and A. Chénedé, Stabilized 2-Iodoxybenzoic Acid (SIBX), in Encycl. Reagents Org. Synth, John Wiley & Sons, Ltd, Chichester, UK, 2012, pp. 1–3. DOI:10.1002/047084289X.rn01376.
- E. Adler, S. Hernestam, E. Boss and A. Caglieris, Estimation of Phenolic Hydroxyl Groups in Lignin. I. Periodate Oxidation of Guaiacol Compounds., Acta Chem. Scand., 1955, 9, 319–334, DOI:10.3891/acta.chem.scand.09-0319.
- E. Adler, R. Magnusson, S. E. Hansen, R. Sömme, E. Stenhagen and H. Palmstierna, Periodate Oxidation of Phenols. I. Monoethers of Pyrocatechol and Hydroquinone, Acta Chem. Scand., 1959, 13, 505–519, DOI:10.3891/acta.chem.scand.13-0505.
- G. I. Feutrill and R. N. Mirrington, Demethylation of aryl methyl ethers with thioethoxide ion in dimethyl formamide, Tetrahedron Lett., 1970, 11, 1327–1328, DOI:10.1016/S0040-4039(01)97960-2.
- G. I. Feutrill and R. N. Mirrington, Reactions with thioethoxide ion in dimethylformamide: I. Selective demethylation of aryl methyl ethers, Aust. J. Chem., 1972, 25, 1719–1729, DOI:10.1071/CH9721719.
- J. Magano, M. H. Chen, J. D. Clark and T. Nussbaumer, 2-(Diethylamino)ethanethiol, a new reagent for the odorless deprotection of aromatic methyl ethers, J. Org. Chem., 2006, 71, 7103–7105, DOI:10.1021/jo0611059.
- J. Chae, Practical demethylation of aryl methyl ethers using an odorless thiol reagent, Arch. Pharmacal Res., 2008, 31, 305–309, DOI:10.1007/s12272-001-1156-y.
- M. S. Lamba and J. K. Makrandi, Demethylation of aryl methyl ethers using pyridinium p-toluenesulfonate under microwave irradiation, J. Chem. Res., 2007, 153, 585–586, DOI:10.3184/030823407X255551.
- G. Driver and K. E. Johnson, 3-Methylimidazolium bromohydrogenates(I): A room-temperature ionic liquid for ether cleavage, Green Chem., 2003, 5, 163–169, 10.1039/b211548d.
- S. K. Boovanahalli, D. W. Kim and D. Y. Chi, Application of Ionic Liquid Halide Nucleophilicity for the Cleavage of Ethers: A Green Protocol for the Regeneration of Phenols from Ethers, J. Org. Chem., 2004, 69, 3340–3344, DOI:10.1021/jo035886e.
- J. Park and J. Chae, Microwave-assisted demethylation of methyl aryl ethers using an ionic liquid, Synlett, 2010, 1651–1656, DOI:10.1055/s-0030-1258087.
- L. Liu, Z. Li, C. Chen, H. Li, L. Xu and Z. Yu, Efficient dealkylation of aryl alkyl ethers catalyzed by Cu2O, Tetrahedron, 2018, 74, 2447–2453, DOI:10.1016/j.tet.2018.03.070.
- A. F. Duprat, P. Capdevielle and M. Maumy, Aromatic O-demethylation with the [Fe0/acetic acid/O2] system, J. Mol. Catal., 1992, 77, L7–L11, DOI:10.1016/0304-5102(92)80190-R.
- A. Maercker, Ether Cleavage with Organo–Alkali–Metal Compounds and Alkali Metals, Angew. Chem., Int. Ed. Engl., 1987, 26, 972–989, DOI:10.1002/anie.198709721.
- S. B. Hawthorne, Y. Yang and D. J. Miller, Extraction of Organic Pollutants from Environmental Solids with Sub- and Supercritical Water, Anal. Chem., 1994, 66, 2912–2920, DOI:10.1021/ac00090a019.
- S. Zhao and M. M. Abu-Omar, Biobased epoxy nanocomposites derived from lignin-based monomers, Biomacromolecules, 2015, 16, 2025–2031, DOI:10.1021/acs.biomac.5b00670.
- D. Landini, F. Montanari and F. Rolla, Cleavage of Dialkyl and Aryl Alkyl Ethers with Hydrobromic Acid in the Presence of Phase-Transfer Catalysts, Synthesis, 1978, 1978, 771–773, DOI:10.1055/s-1978-24888.
- S. B. Waghmode, G. Mahale, V. P. Patil, K. Renalson and D. Singh, Efficient method for demethylation of aryl methyl ether using aliquat-336, Synth. Commun., 2013, 43, 3272–3280, DOI:10.1080/00397911.2013.772201.
- L. Yang, W. Zhou, K. Seshan and Y. Li, Green and efficient synthesis route of catechol from guaiacol, J. Mol. Catal. A: Chem., 2013, 368–369, 61–65, DOI:10.1016/j.molcata.2012.11.024.
- J. Bomon, E. Van Den Broeck, M. Bal, Y. Liao, S. Sergeyev, V. Van Speybroeck, B. F. Sels and B. U. W. Maes, Brønsted Acid Catalyzed Tandem Defunctionalization of Biorenewable Ferulic acid and Derivates into Bio-Catechol, Angew. Chem., Int. Ed., 2020, 59, 3063–3068, DOI:10.1002/anie.201913023.
- L. Yang, Y. Li and P. E. Savage, Hydrolytic cleavage of C-O linkages in lignin model compounds catalyzed by water-tolerant Lewis acids, Ind. Eng. Chem. Res., 2014, 53, 2633–2639, DOI:10.1021/ie403545n.
- X. Wu, Y. Liao, J. Bomon, G. Tian, S. Bai, K. Van Aelst, Q. Zhang, W. Vermandel, B. Wambacq, B. U. W. Maes, J. Yu and B. F. Sels, Lignin–First Monomers to Catechol: Rational Cleavage of C−O and C−C Bonds over Zeolites, ChemSusChem, 2022, 15(7), e202102248, DOI:10.1002/cssc.202102248.
- M. Sainsbury, S. F. Dyke and B. J. Moon, 1,2-Dihydroisoquinolines. Part XIII. Synthesis of sanguinarine chloride, J. Chem. Soc. C, 1970, 1797, 10.1039/j39700001797.
- H. Weingärtner and E. U. Franck, Supercritical water as a solvent, Angew. Chem., Int. Ed., 2005, 44, 2672–2692, DOI:10.1002/anie.200462468.
- J. J. R. Lawson and M. M. T. Klein, Influence of Water on Guaiacol Pyrolysis, Ind. Eng. Chem. Fundam., 1985, 24, 203–208, DOI:10.1021/i100018a012.
- S. H. Townsend, M. A. Abraham, G. L. Huppert, M. T. Klein and S. C. Paspek, Solvent Effects During Reactions in Supercritical Water, Ind. Eng. Chem. Res., 1988, 27, 143–149, DOI:10.1021/ie00073a026.
- G. L. Huppert, B. C. Wu, S. H. Townsend, S. C. Paspek and M. T. Klein, Hydrolysis in Supercritical Water: Identification and Implications of a Polar Transition State, Ind. Eng. Chem. Res., 1989, 28, 161–165, DOI:10.1021/ie00086a006.
- G. J. DiLeo, M. E. Neff and P. E. Savage, Gasification of guaiacol and phenol in supercritical water, Energy Fuels, 2007, 21, 2340–2345, DOI:10.1021/ef070056f.
- Wahyudiono, M. Sasaki and M. Goto, Thermal decomposition of guaiacol in sub- and supercritical water and its kinetic analysis, J. Mater. Cycles Waste Manage., 2011, 13, 68–79, DOI:10.1007/s10163-010-0309-6.
- E. B. Ledesma, C. Campos, D. J. Cranmer, B. L. Foytik, M. N. Ton, E. A. Dixon, C. Chirino, S. Batamo and P. Roy, Vapor-phase cracking of eugenol: Distribution of tar products as functions of temperature and residence time, Energy Fuels, 2013, 27, 868–878, DOI:10.1021/ef3018332.
- E. B. Ledesma, J. N. Hoang, Q. Nguyen, V. Hernandez, M. P. Nguyen, S. Batamo and C. K. Fortune, Unimolecular decomposition pathway for the vapor-phase cracking of eugenol, a biomass tar compound, Energy Fuels, 2013, 27, 6839–6846, DOI:10.1021/ef401760c.
- R. Ceylan and J. B. s. Bredenberg, Hydrogenolysis and hydrocracking of the carbon-oxygen bond. 2. Thermal cleavage of the carbon-oxygen bond in guaiacol, Fuel, 1982, 61, 377–382, DOI:10.1016/0016-2361(82)90054-0.
- J. B. Bredenberg and R. Ceylan, Hydrogenolysis and hydrocracking of the carbon-oxygen bond. 3. Thermolysis in tetralin of substituted anisoles, Fuel, 1983, 62, 342–344, DOI:10.1016/0016-2361(83)90093-5.
- A. I. Afifi, E. Chornet, R. W. Thring and R. P. Overend, The aryl ether bond reactions with H-donor solvents: Guaiacol and tetralin in the presence of catalysts, Fuel, 1996, 75, 509–516, DOI:10.1016/0016-2361(95)00199-9.
- M. Bystrzanowska, P. Petkov and M. Tobiszewski, Ranking of Heterogeneous Catalysts Metals by Their Greenness, ACS Sustainable Chem. Eng., 2019, 7, 18434–18443, DOI:10.1021/acssuschemeng.9b04230.
- J. Xing, L. Song, C. Zhang, M. Zhou, L. Yue and X. Li, Effect of acidity and porosity of alkali-treated ZSM-5 zeolite on eugenol hydrodeoxygenation, Catal. Today, 2015, 258, 90–95, DOI:10.1016/j.cattod.2015.04.014.
- C. C. Zhang, J. Qi, J. Xing, S. F. Tang, L. Song, Y. Sun, C. C. Zhang, H. Xin and X. Li, An investigation on the aqueous-phase hydrodeoxygenation of various methoxy-substituted lignin monomers on Pd/C and HZSM-5 catalysts, RSC Adv., 2016, 6, 104398–104406, 10.1039/c6ra22492j.
- X. Besse, Y. Schuurman and N. Guilhaume, Hydrothermal conversion of lignin model compound eugenol, Catal. Today, 2015, 258, 270–275, DOI:10.1016/j.cattod.2014.12.010.
- L. Nie, B. Peng and X. Zhu, Vapor-Phase Hydrodeoxygenation of Guaiacol to Aromatics over Pt/HBeta: Identification of the Role of Acid Sites and Metal Sites on the Reaction Pathway, ChemCatChem, 2018, 10, 1064–1074, DOI:10.1002/cctc.201701413.
- F. P. Bouxin, X. Zhang, I. N. Kings, A. F. Lee, M. J. H. Simmons, K. Wilson and S. D. Jackson, Deactivation study of the hydrodeoxygenation of p-methylguaiacol over silica supported rhodium and platinum catalysts, Appl. Catal., A, 2017, 539, 29–37, DOI:10.1016/j.apcata.2017.03.039.
- C. Zhang, J. Xing, L. Song, H. Xin, S. Lin, L. Xing and X. Li, Aqueous-phase hydrodeoxygenation of lignin monomer eugenol: Influence of Si/Al ratio of HZSM-5 on catalytic performances, Catal. Today, 2014, 234, 145–152, DOI:10.1016/j.cattod.2014.01.021.
- M.Á González-Borja and D. E. Resasco, Anisole and guaiacol hydrodeoxygenation over monolithic Pt-Sn catalysts, Energy Fuels, 2011, 25, 4155–4162, DOI:10.1021/ef200728r.
- T. Nimmanwudipong, R. C. Runnebaum, S. E. Ebeler, D. E. Block and B. C. Gates, Upgrading of lignin-derived compounds: Reactions of eugenol catalyzed by HY zeolite and by Pt/γ-Al 2O 3, Catal. Lett., 2012, 142, 151–160, DOI:10.1007/s10562-011-0759-z.
- H. Duan, Y. Tian, S. Gong, B. Zhang, Z. Lu, Y. Xia, Y. Shi and C. Qiao, Effects of Crystallite Sizes of Pt/HZSM-5 Zeolite Catalysts on the Hydrodeoxygenation of Guaiacol, Nanomaterials, 2020, 10, 2246, DOI:10.3390/nano10112246.
- R. C. Runnebaum, T. Nimmanwudipong, D. E. Block and B. C. Gates, Catalytic conversion of compounds representative of lignin-derived bio-oils: A reaction network for guaiacol, anisole, 4-methylanisole, and cyclohexanone conversion catalysed by Pt/γ-Al 2O 3, Catal. Sci. Technol., 2012, 2, 113–118, 10.1039/c1cy00169h.
- N. K. G. Silva, R. A. R. Ferreira, R. M. Ribas, R. S. Monteiro, M. A. S. Barrozo and R. R. Soares, Gas-phase hydrodeoxygenation (HDO) of guaiacol over Pt/Al2O3 catalyst promoted by Nb2O5, Fuel, 2021, 287, 119509, DOI:10.1016/j.fuel.2020.119509.
- E. A. Roldugina, A. P. Glotov, A. L. Isakov, A. L. Maksimov, V. A. Vinokurov and E. A. Karakhanov, Ruthenium Catalysts on ZSM-5/MCM-41 Micro-Mesoporous Support for Hydrodeoxygenation of Guaiacol in the Presence of Water, Russ. J. Appl. Chem., 2019, 92, 1170–1178, DOI:10.1134/S1070427219080172.
- Y. Nakagawa, M. Ishikawa, M. Tamura and K. Tomishige, Selective production of cyclohexanol and methanol from guaiacol over Ru catalyst combined with MgO, Green Chem., 2014, 16, 2197–2203, 10.1039/c3gc42322k.
- S. Gyergyek, A. Kocjan, A. Bjelić, M. Grilc, B. Likozar and D. Makovec, Magnetically separable Ru-based nano-catalyst for the hydrogenation/hydro-deoxygenation of lignin-derived platform chemicals, Mater. Res. Lett., 2018, 6, 426–431, DOI:10.1080/21663831.2018.1477847.
- A. A. Dwiatmoko, J. Seo, J. W. Choi, D. J. Suh, J. Jae and J. M. Ha, Improved activity of a CaCO3-supported Ru catalyst for the hydrodeoxygenation of eugenol as a model lignin-derived phenolic compound, Catal. Commun., 2019, 127, 45–50, DOI:10.1016/j.catcom.2019.04.024.
- Z. Luo, Z. Zheng, Y. Wang, G. Sun, H. Jiang and C. Zhao, Hydrothermally stable Ru/HZSM-5-catalyzed selective hydrogenolysis of lignin-derived substituted phenols to bio-arenes in water, Green Chem., 2016, 18, 5845–5858, 10.1039/c6gc01971d.
- M. Ishikawa, M. Tamura, Y. Nakagawa and K. Tomishige, Demethoxylation of guaiacol and methoxybenzenes over carbon-supported Ru-Mn catalyst, Appl. Catal., B, 2016, 182, 193–203, DOI:10.1016/j.apcatb.2015.09.021.
- D. Singh and P. L. Dhepe, An efficient catalytic transfer hydrogenation-hydrodeoxygenation of lignin derived monomers: Investigating catalyst properties-activity correlation, Catal. Commun., 2020, 196, 106220, DOI:10.1016/j.catcom.2020.106220.
- C. R. Lee, J. S. Yoon, Y. W. Suh, J. W. Choi, J. M. Ha, D. J. Suh and Y. K. Park, Catalytic roles of metals and supports on hydrodeoxygenation of lignin monomer guaiacol, Catal. Commun., 2012, 17, 54–58, DOI:10.1016/j.catcom.2011.10.011.
- A. Bjelić, M. Grilc, M. Huš and B. Likozar, Hydrogenation and hydrodeoxygenation of aromatic lignin monomers over Cu/C, Ni/C, Pd/C, Pt/C, Rh/C and Ru/C catalysts: Mechanisms, reaction micro-kinetic modelling and quantitative structure-activity relationships, Chem. Eng. J., 2019, 359, 305–320, DOI:10.1016/j.cej.2018.11.107.
- M. Alda-Onggar, P. Mäki-Arvela, K. Eränen, A. Aho, J. Hemming, P. Paturi, M. Peurla, M. Lindblad, I. L. Simakova and D. Y. Murzin, Hydrodeoxygenation of Isoeugenol over Alumina-Supported Ir, Pt, and Re Catalysts, ACS Sustainable Chem. Eng., 2018, 6, 16205–16218, DOI:10.1021/acssuschemeng.8b03035.
- R. K. M. R. Kallury, W. M. Restivo, T. T. Tidwell, D. G. B. Boocock, A. Crimi and J. Douglas, Hydrodeoxygenation of hydroxy, methoxy and methyl phenols with molybdenum oxide/nickel oxide/alumina catalyst, J. Catal., 1985, 96, 535–543, DOI:10.1016/0021-9517(85)90321-5.
- A. K. Deepa and P. L. Dhepe, Function of metals and supports on the hydrodeoxygenation of phenolic compounds, ChemPlusChem, 2014, 79, 1573–1583, DOI:10.1002/cplu.201402145.
- M. Huš, A. Bjelić, M. Grilc and B. Likozar, First-principles mechanistic study of ring hydrogenation and deoxygenation reactions of eugenol over Ru(0001) catalysts, J. Catal., 2018, 358, 8–18, DOI:10.1016/j.jcat.2017.11.020.
- A. Bjelić, M. Grilc, S. Gyergyek, A. Kocjan, D. Makovec and B. Likozar, Catalytic hydrogenation, hydrodeoxygenation, and hydrocracking processes of a lignin monomer model compound eugenol over magnetic Ru/C–Fe2O3 and mechanistic reaction microkinetics, Catalysts, 2018, 333(8), 240–259, DOI:10.3390/catal8100425.
- A. Bjelić, M. Grilc and B. Likozar, Catalytic hydrogenation and hydrodeoxygenation of lignin-derived model compound eugenol over Ru/C: Intrinsic microkinetics and transport phenomena, Chem. Eng. J., 2018, 333, 240–259, DOI:10.1016/j.cej.2017.09.135.
- A. Bjelić, B. Likozar and M. Grilc, Scaling of lignin monomer hydrogenation, hydrodeoxygenation and hydrocracking reaction micro-kinetics over solid metal/acid catalysts to aromatic oligomers, Chem. Eng. J., 2020, 399, 125712, DOI:10.1016/j.cej.2020.125712.
- Y. C. Lin, C. L. Li, H. P. Wan, H. T. Lee and C. F. Liu, Catalytic hydrodeoxygenation of guaiacol on Rh-based and sulfided CoMo and NiMo catalysts, Energy Fuels, 2011, 25, 890–896, DOI:10.1021/ef101521z.
- X. Liu, W. Jia, G. Xu, Y. Zhang and Y. Fu, Selective Hydrodeoxygenation of Lignin-Derived Phenols to Cyclohexanols over Co-Based Catalysts, ACS Sustainable Chem. Eng., 2017, 5, 8594–8601, DOI:10.1021/acssuschemeng.7b01047.
- R. Ma, K. Cui, L. Yang, X. Ma and Y. Li, Selective catalytic conversion of guaiacol to phenols over a molybdenum carbide catalyst, Chem. Commun., 2015, 51, 10299–10301, 10.1039/c5cc01900a.
- L. Bomont, M. Alda-Onggar, V. Fedorov, A. Aho, J. Peltonen, K. Eränen, M. Peurla, N. Kumar, J. Wärnå, V. Russo, P. Mäki-Arvela, H. Grénman, M. Lindblad and D. Y. Murzin, Production of Cycloalkanes in Hydrodeoxygenation of Isoeugenol Over Pt- and Ir-Modified Bifunctional Catalysts, Eur. J. Inorg. Chem., 2018, 2018, 2841–2854, DOI:10.1002/ejic.201800391.
- J. Qi, S. F. Tang, Y. Sun, C. Xu and X. Li, Nickel Phosphides Supported on HZSM-5 for Catalytic Hydrodeoxygenation of Eugenol: Effect of Phosphorus Content, ChemistrySelect, 2017, 2, 7525–7529, DOI:10.1002/slct.201701391.
- A. Ramesh, P. Tamizhdurai, V. L. Mangesh, K. Palanichamy, S. Gopinath, K. Sureshkumar and K. Shanthi, Mg/SiO2−Al2O3 supported nickel catalysts for the production of naphthenic hydrocarbon fuel by hydro-de-oxygenation of eugenol, Int. J. Hydrogen Energy, 2019, 44, 25607–25620, DOI:10.1016/j.ijhydene.2019.08.024.
- J. Qi, X. Sun, S. F. Tang, Y. Sun, C. Xu, X. Li and X. Li, Integrated study on the role of solvent, catalyst and reactant in the hydrodeoxygenation of eugenol over nickel-based catalysts, Appl. Catal., A, 2017, 535, 24–31, DOI:10.1016/j.apcata.2017.01.020.
- A. Sreenavya, A. Sahu and A. Sakthivel, Hydrogenation of Lignin-Derived Phenolic Compound Eugenol over Ruthenium-Containing Nickel Hydrotalcite-Type Materials, Ind. Eng. Chem. Res., 2020, 59, 11979–11990, DOI:10.1021/acs.iecr.0c01106.
- H. Jahromi and F. A. Agblevor, Hydrotreating of guaiacol: A comparative study of Red mud-supported nickel and commercial Ni/SiO2-Al2O3 catalysts, Appl. Catal., A, 2018, 558, 109–121, DOI:10.1016/j.apcata.2018.03.016.
- S. Gutiérrez-Rubio, I. Moreno, D. P. Serrano and J. M. Coronado, Hydrotreating of Guaiacol and Acetic Acid Blends over Ni2P/ZSM-5 Catalysts: Elucidating Molecular Interactions during Bio-Oil Upgrading, ACS Omega, 2019, 4, 21516–21528, DOI:10.1021/acsomega.9b03221.
- X. Lan, E. J. M. Hensen and T. Weber, Hydrodeoxygenation of guaiacol over Ni2P/SiO2−reaction mechanism and catalyst deactivation, Appl. Catal., A, 2018, 550, 57–66, DOI:10.1016/j.apcata.2017.10.018.
- S. K. Wu, P. C. Lai and Y. C. Lin, Atmospheric hydrodeoxygenation of guaiacol over nickel phosphide catalysts: Effect of phosphorus composition, Catal. Lett., 2014, 144, 878–889, DOI:10.1007/s10562-014-1231-7.
- S. K. Wu, P. C. Lai, Y. C. Lin, H. P. Wan, H. T. Lee and Y. H. Chang, Atmospheric hydrodeoxygenation of guaiacol over alumina-, zirconia-, and silica-supported nickel phosphide catalysts, ACS Sustainable Chem. Eng., 2013, 1, 349–358, DOI:10.1021/sc300157d.
- H. Y. Zhao, D. Li, P. Bui and S. T. Oyama, Hydrodeoxygenation of guaiacol as model compound for pyrolysis oil on transition metal phosphide hydroprocessing catalysts, Appl. Catal., A, 2011, 391, 305–310, DOI:10.1016/j.apcata.2010.07.039.
- J. E. Peters, J. R. Carpenter and D. C. Dayton, Anisole and guaiacol hydrodeoxygenation reaction pathways over selected catalysts, Energy Fuels, 2015, 29, 909–916, DOI:10.1021/ef502551p.
- C. Sepúlveda, K. Leiva, R. García, L. R. Radovic, I. T. Ghampson, W. J. Desisto, J. L. G. Fierro and N. Escalona, Hydrodeoxygenation of 2-methoxyphenol over Mo2N catalysts supported on activated carbons, Catal. Today, 2011, 172, 232–239, DOI:10.1016/j.cattod.2011.02.061.
- P. E. Ruiz, B. G. Frederick, W. J. De Sisto, R. N. Austin, L. R. Radovic, K. Leiva, R. García, N. Escalona and M. C. Wheeler, Guaiacol hydrodeoxygenation on MoS 2 catalysts: Influence of activated carbon supports, Catal. Commun., 2012, 27, 44–48, DOI:10.1016/j.catcom.2012.06.021.
- M. Ferrari, S. Bosmans, R. Maggi, B. Delmon and P. Grange, CoMo/carbon hydrodeoxygenation catalysts: Influence of the hydrogen sulfide partial pressure and of the sulfidation temperature, Catal. Today, 2001, 65, 257–264, DOI:10.1016/S0920-5861(00)00559-9.
- E. Santillan-Jimenez, M. Perdu, R. Pace, T. Morgan and M. Crocker, Activated carbon, carbon nanofiber and carbon nanotube supported molybdenum carbide catalysts for the hydrodeoxygenation of guaiacol, Catalysts, 2015, 5, 424–441, DOI:10.3390/catal5010424.
- L. Petitjean, R. Gagne, E. S. Beach, D. Xiao and P. T. Anastas, Highly selective hydrogenation and hydrogenolysis using a copper-doped porous metal oxide catalyst, Green Chem., 2015, 18, 150–156, 10.1039/c5gc01464f.
- F. Yan, Y. Sang, Y. Bai, K. Wu, K. Cui, Z. Wen, F. Mai, Z. Ma, L. Yu, H. Chen and Y. Li, Guaiacol demethoxylation catalyzed by Re2O7 in ethanol, Catal. Today, 2019, 1–7, DOI:10.1016/j.cattod.2019.07.018.
- K. Bin Jung, J. Lee, J. M. Ha, H. Lee, D. J. Suh, C. H. Jun and J. Jae, Effective hydrodeoxygenation of lignin-derived phenols using bimetallic RuRe catalysts: Effect of carbon supports, Catal. Today, 2018, 303, 191–199, DOI:10.1016/j.cattod.2017.07.027.
- K. Leiva, C. Sepúlveda, R. García, J. L. G. Fierro and N. Escalona, Effect of water on the conversions of 2-methoxyphenol and phenol as bio-oil model compounds over ReS2/SiO2 catalyst, Catal. Commun., 2014, 53, 33–37, DOI:10.1016/j.catcom.2014.04.023.
- H. Lee, Y.-M. Kim, I.-G. Lee, J.-K. Jeon, S.-C. Jung, J. Do Chung, W. G. Choi and Y.-K. Park, Recent advances in the catalytic hydrodeoxygenation of bio-oil, Korean J. Chem. Eng., 2016, 33, 3299–3315, DOI:10.1007/s11814-016-0214-3.
- J. B. s. Bredenberg, M. Huuska, J. Räty and M. Korpio, Hydrogenolysis and hydrocracking of the carbon-oxygen bond. I. Hydrocracking of some simple aromatic O-compounds, J. Catal., 1982, 77, 242–247, DOI:10.1016/0021-9517(82)90164-6.
- E. Blanco, A. B. Dongil and N. Escalona, Synergy between ni and co nanoparticles supported on carbon in guaiacol conversion, Nanomaterials, 2020, 10, 1–18, DOI:10.3390/nano10112199.
- E. Laurent and B. Delmon, Study of the hydrodeoxygenation of carbonyl, carboxylic and guaiacyl groups over sulfided CoMo/γ-Al2O3 and NiMo/γ-Al2O3 catalyst. II. Influence of water, ammonia and hydrogen sulfide, Appl. Catal., A, 1994, 109, 97–115, DOI:10.1016/0926-860X(94)85005-4.
- V. N. Bui, D. Laurenti, P. Delichère and C. Geantet, Hydrodeoxygenation of guaiacol. Part II: Support effect for CoMoS catalysts on HDO activity and selectivity, Appl. Catal., B, 2011, 101, 246–255, DOI:10.1016/j.apcatb.2010.10.031.
- P. Sangnikul, C. Phanpa, R. Xiao, H. Zhang, P. Reubroycharoen, P. Kuchonthara, T. Vitidsant, A. Pattiya and N. Hinchiranan, Role of copper- or cerium-promoters on NiMo/Γ-Al2O3 catalysts in hydrodeoxygenation of guaiacol and bio-oil, Appl. Catal., A, 2019, 574, 151–160, DOI:10.1016/j.apcata.2019.02.004.
- I. D. Mora, E. Méndez, L. J. Duarte and S. A. Giraldo, Effect of support modifications for CoMo/γ-Al2O 3 and CoMo/ASA catalysts in the hydrodeoxygenation of guaiacol, Appl. Catal., A, 2014, 474, 59–68, DOI:10.1016/j.apcata.2013.11.011.
- A. L. Jongerius, R. Jastrzebski, P. C. A. Bruijnincx and B. M. Weckhuysen, CoMo sulfide-catalyzed hydrodeoxygenation of lignin model compounds: An extended reaction network for the conversion of monomeric and dimeric substrates, J. Catal., 2012, 285, 315–323, DOI:10.1016/j.jcat.2011.10.006.
- H. Wang, T. L. Eberhardt, C. Wang, S. Gao and H. Pan, Demethylation of alkali lignin with halogen acids and its application to phenolic resins, Polymers, 2019, 11, 1–16, DOI:10.3390/polym11111771.
- E. Jasiukaitytė-Grojzdek, M. Huš, M. Grilc and B. Likozar, Acid-catalysed α-O-4 aryl-ether bond cleavage in methanol/(aqueous) ethanol: understanding depolymerisation of a lignin model compound during organosolv pretreatment, Sci. Rep., 2020, 10, 1–13, DOI:10.1038/s41598-020-67787-9.
- X. Meng, C. Crestini, H. Ben, N. Hao, Y. Pu, A. J. Ragauskas and D. S. Argyropoulos, Determination of hydroxyl groups in biorefinery resources via quantitative 31P NMR spectroscopy, Nat. Protoc., 2019, 14, 2627–2647, DOI:10.1038/s41596-019-0191-1.
- D. S. Zijlstra, A. de Santi, B. Oldenburger, J. de Vries, K. Barta and P. J. Deuss, Extraction of Lignin with High β-O-4 Content by Mild Ethanol Extraction and Its Effect on the Depolymerization Yield, J. Visualized Exp., 2019, e58575, DOI:10.3791/58575.
- S. Constant, H. L. J. Wienk, A. E. Frissen, P. de Peinder, R. Boelens, D. S. van Es, R. J. H. Grisel, B. M. Weckhuysen, W. J. J. Huijgen, R. J. A. Gosselink and P. C. A. Bruijnincx, New insights into the structure and composition of technical lignins: a comparative characterisation study, Green Chem., 2016, 18, 2651–2665, 10.1039/C5GC03043A.
- R. Vendamme, J. Behaghel de Bueren, J. Gracia-Vitoria, F. Isnard, M. M. Mulunda, P. Ortiz, M. Wadekar, K. Vanbroekhoven, C. Wegmann, R. Buser, F. Héroguel, J. S. Luterbacher and W. Eevers, Aldehyde-Assisted Lignocellulose Fractionation Provides Unique Lignin Oligomers for the Design of Tunable Polyurethane Bioresins, Biomacromolecules, 2020, 21, 4135–4148, DOI:10.1021/acs.biomac.0c00927.
- D. S. Argyropoulos, 31P NMR in wood chemistry: A review of recent progress, Res. Chem. Intermed., 1995, 21, 373–395, DOI:10.1007/BF03052265.
- Y. Pu, S. Cao and A. J. Ragauskas, Application of quantitative 31P NMR in biomass lignin and biofuel precursors characterization, Energy Environ. Sci., 2011, 4, 3154–3166, 10.1039/C1EE01201K.
- T. Ročnik, B. Likozar, E. Jasiukaitytė-Grojzdek and M. Grilc, Catalytic lignin valorisation by depolymerisation, hydrogenation, demethylation and hydrodeoxygenation: Mechanism, chemical reaction kinetics and transport phenomena, Chem. Eng. J., 2022, 448, 137309, DOI:10.1016/j.cej.2022.137309.
- C. Crestini, H. Lange and G. Bianchetti, Detailed Chemical Composition of Condensed Tannins via Quantitative 31P NMR and HSQC Analyses: Acacia catechu, Schinopsis balansae, and Acacia mearnsii, J. Nat. Prod., 2016, 79, 2287–2295, DOI:10.1021/acs.jnatprod.6b00380.
- J. Podschun, B. Saake and R. Lehnen, Catalytic demethylation of organosolv lignin in aqueous medium using indium triflate under microwave irradiation, React. Funct. Polym., 2017, 119, 82–86, DOI:10.1016/j.reactfunctpolym.2017.08.007.
- X. Zhen, H. Li, Z. Xu, Q. Wang, J. Xu, S. Zhu, Z. Wang and Z. Yuan, Demethylation, phenolation, and depolymerization of lignin for the synthesis of lignin-based epoxy resin via a one-pot strategy, Ind. Crops Prod., 2021, 173, 114135, DOI:10.1016/j.indcrop.2021.114135.
- Y. Song, Z. Wang, N. Yan, R. Zhang and J. Li, Demethylation of wheat straw alkali lignin for application in phenol formaldehyde adhesives, Polymers, 2016, 8(6) DOI:10.3390/polym8060209.
- W. Zhao, C. Wei, Y. Cui, J. Ye, B. He, X. Liu and J. Sun, Efficient demethylation of lignin for polyphenol production enabled by low-cost bifunctional protic ionic liquid under mild and halogen-free conditions, Chem. Eng. J., 2022, 443, 136486, DOI:10.1016/j.cej.2022.136486.
- W. Schutyser, T. Renders, S. Van den Bosch, S.-F. Koelewijn, G. T. Beckham and B. F. Sels, Chemicals from lignin: an interplay of lignocellulose fractionation, depolymerisation, and upgrading, Chem. Soc. Rev., 2018, 47, 852–908, 10.1039/C7CS00566K.
- M. Brebu and C. Vasile, Thermal degradation of lignin - A review, Cellul. Chem. Technol., 2010, 44, 353–363 CAS.
- Wahyudiono, M. Sasaki and M. Goto, Recovery of phenolic compounds through the decomposition of lignin in near and supercritical water, Chem. Eng. Process. Process Intensif., 2008, 47, 1609–1619, DOI:10.1016/j.cep.2007.09.001.
- L. Du, Z. Wang, S. Li, W. Song and W. Lin, A comparison of monomeric phenols produced from lignin by fast pyrolysis and hydrothermal conversions, Int. J. Chem. React. Eng., 2013, 11, 135–145, DOI:10.1515/ijcre-2012-0085.
- H.-s. Lee, J. Jae, J. M. Ha and D. J. Suh, Hydro- and solvothermolysis of kraft lignin for maximizing production of monomeric aromatic chemicals, Bioresour. Technol., 2016, 203, 142–149, DOI:10.1016/j.biortech.2015.12.022.
- R. Chaudhary and P. L. Dhepe, Solid base catalyzed depolymerization of lignin into low molecular weight products, Green Chem., 2017, 19, 778–788, 10.1039/c6gc02701f.
- M. J. Hidajat, A. Riaz, J. Park, R. Insyani, D. Verma and J. Kim, Depolymerization of concentrated sulfuric acid hydrolysis lignin to high-yield aromatic monomers in basic sub- and supercritical fluids, Chem. Eng. J., 2017, 317, 9–19, DOI:10.1016/j.cej.2017.02.045.
- H. Pińkowska, P. Wolak and A. Złocińska, Hydrothermal decomposition of alkali lignin in sub- and supercritical water, Chem. Eng. J., 2012, 187, 410–414, DOI:10.1016/j.cej.2012.01.092.
- J. A. Onwudili and P. T. Williams, Catalytic depolymerization of alkali lignin in subcritical water: influence of formic acid and Pd/C catalyst on the yields of liquid monomeric aromatic products, Green Chem., 2014, 16, 4740–4748, 10.1039/C4GC00854E.
- K. Barta, G. R. Warner, E. S. Beach and P. T. Anastas, Depolymerization of organosolv lignin to aromatic compounds over Cu-doped porous metal oxides, Green Chem., 2014, 16, 191–196, 10.1039/c3gc41184b.
- M. L. Stone, E. M. Anderson, K. M. Meek, M. Reed, R. Katahira, F. Chen, R. A. Dixon, G. T. Beckham and Y. Román-Leshkov, Reductive Catalytic Fractionation of C-Lignin, ACS Sustainable Chem. Eng., 2018, 6, 11211–11218, DOI:10.1021/acssuschemeng.8b02741.
- A. F. Ang, Z. Ashaari, S. H. Lee, P. Md Tahir and R. Halis, Lignin-based copolymer adhesives for composite wood panels – A review, Int. J. Adhes. Adhes., 2019, 95, 102408, DOI:10.1016/j.ijadhadh.2019.102408.
- Z. Sun, B. Fridrich, A. De Santi, S. Elangovan and K. Barta, Bright Side of Lignin Depolymerization: Toward New Platform Chemicals, Chem. Rev., 2018, 118, 614–678, DOI:10.1021/acs.chemrev.7b00588.
- C. Gao, M. Li, C. Zhu, Y. Hu, T. Shen, M. Li, X. Ji, G. Lyu and W. Zhuang, One-pot depolymerization, demethylation and phenolation of lignin catalyzed by HBr under microwave irradiation for phenolic foam preparation, Composites, Part B, 2021, 205, 108530, DOI:10.1016/j.compositesb.2020.108530.
- Z. Li, E. Sutandar, T. Goihl, X. Zhang and X. Pan, Cleavage of ethers and demethylation of lignin in acidic concentrated lithium bromide (ACLB) solution, Green Chem., 2020, 22, 7989–8001, 10.1039/d0gc02581j.
- M. Sun, X. Wang, S. Ni, L. Jiao, H. Bian and H. Dai, Structural modification of alkali lignin into higher performance energy storage materials: Demethylation and cleavage of aryl ether bonds, Ind. Crops Prod., 2022, 187, 115441, DOI:10.1016/j.indcrop.2022.115441.
- L. Chai, B. Du, S. Yan, W. Li, X. Chen and R. Sun, Preparation of activated lignin with high hydroxyl content using lewis acid as demethylation reagent, Int. J. Biol. Macromol., 2022, 222, 2571–2580, DOI:10.1016/j.ijbiomac.2022.10.040.
- L. Hu, H. Pan, Y. Zhou, C. Y. Hse, C. Liu, B. Zhang and B. Xu, Chemical groups and structural characterization of lignin via thiol-mediated demethylation, J. Wood Chem. Technol., 2014, 34, 122–134, DOI:10.1080/02773813.2013.844165.
- K. Sawamura, Y. Tobimatsu, H. Kamitakahara and T. Takano, Lignin Functionalization through Chemical Demethylation: Preparation and Tannin-Like Properties of Demethylated Guaiacyl-Type Synthetic Lignins, ACS Sustainable Chem. Eng., 2017, 5, 5424–5431, DOI:10.1021/acssuschemeng.7b00748.
- K. H. Kim, K. Jeong, J. Zhuang, H. J. Jeong, C. S. Kim, B. Koo and C. G. Yoo, Tandem conversion of lignin to catechols via demethylation and catalytic hydrogenolysis, Ind. Crops Prod., 2021, 159, 113095, DOI:10.1016/j.indcrop.2020.113095.
- W. Zhao, C. Wei, Y. Cui, J. Ye, B. He, X. Liu and J. Sun, Efficient demethylation of lignin for polyphenol production enabled by low-cost bifunctional protic ionic liquid under mild and halogen-free conditions, Chem. Eng. J., 2022, 443, 136486, DOI:10.1016/j.cej.2022.136486.
| This journal is © The Royal Society of Chemistry 2023 |