
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelective deconstruction of mixed plastics by a tailored organocatalyst†
Md
Arifuzzaman
a,
Bobby G.
Sumpter
 b,
Zoriana
Demchuk
a,
Changwoo
Do
c,
Mark A.
Arnould
b,
Md Anisur
Rahman
a,
Peng-Fei
Cao
a,
Ilja
Popovs
a,
Robert J.
Davis
d,
Sheng
Dai
a and
Tomonori
Saito
*a
b,
Zoriana
Demchuk
a,
Changwoo
Do
c,
Mark A.
Arnould
b,
Md Anisur
Rahman
a,
Peng-Fei
Cao
a,
Ilja
Popovs
a,
Robert J.
Davis
d,
Sheng
Dai
a and
Tomonori
Saito
*a
aChemical Sciences Division, Oak Ridge National Laboratory, Oak Ridge, TN 37831, USA. E-mail: saitot@ornl.gov
bCenter for Nanophase Materials Sciences, Oak Ridge National Laboratory, Oak Ridge, TN 37831, USA
cNeutron Scattering Division, Oak Ridge National Laboratory, Oak Ridge, TN 37831, USA
dDepartment of Chemical Engineering, The University of Virginia, Charlottesville, VA 22904-4741, USA
First published on 24th July 2023
Abstract
Plastic represents an essential material in our society; however, a major imbalance between their high production and end-of-life management is leading to unrecovered energy, economic hardship, and a high carbon footprint. The adoption of plastic recycling has been limited, mainly due to the difficulty of recycling mixed plastics. Here, we report a versatile organocatalyst for selective glycolysis of diverse consumer plastics and their mixed waste streams into valuable chemicals. The developed organocatalyst selectively deconstructs condensation polymers at a specific temperature, and additives or other polymers such as polyolefin or cellulose can be readily separated from the mixed plastics, providing a chemical recycling path for many existing mixed plastics today. The Life Cycle Assessment indicates that the production of various condensation polymers from the deconstructed monomers will result in a significant reduction in greenhouse gas emissions and energy input, opening a new paradigm of plastic circularity toward a net-zero carbon society.
New conceptsThe recycling of mixed plastics is a complex issue because of varied physical properties among different types of plastics. Chemical recycling by catalytic deconstruction provides a path to convert plastic waste to pristine chemicals, but no single catalyst or technology has ever transformed a mixture of multiple plastics selectively into valuable chemicals. In this research, we demonstrate a new method for the selective deconstruction of mixed plastics by a tailored organocatalyst. The exceptional efficiency of the organocatalyst enables selective and sequential deconstruction of the condensation polymers at a specific temperature, while additives or other plastics such as polyolefin and cellulose are kept intact. The ability of sequential deconstruction coupled with facile separation of the mixed inert components at specific temperatures provides a new recycling path for a variety of currently unrecyclable mixed plastics. This study highlights that the versatile organocatalyst can convert diverse consumer plastics and their mixed waste streams into valuable chemicals. The concept offers highly efficient and low-carbon closed-loop chemical recycling and presents a promising strategy for combating global plastic waste challenges toward a sustainable and carbon neutral society. |
Introduction
Plastics have transformed everyday life because of their robust mechanical properties, lightweight, scalability, versatility, and functional performance.1 10.5 billion metric tons of plastic have been produced worldwide, and the annual production rate reached more than 400 million metric tons (Mt) in 2021.2,3 Due to inadequate recycling paths, plastic-derived waste causes a global challenge for end-of-life management.4–9 Nearly 79% of produced plastics are estimated to enter landfills or accumulate in the natural environment, and 12% are incinerated.1 Only ∼9% of plastic waste has been recycled typically by mechanical recycling, and most recycled plastics are currently down-cycled toward less recyclable and lower-value products.10–13 If the high production and consumption of plastic continue at the current rate, the annual production of plastics is expected to reach more than 1100 Mt by 2050.14 Commodity plastics used yearly in the USA alone account for an estimated 3.2 quadrillion BTUs (quads) of energy and 104 Mt CO2 equivalent of greenhouse gas (GHG) emissions.15,16 One of the most effective strategies to mitigate GHG emissions includes establishing closed-loop circularity for plastics, such as chemical recycling, to exchange the fossil carbon feedstock and minimize the energy and carbon inputs in the entire plastic supply chain.17–20 Significant scientific advancement is needed to achieve the closed-loop circularity of plastics by replacing fossil-based carbon feedstocks with bio-based or waste-sourced feedstocks from the chemical recycling of discarded plastics to a much greater extent. Establishing circular plastic recycling on a global scale is estimated to alleviate energy consumption by an amount equivalent to 3.5 billion barrels of oil, valued at approximately $176 billion annually.8Among various commodity plastics, condensation polymers such as poly(ethylene terephthalate) (PET), poly(carbonate) (PC), poly(urethane) (PU), and poly(amide) (PA) comprise ∼128 Mt, which is equivalent to 32% of the global plastic production, with PETs and PUs being the 5th and 6th most-produced plastics, respectively.13,21 The chemical recycling of condensation polymers by methods such as glycolysis is an evolving and versatile route for creating value-added materials from plastic waste or regenerating essential monomers for use in plastic circularity. Several innovative solutions such as metal-based catalysts, ionic liquids, or deep-eutectic solvents have been recently reported to allow depolymerization of individual polar plastic (PET, PC, PU, and PA) into monomer products (Fig. 1A).7,9,22–25 Despite the advancements, many challenges remain, including the possible presence of metal in the final product, limited monomer yields, requirement of a unique catalyst for deconstructing each type of polymer, laborious sorting (Fig. 1A), and purification procedures that entail high environmental and economic costs.26 A few catalysts have been shown to be effective against both PET and PC,27,28 but a specific individual catalyst has been needed for the deconstruction of PU29 and PA,30 respectively. Until now, no catalyst has been shown to be effective against all four: PC, PET, PU, and PA.
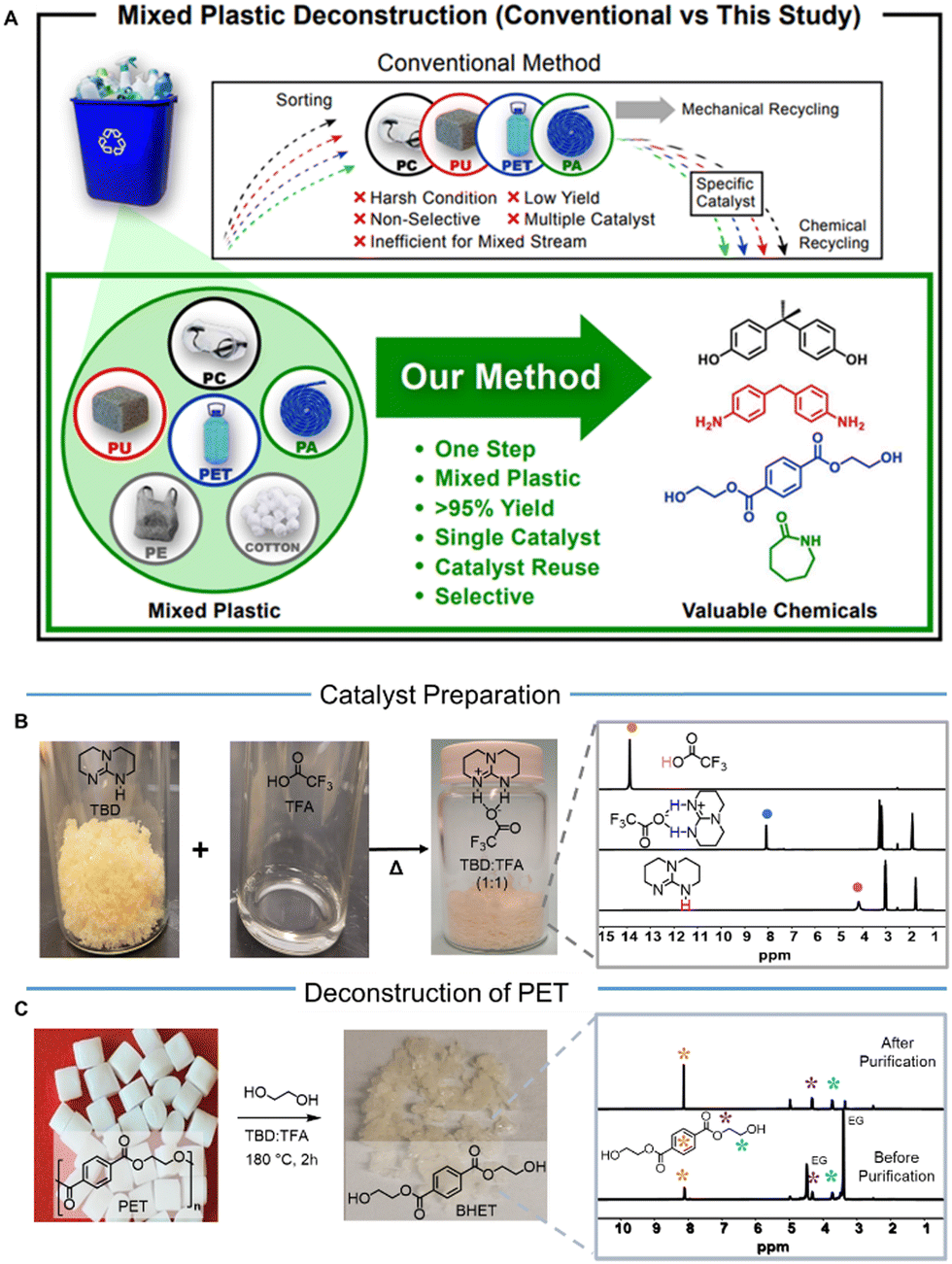 | ||
| Fig. 1 Methods of mixed plastics deconstruction, and the use of synthesized TBD:TFA for PET deconstruction. (A) Conventional methods include sorting individual polymers, followed by mechanical or chemical recycling with a specific catalyst (top arrow). Our method: Efficient deconstruction of mixed plastics (PC, PU, PET, and PA) by a tailored organocatalyst to produce valuable chemicals. (B) Formation of TBD:TFA. 1H NMR spectra show the disappearance of a peak at δ13.84 ppm for TFA (right, top) and shifting from δ4.14 ppm (right, bottom) for TBD to δ8.12 ppm, indicating the formation of the TBD:TFA. (right, middle). (C) Deconstruction of PET by using TBD:TFA to yield a pure BHET crystal through recrystallization in water. 1H NMR spectra before (right, bottom) and after (right, up) purification show pure BHET, and the successful removal of the catalyst and excess EG. | ||
Organocatalysts provide many advantages for deconstructing condensation polymers. Their greener characteristics of good selectivity, thermal stability, non-volatility, and low flammability enable them to substitute traditional heterogeneous catalysts.31–34 Protic ionic salt (PIS) based organocatalysts are ideal candidates for the deconstruction of condensation polymers compared to a conventional organic base or acid that can be readily deactivated or degraded at elevated temperature.35 The catalytic activity of PIS is governed by a dual-activation mechanism, where the anion activates the nucleophile and the cation activates the electrophile (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond).28 One of the PIS catalysts, triazabicyclododecane:methanesulfonic acid (TBD:MSA) has been the most efficient catalyst reported for the deconstruction of PC and PET individually and their mixture.31 The development of TBD:MSA has shed light on the design of PIS organocatalysts, but a significant challenge remains for implementing them in a wide variety of condensation polymers and their mixtures, as well as various other mixed plastics.13,36,37 Mixed plastics are difficult to recycle from post-consumer waste because of the need for sorting and separation before reprocessing. Lack of pre-sorting typically results in the unselective and inefficient deconstruction of mixed plastics, leading to an undesirable complex mixture of products.29,32 From a raw material cost perspective, manufacturing new plastic products from virgin feedstocks is typically more cost-efficient than sorting and reusing reprocessed material. Increased mixed-waste streams of multicomponent plastic, such as composites and packaging, exacerbate the low recycling rate. There are a few recently developed catalysts that can convert mixed PET to chemicals38 as well as mixed PE, PS, and PET into a single product through multiple steps.39 However, there is no single catalyst or technology that has converted multiple types of individual and mixed condensation polymers selectively into valuable chemicals. Furthermore, when mixed plastics are deconstructed to valuable chemicals, their facile separation process needs to be simultaneously developed. One of the most effective routes is to use sequential plastic deconstruction,28 that simultaneously allows facile sequential separation of individual monomers and polymers from the mixed waste. Thus, it is imperative to develop a more robust single catalyst that can sequentially deconstruct wide variety of mixed plastic waste in a single batch and allow recovering valuable individual chemicals consecutively while leaving other unreacted plastics intact.
O bond).28 One of the PIS catalysts, triazabicyclododecane:methanesulfonic acid (TBD:MSA) has been the most efficient catalyst reported for the deconstruction of PC and PET individually and their mixture.31 The development of TBD:MSA has shed light on the design of PIS organocatalysts, but a significant challenge remains for implementing them in a wide variety of condensation polymers and their mixtures, as well as various other mixed plastics.13,36,37 Mixed plastics are difficult to recycle from post-consumer waste because of the need for sorting and separation before reprocessing. Lack of pre-sorting typically results in the unselective and inefficient deconstruction of mixed plastics, leading to an undesirable complex mixture of products.29,32 From a raw material cost perspective, manufacturing new plastic products from virgin feedstocks is typically more cost-efficient than sorting and reusing reprocessed material. Increased mixed-waste streams of multicomponent plastic, such as composites and packaging, exacerbate the low recycling rate. There are a few recently developed catalysts that can convert mixed PET to chemicals38 as well as mixed PE, PS, and PET into a single product through multiple steps.39 However, there is no single catalyst or technology that has converted multiple types of individual and mixed condensation polymers selectively into valuable chemicals. Furthermore, when mixed plastics are deconstructed to valuable chemicals, their facile separation process needs to be simultaneously developed. One of the most effective routes is to use sequential plastic deconstruction,28 that simultaneously allows facile sequential separation of individual monomers and polymers from the mixed waste. Thus, it is imperative to develop a more robust single catalyst that can sequentially deconstruct wide variety of mixed plastic waste in a single batch and allow recovering valuable individual chemicals consecutively while leaving other unreacted plastics intact.
Here, we have developed an efficient PIS organocatalyst for the deconstruction of condensation polymers, which was formed from TBD and trifluoracetic acid (TFA) named TBD:TFA. While the use of TBD:TFA as a catalyst was reported for N-formylation of amines,40 its application to polymer deconstruction has never been explored. TFA is specifically chosen because the TFA anion (TFA−) provides high conjugate basicity41 with weaker ionic interaction which is desirable for enhancing the reactivity of ethylene glycol (EG) in glycolysis. The design and efficiency of TBD:TFA stem from the expected coordination ability of protonated TBD (TBDH+) toward the carbonyl (CO) group, high conjugate basicity of TFA−, and their comparatively weaker ionic interaction, which enhances the reactivity during glycolysis. The designed organocatalyst can deconstruct multiple condensation polymers selectively and sequentially into corresponding monomers, while keeping other polymers, including polyolefins (e.g., polyethylene (PE) and polypropylene (PP)) or cotton intact that allow facile separation from the mixture. The developed chemical deconstruction process can be applied to the majority of existing mixed plastics today, and it provides a viable step toward the closed-loop circularity of plastic and carbon neutrality.
Results and discussion
We have designed a PIS-based organocatalyst formed from an equimolar amount of TBD and TFA, (TBD:TFA) (Fig. 1B and ESI,† Section S3.1.1), where the structure was confirmed by Fourier-transform infrared (FTIR) spectroscopy (Fig. S15, ESI†), proton nuclear magnetic resonance (1H NMR) spectroscopy (Fig. 1B and Fig. S16, ESI†), and carbon nuclear magnetic resonance (13C NMR) spectroscopy (Fig. S17, ESI†). The comparatively higher thermal stability and melting point of TBD:TFA (T50% = 215 °C, Tm = 157 °C) indicate the formation of thermally stable PIS as compared to those of TBD (T50% = 175 °C, Tm = 125 °C) and TFA (T50% = 50 °C, Tm = −15 °C) individually (Fig. S18A and B, ESI†). Using TBD:TFA as a catalyst, we first investigated the heterogeneous glycolysis of PET (Mw 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1) with EG as a reactant and solvent at 180 °C for 2 h to yield bis(2-hydroxyethyl) terephthalate (BHET) (Fig. 1C), which was purified through a successive precipitation method followed by recrystallization in water (ESI,† Section S4.1). 1H NMR spectroscopy was used to track the yields of the respective monomers by using the catalyst peak at 1.88 ppm as an internal standard.28 The PET deconstruction efficiency (ESI,† Section S4.2) by the TBD:TFA catalyst was evaluated as a function of EG content (5 to 20 eq.) (Fig. S2A and S19, ESI†), catalyst amount (0.05 to 0.5 eq.) (Fig. S2B and S20, ESI†), and temperature (150 to 180 °C) (Fig. S2C and S21–S24, ESI†). PET pellets are deconstructed to yield more than 96% of BHET with a small mixture of oligomer (<4%) (Table S1, ESI†) at much lower catalyst loading (5 mol%, 0.05 eq.) and less amount of EG (10 eq.) at 180 °C within 2 h, compared to the reported optimized condition of PET glycolysis by TBD:MSA, where ten times catalyst (50 mol%, 0.5 eq.) and twice the amount of EG (20 eq.) are used to yield ∼90% BHET monomer.31 We compare TBD:TFA and TBD:MSA at the same loading of catalyst (0.05 eq.) and EG (10 eq.) for the deconstruction of PET to BHET at 180 °C in 2 h (ESI,† Section SS4.3). TBD:TFA shows much higher efficiency, exhibiting 100% conversion, while TBD:MSA yields ∼60% conversion (Table S2, ESI†). We also performed the same experiment using TBD:TFA as a catalyst with various molecular weights of PET (ESI,† Section S4.4 and Table S3) as well as a larger scale of PET (10 g, ESI,† Section S4.5), and no significant differences were observed in the efficiency of PET deconstruction.
000 g mol−1) with EG as a reactant and solvent at 180 °C for 2 h to yield bis(2-hydroxyethyl) terephthalate (BHET) (Fig. 1C), which was purified through a successive precipitation method followed by recrystallization in water (ESI,† Section S4.1). 1H NMR spectroscopy was used to track the yields of the respective monomers by using the catalyst peak at 1.88 ppm as an internal standard.28 The PET deconstruction efficiency (ESI,† Section S4.2) by the TBD:TFA catalyst was evaluated as a function of EG content (5 to 20 eq.) (Fig. S2A and S19, ESI†), catalyst amount (0.05 to 0.5 eq.) (Fig. S2B and S20, ESI†), and temperature (150 to 180 °C) (Fig. S2C and S21–S24, ESI†). PET pellets are deconstructed to yield more than 96% of BHET with a small mixture of oligomer (<4%) (Table S1, ESI†) at much lower catalyst loading (5 mol%, 0.05 eq.) and less amount of EG (10 eq.) at 180 °C within 2 h, compared to the reported optimized condition of PET glycolysis by TBD:MSA, where ten times catalyst (50 mol%, 0.5 eq.) and twice the amount of EG (20 eq.) are used to yield ∼90% BHET monomer.31 We compare TBD:TFA and TBD:MSA at the same loading of catalyst (0.05 eq.) and EG (10 eq.) for the deconstruction of PET to BHET at 180 °C in 2 h (ESI,† Section SS4.3). TBD:TFA shows much higher efficiency, exhibiting 100% conversion, while TBD:MSA yields ∼60% conversion (Table S2, ESI†). We also performed the same experiment using TBD:TFA as a catalyst with various molecular weights of PET (ESI,† Section S4.4 and Table S3) as well as a larger scale of PET (10 g, ESI,† Section S4.5), and no significant differences were observed in the efficiency of PET deconstruction.
The catalytic activities of TBD:TFA at different ratios, including TBD:TFA of 1:0, 1:1, 1:3, 3:1, and 0:1 (ESI,† Section S4.6 and Table S4), were evaluated to deconstruct PET in the presence of EG for 2 h at 180 °C. Analysis of the final crude products by 1H NMR spectroscopy reveals that the reactions with TFA alone or excess TFA (3 eq.) deconstruct PET only 0% and 20%, respectively. In contrast, the TBD alone and excess TBD (3 eq.) resulted in 60% and 100% conversion, respectively, but yielded a mixture of products (31% and 42%, respectively) besides the BHET monomer (Table S4 and Fig. S25, ESI†). By using a 1:1 TBD:TFA as a catalyst and EG as reactant and solvent, glycolysis of PET yielded BHET monomer as a major product (>96%) (Fig. 1C and Fig. S26, ESI†). The 1:1 TBD:TFA ratio exhibited the highest efficiency compared with other ratios and will be named TBD:TFA for the rest of the manuscript. The absence of catalyst peaks in 1H NMR spectra of produced BHET indicates (Fig. S27, ESI†) nearly complete removal of the catalyst by facile crystallization of BHET, while there is still a possibility of containing an undetectable trace amount of residual catalyst. The formation of BHET as a major product in the PET deconstruction is also confirmed by Matrix-Assisted Laser Desorption/Ionization-Time Of Flight mass spectrometry (MALDI-TOF MS) (ESI,† Section S1.2 and Fig. S28), and Small-Angle Neutron Scattering (SANS) (ESI,† Section S4.7) and High-Performance Liquid Chromatography (HPLC) (Fig. S53, ESI†).
To understand the mechanism of TBD:TFA catalysis, all-electron density functional theory (DFT) calculations using the hybrid meta functional m06-2x and the aug-cc-pvdz basis set42,43 were performed on a model PET chain of eight monomers (ESI,† Section S4.8) interacting with TBDH+ and TFA−. These calculations are intended to provide useful insight and qualitative mechanisms. First, we note that the TBD:TFA interaction energy in a continuum solvent model44 was calculated to be 13.6 kcal mol−1 (ΔG = 3.4 kcal mol−1), and ab initio molecular dynamics (MD) shows that the TBD:TFA complex can be successfully dissociated into TBDH+ and TFA− (Fig. 2A; step 1, Fig. 2B, Movie S1, ESI†) at a relatively low temperature. Once dissociated into the cation and anion, DFT results show the formation of a stable complex between TBDH+ and PET with an interaction energy of 24.7 kcal mol−1. At the same time, the TFA− prefers forming a complex with EG at an interaction energy of 30.4 kcal mol−1 (Fig. 2A; step 2, Fig. 2C). Successive bond formation (Fig. 2A; step 3, Fig. 2D) and bond-breaking (Fig. 2E) yield oligomers and finally monomers. When we compare these DFT results with those of the TBD:MSA complex, several notable differences are obtained. First, the interaction energy between TBD and MSA is slightly higher (14.8 kcal mol−1). Second, the MSA anion forms a much stronger interaction with PET at 42 kcal mol−1 than that of TFA− at only 13 kcal mol−1. Third, the transition state barriers for TBD:TFA (1st transition (Fig. 2 step 1): 13.6 kcal mol−1 and 2nd transition (Fig. 2 step 3): 14.4 kcal mol−1) are lower as compared to those reported for TBD:MSA (1st transition: 18.8 kcal mol−1 and 2nd transition: 20.5 kcal mol−1).28 We note that a PET chain, e.g., not just a small molecule model, is required to observe this difference, thus indicating a fundamental role of PET conformations. The overall kinetics between TBD:TFA and TBD:MSA for PET deconstruction rely on (i) the interaction between EG and the anion and (ii) the interaction between TBDH+ and PET through hydrogen-bonding to activate the electrophilicity of the O–CO bond. The stronger interaction of the MSA anion with PET (42 kcal mol−1) appears to hinder the activation of both EG and TBDH+ interactions with O–CO on PET, thus decreasing the reaction rate. On the other hand, the smaller interaction between the TFA− and PET (13 kcal mol−1) and strong conjugate basicity accelerates the activation of EG and interaction between TBDH+ and O–CO on PET, thus increasing the overall catalytic activity of TBD:TFA.
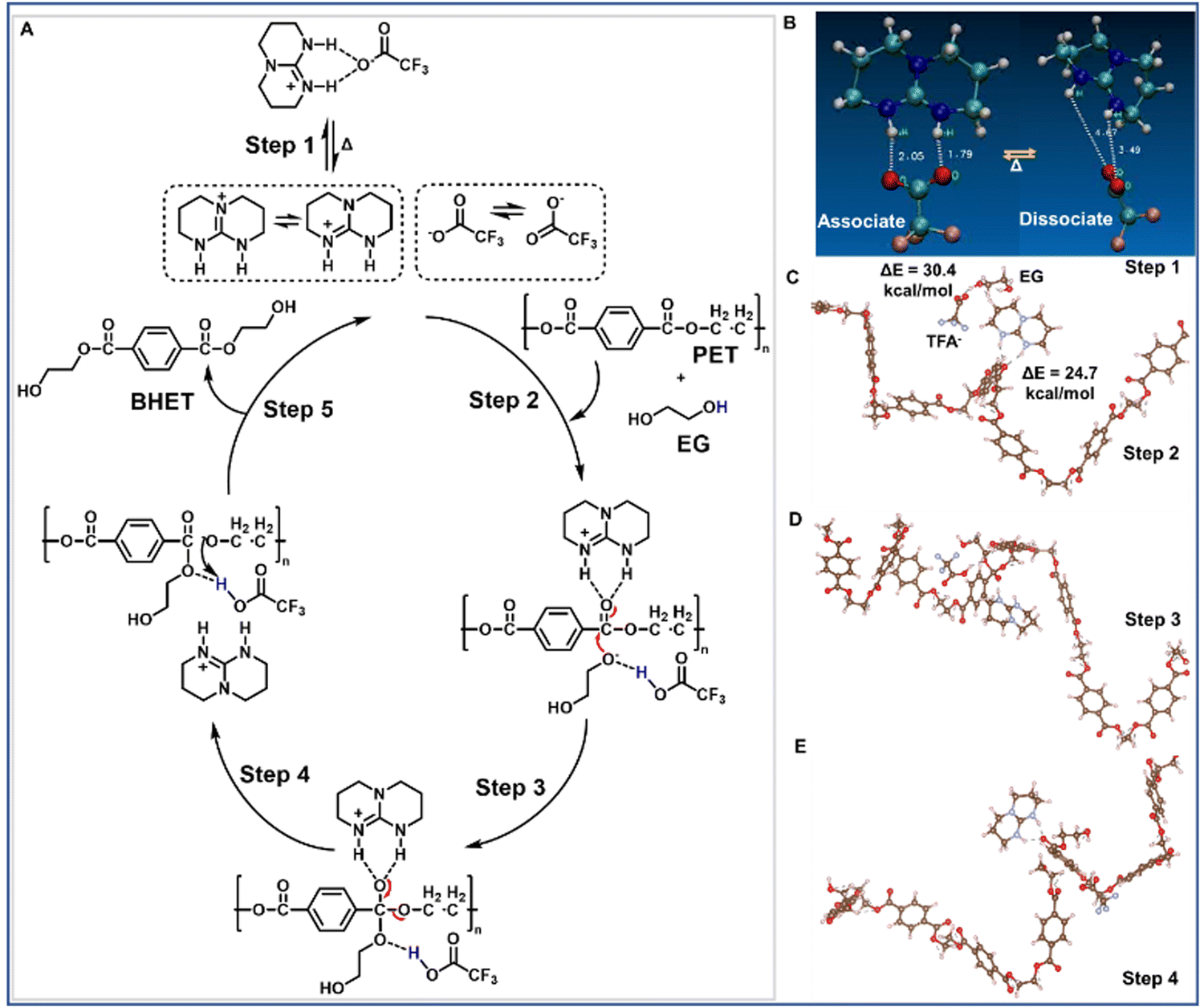 | ||
| Fig. 2 Proposed deconstruction mechanism of PET with EG using TBD:TFA as a catalyst based on the computational analysis. (A) Dual activation mechanism for the deconstruction of PET, using EG as a nucleophile, and TBD:TFA as a catalyst, yielding BHET as a product. (B) Association and dissociation between TBDH+ and TFA− (C) Interaction between TBDH+ with PET (ΔE = 24.7 kcal mol−1) and the TFA− with EG (ΔE = 30.4 kcal mol−1), (D) Simultaneous chemical bond formation between PET and EG and proton abstraction by the TFA−. (E) Cleavage of the PET to form oligomers and finally monomers. | ||
The catalytic activity of TBD:TFA arises from a dual-activation mechanism by activating both EG and the polymer chain (Fig. 2). The TFA− activates the nucleophile to be inserted, while the TBDH+ activates the polymer chain to be broken by coordinating with the CO group of the polymer chain. To further understand the catalytic activity and the mechanism, we synthesized two more catalysts (ESI,† Section S3.1.2) as controls named TBD:mTFA (Fig. S29, ESI†) and mTBD:TFA (Fig. S30, ESI†), where mTFA and mTBD denote methyl-TFA and methyl-TBD, respectively. The kinetic study of PET glycolysis at 180 °C for 0–2 h using TBD:mTFA and mTBD:TFA as well as TBD:TFA, TBD, and TBD:MSA is conducted. (Fig. 3A). After 2 h deconstruction, TBD:mTFA and mTBD:TFA exhibits less conversion (50% and 32%, respectively), and lower BHET yields (65% and 48%, respectively) compared to those of TBD:TFA (100% conversion with >96% BHET yield) (Table S4 and Fig. S31, ESI†), due to the diminished proton accepting capability of mTBD and the absence of a labile proton in mTFA. DFT calculations give ΔG = 23.8 kcal mol−1 for mTBD:TFA to form the mTBD cation and TFA anion, which is significantly higher than 13.6 kcal mol−1 of TBD:TFA, thus corresponding to the diminished yield (Fig. 3A) and further confirming the proposed reaction mechanism (Fig. 2). Overall the high deconstruction efficiency by TBD:TFA compared with TBD:mTFA, mTBD:TFA, TBD, and TBD:MSA further indicates the importance of TBDH+ and TFA− presence in the glycolysis mechanism.
 | ||
| Fig. 3 Catalytic activity of TBD:TFA. (A) Catalytic activity of TBD:TFA (gold) in PET deconstruction compared to TBD:MSA (purple), TBD (black), TBD:mTFA (red), and mTBD:TFA (orange) at 180 °C. (B) Deconstruction of PC (black), PU (red), PET (blue), and PA (green) to yield corresponding monomer BPA, MDA, BHET, and CPL, respectively, (C) Deconstruction of PC, PU, PET, and PA using TBD:TFA for 2 h at specific temperature of 110-210 °C to yield corresponding monomer (D) Recyclability of the catalyst among PC, PU, PET, and PA deconstruction at 180 °C for 2 h, where 1st to 5th indicates the number of deconstruction cycles. *Reagent ratio for A–D; Polymer (1 eq.), EG (10 eq.), Cat. (0.05 eq.). | ||
The efficient deconstruction mechanism of PET by TBD:TFA suggests that TBD:TFA can be applied to cleave all types of functional linkages in condensation polymers (Fig. 3B). The deconstruction of PC (Bisphenol-A based PC, Mw 45000 g mol−1) (ESI,† Section S4.9), thermoplastic PU (Diphenylmethylene-ethylene glycol based PU, Mw 24600 g mol−1) (ESI,† Section S4.10), PET (Mw 40000 g mol−1) and PA (Nylon 6, Mw 35000 g mol−1) (ESI,† Section S4.11) was conducted with the presence of the TBD:TFA catalyst and EG. The structural differences of carbonate, urethane, ester, and amide groups in PC, PU, PET, and PA provide sufficiently different thermodynamics to cause a tailored rate of deconstruction. PC, PU, PET, and PA were first deconstructed individually using 10 eq. of EG and 0.05 eq. of TBD:TFA catalyst for 2 h at a specific temperature between 110–210 °C (Fig. 3C). The complete deconstruction within 2 h was demonstrated in PC to bisphenol A (BPA) at 130 °C (Fig. 3C and Fig. S32–S36, Table S5, ESI†), PU to 4,4′-methylenedianiline (MDA) at 160 °C (Fig. 3C and Fig. S37–S41, Table S6, ESI†), PET to BHET at 180 °C (Fig. 3C and Fig. S42–S48, ESI†), and PA to caprolactam (CPL) at 210 °C (Fig. 3C and Fig. S49–S52, Table S7, ESI†). In addition to the 1H NMR spectra, HPLC chromatograms further confirmed the formation of respective monomers as major products (Fig. S53, ESI†). The deconstruction ability of the TBD:TFA catalyst for multiple condensation polymers provides versatility in their chemical recycling.
Reusing catalysts in polymer deconstruction processes provides benefits in reduced costs, increased efficiency, less environmental impact, and greater flexibility in production. Thus, we investigated recyclability of TBD:TFA catalyst and EG in PC, PU, PET, and PA deconstruction using two techniques (Fig. 3D and ESI† Section S4.12). In the first method, we separated the mixture of EG and the catalyst after each cycle and used it again for the following deconstruction. In the second method, we added a new batch of polymer (e.g., PET) without separating the catalyst. Since the deconstructed monomers, EG, and catalyst are soluble in water, the deconstruction solution is first transferred into water and any impurities such as pigments can be readily removed by filtration. Then, the monomer is separated at a high yield (>90%) by recrystallization or extraction at each 1st to 5th cycle, leaving EG and the catalyst in the water. The mixture of EG and the catalyst (>95%) is recovered by evaporating water and directly used for the following catalytic deconstruction cycle (Fig. S54–S57, ESI†). In addition to reusability, our TBD:TFA catalyst exhibits high stability under kinetically controlled conditions over five cycles (Fig. S8C, ESI†), which also manifests high activity of TBD:TFA catalyst. Comparatively, when the recyclability of a TBD catalyst is evaluated for PET deconstruction under the same conditions, the yield of BHET drops from 45% to 10% from the 1st to the 5th cycle (ESI,† Section S4.12) most likely due to the limited thermal stability of the TBD (Fig. S18A, ESI†). In addition, since water tends to harm the efficiency of organocatalysts, we performed the deconstruction of PET in the presence of water to understand the impact of water on the TBD:TFA catalyst efficiency (ESI,† Section S4.12). The results from 1H NMR (Fig. S58–S61, ESI†) and HPLC (Fig. S62, ESI†) also indicate that the deconstruction occurs via glycolysis over hydrolysis even with the presence of 30% water likely due to the presence of more EG (10 equivalents) than water (3 equivalents) in the reaction.
The structural stability among carbonate, urethane, ester, and amide functional groups in PC, PU, PET, and PA, respectively, provides a distinctively different rate of deconstruction that allows selective and sequential deconstruction of mixed PC, PU, PET, and PA with the TBD:TFA catalyst. Thus, the concurrent glycolysis of assorted PC, PU, PET, and PA was investigated (Fig. 4A–E and ESI† Section S4.13). All polymer pellets (1.3 mmol of each, equivalent to 0.33 g PC, 0.41 g PU, 0.25 g PET, and 0.15 g PA) were mixed in the same vial with EG (10 eq.) and TBD:TFA (0.05 eq.), and the successive deconstruction was monitored at different temperatures. The kinetics on the degree of deconstruction was quantified by 1H NMR spectroscopy using each yielded molecule's characteristic signals (Fig. 4B′–E′). At 130 °C, only PC was deconstructed, yielding 97% of BPA after 2 h (Fig. 4B, B′ and Fig. S63, ESI†), which demonstrates the capability to selectively deconstruct PC in the co-presence of PU, PET, and PA (Fig. 4B). By increasing the temperature to 160 °C, PU is deconstructed to MDA, and a small quantity of PET is also deconstructed to BHET (Fig. 4C, C′ and Fig. S64, ESI†). Further elevation of the temperature to 180 °C deconstructs the rest of the PET to yield BHET (Fig. 4D, D′ and Fig. S65, ESI†). Finally, at 210 °C, PA is deconstructed into CPL (Fig. 4E, E′ and Fig. S66, ESI†). Alternately, direct heating of combined PC, PU, PET, and PA at 210 °C deconstructs all the polymers, resulting in a mixture of respective monomers (Fig. 4V), that could be separated through known separation methods such as fractional distillation or CombiFlash®. The ability to deconstruct these mixed condensation polymers provides a clear path to eliminate costly sorting and allows for recovering valuable chemicals from mixed plastics waste.
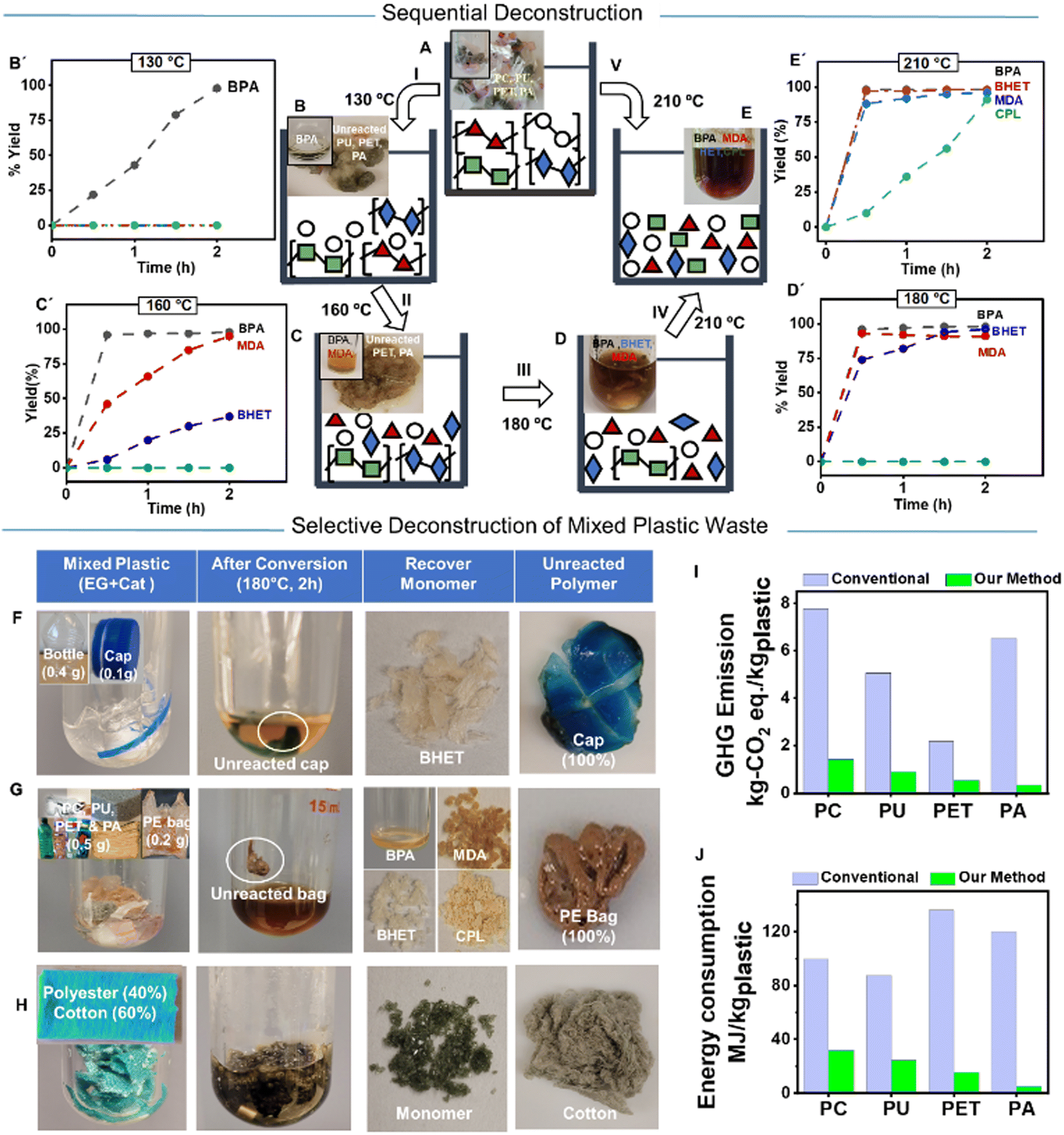 | ||
| Fig. 4 Sequential and selective deconstruction of mixed plastics. (A)–(E) Sequential deconstruction of mixed PC, PU, PET, and PA using TBD:TFA (0.05 eq.) and EG (10 eq.) for 2 h. At 130 °C (I), PC fully converts to BPA while PU, PET, and PA are kept intact (B & B'). At 160 °C (II), PU fully converts to MDA with a small conversion of PET while PA is kept intact (C & C'). At 180 °C (III), PET fully converts to BHET while keeping PA unreacted (D & D'). Finally, at 210 °C (IV), the remaining unreacted PA fully converts to CPL (E & E′). Alternatively, directly heating at 210 °C (V) can deconstruct PC, PU, PET, and PA once to yield the mixture of corresponding BPA, MDA, BHET, and CPL. (F) Deconstruction of a PET bottle and a PP cap mixture produces BHET with keeping PP intact. (G) The combination of PC, PU, PET, and PA consumer products with a piece of PE bag is deconstructed to produce corresponding monomer BPA, MDA, BHET, and CPL, respectively, while keeping PE intact, (H) Deconstruction of fabric based on polyester (40%) and cotton (60%) to produce BHET monomer, while unreacted cotton can be readily separated. (I) Carbon footprint and (J) energy footprint of PC, PU, PET, and PA production by conventional petroleum-based approach against the reconstruction of PC, PU, PET, and PA using the deconstructed monomer by TBD:TFA-based process. | ||
The properties of plastics are often tailored to the respective application by blending different additives during their manufacturing, and the mixed state makes the deconstruction of discarded plastics an even greater challenge. Thus, during our investigation, the efficacy of the TBD:TFA catalyst was further evaluated by deconstructing selected consumer products based on PC, PU, PET, and PA (ESI,† Section S5.1 and Table S8). First, commercially available colorful PET water bottles were cut into small pieces (0.5 g) and deconstructed by loading EG (10 eq.) and TBD:TFA (0.05 eq.) at 180 °C for 2 h (Movie S2, ESI†). The water bottle was fully converted to BHET (Table S8A and Fig. S67, ESI†). Both a polyester-based carpet (Table S8B and Fig. S68, ESI†) and cloth (Table S8C, ESI†) were deconstructed in the same deconstruction condition and exhibited high or complete conversion, though the precise degree of conversion is difficult to determine due to the unknown composition of the starting material. Similarly, a PC-based consumer product, safety goggles, were fully converted to yield pure BPA (Table S8D and Fig. S69, ESI†). Thermoset PU foam (Table S8E, ESI†) and a nylon rope (Table S8F, ESI†), as consumer products of PU and PA, were also thoroughly deconstructed to yield the corresponding monomers (Fig. S70 and S71, respectively, ESI†). These results indicate the broad applicability of TBD:TFA for deconstructing various consumer products and the negligible influence of polymer additives or color pigments on the catalyst activity.
The deconstruction of mixed consumer plastics waste via selective glycolysis was further investigated (ESI,† Section S5.2). A mixture of safety goggles (PC), PU foam, PET water bottle, and nylon rope (PA) (0.5 g each) from customary sources was placed in a reaction vessel, and successive heat was applied by using EG (10 eq.) and TBD:TFA (0.05 eq.) for 2 h to validate the catalyst efficiency in mixed consumer plastics waste. Reaction temperature of 130 °C led to the complete conversion of PC waste to BPA, while PU, PET, and PA waste remained unconverted. Because of its insolubility in EG, the unreacted PU, PET, and PA waste can be easily filtered off from the reaction solution and proceed with deconstruction in a subsequent reaction. Increasing the temperature to 160 °C resulted in the complete conversion of PU foam to MDA. At an elevated temperature of 180 °C, the PET water bottle is fully converted to BHET. The nylon rope is intact after 160 °C or 180 °C deconstruction conditions; thus, it can be easily separated. When the residue is heated to 210 °C, the nylon rope is fully deconstructed to CPL. Alternatively, we used the TBD:TFA catalyst to deconstruct mixed plastics waste of the same consumer products in the same reactor at 210 °C for 2 h, yielding a mixture of BPA, MDA, BHET, and CPL.
Our organocatalyst, TBD:TFA, can deconstruct specific plastics (PC, PU, PET, and PA) and leave other plastics intact through their reactivity differences in the polymer structure, solvation, as well as physical and mass transport properties. To evaluate the efficiency and selectivity of TBD:TFA as a catalyst for multiple mixed wastes (ESI,† Section S5.3), we performed TBD:TFA-based glycolysis by using (i) a PET bottle with a PP cap (Fig. 4F), (ii) a PET bottle with PE bags (Fig. S14, ESI†), (iii) a mixture of PC, PU, PET, and PA based consumer products with PE bags (Fig. 4G), and (iv) fabrics consisting of 40% polyester and 60% cotton (Fig. 4H). Due to the presence of labile linkage (O–CO, ester, or –N–CO, amide), condensation polymers exhibit complete conversion to yield the respective monomer while keeping unreacted PP (100%), PE (100%), and cotton (100%). This selective deconstruction method using TBD:TFA can eliminate the need for upfront separation of mixed plastics and could also apply to multicomponent plastics, such as multi-layer packaging or textiles.
Evaluating energy and carbon inputs is critical for understanding their circular plastic and environmental impacts.18,45 Commonly, the production of virgin polymers involves a long list of processes, from raw material extraction to waste management, which has a considerable impact on embodied carbon and energy demand in the final plastic product, ranging from 1.9 to 5.8 kg CO2 eq. per kgpolymer and 51 to 176 MJ/kg, respectively.15 Recycling polymer waste and reconstructing new polymeric materials has decreased the impact on embodied carbon by a factor of 2–6 in some cases.46 Life Cycle Assessment (LCA) modeling is an integral tool to evaluate the environmental impact of the developed approach in the deconstruction of each polymer individually and the production of reconstructed polymers from the recycled monomer. We conducted a cradle-to-gate LCA to estimate the carbon and energy footprint in the chemical recycling of PC, PET, PU, and PA waste, followed by the production of PC, PET, PU, and PA (ESI,† Section S6 and Tables S9, S10). Since the use of BHET for PET synthesis, BPA for PC synthesis, MDA for PU synthesis, and CPL for PA synthesis is well established in the industry, the recovered pure monomers can be readily incorporated into the currently used plastic production process. The analysis considers any options that influence the environment by consuming resources or releasing emissions, including resource use, human health, and ecological impacts. EG and electricity are the main contributors to the environmental impacts of the polymer deconstruction process mainly affecting ozone depletion, global warming, and acidification. Due to the efficient deconstruction process of each condensation polymer with the TBD:TFA catalyst, the resulting deconstructed monomers produce the corresponding polymers with low embodied carbon values (0.34–1.41 kg CO2 eq.) and cumulative energy demand (4.96–32.0 MJ kg−1). The comparative LCA analysis shows that the synthesis of PC, PU, PET, and PA from the deconstructed monomers results in 82%, 81%, 75%, and 95% less GHG emissions (Fig. 4I and Table S11, ESI†), as well as 68%, 72%, 84%, and 94% less energy input (Fig. 4J and Table S11, ESI†) than that from the traditional petroleum-based monomers, respectively. Moreover, the comparative LCA model for mixed plastics waste of PC, PU, PET, and PA shows a reduction of 51% in fossil energy consumption compared to the combined energy demand for each polymer individually. The significant decrease in carbon and energy footprints in the reconstructed polymers using the deconstructed monomers from the TBD:TFA-based process is attributed to the combination of several factors, including the effectiveness of the catalyst, the ability to recycle the excess of EG, fast kinetics, and high yields of polymer deconstruction. Furthermore, the simultaneous recovery of intact plastics such as polyolefins and cellulose (Fig. 4F, G and H) is expected to further reduce GHG and energy input for the overall circularity of multiple plastics.
Conclusions
We have demonstrated the selective and sequential deconstruction of condensation polymers, PC, PET, PU, and PA, and their various mixed plastics to address the global challenge of plastic recycling. A wide range of post-consumer plastic waste, such as bottles, packaging, foam, carpet, etc. is readily deconstructed into monomers with high efficiency. Our approach using the TBD:TFA organocatalyst enables efficient and versatile conversion of those plastics' waste to high-value chemicals that can be readily used to reproduce high-value plastic or other valuable materials. The tailored catalyst design allows for 100% conversion of polymers to corresponding monomers within 2 h using 1/10 of the catalyst and half of EG compared to the state-of-the-art organocatalyst. Furthermore, the reagent and the catalyst are easily recyclable, demonstrating an equivalent catalytic activity even after 5 repeated reactions with the same catalyst. Moreover, the developed organocatalyst accomplished the deconstruction of various consumer plastics mixtures, enabling the selective glycolysis of condensation polymers and a facile separation path for the other intact polymers. The selective deconstruction of mixed plastics by TBD:TFA provides a feasible path for implementing in a practical large pilot scale deconstruction plant due to its high efficiency, good reproducibility, straightforward process, cost-effectiveness, and environmental viability. The approach offers a highly efficient closed-loop chemical recycling of mixed plastics by facile deconstruction and separation, leading to a more than 80% reduction in energy and carbon footprint.Author contributions
Conceptualization: MA and TS. Methodology: MA, TS, and BS. Investigation: MA, TS, BS, ZD, CD, MAR, and MAA. Visualization: TS, and MA. Funding acquisition: TS, PC, IP, BS, CD, RJD, and SD. Project administration: TS. Supervision: TS. Writing – original draft: MA, ZD, BS, and TS. Writing – review & editing: MA, BS, CD, ZD, MAA, MAR, PC, IP, RJD, SD, and TS.Conflicts of interest
M. A. and T. S. are inventors on a patent application related to this work filed by USPTO (no. 18/096,849, filed on 13 January 2023). The authors declare no other competing interests.Acknowledgements
This research was supported by the US Department of Energy, Office of Science, Basic Energy Sciences, Materials Sciences and Engineering Division. MALDI-TOF MS and DFT simulations were conducted at the Center for Nanophase Materials Sciences (CNMS), which is a US Department of Energy, Office of Science User Facility at Oak Ridge National Laboratory. A portion of this research used resources at the Spallation Neutron Source, as appropriate, a DOE Office of Science User Facility operated by the Oak Ridge National Laboratory. We thank Mr Jackie Zheng on assisting GPC measurements, and Dr Chao Guan for assisting DSC measurements.This manuscript has been authored by UT-Battelle, LLC, under contract DE-AC05-00OR22725 with the US Department of Energy (DOE). The US government retains and the publisher, by accepting the article for publication, acknowledges that the US government retains a nonexclusive, paid-up, irrevocable, worldwide license to publish or reproduce the published form of this manuscript, or allow others to do so, for US government purposes. DOE will provide public access to these results of federally sponsored research in accordance with the DOE Public Access Plan (http://energy.gov/downloads/doe-publicaccess-plan).
References
- R. Geyer, J. R. Jambeck and K. L. Law, Sci. Adv., 2017, 3, e1700782 CrossRef PubMed.
- M. Chu, Y. Liu, X. Lou, Q. Zhang and J. Chen, ACS Catal., 2022, 12, 4659–4679 CrossRef CAS.
- Global plastic production 1950-2021, 2023.
- W. W. Y. Lau, Y. Shiran, R. M. Bailey, E. Cook, M. R. Stuchtey, J. Koskella, C. A. Velis, L. Godfrey, J. Boucher, M. B. Murphy, R. C. Thompson, E. Jankowska, A. C. Castillo, T. D. Pilditch, B. Dixon, L. Koerselman, E. Kosior, E. Favoino, J. Gutberlet, S. Baulch, M. E. Atreya, D. Fischer, K. K. He, M. M. Petit, U. R. Sumaila, E. Neil, M. V. Bernhofen, K. Lawrence and J. E. Palardy, Science, 2020, 369, 1455–1461 CrossRef CAS PubMed.
- L. T. J. Korley, T. H. Epps, B. A. Helms and A. J. Ryan, Science, 2021, 373, 66–69 CrossRef CAS PubMed.
- M. MacLeod, H. P. H. Arp, M. B. Tekman and A. Jahnke, Science, 2021, 373, 61–65 CrossRef CAS PubMed.
- G. W. Coates and Y. D. Y. L. Getzler, Nat. Rev. Mater., 2020, 5, 501–516 CrossRef CAS.
- J. M. Garcia and M. L. Robertson, Science, 2017, 358, 870–872 CrossRef CAS PubMed.
- M. Hong and E. Y. X. Chen, Green Chem., 2017, 19, 3692–3706 RSC.
- B. C. Gibb, Nat. Chem., 2019, 11, 394–395 CrossRef CAS PubMed.
- Basic Energy Sciences Roundtable on Chemical Upcycling of Polymers, https://science.osti.gov/-/media/bes/pdf/reports/2020/Chemical_Upcycling_Polymers.pdf.
- S. Billiet and S. R. Trenor, ACS Macro Lett., 2020, 9, 1376–1390 CrossRef CAS PubMed.
- J. C. Worch and A. P. Dove, ACS Macro Lett., 2020, 9, 1494–1506 CrossRef CAS PubMed.
- E. M. Foundation, The New Plastics Economy: Rethinking the future of plastics, https://ellenmacarthurfoundation.org/the-new-plastics-economy-rethinking-the-future-of-plastics).
- S. R. Nicholson, N. A. Rorrer, A. C. Carpenter and G. T. Beckham, Joule, 2021, 5, 673–686 CrossRef CAS.
- C. Jehanno, J. W. Alty, M. Roosen, S. De Meester, A. P. Dove, E. Y. X. Chen, F. A. Leibfarth and H. Sardon, Nature, 2022, 603, 803–814 CrossRef CAS PubMed.
- R. Meys, A. Kätelhön, M. Bachmann, B. Winter, C. Zibunas, S. Suh and A. Bardow, Science, 2021, 374, 71–76 CrossRef CAS PubMed.
- N. Vora, P. R. Christensen, J. Demarteau, N. R. Baral, J. D. Keasling, B. A. Helms and C. D. Scown, Sci. Adv., 2021, 7, eabf0187 CrossRef CAS PubMed.
- T. Keijer, V. Bakker and J. C. Slootweg, Nat. Chem., 2019, 11, 190–195 CrossRef CAS PubMed.
- J. Zheng, M. Arifuzzaman, X. Tang, X. C. Chen and T. Saito, Mater. Horiz., 2023, 10, 1608–1624 RSC.
- Plastic Market Size, Share & Trends Report, 2022–2030, 2022.
- B. Geyer, G. Lorenz and A. Kandelbauer, eXPRESS Polym. Lett., 2016, 10, 559–586 CrossRef CAS.
- Q. Hou, M. Zhen, H. Qian, Y. Nie, X. Bai, T. Xia, M. Laiq Ur Rehman, Q. Li and M. Ju, Cell Rep. Phys. Sci., 2021, 2, 100514 CrossRef CAS.
- R. Heiran, A. Ghaderian, A. Reghunadhan, F. Sedaghati and S. Thomas, J. Polym. Res., 2021, 28, 1–19 CrossRef.
- L. D. Ellis, N. A. Rorrer, K. P. Sullivan, M. Otto, J. E. McGeehan, Y. Román-Leshkov, N. Wierckx and G. T. Beckham, Nat. Catal., 2021, 4, 539–556 CrossRef CAS.
- T. Uekert, A. Singh, J. S. DesVeaux, T. Ghosh, A. Bhatt, G. Yadav, S. Afzal, J. Walzberg, K. M. Knauer, S. R. Nicholson, G. T. Beckham and A. C. Carpenter, ACS Sustainable Chem. Eng., 2023, 11, 965–978 CrossRef CAS.
- R. Yang, G. Xu, B. Dong, X. Guo and Q. Wang, ACS Sustainable Chem. Eng., 2022, 10, 9860–9871 CrossRef CAS.
- C. Jehanno, J. Demarteau, D. Mantione, M. C. Arno, F. Ruipérez, J. L. Hedrick, A. P. Dove and H. Sardon, Angew. Chem., Int. Ed., 2021, 60, 6710–6717 CrossRef CAS PubMed.
- L. Gausas, S. K. Kristensen, H. Sun, A. Ahrens, B. S. Donslund, A. T. Lindhardt and T. Skrydstrup, JACS Au, 2021, 1, 517–524 CrossRef CAS PubMed.
- V. Zubar, A. T. Haedler, M. Schütte, A. S. K. Hashmi and T. Schaub, ChemSusChem, 2022, 15, e202101606 CrossRef CAS PubMed.
- C. Jehanno, I. Flores, A. P. Dove, A. J. Müller, F. Ruipérez and H. Sardon, Green Chem., 2018, 20, 1205–1212 RSC.
- B. D. Vogt, K. K. Stokes and S. K. Kumar, ACS Appl. Polym. Mater., 2021, 3, 4325–4346 CrossRef CAS.
- C. Jehanno, M. M. Pérez-Madrigal, J. Demarteau, H. Sardon and A. P. Dove, Polym. Chem., 2019, 10, 172–186 RSC.
- S. C. Kosloski-Oh, Z. A. Wood, Y. Manjarrez, J. P. De LosRios and M. E. Fieser, Mater. Horiz., 2021, 8, 1084–1129 RSC.
- A. Basterretxea, C. Jehanno, D. Mecerreyes and H. Sardon, ACS Macro Lett., 2019, 8, 1055–1062 CrossRef CAS PubMed.
- S. R. Nicholson, J. E. Rorrer, A. Singh, M. O. Konev, N. A. Rorrer, A. C. Carpenter, A. J. Jacobsen, Y. Román-Leshkov and G. T. Beckham, Annu. Rev. Chem. Biomol. Eng., 2022, 13, 301–324 CrossRef PubMed.
- P. S. Roy, G. Garnier, F. Allais and K. Saito, ChemSusChem, 2021, 14, 4007–4027 CrossRef CAS PubMed.
- M. W. Guzik, T. Nitkiewicz, M. Wojnarowska, M. Sołtysik, S. T. Kenny, R. P. Babu, M. Best and K. E. O'Connor, Waste Manage., 2021, 135, 60–69 CrossRef CAS PubMed.
- K. P. Sullivan, A. Z. Werner, K. J. Ramirez, L. D. Ellis, J. R. Bussard, B. A. Black, D. G. Brandner, F. Bratti, B. L. Buss, X. Dong, S. J. Haugen, M. A. Ingraham, M. O. Konev, W. E. Michener, J. Miscall, I. Pardo, S. P. Woodworth, A. M. Guss, Y. Román-Leshkov, S. S. Stahl and G. T. Beckham, Science, 2022, 378, 207–211 CrossRef CAS PubMed.
- S. M. Baghbanian and M. Farhang, J. Mol. Liq., 2013, 183, 45–49 CrossRef CAS.
- Acidity-Basicity Data (pKa Values) in Nonaqueous Solvents, 2022.
- M. Valiev, E. J. Bylaska, N. Govind, K. Kowalski, T. P. Straatsma, H. J. J. Van Dam, D. Wang, J. Nieplocha, E. Aprà and T. L. Windus, Comput. Phys. Commun., 2010, 181, 1477–1489 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, J. Chem. Phys., 2006, 125, 194101 CrossRef PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- S. Das, C. Liang and J. B. Dunn, Circular Economy of Polymers: Topics in Recycling Technologies, American Chemical Society, 2021, vol. 1391, ch. 8, pp. 143–170 Search PubMed.
- S. Arvin, S. Seunghwan and K. Do Hyun, Advanced Catalytic Materials, ed. N. Luis Enrique and W. Jin-An, IntechOpen, Rijeka, 2016, ch. 6 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3mh00801k |
| This journal is © The Royal Society of Chemistry 2023 |