
Alkyl-C60 liquid electrets as deformable mechanoelectric generators†
Ravindra Kumar
Gupta
 a,
Manabu
Yoshida
b,
Akinori
Saeki
c,
Zhenfeng
Guo
ad and
Takashi
Nakanishi
*ad
a,
Manabu
Yoshida
b,
Akinori
Saeki
c,
Zhenfeng
Guo
ad and
Takashi
Nakanishi
*ad
aResearch Center for Materials Nanoarchitectonics (MANA), National Institute for Materials Science (NIMS), 1-1 Namiki, Tsukuba 305-0044, Japan. E-mail: nakanishi.takashi@nims.go.jp
bFlexible Electronic Research Center, National Institute of Advanced Industrial Science and Technology (AIST), 1-1-1 Umezono, Tsukuba 305-8565, Japan
cDepartment of Applied Chemistry, Graduate School of Engineering, Osaka University, 2-1 Yamadaoka, Suita, Osaka 565-0871, Japan
dDivision of Soft Matter, Graduate School of Life Science, Hokkaido University, Kita 10, Nishi 8, Kita-ku, Sapporo 060-0810, Japan
First published on 15th June 2023
Abstract
Special attention is being paid to the potential applicability of various soft electronics in deformable/wearable devices. These devices must be constantly connected to energy sources to ensure their uninterrupted operation. Electrets, which are capable of retaining quasi-permanent electric charges inside or on the surface of materials, are expected to be a battery-less power source. Here, we present a strategy for harvesting the charges in alkyl-C60 liquids. Suitable substitution of bulky yet flexible branched long-alkyl chains generated C60-mono-adducts and regioisomeric bis-adducts as room-temperature solvent-free liquids. These alkyl–C60 liquids were negatively poled by the corona-discharging and soaked in nylon fabric. The liquid of the C60 bis-adduct exhibited better charge retention in comparison to the liquid of the C60 mono-adduct. This suggests that the bulky long-alkyl chains provided proper insulation for the C60 core and charge trapping in the liquid. This charge-trapping behaviour and the inherent fluidity of the alkyl-C60 liquids enabled their fabrication into deformable mechanoelectric generator (MEG) devices. The MEG exhibited applicability as a deformable micropower source or vibration sensor by generating output voltage pulses even under folded/twisted/rolled conditions. The alkylated-liquid-based MEGs worked at frequencies similar to human body motion, showing promising potential for body motion sensors and healthcare applications.
New conceptsMechanical flexibility and toughness are beneficial advantages seen in certain polymer materials, such as polymer electrets, which act as mechanoelectric generators (MEGs) and vibration sensors for healthcare applications. However, their applicability in free-deformable and foldable devices needs to be improved by adopting an innovative alternative approach. The fluid nature of liquid matter guarantees excellent deformability; the choice falls on liquid electrets in which the π-conjugated moiety is shielded and liquefied by bulky yet flexible branched alkyl chains. An electron-deficient fullerene C60 moiety was used to develop negatively charged liquid electrets. We stabilised the charges in the alkylated C60 liquid by applying corona charging and demonstrated the effect of alkyl chain wrapping on the C60 moiety by controlling the number of attachments and substituent units possessing two branched alkyl chains. The excellent charge retention of the alkylated C60 liquid based electret allowed us to fabricate MEGs with ultimate deformability such as folding, twisting, and rolling. Importantly, the MEGs work at frequencies similar to those of human body motions; understanding the output signal width under deformed conditions would help to explain body motions. The current results pave the way for future motion sensors and healthcare applications in developing wearable energy harvesters. |
1. Introduction
Flexible, lightweight, deformation-free energy harvesting devices that convert small-amplitude, low-frequency vibrations into electrical energy, namely mechanoelectric generators (MEGs), are an attractive and demanding challenge in today's wearable electronics.1,2 Self-powered wearable and portable systems are able to support information processing and artificial intelligence technologies.3 Fibre-based wearable systems were also developed for powering wearable microelectronics.4 Self-powered wearable vibration sensors were also developed which analyse the vibration effect.5 MEG devices are usually constructed with “electrets,” which have quasi-permanent electric charges or molecular dipoles.6 Such MEGs are promising for diverse applications such as healthcare sensors and muscle-driven scavengers.7 However, the most commonly utilised electrets are prepared with non-flexible materials, mainly inorganic metal oxides, ceramics, and polymeric hard films.8,9 Thus, for MEGs to be used as shape-independent energy harvesters in wearables of the future, they must be able to work under such conditions as bending, twisting, and excessive deformation.10 A self-powered wireless monitoring system was developed11 as well for fluid energy harvesting and self-powered flow monitoring systems12 and a tactile sensor was developed to predict the contacting objects in an open environment.13 In the current scenario, lithium-ion batteries are utilised in various electronic devices,14 which are not suitable for wearable healthcare applications, especially for long-term treatments inside or outside the human body.15 The utilisation of electrets as alternative power sources for wearable and wireless electronics represents a promising step in this direction. The electric field induced in electret materials originates from the net orientation of the dipolar molecules or the net macroscopic charge on the material.9 A model system for a self-assembled electret has been demonstrated by utilising the spontaneous orientation polarisation of 1,3,5-tris(1-phenyl-1H-benzimidazole-2-yl)benzene.16 This self-assembled electret was developed as an electret-based microelectromechanical system (MEMS) vibrational energy harvester.17 An organic molecule-based electret utilising perylene diimide also showed applicability to phototransistor memory devices.18 However, these examples are without deformation capability. As examples utilizing fluid matter for the development of energy harvesting devices, a prototype of energy harvester made-up of conducting solutions saturated with K+ and Cl− was applied as an electrostatic energy harvester using a vibration exciter that efficiently generates power from body movements without any mechanical springs.19,20 The capacitance of the harvester is periodically changed by the contacts of liquid to the insulating layer. In addition, a liquid crystal (4-cyano-4′-n-pentylbiphenyl, 5CB) was also utilised as a molecular-based electret leading to power generation under the influence of vibration.21 However, the lack of deformability of these electret devices makes them difficult to use in practice as wearable/deformable MEGs. Charge stabilising and deformability (fluidity) would benefit deformable electret devices using small molecules. Initially, our group demonstrated a liquid electret-based MEG featuring an alkyl–porphyrin liquid as the π-electret where insulating flexible alkyl chains were wrapped around the porphyrin core.22 Alkylated porphyrins were also explored as self-assembled highly ordered nanostructured thin films23 as well in photovoltaic applications.24 Self-healable elastomers acted as wearable microwave transmissions.25 Corona discharging is the most common technique for poling dielectric materials either positively or negatively.26 Positively poled porphyrin-based liquid electrets have been constructed, but the electret performance has not yet been explored under the negative-poling conditions of alkyl-π liquids containing electron-deficient π-conjugated core units and under excessively deformed states such as folding, twisting, and rolling.Motivated by our recent development in liquid electret science22 and the current ongoing research on deformable device technology, we came up with novel liquid materials based on an electron-deficient fullerene (C60).27 The present article describes the synthesis of alkyl-C60 liquids, mainly focusing on the C60 bis-adduct and its potential application in MEGs (Fig. 1). The better charge retention in the liquid electret was achieved by properly shielding the C60 unit with multiple, bulky, and flexible branched-alkyl chains (Fig. 1(c)). Its mono-adduct of C60 liquid, which has a less shielded C60 core, and a previous liquid featuring alkylated porphyrin were utilized for comparison. The MEGs fabricated with the bis-adduct of C60 liquid demonstrated applications even under folded/twisted/rolled conditions. We have also shown its working applicability at frequencies similar to human body motions with its possible future diverse healthcare and muscle-driven applications (Scheme 1).
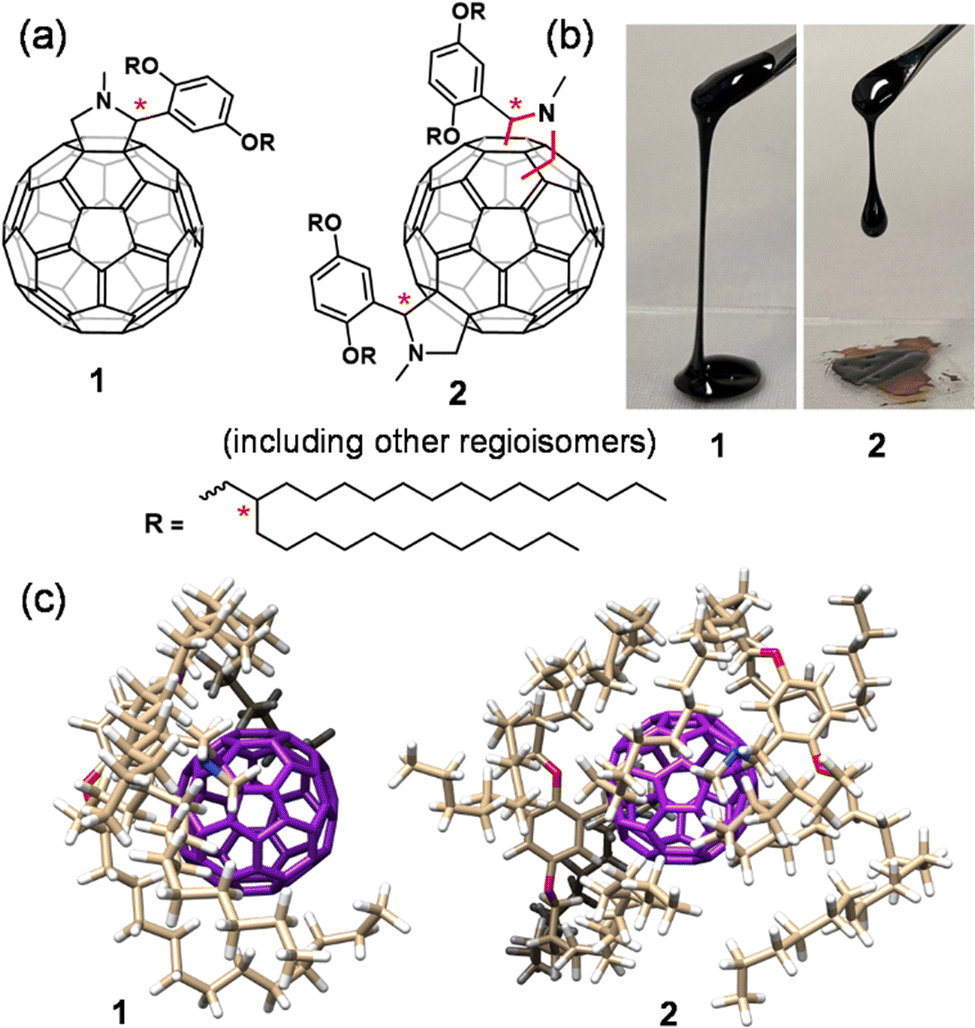 | ||
| Fig. 1 (a) Chemical structure of alkyl-C60 mono- (1) and bis-adducts (2) with 2,5-di(2-dodecylhexadecyloxy)phenyl-N-methylpyrrolidine. Asterisks show the chiral carbon centres. (b) Photographs are the corresponding solvent-free liquids at room temperature under daylight. (c) 3D structures of 1 and 2 demonstrating the shielding effect of the alkyl chain over the C60-core. | ||
 | ||
| Scheme 1 Schematic drawing of deformable MEGs utilized in this study showing their future diverse applications, such as in healthcare medical sensors and muscle-driven scavengers. | ||
2. Results and discussion
Molecular design and characterization
The synthesis of alkyl–C60 compounds (1 and 2) was followed by the procedure previously adopted (ESI,† Scheme S1).28–30 The compounds were designed by reacting N-methylglycine and (2,5-di(2-dodecylhexadecyloxy)benzaldehyde), generating a ylide, which undergoes 1,3-dipolar cycloaddition with a double bond at the [6,6] ring position of C60 (Prato reaction).31 The mono- (1) and bis-adduct (2) of 2,5-di(2-dodecylhexadecyloxy)phenyl-N-methylfulleropyrrolidines, were obtained at a yield of 69% and 25%, respectively (based on C60 conversion) and appeared as dark black viscous liquids (Fig. 1(b) and Scheme S1, ESI†). It is noted that 2 contained several regioisomers depending on the pyrrolidine ring modification point on the C60 unit. Generally, asymmetric substitution on C60 yields thirty-seven regioisomers.32 In our case, 2 had an asymmetric substitution along with six chiral centres (two from the pyrrolidine unit and four from the branched alkyl chain) (Fig. 1), since 1 existed in eight isomeric forms (two chains times four forms (RR, RS, SS, SR)); therefore, 2 existed in approximately 296 (eight times thirty-seven) regioisomers. The isolated 1 (including eight regioisomers) and 2 (mixture of 296 regioisomers) were characterised by nuclear magnetic resonance (NMR) and matrix-assisted laser desorption/ionisation time-of-flight (MALDI-TOF) mass spectrometry (Fig. S1–S4, ESI†). 1H NMR (Fig. S1 and S2, ESI†) and thermogravimetric analysis (TGA, Fig. S5, ESI†) of 1 and 2 were performed to ensure the absence of the residual solvent after drying the liquids under vacuum at 70 °C overnight. 1 and 2 demonstrate the effect of alkyl substitution on the C60 core, influencing the thermal, rheological, and charge retention properties in the solvent-free liquid state.Liquid physical properties
1 and 2 in the solvent-free neat state were obtained as a viscous liquid at room temperature. The thermal properties of these liquids were investigated utilizing differential scanning calorimetry (DSC, Fig. S6, ESI†) and TGA (Fig. S5, ESI†). The glass transition (Tg) observed for 1 and 2 was −28.4 °C (heat capacity (Cp) = 241.6 J mol−1 K−1) and −36.5 °C (Cp = 800.0 J mol−1 K−1), respectively, and no other transitions were observed at temperatures above Tg. The low Tg and high thermal stability confirmed by TGA (Td,95% = 381 °C for 1 and 363 °C for 2) allowed them to exist as solvent-free liquids over a wide range of temperatures. The amorphous nature of 1 and 2 was investigated with the help of small- and wide-angle X-ray scattering (SWAXS) analysis (Fig. 2(a)). Both 1 and 2 exhibited mainly two broad halos. This is a feature commonly found in alkylated π-conjugated molecular liquids.33,34 The halo at the lower q region was assigned to the distributed distance between randomly oriented π-rich C60 units, separated by substituted alkyl chains. The average C60–C60 (centre-to-centre) distance for 1 was 23.9 Å, along with a shoulder at 5.36 nm−1 (d = 11.7 Å), suggesting partial aggregation formation. In contrast, the absence of secondary reflection in 2 ensured its somewhat disordered fluid phase with an average C60–C60 distance of 23.7 Å.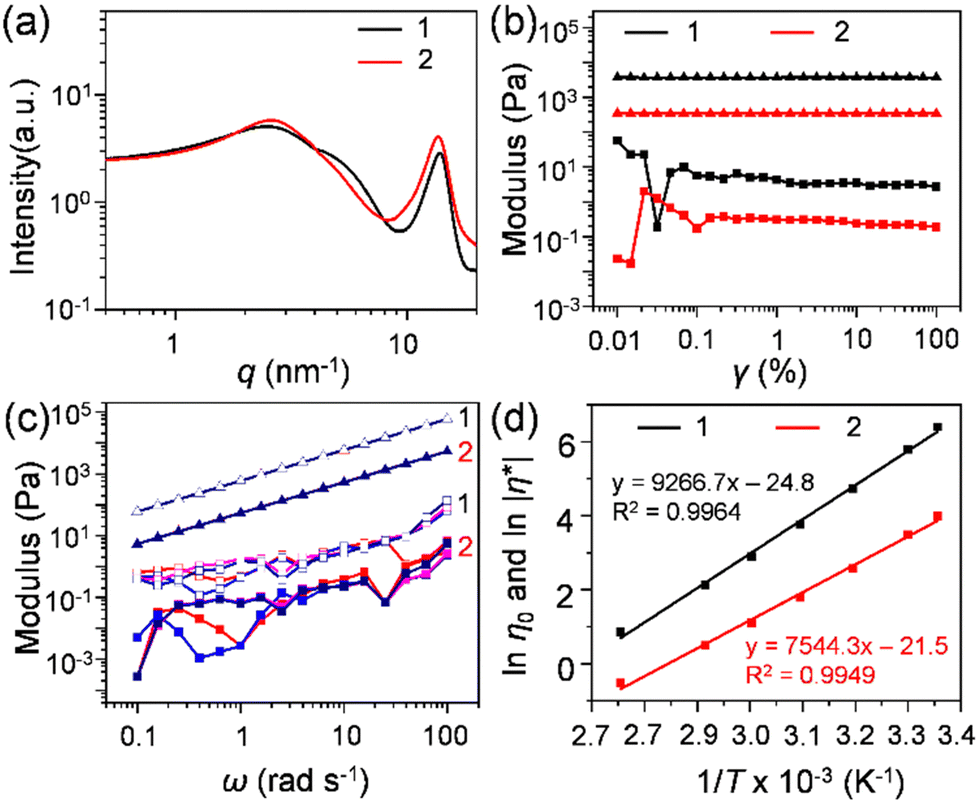 | ||
| Fig. 2 (a) SWAXS profiles of 1 (black line) and 2 (red line) at 25 °C. (b) Storage modulus (G′, squares) and loss modulus (G′′, triangles) of 1 (black colour) and 2 (red colour) as a function of the strain amplitude (γ); (c) G′ and G′′ of 1 (open markers) and 2 (closed markers) as a function of the angular frequency (ω) with γ of 0.01 (red), 0.05 (blue), 0.10 (pink), and 0.25 (navy) at 25 °C. (d) Arrhenius plot of ln η0 for 1 (black) and ln |η*| for 2 (red) versus 1/T under γ = 0.25, ω = 10 rad s−1 from 25 to 90 °C. | ||
The viscoelastic behaviour of 1 and 2 was investigated by rheological measurement. The obtained higher loss moduli (G′′) over the storage loss moduli (G′) throughout the measured range for the amplitude (γ) sweep (Fig. 2(b)), and frequency (ω) (Fig. 2(c)) confirmed their liquid nature at room temperature. The complex viscosity (|η*|) of 1 displayed dependence on ω measured at 25 °C at various shear strains (γ = 0.01 to 0.25) (Fig. S7a, ESI†). The higher |η*| at lower frequencies than at higher frequencies signified its shear thinning and non-Newtonian liquid behaviour, indicating the presence of intermolecular interactions at low frequencies. The presence of such interactions in bulk liquid 1 was elucidated by the deviation observed in the G′ curves at the high-frequency region by varying the strain amplitude (Fig. 2(c)). Using the onset frequency of the deviation (39.8 rad s−1) as well as the absolute value of |η*| (598 Pa s), the effective hydrodynamic radius was estimated to be approximately 7 nm,35,36 which corresponds to the size of a cluster consisting of about 50 molecules of 1. In addition, the slope of G′′ as a function of ω was found to be unity for both liquids (Δ = 1), which suggests Newtonian-like liquid behaviour. However, G′ showed some structural change under the measured frequency range, which might reveal the presence of C60–C60 interactions in C60 liquids. In contrast, 2 showed an independency of |η*| with 54.3 Pa s, over a varied range of ω suggesting its Newtonian-type fluid behaviour (Fig. S7b, ESI†). In general, the zero-shear viscosity (η0, η0 = limω∼0 |η*|) indicates the viscosity of a fluid material at rest. In the case of shear-thinning liquids, it can be obtained by reaching a constant complex viscosity after shear-thinning.37,38 The obtained η0 value for 1 was 598 Pa s (γ = 0.05) at 25 °C (Fig. S7a, ESI†). Liquids 1 and 2 had lower viscosities (η0 for 1 and |η*| for 2) with an increase in temperature (Fig. S8a and b, ESI†). These lower viscosity samples at a higher temperature, e.g., 100 °C, were used for the corona charging experiment (vide infra). The activation energy (Ea) of both liquids was calculated based on the Arrhenius plot of ln |η0| for 1 and ln |η*| for 2 as a function of 1/T (Fig. 2(d)). The obtained Ea for 1 and 2 was 77.0 and 62.7 kJ mol−1, respectively. These values are higher than those reported for polymer fluids such as molten low-density polyethylene38,39 and poly(p-dioxanone),40 and typical ionic liquids (ILs), e.g., phosphonium41 and ammonium nitrate-based ILs.42 The value of Ea is related to the viscosity of fluids and determines the temperature sensitivity of the flow process. Therefore, the lower value of Ea for 2 shows a low sensitivity under temperature variation compared to 1. Moreover, 1 exhibited an 11-fold higher viscosity than 2, indicating proper isolation of the C60 core by multiple bulky alkyl chains in the case of 2. A similar trend was seen for previously studied alkyl-pyrene liquids.33 This also confirmed that the number of bulky branched alkyl chains could generate a higher entropy from the higher Cp at the glassy transition (vide supra, Fig. S6, ESI†), leading to low viscosity liquids as observed in 2. Racemisation and coexisting regioisomers also play an essential role in reducing the viscosity of liquids.43 Here, 1 and 2 possessed stereogenic chiral centres at their fulleropyrolidine unit44–46 and the branching point of the 2-dodecylhexadecyloxy chains (Fig. 1). These multiple factors could have reduced the viscosity of the C60 liquids in comparison to their linear chain counterpart.35,36
To further understand the aggregation tendency, the photophysical behaviour of 1 and 2 were studied by UV-Visible absorption spectroscopy in the solvent-free liquid state and their solvated state (dilute 5, 10, and 20 μM n-hexane solutions) (Fig. S9 and S10, ESI†). The reduced absorbance in the liquid form compared to the solvated state gave the signature of the C60–C60 interactions (Fig. S10, ESI†). At longer wavelengths of around 710–725 nm, 1 showed a 7 nm red-shift in absorption compared to that in n-hexane solution along with broadening of the peak, whereas 2 only indicated a slight red shift of 4 nm without the broadening of the peaks (Fig. S10, ESI† inset). This suggests that C60–C60 interactions were more prominent in 1 than in 2, which influenced the viscosity and shielding of the C60 unit. However, C60–C60 interactions are insufficient to generate the intensive photoconductivity known for organic semiconductors.47,48 The photoconductivity maximum (φΣμmax) values were obtained by the flash-photolysis time-resolved microwave conductivity (FP-TRMC) technique (Fig. S11, ESI†).49 The low values around 10−5 cm2 V−1 s−1 for both 1 and 2 obtained for φΣμmax are similar to those obtained for alkyl-porphyrin,22 -pyrene,33 and -anthracene liquids50 corresponding to the absence of long-range ordering of the C60 units in their liquids.
Mechanoelectric generator (MEG) performance
The performance of MEGs was investigated by initially testing the intrinsic charge-trapping ability of 1 and 2. The previously reported alkyl-porphyrin liquid 3 (Fig. 3(a)) was also tested for comparison.22 The liquids soaked in nylon fabric were negatively poled (−80 kV) under corona discharge at a maintained temperature of 100 °C for a period of (th) 30 min. Then the heating was turned off, and the sample was continuously charged until the hot plate temperature reached room temperature for 60 min (total poling time, tp = 90 min) (Fig. S12a, ESI†). After charging, the blank nylon fabric showed a nominal charge density of 0.03 C m−3, which dropped within 100 min. In contrast, the liquids soaked in nylon fabric offered better charging capabilities. Liquid 2 showed the highest charge retention among the tested samples with a charge retention time of approximately 900 min, as well as the highest volume charge density of 0.28 C m−3, whereas 1 and 3 showed 0.17, and 0.09 C m−3, respectively (Fig. 3(b)). The lower volume charge density of 3 could be understood by its symmetrical chemical structure with a negligible dipole moment, whereas 1 and 2 had asymmetrical chemical structures leading to the formation of dipoles, which may have helped to enhance their volume charge density. Another factor is that the shielding of 2 with alkyl chains was better than that of 1, which might be the reason for the higher volume charge density of 2 over 1. Polydimethylsiloxane (PDMS) composited with Si nanoparticles is a typical example of a stretchable electret, but it possesses a lower volume charge density of 0.04 C m−3.51 The volume charge density of 2 was 7 times larger than that of the PDMS-Si nanoparticle composite. As discussed above, according to SWAXS, UV-Vis absorption, and the rheology results, liquid 2 had better isolation of the C60 unit via a two-fold number of alkyl chains in comparison to 1, confirming that proper insulation of the C60 core helped to retain the charge for a longer time. Furthermore, a control experiment was also performed with non-alkylated phenyl-N-methylfulleropyrrolidine (PhC60) to understand the effect of alkyl chain shielding over the charging capability of PhC60 in comparison to the C60 liquids (1 and 2). The obtained volume charge density for PhC60 is 0.15 C m−3, which is lower than the volume charge density obtained for 1 and 2. Therefore, it is evident that the presence of bulky yet flexible branched-alkyl chains in 1 and 2 helps to shield the C60 core, which could help to stabilize the trapped charges in the C60 liquids.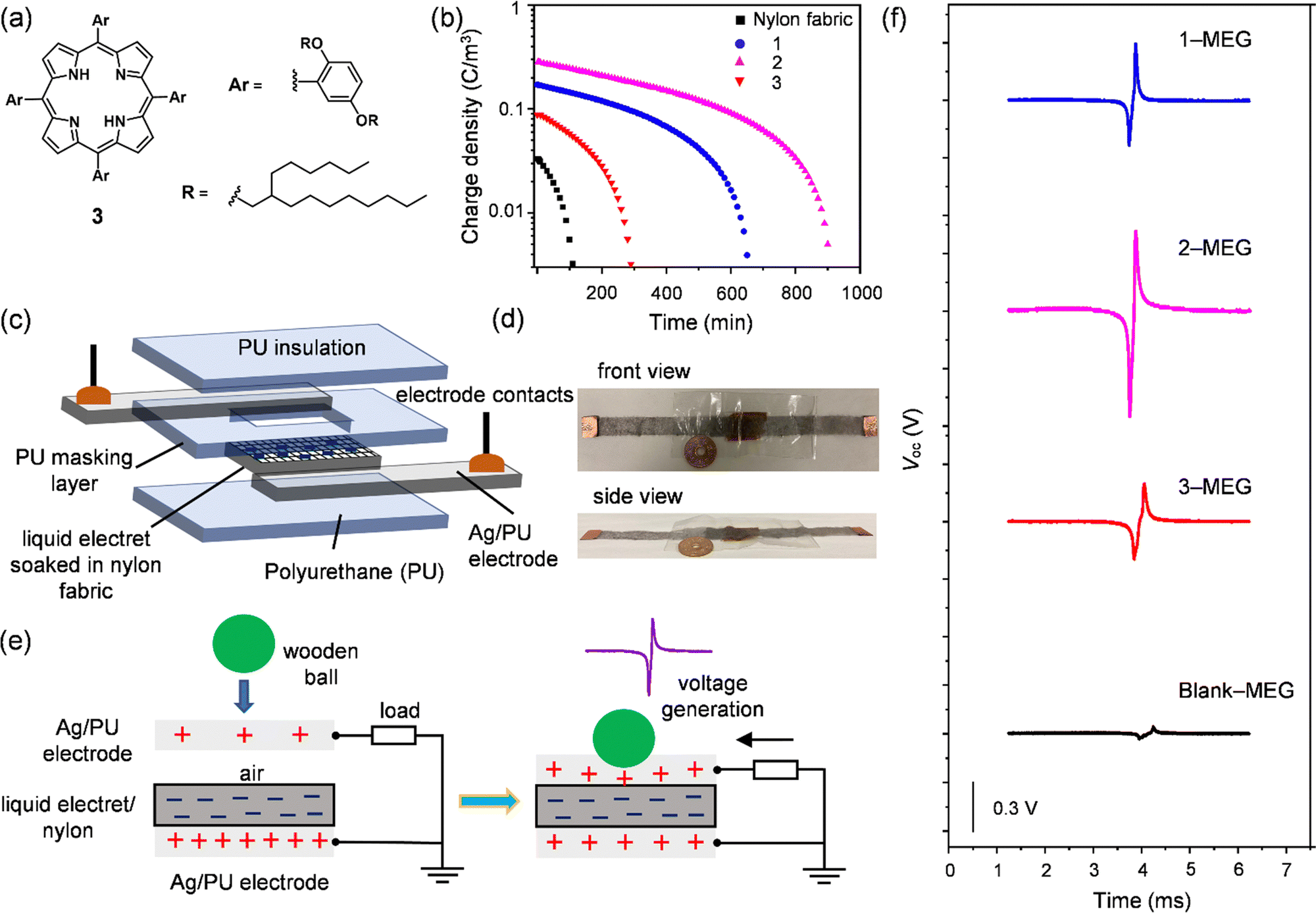 | ||
| Fig. 3 (a) Chemical structure of alkylated porphyrin-based liquid 3; (b) electrostatic charge density decay of negatively poled liquids 1, 2, 3, and a blank (Nylon fabric, 2 × 3 cm2) over time after poling (assuming the density of alkylated liquids as 1.0 and for nylon fabric (nylon 66) 1.14); (c) schematic illustration of deformable MEG device structure; (d) front and side view of the fabricated MEG (device thickness: approximately 200 μm; PU film: 24 μm; Ag electrode coated on PU: 30 μm; nylon fabric: 70 μm) with poled 2, where a Japanese coin with a thickness of 1.4 mm was included to show the comparable thickness of the MEG; (e) schematic illustration of the ball-dropping experiment; (f) open-circuit voltage (Voc) characteristics of the fabricated MEGs with 1 (blue), 2 (pink), 3 (red) and a blank (black) from the ball-dropping experiment (four days after device fabrication). | ||
The superior charge retention of liquid 2 enabled us to investigate the deformable liquid electret behaviour in fabricating a foldable MEG. We fabricated the MEG with 2, 1, and 3 for comparison. The deformable MEG was composed of several layers as follows (from the bottom); polyurethane film (24 μm, PU)/Ag coated PU electrode (30 μm)/liquid electret soaked in nylon fabric (70 μm)/PU masking layer (24 μm)/Ag coated PU electrode (30 μm)/PU film (24 μm). The total thickness of the fabricated MEG was approximately 200 μm (Fig. 3(c)). The fabricated MEGs were highly deformable and thin (Fig. 3(d) and ESI,† Video file-1). Negative poling was applied as per the conditions mentioned above. The active area of the liquid electret in the MEGs was 1.5 × 2.5 cm2.
To evaluate the output characteristics of the fabricated MEGs, we tested the potential difference between the top and bottom electrodes using a ball-dropping experiment52 (Fig. 3(e) and Fig. S13, ESI†). A wooden ball (diameter = 16 mm; weight m = 1.57 g, mgh = mv = 0.31 mJ for initial height h = 200 mm, g and v are the gravitational acceleration and impact rate, respectively) was dropped on the fabricated MEGs. As a result, a single induction of a bipolar open circuit voltage (Voc) was obtained following the principle of the contact-separation mode (Fig. 3(e)).53 The induced (compensation) charge in the MEGs formed with negatively poled liquid electrets moved from the bottom electrode to the top electrode due to a change in distance between the top electrode and the electret layer. During relaxation, the opposite current flowed, and the time integral of the voltage equalled zero. As a result, the output voltage (negative/positive) obtained for the fabricated MEGs increased in the order of blank (−0.03/0.03 V) < 3-MEG (−0.20/0.20 V) < 1-MEG (−0.24/0.30 V) < 2-MEG (−0.55/0.42 V) voltage power generation (Fig. 3(f)).
The MEGs were also evaluated for output voltage generation under continuous vibration at various frequencies (18, 32, and 53 Hz) using the measurement setup shown in Fig. S12b and ESI,† Video file-2. Under continuous vibration at different frequencies generated by a vibration device (Exagun, Fig. S14, ESI†), these MEGs showed a similar trend of output voltage pulse generation (Fig. S15, ESI†) as observed in the ball-dropping experiment. As shown in Fig. 4(a), 2-MEG on continuous vibration at 53 Hz showed a higher output voltage (average positive signal (Vave-p): 2.59 ± 0.05 V, average negative signal (Vave-n): −3.75 ± 0.09 V) fluctuation in comparison to other fabricated MEGs with 1, 3 and blank. Initially, these fabricated devices showed higher output voltages, which gradually decreased due to gradual discharging from the liquid electrets, which are not perfectly sealed by air-permeable PU films. In addition, the time-dependent stability of the fabricated devices was measured to ensure their charge stabilisation. For one week after device fabrication, the output voltage generation continuously dropped and then it stabilised, which might have been caused by the neutralising charges flowing to the ground on the surface charge that is not strongly trapped in the liquid electret. After stabilisation of charges, the fabricated MEGs showed the highest output pulse for 2-MEG (Vave-p: 1.52 ± 0.03, Vave-n −1.59 ± 0.03 V) under continuous vibration at 53 Hz even at 65 days after device fabrication (Fig. 4(b) and Fig. S16, ESI†).
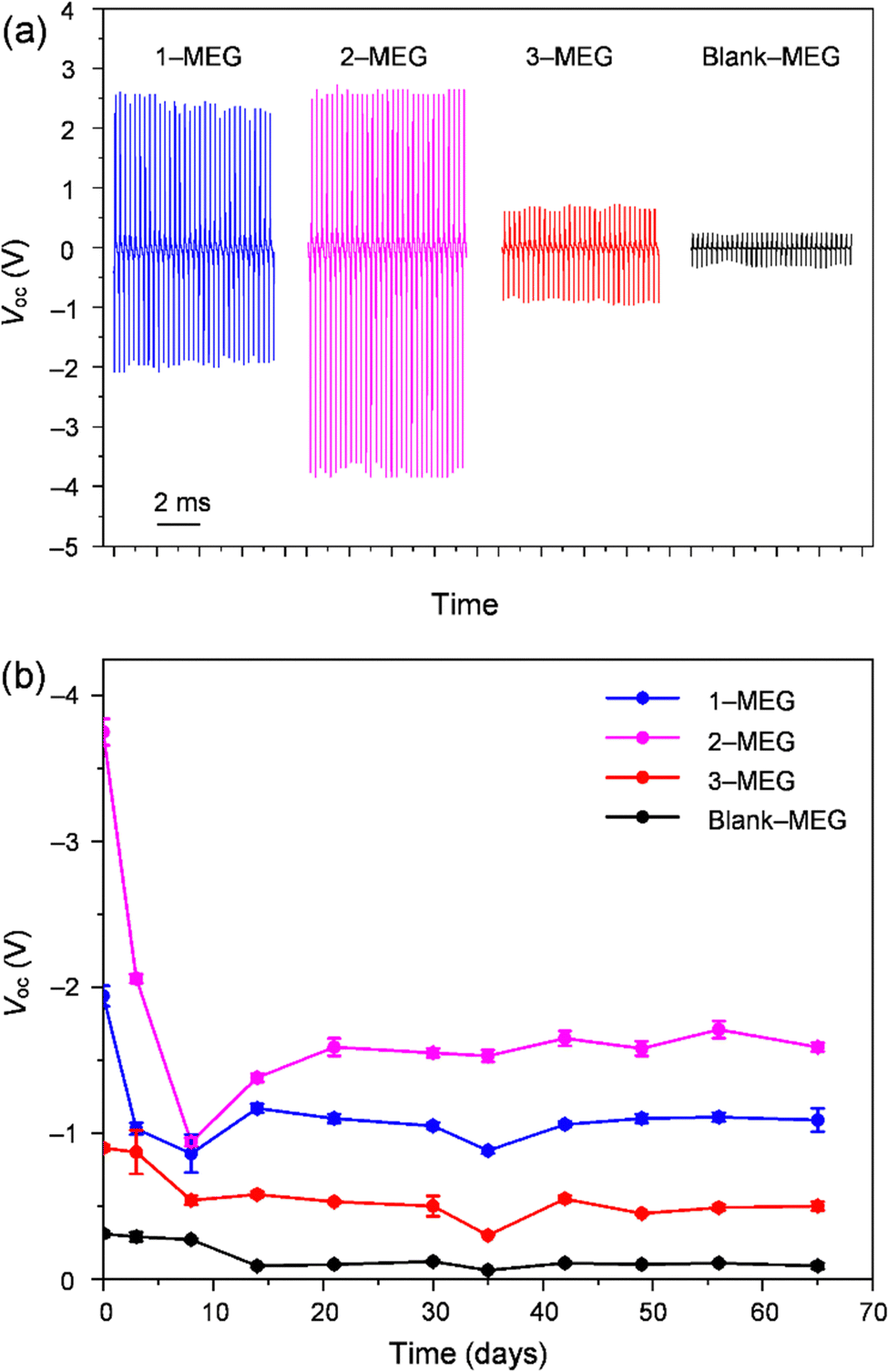 | ||
| Fig. 4 (a) Open-circuit voltage (Voc) characteristics of fabricated mechanoelectric generators (MEGs) with 1, 2, 3, and a blank under continuous vibration at 53 Hz; (b) decay graph of MEG devices showing the stability of the average negative output voltage (Vave-n) of the devices under continuous vibration at 53 Hz on the time scale (days). | ||
The reliability of 2-MEG under deformation was confirmed by various conditions such as folding, twisting, and rolling and was tested for output voltage generation under continuous vibration of 53 Hz (Fig. 5(a) and ESI,† Video files 3–5). The Voc values (negative/positive signal maximum) under various deformed conditions were as follows: standard (−1.12/0.84 V), standard reversed (−1.32/2.20 V), up folded (−0.84/0.60 V), down folded (−0.84/1.56 V), clockwise twisted (−0.88/0.76 V), anti-clockwise twisted (−0.80/0.72 V), and rolled (−0.92/1.64 V). Folding and rolling gave similar Voc, but a significant change was observed under twisting conditions. More importantly, the gap in the compression and relaxation signals increased under the deformed conditions (6.0 ms for standard reversed, 5.4 ms for up folded, 6.6 ms for down folded, 5.0 ms for clockwise twisted, 5.0 ms for anti-clockwise twisted, and 8 ms for rolled) compared to the standard one (3.7 ms) (Fig. 5(a), green dotted line). This widening of the gap in signals might be due to the tapping that happened horizontally as well as vertically to the MEG under deformed conditions. Moreover, the clockwise and anti-clockwise twisting directions do not have significant differences with similar output voltages, and the crooked part of the MEG under deformation might also affect the signal widening specifically under rolled conditions. To test the working mechanism, the device was subjected to 53 Hz continuous vibration on its front and reverse faces. When the pressure was applied to the front of 2-MEG, the top electrode came closer to the electret layer, increasing the positive counter charge on the top electrode induced by the negatively charged liquid electret, giving a compression signal of −1.60 V. After removal of the pressure, the charge decreased, giving a relaxation signal of 1.48 V, following the principle of the contact-separation process (Fig. 3(e) and 5(b)). On the other hand, when the pressure was applied to the reverse, the liquid electret with the bottom electrode coming closer to the top electrode induced a positive counter charge on the top electrode, generating a compression signal of 1.28 V and a subsequent relaxation signal of −1.12 V after removal of the pressure (Fig. 5(c)). Moreover, reverse tapping showed reverse output voltage signals concerning obverse tapping with positive Voc in compression and negative Voc in relaxation. The observed difference in the compression and relaxation signals under the front and reverse tapping might be due to the extent of deformation occurring on the electrode. In the front tapping condition, the deformation of the top electrode was more in comparison to deformation under the reverse situation (Fig. 5(b), inset and Fig. 5(c), inset). The signal width was also affected under the front and reverse tapping (3.7 ms for front tapping and 6 ms for reverse tapping), leading to wider, sharp signals. This might be due to the required recovery time for the deformed top electrode being longer than the less deformed bottom electrode with liquid electrets under the front and reverse tapping conditions, respectively. Interestingly, the up folded MEG shows the same signal directions as the standard whereas the down folded MEG shows reversed signal directions. These working phenomena of MEGs might help identify the direction of the pulsation or pressure occurring during human body motion. Highly deformable MEGs developed based on alkyl-C60 liquids will open up new horizons for future soft electronics in healthcare applications, although they require further sophistication. Overall, these MEGs are able to distinguish the direction of pressure application based on their signal gap and it is also noted that the top-outer surface of the device is the most sensitive to detect the vibration (pressure) applied (Fig. 5(a)).
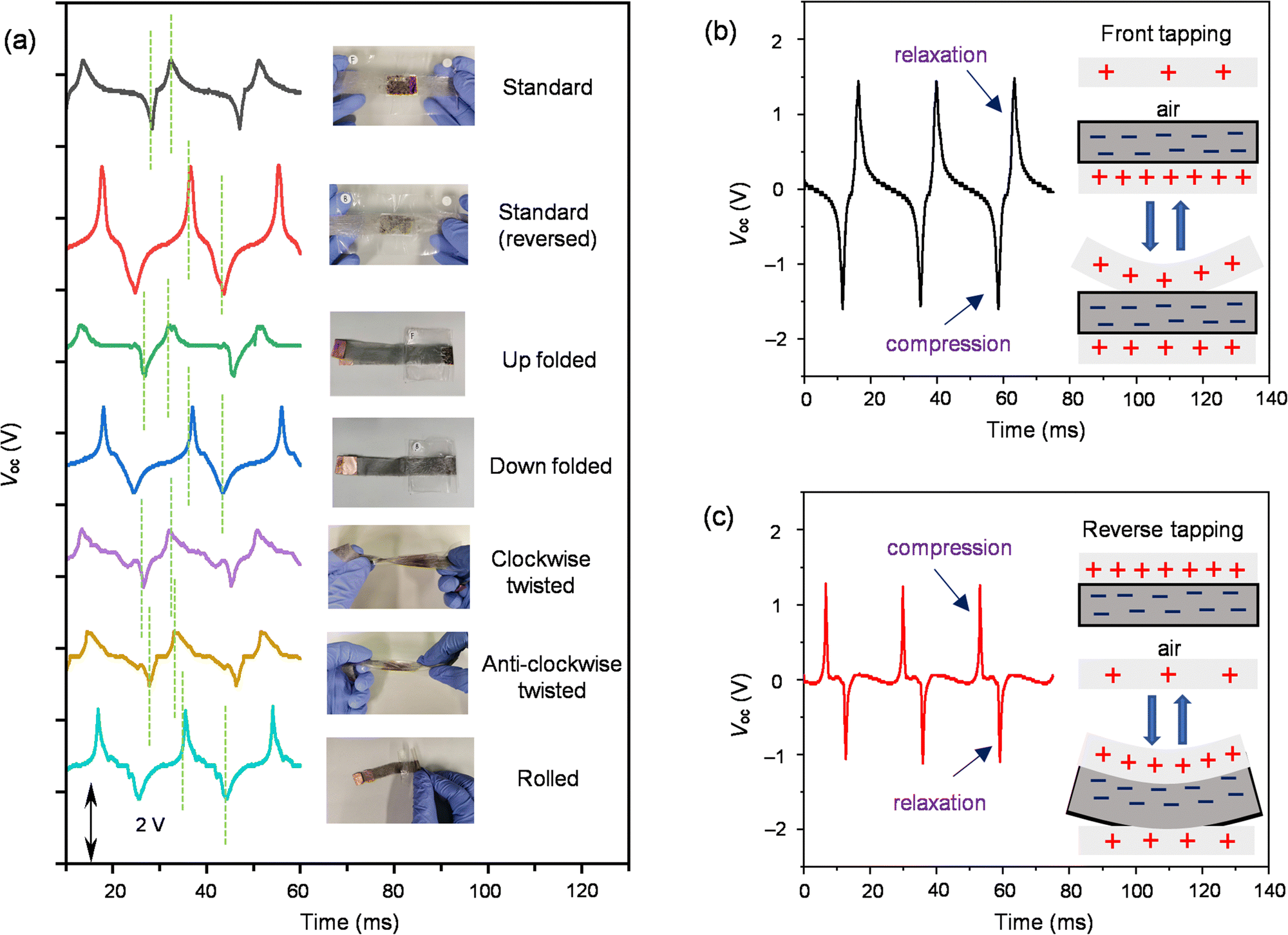 | ||
| Fig. 5 (a) Open-circuit voltage (Voc) characteristics of the fabricated 2-MEG under standard (black), standard reversed (red), up folded (green), down folded (blue), clockwise twisted (purple), anticlockwise twisted (brown) and rolled (sky blue) conditions from continuous vibration at 53 Hz with photo images of the 2-MEG under standard, standard reversed, up folded, down folded, clockwise twisted, anti-clockwise twisted and rolled conditions (green dotted lines show a widening of the signal-gap (6.0 ms for standard reversed, 5.4 ms for up folded, 6.6 ms for down folded, 5.0 ms for clockwise twisted, 5.0 ms for anticlockwise twisted, and 8 ms for rolled) under deformed conditions in comparison to the standard one (3.7 ms)); (b) Voc characteristics of the fabricated 2-MEG from constant vibration at 53 Hz after three months of device fabrication with front tapping with initial compression and relaxation (inset shows a working mechanism for the extent of deformation) and (c) with reverse tapping with the compression and relaxation peaks in a reverse manner (inset shows a working mechanism for the extent of deformation). | ||
3. Conclusions
We synthesised alkyl-C60 liquids with the mono-adduct (1) and regioisomeric bis-adduct (2) of 2,5-di(2-dodecylhexadecyloxy)phenyl-N-methylpyrrolidine and demonstrated their liquid electret performance as deformable mechanoelectric generators (MEGs). Interestingly, out of both alkyl-C60 liquids, poled 2 exhibited better charge retention performance, confirming proper insulation/isolation of the C60 core with bulky-branched alkyl chains. Furthermore, the 2-based MEG displayed approximately three times larger output voltages (Vave-n = −1.59 ± 0.03 V, after charge stabilisation) than the first reported alkyl-porphyrin liquid electret device under a continuous vibration of 53 Hz. Moreover, the 2-MEG generated output voltage pulses of −0.84, −0.88, and −0.92 V under highly deformable folded, twisted, and rolled conditions, respectively, which has never been demonstrated in molecule- and polymer-based electret devices.The alkyl-π engineering strategy in π-conjugated systems54–62 leads to the isolation of π-units. Thus, the trapping of charges could be stabilised in these room-temperature alkyl-π molecular liquids. The superior deformable electret device performance of alkyl-C60 liquids provides an excellent opportunity for future development toward healthcare applications. Notably, the fabricated MEGs work on frequencies equivalent to those of human body motion lower than 100 Hz. Therefore, liquid electret science utilising alkyl-π liquids has a bright future, particularly in future body motion sensors, electricity generation toward wireless technology, muscle-driven energy scavengers, and more.
Author contributions
R. K. G. performed the synthesis, characterization, and fabrication of MEG devices, measured all MEG-related data, and wrote the original manuscript. M. Y. guided the electret experiments. A. S. performed the TRMC studies. Z. G. carried out MEG experiments under various deformed conditions. T. N. conceived the central concept, and supervised, reviewed and polished the manuscript. All authors discussed the results.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the financial support by Grants-in-Aid for Scientific Research (JSPS KAKENHI Grant Number JP18H03922 and 20F40041). R. K. G. thanks the JSPS postdoctoral fellowship (P20041) and NIMS postdoctoral fellowship. This work was also supported by World Premier International Research Center Initiative (WPI), MEXT, Japan. We thank Dr Akira Shinohara and Mr Kunihito Tsumo (NIMS) for the illustration preparation.Notes and references
- T. Miyoshi, M. Adachi, K. Suzuki, Y. Liu and Y. Suzuki, IEEE Micro Electro Mech. Syst., 2018, 230–232 Search PubMed.
- J. Tan, K. Chen, J. Cheng, Z. Song, J. Zhang, S. Zheng, Z. Xu and S. E, Nanomaterials, 2023, 13, 158 CrossRef CAS PubMed.
- C. Xu, Y. Song, M. Han and H. Zhang, Microsyst. Nanoeng., 2021, 7, 25 CrossRef PubMed.
- X. Tao, Acc. Chem. Res., 2019, 52, 307–315 CrossRef CAS PubMed.
- F. Mokhtari, Z. Cheng, C. H. Wang and J. Foroughi, Global Challenges, 2023, 7, 2300019 CrossRef PubMed.
- S. Wang, Y. Xie, S. Niu, L. Lin, C. Liu, Y. S. Zhou and Z. L. Wang, Adv. Mater., 2014, 26, 6720–6728 CrossRef CAS PubMed.
- Z. Guo, Y. Patil, A. Shinohara, K. Nagura, M. Yoshida and T. Nakanishi, Mol. Syst. Des. Eng., 2022, 7, 537–552 RSC.
- G. M. Sessler, J. Electrostat., 2001, 51–52, 137–145 CrossRef CAS.
- N. Wada, K. Mukougawa, N. Horiuchi, T. Hiyama, M. Nakamura, A. Nagai, T. Okura and K. Yamashita, Mater. Res. Bull., 2013, 48, 3854–3859 CrossRef CAS.
- W. Deng, Y. Zhou, A. Libanori, G. Chen, W. Yang and J. Chen, Chem. Soc. Rev., 2022, 51, 3380–3435 RSC.
- F. Zheng, Y. Zhou, S. Hu, R. Li, Z. L. Wang and Z. Wu, Adv. Energy Mater., 2022, 12, 2201966 CrossRef CAS.
- Y. Zhong, H. Zhao, Y. Guo, P. Rui, S. Shi, W. Zhang, Y. Liao, P. Wang and Z. L. Wang, Adv. Mater. Technol., 2019, 4, 1900741 CrossRef.
- X. Wei, B. Wang, Z. Wu and Z. L. Wang, Adv. Mater., 2022, 34, 2203073 CrossRef CAS PubMed.
- R. Marom, S. F. Amalraj, N. Leifer, D. Jacob and D. Aurbach, J. Mater. Chem., 2011, 21, 9938–9954 RSC.
- S. Mukherjee, A. Albertengo and T. Djenizian, Energy Storage Mater., 2021, 42, 773–785 CrossRef.
- Y. Tanaka, N. Matsuura and H. Ishii, Sci. Rep., 2020, 10, 6648 CrossRef CAS PubMed.
- D. Yamane, H. Kayaguchi, K. Kawashima, H. Ishii and Y. Tanaka, Appl. Phys. Lett., 2021, 119, 254102 CrossRef CAS.
- C.-H. Ho, Y.-C. Lin, W.-C. Yang, E. Ercan, Y.-C. Chiang, B.-H. Lin, C.-C. Kuo and W.-C. Chen, ACS Appl. Mater. Interfaces, 2022, 14, 15468–15477 CrossRef CAS PubMed.
- D.-H. Choi, C.-H. Han, H.-D. Kim and J.-B. Yoon, Smart Mater. Struct., 2011, 20, 125012 CrossRef.
- Y. C. Chen, K. Y. Song, K. Morimoto and Y. Suzuki, 28th IEEE International Conference on Micro Electro Mechanical Systems (MEMS), 2015, 1086–1089 DOI:10.1109/MEMSYS.2015.7051152.
- K. Kittipaisalsilpa, T. Kato and Y. Suzuki, 29th IEEE International Conference on Micro Electro Mechanical Systems (MEMS), 2016, 37–40 DOI:10.1109/MEMSYS.2016.7421551.
- A. Ghosh, M. Yoshida, K. Suemori, H. Isago, N. Kobayashi, Y. Mizutani, Y. Kurashige, I. Kawamura, M. Nirei, O. Yamamuro, T. Takaya, K. Iwata, A. Saeki, K. Nagura, S. Ishihara and T. Nakanishi, Nat. Commun., 2019, 10, 4210 CrossRef PubMed.
- X. Zhou, S.-W. Kang, S. Kumar, R. R. Kulkarni, S. Z. D. Cheng and Q. Li, Chem. Mater., 2008, 20, 3551–3553 CrossRef CAS.
- L. Li, S.-W. Kang, J. Harden, Q. Sun, X. Zhou, L. Dai, A. Jakli, S. Kumar and Q. Li, Liq. Cryst., 2008, 3, 233–239 CrossRef.
- B.-Y. Yu, D.-W. Yue, K.-X. Hou, L. Ju, H. Chen, C. Ding, Z.-G. Liu, Y.-Q. Dai, H. K. Bisoyi, Y.-S. Guan, W.-B. Lu, C.-H. Li and Q. Li, Light: Sci. Appl., 2022, 11, 307 CrossRef CAS PubMed.
- R. Kou, Y. Zhong and Y. Qiao, Langmuir, 2020, 36, 9571–9577 CrossRef CAS PubMed.
- P. W. Fowler and A. Ceulemans, J. Phys. Chem., 1995, 99, 508–510 CrossRef CAS.
- H. Li, S. S. Babu, S. T. Turner, D. Neher, M. J. Hollamby, T. Seki, S. Yagai, Y. Deguchi, H. Möhwald and T. Nakanishi, J. Mater. Chem. C, 2013, 1, 1943–1951 RSC.
- L. Bianchi, X. Zhang, Z. Chen, P. Chen, X. Zhou, Y. Tang, B. Liu, X. Guo and A. Facchetti, Chem. Mater., 2019, 31, 6519–6529 CrossRef CAS.
- X. Zheng, K. Nagura, T. Takaya, K. Hashi and T. Nakanishi, Chem. – Eur. J., 2023, 29, e202203775 CrossRef CAS PubMed.
- M. Maggini, G. Scorrano and M. Prato, J. Am. Chem. Soc., 1993, 115, 9798–9799 CrossRef CAS.
- S. B. Beil and M. von Delius, Org. Mater., 2021, 03, 146–154 CrossRef CAS.
- F. Lu, T. Takaya, K. Iwata, I. Kawamura, A. Saeki, M. Ishii, K. Nagura and T. Nakanishi, Sci. Rep., 2017, 7, 3416 CrossRef PubMed.
- B. Narayan, K. Nagura, T. Takaya, K. Iwata, A. Shinohara, H. Shinmori, H. Wang, Q. Li, X. Sun, H. Li, S. Ishihara and T. Nakanishi, Phys. Chem. Chem. Phys., 2018, 20, 2970–2975 RSC.
- T. Michinobu, K. Okoshi, Y. Murakami, K. Shigehara, K. Ariga and T. Nakanishi, Langmuir, 2013, 29, 5337–5344 CrossRef CAS PubMed.
- T. Michinobu, T. Nakanishi, J. P. Hill, M. Funahashi and K. Ariga, J. Am. Chem. Soc., 2006, 128, 10384–10385 CrossRef CAS PubMed.
- Y. Chino, A. Ghosh, T. Nakanishi, N. Kobayashi, K. Ohta and M. Kimura, Chem. Lett., 2017, 46, 1539–1541 CrossRef CAS.
- B. A. Wolf, Macromol. Chem. Phys., 2020, 221, 2000130 CrossRef CAS.
- M. Zatloukal, Polymer, 2016, 104, 258–267 CrossRef CAS.
- C. Liu, S. Andjelić, J. Zhou, Y. Xu, C. Vailhe and R. Vetrecin, J. Mater. Sci.: Mater. Med., 2008, 19, 3481–3487 CrossRef CAS PubMed.
- M. D. Green, C. Schreiner and T. E. Long, J. Phys. Chem. A, 2011, 115, 13829–13835 CrossRef CAS PubMed.
- S. B. Capelo, T. Méndez-Morales, J. Carrete, E. López Lago, J. Vila, O. Cabeza, J. R. Rodríguez, M. Turmine and L. M. Varela, J. Phys. Chem. B, 2012, 116, 11302–11312 CrossRef CAS PubMed.
- C. Schäfer, S. Hultmark, Y. Yang, C. Müller and K. Börjesson, Chem. Mater., 2022, 34, 9294–9302 CrossRef.
- Y. Hizume, K. Tashiro, R. Charvet, Y. Yamamoto, A. Saeki, S. Seki and T. Aida, J. Am. Chem. Soc., 2010, 132, 6628–6629 CrossRef CAS PubMed.
- R. M. Girón, J. Ouyang, L. Favereau, N. Vanthuyne, J. Crassous, S. Filippone and N. Martín, Org. Lett., 2018, 20, 1764–1767 CrossRef PubMed.
- E. E. Maroto, S. Filippone, M. Suárez, R. Martínez-Álvarez, A. de Cózar, F. P. Cossío and N. Martín, J. Am. Chem. Soc., 2014, 136, 705–712 CrossRef CAS PubMed.
- S. Seki, A. Saeki, T. Sakurai and D. Sakamaki, Phys. Chem. Chem. Phys., 2014, 16, 11093–11113 RSC.
- A. Saeki, Y. Koizumi, T. Aida and S. Seki, Acc. Chem. Res., 2012, 45, 1193–1202 CrossRef CAS PubMed.
- A. Saeki, S. Seki, T. Takenobu, Y. Iwasa and S. Tagawa, Adv. Mater., 2008, 20, 920–923 CrossRef CAS.
- S. S. Babu, M. J. Hollamby, J. Aimi, H. Ozawa, A. Saeki, S. Seki, K. Kobayashi, K. Hagiwara, M. Yoshizawa, H. Möhwald and T. Nakanishi, Nat. Commun., 2013, 4, 1969 CrossRef PubMed.
- S. Zhang, Y. Wang, X. Yao, P. Le Floch, X. Yang, J. Liu and Z. Suo, Nano Lett., 2020, 20, 4580–4587 CrossRef CAS PubMed.
- A. Shinohara, M. Yoshida, C. Pan and T. Nakanishi, Polym. J., 2023, 55, 529–535 CrossRef CAS.
- R. Liu, Z. L. Wang, K. Fukuda and T. Someya, Nat. Rev. Mater., 2022, 7, 870–886 CrossRef.
- X. Zheng, R. K. Gupta and T. Nakanishi, Curr. Opin. Colloid Interface Sci., 2022, 62, 101641 CrossRef CAS.
- T. Machida, R. Taniguchi, T. Oura, K. Sada and K. Kokado, Chem. Commun., 2017, 53, 2378–2381 RSC.
- K. Kushwaha, L. Yu, K. Stranius, S. K. Singh, S. Hultmark, M. N. Iqbal, L. Eriksson, E. Johnston, P. Erhart, C. Müller and K. Börjesson, Adv. Sci., 2019, 6, 1801650 CrossRef PubMed.
- F. Lu, K. Jang, I. Osica, K. Hagiwara, M. Yoshizawa, M. Ishii, Y. Chino, K. Ohta, K. Ludwichowska, K. J. Kurzydłowski, S. Ishihara and T. Nakanishi, Chem. Sci., 2018, 9, 6774–6778 RSC.
- A. Nowak-Król, D. Gryko and D. T. Gryko, Chem. – Asian J., 2010, 5, 904–909 CrossRef PubMed.
- K. Kubota, S. Hirata, Y. Shibano, O. Hirata, M. Yahiro and C. Adachi, Chem. Lett., 2012, 41, 934–936 CrossRef CAS.
- K. Chung, M. S. Kwon, B. M. Leung, A. G. Wong-Foy, M. S. Kim, J. Kim, S. Takayama, J. Gierschner, A. J. Matzger and J. Kim, ACS Cent. Sci., 2015, 1, 94–102 CrossRef CAS PubMed.
- M. J. Hollamby, M. Karny, P. H. H. Bomans, N. A. J. M. Sommerdjik, A. Saeki, S. Seki, H. Minamikawa, I. Grillo, B. R. Pauw, P. Brown, J. Eastoe, H. Möhwald and T. Nakanishi, Nat. Chem., 2014, 6, 690–696 CrossRef CAS PubMed.
- E. A. Neal and T. Nakanishi, Bull. Chem. Soc. Jpn., 2021, 94, 1769–1788 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3mh00485f |
| This journal is © The Royal Society of Chemistry 2023 |