
Self-healing polymers through hydrogen-bond cross-linking: synthesis and electronic applications
Long
Chen†
,
Jianhua
Xu†
 ,
Miaomiao
Zhu
*,
Ziyuan
Zeng
,
Yuanyuan
Song
,
Yingying
Zhang
,
Xiaoli
Zhang
,
Yankang
Deng
,
Ranhua
Xiong
* and
Chaobo
Huang
*
,
Miaomiao
Zhu
*,
Ziyuan
Zeng
,
Yuanyuan
Song
,
Yingying
Zhang
,
Xiaoli
Zhang
,
Yankang
Deng
,
Ranhua
Xiong
* and
Chaobo
Huang
*
Joint Laboratory of Advanced Biomedical Materials (NFU-UGent), Jiangsu Co-Innovation Center of Efficient Processing and Utilization of Forest Resources, Nanjing Forestry University, Nanjing 210037, P. R. China. E-mail: chaobo.huang@njfu.edu.cn; ranhua.xiong@njfu.edu.cn; miaomiao.zhu@njfu.edu.cn
First published on 8th July 2023
Abstract
Recently, polymers capable of repeatedly self-healing physical damage and restoring mechanical properties have attracted extensive attention. Among the various supramolecular chemistry, hydrogen-bonding (H-bonding) featuring reversibility, directionality and high per-volume concentration has become one of the most attractive directions for the development of self-healing polymers (SHPs). Herein, we review the recent advances in the design of high-performance SHPs based on different H-bonding types, for example, H-bonding motifs and excessive H-bonding. In particular, the effects of the structural design of SHPs on their mechanical performance and healing efficiency are discussed in detail. Moreover, we also summarize how to employ H-bonding-based SHPs for the preparation of self-healable electronic devices, focusing on promising topics, including energy harvesting devices, energy storage devices, and flexible sensing devices. Finally, the current challenges and possible strategies for the development of H-bonding-based SHPs and their smart electronic applications are highlighted.
Wider impactIn recent years, significant advancements have been made in the area of self-healing polymers (SHPs) through hydrogen-bond cross-linking. Herein, we extensively discuss the advances and design strategies of high-performance SHPs based on different H-bonding types, highlighting the importance of manipulating the H-bond interactions. Moreover, the novel electronic applications of these materials are explored, including energy harvesting/storage devices and flexible electronics. SHPs can revolutionize the manufacturing industry by reducing material waste and enhancing the product lifespan, bringing immense value to engineering applications. Additionally, the development of self-healing electronics holds promise to create more durable and robust devices, contributing to the growing field of wearable technology. Continued research and development in SHPs and their electronic applications will lead to even more advanced materials with improved healing efficiency and mechanical properties. The insights discussed in this review will provide valuable guidance for materials scientists to design and synthesize novel SHPs. Consequently, this will facilitate the development of cutting-edge materials with applications in diverse industries, shaping the future of materials science towards more sustainable and resilient solutions. |
1 Introduction
Synthetic polymers have become an essential part of modern engineering materials since their commercialization in the early 20th century.1–4 Over the last decades, high-performance polymeric composites with desirable mechanical properties, toughness, and thermal stability have been widely used in aerospace engineering,5–7 smart electronics,8–14 intelligent architecture,15 and other high-tech fields.16,17 Although the polymers used in energy and flexible electronics devices are designed to have mechanical properties to accommodate large stress/strain or geometrical deformation, most of them inevitably suffer from damages, such as mechanical fatigue/failure, micro-cracks, punctures, and electrochemical corrosion during their long service life. Such damages may lead to unexpected failures or paralysis of the device during operation, which requires regular maintenance or repair, resulting in additional costs and consumption. Therefore, it is desirable to fabricate polymeric polymers that can restore mechanical properties and original functionalities after physical or chemical damage to satisfy practical application requirements in various emerging fields.18,19In nature, biological systems can heal themselves autonomously and regenerate their structures and original properties after being subjected to external mechanical damage. Inspired by natural systems, endowing high-performance polymers with self-healing properties is considered a promising approach owing to the potential to significantly extend their durability, safety, and lifetime.20–23 According to the self-healing mechanism, SHPs are classified as extrinsic healing type and intrinsic healing type.4,24 Extrinsic SHPs relies on pre-embedded microcapsules/nanocapsules or vascular network in the polymer matrix, which only allow the polymer to be repaired for a limited number of times. More importantly, due to the inherent differences between the polymer matrix and the repair agents, extrinsic SHPs cannot address the functional degradation resulting from structural damage.25 By contrast, intrinsic SHPs based on dynamic covalent bonding self-assembly strategies or supramolecular dynamic chemistry have attracted considerable attention because they not only avoid the complicated integration and compatibility considerations of repair agents but also offer repeatable self-healing.2,26,27 Although dynamic covalent interactions often provide additional thermal stability, heating to relatively high temperatures (≥80 °C) is usually required to achieve healing.28,29 For example, a self-healable vitrimer elastomer based on dynamic carbamate bonds was recently reported by He et al., which required 60 h of healing at 100 °C to completely recover its mechanical performance.30 Alternatively, most of the non-covalent bonds are easily exchanged at much lower temperatures, which can facilitate healing under environmental conditions. Therefore, self-healing systems based on non-covalent interactions usually demonstrate exceptional healing characteristics and can satisfy the stringent requirements of industrially advanced polymeric materials.
Supramolecular dynamic chemistry is non-covalent bonding represented by hydrophobic interaction,31 metal coordination,32 electrostatic,33 π–π,34 guest–host,35 van der Waals36 and H-bonding interactions.37–39 Although these interactions are relatively weak compared with covalent bonding, generally they can form dynamic and mechanically strong systems. Among the various supramolecular dynamic chemistry, H-bonding interactions have attracted the most interest due to their reversibility, directionality and high per-volume concentration, resulting in acceptable mechanical strength. The basic structure of H-bonds can be characterized as X–H⋯Y, which consists of two main parts, i.e., proton donor (X–H) and proton acceptor (a Y atom with lone pair electrons). The energy of the H-bond is determined by various factors such as intermolecular distance, compound structure and environment, which is often considered to be a complex non-covalent interaction energy. In general, the strength of a single H-bond is relatively weak, with a bond energy (∼5–30 kJ mol−1) of about one-tenth that of covalent bonds (∼345 kJ mol−1 for C–C bonds), but it can still have an effect on the viscoelasticity, crystallinity, and phase separation structure of the polymer. In 2008, Leibler and co-workers40 pioneered the design of intrinsic SHPs by utilizing supramolecular H-bonding self-assembly techniques. The final plasticized polymer showed up to 500% elongation before breaking and unique room temperature (RT) self-healing capabilities.41–43 Alternatively, the binding of multiple H-bonds can offer high association constants to provide robust assembly bonding, while allowing tunable dynamic behavior including self-healing.44,45 With the cyclic 2-ureido-4-pyrimidone (UPy) as the repeating unit, Guan et al. prepared a biomimetic supramolecular polymer possessing a unique combination of self-healable, high toughness, and high strength. Following the synthesis of this titin-mimicking modular polymer containing UPy motifs, researchers further designed and synthesized many SHPs based on UPy motifs.46–49 With the continuous exploration of the self-healing mechanism, SHPs with microphase separation21,50,51/homogeneous52–54 structure have also been increasingly designed and developed. The rapid increase in novel design methods to significantly improve the mechanical properties and healing efficiency of SHPs opens the door for these polymers to be explored in a broad range of intelligent electronic fields, such as energy harvesting,55–59 energy storage41,60–63 and flexible sensing.50,64–67
However, although various reviews on SHPs have emerged in the last few years,68–72 an assessment of the design strategies and performances of H-bonding-based SHPs is lacking. Herein, we provide a comprehensive review of the recent progress in the structural design of high-performance SHPs based on different H-bonding types and how to employ these polymers for the creation of emerging self-healable electronic devices (Scheme 1). In the first part, the design strategies, mechanical properties and self-healing performances of various H-bonding-based SHPs are summarized. Subsequently, representative examples of these polymers in energy harvesting, energy storage and flexible electronic sensing applications, together with their design, performances and functional healing are presented. Finally, conclusions and the future outlook for the design of H-bonding-based SHPs and self-healing devices are discussed.
 | ||
| Scheme 1 Classification of H-bonding-based SHPs and their electronic applications. | ||
2 Design of H-bond cross-linked self-healing polymers
Among the various supramolecular interactions, H-bonding has become one of the most attractive directions for the development of SHPs owing to its dynamic nature, tunable strength and responsiveness to externally generated stimuli.37 Although a single H-bond is not strong enough to induce supramolecular self-assembly behavior, both the directionality and strength can be increased when multiple H-bonds are arrayed to create H-bonding arrays.73 In addition, composites can commonly possess both self-healable and mechanical strength when the internal structure of the polymer provides a sufficient amount of H-bonding interactions. Hence, the design of SHPs with the advantages of desirable self-healing capabilities by H-bonding has become a research hotspot in recent years. Herein, we systematically review SHPs based on different H-bonding types, including H-bonding motifs and excessive H-bonding.2.1 H-Bonding Motifs
The introduction of H-bonded cross-linking motifs in polymer structures through chemical reactions is one of the most commonly used strategies for the preparation of SHPs. Currently, H-bonding cross-linking in the internal structure of polymers can be achieved by specific H-bond cross-linking motifs or self-conjugation of interchain multiple H-bonding interactions, as well as the addition of external cross-linkers.70 In this section, we focus on the preparation of H-bonding-based SHPs by introducing specific H-bond cross-linking motifs. Multiple H-bonding of these motifs typically involve triple,74 quadruple,75 and sextuple76 H-bonding. Among them, UPy units with self-complementary quadruple H-bonds have been proven to be one of the most promising motifs for the construction of dynamic polymers and biomaterials due to their moderate association–dissociation constant. Therefore, to date, many SHPs with desirable mechanical properties have been achieved through chemically bonding UPy units to the polymer main chains,20,77–80 side chains,47,48,81–83 and chain ends.5,46,49,84,85 | ||
| Fig. 1 (a) Synthesis of the titin-mimicking modular polymer (polymer 1) and control polymer (polymer 2) and (b) their stress–strain curves. (c) Chemical structures of SPMs with different amounts of UPy units in the main chain. (d) Cartoon presentation of the proposed mechanism for the suprastretchable SPMs. (e) Stress–strain curves of the resultant SPMs with varying amounts of UPy. (f) Optical microscopy image of SPM-2 film scratch repair. | ||
Integrating UPy units in the backbone of linear polyurethane (PU) or polyurea is the mainstream due to the fact that the reversible association–dissociation dynamics of H-bonds and the entanglement among linear chains confer high toughness and strength. For example, Sun and co-workers77 fabricated a high-performance healable cross-linked PU by incorporating UPy motifs in the main chains of PUs using 5-(2-hydroxyethyl)-6-methyl-2-aminouracil as a chain extender. The UPy motifs could form self-complementary quadruple H-bonding interactions with each other, resulting in denser assemblies of H-bonds, overcoming the inherent strength limitations of H-bonds, and thus provide enhanced mechanical strength. Moreover, the H-bonds could be reversibly fractured/formed during tensile deformation, enabling effective stress energy dissipation, and thus improved toughness. Specifically, the PUU4B0 sample with artificial cracks was almost completely healed and its mechanical properties were fully restored after heating at 135 °C for 20 min. Similar work was also observed in Yang's group.78 In this study, flexible polypropylene glycol (PPG) diol and dynamic UPy motifs were coupled with iso-phorone diisocyanate (IPDI) to prepare a series of multi-block copolymer PPG-mUPy. Similar to the structure of polyurethane prepared by Sun et al.,77 the high content of mUPy in the backbone could form microcrystals with the help of stacking effects, thus improving the mechanical strength of PU at RT. For example, PPG1000-mUpy100% exhibited the greatest tensile strength (49.64 ± 3.54 MPa), Young's modulus (7.82 ± 1.28 MPa) and toughness (215.73 ± 8.89 MJ m−3). Balancing the two factors of polymer chain fluidity and H-bond density, PPG1000-mUPy50% showed the best self-healing capability. The self-healing performance quantitative experiments indicated that the tensile strength of the restored PPG1000-mUPy50% reached 19.22 MPa, showing a high repair efficiency of 93%.
However, despite their promising prospects, the above-mentioned SHPs still have one significant disadvantage, namely, external energy (either heat or light) is required to achieve self-healing, which severely limits their application (especially in the field of smart electronics). Therefore, it is of great significance to design and develop SHPs that can heal under ambient conditions. Considering this challenge, Bao et al.79 reported a novel chemical design for supramolecular polymeric materials (SPMs) with different UPy motif quantities (from 0 to 30 mol%), which were synthesized by quadruple H-bonding cross-linkers and soft polymeric chains. The chemical structure of SPMS and a cartoon presentation of the proposed mechanism are shown in Fig. 1c and d, respectively. Thanks to the attractive combination of rapid kinetic reversibility and high thermodynamic stability, the existence of self-complementary UPy motifs was not expected to disrupt the PTMG-based soft domains, thus making the SPMs stretchable, soft, self-healing, and tough. SPM-2 containing 20 mol% UPy motifs had a moderate crosslinking density, showing the maximum breaking strain (17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000%), Young's modulus compatible with human tissue (375 kPa), and relatively high tensile stress (0.91 MPa) (Fig. 1e). Surprisingly, SPMs exhibited a promising RT self-healing performance due to their multiple intra- and interchain H-bonding in the polymeric networks. Scratches on the SPM-2 film were almost completely healed after healing for 12 h at RT (Fig. 1f).
000%), Young's modulus compatible with human tissue (375 kPa), and relatively high tensile stress (0.91 MPa) (Fig. 1e). Surprisingly, SPMs exhibited a promising RT self-healing performance due to their multiple intra- and interchain H-bonding in the polymeric networks. Scratches on the SPM-2 film were almost completely healed after healing for 12 h at RT (Fig. 1f).
To date, although a series of highly tough SHPs has been constructed by molecular-scale composite dynamic cross-linking strategies, achieving high toughness and autonomous self-healable performance at RT remains a challenge. Recently, Jia et al.86 prepared a dual-crosslinked supramolecular elastomer by combining UPy motifs and slidable cross-linkers (polyrotaxane) in one system, conferring the elastomer with both high toughness and autonomous self-healable properties at RT. On the one hand, polyrotaxane could effectively dissipate energy and lower the stress via the sliding behaviour of cyclodextrins during the stretching process, leading to high toughness. On the other hand, the quadruple H-bonding afforded autonomous self-healability at RT owing to the dynamic and fast bond exchange rate. Specifically, the representative sample PUPR-5 exhibited a high fracture energy (≈127.2 kJ m−2), toughness (77.3 MJ m−3), and good healing efficiency (91% after 24 h) at RT.
Thus, the UPy motif located on the backbone was well-positioned to act as a cross-linker to react with other functional monomers, leading to the design of high-performance SHPs for meeting various fields. Meanwhile, the random distribution of multiple H-bond complementarity and steric spacing endow the resultant polymer an efficient self-healing performance, this combination of properties allows these advanced composites to find numerous important cutting-edge applications.
Although some biocompatible materials exhibit mechanical properties similar to that of natural tissues,87 their ability to simulate natural functional dynamics remains limited. Thus, to overcome this challenge, Guo and co-workers82 synthesized similar supramolecular bio-elastomers that are highly tunable mechanical by grafting UPy units in the elastic PGS backbone (Fig. 2a). As shown in Fig. 2b, the mechanical properties of the PGS-U elastomer increased significantly with an increase in the content of UPy units. Due to the dynamic supramolecular interactions, the resultant polymers exhibited efficient self-healing. For instance, the scar at the damaged interface of the PGS-3U polymer film was completely healed after 1 h at 55 °C (Fig. 2c). However, the construction of SHPs with desirable mechanical properties remains a significant challenge due to the contradiction between the properties of fully dynamic and mobile supramolecular chains required for self-healable and strong interactions required for robustness. Inspired by the molecular structure of the titin protein, Li and co-workers48 designed a biomimetic transparent and healable elastomer featuring UPy units as pendant groups (Fig. 2d). The hierarchical H-bonding moieties resulted in the formation of a durable network structure, endowing the elastomer with remarkable mechanical properties and fast self-healable ability (Fig. 2e). In addition, a healed PT-HM-U20 sample (37 mm diameter and 0.7 mm thickness) could lift a 10 kg dumbbell with only slight deformation (Fig. 2f).
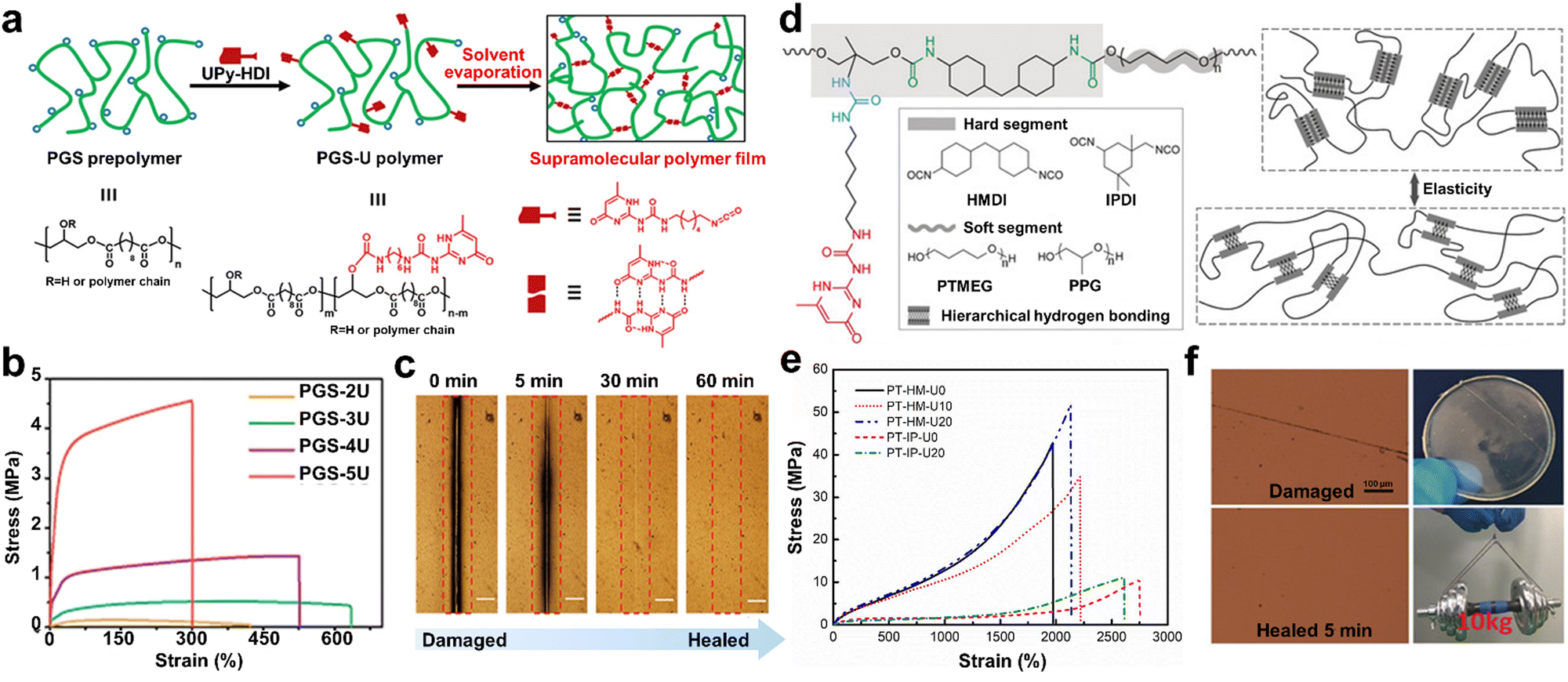 | ||
| Fig. 2 (a) Schematic of PGS-U polymer with UPy unit in its side chains and the preparation of supramolecular elastic film. (b) Stress–strain curves of PGS-U film. (c) Optical microscopy images of scratch healing process of the PGS-U samples at 55 °C. (d) Modular structure of the composites (PT-HM-Ux) with UPy unit in their side chains and illustration of their healing process and (e) typical stress–strain curves of the resulting elastomers. (f) Optical microscopy images of PT-HM-U20 film before and after recovering at 90 °C for 5 min. The digital picture on the right shows that a healed PT-HM-U20 sample can lift a 10 kg dumbbell. | ||
Besides the above-mentioned work, researchers also improved both the self-recoverability and toughness of SHPs by carefully designing block copolymers containing H-bonded units. Inspired by the structure of mussel cuticle, Yoshie et al.47 designed an ABA-type triblock copolymer (PU-b-P(D-r-U)-b-PU) with high toughness and superior self-recovery properties. The A-block was a short homopolymer of UPy-functionalized norbornene and the B-block was a long random copolymer formed by a flexible dodecanyl norbornene and UPy-functionalized norbornene. With the protection of UPy units, this structure could effectively enhance the toughness and strength of the final polymer. Specifically, the PU20-b-P(D320-r-U40)-b-PU20 sample exhibited high toughness (≈62 MJ m−3), strength (16.3 MPa) and outstanding self-recovery. Similar bionic SHPs with ultra-high toughness were also prepared in another work, in which environmentally adaptable SHPs were prepared using PU and dopamine-modified graphene oxide.83 Due to the dynamic and rapid reconstruction of the quadruple H-bonds, the SHPs with inverse artificial nacre structure had excellent toughness (37.8 MJ m−3) and high tensile strength (10.3 MPa). Moreover, dopamine, as a binder, not only improved the interfacial compatibility between the substrates, but also increased the H-bond density to obtain an interwoven network structure, which endowed the obtained SHPs with excellent self-healable ability at RT (90%, 25 °C for 1 h).
As is known, the stiffness-ductility trade-off for polymeric materials such as elastomers is a long-standing challenge that has restricted the overall mechanical performance of soft matter. Although there are some reports on avoiding this trade-off, such as increasing the Young's modulus without sacrificing ductility, the design principles that can improve both stiffness and ductility remain undeveloped. Recently, by introducing a rational arrangement of UPy H-bonding units on the side chains of a poly(dimethylsiloxane)-based model elastic chemical network, Zhang et al.88 reported a novel elastomer design that can surpass the stiffness-extensibility trade-off and provide significant improvement in both parameters. Unfortunately, the authors did not further investigate the self-healing properties of the elastomer. The authors believed that the introduction of UPy units and their microscopic distribution is the key to simultaneously improving the extensibility and stiffness. Specifically, H-bonding increased the crosslink density, and the H-bond clusters could act as nanoparticles, thus effectively improving the modulus of the designed network. Alternatively, due to the dynamic properties of hydrogen bonding itself, the clusters could be rearranged during deformation, thus providing high ductility without damaging the macroscopic sample. This newly designed guideline greatly improves the understanding of how to tune the properties of polymers, overcomes the limitations of existing methods, and also opens up several areas for the future exploration and enhancement of polymeric material performance.89 We predict that other fascinating and even more complex examples will emerge in the near future.
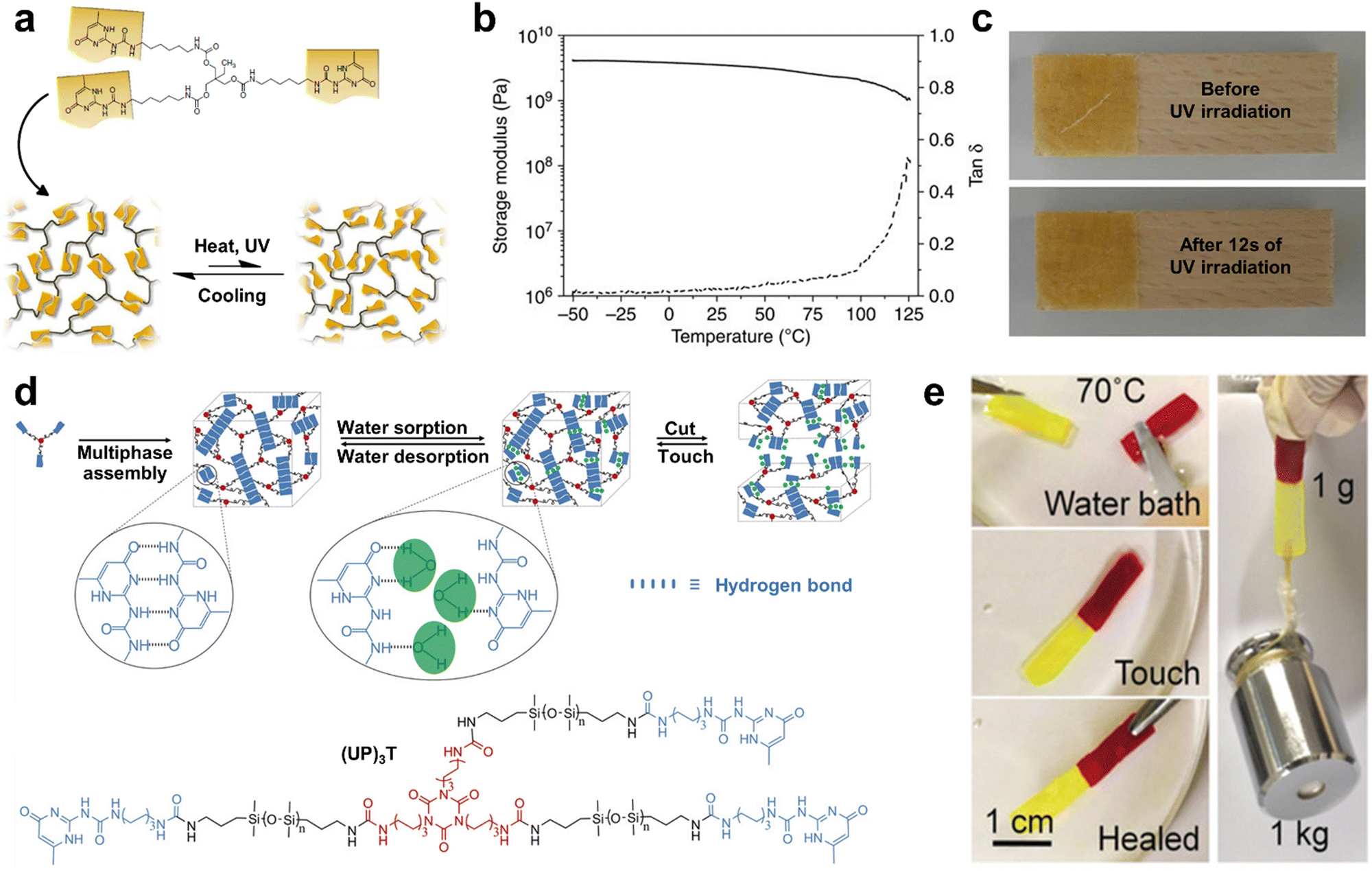 | ||
| Fig. 3 (a) Schematic illustration of the disordered supramolecular network structure of (UPyU)3TMP and its reversible, heat or light-induced dissociation. (b) Representative dynamic mechanical analysis trace of (UPyU)3TMP, where the solid line represents the storage modulus and the dashed line represents tanδ. (c) Optical healing process of a 300 mm-thin amorphous (UPyU)3TMP layer. (d) Multiphase design of self-healable siloxane oligomer (UP)3T and its molecular structure. Upon absorbing water, the UPy units can be exchanged with water molecules (green dots) by the dissociation of UPy dimers. (e) Photographs showing that the 1 g (UP)3T film can withstand a 1 kg counterweight after 5 min of contact healing in a 70 °C water bath. | ||
It was confirmed that the (UPyU)3TMP film had a very high stiffness via dynamic mechanical analysis, including high room-temperature storage modulus of 3.65 ± 0.51 GPa and flexural modulus of 3.04 ± 0.26 GPa (Fig. 3b). Additionally, the high UPy unit content also renders the supramolecular (UPyU)3TMP glass with an excellent optical healing performance. As shown in Fig. 3c, the damaged 300 μm-thin amorphous (UPyU)3TMP coating layer was completely healed after exposure to ultraviolet light within a short time of 12 s. However, the most important limitations of (UPyU)3TMP are its high brittleness and limited toughness, which should be improved in subsequent studies.
Inspired by the structure of neurons, Mao et al.90 reported the synthesis of a series of three-arm telechelic polyurea elastomers with hierarchical H-bonds networks. Interestingly, UPy terminates each arm and its length could be controlled within a small range, and the mechanical properties of SHPs could be readily tuned by adjusting the length of their arm. Consequently, a thermoplastic telechelic polymer with excellent notch resistance (187 kJ m−2), high Young's modulus of 97.9 MPa, and rapid self-healable ability (92%, 0.5 h) was achieved. Briefly, this telechelic polymer presented high stiffness and exceptional self-healing performances, which is attributed to the hierarchical H-bonds networks constructed by UPy motifs and urea groups.
It is well-known that H-bonds are susceptible to water molecules, which can lead to a gradual decrease in mechanical strength or even loss of self-healing capabilities of H-bonding-based SHPs under high humidity conditions. Taking advantage of the broad and versatile chemistry of noncovalent interactions, Yao et al.49 synthesized a novel silica-based material composed of polyphase-assembled siloxane oligomers through multivalent H-bonding (Fig. 3d). A high content of UPy units resulted in a high cross-linking density, enabling the resultant (UP)3T film to exhibit a tensile strength of >5.0 MPa and high elastic modulus of 47.39 MPa. The crystallizable and strong multiple H-bonds were not only responsible for the mechanical properties of the resultant silicone material, but also provided a unique water-enhanced healing feature. Specifically, water molecules in the environment are beneficial for the dissociation of the multivalent H-bonds by travel through the permeable siloxane network to achieve rapid healing. Consequently, two pieces of samples could be firmly reunited in a water bath (70 °C) and could withstand a weight of 1 kg without fracture (Fig. 3e). Due to the dynamic high crosslink density and semi-crystalline nature of the polymer, the mechanical strength of the (UP)3T films could be recovered even after 5 generation cycles in water at 70 °C.
In conclusion, UPy motifs are the most commonly used H-bond cross-linking units for the synthesis of H-bonding-based SHPs due to their moderate association–dissociation constant and easy synthesis. As discussed above, the formation of UPy dimers leads to effective cross-linking and enhanced mechanical performance in SHPs. However, it should be noted that the introduction of UPy motifs does not ensure high mechanical strength, ductility and toughness in the final polymer, which is related to various factors, including the self-assembly manner and conditions of the UPy motifs, as well as their crosslinking density. For example, by incorporating small amounts of UPy as reversible H-bonding side groups integrated with short spacers to prevent aggregation, Anthamatten et al.91 reported the synthesis of a biocompatible soft material with a highly tunable viscoelastic relaxation timescale. Therefore, when designing H-bonding-based SHPs, multiple factors need to be considered to simultaneously enhance both the toughness and strength under the premise of ensuring self-healing in polymers.
2.2 Excessive H-bonding
Besides UPy units, currently, the incorporation of H-bond clusters through chemical reactions is another essential strategy for the preparation of H-bonding-based SHPs. Unlike the above-discussed UPy functionalized cross-linked SHPs made of macromolecules, these systems are composed of a mixture of oligomers with various associating groups, which enable the formation of an H-bond cluster network by excessive H-bonds. For example, when low-molecular-weight polymers were cross-linked by dense H-bonds, mechanically robust and readily repairable materials could also be produced.4 Thus far, most cross-linked polymers with H-bond clusters have been designed by microphase separation21,51,92,93 or homogeneous methods.4,40,53,94Generally, most PUs composed of alternating soft segments are formed by low Tg polymers such as PDMS, PPG and polytetramethylene glycol (PTMG), whereas hard segments formed by the reaction of short-chain diols with isocyanates, leading to a microphase-separated structure due to their great difference in solubility parameters. The soft phase usually exhibits high variability and plasticity, which means that they can more readily form dynamic reversible H-bonds. The importance of structural design can be observed in the research by Guan's group.21,98,99 For instance, they pioneered a TPE based on novel multiphase design,21 in which poly(acrylate amide) (PA-amide) containing H-bonds as the soft segment was responsible for the self-healing action and polystyrene (PS) as the hard segment provided mechanical strength (Fig. 4a). Thus, the Young's moduli of resultant polymers were two orders of magnitude higher than that of previously reported self-healing thermally reversible rubber systems,40 showing mechanical properties comparable to classic rubber. In addition, the H-bonding brush polymer could self-heal spontaneously under ambient conditions without introducing any external stimuli or healing agents (optimal healing of up to 92% recovery) because of the reversible H-bonding in the soft matrix (Fig. 4b). Subsequently, by replacing the PS hard phase with polymethylmethacrylate (PMMA), they synthesized another self-healing TPE brush copolymer via atom transfer radical polymerization.98 Notably, the PA-amide brush of the resultant co-polymer was rich in dynamic H-bonding amide functional groups and highly mobile, which endowed a good self-healable performance to the materials at RT. Based on this multiphase design, a series of triblock copolymers having glassy PMMA blocks and dynamic H-bonding blocks was prepared by sequential ATRP.99 Compared to the above-mentioned PS brush copolymer system, the PMMA triblock copolymer system exhibited better mechanical properties with a higher tensile strength and Young's modulus, while maintaining a good self-healing performance, which can be ascribed to the stronger interactions between the hard PMMA microphases and soft PA-amide.
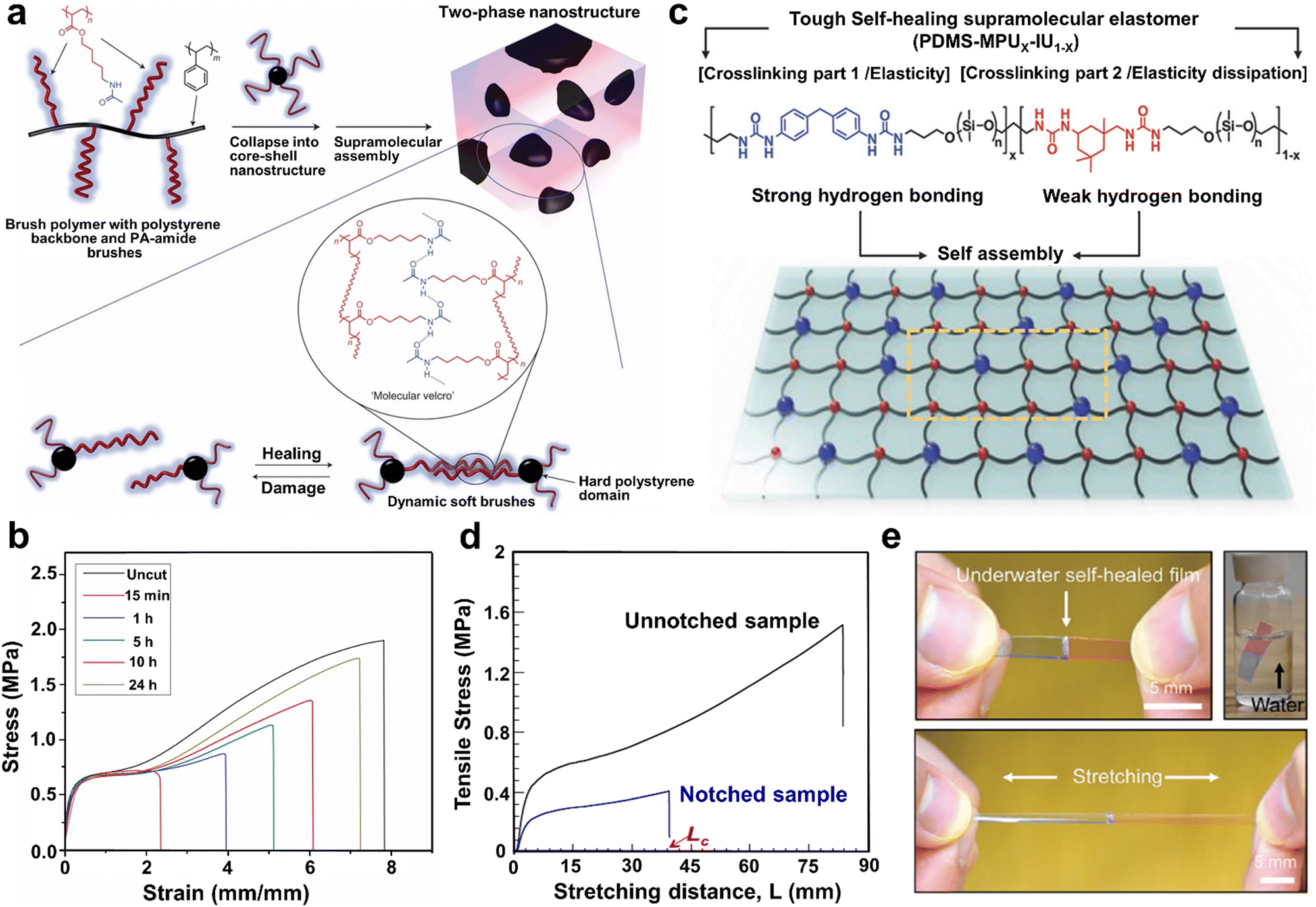 | ||
| Fig. 4 (a) Schematic diagram of the design of multiphase brush polymer and its self-healing mechanism. (b) Tensile stress–strain curves of original and healed samples after healing at 25 °C for different times. (c) Proposed ideal supramolecular network structure and the molecular structure of PDMS-MPUx-IU1−x. (d) Stress-extension curves of the notched and unnotched sample film (PDMS-MPU0.4-IU0.6). Lc is the distance between the clips when the crack starts to propagate. (e) Underwater self-healable process of the PDMS-MPU0.4-IU0.6 (top). The film can be successfully stretched when it has healed for 24 h (bottom). | ||
In addition to the unique design in the above-mentioned studies, H-bonds can be also programmed into the hard phase of a hard–soft multiphase system, which can be seen in PU or polyurea. The programming of dynamic H-bonds into the hard phase not only provides the necessary mechanical strength and elasticity, but also exhibits autonomous self-healable and exceptional toughness via the rapid rearrangement of the molecular network. For example, Bao et al.50 reported the synthesis of a tough and water-insensitive SHP through a one-pot polycondensation reaction between a mixture of IPDI and 4,4′-methylenebis(phenyl isocyanate) and bis(3-aminopropyl)-terminated PDMS (Fig. 4c). In this system, both strong and weak cross-linking H-bonds existed in the hard segment, conferring elastomer robustness, elasticity and self-healing performance. Consequently, the resultant polymer film showed exceptional mechanical properties, including high toughness of 12000 J m−2 and notch-insensitive high stretchability (1200%) (Fig. 4d). Remarkably, the healing process of the elastomer could also occur in water, which is the first report of an H-bonding-based elastomer that can self-heal in sweat or water. As shown in Fig. 4e, the resulting polymer film could be still stretched to 1100% strain after healing in water for 24 h. In general, although the frozen hard domains endow the polymer with elasticity and robustness, their also prevent the autonomous RT self-healing performance. Meanwhile, a regular packing structure of bis-urea units in the hard segment also results in semi-crystallization, which impedes the activity of the H-bonds. Considering this dilemma, Xu and co-workers93 designed a reasonable preparation strategy, which could destroy the crystalline behavior and active the H-bonds within the hard segments at the molecular level. More importantly, the irregular self-sorting of the hexamethylene bis-urea units and symmetric 4,4′-methylenebis(phenylurea) could mutually disrupt their linear arrays, which contributed to the formation of high-dynamic hard domains with amorphous structures. Consequently, the PU elastomer not only showed an optical transparency of over 94% and high tear-resistant stretchability of 800%, but also possessed ultra-high autonomous self-healable capability, which could completely heal a scratch within 100 s under ambient conditions.
In addition, the dynamic hard domain design concept can also be observed in the research by Fu's group.51,100,101 With isophorone diamine (IDA) as the hard phase, they fabricated a polyurethane-urea supramolecular composite (PPGTD–IDA) via a one-step polycondensation route (Fig. 5a).51 The resulting supramolecular elastomers possessed a unique combination of ideal properties due to the integration of dynamic hard domains consisting of hierarchical H-bonds into the supramolecular network. For example, PPGTD–IDA displayed extreme toughness of 42650 J m−2 (exceeding values compared to previously reported values50,79) and outstanding notch-insensitiveness. The damaged PPGTD–IDA film could completely recover its mechanical properties after healing for 48 h under ambient conditions (Fig. 5b), showing repeatable healing of its mechanical properties. In comparison, the healing efficiencies of the reference polymers were only 11.63% (PPGTD-HDA) and 0.1% (PPGTD-PDA), respectively, which further proves the importance of the dynamic hard domain design in the self-healing performance.
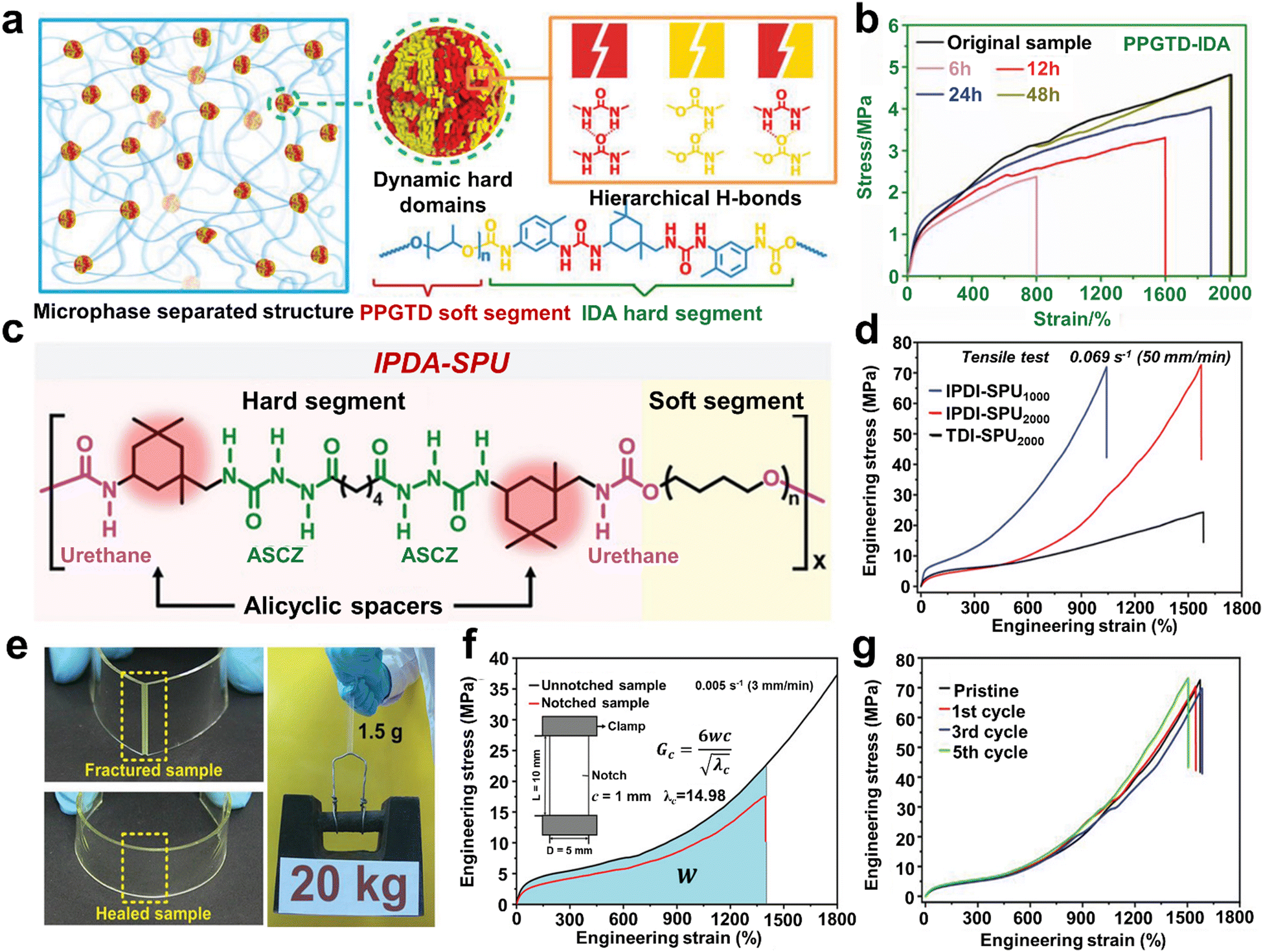 | ||
| Fig. 5 (a) Design concept of the microphase separation structures and dynamic hard domains for the synthesis of PPGTD–IDA polymer networks. (b) Typical stress–strain curves of healed and original PPGTD–IDA films after healing for different times under ambient conditions. (c) Chemical structure of the IPDI-SPU polymer. (d) Typical engineering stress–strain curves of the resultant elastomers measured at a strain rate of 0.069 s−1 (i.e., IPDI-SPU1000, IPDI-SPU2000 and TDI-SPU2000). (e) Optical images of the damaged and healed IPDI-SPU2000 elastomer (left). Healed IPDI-SPU2000 sample (1.5 g) can bear a weight of 20 kg (right). (f) Engineering stress–strain curves of the IPDI-SPU2000 sample with and without notching. (g) Engineering stress–strain curves of the IPDI-SPU2000 sample that underwent 1, 3 and 5 cycles of reprocessing. | ||
In the design of SHP structures, the asymmetric alicyclic structure of IPDI is capable of forming loosely stacked hard domains, which can improve the dynamic exchange of H-bonds. Nevertheless, the amorphous hard domains in SHPs usually do not provide satisfactory mechanical strength, which greatly limits their practical application. The preparation of PPGTD–IDA was used as a basis, and a novel hard domain reinforcement strategy (at the molecular level) was further proposed.100 In this strategy, IDPI is introduced in a mixture of PPGTD and IPDA as the key reactant, and the mechanical performance of the composites can be switched on demand by merely changing the molar ratio of the reactants. Consequently, the representative elastomer PPGTD0.4–IPDA1.0–IPDI0.6 possessed a remarkable fracture energy of 113,560 J m−2 and a record-high toughness of 503.3 MJ m−3, which is much higher than that of PPGTD–IDA. All these prominent improvements are primarily responsible for the unique structure of the IDPI-modified hard domains. Regrettably, PPGTD0.4–IPDA1.0–IPDI0.6 did not show a satisfactory self-healing performance at RT or moderate temperatures, but it could conveniently heal mechanical scratches with the help of trace amounts of solvent.
Moreover, the incorporation of a more flexible alicyclic hexa-atomic spacer rather than a rigid aromatic spacer in the design of the SHP structure facilitated the formation of a higher density of distinctive hard domains. For example, inspired by spider silk, Sun et al.102 demonstrated a healable supramolecular PU elastomer formed by diisocyanates, hydroxyl-terminated PTMG, and a dihydrazide chain extender.
The meticulously designed H-bonding segments were combined between extendable polymer chains that polymerise to form geometrically restricted arrays, similar to spider silk. As illustrated in Fig. 5c, the hard segments consisted of urethane moieties and multiple acylsemicarbazide, which were connected by alicyclic hexatomic IPDI or aromatic tolylene-2,4-diisocyanate (TDI). Compared with TDI, the structure of IPDI was more flexible, facilitating the formation of a higher density H-bond arrays, and thus more efficient dissipation of energy. Consequently, the IPDI-SPU2000 elastomer possesses a tensile strength of up to 75.6 MPa, while still maintaining an elongation at break of ≈1520%, which is approximately two-times higher compared to that of TDI-SPU2000 (Fig. 5d). The pre-damaged IPDI-SPU2000 sample could even be stretched to >14 times its originally length, exhibiting superb crack tolerance (Fig. 5f). However, the cleavage/reformation of the dynamic H-bond crosslinks relies on relatively high temperatures for activation (70 °C). As shown in Fig. 5e, the fractured IPDI-SPU2000 elastomer could be conveniently repaired at 70 °C for 48 h. Due to the dynamic cleavage/reformation of the H-bonds, the elastomer retained its mechanical robustness even after 5 cycles of hot-press processing (Fig. 5g).
In recent years, the concept of bionics has attracted increasing interest from materials engineers. The feasibility of the bionic structure design can be observed in the research by Fu's group.101,103,104 For example, some biological tissues, such as the reticular layer of the human skin dermis, provide promising ideas for researchers to the address conflict between the self-healability and fatigue resistance of SHPs. Employing UPy supramolecular motifs as phase regulators, they proposed a molecular engineering strategy that transforms inherently fragile materials with an island-like structure into soft self-healable elastic materials with a bicontinuous nanophase separation structure.103 Notably, the UPy-modified dynamic and continuous hard domains formed a repairable interlaced nanofibrous network, similar to the reticular layer of animal dermis, allowing the SHPs to exhibit tremendous improvement in the fatigue threshold (34.8 times that of the pristine material), thermodynamic stability (4 orders of magnitude enhancement), and elastic restorability (maximum elongation for full dimensional recovery increasing from 6 times to 13 times), while maintaining autonomous self-healing properties (∼99% at RT). This universal strategy can be applied to other PU/polyurea, providing advanced guidance for the further design of high-performance self-healing soft materials. Inspired by the structure of vascular smooth muscles, the same group developed a soft but tough self-healable polyurea (SSPUGIT) with ultra-high dielectric permittivity (∼14.57). By introducing gallium–indium–tin eutectic alloy (Galinstan) micro-droplets with a core-shell structure through molecular interfacial metal-coordinated assembly, this biomimetic strategy overcomes the trade-off between high fracture toughness and soft self-healing. Consequently, the fracture toughness and crack-resistant strain increased by 34.9 and 12.2 times, respectively, without sacrificing the softness of SSPUGIT.
Besides, implanting dynamic reversible H-bonds in both a dispersed hard phase and continuous macromolecular soft phase to prepare SHPs has also been proposed by researchers.96 Using 1,1-thiocarbonyldiimidazole, PDMS, and 4,4′-methylenebis-(phenyl isocyanate) (MPI) as raw materials, Fu and co-workers105 synthesized an ultra-stretchable and efficient SHP. The introduction of thiourea moieties effectively disrupted the crystallization of hard domains and the insertion of reversible H-bonds in both the hard and soft segments, conferring excellent self-healing properties to the SHP. Upon damage, all the mechanical performances of the PDMS-MPI-TM could completely be recovered after healing for 4 h at RT. Encouragingly, the healing progress could also be carried out in water or at low temperature (−20 °C). Additionally, benefitting from the fast rupture/reformation of the multiphase H-bond pairs, the PDMS-MPI-TM film could be stretched to >315 times its original length without fracture and reached a notch-insensitive stretching of up to 18000%.
Based on fatty di-acid/tri-acid and diethylene tri-amine/urea, Leibler et al.40 prepared a self-healable and thermoreversible rubber via a sequential reaction (Fig. 6a). In this system, a mixture of oligomers formed chains and cross-links by H-bonds. It is noteworthy that the existence of different molecular structures limited the crystallization of the final polymer chain. Consequently, the properties of the plasticized polymers were very similar to rubber, exhibiting a breaking strain exceeding 500% and tensile strength of 3.55 MPa (Fig. 6b). Significantly, this cross-linked polymer also exhibited unique RT self-healing properties due to the presence of multiple H-bond interactions (Fig. 6c).
 | ||
| Fig. 6 (a) Synthesis pathway. A mixture of oligomers (i.e., compound A) possessing complementary H-bonding groups (H-bond donors are shown in green and acceptors in red) is prepared via the condensation between a mixture of fatty triacid and diacid with diethylene triamine, and then reacted with urea. (b) Typical stress–strain curves of as-prepared compound A rubber after plasticizing with 11% w/w dodecane (i.e., compound B). Inset shows the linear relationship between the cross-sectional area and inverse of the tensile strain. (c) stress–strain curves of compound B at 40 °C after different healing times. (d) Chemical structure of poly(ether-thioureas) with different glycols and long-chain alkanes as spacers. (e) Scheme of the different H-bonding patterns of urea and thiourea groups. (f) Stress–strain curves of TUEG2, TUEG3, TUC8, TUC12, and UEG3. | ||
Although supramolecular rubber containing numerous physical cross-links shows an excellent self-healing performance, their resistance to creep and solvent is limited by the absence of chemical cross-links. Accordingly, increasing both H-bonding and chemical cross-links has proven to be an ideal strategy to solve this problem. By introducing two epoxy monomers, Tournilhac and co-workers106 reported the synthesis of a fused chemical platform, which could provide an increased quantity of chemical crosslinks without altering the concentration of H-bonding groups. The results showed that the incorporation of a tetrafunctional epoxide in the presence of 2-methylimidazole could significantly improve the creep resistance of the material. In addition, all the supramolecular elastomers exhibited a good self-healing performance (>50%). Nevertheless, these self-healing supramolecular rubbers did not perform well in terms of mechanical strength. Here, Weitz and co-workers54 demonstrated a self-healable, tough dry elastomer, which successfully integrated reversible H-bonds and permanent covalent crosslinks into a hybrid polymer network. Randomly branched polymers linked these two types of bonds, forcing them to mix at the molecular level without co-solvents. Consequently, this integration enabled the hybrid network to dissipate energy at multiple length scales, showing excellent toughness with a tensile strength of ≈12 MPa and fracture energy of 13500 J m−2. Therefore, the appropriate introduction of covalent crosslinks in the reversible network is an ideal strategy for improving the mechanical strength of SHPs.
Besides supramolecular rubbers and elastomers, mechanically robust materials with homogeneous systems have also been developed by researchers. For example, Aida and et al.4 designed a series of low-molecular weight polymers by implanting numerous nonlinear zigzag thiourea units in a poly(ether-thiourea) network (Fig. 6d and e). The in-depth crystallographic study showed that the thiourea groups as H-bonding motifs mostly formed nonlinear zigzag H-bonded arrays in both trans/trans and cis/trans conformations, while the linear arrays of H-bonded urea units formed only a trans/trans conformation. Although TUEG2 and TUEG3 contained dense H-bonding thiourea units, they could still anomalously form amorphous materials. Compared with its reference polymers (TUC8, TUC12, and UEG3), TUEG3 demonstrated the best mechanical performance, including a breaking strain of up to 393% ± 5% and a high elastic modulus of 1.4 GPa (Fig. 6f). In addition, the mechanical strength of TUGE3 could be completely recovered after healing at RT.
However, the self-healing process of most SHPs cannot take place below the Tg due to the restricted diffusion of molecules in their cross section. Although Aida and co-workers demonstrated that self-repairing below Tg is feasible for SHPs,4 the lack of substitution strategies severely limits the range of glassy polymers with RT ductility. Accordingly, Wu and co-workers94 prepared a class of self-healable glassy polymers with hyperbranched structure (Fig. 7a), which possessed high density of multiple H-bonds. The dynamic mechanical analysis showed that the resultant polymer (RHP-1) possessed the highest storage modulus (2.7 GPa) at 25 °C (Fig. 7b). The hyperbranched structure endowed the branched units and terminal groups with high mobility and effectively avoided the ordered build-up of molecular chains. Consequently, the glassy polymers showed amazing room-temperature self-healing ability. In particular, the instantaneous self-repair within 1 min allowed the tensile strength to be recovered to 5.5 MPa without any intervention from external stimuli. Therefore, this unique strategy has guiding significance for the design of other branched polymers and dynamic bonds. Here, Fu et al.53 synthesized a new type of glassy polyurethane (GPU) consisting of low-molecular weight oligomers containing numerous loose, weak H-bonds (Fig. 7c). The existence of abundant H-bonds within the network endowed GPU with a tensile Young's modulus of about 1.56 GPa and Tg value of 36.8 °C under ambient conditions (Fig. 7d). In addition, owing to the high steric hindrance of the IPDI asymmetric alicyclic structure, their urethane parts possess high molecular mobility in the glassy state, even at temperatures below Tg. Consequently, the tensile strength of GPU could recover to7.74 MPa after healing for only 10 min, showing a fast RT self-healable performance. Remarkably, the GPU specimen could fully recover when the healing time was extended to 60 min (Fig. 7e). Thus, the proposed design concept paves the way for the development of room-temperature SHP glasses.
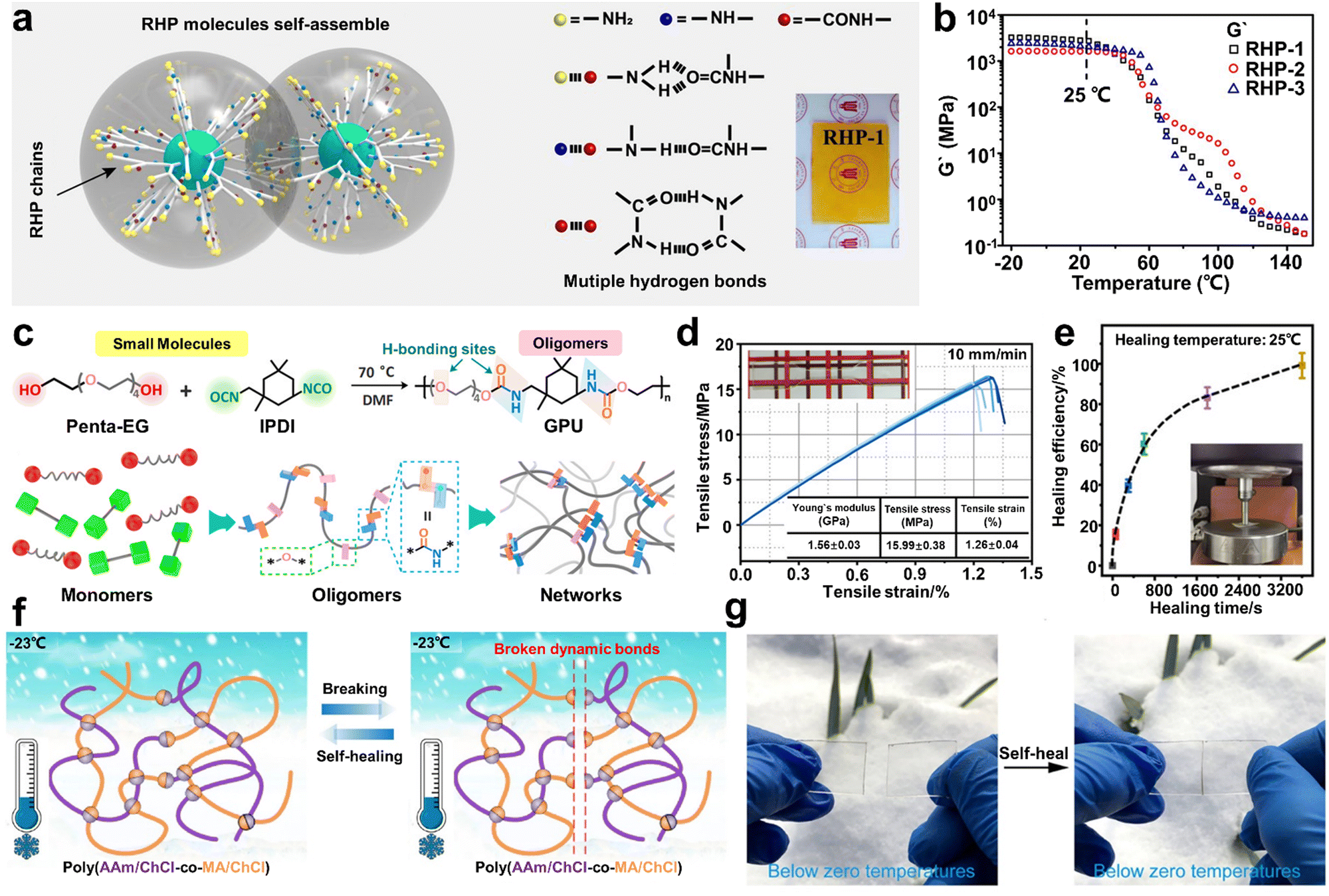 | ||
| Fig. 7 (a) Schematic illustration of the self-assembly of random hyperbranched polymers (RHP) and their intermolecular interactions, with the insert showing a photograph of RHP-1. (b) Temperature dependence of storage modulus of RHP. (c) Synthetic route, chemical structure and illustration of GPU polymer. (d) Summary of mechanical properties and stress–strain curves of GPU sample, with the inset photograph showing the standard spline of the tensile test. (e) Healing efficiency of resulting GPU sample prepared by compression (under 1 MPa constant stress) for different times at 25 °C, with the insert showing the healing process of the GPU sample. (f) Molecular model of poly(AAm/ChCl-co-MA/ChCl) system and demonstration of its healing process. (g) Optical images of healed poly(AAm/ChCl-co-MA/ChCl) film before and after being cut at ambient temperature as low as −23 °C. | ||
In addition to the aforementioned condensation reactions, copolymerization reactions such as free radical polymerization58,107 and photoinitiated copolymerization108–110 have been extensively used for the preparation of SHPs. For example, by introducing N-(hydroxymethyl)acrylamide as a thermocatalytic self-crosslinker and n-butyl acrylate (BA) as a soft hydrophobic material in a soft polymer system, Chiu and co-workers designed an elastic, transparent and autonomously self-healing random copolymer.107 A highly efficient self-healing performance and stronger mechanical properties were obtained by simply tuning the monomer ratios and varying the self-cross-linking reaction conditions. Remarkably, the self-healing properties could still be maintained underwater due to the hydrophobic nature of PBA in the copolymer system. Similarly, Dong et al.58 designed and synthesized a self-healable supramolecular adhesive via the random copolymerization of BA and acrylamide. While retaining the good leveling properties of polyacrylamide in solution, the quantitative introduction of BA controllably tuned the Tg of the copolymer and created a high density of H-bond donors and receptors, enabling supramolecular repair to proceed at a relatively low temperature.
As a new type of polymerizable ionic liquid with the advantages of green fabrication, excellent biocompatibility and post-treatment-free synthesis, polymerizable deep eutectic solvents (PEDSs) are considered promising polymerizable monomers for the preparation of SHPs. Based on the in situ photopolymerization of maleic acid (MA)/ChCl-type and acrylic amide (AAm)/choline chloride (ChCl)-type polymerizable PDES commoners, He and co-workers108 reported the synthesis of a transparent, anti-freezing, and self-healable elastomer (Fig. 7f). To introduce H-bonds in the PDES networks, AAm and MA molecules could serve as H-bond donors, while ChCl was used as the H-bond acceptor.
Indeed, the carboxyl group of the MA molecule terminal could build H-bonds with the hydroxyl group and amino groups on the monomer molecule, respectively. Consequently, the resultant elastomer displayed excellent autonomous self-healing capacity, which could occur in a wide temperature range of −23 °C to 60 °C (Fig. 7g). It should be noted that the hypermolecular network elastomer also demonstrated a fast self-healing performance with an efficiency of up to 94% within only 2 s.
Besides low-temperature resistance,111 researchers also made pioneering efforts for the development of adaptive SHPs in acidic and alkali environments.112 However, these SHPs rely on synthetic chemistry often involving a large number of organic solvents such as diethyl ether, chloroform and acetone.111,113 In addition, small organic molecules can easily access the interior of the polymer networks, leading to inevitable dissolution or swelling and destruction of their physical and chemical properties in an organic solvent environment. Therefore, it is necessary to prepare SHPs with multifunctionality and enhanced adaptability in harsh organic solvent environments. Similar to previous work,108 He's group109 designed another type of supramolecular elastomer with remarkable organic solvent insensitivity via the photoinitiated copolymerization of MA/ChCl and acrylic acid (AA)/ChCl-type PDESs. After 2 min of in situ photopolymerization, the prepared elastomer showed high optical transparency (≈93%) and stretchability (strain up to 1450%). Moreover, the hydroxyl and carboxyl groups among the binary building blocks endowed the obtained elastomer with highly reversible synergistic H-bonding interactions, showing self-healing efficiency up to 91.5%. Notably, this multiple H-bonding interaction could also withstand harsh conditions, and thus the healing process could even be carried out in various organic solvents.
3 Electronic applications of H-bond cross-linked self-healing polymers
With the inherent benefits of self-healing, mechanical flexibility and lightweight, SHPs have received widespread attention from academia and industry as active materials in energy devices and flexible electronics.114,115 These material innovations have enabled the rapid development of various types of self-healing electronic devices. To date, scientists have devoted great efforts to integrating self-healing features (for repairing mechanical integrity and restoring their functions and device performance) in flexible electronics to considerably increase the durability and prolong the lifespan of these devices, thereby reducing the economic cost and electronic waste.116,117 Benefiting from their prominent self-healable and mechanical robustness features, H-bond cross-linked SHPs have been extensively exploited as self-healing materials in a variety of energy and electronic devices. In this section, we summarize the discussion by dividing self-healing devices into three categories according to their different functions including energy harvesting devices,55–59 energy storage devices41,60–63 and flexible sensing devices.50,64–673.1 H-Bond cross-linked SHPs for energy conversion devices
Integrating self-healing technology into energy conversion devices can provide devices with the ability to operate independently and sustainably, which is of great significance for the next generation of smart electronics. In this section, we overview the application of H-bond cross-linked SHPs in energy conversion devices, including TENGs, perovskite solar cells and thermoelectric generators (Table 1).| Self-healing matrix | SHP type | Format of damage | Healing conditions | Usage | Characteristics | Ref. |
|---|---|---|---|---|---|---|
| Self-healing TENGs | ||||||
| Extremely stretchable PU | Microphase separation system | Mechanical damage (complete bifurcation) | 100 °C, 24 h | Friction layer | Good conductivity (6250 S cm−1), high stretchability of 2500%, | 55 |
| Linear silicone modified PU | Microphase separation system | Cut | RT, 30 min | Friction layer | With a short-circuit current of 31.9 μA and output voltage of 517.5 V | 56 |
| uPDMS | Microphase separation system | Cut | 90 °C, 1 h | Friction layer | Triboelectric current (12.1 ± 1.5 μA), triboelectric voltage (403.9 ± 46.2 V) | 57 |
| PDMS elastomer | Microphase separation system | Cut | RT, 72 h | Friction layer | Transparent, tough (16500 J m−2) |
118 |
| Fluorinated poly (urethane urea) | Microphase separation system | Cut | 60 °C, 48 h | Friction layer | With a high output power density of 2.75 W m−2 | 119 |
| Telechelic polyurea | UPy units at chain ends | Cut | 100 °C, 30 min | Friction layer | With a Voc of 52 V and circuit current of 0.5 μA (3 × 3 cm2) | 90 |
| Self-healing perovskite solar cells | ||||||
| TUEG3 | Homogeneous system | Scratch | 100 °C, 1 h | Self-healable matrix | Retains 94% PCE even after 3000 cycles | 120 |
| AD-23 | Homogeneous system | Crack | 70 °C, 5 min | Supramolecular adhesive | PCE on flexible substrates >20%, PCE on rigid substrates ∼21.99% | 58 |
| Self-healing thermoelectric generator | ||||||
| Ternary composite | — | Cut | RT, — | Thermoelectric composite | With a power output of 12.2 nW, retains >85% initial power output after repetitive self-healing cycles | 59 |
| Photothermal conversion elastomer | — | Cut | RT, 5 min | Photothermal conversion materials | With 68.08 mV voltage under a solar power of 200 mW cm−2 | 121 |
As mentioned above, PU is a multi-segmented polymer with soft-hard alternating chain units in which dynamic reversible H-bonds responsible for self-healing can be designed in either hard or soft phases. Its appropriate medium friction electronegativity (∼−1.87 V) allows it to be used as a positive/negative friction layer, providing a flexible alternative for the fabrication of self-healable friction layers.55,56 For example, Lee et al.55 designed an extremely stretchable (with tensile strength of >1.5 MPa and high strain of 2500%) and healable TENG by using polyurethane acrylate (PUA) elastomers as the triboelectric layer (Fig. 8a). To address the problem of interfacial compatibility between the triboelectric layer and conductor and weaken the modulus mismatch between them, silver flakes and liquid metal were doped in the PUA matrix for conductive self-healing PU. Liquid metal particles are distributed throughout the PUA network to provide the required electrical anchor among the separated silver flakes, resulting in good electrical conductivity under extreme stretching. Consequently, the prepared self-healing TENG could obtain electrical properties even at high strains of 2500% (Fig. 8b) and also withstand 500 cycles of 1000% strain, showing outstanding tensile and tensile fatigue resistance. Benefiting from the reversible breaking and reorganization of dynamic multivalent H-bonds in the PUA network, the TENG could recover its energy-harvesting properties after severe mechanical damage (Fig. 8c). In addition, by utilizing a linear silicone-modified PU coating as a positive triboelectric layer, Wang et al.56 reported a novel self-healing TENG. Upon suffering abrasion, the broken PU polymer chains in the friction layer would gradually be cross-linked again via H-bonding to achieve self-healing. Specifically, the output performance of the self-healing TENG could recover its initial value even after 4 cycles of cutting–repairing procedures (30 min at RT).
 | ||
| Fig. 8 (a) Schematic diagram of the stack of the stretchable and self-healable TENG, where PUA is used as the negative friction layer. (b) Output voltage of stretchable and self-healable TENG under different uniaxial strains and (c) before and after healing. (d) Schematic of interactions between TUEG3 and MAPbI3 perovskite (left). Optical images of the MAPbI3/TUEG3 composite film before and after healing (right). (e) Normalized PCE of the self-healable perovskite solar cell in a bending cycle with a radius of 5 mm and (f) in a bending step with a radius of 1.5 mm for 50 times and repairing steps at 85 °C for 30 min. (g) Schematic of self-healable thermoelectric generator device comprised with six legs of ternary composite film. (h) Changes in σS2 for the ternary composite film under different tensile strains before cutting and after cutting and reattachment. (i) Normalized power output of healable thermoelectric generator versus number of cuts. | ||
Besides PU, PDMS is also a promising candidate for the preparation of friction materials owing to its good flexibility, excellent wear resistance and strong electron attraction ability. In Section 2.2.1 we mentioned that PDMS with low Tg is usually incorporated as soft segments in H-bonding-based SHPs. Ko et al.57 developed a single-electrode self-healing TENG with triple-layer configuration using self-healable PDMS polymer films prepared via the urea bonding reaction of amino-functionalized PDMS with IPDI as a negative friction layer. Based on the dynamic reversible H-bonding in the polymer network, the proposed TENG not only exhibited excellent frictional electrical properties, but also showed good self-healing ability to ensure continuous device operation even after multiple scratches on its surface. After healing at 90 °C for 1 h, the frictional damage caused by sandpaper (with 1 N loading force) on the TENG surface could completely recover and maintain the original output performance. However, the nature of siloxane bonds and low inter-chain interactions led to low strength and low fracture energy of PDMS-based composites under external pressure. Given that the persistent operation of TENGs requires continuous mechanical interaction of dielectric materials, it is necessary to develop self-healing friction layer materials with high strength, stretchability and toughness. He et al.118 fabricated a series of ultra-high toughness, transparent and autonomous self-healable PDMS-based elastomers by adopting a strategy of combining hierarchical H-bonding kinetics with phase-separation-like structures. The mechanical properties could be tuned by simply changing the mole ratio of bis(hydroxyalkyl)-terminated PDMS and amino-functionalized PDMS during the synthesis process. On the one hand, the H-bond-rich domain ensured the strength and rigidity of the material given that it required more energy for dissociation. On the other hand, the H-bond-poor domain could rapidly dissipate stress through reversible bond breakage and reorganization, leading to high stretchability and toughness. Finally, a transparent, tough (16500 J m−2) and self-healable TENG (healing efficiency up to 97%) was achieved by combining the prepared PDMAS-U10 as a conductive layer.
Although the integration of self-healing capabilities has improved the damage tolerance and durability of TENGs to a certain extent, from the perspective of practical applications, the fabrication of self-healing friction layers with high frictional electrical properties, high mechanical strength, high healing efficiency and high chemical stability is still a pressing challenge to be solved. Herein, using hexadecafluoro-decanediol (HFDD) as a chain extender to react with isocyanate-terminated PDMS prepolymer, Sun et al.119 designed and synthesized a self-healing fluorinated poly(urethane urea). The use of HFDD not only improved the mechanical strength (tensile stress of 4.18 MPa) of the composite, but also exhibited a better triboelectric performance than PDMS and PTFE. Based on the abundance of dynamic reversible H-bonds in the network, the polymer films could repeatedly repair damages under heating at 60 °C, showing excellent self-healing ability. Finally, a completely self-healing TENG with an output power density of 2.75 W m−2 was achieved by sandwiching a self-healable ionogel with the self-healing fluorinated poly(urethane urea). Notably, the TENG exhibited a reliable electrical output performance even under extreme environments.
To address the issue of stability, SHPs are introduced into PSCs to repair their mechanical damage and enhance their resistance to environmental degradation. However, combining perovskite materials with an ordered crystalline or semi-crystalline structure with amorphous SHPs is a significant challenge for researchers.126 As mentioned before, Aida et al.4 designed a series of low-molecular weight polymers (i.e., TUEG3) by implanting numerous nonlinear zigzag thiourea units into a poly(ether-thiourea) network. Based on this, Dou et al.120 prepared a self-healing hybridized perovskite via the slight modification of TUEG3 using amine termination and incorporating it into polycrystalline methylammonium lead iodide (MAPbI3) using secondary-bonding interactions (Fig. 8d). The nonlinear zigzag array of H-bonds formed between the thiourea groups made TUEG3 mechanically robust but readily repairable. Moreover, the secondary-bonding interactions could effectively promote the re-joining of distant perovskite grains and facilitate the healing of large cracks. As shown in Fig. 8d, the hybrid perovskite film with a polymer concentration of ≥2.1 wt% could repair a 6 μm-wide crack after healing (100 °C) for 1 h under a nitrogen atmosphere. Flexible solar cells using the TUEG3 (6.0 wt%)-MAPbI3 composite as the active layer could achieve a PCE of 8.9% and retained 94% of its initial performance after 3000 bending cycles at a 5 mm radius (Fig. 8e). Notably, even after 50 bending cycles at a 1.5 mm radius, the PCE of the TUEG3-6.0 wt% device could be recovered to 80% of its initial value after annealing at 85 °C (Fig. 8f). This self-healable perovskite solar cell design strategy paves a new path for the development of wearable energy harvesting devices.
Meanwhile, the use of thermal dynamic self-healing supramolecular dopants has also been proposed as an alternative solution for improving the performance and stability of flexible PSCs. Dong et al.58 reported the synthesis of a self-healable functional polymeric adhesive (AD-23) via the random copolymerization of acrylamide and BA, which had a high density of H-bond donors and receptors. Benefiting from the supramolecular passivation effects and amphiphilic nature of AD-23, the flexible PSCs containing this functional adhesive achieved a PCE of more than a 20% on flexible substrates, with improved electron mobility and bending resistance. In addition, the damaged grain boundary cracks of the AD-23-modified PSC samples could be greatly repaired after heating at 70 °C for 5 min. Thus, this low-temperature self-healing effect could ensure their high stability even under the aging conditions of 85 °C/85% relative humidity.
To date, the main direction for the preparation of self-healing thermoelectric devices is the combination of thermoelectric materials and SHP substrates. For example, Baran and co-workers59 demonstrated a stretchable and self-healable thermoelectric generator based on a ternary composite (Fig. 8g). The ternary composite was comprised of dimethyl sulfoxide-mixed poly(3,4-ethylenedioxythiophene)/polystyrene sulfonate with polymeric surfactant (Triton X-100) as the soft matrix. Interestingly, the free-standing composite film still provided stable thermoelectric properties during 35% tensile strain without affecting the conductivity and Seebeck coefficient (Fig. 8h). Based on the combination of H-bonding in Triton X-100 and viscoelasticity of the composite film, the damaged areas on the surface of the thermoelectric generator could be effectively healed and reattached, recovering its thermoelectric performance autonomously. Consequently, the prepared 3D-printed thermoelectric generator maintained more than 85% of its initial output power even after repeated cuts (Fig. 8i).
Recently, photothermal conversion elastomer composites have shown great potential for applications in thermoelectric conversion. The use of PU or natural rubber (NR) as the matrix combined with functional fillers is one of the facile strategies for the preparation of photothermal conversion composites. Lu et al.121 prepared a fast self-healable, photothermal elastomer through multiple H-bonding interactions among NR, polydopamine, and cellulose nanocrystal. The formation of multiple H-bonding networks endowed the elastomer with rapid and spontaneous self-healing capability, with a strain recovery rate of 92.2% within 5 min. Meanwhile, the elastomer also exhibited good and stable photothermal conversion performance (49.7%) due to the introduction of the photothermal agent PDA. Encouraged by these features, the authors demonstrated a solar thermoelectric generator by combining the elastomer composite film with a thermoelectric module and a cooling system. As the power density of solar energy increased from 50 mW cm−2 to 200 mW cm−2, the voltage generated by the thermoelectric generator increased from 36.33 mV to 68.08 mV.
3.2 H-Bond cross-linked SHPs for energy storage devices
With the rapid development of novel electronic devices, energy electronic devices with high-performance and high-stability have attracted increasing interests. In this section, we focus on two different types of energy storage devices, i.e., lithium-ion batteries (Li-ion batteries) and supercapacitors (SCs) (Table 2).| Polymer matrix | SHP type | Format of damage | Healing conditions | Usage | Performance | Performance after healing | Ref. |
|---|---|---|---|---|---|---|---|
| Self-healing Li-ion batteries | |||||||
| SHP | Homogeneous system | Cut | RT, <60 s | Binders | 2617 mA h g−1 at 0.1C | — | 41 |
| 80% after 90 cycles | |||||||
| SHP | Homogeneous system | Binders | 2620 mA h g−1 at 0.05C | — | 60 | ||
| 80% after 500 cycles | |||||||
| SHP | Homogeneous system | Scratch | RT, 3 h | Binders | 1625 mA h g−1 at 0.5C | — | 61 |
| 80% after 150 cycles | |||||||
| SHP | Homogeneous system | Scratch | RT, <1 h | Binders | 2212 mA h g−1 at 0.1C | 1924.4 mA h g−1 after 25th cycle after 4th healing | 128 |
| 91.8% after 100 cycles | |||||||
| PAA-P(HEA-co-DMA) | — | Cut | RT, 10 min | Binders | 2552 mA h g−1 at 0.1 A g−1 | — | 129 |
| 93.8% after 220 cycles | |||||||
| SHP | Homogeneous system | Cut | RT, 30 min | Binders | 719 mA h g−1 at 0.1 A g−1 | — | 42 |
| 81% after 100 cycles | |||||||
| PAA-UPy | UPy units at side chains | Cut | RT, — | Binders | 4194 mA h g−1 at 0.2C | — | 130 |
| 62.89% after 110 cycles | |||||||
| PVP-PEI | — | Scratch | RT, 24 h | Binders | 898.6 mA h g−1 at 1C | — | 131 |
| 67.68% after 450 cycles | |||||||
| PDMS-MPD-TM | Microphase separation system | Scratch | RT, 4 h | Interlayer | 722.7 mA h g−1 at 0.2C | — | 105 |
| 67.1% after 100 cycles | |||||||
| SHP | Homogeneous system | Artificially created pinhole | RT, 60 s | Protective coating | 1 mA h cm−2 at 1 mA cm−2 for 180 cycles | 132 | |
| PEO-UPy | UPy units at side chains | Cut | RT, few minutes | Protective layer | 10 mA h cm−2 at 5 mA cm−2 for 1000 h | — | 133 |
| PEGMA-UPyMA | UPy units at side chains | Cut | RT, 2 h | Electrolyte | 157 mA h g−1 at 0.1C at 60 °C | 155 mA h g−1 at 0.1C at 60 °C; 91.6% after 80 cycles | 134 |
| 91% after 100 cycles | |||||||
| P(NH2-PEG-NH2-co-TPU) | Microphase separation system | Cut | RT, 60 s | Electrolyte | 147.9 mA h g−1 at 0.1C | — | 135 |
| 90% after 100 cycles | |||||||
| P(PEGMA-co-UPyMA)-SiO2-UPy | UPy units at side chains | Cut | RT, 30 min | Electrolyte | 145 mA h g−1 at 0.2C at 60 °C | — | 136 |
| 95.8% after 60 cycles | |||||||
| SHP | Homogeneous system | Crack | RT, 1 h | Electrolyte | 1 mA h cm−2 at 20 mA cm−2 for 1500 cycles | — | 137 |
| 157 mA h g−1 at 0.2C | |||||||
| 99.4% after 120 cycles | |||||||
| Self-healing SCs | |||||||
| SHP | Homogeneous system | Cut | RT, 5 min | Substrate | 35 F g−1 at 1 A g−1 | ∼85.7% (30 F g−1) after 5th healing; 96.4% after 1000 cycles at 5 A g−1 | 62 |
| ∼100% after 1000 cycles at 5 A g−1 | |||||||
| SHP | Homogeneous system | Cut | RT, — | Substrate | 140 F g−1 at 0.33 A g−1 | 92% (128.8 F g−1) after 1st healing | 63 |
| Self-healing PU | Microphase separation system | Cut | RT, — | Protective shell | 25.9 mF cm−2 at 10 mV s−1 | 54.2% after 3rd healing | 138 |
| 82.4% after stretching 100% strain | |||||||
| Self-healing PU | Microphase separation system | Cut | RT, — | Protective shell | 34.6 mF cm−2 at 1 mV s−1 | 81.7% after 5th healing | 139 |
| 91% after 150000 cycles |
|||||||
| VSNP-PAA | — | Cut | RT, — | Electrolyte | >200 F g−1 at 10 mV s−1 | ∼100% after 20th healing | 140 |
| MGO-PAA | — | Cut | RT, — | Electrolyte | 56 F g−1 at 1 A g−1 | 80% after 3rd healing | 141 |
| PVA–H2SO4 | — | Cut | RT, 1 h | Electrolyte | 15.8 mF cm−2 at 0.044 mA cm−2 | ∼80% after 5th healing | 142 |
3.2.1.1 Si Anodes. Binders integrating desired mechanical properties, remarkable adhesion strength and good electrochemical stability play an extremely important role in Li batteries, which bind the electrode components to build a strong electrode and adhering the electrode to the current collector.143 In the case of Si, it possesses high theoretical capacity and can charge at an extremely low operating voltage (0.4 vs. Li/Li+), showing great promise to replace graphite as a next-generation high-energy density Li battery anode material. Nevertheless, the major issue associated with Si anodes is the drastic volume change during repeated lithiation/delithiation processes, resulting in the formation of cracks or even pulverization. Hence, it is of great significance to explore self-healing elastic adhesives that can adapt to volume changes.128
For instance, Bao et al.41 first used an H-bonding-based SHP to alleviate the volumetric expansion of electrodes. Specifically, a self-healable Si anode was prepared by coating a thin layer of self-healable supramolecular rubber on Si microparticles. This cross-linked reversible network endowed the SHP with excellent mechanical stretchability, allowing the polymer to adapt to the bulk expansion of the Si particles, thus avoiding irreversible damage (Fig. 9a). Unlike traditional binders (alginate, sodium hydroxymethyl cellulose and polyvinylidene fluoride), this healable binder could repair micro-cracks by eliminating mechanical stresses arising from volume changes through the synergistic action of internal urea-based H-bonds. Meanwhile, it presented a specific capacity as high as 2617 mA h g−1 and excellent cycling performance at 0.1C (Fig. 9b). Besides the self-healable performance, interfacial adhesion between Si particles is another important parameter in maintaining battery integrity during cycling processes. However, directly coating the SHP on the surface of the Si particle layer cannot achieve this goal. Therefore, a high Si mass loading and long-term cycling stability have not been obtained to date. Based on the above-mentioned issues, various self-healing Si electrodes have been further systematically investigated and developed by Bao and co-workers.42,43,60,61 A novel strategy that distributed the three-dimensional (3D) SHP in the Si particle layer was proposed to maintain low cost and high cycling stability (Fig. 9c).60 After blading repeatedly under heating conditions, the 3D spatial distribution of CB/SHP in the Si electrode can be clearly seen. Unlike coating the SHP only on the top of Si electrodes, the 3D spatial distribution provides a faster repairing response due to the relatively short diffusion distance of the SHP into the resulting fracture sites. Additionally, the SHP was used as a binder between the current collector and active materials to ensure good electrochemical contact throughout the composite electrode. Consequently, the 3D distribution mode could form a solid interface between the SHP and Si particles, resulting in the more prominent cycling stability of the composite electrode.
 | ||
| Fig. 9 (a) Schematic illustration of the behavior of conventional Si electrode binders (blue), where the electrodes fail due to cracking of the particles and polymer binder. Schematic illustration of the behavior of stretchable self-healable Si electrode binders (pink). (b) Capacity retention of SHP/SiMP/CB electrode cycled at different current densities. (c) Schematic illustration of electrode design toward adjusting the spatial distribution of CB/SHP in Si layer (top) and scanning electron microscopy images of Si and CB/Si-SHP electrodes (bottom). (d) Chemical structure of the PAA-UPy polymer and scheme of the charge/discharge cycle of Si anodes using the PAA-UPy polymer as the binder. (e) Molecular structure of PAA-P(HEA-coDMA) and PAA, and the spring expander model of their complex. (f) Cycling performance of SiMP electrodes using different binders. | ||
In addition to excellent cycle stability and high capacity, a high rate performance (>0.5C) is also extremely relevant for the practical application of Si anodes. Although some new polymer binders such as cross-linked polymers,144,145 alginate binders146 and natural gums147 have also been shown to enhance the cycle life of Si nanoparticle electrodes, their poor mechanical stretchability and relatively weak interaction with Si particles cannot provide stable electrode operation for Si microparticles. By incorporating PEG in the SHP, the same group61 described a novel self-healing polymeric binder for the preparation of low-cost Si particle electrodes. This viscoelastic and highly stretchable SHP contained a large number of H-bonding sites, which allowed it to mechanically heal at RT. The H-bonds also facilitated better adhesion between the Si microparticles due to the interaction with the hydroxyl groups on the Si surface. Moreover, the introduction of PEG groups could effectively promote the conduction of Li ions given that the lone pair of electrons on oxygen can coordinate and aid the transport of Li ions in the matrix. Hence, the polymeric binder achieved a high discharging capacity (≈2600 mA h g−1) and reasonable rate performance, especially when >1C.
Polyacrylic acid (PAA) is also a commonly used adhesive in Si electrodes, in which the abundant carboxylic acid groups can form strong H-bonds with themselves and Si particles, thus enhancing the electrochemical stability of the Si electrode.148 For instance, Deng et al.130 reported a self-healable PAA-UPy binder prepared by copolymerizing tert-butyl acrylate and UPy monomers in dioxane, followed by hydrolysis with trifluoracetic acid (Fig. 9d). Due to the cooperation interaction between quadruple H-bonds and carboxyl single H-bonds, the PAA-UPy binders exhibited an excellent self-healable performance. Based on this feature, the interfacial tension induced by bulk expansion could be effectively compensated by the binder inside the Si electrodes. Therefore, the composite anode prepared with PAA-UPy as the binder displayed a high initial discharging capacity of 4194 mA h g−1. Additionally, it still maintained a capacity of up to 2638 mA h g−1 after 110 cycles, revealing outstanding cycling stability.
Besides Si nanoparticles, Si microparticle (SiMP) powders are also a prospective electrode material for modern industrial applications. However, given that SiMPs possess a greater size change (micrometer-level) and stronger pulverization tendency during cycling, stabilizing the cycling stability of SiMP electrodes is a formidable challenge. Hence, Hirano and co-workers129 designed and synthesized another type of self-healable binder (Fig. 9e) by mixing P(HEA-co-DMA) and PAA in a weight ratio of 1:4. Specifically, the obtained binders had high reversible elasticity and could withstand a tensile strain of 400%, thus effectively suppressing the bulk expansion of Si microparticles. In addition, the covalently crosslinking of soft P(HEA-co-DMA) chain and rigid PAA chain in the polymer network remarkably improved the electrochemical performance of the Si microparticle anode. Consequently, no decay was observed following 200 cycles with 0.4 A g−1, showing excellent cycling stability. In comparison, the contrast electrode with commercial PAA binders resulted in rapid capacity fading over 100 cycles (Fig. 9f). Significantly, the SiMP electrode using the prepared binder retained its surface morphology even after 100 cycles, whereas a great deal of cracks was observed in the whole electrode surface with PAA binder, revealing the good self-healable capacity of the prepared binder. Thus, based on its strong binding and self-healing feature, this binder could be used to produce stable Si-based electrodes with long-term stable cycling and high energy density, meeting the demands of commercial Li-ion batteries.
3.2.1.2 S Cathode. S cathodes have developed into hopeful alternatives due to the high theoretical capacity and low-cost of their raw materials. Unfortunately, Li–S batteries still exhibit some limitations during their practical application, such as the cathode pulverization caused by volume expansion during cycling149–152 and formation of dendritic crystallites on the Li metal anode.153 Moreover, another challenge is the generation of the reduction compound called intermediate lithium polysulfide species, which can be dissolved in the organic liquid electrolyte and deposited on the Li–metal anode via the shuttle mechanism. Therefore, simultaneously achieving S cathodes with a stable structure and effectively inhibiting the diffusion of soluble polysulfides in the electrolyte is the main direction of current research.
Using diamines and polybasic acids as raw materials, Yang and co-workers154 synthesized a supramolecular SHP, which was employed as a multi-functional binder in Li–S batteries. Specifically, 10 wt% amine groups in the SHP networks provided extensive chemical adsorption sites for polysulfides, thereby effectively inhibiting polysulfide shuttling and maintaining the sulfur content in the cathode. Furthermore, the introduction of polar groups in the molecular structure design of self-healing binders is a promising strategy for improving the performance of Li–S batteries. The polar groups can immobilize polysulfides, thus effectively inhibiting the shuttle effect. By exploiting the cross-linking effect between polyvinylpyrrolidone (PVP) and polyethyleneimine (PEI), Zhou et al.131 reported a novel polysulfide-trapping and self-healing binder. On the one hand, its numerous polar groups such as the carbonyl groups (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) and amino groups (–NH2) in PVP can interact effectively with the Li-ion in lithium polysulfides, and thus suppress the shuttle effect. On the other hand, PEI with a highly branched structure is conducive to the formation of dynamic H-bond networks between –NH2 and CO, thereby endowing the binders with good self-healing ability. Consequently, the flexible Li–S pouch battery with this binder exhibited more than 95% capacity retention following 2800 bending cycles. This type of polysulfide-trapping, water-soluble, and self-healing binder paves the way for further promoting the development of advanced flexible Li–S batteries.
O) and amino groups (–NH2) in PVP can interact effectively with the Li-ion in lithium polysulfides, and thus suppress the shuttle effect. On the other hand, PEI with a highly branched structure is conducive to the formation of dynamic H-bond networks between –NH2 and CO, thereby endowing the binders with good self-healing ability. Consequently, the flexible Li–S pouch battery with this binder exhibited more than 95% capacity retention following 2800 bending cycles. This type of polysulfide-trapping, water-soluble, and self-healing binder paves the way for further promoting the development of advanced flexible Li–S batteries.
In addition to self-healing binders, self-healing interlayers are also a common strategy for elevating the electrochemical stability of S cathodes. The self-healing interlayer allowed rapid repair of morphological defects and restricted the movement of high-soluble compounds to the anode area, thereby considerably enhancing the cycling stability of S cathodes. Thus, to further overcome the “shuttle effect” and promote the healing efficiency of the S cathode interlayer, Fu and co-workers105 proposed a novel strategy of multiphase active H-bonds, as detailed in Section 2.2.1. The obtained composite (PDMSMPI-TM/MWCNT) could restore the air conductivity under environmental conditions as well as repair man-made microcracks in Li–S battery electrolytes. Moreover, the composite also exhibited outstanding impermeability. In contrast to polypropylene diaphragms, the repaired PDMSMPI-TM/MWCNT composite could still significantly suppress the migration of polysulfides, revealing the recovery of the natural barrier properties (Fig. 10a). The as-prepared Li–S battery still provided a discharging capacity of 722.7 mA h g−1 after 100 cycles at 0.2C, which is much higher than the electrode without a composite interlayer (446.1 mA h g−1).
 | ||
| Fig. 10 (a) Schematic diagram of the comparison between the working mechanisms of traditional and prepared functional Li–S batteries, where the electrical healing process is demonstrated with LEDs in series with PDMS-MPI-TM/MWCNT composite. (b) Schematic illustration of highly adaptive polymer coating inhibiting dendrite growth. (c) Cycling life and CE of the Li metal electrode with and without the adaptive polymer coating at a capacity of 1 mA h cm−2. (d) Supramolecular chemical structure and dynamic H-bonding for the SHPE. (e) Optical microscopy images of the healing process of SHPE. (f) Cycling performances of a solid-state battery cycled at 0.2C. | ||
3.2.1.3 Li metal anode. Among the possible electrode candidates, Li metal possesses the lowest negative electrochemical potential (−3.04 V) and a remarkably high theoretical specific capacity (3860 mA h g−1).155,156 However, Li-metal anode batteries have not become viable technology due to their safety and cycle stability, where the greatest obstacles are the large volume changes and the growth of dendritic lithium during the charging/discharging processes, leading to rapid capacity deterioration, short-circuiting of the batteries, and even explosion hazards.157,158 Additionally, another critical challenge for improving the performance of Li-metal electrodes is how to form a uniform, stable SEI layer on the surface of Li that could adapt to the vast volumetric changes during cycling.
To date, various techniques have been adopted to address these challenges, such as the construction of interfacial layers to inhibit harmful reactions with the electrolyte159,160 and using other types of additives to get uniform Li deposition,161 and others.162,163 Remarkably, each strategy is a trade-off between the pros and cons, and the aforementioned problems with Li metal anodes have not yet been fully resolved. For example, previous works164,165 showed that interfacial engineering is capable of uniformly depositing Li metal at a current density of approximately 1 mA cm−2, but improving the cycling stability of Li metal electrodes at high current densities remains a challenge. Therefore, Cui and co-workers132 demonstrated a suitable method to suppress the formation of Li dendrites and promote the electrochemical performance of Li-metal anodes (Fig. 10b). Due to the existence of weaker H-bonding crosslinks in the network, the prepared SHP possessed high stretchability and behaved as a viscoelastic material. Meanwhile, it also had a low Tg (about −26 °C), and thus the spontaneous reorganisation of the bonding groups and movement of the polymer chains could occur at RT. These properties enabled the SHP coating to flow slowly over the electrode surface, repairing pinholes and cracks in the SEI layer and preventing the rapid formation of dendrites at SEI hotspots. Consequently, the electrode coated with SHP could maintain a CE of close to 97% over 180 cycles, while the CE of the bare metal electrode dropped below 90% after 145 cycles (Fig. 10c).
In addition, the adhesion between Li metal and the SEI layer is also an important factor affecting the electrochemical properties of self-healable artificial SEI film-based Li-metal anodes.166,167 For example, a strongly adhesive and self-healable polymer (LiPEO-UPy) was designed as a protective layer for Li anodes by Xiong and co-workers,133 which successfully achieved long-term lithium plating/stripping cycles. The self-healing supramolecular PEO-UPy matrix was synthesized via reversible addition–fragmentation chain transfer (RAFT) polymerization. Due to the quadruple-H-bonding interactions between UPy dimers, the PEO-UPy polymer possessed a prominent self-healable and stretchable performance. Further, the LiPEO-UPy coating could also effectively eliminate the formation of Li dendrites, and also guarantee that it could withstand huge interface fluctuations without breakdown. Consequently, the LiPEO–UPy-coated Li anode realized a long cycle life of more than 1000 h at a high areal-capacity of 10 mA h cm−2 and could work at an ultrahigh current density (20 mA h cm−2) in more than 4000 cycles in a symmetric battery.
3.2.1.4 Polymer electrolytes. Besides as a binder and protective coating, SHPs can also be used in polymer electrolytes to enhance the electrochemical stability of batteries and prolong their lifetime. In Li batteries, the electrolyte is an essential component for ion transport, which need to maintain reliability and high performance. In general, electrolytes can be classified into solid electrolytes and liquid electrolytes according to their physical state. Although liquid electrolytes can facilitate the rapid conduction of Li+, they also exhibit serious safety concerns (e.g., flammability, explosions, and leaks). Compared to liquid electrolytes, polymer electrolytes possess several advantages, such as inherent thermal stability, excellent flexibility and processability, and do not have the troublesome leakage dilemma.168–171 Therefore, the use of polymer electrolytes is considered a practical method to address the safety problems of conventional electrolytes.
Nevertheless, the poor ionic conductivity and inherent brittleness (easy to deform or crack) of most polymer electrolytes lead to a meagre cycling performance and short lifetime of batteries.172–174 The introduction of a dynamic H-bonding cross-linked network in a polymer electrolyte matrix is a feasible synthetic strategy.21,99,175 For example, Xue et al.134 recently prepared a novel stretchable polymer electrolyte material for Li-based batteries through quadruple H-bonding, providing rapid intrinsic self-healing and high stretchability to address problems such as cracking and fracture. However, to further improve the safety performance and lifespan of lithium metal batteries, the mechanical properties of the self-healing system need to be enhanced by physically cross-linking the network. In the same year, they further fabricated a flexible SHPE with excellent self-healing ability and enhanced mechanical properties,176 which could withstand the volume expansion of the electrode material during cycling.
The cell assembled using the SHPE exhibited an initial capacity of 137.9 mA h g−1 and maintained a capacity of 128 mA h g−1 after 120 cycles, showing a very stable cycling performance.
In the case of solid-state electrolytes, the supramolecular structure design is in high demand, which needs to combine high ion conductivity, fast self-healing performance, and high mechanical stability, given that short-circuiting may occur if the self-healing rate is slow. Using a simple condensation polymerization approach, Guo and co-workers135 reported a solid polymeric electrolyte with rapid self-healing and high ionic conductivity. In this system, amino-terminated PET acted as the supramolecular backbone, while thermoplastic polyurethane worked as a physical cross-linked agent (Fig. 10d). The former provided dynamic H-bonds and high ionic conductivity, and the latter improved the mechanical strength of the SHPE. Hence, the optimized SHPE sample exhibited an outstanding self-healing performance, which could completely heal a deep cut within 60 s (Fig. 10e). Based on this freely adjustable SHPE, the constructed solid Li metal full batteries exhibited superior cycling stability, where the discharge capacity still remained at 90% of the initial capacity after 100 cycles at 0.2C (Fig. 10f).
Despite their numerous advantages, the ionic conductivity of polymer electrolytes at RT is relatively low (10−6 to 10−8 S cm−1) due to the influence of the crystalline region of the polymer matrix.177 Composite polymer electrolytes (CPEs) formed by mixing ceramic fillers in the polymer matrix appear to be a promising alternative strategy because ceramic fillers can change the recrystallization kinetics of polymer chains and enhance the ionic conductivity. However, they also suffer from the problem that the cross-linked structure in the polymer may induce cracking during the charging/discharging of Li-ion batteries. In addition to the solvent-free polymer electrolytes discussed above, SHPs have also been introduced in composite polymer electrolytes (CPEs) to avoid battery failure in case a crack is formed during cycling.136,137,178 As an extension of their work (PEG-UPy),134 Xue and co-workers reported the fabrication of a CPE with better self-healing properties and ionic conductivity, which was fabricated by incorporating UPy-functionalized SiO2 nanoparticles (SiO2-UPy) in PEG-UPy.136 The SiO2-UPy filler could be uniformly dispersed in the polymer matrix because of the interaction of the UPy dimer, thereby increasing the interphase conduction between them. Therefore, compared with the pristine SiO2-doped sample, the CPE had a better ionic conductivity level, reaching 8.01 × 10−5 S cm−1. Moreover, the mechanical and self-healable properties of SHPE were also enhanced due to the increase in physical cross-linking sites in the polymer matrix. Consequently, the Li batteries using this SHPE exhibited a stable cycling performance, where its discharge capacity still remained at 139 mA h g−1 with a CE of 97.9% after 60 cycles.
However, the performance of high area capacity and high current density during long cycles has not yet been fully explored due to the safety concern of dendrite-related failure. Thus, to address this, a solid–liquid hybrid electrolyte using organic solvents as plasticizers was developed by Liu et al.,137 in which a fatty-acid based SHP worked as the SHP matrix. In addition, Ga-doped Li7La3Zr2O12 nanoparticles acted as an ionic conductor and dispersed homogenously in the SHP matrix. Compared with the dendritic and pulverized structure of conventional separators, the hybrid electrolyte could achieve the smooth, uniform and dense electrodeposition of lithium metal. Consequently, the obtained Li|Li4Ti5O12 cells delivered a higher specific capacity and longer cycling performance than that using conventional separators, which is a significant step toward the realization of high-cost, large-scale Li batteries.
3.2.2.1 Electrodes. The integration of SHPs in electrodes can repair mechanical damage-induced cracks, thereby restoring the electrical performance of the device. By spreading single-walled CNT films on self-healing substrates, Chen et al. reported the first example of mechanically and electrically self-healing SCs (Fig. 11a).62 The substrate was composed of a supramolecular network reinforced by TiO2 nanospheres. Due to the large number of H-bonding interactions between the supramolecular and nanospheres, the composites exhibited excellent healing capability at RT. Once encountering break-down, the separated single-walled CNTs could restore and contact each other by the sliding movement of the self-healing matrix. Therefore, the specific capacitance was still restored as much as 85.7% even after the fifth healing cycle. This pioneering work has become a stepping stone for the design of various self-healing electronic devices in the future.
 | ||
| Fig. 11 (a) Schematic illustration of the design and assembly process of the self-healable SC composed of single-walled CNT films and self-healable substrate. (b) Schematic diagram of the high-performance self-healing conducting wire and its healing process. (c) Galvanostatic charge–discharge curves of the linear self-healing SC before fracturing and after healing. (d) Manufacturing process of the self-healable and stretchable SC-based polypyrrole/graphene oxide/MWCNT electrodes. (e) Typical cyclic voltammogram curves of the resulting SC before and after stretching to 100%. (f) Typical cyclic voltammogram curves of the resulting SC before healing and after several self-healing. (g) Preparation of flexible healable all-in-one configured SC and demonstration of its healing process. (h) GCD curves of the assembled all-in-one configured SC before and after self-healing. (i) Healing efficiency of different self-healing cycles calculated from GCD curves. | ||
With the ability to be woven into textile devices, wire-shaped or yarn-shaped SCs show great potential for the fabrication of flexible, light-weight, wearable electronic devices.181 However, yarn electrodes are susceptible to bending or other irreversible damage during use, resulting in the failure of the entire device or its function with low efficiency. Thus, the lifetime of most wire-shaped devices is very low in practical applications. To address this problem, Peng and co-workers63 reported for the first time the preparation of a novel wire-shaped SC based on a high-performance self-healing conducting wire by wrapping aligned electrically conducting nanomaterials around SHP fibers (Fig. 11b). Due to the abundance of reversible H-bonding interactions in SHPs, multiple self-healing of structure and electrical conductivity could occur in the core-sleeve wire after breaking. As a demonstration, a wire-shaped SC was fabricated by twisting two healable wire electrodes (CNT/SHP fiber) with PVA-H2SO4 gel electrolyte. The specific capacitance reached 82.6% after the fifth healing cycle. Furthermore, the electrochemical properties of the wire-shaped SC could be further improved after introducing a second active material, i.e., polyaniline, whose specific capacitance was 140.0 F g−1 and its capacitance was recovered up to 92% after self-healing (Fig. 11c).
Self-healing coating shells can heal themselves after mechanical damage, thereby restoring the integrity and mechanical properties of the device. Additionally, they can also prevent the decomposition or corrosion of the underlying matrix materials, as well as the leakage of toxic substances. By wrapping spring-like fiber electrodes with an SHP outer shell, Gao and co-workers successfully fabricated a stretchable and self-healable SC (Fig. 11d).138 The stretchable property originated from the spring-like fiber and PU; meanwhile, the mechanically self-healing performance originated from the carboxylated PU shell. As illustrated in Fig. 11e, the enclosed area of the CV curve hardly shrunk when the SC was stretched from 0 to 100%, indicating that the self-healing SC could withstand large stresses without affecting its electronic performance. Once the spring-like fiber electrodes were broken, by simply aligning the detached sections with the links, the self-healing carboxylated PU could guide the reconnection process, and thus restore the electrical properties. During the cut/heal cycle, the CV curve of the assembled SC remained a rectangular shape even after the third cycle (Fig. 11f), indicating that the capacitance of the healed SC still remained at a high level. In addition, the three-time healed device still retained 84% capacitance even after 3000 galvanostatic discharge. Utilizing the same SHP for the electrode outer shell, they further reported a high self-healable 3D micro-supercapacitor (MSC).139 The 3D MSC consisted of an MXene (Ti3C2Tx)-graphene composite aerogel electrode, combining the large specific surface area of reduced graphene oxide and high conductivity of the MXene. Consequently, it delivered a large area specific capacitance of 34.6 mF cm−2 and an excellent cycling performance with a capacitance retention of up to 91% over 15000 cycles. Due to the rich interfacial H-bonds in the carboxylated PU supramolecular network, this 3D MSC showed satisfactory self-healing ability in capacitance with a specific capacitance retention of 81.7% after the fifth healing.
3.2.2.2 Polyelectrolytes. In addition to the electrodes, the self-healable electrolyte is another key component that determines the performance of flexible SCs. The purpose of self-healing electrolytes is to restore ionic conductivity. Compared with solid-state electrolytes, polyelectrolytes are ideal materials for constructing self-healing electrolytes because of their high ionic conductivity, adjustable mechanical properties, and good chemical compatibility with electrode components. For example, Huang and co-workers140 designed a dual cross-linked polyelectrolyte for self-healable and highly stretchable SC.
The self-healing polyelectrolyte was prepared by PAA dual cross-linked H-bonds and vinyl hybrid silica nanoparticles, which addressed the intrinsic self-healability and high stretchability issues of an SC. Specifically, it could be easily self-repaired at RT and maintained similar ionic properties to that of the original samples after healing cycles. Meanwhile, the polyelectrolyte could be stretched over 3700% without any cracks. It is worth noting that the assembled SC with this polyelectrolyte completely retained its capacitance even after 20 breaking/healing cycles, which significantly outperformed all the self-healing devices reported at that time. Nevertheless, it only had a narrow electro-chemical window of 0–0.6 V because of the weak ionic conductivity of the polyelectrolyte. This low maximum operating voltage significantly limits its application in the field of high energy density energy storage devices. By introducing methacrylated graphene oxide (MGO) in PAA, Qu and co-workers141 reported the synthesis of a highly conductive and self-healable polyelectrolyte. The addition of MGO not only effectively promoted the formation of H-bonded networks among PAA chains, but also acted as a cross-linking mediator to improve the mechanical properties of the polyelectrolyte (950% stain). Furthermore, the MGO-PAA polyelectrolyte exhibited a high ionic conductivity (∼7.16 S m−1). Consequently, the prepared SC using this polyelectrolyte demonstrated a wider electrochemical window of 0–0.8 V, expectedly promoting the development of high-energy density healable SCs.
Despite the significant progress of self-healing SCs with sandwich-like laminated structures, the various performance drawbacks arising from their structures have not been well addressed. For example, the laminated configuration shows large interfacial contact resistance due to the multiple interfaces among the layers. Moreover, the inevitable relative displacement between multi-layers during continuous stretching bending or bending cycles may lead to the delamination of these multi-layers, which will undoubtedly degrade the SC performance. Compared to the multilayer laminated configuration, all-in-one configured SCs can simplify the tedious assembly process, improve their interfacial contact resistance and energy densities, and prolong the lifetime.182 Here, Wang and co-workers142 reported the preparation of a healable all-in-one configured SC via the in situ polymerization and deposition of nanocomposite electrode materials on a self-healing hydrogel electrolyte (Fig. 11g). The self-healing hydrogel film was synthesized from a physically cross-linked PVA hydrogel with abundant H-bonds, conferring self-healing capability through dynamic H-bonding. Benefiting from the excellent mechanical and electrical performance of the hydrogel, the assembled SC exhibited reliable self-healing capability, good cycle stability, and high capacitance performance. As shown in Fig. 11h, the galvanostatic charge/discharge (GCD) curves of the assembled all-in-one configured SC showed only a slight decrease at different self-healing times after five breaking/healing cycles. Additionally, the self-healing efficiency of this all-in-one SC could still be well maintained within the range of 80% after the 5th healing cycle (Fig. 11i), exhibiting favorable self-healing ability. Benefit from these advantages, this SC is expected to be coupled with wearable devices for use as a portable energy storage device for electronic devices.
3.3 H-Bond cross-linked SHPs for flexible sensing devices
Flexible sensors capable of effectively detecting and perceiving various different stimuli have attracted widespread research interest given that they show great potential for applications in wearable electronics, soft robotics, and the IoT.116,183 However, flexible sensors are highly sensitive to structural and mechanical damage, such as cracks and rupture. Therefore, to extend the lifespan of smart electronics, polymers with high effective self-healing ability and excellent mechanical performance are needed to address these issues. Here, we summarize the main advances in self-healing flexible sensors that target varying stimulus signals including strain/pressure, temperature, and chemical parameters (Table 3).| Polymer matrix | SHP type | Format of damage | Healing conditions | Usage | Characteristics | Ref. |
|---|---|---|---|---|---|---|
| Self-healing strain/pressure sensors | ||||||
| PTMEG-TDI-IP | Homogeneous system | Cut | RT, 6 h | Self-healing substrate | Remains stable over 2000 stretching-releasing cycles | 184 |
| Self-healing polyurea | Microphase separation system | Cut | RT, 15 h/NIR irradiation, 1 min | Dielectric and packaging layer | With a gauge factor of 1.003, response time as low as 32 ms at 1% strain | 101 |
| SHP | Homogeneous system | Cut | RT, 15 s (conductivity) RT, 10 min (mechanical properties) | Self-healing substrate | High conductivity (40 S cm−1), fast conductivity healing (15 s) | 64 |
| C-CNC@CT/ENR | — | Cut | RT, 15 s | Sensing layer | Low strain detection limit of 0.2% high stability (even after healing and bending over 20000 cycles) |
185 |
| CNT/PDMS-MPU0.4-IU0.6 | Microphase separation system | Scratch | RT, 12 h/artificial sweat | Sensing layer | Reliable waterproof operation, can be used as active components in devices | 65 |
| PDMS-MPU0.4-IU0.6 | Microphase separation system | Scratch | RT, 3 d/underwater, 24 h | Dielectric layer | High stretchability (500%) with stable (100 cycles) and reversible low resistance (0–10 Ω) | 50 |
| Self-healing PU | Microphase separation system | Cut | — | Self-healing substrate | Ultrahigh sensitivity (509.8 kPa−1), fast response (67.3 ms), good stability (10000 cycles) |
186 |
| Self-healing temperature sensors | ||||||
| SHP | Homogeneous system | Scratch | RT, 1 h | Self-healing substrate | Resistance decreased ∼225 Ω with increasing temperature for only one degree | 187 |
| SHP | Homogeneous system | Cut | RT, 10 min/50 °C, 5 min | Self-healing encapsulation layer | Conductivity change ∼7% with increasing temperature for only one degree (35–45 °C) | 66 |
| Self-healing PU | Microphase separation system | Cut | 100 °C, — | Self-healing substrate | Bifunctional monitoring of pressure and temperature | 188 |
| Self-healing sweat sensors | ||||||
| Bio-based supramolecular polymer | Homogeneous system | Scratch | RT, 24 h | Self-healing coating | Real-time non-invasive monitoring of Na+ and K+ in human sweat | 67 |
| PDES ionic elastomers | — | Crack | RT, 24 h | Sensing layer | Bimodal detection of human motion and sweat physiological information | 189 |
The direct incorporation of SHPs as substrate/dielectric layers or in combination with functional conductive fillers (such as nickel micro-particles, liquid metal, CNT and AgNWs) as sensing layers is the main direction for preparing self-healing strain/pressure sensors currently. For instance, Yang et al.184 reported a stretchable and self-healing strain sensor for the real-time monitoring of various physiological activities. By reasonably tuning the strength of the dynamic crosslinking bond and the hardness of the molecular segment, the resulting polymer (for sensor substrate) featured a large tensile strain (∼2100%) and high RT self-healing efficiency (∼97% within 6 h). Consequently, the amplitude of the resistance change of the sensor remained stable even during 2000 stretch-release cycles, showing excellent cyclic stability. After cutting, the sensor could recover its sensing ability after healing at RT for 12 h.
In the case of soft capacitive strain-sensors, the dielectric permittivity of dielectric material plays a crucial role in the capacitive sensitivity of soft sensors. Although the dielectric properties of soft polymers can be greatly enhanced by the incorporation of various rigid fillers (e.g., ceramic, metal and carbon-based particles), this is always at the expense of inherent compliance and flexibility. As mentioned before, Fu and co-workers101 developed a self-healing polyurea (SSPUGIT) with ultra-high dielectric permittivity (∼14.57) inspired by the structure of vascular smooth muscles.
The excellent photothermal properties of Galinstan droplets under near-infrared laser irradiation endowed the resultant composite with ultra-fast self-healing capabilities (∼98.13% ± 1.93% within 1 min). The unique combination of low modulus, high self-healing efficiency and high dielectric permittivity makes it an outstanding candidate as a robust dielectric material for capacitive strain sensors. Finally, the authors demonstrated a capacitive strain sensor with excellent strain sensitivity (Fig. 12b) (GF = 1.003) and fast self-healing capability by using SSPUGIT-3 as dielectric and packaging layers (Fig. 12a). As an application demonstration, the sensor was mounted on the elbow of a ping-pong player to detect whether the stroke met the training requirements, and thus correct their incorrect catching actions (Fig. 12c).
 | ||
| Fig. 12 (a) Schematic illustration of the self-healable soft strain-sensor based on SSPUGIT-3 composite. (b) Variation in relative capacitance with tensile strain for original, self-healed and notched soft strain sensors (based on SSPUGIT-3). (c) Relative capacitance response of the wrong and standard ball striking motions. (d) Schematic illustration of the interaction between oligomer chains and nanostructured μNi particles. The pink lines represent the linear and branched polymers forming the randomly branched network, and the blue and purple shapes represent the primary H-bonds formed between the polymer chains. (e) Photograph of the compression-moulded healable electronic composite material, showing the flexibility of the material. (f) Time evolution of the typical electrical healing process of the resulting composite material. (g) Application of the RT self-healing electronic composite as electronic sensor skin. | ||
Self-healing elastomers incorporated with inorganic conductive fillers are widely used as sensing layers for resistive strain/pressure sensors due to their simple process and low cost. Bao and co-workers64 pioneered the development of an intrinsically self-healing conductive composite material (Fig. 12e), which was formed with an H-bonded polymer matrix and nickel microparticles (μNi particles). The preparation process preserved the original nano-features of the μNi particles, which were dispersed in the H-bonded polymer network, and then thermally cross-linked with urea (Fig. 12d). The prepared composite had high conductive (up to 40 S cm−1) and showed mechanical and electrical self-healing properties under ambient conditions. As shown in Fig. 12f, 90% of its original conductivity could be restored within 15 s of incurring mechanical damage at RT. Furthermore, the composite material was also pressure- and flexion-sensitive by maintaining the volume loading of the μNi particles just below the percolation threshold (15 vol% loading). To demonstrate the potential of this self-healing composite for application as E-skin, the authors integrated the flexion and tactile sensors into a puppet model. Consequently, they could detect both arm bending and hand shaking when laminated on the elbow and palm, respectively (Fig. 12g).
Despite brilliant advances, real-time (≤30 s) healing remains a hurdle for H-bonding-based SHPs, and their low healing efficiency significantly reduces their reliability in flexible sensors. Here, Yu and co-workers185 created a highly sensitive and self-healing resistive-type strain sensor for human-machine interactions using a conductive supramolecular multiple H-bonding elastomer (MHBE) as the sensing layer. The presence of multiple H-bonding interactions in MHBE provided extremely fast (ca. 15 s) and repeatable self-healing ability with high healing efficiency (93% after the third healing process). Due to the uniform nanostructured conductive network in the sensor, it could precisely detect tiny human movements. The sensor demonstrated highly recognizable and reliable signals even after more than 20000 cycles of cut healing and bending.
In addition, the detection limit of this sensor was as low as 0.2%. Meanwhile, the MHBE-based sensor could help a pronunciation-impaired person to speak in real time when integrated in a signal processing software, showing great potential as a human-machine interaction system.
Unlike solid sensing materials, liquids are inherently self-healable. With the advantages of thermal sensitivity, electrochemical stability, and negligible volatility, ionic liquids are considered as a type of promising element for the fabrication of self-healing temperature sensors. However, the approach of obtaining conductivity by encapsulating ionic liquids is extremely challenging owing to the high risk of leakage. Here, Wang et al.66 reported a self-healing temperature sensor using an SHP that rapidly heals under mild conditions as an encapsulated channel to load thermally sensitive ionic liquids (Fig. 13a). The SHP was synthesized by a modified procedure derived from Leibler's method40 and exhibited excellent self-healing properties at RT. Based on the capillary effect, liquid leakage could be effectively avoided by optimizing the size of the SHP microchannel (Fig. 13b). Given that the conductivity of ionic liquids increases at high temperatures, the sensor exhibited outstanding thermal sensitivity in the temperature range of 35–45 °C. Additionally, the conductivity change could be increased by ∼7% for a temperature increase of only one degree, demonstrating promising clinical diagnostic potential. It is worth noting that the temperature sensor still exhibited repeatable and stable thermal sensitivity after damage and self-healing (Fig. 13c).
 | ||
| Fig. 13 (a) Schematic illustration of SHP based on H-bonding interaction (left). The blue lines represent the linear and branched polymers forming the randomly branched network, and the red lines represent the primary H-bonds formed between amide moieties. Photograph of the SHP and a damaged polymer coupon after healing for 5 min at room temperature (right). (b) Force analysis diagram of the ionic liquid in the vertical microchannel. (c) Thermal sensing performance of the self-healable sensor before and after healing. (d) Self-healable bimodal sensor based on bioderived PDES ionic elastomer. (e) Real-time current healing process of the bimodal sensor during repetitive self-healing cycles. | ||
Given that most sensing platforms are also sensitive to temperature, we predict that the integration of highly sensitive and accurate temperature sensors will be essential in future platforms. For intelligent E-skin, combining the ability to detect and recognize multiple complex stimuli in the environment with self-healing functions remains a great challenge. With PU and PU@CNTs as the substrate and sensing layer, respectively, Shen et al.188 developed a self-healable bifunctional sensing platform by integrating pressure and temperature sensor. Due to the presence of a large number of reversible dynamic H-bonds in the matrix, the as-prepared devices exhibited stable self-healing ability after multiple damage. Even after the third self-healing process, both the pressure response (relative capacitance) and temperature response current of the single-unit device remained basically unchanged. Notably, the sensors in the device were sensitive only to their associated external stimuli and were not affected by other stimuli, which further improved their stability in practical applications. The fabricated E-skin could simultaneously image the pressure and temperature distribution, creating a new opportunity in the field of machine interfaces and biomonitoring devices.
Owing to the need for close contact with human skin, the biocompatibility of SHPs (as self-healing substrates/sensing layer for wearable chemical sensors) is a crucial consideration. Based on the biomass-derived and nontoxic monomers citric acid and succinic acid, Choi and co-workers67 synthesized a bio-based supramolecular SHP (PCSC) through a one-step melt polycondensation process. The robust PCSC exhibited instantaneous healing (60 s) at RT together with remarkable self-healing efficiency (∼92%). Taking advantage of these features, a flexible and weaveable self-healable sweat sensing yarn was realized by coating carbon fiber threads with PCSC as a self-healing coating. Moreover, the PCSC could also be used as an insulation layer between sweat electrolytes and conductive carbon fiber thread wires to ensure a reliable sensor performance. Consequently, the self-healing sweat sensors enabled reliable and accurate measurements of Na+ and K+ with an impressive healing efficiency (>97% after healing 30 s). Finally, a wearable sweat-sensor system was achieved by integrating the multi-ion sensors with a wireless flexible circuit board, which was capable of non-invasively monitoring electrolyte ions in real time and transmitting the data to a smart phone. Based on the dynamic reversibility of the H-bonds, the damaged sweat sensors could be operated normally after healing within about 20 s at RT.
The combination of self-healing capability, multimodal response, and biocompatibility is an objective requirement for the future development of wearable and implantable flexible sensor devices.
Although several stretchable bimodal sensors have been successfully developed by coating, wrapping, or embedding the working electrodes (e.g., metal nanowires/nanosheets) in polymer substrates,191,192 the mechanical mismatch between the substrate and electrode reduces the stability and reliability of the multifunctional responses. Moreover, the required petroleum-based materials and complex chemical synthesis processes may greatly compromise the biocompatibility of sensing devices. Based on bio-derived monomer itaconic acid, a PDES ionic elastomer was demonstrated for the preparation of fully bio-based self-healing bimodal sensors189 (Fig. 13d). The PDES composite was stretchable and biocompatible (cell viability of above 87%) and could self-heal without other conditions at RT because of the synergistic physical network and H-bonding network constructed by polydopamine-coated cellulose nanocrystal. In addition, the ionic migration of ChCl inside PDES conferred the bimodal sensors with stress-electric sensing and strain detection capabilities without additional working electrodes, thus effectively solving the problem of mechanical mismatch. As shown in Fig. 13e, after repeated healing cycles, the current value still rapidly recovered to the initial value, showing that the sensor was capable of real-time electrical self-healing. Combined with the sensitivity of anthocyanin to pH, this self-healing sensor achieved the bimodal detection of physiological information of human motion and sweat.
4 Conclusions and prospects
Inspired by the self-healable phenomenon of biological organisms, SHPs have developed rapidly over the past few decades, especially H-bonding-based SHPs. In this review, we summarized the advances and design strategies of high-performance SHPs based on different H-bonding types, including H-bonding motifs and excessive H-bonding. Meanwhile, the effects of structural design strategies of SHPs on their mechanical performance and healing efficiency are described in detail. In the last part, we also discussed how to make use of these SHPs for the fabrication of self-healable electronic devices, with the focus on promising topics, including energy harvesting devices, energy storage devices, and flexible sensing devices.It is evident that increasing efforts have been devoted to SHPs based on H-bond during the past few decades. However, this field is still far from maturity. Firstly, these polymers often require complex synthetic pathways, partly involving a large amount of organic solvents, which not only limits their scalable preparation but also poses irreversible damage to future global environmental sustainability. Therefore, SHPs using green, bio-friendly-based materials and simple synthetic processes should be further explored in the future. Secondly, another drawback of H-bonding-based SHPs is that H-bonds are susceptible to water molecules, resulting in a gradual decrease in mechanical strength or even loss of self-healing capabilities under high humidity conditions. Thus, designing and developing reversible, robust bond underwater SHPs, and how to introduce bonds in SHPs remains a long-term issue. Thirdly, many studies only focus on the synthesis of SHPs and characterization of their self-healing performance, and only a few studies attempt to derive consistent and quantitative relations between the H-bonding cluster distribution/topology and mechanical/self-healing properties. Thus, a more profound understanding of the structure–property relationships of SHPs is urgently needed to explore more advanced “smart” SHPs.
In terms of self-healable electronic devices, a significant challenge is balancing and optimizing multiple properties for different applications. For energy conversion/storage devices, self-healable TENGs, perovskite solar cells, Li-ion batteries, and SCs have been successfully shown by applying the self-healable concept in the design of friction layers, binders, electrodes, and electrolytes. However, it should be noted that most of the reported SHPs thus far are far from practical applications. Specifically, most SHPs require external energy such as heat to trigger or speed up the healing process, which is difficult to achieve during the actual operation of the devices. Therefore, polymeric materials capable of self-healing under mild conditions or RT are highly desirable. In addition, another challenge lies in the design of SHPs with suitable self-healing capabilities, while possessing high electrical conductivity, which is certainly a major research direction for self-healing electronics in the future. Alternatively, SHPs for energy storage conversion/devices are usually more expensive than commercial polymers due to the need for more synthesis steps and chemical modification processes. To further shorten the commercialization process, reducing the manufacturing cost, while ensuring a high performance is also a key issue that needs to be addressed urgently.
In the case of H-bonding-based healable flexible sensing systems, relatively few works have been reported to date due to the significant challenges in material design and integration. In particular, the patterning of small-sized SHPs required for embedded electronic applications still remains a big challenge, which will directly hinder the further development of the soft electronics field. Moreover, signal transfer methods, calibration and signal interference, selectivity, accuracy and compatibility are some of the main challenges facing the field of self-healable flexible sensors. Accordingly, to address these issues, huge efforts are still needed irrespective of the work in the self-healing area. Considering the need for direct contact with human skin or tissues and long-term monitoring of physiological signals, the next generation of flexible sensing devices requires significant efforts in the development of biocompatibility, comfort, reliability, and cyclic stability. Fortunately, the rapid advances and current trends seen in the development of high-performance sensing materials and components look promising and exhibit great potential in the future market for self-healable flexible sensing devices. Finally, although there are a few reports of H-bonding-based SHPs being used in emerging fields such as aerospace and biomedicine, more promising applications should be explored in the future. We believe that these advanced SHPs may find valuable applications such as drug-delivery carriers, artificial muscles, tissue engineering, and other biomedical applications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
National Natural Science Foundation of China (No. 22275094, 22275093, 52203059), Natural Science Foundation of Jiangsu Province (No. BK20220420) and Postgraduate Research & Practice Innovation Program of Jiangsu Province (KYCX23_1159, SJCX23_0328) are acknowledged with gratitude.References
- M. Burnworth, L. Tang, J. R. Kumpfer, A. J. Duncan, F. L. Beyer, G. L. Fiore, S. J. Rowan and C. Weder, Nature, 2011, 472, 334–337 CrossRef CAS PubMed.
- Y. Yang and M. W. Urban, Chem. Soc. Rev., 2013, 42, 7446–7467 RSC.
- B. Ghosh and M. W. Urban, Science, 2009, 323, 1458–1460 CrossRef CAS PubMed.
- Y. Yanagisawa, Y. Nan, K. Okuro and T. Aida, Science, 2018, 359, 72–76 CrossRef CAS PubMed.
- X. Wang, Y. Li, Y. Qian, H. Qi, J. Li and J. Sun, Adv. Mater., 2018, 30, 1803854 CrossRef PubMed.
- W. Guo, Y. Jia, K. Tian, Z. Xu, J. Jiao, R. Li, Y. Wu, L. Cao and H. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 21046–21054 CrossRef CAS PubMed.
- Y. Zhu, K. Cao, M. Chen and L. Wu, ACS Appl. Mater. Interfaces, 2019, 11, 33314–33322 CrossRef CAS PubMed.
- W. Mai, Q. Yu, C. Han, F. Kang and B. Li, Adv. Funct. Mater., 2020, 30, 1909912 CrossRef CAS.
- J. C. Yang, J. Mun, S. Y. Kwon, S. Park, Z. Bao and S. Park, Adv. Mater., 2019, 31, 1904765 CrossRef CAS PubMed.
- D. Chen, D. Wang, Y. Yang, Q. Huang, S. Zhu and Z. Zheng, Adv. Energy Mater., 2017, 7, 1700890 CrossRef.
- Q. Li, C. Ding, W. Yuan, R. Xie, X. Zhou, Y. Zhao, M. Yu, Z. Yang, J. Sun, Q. Tian, F. Han, H. Li, X. Deng, G. Li and Z. Liu, Adv. Fiber Mater., 2021, 3, 302–311 CrossRef CAS.
- S. Terryn, J. Langenbach, E. Roels, J. Brancart, C. Bakkali-Hassani, Q.-A. Poutrel, A. Georgopoulou, T. George Thuruthel, A. Safaei, P. Ferrentino, T. Sebastian, S. Norvez, F. Iida, A. W. Bosman, F. Tournilhac, F. Clemens, G. Van Assche and B. Vanderborght, Mater. Today, 2021, 47, 187–205 CrossRef CAS.
- S. Bonardd, M. Nandi, J. I. Hernandez Garcia, B. Maiti, A. Abramov and D. Diaz Diaz, Chem. Rev., 2023, 123, 736–810 CrossRef CAS PubMed.
- C. Shi, Z. Zou, Z. Lei, P. Zhu, W. Zhang and J. Xiao, Sci. Adv., 2022, 6, eabd0202 CrossRef PubMed.
- Y. Huang, P. Wang, W. Tan, W. Hao, L. Ma, J. Wang, T. Liu, F. Zhang, C. Ren, W. Liu and D. Zhang, J. Mater. Sci. Technol., 2022, 107, 34–42 CrossRef CAS.
- Y. Yang, X. Ding and M. W. Urban, Prog. Polym. Sci., 2015, 49–50, 34–59 CrossRef CAS.
- H. Jinno, T. Yokota, M. Koizumi, W. Yukita, M. Saito, I. Osaka, K. Fukuda and T. Someya, Nat. Commun., 2021, 12, 2234 CrossRef CAS PubMed.
- W. Ma, Y. Li, S. Gao, J. Cui, Q. Qu, Y. Wang, C. Huang and G. Fu, ACS Appl. Mater. Interfaces, 2020, 12, 23644–23654 CrossRef CAS PubMed.
- W. Ma, Y. Ding, Y. Li, S. Gao, Z. Jiang, J. Cui, C. Huang and G. Fu, J. Membr. Sci., 2021, 634, 119402 CrossRef CAS.
- J. D. V. Aaron, M. Kushner, G. A. Williams and Z. Guan, J. Am. Chem. Soc., 2009, 131, 8766–8768 CrossRef PubMed.
- Y. Chen, A. M. Kushner, G. A. Williams and Z. Guan, Nat. Chem., 2012, 4, 467–472 CrossRef CAS PubMed.
- M. J. Webber, E. A. Appel, E. W. Meijer and R. Langer, Nat. Mater., 2016, 15, 13–26 CrossRef CAS PubMed.
- H. Wu, W. Li, M. Zhao, S. Lu, L. Huang and L. Chen, J. For. Eng., 2020, 5, 11–17 CrossRef.
- N. Roy, B. Bruchmann and J. M. Lehn, Chem. Soc. Rev., 2015, 44, 3786–3807 RSC.
- J. F. Patrick, M. J. Robb, N. R. Sottos, J. S. Moore and S. R. White, Nature, 2016, 540, 363–370 CrossRef CAS PubMed.
- X. Fan, L. Zhang, F. Dong, H. Liu and X. Xu, Carbohydr. Polym., 2023, 308, 120654 CrossRef CAS PubMed.
- X. Fan, X. Yang, S. Wang, S. Wang, X. Xu, J. Jiang, S. Shang and Z. Song, Carbohydr. Polym., 2021, 273, 118529 CrossRef CAS PubMed.
- Y. Guan, H. Li, S. Zhang and W. Niu, Adv. Funct. Mater., 2023, 33, 2215055 CrossRef CAS.
- Z. W. An, R. Xue, K. Ye, H. Zhao, Y. Liu, P. Li, Z. M. Chen, C. X. Huang and G. H. Hu, Nanoscale, 2023, 15, 6505–6520 RSC.
- W. Niu, X. Cao, Y. Wang, B. Yao, Y. Zhao, J. Cheng, S. Wu, S. Zhang and X. He, Adv. Funct. Mater., 2021, 31, 2009017 CrossRef CAS.
- Y. Deng, I. Hussain, M. Kang, K. Li, F. Yao, S. Liu and G. Fu, Chem. Eng. J., 2018, 353, 900–910 CrossRef CAS.
- Y. Shi, M. Wang, C. Ma, Y. Wang, X. Li and G. Yu, Nano Lett., 2015, 15, 6276–6281 CrossRef CAS PubMed.
- T. L. Sun, T. Kurokawa, S. Kuroda, A. B. Ihsan, T. Akasaki, K. Sato, M. A. Haque, T. Nakajima and J. P. Gong, Nat. Mater., 2013, 12, 932–937 CrossRef CAS PubMed.
- J. Fox, J. J. Wie, B. W. Greenland, S. Burattini, W. Hayes, H. M. Colquhoun, M. E. Mackay and S. J. Rowan, J. Am. Chem. Soc., 2012, 134, 5362–5368 CrossRef CAS PubMed.
- X. Li, H. Zhang, P. Zhang and Y. Yu, Chem. Mater., 2018, 30, 3752–3758 CrossRef CAS.
- M. W. Urban, D. Davydovich, Y. Yang, T. Demir, Y. Zhang and L. Casabianca, Science, 2018, 362, 220–225 CrossRef CAS PubMed.
- S. Wang and M. W. Urban, Nat. Rev. Mater., 2020, 5, 562–583 CrossRef CAS.
- D. Hua, S. Gao, M. Zhang, W. Ma and C. Huang, Carbohydr. Polym., 2020, 247, 116743 CrossRef CAS PubMed.
- S. Li, J. Liu, Z. Wei, Q. Cui, X. Yang, Y. Yang and X. Zhang, Adv. Funct. Mater., 2022, 2210441–2210450 Search PubMed.
- P. Cordier, F. Tournilhac, C. Soulie-Ziakovic and L. Leibler, Nature, 2008, 451, 977–980 CrossRef CAS PubMed.
- C. Wang, H. Wu, Z. Chen, M. T. McDowell, Y. Cui and Z. Bao, Nat. Chem., 2013, 5, 1042–1048 CrossRef CAS PubMed.
- Y. Sun, J. Lopez, H. W. Lee, N. Liu, G. Zheng, C. L. Wu, J. Sun, W. Liu, J. W. Chung, Z. Bao and Y. Cui, Adv. Mater., 2016, 28, 2455–2461 CrossRef CAS PubMed.
- J. Lopez, Z. Chen, C. Wang, S. C. Andrews, Y. Cui and Z. Bao, ACS Appl. Mater. Interfaces, 2016, 8, 2318–2324 CrossRef CAS PubMed.
- R. P. Sijbesma, F. H. Beijer, L. Brunsveld, B. J. B. Folmer, J. H. K. K. Hirschberg, R. F. M. Lange, J. K. L. Lowe and E. W. Meijer, Science, 1997, 278, 1601–1604 CrossRef CAS PubMed.
- T. Aida, E. W. Meijer and S. I. Stupp, Science, 2012, 335, 813–817 CrossRef CAS PubMed.
- D. W. Balkenende, C. A. Monnier, G. L. Fiore and C. Weder, Nat. Commun., 2016, 7, 10995 CrossRef PubMed.
- S. Yoshida, H. Ejima and N. Yoshie, Adv. Funct. Mater., 2017, 27, 1701670 CrossRef.
- Y. Song, Y. Liu, T. Qi and G. L. Li, Angew. Chem., Int. Ed., 2018, 57, 13838–13842 CrossRef CAS PubMed.
- M. Liu, P. Liu, G. Lu, Z. Xu and X. Yao, Angew. Chem., Int. Ed., 2018, 57, 11242–11246 CrossRef CAS PubMed.
- J. Kang, D. Son, G. N. Wang, Y. Liu, J. Lopez, Y. Kim, J. Y. Oh, T. Katsumata, J. Mun, Y. Lee, L. Jin, J. B. Tok and Z. Bao, Adv. Mater., 2018, 30, 1706846 CrossRef PubMed.
- D. Wang, J. Xu, J. Chen, P. Hu, Y. Wang, W. Jiang and J. Fu, Adv. Funct. Mater., 2019, 30, 1907109 CrossRef.
- M. W. M. Tan, G. Thangavel and P. S. Lee, Adv. Funct. Mater., 2021, 31, 2103097 CrossRef CAS.
- J. Xu, J. Chen, Y. Zhang, T. Liu and J. Fu, Angew. Chem., Int. Ed., 2021, 60, 7947–7955 CrossRef CAS PubMed.
- J. Wu, L. H. Cai and D. A. Weitz, Adv. Mater., 2017, 29, 1702616 CrossRef PubMed.
- K. Parida, G. Thangavel, G. Cai, X. Zhou, S. Park, J. Xiong and P. S. Lee, Nat. Commun., 2019, 10, 2158 CrossRef PubMed.
- W. Sun, N. Luo, Y. Liu, H. Li and D. Wang, ACS Appl. Mater. Interfaces, 2022, 14, 10498–10507 CrossRef CAS PubMed.
- M. P. Kim, Y.-R. Kim and H. Ko, Nano Energy, 2022, 92, 106704 CrossRef CAS.
- C. Ge, X. Liu, Z. Yang, H. Li, W. Niu, X. Liu and Q. Dong, Angew. Chem., Int. Ed., 2022, 61, 202116602 Search PubMed.
- S. Kee, M. A. Haque, D. Corzo, H. N. Alshareef and D. Baran, Adv. Funct. Mater., 2019, 29, 1905426 CrossRef CAS.
- Z. Chen, C. Wang, J. Lopez, Z. Lu, Y. Cui and Z. Bao, Adv. Energy Mater., 2015, 5, 1401826 CrossRef.
- T. Munaoka, X. Yan, J. Lopez, J. W. F. To, J. Park, J. B. H. Tok, Y. Cui and Z. Bao, Adv. Energy Mater., 2018, 8, 1703138 CrossRef.
- H. Wang, B. Zhu, W. Jiang, Y. Yang, W. R. Leow, H. Wang and X. Chen, Adv. Mater., 2014, 26, 3638–3643 CrossRef CAS PubMed.
- H. Sun, X. You, Y. Jiang, G. Guan, X. Fang, J. Deng, P. Chen, Y. Luo and H. Peng, Angew. Chem., Int. Ed., 2014, 53, 9680–9685 CrossRef.
- B. C. Tee, C. Wang, R. Allen and Z. Bao, Nat. Nanotechnol., 2012, 7, 825–832 CrossRef CAS PubMed.
- D. Son, J. Kang, O. Vardoulis, Y. Kim, N. Matsuhisa, J. Y. Oh, J. W. To, J. Mun, T. Katsumata, Y. Liu, A. F. McGuire, M. Krason, F. Molina-Lopez, J. Ham, U. Kraft, Y. Lee, Y. Yun, J. B. Tok and Z. Bao, Nat. Nanotechnol., 2018, 13, 1057–1065 CrossRef CAS PubMed.
- Y. He, S. Liao, H. Jia, Y. Cao, Z. Wang and Y. Wang, Adv. Mater., 2015, 27, 4622–4627 CrossRef CAS PubMed.
- J. H. Yoon, S.-M. Kim, Y. Eom, J. M. Koo, H.-W. Cho, T. J. Lee, K. G. Lee, H. J. Park, Y. K. Kim, H.-J. Yoo, S. Y. Hwang, J. Park and B. G. Choi, ACS Appl. Mater. Interfaces, 2019, 11, 46165–46175 CrossRef CAS PubMed.
- D. Y. Wu, S. Meure and D. Solomon, Prog. Polym. Sci., 2008, 33, 479–522 CrossRef CAS.
- M. Zhang and M. Rong, Sci. China: Chem., 2012, 55, 648–676 CrossRef CAS.
- P. Song and H. Wang, Adv. Mater., 2020, 32, 1901244 CrossRef CAS PubMed.
- L. Zhai, A. Narkar and K. Ahn, Nano Today, 2020, 30, 100826 CrossRef CAS.
- M. Zhu, J. Yu, Z. Li and B. Ding, Angew. Chem., Int. Ed., 2022, 134, e202208949 Search PubMed.
- L. Yang, X. Tan, Z. Wang and X. Zhang, Chem. Rev., 2015, 115, 7196–7239 CrossRef CAS PubMed.
- N. Zhang, Z. Pan, C. Li, J. Wang, Y. Jin, S. Song, M. Pan and J. Yuan, Polymer, 2022, 246, 124778 CrossRef CAS.
- M. Guo, L. M. Pitet, H. M. Wyss, M. Vos, P. Y. Dankers and E. W. Meijer, J. Am. Chem. Soc., 2014, 136, 6969–6977 CrossRef CAS PubMed.
- J. Tellers, S. Canossa, R. Pinalli, M. Soliman, J. Vachon and E. Dalcanale, Macromolecules, 2018, 51, 7680–7691 CrossRef CAS.
- M. Li, R. Zhang, X. Li, Q. Wu, T. Chen and P. Sun, Polymer, 2018, 148, 127–137 CrossRef CAS.
- C.-J. Fan, Z.-C. Huang, B. Li, W.-X. Xiao, E. Zheng, K.-K. Yang and Y.-Z. Wang, Sci. China: Mater., 2019, 62, 1188–1198 CAS.
- X. Yan, Z. Liu, Q. Zhang, J. Lopez, H. Wang, H. C. Wu, S. Niu, H. Yan, S. Wang, T. Lei, J. Li, D. Qi, P. Huang, J. Huang, Y. Zhang, Y. Wang, G. Li, J. B. Tok, X. Chen and Z. Bao, J. Am. Chem. Soc., 2018, 140, 5280–5289 CrossRef CAS PubMed.
- B. Wu, Z. Liu, Y. Lei, Y. Wang, Q. Liu, A. Yuan, Y. Zhao, X. Zhang and J. Lei, Polymer, 2020, 186, 122003 CrossRef CAS.
- S. Chen, X. Bi, L. Sun, J. Gao, P. Huang, X. Fan, Z. You and Y. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 20591–20599 CrossRef CAS PubMed.
- Y. Wu, L. Wang, X. Zhao, S. Hou, B. Guo and P. X. Ma, Biomaterials, 2016, 104, 18–31 CrossRef CAS PubMed.
- X. Zhu, W. Zheng, H. Zhao and L. Wang, J. Mater. Chem. A, 2021, 9, 20737–20747 RSC.
- J. Hentschel, A. M. Kushner, J. Ziller and Z. Guan, Angew. Chem., Int. Ed., 2012, 51, 10561–10565 CrossRef CAS PubMed.
- M. Wei, M. Zhan, D. Yu, H. Xie, M. He, K. Yang and Y. Wang, ACS Appl. Mater. Interfaces, 2015, 7, 2585–2596 CrossRef CAS PubMed.
- Q. Jin, R. Du, H. Tang, Y. Zhao, W. Peng, Y. Li, J. Zhang, T. Zhu, X. Huang, D. Kong, Y. He, T. Bao, D. Kong, X. Wang, R. Wang, Q. Zhang and X. Jia, Angew. Chem., Int. Ed., 2023, 135, e202305282 CrossRef.
- B. Joseph, V. K. Sagarika, C. Sabu, N. Kalarikkal and S. Thomas, J. Bioresour. Bioprod., 2020, 5, 223–237 CrossRef CAS.
- Z. Zhang, J. Luo, S. Zhao, S. Ge, J.-M. Y. Carrillo, J. K. Keum, C. Do, S. Cheng, Y. Wang, A. P. Sokolov and P.-F. Cao, Matter, 2022, 5, 237–252 CrossRef CAS.
- P. Wei and E. B. Pentzer, Matter, 2022, 5, 2479–2481 CrossRef CAS.
- H. Chen, Z. Sun, H. Lin, C. He and D. Mao, Adv. Funct. Mater., 2022, 32, 2204263 CrossRef CAS.
- Y. Meng, W. Xu, M. R. Newman, D. S. W. Benoit and M. Anthamatten, Adv. Funct. Mater., 2019, 29, 1903721 CrossRef.
- Y. Yao, Z. Xu, B. Liu, M. Xiao, J. Yang and W. Liu, Adv. Funct. Mater., 2020, 31, 2006944 CrossRef.
- F. Sun, J. Xu, T. Liu, F. Li, Y. Poo, Y. Zhang, R. Xiong, C. Huang and J. Fu, Mater. Horiz., 2021, 8, 3356–3367 RSC.
- H. Wang, H. Liu, Z. Cao, W. Li, X. Huang, Y. Zhu, F. Ling, H. Xu, Q. Wu, Y. Peng, B. Yang, R. Zhang, O. Kessler, G. Huang and J. Wu, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 11299–11305 CrossRef CAS PubMed.
- R. Hoogenboom, Angew. Chem., Int. Ed., 2012, 51, 11942–11944 CrossRef CAS PubMed.
- Y. Yang, D. Davydovich, C. C. Hornat, X. Liu and M. W. Urban, Chem., 2018, 4, 1928–1936 CAS.
- S. M. Kim, H. Jeon, S. H. Shin, S. A. Park, J. Jegal, S. Y. Hwang, D. X. Oh and J. Park, Adv. Mater., 2018, 30, 1705145 CrossRef PubMed.
- Y. Chen and Z. Guan, Polymer, 2015, 69, 249–254 CrossRef CAS.
- Y. Chen and Z. Guan, Chem. Commun., 2014, 50, 10868–10870 RSC.
- D. Wang, Z. Wang, S. Ren, J. Xu, C. Wang, P. Hu and J. Fu, Mater. Horiz., 2021, 8, 2238–2250 RSC.
- F. Sun, L. Liu, T. Liu, X. Wang, Q. Qi, Z. Hang, K. Chen, J. Xu and J. Fu, Nat. Commun., 2023, 14, 130 CrossRef CAS PubMed.
- Z. Li, Y. L. Zhu, W. Niu, X. Yang, Z. Jiang, Z. Y. Lu, X. Liu and J. Sun, Adv. Mater., 2021, 33, 2101498 CrossRef CAS PubMed.
- T. Liu, C. Li, H. Yao, F. Sun, L. Wang, B. Yao, J. Xu and J. Fu, Mater. Horiz., 2023 10.1039/d3mh00358b.
- J. Xu, Y. Li, T. Liu, D. Wang, F. Sun, P. Hu, L. Wang, J. Chen, X. Wang, B. Yao and J. Fu, Adv. Mater., 2023, 2300937, DOI:10.1002/adma.202300937.
- J. Xu, P. Chen, J. Wu, P. Hu, Y. Fu, W. Jiang and J. Fu, Chem. Mater., 2019, 31, 7951–7961 CrossRef CAS.
- F. Sordo, S.-J. Mougnier, N. Loureiro, F. Tournilhac and V. Michaud, Macromolecules, 2015, 48, 4394–4402 CrossRef CAS.
- L. Laysandra, C.-H. Chuang, S. Kobayashi, A.-N. Au-Duong, Y.-H. Cheng, Y.-T. Li, M. M. Mburu, T. Isono, T. Satoh and Y.-C. Chiu, ACS Appl. Polym. Mater., 2020, 2, 5432–5443 CrossRef CAS.
- R. a Li, T. Fan, G. Chen, K. Zhang, B. Su, J. Tian and M. He, Chem. Mater., 2020, 32, 874–881 CrossRef CAS.
- R. A. Li, G. Chen, T. Fan, K. Zhang and M. He, J. Mater. Chem. A, 2020, 8, 5056–5061 RSC.
- Z. Zhou, S. Chen, X. Xu, Y. Chen, L. Xu, Y. Zeng and F. Zhang, Prog. Org. Coat., 2021, 154, 106213 CrossRef CAS.
- C. H. Li, C. Wang, C. Keplinger, J. L. Zuo, L. Jin, Y. Sun, P. Zheng, Y. Cao, F. Lissel, C. Linder, X. Z. You and Z. Bao, Nat. Chem., 2016, 8, 618–624 CrossRef CAS PubMed.
- Y. Cao, H. Wu, S. I. Allec, B. M. Wong, D.-S. Nguyen and C. Wang, Adv. Mater., 2018, 30, 1804602 CrossRef PubMed.
- Y. Cao, T. G. Morrissey, E. Acome, S. I. Allec, B. M. Wong, C. Keplinger and C. Wang, Adv. Mater., 2017, 29, 1605099 CrossRef PubMed.
- F. Dong, X. Yang, L. Guo, Y. Qian, P. Sun, Z. Huang, X. Xu and H. Liu, J. Colloid Interface Sci., 2023, 631, 239–248 CrossRef CAS PubMed.
- X. Yang, B. Zhang, J. Li, M. Shen, H. Liu, X. Xu and S. Shang, Carbohydr. Polym., 2023, 313, 120813 CrossRef CAS PubMed.
- M. Khatib, O. Zohar and H. Haick, Adv. Mater., 2021, 33, 2004190 CrossRef CAS PubMed.
- Y. Zhou, L. Li, Z. Han, Q. Li, J. He and Q. Wang, Chem. Rev., 2023, 123, 558–612 CrossRef CAS PubMed.
- H. Chen, J. J. Koh, M. Liu, P. Li, X. Fan, S. Liu, J. C. C. Yeo, Y. Tan, B. C. K. Tee and C. He, ACS Appl. Mater. Interfaces, 2020, 12, 31975–31983 CrossRef CAS PubMed.
- H. Li, F. Xu, J. Wang, J. Zhang, H. Wang, Y. Li and J. Sun, Nano Energy, 2023, 108, 108243 CrossRef CAS.
- B. P. Finkenauer, Y. Gao, X. Wang, Y. Tian, Z. Wei, C. Zhu, D. J. Rokke, L. Jin, L. Meng, Y. Yang, L. Huang, K. Zhao and L. Dou, Cell Rep. Phys. Sci., 2021, 2, 100320 CrossRef CAS.
- C. Lu, Z. Ling, C. Wang, J. Wang, Q. Yong and F. Chu, Composites, Part B, 2022, 228, 109428 CrossRef CAS.
- C. Li, P. Wang and D. Zhang, Nano Energy, 2023, 109, 108285 CrossRef CAS.
- M. Feng, Y. Wu, Y. Feng, Y. Dong, Y. Liu, J. Peng, N. Wang, S. Xu and D. Wang, Nano Energy, 2022, 93, 106835 CrossRef CAS.
- J. J. Yoo, G. Seo, M. R. Chua, T. G. Park, Y. Lu, F. Rotermund, Y. K. Kim, C. S. Moon, N. J. Jeon, J. P. Correa-Baena, V. Bulovic, S. S. Shin, M. G. Bawendi and J. Seo, Nature, 2021, 590, 587–593 CrossRef CAS PubMed.
- G.-H. Kim and D. S. Kim, Joule, 2021, 5, 1033–1035 CrossRef.
- J. Y. Oh, D. Son, T. Katsumata, Y. Lee, Y. Kim, J. Lopez, H.-C. Wu, J. Kang, J. Park, X. Gu, J. Mun, N. G.-J. Wang, Y. Yin, W. Cai, Y. Yun, J. B.-H. Tok and Z. Bao, Sci. Adv., 2019, 5, eaav3097 CrossRef CAS PubMed.
- Y. Jia, Q. Jiang, H. Sun, P. Liu, D. Hu, Y. Pei, W. Liu, X. Crispin, S. Fabiano, Y. Ma and Y. Cao, Adv. Mater., 2021, 33, 2102990 CrossRef CAS PubMed.
- D. Kim, S. Hyun and S. M. Han, J. Mater. Chem. A, 2018, 6, 11353–11361 RSC.
- Z. Xu, J. Yang, T. Zhang, Y. Nuli, J. Wang and S.-I. Hirano, Joule, 2018, 2, 950–961 CrossRef CAS.
- G. Zhang, Y. Yang, Y. Chen, J. Huang, T. Zhang, H. Zeng, C. Wang, G. Liu and Y. Deng, Small, 2018, 14, 1801189 CrossRef PubMed.
- R. Gao, Q. Zhang, Y. Zhao, Z. Han, C. Sun, J. Sheng, X. Zhong, B. Chen, C. Li, S. Ni, Z. Piao, B. Li and G. Zhou, Adv. Funct. Mater., 2021, 32, 2110313 CrossRef.
- G. Zheng, C. Wang, A. Pei, J. Lopez, F. Shi, Z. Chen, A. D. Sendek, H.-W. Lee, Z. Lu, H. Schneider, M. M. Safont-Sempere, S. Chu, Z. Bao and Y. Cui, ACS Energy Lett., 2016, 1, 1247–1255 CrossRef CAS.
- G. Wang, C. Chen, Y. Chen, X. Kang, C. Yang, F. Wang, Y. Liu and X. Xiong, Angew. Chem., Int. Ed., 2020, 59, 2071–2076 CrossRef.
- B. Zhou, D. He, J. Hu, Y. Ye, H. Peng, X. Zhou, X. Xie and Z. Xue, J. Mater. Chem. A, 2018, 6, 11725–11733 RSC.
- N. Wu, Y. R. Shi, S. Y. Lang, J. M. Zhou, J. Y. Liang, W. Wang, S. J. Tan, Y. X. Yin, R. Wen and Y. G. Guo, Angew. Chem., Int. Ed., 2019, 58, 18146–18149 CrossRef CAS PubMed.
- B. Zhou, Y. H. Jo, R. Wang, D. He, X. Zhou, X. Xie and Z. Xue, J. Mater. Chem. A, 2019, 7, 10354–10362 RSC.
- S. Xia, J. Lopez, C. Liang, Z. Zhang, Z. Bao, Y. Cui and W. Liu, Adv. Sci., 2019, 6, 1802353 CrossRef PubMed.
- S. Wang, N. Liu, J. Su, L. Li, F. Long, Z. Zou, X. Jiang and Y. Gao, ACS Nano, 2017, 11, 2066–2074 CrossRef CAS PubMed.
- Y. Yue, N. Liu, Y. Ma, S. Wang, W. Liu, C. Luo, H. Zhang, F. Cheng, J. Rao, X. Hu, J. Su and Y. Gao, ACS Nano, 2018, 12, 4224–4232 CrossRef CAS PubMed.
- Y. Huang, M. Zhong, Y. Huang, M. Zhu, Z. Pei, Z. Wang, Q. Xue, X. Xie and C. Zhi, Nat. Commun., 2015, 6, 10310 CrossRef CAS PubMed.
- X. Jin, G. Sun, H. Yang, G. Zhang, Y. Xiao, J. Gao, Z. Zhang and L. Qu, J. Mater. Chem. A, 2018, 6, 19463–19469 RSC.
- Y. Guo, K. Zheng and P. Wan, Small, 2018, 14, 1704497 CrossRef PubMed.
- Y. Wang, Q. Qu, S. Gao, G. Tang, K. Liu, S. He and C. Huang, Carbon, 2019, 155, 706–726 CrossRef CAS.
- T. W. Kwon, Y. K. Jeong, I. Lee, T. S. Kim, J. W. Choi and A. Coskun, Adv. Mater., 2014, 26, 7979–7985 CrossRef CAS PubMed.
- B. Koo, H. Kim, Y. Cho, K. T. Lee, N. S. Choi and J. Cho, Angew. Chem., Int. Ed., 2012, 124, 8892–8897 CrossRef.
- I. Kovalenko, B. Zdyrko, A. Magasinski, B. Hertzberg, Z. Milicev, R. Burtovyy, I. Luzinov and G. Yushin, Science, 2011, 334, 75–79 CrossRef CAS PubMed.
- Y. Bie, J. Yang, Y. Nuli and J. Wang, J. Mater. Chem. A, 2017, 5, 1919–1924 RSC.
- Y. Wang, H. Xu, X. Chen, H. Jin and J. Wang, Energy Storage Mater., 2021, 38, 121–129 CrossRef.
- X. Ji and L. F. Nazar, J. Mater. Chem., 2010, 20, 9821 RSC.
- H. Wang, Y. Yang, Y. Liang, J. T. Robinson, Y. Li, A. Jackson, Y. Cui and H. Dai, Nano Lett., 2011, 11, 2644–2647 CrossRef CAS PubMed.
- A. Rosenman, E. Markevich, G. Salitra, D. Aurbach, A. Garsuch and F. F. Chesneau, Adv. Energy Mater., 2015, 5, 1500212 CrossRef.
- J. Wang, J. Zhang, X. Yu, Y. Zhang, X. Zhu, H. Yue, Z. Wang and W. Zhou, J. For. Eng., 2019, 4, 84–91 Search PubMed.
- D. Lin, Y. Liu and Y. Cui, Nat. Nanotechnol., 2017, 12, 194–206 CrossRef CAS PubMed.
- Z. Yu, T. Gao, T. Le, W. Wang, L. Wang and Y. Yang, J. Mater. Sci.: Mater. Electron., 2019, 30, 5536–5543 CrossRef CAS.
- W. Xu, J. Wang, F. Ding, X. Chen, E. Nasybulin, Y. Zhang and J.-G. Zhang, Energy Environ. Sci., 2014, 7, 513–537 RSC.
- Q. Ma, X. Sun, P. Liu, Y. Xia, X. Liu and J. Luo, Angew. Chem., Int. Ed., 2019, 58, 6200–6206 CrossRef CAS PubMed.
- X. Xin, K. Ito, A. Dutta and Y. Kubo, Angew. Chem., Int. Ed., 2018, 57, 13206–13210 CrossRef CAS PubMed.
- X. Zhang, R. Lv, A. Wang, W. Guo, X. Liu and J. Luo, Angew. Chem., Int. Ed., 2018, 57, 15028–15033 CrossRef CAS PubMed.
- S. Choudhury and L. A. Archer, Adv. Electron. Mater., 2016, 2, 1500246 CrossRef.
- J. Zheng, M. H. Engelhard, D. Mei, S. Jiao, B. J. Polzin, J.-G. Zhang and W. Xu, Nat. Energy, 2017, 2, 17012 CrossRef CAS.
- Y. Lu, Z. Tu and L. A. Archer, Nat. Mater., 2014, 13, 961–969 CrossRef CAS PubMed.
- W. Zhou, Z. Wang, Y. Pu, Y. Li, S. Xin, X. Li, J. Chen and J. B. Goodenough, Adv. Mater., 2019, 31, 1805574 CrossRef PubMed.
- X. Fan, L. Chen, X. Ji, T. Deng, S. Hou, J. Chen, J. Zheng, F. Wang, J. Jiang, K. Xu and C. Wang, Chem., 2018, 4, 174–185 CAS.
- K. Yan, H. W. Lee, T. Gao, G. Zheng, H. Yao, H. Wang, Z. Lu, Y. Zhou, Z. Liang, Z. Liu, S. Chu and Y. Cui, Nano Lett., 2014, 14, 6016–6022 CrossRef CAS PubMed.
- G. Zheng, S. W. Lee, Z. Liang, H.-W. Lee, K. Yan, H. Yao, H. Wang, W. Li, S. Chu and Y. Cui, Nat. Nanotechnol., 2014, 9, 618–623 CrossRef CAS PubMed.
- B. Xu, Z. Liu, J. Li, X. Huang, B. Qie, T. Gong, L. Tan, X. Yang, D. Paley, M. Dontigny, K. Zaghib, X. Liao, Q. Cheng, H. Zhai, X. Chen, L.-Q. Chen, C.-W. Nan, Y.-H. Lin and Y. Yang, Nano Energy, 2020, 67, 104242 CrossRef CAS.
- H. Duan, Y. X. Yin, Y. Shi, P. F. Wang, X. D. Zhang, C. P. Yang, J. L. Shi, R. Wen, Y. G. Guo and L. J. Wan, J. Am. Chem. Soc., 2018, 140, 82–85 CrossRef CAS PubMed.
- L. Long, S. Wang, M. Xiao and Y. Meng, J. Mater. Chem. A, 2016, 4, 10038–10069 RSC.
- I. Osada, H. de Vries, B. Scrosati and S. Passerini, Angew. Chem., Int. Ed., 2016, 55, 500–513 CrossRef CAS PubMed.
- A. Manthiram, X. Yu and S. Wang, Nat. Rev. Mater., 2017, 2, 16103 CrossRef CAS.
- J. Mindemark, M. J. Lacey, T. Bowden and D. Brandell, Prog. Polym. Sci., 2018, 81, 114–143 CrossRef CAS.
- Y. Wang, W. D. Richards, S. P. Ong, L. J. Miara, J. C. Kim, Y. Mo and G. Ceder, Nat. Mater., 2015, 14, 1026–1031 CrossRef CAS PubMed.
- Y. Zhao, Y. Zhang, H. Sun, X. Dong, J. Cao, L. Wang, Y. Xu, J. Ren, Y. Hwang, I. H. Son, X. Huang, Y. Wang and H. Peng, Angew. Chem., Int. Ed., 2016, 55, 14384–14388 CrossRef CAS PubMed.
- Y. J. Nam, S. J. Cho, D. Y. Oh, J. M. Lim, S. Y. Kim, J. H. Song, Y. G. Lee, S. Y. Lee and Y. S. Jung, Nano Lett., 2015, 15, 3317–3323 CrossRef CAS PubMed.
- P. Guo, A. Su, Y. Wei, X. Liu, Y. Li, F. Guo, J. Li, Z. Hu and J. Sun, ACS Appl. Mater. Interfaces, 2019, 11, 19413–19420 CrossRef CAS PubMed.
- B. Zhou, C. Zuo, Z. Xiao, X. Zhou, D. He, X. Xie and Z. Xue, Chem. – Eur. J., 2018, 24, 19200–19207 CrossRef CAS PubMed.
- Z. Xue, D. He and X. Xie, J. Mater. Chem. A, 2015, 3, 19218–19253 RSC.
- J. M. Whiteley, P. Taynton, W. Zhang and S.-H. Lee, Adv. Mater., 2015, 27, 6922 CrossRef CAS PubMed.
- Z. Zhao, K. Xia, Y. Hou, Q. Zhang, Z. Ye and J. Lu, Chem. Soc. Rev., 2021, 50, 12702–12743 RSC.
- X. Wang, X. Li, W. Ge and Y. Yang, J. For. Eng., 2019, 4, 1–10 CrossRef CAS.
- Y. Huang, H. Hu, Y. Huang, M. Zhu, W. Meng, C. Liu, Z. Pei, C. Hao, Z. Wang and C. Zhi, ACS Nano, 2015, 9, 4766–4775 CrossRef CAS PubMed.
- K. Wang, X. Zhang, C. Li, X. Sun, Q. Meng, Y. Ma and Z. Wei, Adv. Mater., 2015, 27, 7451–7457 CrossRef CAS PubMed.
- S. T. Han, H. Peng, Q. Sun, S. Venkatesh, K. S. Chung, S. C. Lau, Y. Zhou and V. A. L. Roy, Adv. Mater., 2017, 29, 1700375 CrossRef PubMed.
- X. Wu, J. Wang, J. Huang and S. Yang, J. Colloid Interface Sci., 2020, 559, 152–161 CrossRef CAS PubMed.
- J. Cao, C. Lu, J. Zhuang, M. Liu, X. Zhang, Y. Yu and Q. Tao, Angew. Chem., Int. Ed., 2017, 56, 8921–8926 CrossRef.
- M. Yang, Y. Cheng, Y. Yue, Y. Chen, H. Gao, L. Li, B. Cai, W. Liu, Z. Wang, H. Guo, N. Liu and Y. Gao, Adv. Sci., 2022, 9, e2200507 CrossRef PubMed.
- H. Yang, D. Qi, Z. Liu, B. K. Chandran, T. Wang, J. Yu and X. Chen, Adv. Mater., 2016, 28, 9175–9181 CrossRef CAS PubMed.
- Z. Gao, Z. Lou, W. Han and G. Shen, ACS Appl. Mater. Interfaces, 2020, 12, 24339–24347 CrossRef CAS PubMed.
- Q. Cui, X. Huang, X. Dong, H. Zhao, X. Liu and X. Zhang, Chem. Mater., 2022, 34, 10778–10788 CrossRef CAS.
- Y. Liu, M. Pharr and G. A. Salvatore, ACS Nano, 2017, 11, 9614–9635 CrossRef CAS PubMed.
- S. Wang, Y. Bai, X. Yang, L. Liu, L. Li, Q. Lu, T. Li and T. Zhang, Talanta, 2020, 214, 120869 CrossRef CAS PubMed.
- W. D. Li, K. Ke, J. Jia, J. H. Pu, X. Zhao, R. Y. Bao, Z. Y. Liu, L. Bai, K. Zhang, M. B. Yang and W. Yang, Small, 2022, 18, 2103734 CrossRef CAS PubMed.
- L. Yang, Z. Wang, H. Wang, B. Jin, C. Meng, X. Chen, R. Li, H. Wang, M. Xin, Z. Zhao, S. Guo, J. Wu and H. Cheng, Adv. Mater., 2023, 35, 2207742 CrossRef CAS PubMed.
- X. Zeng, L. Xu, X. Xia, X. Bai, C. Zhong, J. Fan, L. Ren, R. Sun and X. Zeng, Small, 2023, 19, 2207409 CrossRef CAS PubMed.
Footnote |
| † L. C. and J. H. X. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |