
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A comparative study of palladium-gold and palladium-tin catalysts in the direct synthesis of H2O2†
Dávid
Kovačič‡
a,
Richard J.
Lewis‡
 *a,
Caitlin M.
Crombie
a,
David J.
Morgan
ab,
Thomas E.
Davies
a,
Ángeles
López-Martín
a,
Tian
Qin
c,
Christopher S.
Allen
fg,
Jennifer. K.
Edwards
d,
Liwei
Chen
c,
Martin Skov
Skjøth-Rasmussen
e,
Xi
Liu
*c and
Graham J.
Hutchings
*a
*a,
Caitlin M.
Crombie
a,
David J.
Morgan
ab,
Thomas E.
Davies
a,
Ángeles
López-Martín
a,
Tian
Qin
c,
Christopher S.
Allen
fg,
Jennifer. K.
Edwards
d,
Liwei
Chen
c,
Martin Skov
Skjøth-Rasmussen
e,
Xi
Liu
*c and
Graham J.
Hutchings
*a
aMax Planck–Cardiff Centre on the Fundamentals of Heterogeneous Catalysis FUNCAT, Cardiff Catalysis Institute, School of Chemistry, Cardiff University, Cardiff, CF24 4HQ, UK. E-mail: LewisR27@cardiff.ac.uk; LiuXi@sjtu.edu.cn; Hutch@cardiff.ac.uk
bHarwell XPS, Research Complex at Harwell (RCaH) Didcot, OX11 0FA, UK
cIn-situ Centre for Physical Sciences, School of Chemistry and Chemical, Frontiers Science Centre for Transformative Molecules, Shanghai 200240, P. R. China
dCardiff Catalysis Institute, School of Chemistry, Cardiff University, Main Building, Park Place, Cardiff, CF10 3AT, UK
eHaldor Topsøe A/S, Haldor Topsøes Allé 1, DK-2800 Kongens Lyngby, Denmark
fElectron Physical Sciences Imaging Centre, Diamond Light Source Ltd, Didcot, OX11 0DE, UK
gDepartment of Materials, University of Oxford, Oxford, OX1 3PH, UK
First published on 14th November 2023
Abstract
Herein we evaluate the promotive effect of Au and Sn incorporation into supported Pd nanoparticles for the direct synthesis of H2O2 from molecular H2 and O2. The introduction of both secondary metal modifiers was found to result in a significant enhancement in catalytic performance, although, in the case of the PdSn system, it was identified that relatively large quantities of the secondary metal were required to rival the activity observed over optimal Au-containing formulations, with the 0.25%Pd–2.25%Sn/TiO2 catalyst offering comparable H2O2 synthesis rates to the optimised 0.25%Pd–0.25%Au/TiO2 formulation. The introduction of Sn was found to considerably improve Pd dispersion, correlating with an improvement in selective H2 utilisation. Notably, the optimal PdSn catalyst identified in this work achieves superior H2O2 selectivities compared to the PdAu analogue and is able to rival the performance of state-of-the-art materials.
Introduction
Hydrogen peroxide (H2O2) is a highly efficacious, environmentally friendly oxidant, used widely in sectors ranging from textile and paper manufacture, where its strong bleaching properties are sought,1 to chemical synthesis,2,3 where H2O2 is superseding traditional reagents, such as perchlorate or permanganate. Indeed, the use of H2O2 in chemical valorisation is particularly attractive, given that the only by-product of its use is water, reducing separation costs associated with stoichiometric oxidants and typically allowing for the use of lower reaction temperatures compared to alternative aerobic routes.4,5The current demand for H2O2 is met almost entirely via the anthraquinone oxidation (AO) process. Although, highly efficient there are several concerns associated with the industrial route, in particular around its atom efficiency, with the over-hydrogenation of the anthraquinone H2 carrier necessitating its periodic replacement.6 This coupled with the overall complexity of the process has typically precluded the production of H2O2 at the point of final application. As such H2O2 is shipped at concentrations far greater than that required by the end user, with the resulting dilution of the oxidant prior to use effectively wasting the energy utilised in its distillation and concentration before transit. Furthermore, the instability of H2O2, with its rapid decomposition to H2O in the presence of relatively mild temperatures or weak bases requires the use of acidic stabilizing agents to prolong shelf-life,7 with such species often promoting reactor corrosion and catalyst deactivation.8
Alternatively, the direct synthesis of H2O2 from its constituent elements would allow for on-site production, at appropriate concentrations, while avoiding the use of additives and the costs associated with the transport and storage of the oxidant.9,10 Indeed there is a growing interest in the use of H2O2 generated in situ for use in chemical transformations.11–13 While Pd-based catalysts have been well reported to offer high activity towards H2O2 production via the direct route,14,15 limited catalytic selectivity has typically necessitated the use of acidic or halogenated stabilising agents.16–19 Alternatively, there has been extensive investigation into the alloying of Pd with secondary metals, primarily other precious metals20–24 to improve catalytic performance. The alloying of Pd with Au in particular has been shown to greatly enhance catalytic selectivity towards H2O2, through isolation and electronic effects, while also avoiding the need for the stabilising agents often required by Pd catalysts.21,25,26
In recent years numerous studies have reported that enhancements in catalytic performance similar to those achieved through the alloying of Pd with Au can be achieved via the incorporation of a range of Earth-abundant metals.27–31 The use of such secondary metal modifiers for Pd represents a key step towards an industrially viable route to the direct synthesis of H2O2. In particular, the use of PdSn formulations has received growing attention, with a number of catalyst formulations reported to offer high selectivity and activity towards H2O2.27,32–34 With these earlier studies in mind and with the aim of gaining further understanding of this relatively new class of catalytic materials we now investigate the efficacy of a series of bimetallic PdSn and PdAu catalysts for the direct synthesis of H2O2.
Experimental
Catalyst preparation
Mono- and bi-metallic PdX/TiO2 (X = Au, Sn) catalysts, were prepared on a weight basis, via a wet co-impregnation procedure, based on a methodology previously reported in the literature.35 With catalysts produced via such a procedure widely studied for the direct synthesis of H2O2 due to the simplicity and ease with which this approach can be scaled to meet industrial application. The procedure to produce the 0.25%Pd–0.25%Au/TiO2 (2 g) catalyst is detailed below, with a similar approach utilized for all mono- and bi-metallic catalysts studied, using chloride-based metal precursors in all cases.Aqueous PdCl2 solution (0.833 mL, [Pd] = 6 mg mL−1, Merck) and aqueous HAuCl4·3H2O solution (0.408 mL, [Au] = 12.25 mg mL−1, Strem Chemicals) were mixed in a 50 mL round-bottom flask and heated to 65 °C with stirring (1000 rpm) in a thermostatically controlled oil bath, with total volume fixed to 16 mL using H2O (HPLC grade). Upon reaching 65 °C, TiO2 (1.98 g, Degussa, P25) was added over the course of 5 minutes, with constant stirring. The resulting slurry was stirred at 65 °C for a further 15 minutes, following this the temperature was raised to 95 °C for 16 h to allow for complete evaporation of water. The resulting solid was ground prior to an oxidative heat treatment (static air, 400 °C, 3 h, 10 °C min−1).
In the case of the PdSn/TiO2 catalyst series SnCl4·5H2O ([Sn] = 5.0 mgmL−1, Merck) was utilised as the Sn precursor.
The surface area of key catalysts studied, as determined by 5-point BET analysis is reported in Table S.1.†
Catalyst testing
Note 1: The reaction conditions used within this study operate under the flammability limits of gaseous mixtures of H2 and O2.36Note 2: The conditions used within this work for H2O2 synthesis and degradation have previously been investigated, with the presence of CO2 as a diluent for reactant gases and a methanol co-solvent identified as key to maintaining high catalytic efficacy towards H2O2 production.37 In particular the CO2 gaseous diluent, has been found to act as an in situ promoter of H2O2 stability through dissolution in the reaction medium and the formation of carbonic acid. We have previously reported that the use of the CO2 diluent has a comparable promotive effect to that observed when acidifying the reaction solution to a pH of 4 using HNO3.38
Direct synthesis of H2O2
Hydrogen peroxide synthesis was evaluated using a Parr Instruments stainless steel autoclave with a nominal volume of 50 mL, equipped with a PTFE liner (so that the total volume is reduced to 33 mL), and a maximum working pressure of 2000 psi. To test each catalyst for H2O2 synthesis, the autoclave liner was charged with catalyst (0.01 g) and solvent (methanol (5.6 g, HPLC grade, Fischer Scientific) and H2O (2.9 g, HPLC grade, Fischer Scientific)). The charged autoclave was then purged three times with 5%H2/CO2 (100 psi) before filling with 5%H2/CO2 to a pressure of 420 psi, followed by the addition of 25%O2/CO2 (160 psi), with the pressure of 5%H2/CO2 and 25%O2/CO2 given as gauge pressures. The reaction was conducted at a temperature of 20 °C for 0.5 h with stirring (1200 rpm). H2O2 productivity was determined by titrating aliquots of the final solution after reaction with acidified Ce(SO4)2 (0.0085 M) in the presence of ferroin indicator. Catalyst productivities are reported as molH2O2 kgcat−1 h−1.The catalytic conversion of H2 and selectivity towards H2O2 were determined using a Varian 3800 GC fitted with TCD and equipped with a Porapak Q column.
H2 conversion (eqn (1)) and H2O2 selectivity (eqn (2)) are defined as follows:
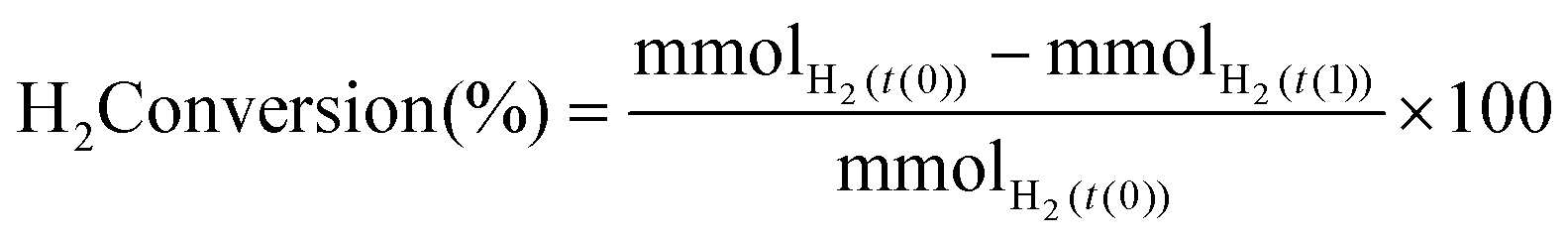 | (1) |
 | (2) |
The total autoclave capacity was determined via water displacement to allow for accurate determination of H2 conversion and H2O2 selectivity.
Gas replacement experiments for the direct synthesis of H2O2
An identical procedure to that outlined above for the direct synthesis reaction was followed for a reaction time of 0.5 h. After this, stirring was stopped and the reactant gas mixture was vented prior to replacement with the standard pressures of 5%H2/CO2 (420 psi) and 25%O2/CO2 (160 psi). The reaction mixture was then stirred (1200 rpm) for a further 0.5 h. To collect a series of data points, as in the case of Fig. 3, it should be noted that individual experiments were carried out and the reactant mixture was not sampled on-line.Degradation of H2O2
Catalytic activity towards H2O2 degradation was determined in a similar manner to the direct synthesis activity of a catalyst. The autoclave liner was charged with solvent (methanol (5.6 g, HPLC grade, Fischer Scientific) and H2O (2.22 g, HPLC grade, Fischer Scientific)) and H2O2 (50 wt% 0.68 g, Sigma Aldrich), with the solvent composition equivalent to a 4 wt% H2O2 solution. From the solution, two 0.05 g aliquots were removed and titrated with acidified Ce(SO4)2 solution using ferroin as an indicator to determine an accurate concentration of H2O2 at the start of the reaction. Subsequently catalyst (0.01 g) was added to the reaction media and the autoclave was purged with 5%H2/CO2 (100 psi) prior to being pressurised with 5% H2/CO2 (420 psi). The reaction medium was conducted at a temperature of 20 °C and stirred at 1200 rpm for 0.5 h. After the reaction was complete the catalyst was removed from the reaction mixture and two 0.05 g aliquots were titrated against the acidified Ce(SO4)2 solution using ferroin as an indicator. The degradation activity is reported as molH2O2 kgcat−1 h−1.Catalyst reusability in the direct synthesis and degradation of H2O2
To determine catalyst reusability, a similar procedure to that outlined above for the direct synthesis of H2O2 is followed utilising 0.05 g of catalyst. Following the initial test, the catalyst was recovered by filtration and dried (30 °C, 16 h, under vacuum); from the recovered catalyst sample 0.01 g was used to conduct a standard H2O2 synthesis or degradation test.Note 3: In all cases, the reactor temperature was controlled using a HAAKE K50 bath/circulator using an appropriate coolant.
Note 4: In all cases, reactions were run multiple times, over multiple batches of catalyst, with the data being presented as an average of these experiments.
Characterisation
X-ray photoelectron spectroscopy (XPS) analyses were made on a Kratos Axis Ultra DLD spectrometer. Samples were mounted using double-sided adhesive tape and binding energies were referenced to the C(1s) binding energy of adventitious carbon contamination that was taken to be 284.8 eV. Monochromatic AlKα radiation was used for all measurements; an analyser pass energy of 160 eV was used for survey scans, while 40 eV was employed for more detailed regional scans. The intensities of the Au(4f), Pt(4f) and Pd(3d) features were used to derive the Pd/Pt and Au/Pt surface composition ratios.Aberration-corrected scanning transmission electron microscopy (AC-STEM) was performed using a probe-corrected Hitachi HF5000 S/TEM, operating at 200 kV. The instrument was equipped with bright field (BF) and annular dark field (ADF) detectors for high spatial resolution STEM imaging experiments. This microscope was also equipped with dual Oxford Instruments XEDS detectors (2 × 100 mm2) having a total collection angle of 2.02 sr. Additional AC-STEM was performed using a probe-corrected ThermoFisher Scientific SPECTRA 200 operating at 200 kV and a JEOL ARM200F operating at 200 kV. In all cases, the samples were prepared by dry dispersion of the powder over 300 mesh holey carbon film copper grids.
Total metal leaching from the supported catalyst was quantified via inductively coupled plasma mass spectrometry (ICP-MS). Post-reaction solutions were analysed using an Agilent 7900 ICP-MS equipped with I-AS auto-sampler. All samples were diluted by a factor of 10 using HPLC grade H2O (1%HNO3 and 0.5% HCl matrix). All calibrants were matrix-matched and measured against a five-point calibration using certified reference materials purchased from PerkinElmer and certified internal standards acquired from Agilent.
Brunauer–Emmett–Teller (BET) surface area measurements were conducted using a Quadrasorb surface area analyzer. A five-point isotherm of each material was measured using N2 as the adsorbate gas. Samples were degassed at 250 °C for 2 h prior to the surface area being determined by five-point N2 adsorption at −196 °C, and data were analyzed using the BET method.
Results and discussion
Our initial studies investigated the efficacy of bimetallic PdAu and PdSn catalysts, prepared by a wet co-impregnation procedure, towards the direct synthesis and subsequent degradation of H2O2 (Tables 1 and 2), under conditions considered detrimental towards the stability of the desired product, namely through the use of ambient reaction temperatures.37 In keeping with a large body of literature20,21,26,39 the alloying of Pd with Au resulted in a significant enhancement in H2O2 synthesis activity (Table 1). Investigation of the role of Pd![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Au ratio on catalytic performance, identified an optimal formulation of 0.25%Pd–0.25%Au/TiO2, with rates of H2O2 synthesis (90 molH2O2 kgcat−1 h−1), particularly noteworthy given the relatively low total metal loading utilised and the use of ambient reaction temperatures. Indeed the activity of the 0.25%Pd–0.25%Au/TiO2 catalyst significantly exceeds that previously reported for analogous formulations consisting of ten times the total metal loading used in this study, when evaluated under conditions more conducive to H2O2 stability.40
:Au ratio
:Au ratio on catalytic performance, identified an optimal formulation of 0.25%Pd–0.25%Au/TiO2, with rates of H2O2 synthesis (90 molH2O2 kgcat−1 h−1), particularly noteworthy given the relatively low total metal loading utilised and the use of ambient reaction temperatures. Indeed the activity of the 0.25%Pd–0.25%Au/TiO2 catalyst significantly exceeds that previously reported for analogous formulations consisting of ten times the total metal loading used in this study, when evaluated under conditions more conducive to H2O2 stability.40
:Au ratio
| Catalyst | Productivity/molH2O2 kgcat−1 h−1 | H2O2 Conc. /wt. % | H2 Conv.% | H2O2 Selectivity/% | Rate of reaction/mmolH2O2 mmolmetal−1 h−1 | Degradation/molH2O2 kgcat−1 h−1 |
|---|---|---|---|---|---|---|
| H2O2 direct synthesis reaction conditions: catalyst (0.01 g), H2O (2.9 g), MeOH (5.6 g), 5% H2/CO2 (420 psi), 25% O2/CO2 (160 psi), 0.5 h, 20° C, 1200 rpm. H2O2 degradation reaction conditions: catalyst (0.01 g), H2O2 (50 wt% 0.68 g) H2O (2.22 g), MeOH (5.6 g), 5% H2/CO2 (420 psi), 0.5 h, 20 °C 1200 rpm. BDL: below the accurate detection limit. *TiO2 used as received. Note: reaction rates are based on theoretical metal loading. | ||||||
| 0.5%Pd/TiO2 | 68 | 0.140 | 57 | 44 | 1.49 × 103 | 0 |
| 0.375%Pd–0.125%Au/TiO2 | 80 | 0.170 | 61 | 50 | 2.02 × 103 | 110 |
| 0.25%Pd–0.25%Au/TiO2 | 90 | 0.180 | 53 | 59 | 2.49 × 103 | 119 |
| 0.125%Pd–0.375%Au/TiO2 | 74 | 0.150 | 40 | 76 | 2.43 × 103 | 108 |
| 0.5%%Au/TiO2 | 3 | 0.010 | BDL | BDL | 1.06 × 102 | 0 |
| 0.25%Pd/TiO2 | 58 | 0.120 | 31 | 69 | 2.47 × 103 | 0 |
| TiO2* | 0 | — | — | — | 0 | 0 |
:Sn ratio
| Catalyst | Productivity/molH2O2 kgcat−1 h−1 | H2O2 Conc. /wt. % | H2 Conv.% | H2O2 selectivity/% | Rate of reaction/mmolH2O2 mmolmetal−1 h−1 | Degradation/molH2O2 kgcat−1 h−1 |
|---|---|---|---|---|---|---|
| H2O2 direct synthesis reaction conditions: catalyst (0.01 g), H2O (2.9 g), MeOH (5.6 g), 5% H2/CO2 (420 psi), 25% O2/CO2 (160 psi), 0.5 h, 20° C, 1200 rpm. H2O2 degradation reaction conditions: catalyst (0.01 g), H2O2 (50 wt% 0.68 g) H2O (2.22 g), MeOH (5.6 g), 5% H2/CO2 (420 psi), 0.5 h, 20 °C 1200 rpm. *TiO2 used as received. Note: reaction rates are based on theoretical metal loading. | ||||||
| 0.5%Pd/TiO2 | 68 | 0.14 | 57 | 44 | 1.49 × 103 | 0 |
| 0.375%Pd–0.125%Sn/TiO2 | 64 | 0.13 | 46 | 50 | 1.40 × 103 | 0 |
| 0.25%Pd–0.25%Sn/TiO2 | 62 | 0.12 | 31 | 71 | 1.35 × 103 | 0 |
| 0.125%Pd–0.375%Sn/TiO2 | 22 | 0.08 | 16 | 89 | 9.23 × 102 | 0 |
| 0.5%Sn/TiO2 | 0 | — | — | — | — | 0 |
| 0.25%Pd/TiO2 | 58 | 0.12 | 31 | 69 | 2.47 × 103 | 0 |
| TiO2* | 0 | — | — | — | 0 | 0 |
While the enhanced H2O2 synthesis activity of the PdAu formulation, compared to the Pd-only analogue, may have been expected based on numerous previous studies, the inability of the 0.5%Pd/TiO2 catalyst to degrade H2O2 is perhaps surprising and may lead to the inference that this material is highly selective towards H2O2. However, it is important to note that the determination of H2O2 selectivity under direct synthesis conditions contradicts this assumption. While the selectivity of the 0.25%Pd–0.25%Au/TiO2 catalyst (59%) is significantly improved compared to the Pd-only analogue (44%), this metric is far from the total selectivity implied by our degradation studies. Notably, such selectivity comparisons were made at relatively similar rates of H2 conversion (53 and 57% for the 0.25%Pd–0.25%Au/TiO2 and 0.5%Pd/TiO2 catalysts respectively). Such a discrepancy between H2O2 degradation rates and H2O2 selectivity, during H2O2 synthesis, may be associated with the relatively high concentration of preformed oxidant used to conduct the H2O2 degradation experiments (4 wt%), and the potential for metallic Pd species to be oxidised under such conditions. Indeed, the high H2O2 selectivity of PdO, in comparison to Pd0 species, has been well reported.41 While the requirement to use such high concentrations of H2O2 is evident, allowing for differences in catalytic activity to be more easily discerned, we consider that these observations highlight the need for the determination of H2O2 selectivities under direct synthesis conditions in order to achieve a comprehensive understanding of catalytic performance.
By comparison to PdAu formulations, the application of PdSn based catalysts for the direct synthesis of H2O2 has only recently begun to receive attention in the literature,27,31,33 although it is clear that such catalysts can rival the performance of state-of-the-art materials.27 Investigation into the Pd:Sn ratio revealed that, unlike the PdAu system, the introduction of small quantities of the secondary metal did not significantly improve catalytic performance compared to the 0.5%Pd/TiO2 catalyst (68 molH2O2 kgcat−1 h−1) (Table 2). A comparison of the 0.25%Pd/TiO2 and 0.25%Pd–0.25%Sn/TiO2 catalysts (i.e. formulations with an equivalent Pd loading), further indicates that the introduction of low concentrations of Sn does not result in a meaningful enhancement in catalytic performance, compared to Pd-analogues. Indeed, the performance of the 0.25%Pd/TiO2 (58 molH2O2 kgcat−1 h−1 and 69% H2O2 selectivity) and 0.25%Pd–0.25%Sn/TiO2 (62 molH2O2 kgcat−1 h−1 and 71% H2O2 selectivity) catalysts were found to be nearly identical.
We were subsequently motivated to investigate a subset of catalyst formulations, namely the 0.5%Pd/TiO2, 0.25%Pd–0.25%Au/TiO2 and 0.25%Pd–0.25%Sn/TiO2 catalysts, in order to gain further insight into these materials.
The activity of Pd-based catalysts towards H2O2 production is well known to be highly influenced by Pd oxidation state. In particular, the presence of PdO has been reported to aid the suppression of O–O bond dissociation and the unselective formation of H2O, in addition to promoting the rapid desorption of H2O2 from catalytic surfaces, with both factors resulting in improved catalytic selectivity.42 While the identity of the true active site for H2O2 synthesis is still debated, there is a consensus that a proportion of the Pd must exist in the reduced state to form H2O2. However, it is not clear if the active sites that bind and selectively reduce O2 to H2O2 are fully or partially reduced or indeed fully oxidized Pd atoms, for a detailed discussion of this we direct the reader to the excellent review on the topic by D.W. Flaherty.14 Analysis of the as-prepared materials by X-ray photoelectron spectroscopy (XPS) is reported in Fig. 1 and reveals clear differences in Pd speciation across the catalytic series. Despite exposure to a high-temperature oxidative heat treatment (static air, 400 °C, 3 h, 10 °C min−1), all catalysts were found to consist of mixed Pd oxidation states (i.e. Pd2+ and Pd0). However, the introduction of Au and Sn was found to significantly shift Pd speciation towards Pd2+. Notably, our observations align well with earlier works which have identified the ability of both metals to modify the electronic state of Pd which, at least in the case of the PdAu catalyst, may be partly responsible for the improved catalytic activity, when compared to the Pd-only analogue.33,43 Although it should be noted that the Pd oxidation state of the as-prepared materials may not be fully representative of that under reaction conditions.
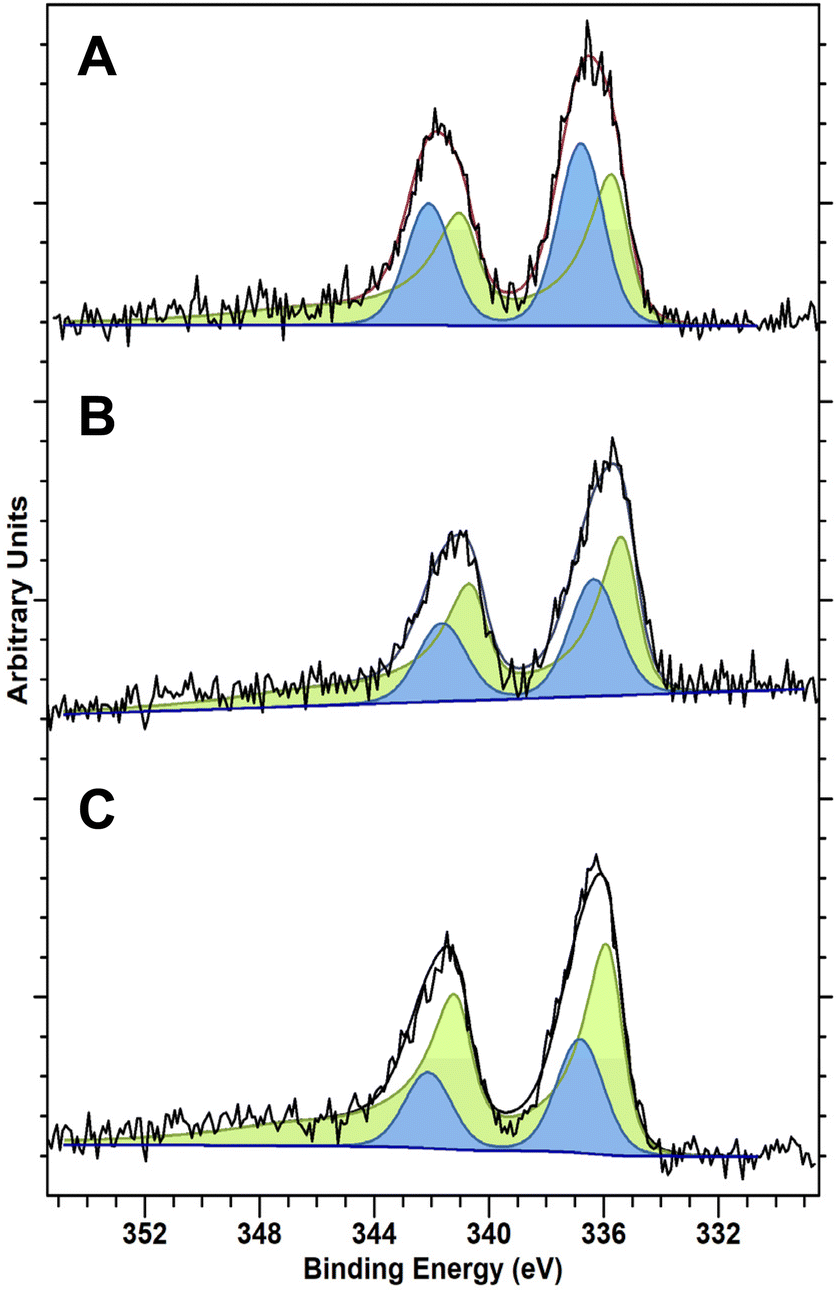 | ||
| Fig. 1 XPS spectra of Pd(3d) regions of the as-prepared (A) 0.25%Pd–0.25%Sn/TiO2, (B) 0.25%Pd–0.25%Au/TiO2 and (C) 0.5%Pd/TiO2 catalysts. Key: Pd0 (green) Pd2+ (blue). | ||
A comparison of the initial rate of reaction, determined at a reaction time of 5 minutes, where the contribution of competitive H2O2 degradation pathways can be considered to be negligible, is reported in Table 3 and further reveals the enhanced activity of the 0.25%Pd–0.25%Au/TiO2 catalyst.
| Catalyst | Rate of reaction/mmolH2O2 mmolmetal−1 h−1 | |
|---|---|---|
| 5 min | 30 min | |
| H2O2 direct synthesis reaction conditions: catalyst (0.01 g), H2O (2.9 g), MeOH (5.6 g), 5% H2/CO2 (420 psi), 25% O2/CO2 (160 psi), 20 °C 1200 rpm. Note: reaction rates are based on theoretical metal loading. | ||
| 0.5%Pd/TiO2 | 4.21 × 103 | 1.49 × 103 |
| 0.25%Pd–0.25%Au/TiO2 | 5.87 × 103 | 2.49 × 103 |
| 0.25%Pd–0.25%Sn/TiO2 | 5.21 × 103 | 1.35 × 103 |
Time-on-line studies comparing H2O2 synthesis rates over the catalytic series are reported in Fig. 2. The enhanced activity of the 0.25%Pd–0.25%Au/TiO2 catalyst is again apparent, particularly at extended reaction times, with the concentration of H2O2 (0.21 wt%) significantly greater than that achieved by either the 0.25%Pd–0.25%Sn/TiO2 (0.15 wt%) or 0.5%Pd/TiO2 (0.12 wt%) analogues. Comparison of catalytic selectivity towards H2O2, at near iso-conversion further identifies the greater efficacy of the PdAu formulation, particularly at relatively high rates of H2 conversion (Table S.2†). However, the improved selectivity of the 0.25%Pd–0.25%Sn/TiO2 catalyst (91%) at relatively low rates of H2 conversion (approx. 15%), is notable, outperforming both the 0.25%Pd–0.25%Au/TiO2 (83% selectivity at 14% H2 conversion) and 0.5%Pd/TiO2 (78% selectivity at 16% H2 conversion) analogues. Assessment of the catalysts by XPS, over the course of the H2O2 synthesis reaction, reveals a clear shift in Pd oxidation state, towards Pd0, which may be expected given the reductive atmosphere used and correlates well with the observed loss of selectivity over all three formulations on-stream, with the lower selectivity of Pd0 towards H2O2, compared to Pd2+ species well reported (Fig. S.1†).44 Notably, in the case of the 0.25%Pd–0.25%Sn/TiO2 catalyst our XPS analysis reveals a significant reduction of Pd2+ at extended reaction times (Fig. S.1†), which aligns well with the observed loss of selectivity.
 | ||
| Fig. 2 Comparison of catalytic activity towards the direct synthesis of H2O2, as a function of reaction time. (A) Catalytic activity based on net H2O2 concentration. Determination of H2 conversion and H2O2 selectivity for the (B) 0.5%Pd/TiO2 (C) 0.25%Pd–0.25%Au/TiO2 and (D) 0.25%Pd–0.25%Sn/TiO2 catalysts Key: 0.5%Pd/TiO2 (black squares), 0.25%Pd–0.25%Au/TiO2 (red circles), 0.25%Pd–0.25%Sn/TiO2 catalysts (blue triangles) H2 conversion (purple inverted triangles), H2O2 selectivity (green diamonds). H2O2 direct synthesis reaction conditions: catalyst (0.01 g), H2O (2.9 g), MeOH (5.6 g), 5% H2/CO2 (420 psi), 25% O2/CO2 (160 psi), 20 °C, 1200 rpm. | ||
Evaluation of catalytic performance over successive H2O2 synthesis experiments, where the gaseous reagents were replenished at 30-minute intervals, was subsequently conducted (Fig. 3). After five consecutive H2O2 reactions the improved performance of the 0.25%Pd–0.25%Au/TiO2 catalyst is again clear (0.52 wt%). However, based on catalytic activity measurements over our standard reaction time (0.5 h), one may not have expected the relatively high H2O2 concentration achieved by the 0.25%Pd–0.25%Sn/TiO2 catalyst (0.49 wt%). Indeed, the similar performance of the two bimetallic catalysts is striking, with both catalysts achieving concentrations of H2O2 comparable to that we have previously reported using a near 100% selective 3%Pd–2%Sn/TiO2 catalyst,27 this is despite the less conducive reaction conditions and significantly lower active metals loading of the catalysts reported within this work.
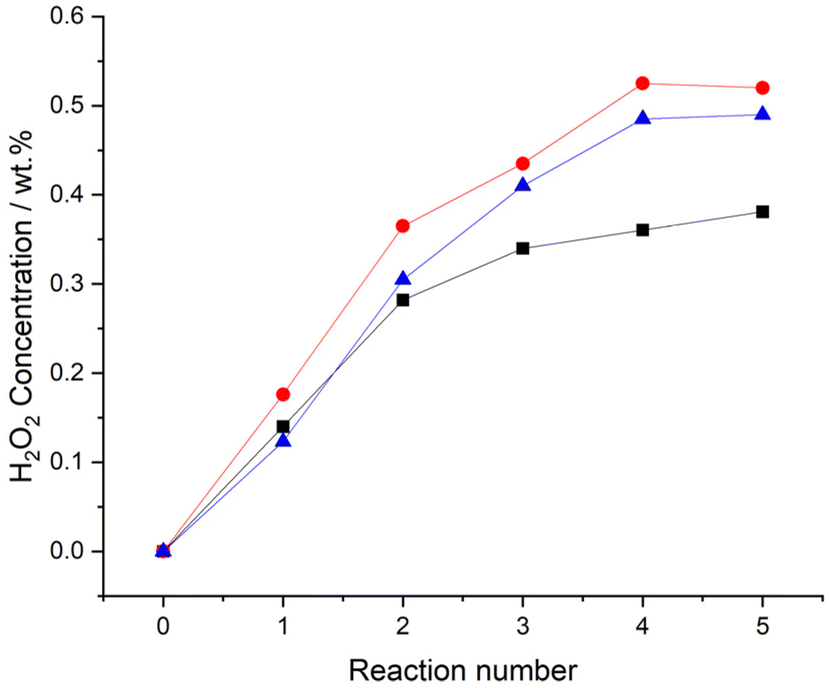 | ||
| Fig. 3 Comparison of catalytic activity, over sequential H2O2 synthesis reactions. Key: 0.5%Pd/TiO2 (black squares), 0.25%Pd–0.25%Au/TiO2 (red circles), 0.25%Pd–0.25%Sn/TiO2 catalysts (blue triangles) H2O2 direct synthesis reaction conditions: catalyst (0.01 g), H2O (2.9 g), MeOH (5.6 g), 5% H2/CO2 (420 psi), 25% O2/CO2 (160 psi), 0.5 h, 20 °C, 1200 rpm. | ||
With the requirement to successfully reuse a catalyst at the heart of green chemistry, we next evaluated catalytic activity towards H2O2 synthesis and H2O2 degradation pathways, upon re-use (Table 4). It was found that for all formulations studied, catalytic activity towards H2O2 production decreased considerably upon second use. Determination of metal leaching during the direct synthesis reaction via ICP-MS analysis of post-reaction solutions is also reported in Table 4, a degree of metal leaching was observed for all catalysts, although we should highlight no Au leaching was detected, and in all cases the degree of metal leaching is relatively low (<2%). As such it is considered that the loss in catalytic performance upon reuse does not result from the leaching of active metals. Rather, we consider that changes in catalytic selectivity explain the observed variation in catalytic performance between first and second use. For all catalysts studied, H2O2 degradation rates were found to increase considerably upon reuse. This is particularly noteworthy for the 0.5%Pd/TiO2 (321 molH2O2 kgcat−1 h−1) and 0.25%Pd–0.25%Sn/TiO2 (88 molH2O2 kgcat−1 h−1) catalysts given that neither offered any activity towards H2O2 degradation upon initial use (Tables 1 and 2). Our analysis of the catalysts after use in the direct synthesis of H2O2via XPS (Fig. S.2†) revealed a clear shift towards Pd0 for all formulations. With the enhanced activity of Pd0 species towards H2O2 degradation well known44 it is possible to, at least in part, attribute the decreased H2O2 selectivity of these catalysts to the in situ reduction of Pd2+ to Pd0 species.
| Catalyst | Productivity/molH2O2 kgcat−1 h−1 | H2 Conv. /% | H2O2 selectivity/% | Degradation/molH2O2 kgcat−1 h−1 | Metal leaching/% | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Use 1 | Use 2 | Use 1 | Use 2 | Use 1 | Use 2 | Use 1 | Use 2 | Pd | Au | Sn | |
| H2O2 direct synthesis reaction conditions: catalyst (0.01 g), H2O (2.9 g), MeOH (5.6 g), 5% H2/CO2 (420 psi), 25% O2/CO2 (160 psi), 0.5 h, 20 °C, 1200 rpm. | |||||||||||
| 0.5%Pd/TiO2 | 68 | 44 | 57 | 62 | 44 | 25 | 0 | 321 | 1.90 | — | — |
| 0.25%Pd–0.25%Au/TiO2 | 90 | 53 | 53 | 60 | 59 | 32 | 100 | 148 | 1.57 | 0 | — |
| 0.25%Pd–0.25%Sn/TiO2 | 62 | 34 | 31 | 39 | 71 | 30 | 0 | 88 | 1.45 | — | 1.26 |
We have previously demonstrated that the introduction of large quantities of base metals (including Sn, Ni, Ga, In, Zn and Co) into a supported Pd catalyst can significantly improve catalyst performance towards H2O2 production.27,28 Notably, while all formulations were found to be highly selective towards H2O2, the reactivity of the optimal PdSn catalyst was two to three times greater than analogous materials containing alternative transition metals.27 Motivated by these earlier works we subsequently investigated the effect of varying Au and Sn loading on catalytic reactivity and selectivity, while maintaining Pd content at 0.25 wt% (Table 5).
| Catalyst | Productivity/molH2O2 kgcat−1 h−1 | H2O2 Conc. /wt. % | H2 Conv.% | H2O2 selectivity/% | Rate of reaction/mmolH2O2 mmolmetal−1 h−1 | Degradation/molH2O2 kgcat−1 h−1 |
|---|---|---|---|---|---|---|
| H2O2 direct synthesis reaction conditions: catalyst (0.01 g), H2O (2.9 g), MeOH (5.6 g), 5% H2/CO2 (420 psi), 25% O2/CO2 (160 psi), 0.5 h, 20° C, 1200 rpm. H2O2 degradation reaction conditions: catalyst (0.01 g), H2O2 (50 wt% 0.68 g) H2O (2.22 g), MeOH (5.6 g), 5% H2/CO2 (420 psi), 0.5 h, 20 °C 1200 rpm. | ||||||
| 0.25%Pd/TiO2 | 58 | 0.120 | 31 | 69 | 2.55 × 103 | 0 |
| 0.25%Pd–0.25%Au/TiO2 | 90 | 0.180 | 53 | 59 | 2.71 × 103 | 119 |
| 0.25%Pd–0.5%Au/TiO2 | 70 | 0.140 | 38 | 65 | 1.42 × 103 | 90 |
| 0.25%Pd–0.75%Au/TiO2 | 65 | 0.130 | 33 | 70 | 1.05 × 103 | 79 |
| 0.25%Pd–1.5%Au/TiO2 | 52 | 0.110 | 24 | 78 | 5.26 × 102 | 40 |
| 0.25%Pd–2.25%Au/TiO2 | 44 | 0.080 | 18 | 83 | 3.05 × 102 | 6 |
| 0.25%Pd–0.25%Sn/TiO2 | 62 | 0.120 | 31 | 71 | 1.35 × 103 | 0 |
| 0.25%Pd–0.5% Sn /TiO2 | 75 | 0.150 | 31 | 84 | 1.11 × 103 | 0 |
| 0.25%Pd–0.75% Sn /TiO2 | 83 | 0.160 | 32 | 90 | 9.22 × 102 | 0 |
| 0.25%Pd–1.5% Sn/TiO2 | 88 | 0.180 | 34 | 93 | 5.93 × 102 | 0 |
| 0.25%Pd–2.25% Sn /TiO2 | 92 | 0.185 | 35 | 94 | 4.34 × 102 | 0 |
As discussed above, the introduction of relatively small quantities of Au (Pd:Au = 1 (wt/wt)), led to a considerable increase in the rate of H2O2 production (90 molH2O2 kgcat−1 h−1). However, increasing Au content further resulted in a significant loss in catalytic performance, with the activity of the 0.25%Pd–2.25%Au/TiO2 catalyst (44 molH2O2 kgcat−1 h−1 and 18% H2 conversion) less than half that observed over the optimal 0.25%Pd–0.25%Au/TiO2 formulation (90 molH2O2 kgcat−1 h−1 and 53% H2 conversion). In contrast to the PdAu system, the introduction of high loadings of Sn was found to significantly enhance catalytic activity, with the H2O2 synthesis rate of the 0.25%Pd–2.25%Sn/TiO2 catalyst (92 molH2O2 kgcat−1 h−1) 1.6 times greater than the 0.25%Pd/TiO2 analogue (58 molH2O2 kgcat−1 h−1) and comparable to that offered by the optimal 0.25%Pd–0.25%Au/TiO2 formulation (90 molH2O2 kgcat−1 h−1). Notably, we did not extend our study beyond a Pd:Sn ratio of 1:10 and so further improvements may be obtained through further optimisation of metal loading. Interestingly, the observed catalytic improvement upon the introduction of high concentrations of Sn was found to result from an increase in the selective utilisation of H2, rather than an increase in the rate of H2O2 production, as indicated by H2 conversion measurements, with this metric varying little across the catalytic series. Indeed, the H2O2 selectivity of the 0.25%Pd–2.25%Sn/TiO2 catalyst (94%), was found to be comparable to that offered by state-of-the-art formulations.
Focussing on the 0.25%Pd–2.25%Sn/TiO2 catalyst and in an attempt to improve the net concentration of H2O2, we conducted a series of sequential H2O2 direct synthesis experiments (Fig. S.3†). After five consecutive reactions, the net concentration of H2O2 increased to a value of 0.69 wt%, which is far superior to the yields of H2O2 achieved over the 0.5%Pd/TiO2 (0.38 wt% H2O2), 0.25%Pd–0.25%Au/TiO2 (0.52 wt% H2O2) or 0.25%Pd–0.25%Sn/TiO2 (0.49 wt% H2O2) catalysts over the same number of reactions (Fig. 3).
HAADF/ADF-STEM (Fig. 4) and corresponding EDX (Fig. 5) analysis of optimal PdAu and PdSn formulations (i.e. the 0.25%Pd–0.25%Au/TiO2 and 0.25%Pd–2.25%Sn/TiO2 catalysts), in addition to the parent 0.25%Pd/TiO2 catalyst (Fig. S.4†), was conducted in order to gain further insight into underlying cause for the promotive effect observed through secondary metal introduction. A considerable variation in nanoparticle size across the catalytic series was observed. Perhaps as expected given the low metal loading of the 0.25%Pd/TiO2 catalyst Pd was found to be present primarily as sub-nanometre clusters, with a very limited number of nanoparticles in the 3–5 nm range also observed (Fig. S.4†). The alloying of Pd with Au resulted in the bifurcation of particle size, with a proportion of larger (10–15 nm) nanoparticles observed in addition to smaller clusters (Fig. 4A and B). Subsequent STEM-XEDS mapping of individual nanoparticles, as presented in Fig. 5A, confirmed that the larger particles consisted of random alloys of Pd and Au, while the smaller clusters were found to consist of a mixture of Pd-only and AuPd alloys (Fig. S.5†), which is in keeping with our previous investigations into similar materials.45 By comparison the introduction of large quantities of Sn resulted in an improved dispersion of Pd and the formation of single atoms of Pd, surrounded by Sn/SnOx domains (Fig. 4C and D, with additional data reported in Fig. S.6†). Notably, our analysis of the PdSn catalytic series by XPS (Fig. S.7†) indicated that, regardless of Sn content, Pd speciation remained broadly similar for all formulations. As such, it is possible to conclude that the improved catalytic performance of the 0.25%Pd–2.25%Sn/TiO2 catalyst is not a result of the electronic modification of Pd species. Rather, we consider that the primary cause for the enhanced activity is a result of the improved dispersion of Pd species with the introduction of Sn. Such observations would align well with the numerous studies that have demonstrated the dependency of catalytic performance on nanoparticle size, and that in particular highly dispersed Pd species can offer exceptional selectivity towards H2O2.46–48
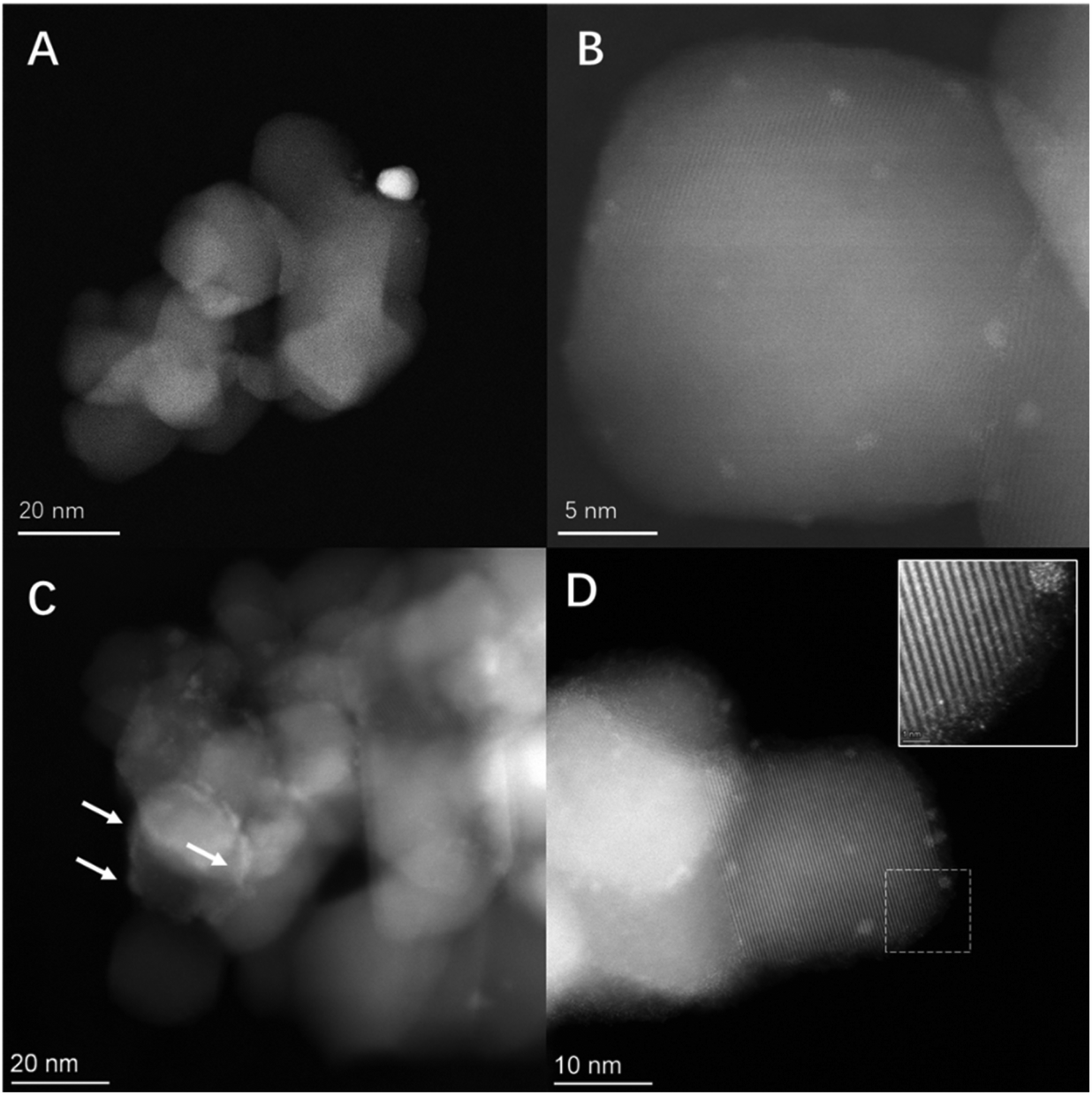 | ||
| Fig. 4 Aberration corrected STEM images of 0.25%Pd–0.25%Au/TiO2 and 0.25%Pd–2.25%Sn/TiO2 catalysts. (A) HAADF image of the 0.25%Pd–0.25%Au/TiO2 catalyst showing larger Au–Pd alloyed particles and (B) HAADF image of the 0.25%Pd–0.25%Au/TiO2 catalyst showing smaller <1 nm Pd-rich clusters. (C) HAADF image of the 0.25%Pd–2.25%Sn/TiO2 catalyst showing islands of SnOx over the TiO2 support surface and (D) HAADF image of 0.25%Pd–2.25%Sn/TiO2 showing smaller <3 nm Pd particles along with (inset) magnified image of highlighted area showing atomically dispersed Pd. Note: In all cases, active metallic species are identified by high Z-contrast (i.e. brighter regions of the micrograph). | ||
 | ||
| Fig. 5 HAADF-STEM and XEDS maps of (A) 0.25%Pd–0.25%Au/TiO2 showing the formation of AuPd alloys in the larger particles and (B) 0.25%Pd–2.25%Sn/TiO2 showing the presence of SnOx islands and well dispersed Pd species. Note: In all cases, active metallic species are identified by high Z-contrast (i.e. brighter regions of the micrograph). | ||
Conclusions
The efficacy of a series of PdAu and PdSn catalysts prepared by a wet co-impregnation methodology for the direct synthesis of H2O2 has been compared, with the introduction of both Au and Sn into supported Pd nanoparticles found to result in an improved catalytic performance. While the introduction of relatively low concentrations of Au resulted in a considerable synergistic enhancement in catalyst activity (Pd:Au = 1:1 (wt/wt)), significantly larger Sn loadings were required to rival the H2O2 synthesis rates offered by the optimal PdAu formulation (Pd:Sn = 1:10 (wt/wt)). A 0.25%Pd–2.25%Sn/TiO2 catalyst was found to be highly selective towards H2O2, with this metric comparable to that offered by state-of-the-art materials, due to improved Pd dispersion. While we consider that these catalysts represent a promising basis for further exploration, additional efforts are required to improve catalyst stability if they are to fully rival leading formulations.
Author contributions
D. K. and R. J. L. conducted catalytic synthesis, testing and data analysis. D. K., R. J. L., C. C., D. J. M., T. E. D, A. L., T. Q., C. S. A. and X. L. conducted catalyst characterisation and corresponding data processing. R. J. L. and G. J. H. contributed to the design of the study. R. J. L., A. L., J. K. E., L. C., M. S. R., X. L. and G. J. H. provided technical advice and result interpretation. R. J. L. wrote the manuscript and ESI with all authors commenting on and amending both documents. All authors discussed and contributed to the work.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
The authors wish to thank the financial support and detailed discussion with Haldor Topsøe.Additionally, the Cardiff University electron microscope facility (CCI-EMF) and Diamond Light Source electron Physical Science Imaging Centre (ePSIC proposal number MG27777) is acknowledged for the transmission electron microscopy. XPS data collection was performed at the EPSRC National Facility for XPS (‘HarwellXPS’), operated by Cardiff University and University College London, under contract No. PR16195. D. K. and C. C. acknowledge Haldor Topsøe for financial support. R. J. L. and G. J. H. gratefully acknowledge the Max Planck Centre for Fundamental Heterogeneous Catalysis (FUNCAT) for financial support. X. L. acknowledges financial support from National Key R&D Program of China (2021YFA1500300 and 2022YFA1500146) and National Natural Science Foundation of China (22072090 and 22272106). L. C. acknowledges the financial support from National Natural Science Foundation of China (21991153 and 21991150).
References
- S. H. Zeronian and M. K. Inglesby, Cellulose, 1995, 2, 265–272 CrossRef CAS.
- V. Russo, R. Tesser, E. Santacesaria and M. Di Serio, Ind. Eng. Chem. Res., 2013, 52, 1168–1178 CrossRef CAS.
- M. Lin, C. Xia, B. Zhu, H. Li and X. Shu, Chem. Eng. J., 2016, 295, 370–375 CrossRef CAS.
- C. M. Crombie, R. J. Lewis, D. Kovačič, D. J. Morgan, T. J. A. Slater, T. E. Davies, J. K. Edwards, M. S. Skjøth-Rasmussen and G. J. Hutchings, Catal. Lett., 2021, 151, 2762–2774 CrossRef CAS.
- Y. Du, Y. Xiong, J. Li and X. Yang, J. Mol. Catal. A: Chem., 2009, 298, 12–16 CrossRef CAS.
- G. Goor, J. Glenneberg, S. Jacobi, J. Dadabhoy and E. Candido, Ullmann's Encyclopedia of Industrial Chemistry, 2019 Search PubMed.
- J. R. Scoville and I. A. Novicova, US5900256, 1996, Cottrell Ltd Search PubMed.
- G. Gao, Y. Tian, X. Gong, Z. Pan, K. Yang and B. Zong, Chin. J. Catal., 2020, 41, 1039–1047 CrossRef CAS.
- R. J. Lewis and G. J. Hutchings, ChemCatChem, 2019, 11, 298–308 CrossRef CAS.
- J. M. Campos-Martin, G. Blanco-Brieva and J. L. G. Fierro, Angew. Chem., Int. Ed., 2006, 45, 6962–6984 CrossRef CAS PubMed.
- R. J. Lewis, K. Ueura, X. Liu, Y. Fukuta, T. Qin, T. E. Davies, D. J. Morgan, A. Stenner, J. Singleton, J. K. Edwards, S. J. Freakley, C. J. Kiely, L. Chen, Y. Yamamoto and G. J. Hutchings, ACS Catal., 2023, 13, 1934–1945 CrossRef CAS.
- C. M. Crombie, R. J. Lewis, R. L. Taylor, D. J. Morgan, T. E. Davies, A. Folli, D. M. Murphy, J. K. Edwards, J. Qi, H. Jiang, C. J. Kiely, X. Liu, M. S. Skjøth-Rasmussen and G. J. Hutchings, ACS Catal., 2021, 11, 2701–2714 CrossRef CAS.
- Z. Jin, L. Wang, E. Zuidema, K. Mondal, M. Zhang, J. Zhang, C. Wang, X. Meng, H. Yang, C. Mesters and F. Xiao, Science, 2020, 367, 193–197 CrossRef CAS PubMed.
- D. W. Flaherty, ACS Catal., 2018, 8, 1520–1527 CrossRef CAS.
- R. J. Lewis, E. N. Ntainjua, D. J. Morgan, T. E. Davies, A. F. Carley, S. J. Freakley and G. J. Hutchings, Catal. Commun., 2021, 161, 106358 CrossRef CAS.
- G. Gallina, J. García-Serna, T. O. Salmi, P. Canu and P. Biasi, Ind. Eng. Chem. Res., 2017, 56, 13367–13378 CrossRef CAS.
- Y. Han and J. H. Lunsford, J. Catal., 2005, 230, 313–316 CrossRef CAS.
- Q. Liu, J. C. Bauer, R. E. Schaak and J. H. Lunsford, Appl. Catal., A, 2008, 339, 130–136 CrossRef CAS.
- P. Priyadarshini, T. Ricciardulli, J. S. Adams, Y. S. Yun and D. W. Flaherty, J. Catal., 2021, 399, 24–40 CrossRef CAS.
- J. K. Edwards, B. Solsona, E. N. Ntainjua, A. F. Carley, A. A. Herzing, C. J. Kiely and G. J. Hutchings, Science, 2009, 323, 1037–1041 CrossRef CAS PubMed.
- T. Ricciardulli, S. Gorthy, J. S. Adams, C. Thompson, A. M. Karim, M. Neurock and D. W. Flaherty, J. Am. Chem. Soc., 2021, 143, 5445–5464 CrossRef CAS PubMed.
- M. Kim, G.-H. Han, X. Xiao, J. Song, J. Hong, E. Jung, H. Kim, J. Ahn, S. S. Han, K. Lee and T. Yu, Appl. Surf. Sci., 2021, 562, 150031 CrossRef CAS.
- Y. Zhang, Q. Sun, G. Guo, Y. Cheng, X. Zhang, H. Ji and X. He, Chem. Eng. J., 2023, 451, 138867 CrossRef CAS.
- G. Han, X. Xiao, J. Hong, K. Lee, S. Park, J. Ahn, K. Lee and T. Yu, ACS Appl. Mater. Interfaces, 2020, 12, 6328–6335 CrossRef CAS PubMed.
- N. M. Wilson, P. Priyadarshini, S. Kunz and D. W. Flaherty, J. Catal., 2018, 357, 163–175 CrossRef.
- J. Li, T. Ishihara and K. Yoshizawa, J. Phys. Chem. C, 2011, 115, 25359–25367 CrossRef CAS.
- S. J. Freakley, Q. He, J. H. Harrhy, L. Lu, D. A. Crole, D. J. Morgan, E. N. Ntainjua, J. K. Edwards, A. F. Carley, A. Y. Borisevich, C. J. Kiely and G. J. Hutchings, Science, 2016, 351, 965–968 CrossRef CAS PubMed.
- D. A. Crole, R. Underhill, J. K. Edwards, G. Shaw, S. J. Freakley, G. J. Hutchings and R. J. Lewis, Philos. Trans. R. Soc., A, 2020, 378, 20200062 CrossRef CAS PubMed.
- H. Xu, D. Cheng and Y. Gao, ACS Catal., 2017, 7, 2164–2170 CrossRef CAS.
- Y. Wang, H. Pan, Q. Lin, Y. Shi and J. Zhang, Catalysts, 2020, 10, 303 CrossRef.
- Z. Yang, Z. Hao, S. Zhou, P. Xie, Z. Wei, S. Zhao and F. Gong, ACS Appl. Mater. Interfaces, 2023, 15(19), 23058–23067 CrossRef CAS PubMed.
- H. Li, Q. Wan, C. Du, J. Zhao, F. Li, Y. Zhang, Y. Zheng, M. Chen, K. H. L. Zhang, J. Huang, G. Fu, S. Lin, X. Huang and H. Xiong, Nat. Commun., 2022, 13, 6072 CrossRef CAS PubMed.
- D. E. Doronkin, S. Wang, D. I. Sharapa, B. J. Deschner, T. L. Sheppard, A. Zimina, F. Studt, R. Dittmeyer, S. Behrens and J. Grunwaldt, Catal. Sci. Technol., 2020, 10, 4726–4742 RSC.
- W. Tian, Y. Dong, H. Qin, Z. Luo, Q. Lin, Z. Chen, H. Pan, Y. Shi and K. Wang, Mol. Catal., 2023, 547, 113376 CrossRef CAS.
- R. J. Lewis, K. Ueura, Y. Fukuta, S. J. Freakley, L. Kang, R. Wang, Q. He, J. K. Edwards, D. J. Morgan, Y. Yamamoto and G. J. Hutchings, ChemCatChem, 2019, 11, 1673–1680 CrossRef CAS.
- X. Hu, Q. Xie, J. Zhang, Q. Yu, H. Liu and Y. Sun, Int. J. Hydrogen Energy, 2020, 45, 27837–27845 CrossRef CAS.
- A. Santos, R. J. Lewis, G. Malta, A. G. R. Howe, D. J. Morgan, E. Hampton, P. Gaskin and G. J. Hutchings, Ind. Eng. Chem. Res., 2019, 58, 12623–12631 CrossRef CAS.
- J. K. Edwards, A. Thomas, A. F. Carley, A. A. Herzing, C. J. Kiely and G. J. Hutchings, Green Chem., 2008, 10, 388–394 RSC.
- S. Kanungo, L. van Haandel, E. J. M. Hensen, J. C. Schouten and M. Fernanda Neira d’Angelo, J. Catal., 2019, 370, 200–209 CrossRef CAS.
- J. Edwards, B. Solsona, P. Landon, A. Carley, A. Herzing, C. Kiely and G. Hutchings, J. Catal., 2005, 236, 69–79 CrossRef CAS.
- R. Burch and P. R. Ellis, Appl. Catal., B, 2003, 42, 203–211 CrossRef CAS.
- V. R. Choudhary, A. G. Gaikwad and S. D. Sansare, Catal. Lett., 2002, 83, 235–239 CrossRef CAS.
- X. Gong, R. J. Lewis, S. Zhou, D. J. Morgan, T. E. Davies, X. Liu, C. J. Kiely, B. Zong and G. J. Hutchings, Catal. Sci. Technol., 2020, 10, 4635–4644 RSC.
- F. Wang, C. Xia, S. P. de Visser and Y. Wang, J. Am. Chem. Soc., 2019, 141, 901–910 CrossRef CAS PubMed.
- J. Brehm, R. J. Lewis, T. Richards, T. Qin, D. J. Morgan, T. E. Davies, L. Chen, X. Liu and G. J. Hutchings, ACS Catal., 2022, 12(19), 11777–11789 Search PubMed.
- S. Yu, X. Cheng, Y. Wang, B. Xiao, Y. Xing, J. Ren, Y. Lu, H. Li, C. Zhuang and G. Chen, Nat. Commun., 2022, 13, 4737 CrossRef CAS PubMed.
- P. Tian, D. Ding, Y. Sun, F. Xuan, X. Xu, J. Xu and Y. Han, J. Catal., 2019, 369, 95–104 CrossRef CAS.
- C. Williams, J. H. Carter, N. F. Dummer, Y. K. Chow, D. J. Morgan, S. Yacob, P. Serna, D. J. Willock, R. J. Meyer, S. H. Taylor and G. J. Hutchings, ACS Catal., 2018, 8, 2567–2576 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3gc03706a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |