
Developing extended visible light responsive polymeric carbon nitrides for photocatalytic and photoelectrocatalytic applications†
Sanjit
Mondal
a,
Gabriel
Mark
a,
Liel
Abisdris
a,
Junyi
Li
a,
Tirza
Shmila
a,
Jonathan
Tzadikov
a,
Michael
Volokh
 a,
Lidan
Xing
b and
Menny
Shalom
*a
a,
Lidan
Xing
b and
Menny
Shalom
*a
aDepartment of Chemistry and Ilse Katz Institute for Nanoscale Science and Technology, Ben-Gurion University of the Negev, Beer-Sheva 8410501, Israel. E-mail: mennysh@bgu.ac.il
bSchool of Chemistry, South China Normal University, Guangzhou 510006, China
First published on 26th January 2023
Abstract
Polymeric carbon nitride (CN) has emerged as an attractive material for photocatalysis and photoelectronic devices. However, the synthesis of porous CNs with controlled structural and optical properties remains a challenge, and processable CN precursors are still highly sought after for fabricating homogenous CN layers strongly bound to a given substrate. Here, we report a general method to synthesize highly dispersed porous CN materials that show excellent photocatalytic activity for the hydrogen evolution reaction and good performance as photoanodes in photoelectrochemical cells (PEC): first, supramolecular assemblies of melem and melamine in ethylene glycol and water are prepared using a hydrothermal process. These precursors are then calcined to yield a water-dispersible CN photocatalyst that exhibits beneficial charge separation under illumination, extended visible-light response attributed to carbon doping, and a large number of free amine groups that act as preferential sites for a Pt cocatalyst. The optimized CN exhibits state-of-the-art HER rates up to 23.1 mmol h−1 g−1, with an AQE of 19.2% at 395 nm. This unique synthetic route enables the formation of a homogeneous precursor paste for substrate casting; consequently, the CN photoanode exhibits a low onset potential, a high photocurrent density and good stability after calcination.
New conceptsPhotocatalytic and photoelectronic devices, such as photoelectrochemical cells (PEC), require complex materials that combine a range of structural (including morphology and specific surface area) and optoelectronic properties (such as band gap, electronic band positions, and charge separation under illumination). Polymeric carbon nitride (CN) has emerged as an attractive material for these applications thanks to its low cost its overall desirable and highly tunable properties. Here we introduce a general approach for the rational synthesis of porous CN materials with state-of-the-art photocatalytic activity for the hydrogen evolution reaction (HER) and excellent performance as photoanode materials in photoelectrochemical cells: we integrated strategies of supramolecular chemistry, hydrothermal treatment, and solvent mixtures to prepare a paste composed of monomers that affords CN after calcination. The presence of EG in the condensation process leads to C-doping, high surface area, and a large number of free NH2 groups in the final CNs; as a result, these materials exhibit a unique electronic structure (including homojunction and C-doping), extended light-harvesting properties, excellent dispersibility in water, and high cocatalyst loading capacity. The as-obtained homogenous paste precursor can be directly used for electrode preparation. |
Introduction
Polymeric carbon nitride (CN) has emerged as an attractive material for photocatalytic and photoelectronic devices, such as photoelectrochemical cells (PEC), light-emitting diodes (LED), and biosensors.1–5 Some of the key factors determining the photoactivity of CN materials are their structural properties (including morphology and specific surface area) and their optical and electronic properties (such as band gap, electronic band positions, and charge separation under illumination).6–11 For heterogeneous photocatalysis, the CN powders must also have good dispersibility in water, which is both the dispersant and the reactant in the hydrogen evolution reaction (HER); an additional pivotal requirement is the availability of free sites favoring the anchoring of co-catalysts.12,13 As for photoelectronic devices, the precursors should be amenable to uniform casting and must remain bound to the substrate during the ensuing thermal polymerization. Finding a simple synthetic path toward a material that encompasses all these features is highly desired. Recently, 2,5,8-triamino-tri-s-triazine (melem), the repeating unit in CN, has been used for the synthesis of CNs for its high thermal stability and low sublimation.14–16 The reaction of melem with other small monomers allows the CN properties to be adjusted.17,18 However, the insolubility of melem in most solvents hinders the construction of supramolecular structures in high yield.The design of supramolecular assemblies composed of CN monomers as reactants is a powerful tool for synthesizing tailored CN materials.19–22 The supramolecular approach enables good control over the morphology of CN and over its optical, electronic, and catalytic properties.23–26 The formation, uniformity, and reproducibility of the self-assembly rely strongly on the monomers having adequate solubility in a given solvent: the solubility of the monomers will direct their interactions, thus allowing a molecular ‘blueprint’ to evolve into the morphology on the macroscale. To date, most studies have been done in water, where monomers have limited solubility.18,27,28 The construction of complex structures of several monomers and solvents may pave the path toward novel CN materials. As for photoelectronic devices, the reproducible preparation of a paste with high control over its composition remains challenging.
Here we report a general approach to synthesizing a highly photoactive CN as a photocatalyst for HER and as a photoanode in photoelectrochemical cells. To do so, we construct supramolecular assemblies based on a combination of melem and melamine in a mixture of ethylene glycol (EG) and water. We hypothesized that the hydrogen-bonding EG would favor the self-assembly of the monomers and also allow casting via doctor-blading.
To optimize the assembling the monomers and the organization process, we used a hydrothermal technique to improve solubility. Theoretical and experimental data show that EG strongly improves the solubility of the monomers by binding to the melem and melamine units, leading to the formation of a homogeneous precursor paste of carbon-rich monomers. Upon calcination, a highly porous CN with extended light absorption and high concentration of NH2 surface groups is obtained. The homogeneous precursor paste may be directly used to fabricate CN films on conductive substrates for use as photoelectrochemical water-splitting photoanodes.
Results and discussion
Nanostructured polymeric carbon nitrides (CNs) were synthesized in a three-step synthesis procedure as shown in Fig. 1 (a detailed synthesis procedure is provided in the ESI†).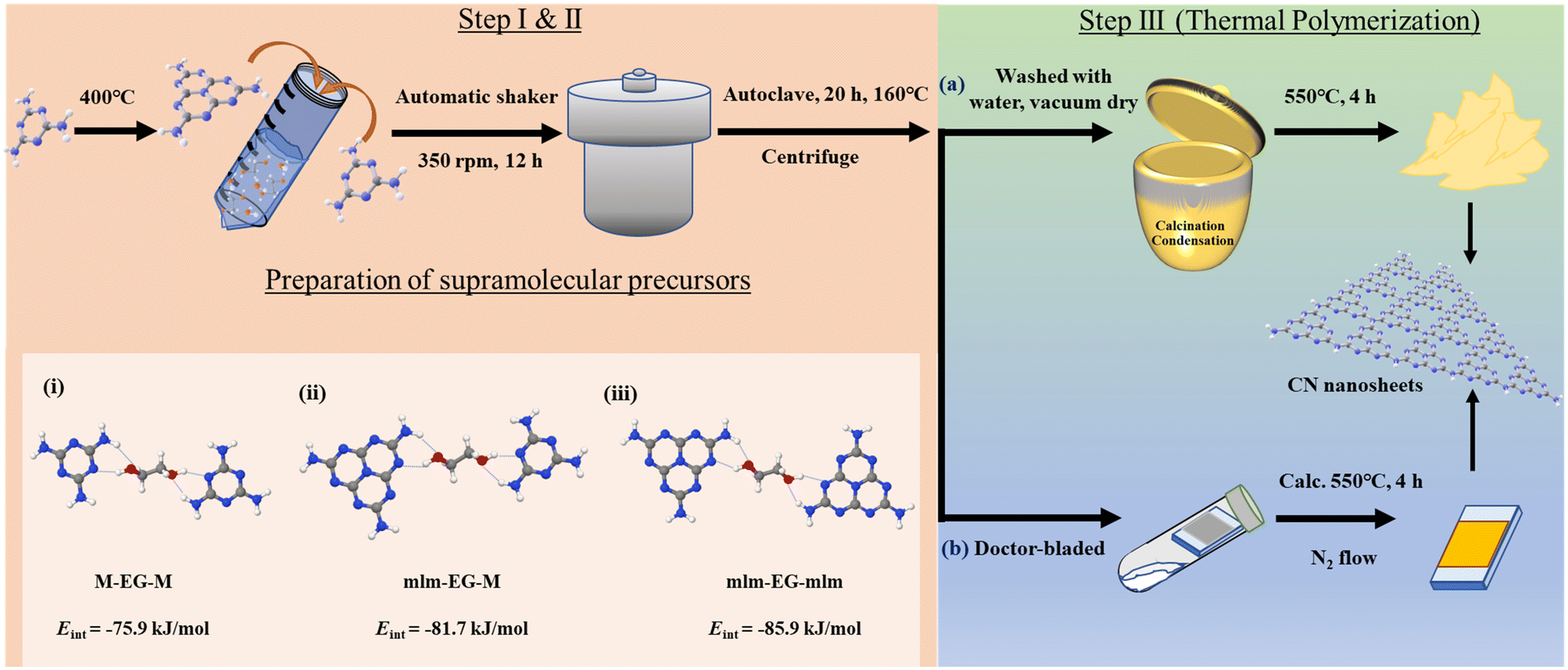 | ||
| Fig. 1 Procedure for the preparation of CN powder photocatalysts and photoelectrodes via doctor-blading. First, melem is synthesized from melamine in one step. Second, a two-step process is used to prepare supramolecular assemblies. Then, they are calcined in a crucible (photocatalyst powder) or deposited on a transparent conductive substrate and calcined (photoelectrode with a CN film). The inset represents the calculated interaction energy values between ethylene glycol (EG) and (i) two melamine molecules (M–EG–M), (ii) one melem (mlm) and one M (mlm–EG–M), and (iii) two mlm molecules (mlm–EG–mlm); all three options can serve as precursors in CN preparation. | ||
Synthesis of CN precursors
The first two synthetic steps involve the preparation of the precursors that will be thermally polymerized into the final CN materials, as discussed later. First, melem (mlm) was synthesized by the thermal polymerization of melamine (M) for 12 h at 400 °C in air.29 Supramolecular assemblies of melamine and melem at a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio were prepared by dispersing these two monomers in a water/EG mixture, followed by overnight shaking. To improve the solubility of the monomers and optimize the formation of supramolecular assemblies, the dispersion was heated at 160 °C for 20 h in a PTFE-lined autoclave. After that, the final supramolecular assemblies were collected by centrifugation; they are labeled as PM+mlm (control precursors were prepared using only melamine and melem and denoted as PM and Pmlm respectively). These supramolecular assemblies were directly used as precursors for (a) the rational synthesis of a powder photocatalyst or (b) the preparation of a photoelectrode. Density functional theory (DFT) simulations reveal that the interaction energy (Eint) in M–EG–mlm structures is more negative than that in M–EG–M structures (−81.7 vs. −75.9 kJ mol−1, respectively), indicating that EG favors the formation of a stable supramolecular structure including both M and mlm (Fig. 1, structure i and ii, Table S1, ESI†). Besides, the calculated Eint for mlm–EG–mlm is −85.9 kJ mol−1, suggesting that EG easily binds to two melem units (Fig. 1, structure iii). In all three cases, the hydrogen bonding EG facilitates supramolecular assembly of two melem unit and melem-melamine.
:1 molar ratio were prepared by dispersing these two monomers in a water/EG mixture, followed by overnight shaking. To improve the solubility of the monomers and optimize the formation of supramolecular assemblies, the dispersion was heated at 160 °C for 20 h in a PTFE-lined autoclave. After that, the final supramolecular assemblies were collected by centrifugation; they are labeled as PM+mlm (control precursors were prepared using only melamine and melem and denoted as PM and Pmlm respectively). These supramolecular assemblies were directly used as precursors for (a) the rational synthesis of a powder photocatalyst or (b) the preparation of a photoelectrode. Density functional theory (DFT) simulations reveal that the interaction energy (Eint) in M–EG–mlm structures is more negative than that in M–EG–M structures (−81.7 vs. −75.9 kJ mol−1, respectively), indicating that EG favors the formation of a stable supramolecular structure including both M and mlm (Fig. 1, structure i and ii, Table S1, ESI†). Besides, the calculated Eint for mlm–EG–mlm is −85.9 kJ mol−1, suggesting that EG easily binds to two melem units (Fig. 1, structure iii). In all three cases, the hydrogen bonding EG facilitates supramolecular assembly of two melem unit and melem-melamine.
Structural characterization of the prepared supramolecular precursors
The powder X-ray diffraction (XRD) pattern of the PM+mlm precursor after hydrothermal treatment is shown in Fig. S1 (ESI†). Fourier transform infrared spectroscopy (FTIR) inspection of the hydrothermally treated CN precursors (PM, Pmlm, and PM+mlm) and pristine M and mlm in Fig. 2a show characteristic stretching vibrations related to the breathing mode of triazine units centered at 790 cm−1 and vibrations of the heptazine units at 1220–1610 cm−1. Furthermore, the hydrothermally treated samples additional vibrational bands at 2920 cm−1 and 2865 cm−1, originating from C–H stretching, and other bands at 1070 cm−1, 1034 cm−1, and 883 cm−1, corresponding to the C–O stretching, C–C–O asymmetric, and C–C–O symmetric stretching modes, respectively, indicating the presence of the EG linker in the final precursor.30,31 For the hydrothermally treated samples, an additional weak band is observed at 1682 cm−1, which is attributed to the O–H bending mode.32 In general, the broad bands (3000–3600 cm−1) are attributed to the stretching modes of primary and secondary amines or their intermolecular H-bonding interactions; the peak intensities and positions (∼3070 cm−1) of the hydrothermally treated samples are slightly different from those of pristine melamine and melem precursors, verifying the H-bonding between monomers and EG molecules. Finally, the FTIR spectra of the PM+mlm precursors during hydrothermal treatment (Fig. S2, ESI†) show that increasing amounts of EG lead to an increased intensity of the bands at 1070 cm−1 and 1034 cm−1, ascribed to the C–O stretching and C–C–O asymmetric modes, respectively, along with a minor shift of their positions. These changes in the FTIR spectra confirm our hypothesis that bonding interactions between EG and the precursors are present during hydrothermal treatment.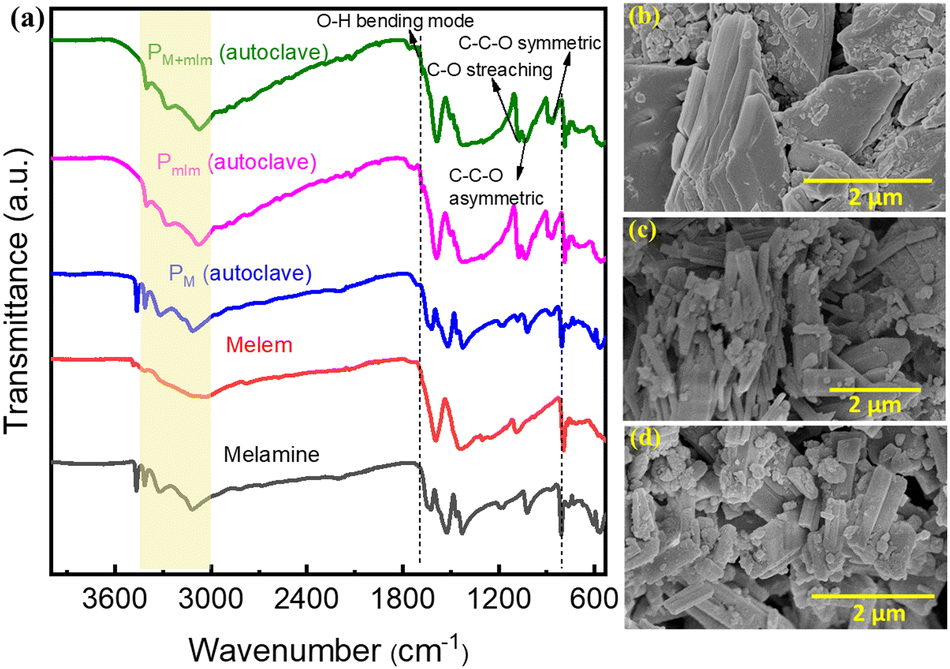 | ||
| Fig. 2 Precursors characterization. (a) FTIR spectra of the precursors before (melem and melamine) and after (PM for melamine, Pmlm for melem, and PM+mlm for the mixture) hydrothermal treatment in an EG/water mixture. SEM images of (b) PM, (c) Pmlm, and (d) PM+mlm. The spectra in (a) are offset for clarity. | ||
Scanning electron microscopy (SEM) images of the precursors indicate that the morphologies of the hydrothermally treated PM, Pmlm, and PM+mlm precursors in the presence of EG are entirely different from those of untreated M and mlm (Fig. 2b–d and Fig. S3, ESI†). The hydrothermal treatment of PM+mlm without EG induces the formation of ordered 1D microstructures as shown in Fig. S4a (ESI†). In contrast, the SEM images of PM+mlm samples treated with various amounts of EG are presented in Fig. S4b–d (ESI†) and show a significant change in their morphology with increasing EG incorporation. The mixed morphology, both 1D microstructure and macroaggregates particles, was observed in the precursors obtained from 16, 32, and 50 v% EG. However, the changes in their morphology were random with the increase in EG concentration.
Structural characterization of the nanostructured CNs
The use of EG in the hydrothermal process has several advantages: (i) the sufficiently high solubility of the CN precursors in EG enables the establishment of highly ordered supramolecular assemblies; (ii) moderate carbon doping shifts the optical and photocatalytic response to longer wavelengths, as shown later; (iii) the prepared precursor paste is readily applicable onto substrates for optoelectronic devices—the precursors, after centrifugation, can be directly used for the preparation of CN photoelectrodes over transparent conductive oxide (TCO) substrates, followed by calcination under N2 atmosphere (as will be shown later). For the preparation of photocatalyst powders, the slurry remaining after centrifugation was washed once with water and then dried overnight in a vacuum oven at 60 °C. The obtained powder was calcined in a muffle furnace at 550 °C for 4 h in air. Light brown CN photocatalysts were obtained; their characterization is described in the following subsections. The resulting polymeric carbon nitride materials are denoted as ‘CN’ with the precursor name in subscript (namely, CNM, CNmlm, and CMM+mlm, which are investigated in detail).The diffraction pattern of CNM+mlm (Fig. 3a) exhibits two significant diffraction signals at 13.2° and 27.7°, corresponding to in-plane tri-s-triazine units (100) and interlayer lattice (002) planes of melon-based CN, respectively.33 The diffraction patterns of CNM and CNmlm similarly exhibit these signature graphitic diffractions. Furthermore, the (002) diffraction plane of CNM+mlm is at a higher angle than CNM and CNmlm, stemming from a decrease in the interlayer-stacking distance in the CN aromatic units. The intensity of the (002) peak is also noticeably diminished relative to those of the CNM and CNmlm samples, indicating a smaller sheet size of the layers.34
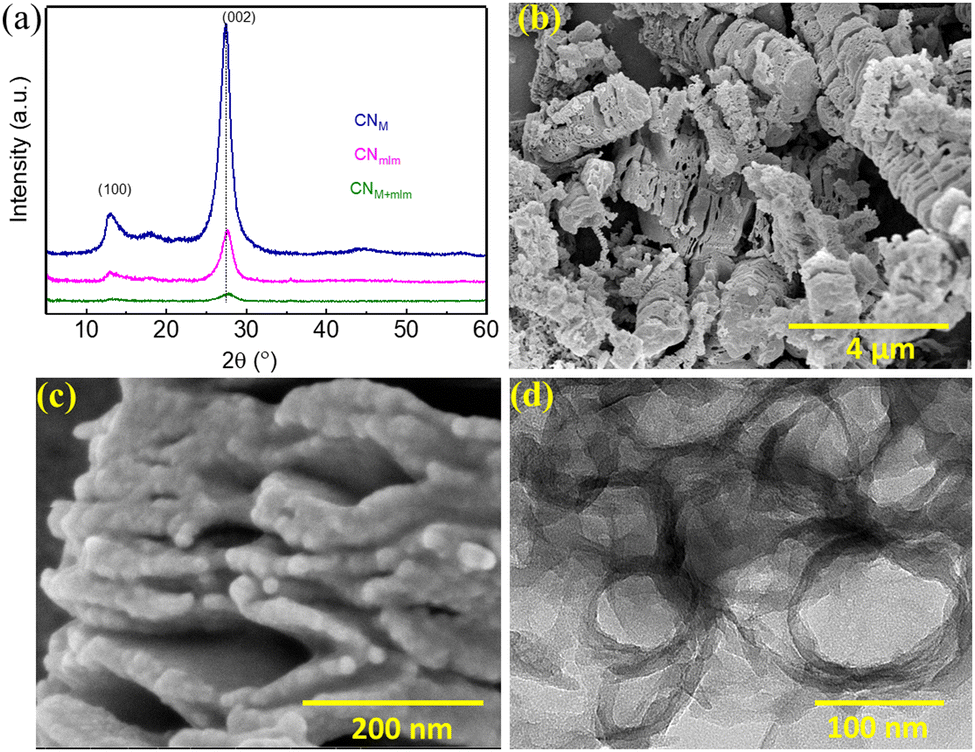 | ||
| Fig. 3 CN characterization. (a) Powder XRD patterns of CNM, CNmlm, and CNM+mlm. SEM images of (b) CNM+mlm, and (c) CNM+mlm at higher magnification. (d) TEM image of CNM+mlm. | ||
The FTIR spectra of the produced CN samples (Fig. S5, ESI†) show the presence of the typical stretching modes of C–N heterocycles at 1220–1610 cm−1 and the breathing mode of the triazine units at 804 cm−1. No noticeable difference is seen between the three spectra, except for a slight change in the broad bands located at 3000–3400 cm−1.35,36 No major changes were observed in the FTIR spectra of the resulting CNM+mlm samples when varying the amount of EG during synthesis.
As observed in the SEM images Fig. 3b and c, CNM+mlm retains the both 1D and particle morphology of the precursor (PM+mlm). Moreover, a careful examination reveals that CNM+mlm is composed of thin nanosheets attached to one another (Fig. 3c). We suggest that the condensation process during calcination of the supramolecular assemblies (1D and microparticles) used as precursors is responsible for this unique morphology. In contrast, CNM shows the formation of dense solid CN agglomerates several micrometers in size (Fig. S6a, ESI†). CNmlm consists of a nanosheet architecture with irregular porous structures (Fig. S6b, ESI†).
Fig. S7 (ESI†) indicates that the morphologies of the precursors and the final CN samples are significantly affected by the amount of EG in the precursor subjected to hydrothermal treatment. The ordered 1D microstructures of the precursor have completely changed and formed nanosheet architecture with irregular porous structures in the case of CN0v%EG (Fig. S7a, ESI†). However, CN obtained from various amounts of EG (16, 32 v%) shows a porous exfoliated 1D morphology composed of nanosheets attached to one another (Fig. S7b and c, ESI†). It can be noted that the CN50v%EG consists of both porous exfoliated 1D and porous particles (Fig. 7d).
To study the role of EG, we varied the EG content in the preparation of CNM+mlm. Fig. S7 (ESI†) indicates that the morphologies of the precursors and the final CN samples are significantly affected by the amount of EG in the precursor subjected to hydrothermal treatment. Specifically, an optimum amount of EG is required to prepare well-textured CN nanostructures. We hypothesize that the amount of EG must be so that it provides sufficient solubility to promote the formation of well-defined supramolecular assemblies while still acting as a binder (Fig. 1, structures i, ii, and iii) that holds the structure together during condensation. Transmission electron microscopy (TEM) images of CNM+mlm (Fig. 3d and Fig. S8, ESI†) disclose a morphology based on porous and thin nanosheets.
The chemical properties of CNM, CNmlm, and CNM+mlm were examined by X-ray photoelectron spectroscopy (XPS). The survey spectra (Fig. S9, ESI†) confirm the presence of C, N, and O; the weak O 1s signal can be ascribed to surface-adsorbed CO2 or H2O, suggesting a negligible amount of impurities in the CN samples. The high-resolution C 1s XPS spectra of CNM, CNmlm, and CNM+mlm are deconvoluted into three peaks (Fig. 4a). The peaks at 284.8, 286.7, and 288.1 eV are ascribed to graphitic carbon (C–C–H), sp2-bonded carbon in the heptazine or triazine rings (N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–N), and sp2 carbon in the aromatic ring attached to an NH2 group (C–(N)3), respectively.37–39 The high-resolution N 1s spectra (Fig. 4b) are deconvoluted into four peaks centered at 398.6, 399.2, 400.1, and 401.1 eV, which are attributed to pyridinic N, tertiary N (N–(C)3), primary N (from –NH2 groups), and quaternary N, respectively. It is worth noting that the apparent peak shifts in the C 1s and N 1s spectra of CNM+mlm with respect to CNmlm and CNM indicate that the local environment (chemical and physical) is significantly different.40–43 The higher binding energy of C 1s and N 1s in the CNM+mlm spectrum can also be described based on the increased C–N bond covalency and the presence of defects. Furthermore, to find out the defects in the CN with various precursors, we have recorded the C 1s XPS (etched for 60 s) for CNM, CNmlm, and CNM+mlm samples (Fig. S10, ESI†). After etching, a new peak appeared for all the samples at 285.9 eV, which is attributed to the presence of a C–C bond, originating through the N-vacancy in the tri-s-triazine.44 In addition, the amount of C–C bonds is higher in the case of CNM+mlm compared to CNM and CNmlm, suggesting that the precursor induces C-defects in the final CN structure.
C–N), and sp2 carbon in the aromatic ring attached to an NH2 group (C–(N)3), respectively.37–39 The high-resolution N 1s spectra (Fig. 4b) are deconvoluted into four peaks centered at 398.6, 399.2, 400.1, and 401.1 eV, which are attributed to pyridinic N, tertiary N (N–(C)3), primary N (from –NH2 groups), and quaternary N, respectively. It is worth noting that the apparent peak shifts in the C 1s and N 1s spectra of CNM+mlm with respect to CNmlm and CNM indicate that the local environment (chemical and physical) is significantly different.40–43 The higher binding energy of C 1s and N 1s in the CNM+mlm spectrum can also be described based on the increased C–N bond covalency and the presence of defects. Furthermore, to find out the defects in the CN with various precursors, we have recorded the C 1s XPS (etched for 60 s) for CNM, CNmlm, and CNM+mlm samples (Fig. S10, ESI†). After etching, a new peak appeared for all the samples at 285.9 eV, which is attributed to the presence of a C–C bond, originating through the N-vacancy in the tri-s-triazine.44 In addition, the amount of C–C bonds is higher in the case of CNM+mlm compared to CNM and CNmlm, suggesting that the precursor induces C-defects in the final CN structure.
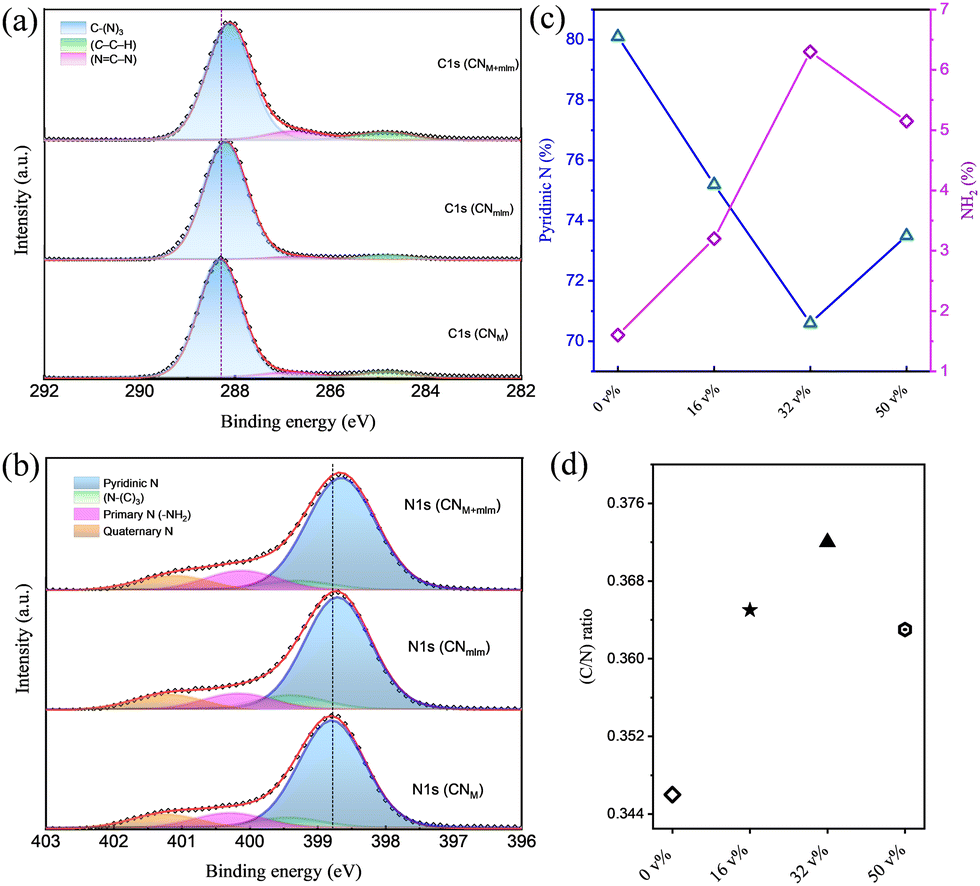 | ||
| Fig. 4 XPS CN analysis. High-resolution (a) C 1s and (b) N 1s XPS spectra of CNM, CNmlm, and CNM+mlm. (c) Calculated percentage of pyridinic N and free NH2 species for various amounts of EG (v%) used in the hydrothermal treatment during CNM+mlm syntheses, calculated from N 1s spectra. (d) Atomic C/N ratio obtained from survey scans of CNM+mlm prepared with varying EG content. The experimental points in (a) and (b) are fitted with a red line, which is deconvoluted into peaks corresponding to the indicated chemical species. The lines connecting the diamonds and triangles in (c) serve as a guide to the eye. | ||
The effect of the EG content in the precursor on the chemical properties of the resulting CNM+mlm was also examined using XPS. The C/N ratio was calculated from the survey spectra of samples prepared with various amounts of EG. The C/N ratio increases with increasing EG content until ∼32 v% as shown in Fig. 4c (and Fig. S11, ESI†). However, the C/N ratio was lowered as the EG content was further increased to 50 v%; a decrease attributed to the excessive solubility of melamine in EG, which disfavors the formation of a supramolecular assembly (Fig. S12, ESI†).
The high-resolution C 1s and N 1s spectra of CNM+mlm samples that were prepared using various EG contents (v% in water) are shown in Fig. S13 (ESI†); they exhibit an increase in the amount of free NH2 and a decrease in the amount of pyridinic N, as the EG content is increased up to 32 v% (Fig. 4d).45–47 EG with its carbon chain creates a large distance between two monomer units through H-bonding; therefore, during the calcination process, some of the EG molecules leave and result in free –NH2 groups rather than polymerization between the two –NH2 moieties. A table of relative ratios of groups in N 1s XPS spectra for CN with various EG content is shown in Table S2, ESI.† Importantly, an increased number of free NH2 groups provides a larger number of electron-rich centers in CN, which favor the adsorption of metal ions (Pt2+) during co-catalyst photodeposition. This interaction, in turn, facilitates the formation of multiple Pt(s) nucleation sites,48 resulting in well-dispersed small Pt nanoparticles, which serve as cocatalysts during photocatalysis. Elemental analysis (EA) of CNM, CNmlm, and CNM+mlm (Table S3, ESI†) indicates that their carbon content also increases with increasing EG content, probably owing to the incorporation of carbon originating from the EG molecules. Solid state 13C NMR spectra of CN0v%EG and CN32v%EG samples indicate that the EG contributes trace amount of –CH2 species to the final CN, as shown in Fig. S14, ESI.†
Optical properties of CN nanostructures
The UV–vis diffuse reflectance spectra of CNM+mlm (Fig. S15, ESI†) show a redshift of the absorption band edge and improved light absorption in the visible range, suggesting the presence of defects states below the conduction band in the CN framework.49,50 The calculated optical direct band gap of CNM+mlm, Eg, is 2.82 eV; higher Eg values of 2.93 eV and 3.01 eV were obtained for CNM and CNmlm, respectively (Fig. 5a).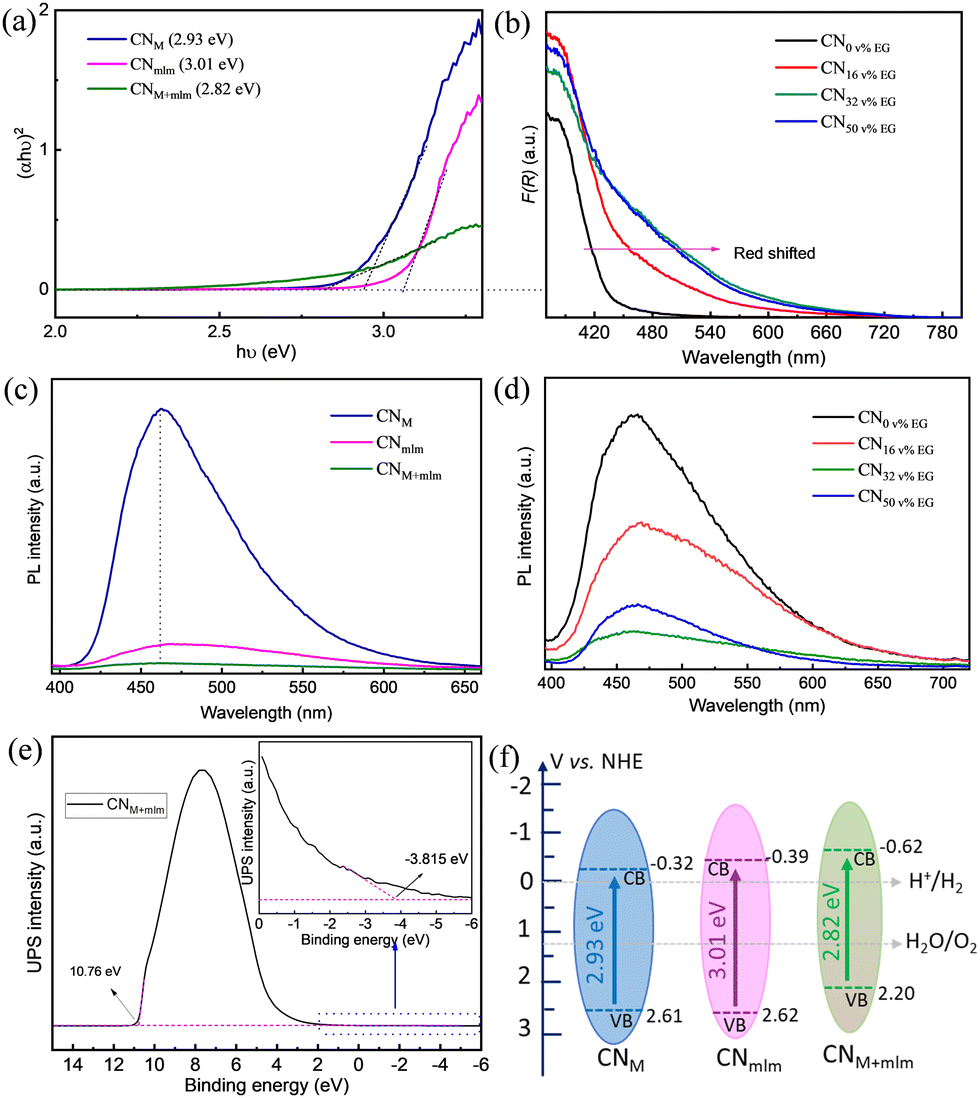 | ||
| Fig. 5 Optical characterization and band positions estimation. (a) Tauc plot analysis of the CN samples assuming a direct Eg. (b) UV-vis diffuse reflectance spectra of CNM+mlm prepared using varying EG content (v%). Photoluminescence emission spectra of (c) CNM, CNmlm, and CNM+mlm, and (d) CNM+mlm prepared using varying EG content in the precursor preparation. (e) UPS valence band spectrum of CNM+mlm. (f) Schematic representation of the electronic band structure (on the normal hydrogen electrode (NHE) scale) of CNM, CNmlm, and CNM+mlm, determined using the UPS-measured VB position and the optical Eg determination. | ||
The amount of EG used in the hydrothermal process has a direct effect on the light-absorption properties of CNM+mlm, as shown in Fig. 5b. A noticeable redshift of the absorption band was observed with increasing EG content, indicating the incorporation of carbon into the CN structure (as confirmed by EA, Table S3, ESI†). The latter observation suggests that even after its removal, residual amounts of EG remain in the precursor, bound within its structure. The photoluminescence (PL) spectra for the CNM, CNmlm, and CNM+mlm samples under an excitation wavelength (λex) of 375 nm are shown in Fig. 5c. CNM shows a strong emission band centered at 462 nm, which is attributed to direct-photogenerated electron–hole recombination. Significant quenching was observed for CNM+mlm as compared with CNM and CNmlm, possibly stemming from an alternative charge recombination process, which suggests that charge separation was improved by the creation of new defect states.51,52 The pronounced decrease in the PL emission of the CN samples with increasing EG amounts (Fig. 5d) is attributed to the higher carbon content. This quenching is maximal for CNM+mlm with 32 v% EG, suggesting an optimum EG content with respect to photogenerated charge recombination.
In addition to having an appropriate Eg, a semiconductor photocatalyst must have conduction band (CB) and valence band (VB) positions that match the redox potentials required for the targeted photocatalytic reactions. To study the VB positions of the CN materials, ultraviolet photoelectron spectroscopy (UPS) was recorded (Fig. 5e and Fig. S16, ESI†). The VB energy (EVB) positions of CNM, CNmlm, and CNM+mlm were calculated to be 2.61, 2.62, and 2.20 V, respectively. A schematic of EVB and ECB for the CN samples is illustrated in Fig. 5f. The ECB of CNM+mlm is more negative (−0.62 V) than that of CNM and CNmlm (−0.33 V and −0.38 V, respectively).38,53 The higher ECB position results in a higher driving force towards the HER. The EVB is positioned below the water oxidation level (H2O to O2), suggesting that CNM+mlm is also suitable for O2 production.
The specific surface area (SA) of the CN materials was analyzed by N2 adsorption–desorption measurements (Fig. S17, ESI†). The introduction of melem into the precursor leads to significant enhancement of the SA of the final material: the SA of CNmlm and CNM+mlm are 13.4 and 9.6 times as high as that of CNM (SA of 187, 134, and 14 m2 g−1, respectively). The introduction of melem in the precursor creates a larger cavity in the supramolecular assembly endowing higher specific surface area in the final CN.54 Moreover, the SA of CNM+mlm is also a function of the amount of EG in its precursor (Fig. S18, ESI†).
Photocatalytic hydrogen evolution reaction (HER) performance
The HER activity under white light illumination of the CNM+mlm samples synthesized using various amounts of EG (0, 16, 32, and 50 v%) discloses an enhancement in activity until 32 v% EG, corresponding to the trends in optical absorbance, PL quenching, NH2 groups content, and SA (Fig. 6a). The corresponding average hydrogen production rates for these photocatalysts (normalized to the total mass of the catalyst) were 10.8, 15.2, 23.1, and 13.0 mmol g−1 h−1, respectively. Because the HER performance of CNM+mlm (32 v% EG) is the highest among all samples, in line with its material properties, all the photoactivity measurements hereinafter are of CN samples from PM+mlm precursors containing 32 v% EG. A comparison table of recently reported results on CN-based HER photocatalysis is displayed in Table S4, ESI,† showing the excellent performance of CNM+mlm (32 v% EG) over other catalysts.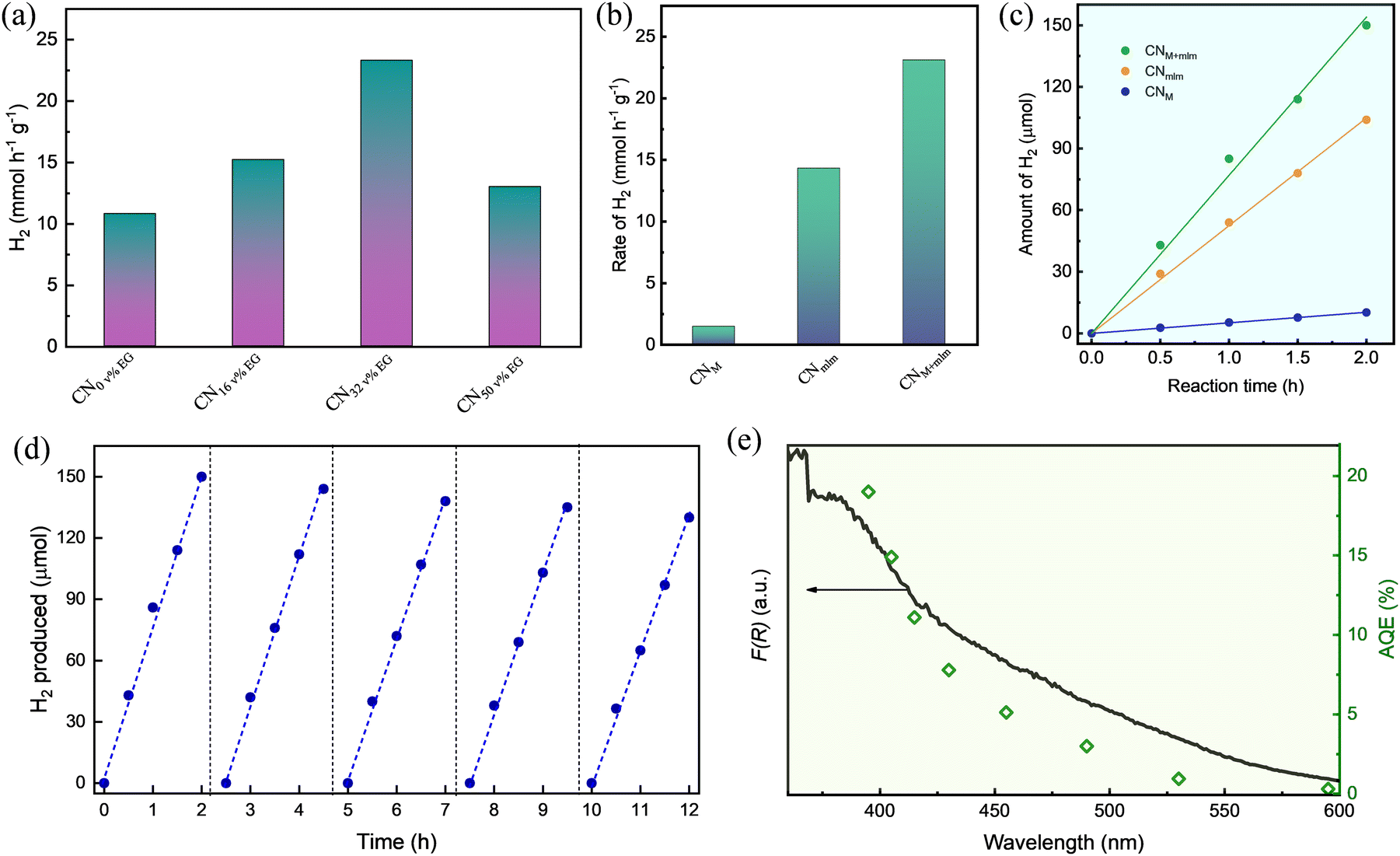 | ||
| Fig. 6 Photocatalytic characterization. (a) HER rates of CNM+mlm prepared using varying EG content, normalized to catalyst mass. HER performance of CNM, CNmlm, and CNM+mlm: (b) photocatalyst mass-normalized HER rate, (c) amount of H2vs. time, (d) evolved H2(g) vs. time in a photocatalytic HER stability test for 5 consecutive cycles, and (e) AQE of CNM+mlm measured at several illumination wavelengths and corresponding absorbance spectrum. | ||
The HER activities of the CNM and CNmlm samples were also determined, as displayed in Fig. 6b. The HER rate of CNM+mlm (23.1 mmol g−1 h−1) is about 1.6 and 17 times as high as those of CNM (14.3 mmol g−1 h−1) and CNmlm (1.34 mmol g−1 h−1), respectively, thanks to its better light adsorption, suitable CB position, and defective nanostructure. The corresponding hydrogen production of the three samples over time (2 h) is shown in Fig. 6c, expressing a linear relationship between the total produced H2 and time.
To investigate the stability of CNM+mlm, cycling tests, that is repeated tests in the same experimental setup and reaction conditions, were performed. Fig. 6d shows the CNM+mlm activity during five consecutive cycles; it exhibits only a 10% decline after the 5th catalytic cycle, illustrating the durability of the material. Post-test characterization of the CNM+mlm catalyst (after 5 catalytic cycles) was performed using TEM (Fig. S19, ESI†) and shows uniformly dispersed Pt nanoparticles on the CN surface, with an average size of 5 ± 3 nm and narrow size distribution, explaining the excellent photocatalytic performance.
The apparent quantum efficiency (AQE) of CNM+mlm was measured at several wavelengths in the 395–595 nm range as a proxy for photocatalytic activity evaluation (Fig. 6e). The maximum AQE of CNM+mlm, ∼19.2%, was obtained at 395 nm. Importantly, CNM+mlm also exhibits good AQE in the visible range, even in the 490–595 nm range, suggesting extended visible light absorption relative to common graphitic carbon nitrides. As shown in Fig. 6e, the photocatalytic performance of CNM+mlm coincides well with its absorbance, indicating that the HER performance is directly linked to the formation of electron–hole pairs by photoexcitation.
The higher HER activity of the 32 v% EG CNM+mlm may be explained by the combination of several factors: (1) high content of free NH2 groups (indicated by N1s XPS, Fig. 4b); these groups facilitate the dispersion of the photodeposited Pt nanoparticle cocatalysts and may serve as ligands for nanoparticles, enhancing their stability during the HER. In addition, the free NH2 groups augment the dispersibility of the CN, as can be seen in Fig. S20 (ESI†), allowing better reactivity in water. (2) Formation of a favorable chemical interaction owing to bridging between melem and melamine units in the precursor; upon calcination, favorable defect sites in the CN matrix are formed, as indicated by the observed PL quenching. Furthermore, the EG-mediated C-bridging might result in the formation of a CN homojunction (CN from melem connected C-doped CN from melamine; the homojunction should improve charge separation and catalytic activity. (3) Enhanced light absorbance in the visible light region, corresponding to non-zero AQE at lower energies. (4) High specific surface area.
Photoelectrochemical performance
To show the advantage of the as-prepared precursor containing EG for optoelectronic device fabrication, we prepared photoelectrodes by doctor-blading the obtained slurry directly after the hydrothermal process of the precursors (PM, Pmlm, and PM+mlm, all using 32 v% EG), on fluorine-doped tin oxide (FTO) coated glass, followed by calcination under N2 at 550 °C. Note that our illustrated method would not obtain an effective CN film under aerial conditions. Due to the thin layer of precursors formed by the doctor blade on FTO, most of the precursor surface is highly exposed, leading to sublimation and degradation of the monomers during the high-temperature calcination process as shown in Fig. S21, ESI.† In this facile procedure, CN photoelectrodes are prepared without using an additional binder, which is typically needed to achieve the required specific viscosity values for the doctor-blade deposition.9,10,55 Thus, it eliminates the time-consuming mixing step and improves reproducibility.The XRD and FTIR spectra of the electrodes (Fig. S22a and b, ESI†) prove the formation of a CN layer on the conductive substrate. UV-vis diffuse reflectance spectroscopy (DRS) spectra of the electrodes (Fig. S22c, ESI†) confirm an extended visible range absorption for the CNM+mlm electrode relative to CNM and CNmlm (Fig. S22c, ESI†), in accordance with powder CNs. In addition, the intensity of the PL emission band is lower for CNM+mlm films (Fig. S22d, ESI†). Fig. 7a shows a photograph of the photoelectrodes obtained after the calcination process. Because the CNmlm film has multiple cracks, hydrothermally treated melem alone is not a suitable precursor to obtain good adhesion between the CN film and the substrate (FTO). We ascribe this incompatibility to partial polymerization, hindering intimate contact with the substrate. In contrast, CNM+mlm exhibits a sheet-like structure on FTO, thanks to the fast sublimation of melamine during calcination, which results in the formation of a continuous film (Fig. 7b). A cross-sectional analysis (Fig. 7c and d) of CNM+mlm reveals a compact CN film with a thickness of 55–60 μm and a strong attachment to the FTO substrate. SEM images of CNM and CNmlm electrodes are displayed in Fig. S23 (ESI†).
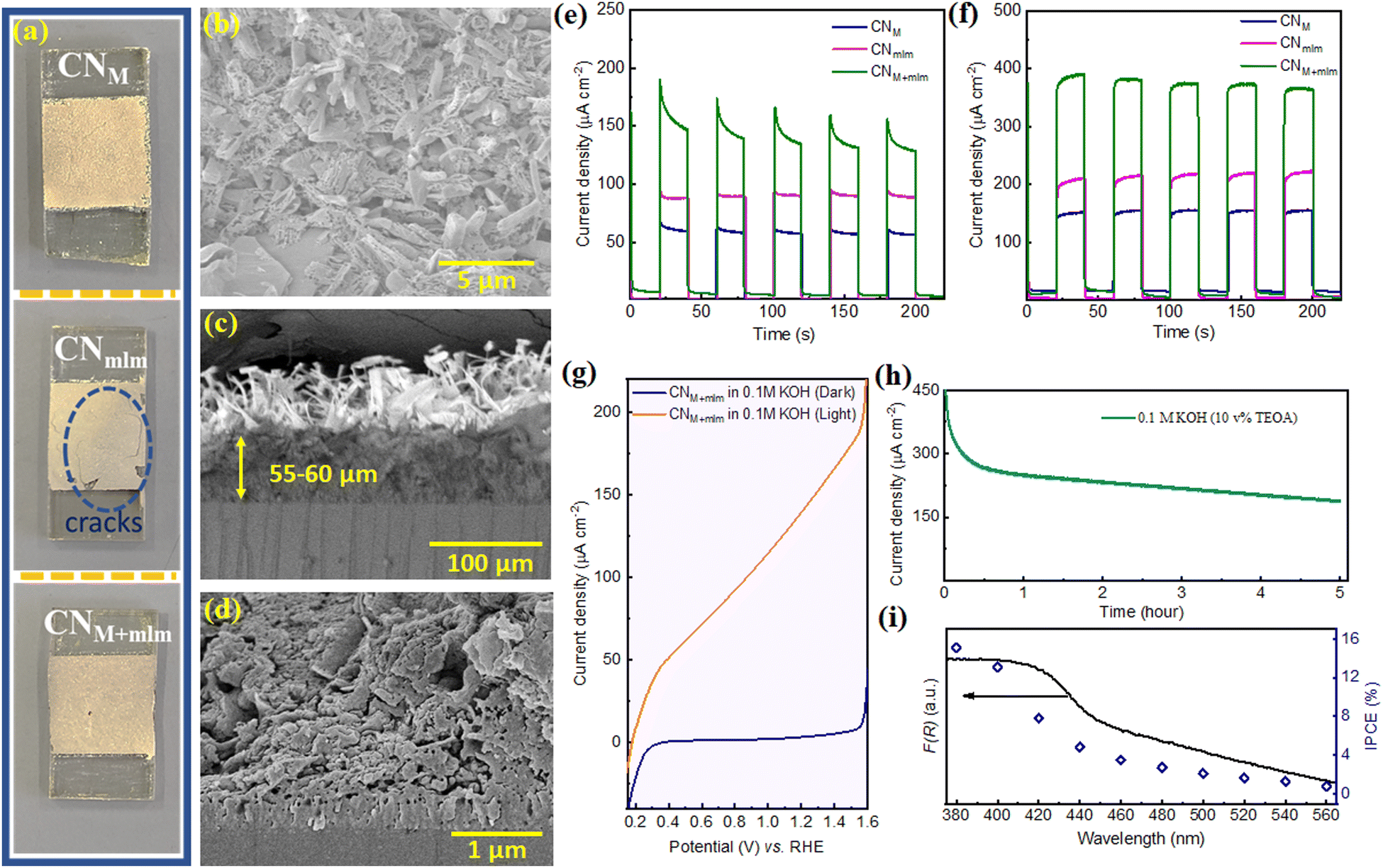 | ||
| Fig. 7 (a) Digital photographs of CNM, CNmlm, and CNM+mlm photoelectrodes on FTO. (b) SEM image (top-view) of CNM+mlm. (c and d) Cross-sectional SEM images of CNM+mlm electrodes at different magnifications. (e) Chronoamperometry (current densities vs. time) of CNM, CNmlm, and CNM+mlm electrodes in 0.1 M KOH at 1.23 VRHE upon on/off 1 sun illumination. (f) Chronoamperometry of CNM, CNmlm, and CNM+mlm electrodes in 0.1 M KOH aqueous solution containing 10 v% TEOA as a hole scavenger at 1.23 VRHE. (g) Linear sweep voltammetry (LSV) of CNM+mlm in 0.1 M KOH in the dark and under 1 sun illumination. (h) Stability measurement (current density under continuous 1 sun illumination) of CNM+mlm electrode at 1.23 VRHE in 0.1 M KOH containing 10 v% TEOA during 5 h. (i) Incident photon-to-current conversion efficiency (IPCE) of the CNM+mlm electrode at different wavelengths (380–560 nm; using band-pass filters) in 0.1 M KOH containing 10 v% TEOA at 1.23 VRHE. | ||
The PEC performance of the CN electrodes was evaluated under 1 sun illumination using an aqueous KOH solution (0.1 M) as the electrolyte in a three-electrode system. The photocurrent upon back illumination gives higher photocurrent densities than front illumination (Fig. S24, ESI†), owing to the shorter distance that the photogenerated electrons have to travel before reaching the FTO. Hence, the photocurrent measurements were performed using back illumination. The CNM+mlm electrode exhibited a photocurrent density (160 ± 10 μA cm−2 at 1.23 V vs. RHE (VRHE)) nearly 2.7 and 1.8 times as high as those of the CNM and CNmlm electrodes (60 ± 5 and 90 ± 5 μA cm−2), respectively (Fig. 7e). The improved photocurrent for the CNM+mlm electrode indicates that the supramolecular precursor, which contains both melamine and melem, plays a vital role in the formation of a compact CN film over FTO.
When triethanolamine (TEOA, 10 v%) was added to the KOH aqueous solution as a hole acceptor, the photocurrent densities of the CNM, CNmlm, and CNM+mlm electrodes were enhanced to 140, 210, and 380 μA cm−2, respectively (Fig. 7f). As expected, it indicates that a sluggish hole extraction is the primary limiting factor in a PEC setup. In addition, CNM+mlm photoanode photocurrent densities were measured over a wide pH range (0.5 M Na2SO4, pH 6.27; and 0.5 M H2SO4, pH 0.27); values of 80 and 60 μA cm−2 were recorded in the neutral and acidic media, respectively (Fig. S25, ESI†).
Linear sweep voltammetry (LSV) curves of CNM+mlm in Fig. 7g correspond to the typical behavior of a photoanode in a PEC setup: no noticeable change in dark current density was measured until applying a voltage bias of ca. 1.55 V, which was sufficient to induce electrocatalytic water splitting, whereas a linear increase in photocurrent density was observed under illumination. The remarkably low onset potential (0.153 V vs. RHE) is complementary evidence of facile charge separation under visible-light illumination of CN.
A stability test for the photoanode was performed under continuous 1 sun illumination for 5 h using similar conditions but in the presence of TEOA as a hole scavenger (pH = 13.1), as shown in Fig. 7h. The current density decreased by nearly 50% after 5 h (∼190 μA cm−2). To examine the reasons for decreased stability after 0.5 h, we have recorded an N 1s XPS spectrum of CN electrode after the stability test (Fig. S26, ESI†). The deconvoluted N 1s spectrum shows that the primary NH2 content decreased after operation and a new peak appears at 403 eV, attributed to N–O species. This suggests that the decrease in the current after 0.5 h is due to (partial) oxidation of the NH2 groups in CN structure. SEM images of the CN electrode after the stability test suggest that the structure of CN remains similar to the unmeasured one (Fig. S27a and b, ESI†). We also noticed that the CN is well attached with the FTO even after a reaction of 5 h, as can be seen from the inset of Fig. S27a, ESI.† In addition, the cross-section image (Fig. S27c, ESI†) indicates that the CN is well attached to the FTO. However, the upper layer thickness of the CN film has been decreased compared to the fresh sample, which could also lead to the decrease stability (Fig. S27c and d, ESI†).
The incident photon-to-current conversion efficiency (IPCE) of the CNM+mlm photoanode was measured from 380 to 560 nm at 1.23 VRHE (Fig. 7i). The IPCE of the CNM+mlm electrode at λ = 380 nm is calculated to be 15.2%. Notably, the IPCE value at λ = 560 nm is ∼0.8%, showing extended activity in the visible range at lower incident photon energies, in line with the measured optical absorption of the films.
With this data in hand, we conclude that the origin of the improved photocurrent and stability of CNM+mlm films as photoanodes stems from their good adhesion and intimate contact with the conductive substrate, extended light absorption, better charge separation, low charge recombination, and enhanced electronic conductivity due to carbon doping.
Conclusions
In summary, we introduced a straightforward method to synthesize polymeric carbon nitride materials and electrodes with an enhanced optical response, high surface area, excellent dispersibility in water, and good activity as photocatalysts for the HER and as photoanodes in photoelectrochemical cells. To do so, we employed a hydrothermal route with an aqueous solution of EG to form highly ordered and thermally stable supramolecular assemblies based on the melem–EG–melamine unit as a precursor. The addition of EG enables the formation of highly organized supramolecular structures thanks to the high solubility of melem and melamine. Moreover, EG binds the precursor molecules, leading to the formation of homogeneous precursor paste with carbon-rich monomers. The obtained paste can be cast on a transparent conductive substrate without further treatment, allowing the easy fabrication of CN photoanodes for photoelectrochemical water splitting.Upon thermal condensation, a highly porous CN with homojunctions and extended light absorption into the visible region is synthesized. The best sample, CNM+mlm, exhibits excellent, state-of-the-art HER rates up to 23.1 mmol h−1 g−1 under a white LED irradiation with an AQE of 19.2% at 395 nm. We also developed an original approach for the fabrication of CN photoelectrodes, where no additional binder is used for doctor-blading, significantly facilitating this process and improving its reproducibility. Compared with the CNM and CMmlm photoanodes, the constructed CNM+mlm photoanodes exhibit a low onset potential of ∼0.153 V vs. RHE, a high photocurrent density of 160 ± 10 μA cm−2 at 1.23 V in 0.1 M KOH (380 μA cm−2 in the presence of a hole scavenger) and high IPCE values ranging from 15.2% at λ = 380 nm to ∼0.8% at λ = 560 nm, and their activity is extended to longer wavelengths.
Author contributions
S. M. performed most of the experiments, analyzed the data, and wrote the initial draft of the manuscript. G. M. helped in photocatalysis studies. L. A. performed XPS characterization. J. L. measured elemental analysis. J. T. has performed TEM analysis. T. S. helped in IPCE measurements. L. X. performed the DFT calculations. M. V. took part in analysis, SEM imaging, and manuscript writing and review. M. S. supervised the study, co-wrote and reviewed the paper, and acquired funding. All the authors discussed the results and reviewed the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 Research and Innovation Programme (Grant Agreement No. 849068). This work was partially supported by the Israel Science Foundation, Grant No. 601/21. The authors thank Dr Chabanne for fruitful discussions.References
- G. Zhang, Z.-A. Lan and X. Wang, Chem. Sci., 2017, 8, 5261–5274 RSC.
- J. Safaei, N. A. Mohamed, M. F. Mohamad Noh, M. F. Soh, N. A. Ludin, M. A. Ibrahim, W. N. R. W. Isahak and M. A. M. Teridi, J. Mater. Chem. A, 2018, 6, 22346–22380 RSC.
- M. Volokh, G. Peng, J. Barrio and M. Shalom, Angew. Chem., Int. Ed., 2019, 58, 6138–6151 CrossRef CAS PubMed.
- J. Liu, H. Wang and M. Antonietti, Chem. Soc. Rev., 2016, 45, 2308–2326 RSC.
- Y. Xiao, G. Tian, W. Li, Y. Xie, B. Jiang, C. Tian, D. Zhao and H. Fu, J. Am. Chem. Soc., 2019, 141, 2508–2515 CrossRef CAS PubMed.
- B. Ma, G. Chen, C. Fave, L. Chen, R. Kuriki, K. Maeda, O. Ishitani, T.-C. Lau, J. Bonin and M. Robert, J. Am. Chem. Soc., 2020, 142, 6188–6195 CrossRef CAS PubMed.
- F. Yang, H. Li, K. Pan, S. Wang, H. Sun, Y. Xie, Y. Xu, J. Wu and W. Zhou, Sol. RRL, 2021, 5, 2000610 CrossRef CAS.
- S. Mondal, L. Sahoo, Y. Vaishnav, S. Mishra, R. S. Roy, C. P. Vinod, A. K. De and U. K. Gautam, J. Mater. Chem. A, 2020, 8, 20581–20592 RSC.
- J. Xia, N. Karjule, L. Abisdris, M. Volokh and M. Shalom, Chem. Mater., 2020, 32, 5845–5853 CrossRef CAS.
- G. Peng, J. Qin, M. Volokh, C. Liu and M. Shalom, J. Mater. Chem. A, 2019, 7, 11718–11723 RSC.
- Z. Fang, D. Li, R. Chen, Y. Huang, B. Luo and W. Shi, ACS Appl. Mater. Interfaces, 2019, 11, 22255–22263 CrossRef CAS PubMed.
- Y. Zhang, J. Zhao, H. Wang, B. Xiao, W. Zhang, X. Zhao, T. Lv, M. Thangamuthu, J. Zhang, Y. Guo, J. Ma, L. Lin, J. Tang, R. Huang and Q. Liu, Nat. Commun., 2022, 13, 58 CrossRef CAS PubMed.
- H. Ou, S. Ning, P. Zhu, S. Chen, A. Han, Q. Kang, Z. Hu, J. Ye, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2022, 61, e202206579 CrossRef CAS PubMed.
- J. Xia, G. Mark, M. Volokh, Y. Fang, H. Chen, X. Wang and M. Shalom, Nanoscale, 2021, 13, 19511–19517 RSC.
- Z. Teng, W. Cai, S. Liu, C. Wang, Q. Zhang, S. Chenliang and T. Ohno, Appl. Catal., B, 2020, 271, 118917 CrossRef CAS.
- M. Rahman, K. Davey and S.-Z. Qiao, Small, 2017, 13, 1700376 CrossRef PubMed.
- J. Xia, N. Karjule, G. Mark, M. Volokh, H. Chen and M. Shalom, Nano Res., 2022, 15, 10148–10157 CrossRef CAS.
- J. Xia, N. Karjule, B. Mondal, J. Qin, M. Volokh, L. Xing and M. Shalom, J. Mater. Chem. A, 2021, 9, 17855–17864 RSC.
- Y.-S. Jun, E. Z. Lee, X. Wang, W. H. Hong, G. D. Stucky and A. Thomas, Adv. Funct. Mater., 2013, 23, 3661–3667 CrossRef CAS.
- J. Barrio and M. Shalom, ACS Appl. Mater. Interfaces, 2018, 10, 39688–39694 CrossRef CAS PubMed.
- J. Xu, S. Cao, T. Brenner, X. Yang, J. Yu, M. Antonietti and M. Shalom, Adv. Funct. Mater., 2015, 25, 6265–6271 CrossRef CAS.
- Q. Liu, C. Chen, K. Yuan, C. D. Sewell, Z. Zhang, X. Fang and Z. Lin, Nano Energy, 2020, 77, 105104 CrossRef CAS.
- B. Wu, L. Zhang, B. Jiang, Q. Li, C. Tian, Y. Xie, W. Li and H. Fu, Angew. Chem., Int. Ed., 2021, 60, 4815–4822 CrossRef CAS PubMed.
- J. Barrio, L. Lin, X. Wang and M. Shalom, ACS Sustainable Chem. Eng., 2018, 6, 519–530 CrossRef CAS.
- R. Li, X. Cui, J. Bi, X. Ji, X. Li, N. Wang, Y. Huang, X. Huang and H. Hao, RSC Adv., 2021, 11, 23459–23470 RSC.
- L. Yin, S. Wang, C. Yang, S. Lyu and X. Wang, ChemSusChem, 2019, 12, 3320–3325 CrossRef CAS PubMed.
- M. Shalom, S. Inal, C. Fettkenhauer, D. Neher and M. Antonietti, J. Am. Chem. Soc., 2013, 135, 7118–7121 CrossRef CAS PubMed.
- J.-W. Zhang, S. Gong, N. Mahmood, L. Pan, X. Zhang and J.-J. Zou, Appl. Catal., B, 2018, 221, 9–16 CrossRef CAS.
- B. Jürgens, E. Irran, J. Senker, P. Kroll, H. Müller and W. Schnick, J. Am. Chem. Soc., 2003, 125, 10288–10300 CrossRef PubMed.
- J. Wen, R. Li, R. Lu and A. Yu, Chem. – Asian J., 2018, 13, 1060–1066 CrossRef CAS PubMed.
- Y. Wang, N. Wu, C. Liu, M. K. Albolkany, M. Wang, Y. Wang, S. Arooj, W. Zhang and B. Liu, Mater. Horiz., 2020, 7, 149–156 RSC.
- P. Shyam, S. Chaturvedi, K. Karmakar, A. Bhattacharya, S. Singh and S. Kulkarni, J. Mater. Chem. C, 2016, 4, 611–621 RSC.
- Y. Miyake, G. Seo, K. Matsuhashi, N. Takada and K. Kanai, Mater. Adv., 2021, 2, 6083–6093 RSC.
- N. Cheng, P. Jiang, Q. Liu, J. Tian, A. M. Asiri and X. Sun, Analyst, 2014, 139, 5065–5068 RSC.
- V. N. Khabashesku, J. L. Zimmerman and J. L. Margrave, Chem. Mater., 2000, 12, 3264–3270 CrossRef CAS.
- Q. Lv, C. Cao, C. Li, J. Zhang, H. Zhu, X. Kong and X. Duan, J. Mater. Chem., 2003, 13, 1241–1243 RSC.
- J. Fang, H. Fan, M. Li and C. Long, J. Mater. Chem. A, 2015, 3, 13819–13826 RSC.
- Y. Yang, J. Chen, Z. Mao, N. An, D. Wang and B. D. Fahlman, RSC Adv., 2017, 7, 2333–2341 RSC.
- Y. Xu, Y. Gong, H. Ren, W. Liu, L. Niu, C. Li and X. Liu, RSC Adv., 2017, 7, 32592–32600 RSC.
- E. Alwin, W. Nowicki, R. Wojcieszak, M. Zieliński and M. Pietrowski, Dalton Trans., 2020, 49, 12805–12813 RSC.
- H. Liu, H. Wang, F. Zhang, J. Xue, J. Zhang and G. Zhang, Chem. Commun., 2021, 57, 927–930 RSC.
- H. Guo, D.-H. Si, H.-J. Zhu, Q.-X. Li, Y.-B. Huang and R. Cao, eScience, 2022, 2, 295–303 CrossRef.
- Z. Li, B. Li, X. Wu, S. A. Sheppard, S. Zhang, D. Gao, N. J. Long and Z. Zhu, Science, 2022, 376, 416–420 CrossRef CAS PubMed.
- J. Bian, L. Xi, J. Li, Z. Xiong, C. Huang, K. M. Lange, J. Tang, M. Shalom and R.-Q. Zhang, Chem. – Asian J., 2017, 12, 1005–1012 CrossRef CAS PubMed.
- B. Lin, Y. Zhou, B. Xu, C. Zhu, W. Tang, Y. Niu, J. Di, P. Song, J. Zhou, X. Luo, L. Kang, R. Duan, Q. Fu, H. Liu, R. Jin, C. Xue, Q. Chen, G. Yang, K. Varga, Q. Xu, Y. Li, Z. Liu and F. Liu, Mater. Horiz., 2021, 8, 612–618 RSC.
- H. Wang, J. Zhang, X. Jin, X. Wang, F. Zhang, J. Xue, Y. Li, J. Li and G. Zhang, J. Mater. Chem. A, 2021, 9, 7143–7149 RSC.
- F. Zhang, J. Li, H. Wang, Y. Li, Y. Liu, Q. Qian, X. Jin, X. Wang, J. Zhang and G. Zhang, Appl. Catal., B, 2020, 269, 118772 CrossRef CAS.
- M. Volokh and T. Mokari, Nanoscale Adv., 2020, 2, 930–961 RSC.
- S. Guo, Y. Tang, Y. Xie, C. Tian, Q. Feng, W. Zhou and B. Jiang, Appl. Catal., B, 2017, 218, 664–671 CrossRef CAS.
- H. Jiang, J. Xu, S. Zhang, H. Cheng, C. Zang and F. Bian, Catal. Sci. Technol., 2021, 11, 219–229 RSC.
- G. Dong, D. L. Jacobs, L. Zang and C. Wang, Appl. Catal., B, 2017, 218, 515–524 CrossRef CAS.
- C. Cheng, J. Shi, L. Wen, C.-L. Dong, Y.-C. Huang, Y. Zhang, S. Zong, Z. Diao, S. Shen and L. Guo, Carbon, 2021, 181, 193–203 CrossRef CAS.
- J. E. Ellis, D. C. Sorescu, S. I. Hwang, S. C. Burkert, D. L. White, H. Kim and A. Star, ACS Appl. Mater. Interfaces, 2019, 11, 41588–41594 CrossRef CAS PubMed.
- J. Xia, N. Karjule, B. Mondal, J. Qin, M. Volokh, L. Xing and M. Shalom, J. Mater. Chem. A, 2021, 9, 17855 RSC.
- N. Karjule, J. Barrio, L. Xing, M. Volokh and M. Shalom, Nano Lett., 2020, 20, 4618–4624 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed synthetic procedures, additional characterization figures and tables. See DOI: https://doi.org/10.1039/d3mh00016h |
| This journal is © The Royal Society of Chemistry 2023 |