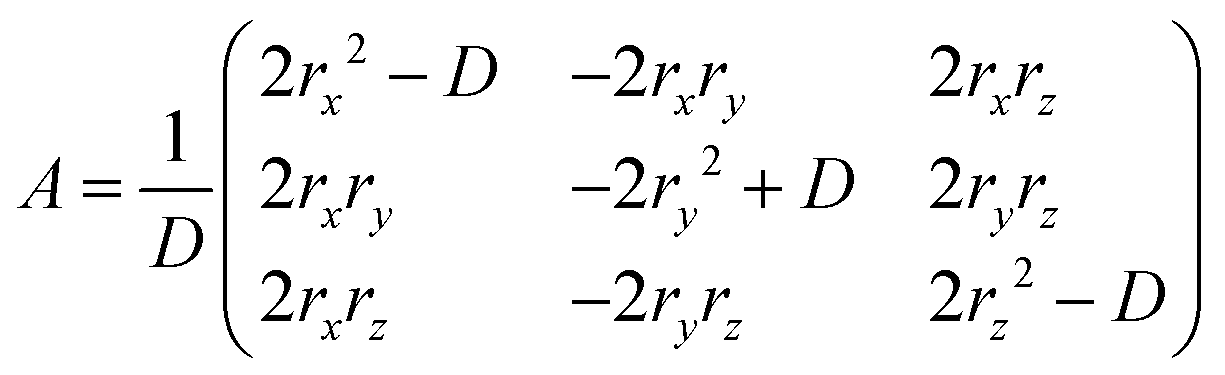DOI:
10.1039/D3MH01055D
(Communication)
Mater. Horiz., 2023,
10, 5492-5499
Crystal-structure simulation of molecular semiconductors: brickwork-related crystal structures of methylthiolated peri-condensed polycyclic aromatic hydrocarbons†
Received
8th July 2023
, Accepted 1st November 2023
First published on 3rd November 2023
Abstract
Despite the critical importance to carrier transport properties, studies on the control and prediction of crystal structures of molecular semiconductors have not been well-matured. To tackle this issue, we have developed “in silico crystallization” (ISC) protocols for simulating the brickwork (BW) crystal structures of methylchalcogenolated polycyclic aromatic hydrocarbons (PAHs). In this study, by carefully analyzing a BW-related polymorph of experimental crystal structures, an inclined brickwork (iBW) structure, we further extend the ISC protocol to simulate various BW-related crystal structures including iBW structures. Rational conditional branching in the simulation not only makes it possible to simulate eight polymorph candidates of methylchalcogenolated PAHs but also helps understand the relationship between the polymorphs. Furthermore, the relative favorability of each polymorphic candidate, i.e., the likelihood of the appearance among the polymorph candidates, can also be evaluated.

Kazuo Takimiya
| It is a great honor to contribute this article to the special issue celebrating ten years of Materials Horizons. Since the launch of the journal in 2013, I have been one of the advisory board members of the journal and enjoyed the cutting-edge sciences in the field of materials science, in particular, new concepts and approaches, on which the journal mainly focuses. Congratulations on the great milestone of ten years and contribution to the community, and I am looking forward to seeing more breakthroughs. |
New concepts
Prediction of crystal structures of organic molecules is one of the critical issues in many fields such as the pharmaceutical industry and materials science. Focusing on the relative simplicity of brickwork (BW) crystal structures of molecular semiconductors based on methylchalcogenolated peri-condensed polycyclic aromatic hydrocarbons (PAHs), we have developed an “in silico crystallization” (ISC) protocol, in which simple calculations of bimolecular intermolecular interaction energies with the Gaussian program package and translation-based grid search to the energy minimum of molecular clusters are the key. This simple yet useful method for simulating the BW structures is further extended, by employing three conditional branching, to simulate different polymorph candidates of BW-related crystal structures. Furthermore, the calculation of the sum of intermolecular interaction energies enables us to evaluate the relative energetic stability, i.e., the likelihood of the appearance, of these polymorph candidates. Although the scope of the ISC protocols is still limited at present, the concepts of the ISC protocols can be further extended to more complicated crystal structures such as herringbone and γ-structures, which would pave the way to simulate crystal structures of various molecular semiconductors prior to their actual synthesis.
|
1. Introduction
The carrier transport properties, one of the most important properties of molecular semiconductors, are heavily dependent on both the molecular electronic structure and the crystal structure.1–3 In contrast to the molecular electronic structure accurately predictable by quantum chemical calculations, the prediction of the crystal structure of molecular semiconductors is still a challenging issue.4–7 In this relationship, the recent progress in crystal structure prediction (CPS) would help predict the crystal structure of molecular semiconductors,8 though the computational methods and protocols are not easily accessible to experimental chemists. Intensive research efforts in the last two decades, on the other hand, have accumulated correlations between the molecular structure and crystal structure and thus carrier transport properties;9–12 the most representative one for promising transport properties is the herringbone structure, represented by pentacene,13 [1]benzothieno[3,2-b][1]benzothiophene (BTBT) derivatives,14,15 and so on.16–18 Other promising crystal structure types are the pitched π-stacking structure realized by rubrene19 and methylthiolated linear arenes,20–25 and the brickwork (BW) structure seen in the crystal structures of (triisopropyl)silylethynyl (TIPS)-pentacene26 and its related compounds.27–30
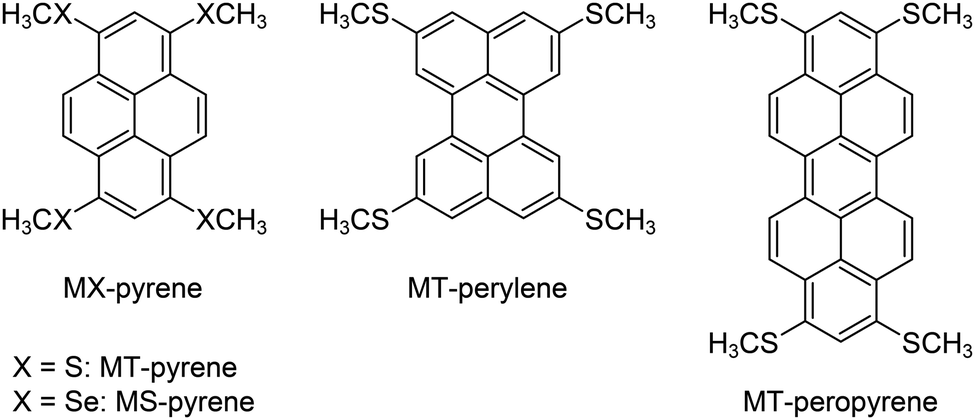
Recently, we found that the BW crystal structure can be realized by methylchalcogenolation on pyrene, the smallest peri-condensed polycyclic aromatic hydrocarbon (PAH).31 Furthermore, methylthiolated pyrene (MT-pyrene)32 showed ultra-high mobility of up to 32 cm2 V−1 s−1 on single-crystal field-effect transistors,33 which reinforced the potential of the BW crystal structure for the development of further outperforming molecular semiconductors. Inspired by these results, we have developed a simple protocol for the simulation of BW crystal structures of unknown methylchalcogenolated peri-condensed PAHs.34 This protocol, which we call “in silico crystallization (ISC)” helped us to select a synthetic target, and in fact, thus selected and then synthesized MT-peropyrene (Fig. 1) turned out to be another ultra-high mobility molecular semiconductor (μ ∼ 30 cm2 V−1 s−1).34
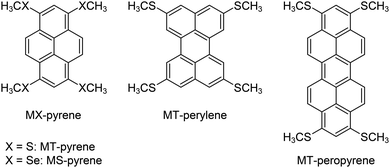 |
| | Fig. 1 Molecular structures of 1,3,6,8-tetrakis(methylthio)- and 1,3,6,8-tetrakis(methylseleno)-pyrene and (MT- and MS-pyrene), 2,5,8,11-tetrakis(methylthio)perylene (MT-perylene), and 1,3,8,10-tetrakis(methylthio)peropyrene (MT-peropyrene). | |
From these results, it is safe to say that methylchalcogenolated peri-condensed PAHs generally afford the BW crystal structure. In fact, recently synthesized methylthiolated perylene (MT-perylene, Fig. 1) also crystallized into a BW structure,35 and the experimental crystal structure was eventually well-matched with the simulated one by the ISC protocol (vide infra). Although the performance of MT-perylene-based SC-FETs was not promising (μ ∼ 0.2 cm2 V−1 s−1), the experimental studies on MT-perylene gave insightful results on the crystal structures of methylchalcogenolated peri-condensed PAHs; MT-perylene afforded a polymorph classified into an inclined brickwork (iBW) structure,35 similar to that of one of the polymorphs observed for methylselenolated pyrene (MS-pyrene).31 Although the relationship between the BW and iBW structures of MS-pyrene was not clear at all, the two polymorphs, BW and iBW structures, of MT-perylene seemed to be closely related to each other, as the molecular arrangement, in other words, the mutual position of molecules, in the respective BW layers, seemed to be identical at a glance.
In this work, we first analyze the relationship between the BW and iBW structures of MT-perylene by using the notation of molecules in the crystal structures developed in the ISC protocol. Then, rigorous analyses of the relationship between a BW structure and an iBW structure are carried out, and then the results are adapted to the extension of the ISC protocol to the iBW structures. Furthermore, these analyses on the iBW structure of MT-perylene hinted to us to rationally describe the iBW structure of MS-pyrene, which further allows us to extend the ISC protocol enabling us to bring eight BW-related polymorph candidates by three conditional branches.
2. Results and discussion
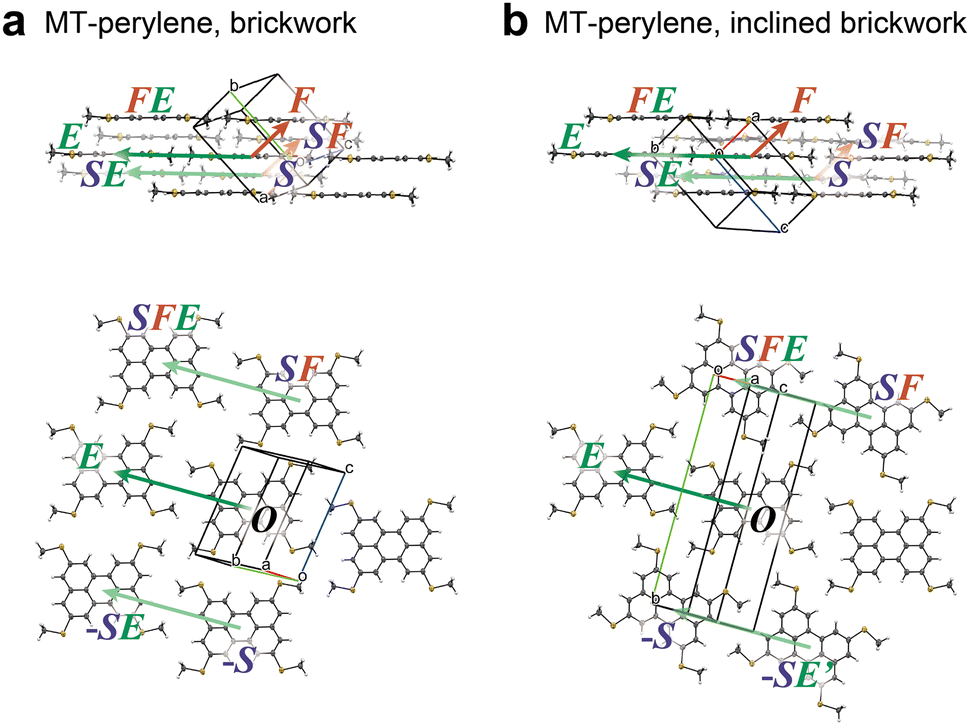
2.1 BW and iBW structures of MT-perylene
The BW and iBW crystal structures of MT-perylene are shown in Fig. 2a and b, respectively. As the BW structure is classified into the triclinic P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group with Z (the number of molecules in the unit cell) = 1, all the molecules in the BW crystal structure are related by the translation operation, which means that the relationship between the neighboring BW layers is also the translation (Fig. 2a bottom).34 On the other hand, in the iBW structure with the monoclinic P21/n space group, the relationship between the neighboring BW layers is the glide operation. Note that all the molecules in each BW layer in the iBW structure are related by the translation operation similar to that of the BW structure.
space group with Z (the number of molecules in the unit cell) = 1, all the molecules in the BW crystal structure are related by the translation operation, which means that the relationship between the neighboring BW layers is also the translation (Fig. 2a bottom).34 On the other hand, in the iBW structure with the monoclinic P21/n space group, the relationship between the neighboring BW layers is the glide operation. Note that all the molecules in each BW layer in the iBW structure are related by the translation operation similar to that of the BW structure.
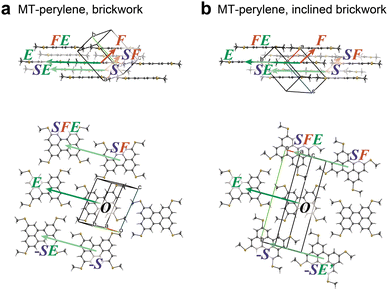 |
| | Fig. 2 Two polymorphs of MT-perylene experimentally elucidated; (a) brickwork (BW) structure and (b) inclined brickwork (iBW) structure. | |
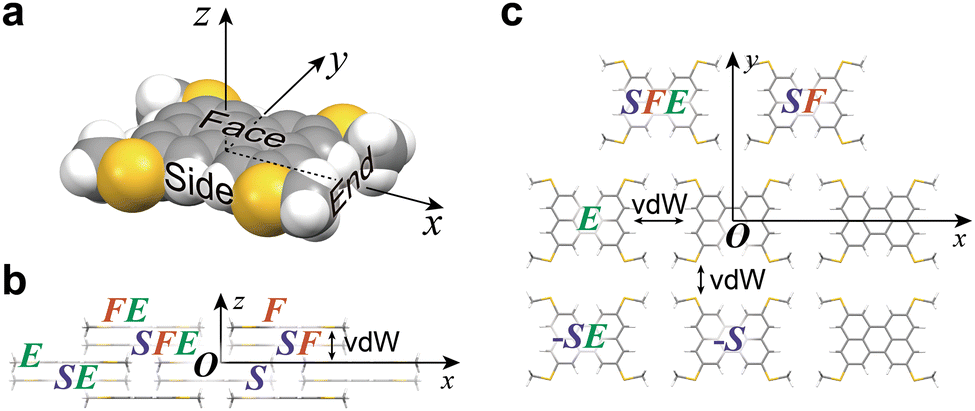
In the protocol of ISC simulation for the BW structure, the molecules in the Cartesian system are designated, as shown in Fig. 3; three directions of the molecule at the origin (designated as molecule O), “face”, “side”, and “end” directions, are first defined (Fig. 3a), and then the neighboring molecules in these directions are defined as F, S, and E, respectively. With these definitions and a combination of the definitions, all the molecules surrounding O are distinguished. For example, in each BW layer, molecule O is surrounded by molecules F, E, and FE (a molecule in the end-to-end direction from F), and their equivalents in the negative directions (−F, −F−E, and −E) (Fig. 3b). Similarly, in the neighboring BW layer, four molecules contacting O, namely, molecule S, (the nearest neighbor to O), SF (a molecule in the face-to-face direction from S), SFE (a molecule in the end-to-end direction from SF), and SE (a molecule in the end-to-end direction from S) are defined (Fig. 3c). Table 1 summarizes the coordinates of three molecules (F, E, and FE) in the BW layer of both the BW and iBW structures experimentally determined. Intriguingly, the coordinates of these molecules are quite similar indicating that the mutual position of molecules in the BW layers in different polymorphs are very similar to each other. It should be noted that by using the Cartesian system, the molecular arrangements in the different polymorphs can be quantitatively compared.
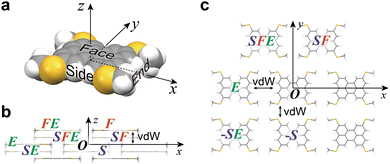 |
| | Fig. 3 (a) Definition of “face”, “side”, and “end” directions. (b) Definition of molecules in the BW layer. (c) Definition of molecules in the neighboring BW layer. | |
Table 1 Cartesian coordinates of molecules (centers of mass) [Å] between the origin (O) and various neighboring molecules in the experimentally determined BW and iBW crystal structures of MT-perylene at 100 K
| MT-perylene, BW structure |
MT-perylene, iBW structure |
| Mol. |
x
|
y
|
z
|
Mol. |
x
|
y
|
z
|
| F |
3.351 |
1.051 |
3.363 |
F |
3.382 |
0.834 |
3.351 |
| E |
−12.787 |
−3.724 |
0.182 |
E |
−12.775 |
−3.574 |
0.213 |
| FE |
−9.436 |
−2.673 |
3.545 |
FE |
−9.393 |
−2.740 |
3.564 |
2.2 Relationship between the BW and iBW structures of MT-perylene
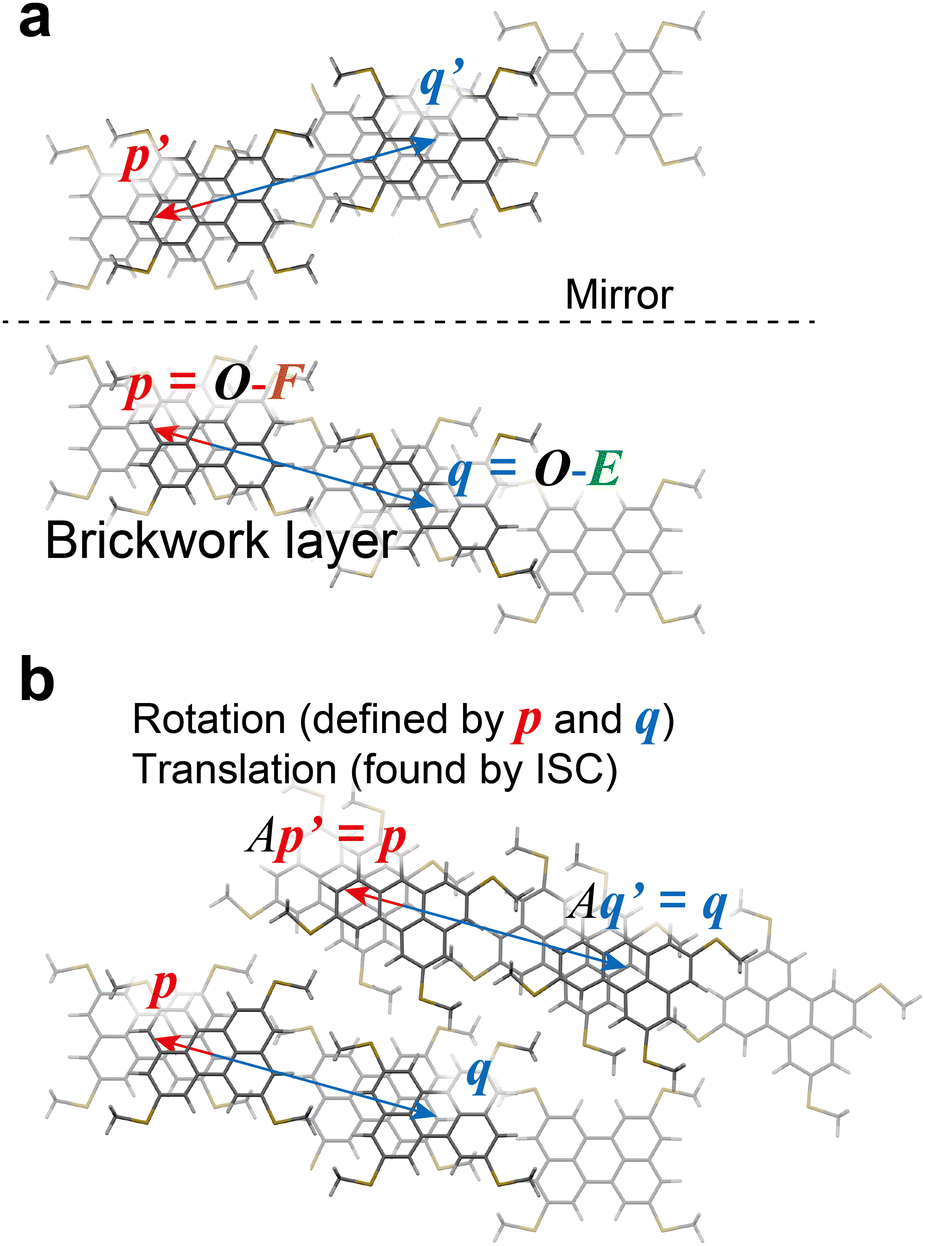
By using the above definition of molecules in the BW layer, the BW layer can be further simply expressed by two vectors, i.e., p = O–F and q = O–E (Fig. 2). For constructing the three-dimensional crystal structures, these two vectors and the corresponding vectors in the neighboring layers, i.e., p′ = S–SF and q′ = S–SE, shall be identical. Obviously, the translation operation, namely, the BW structure, satisfies this condition. In addition to the translation operation, the glide operation also satisfied this condition as experimentally shown in the iBW structure of MT-perylene. In the Cartesian system, this can be expressed by a mirror operation of the initial BW layer against any plane followed by a specific rotation that aligns the mirrored vectors back to being parallel to p and q, and also translation (Fig. 4). The rotation that aligns the mirrored vectors is dependent on the mirror plane; in the case of the mirror plane parallel to the xz plane in the Cartesian system, the mirrored vectors of p = (px, py, pz) and q = (qx, qy, qz) are expressed as p′ = (px, –py, pz) and q′ = (qx, –qy, qz). The rotation matrix A thus shall satisfy Ap′‖p and Aq′‖q. Without loss of generality, equations Ap′ = p and Aq′ = q can be solved to obtain the rotation matrix uniquely based on p and q:| | 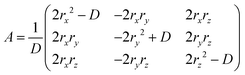 | (1) |
where (rx, ry, rz) = p × q and D = p2q2 − (p·q)2.
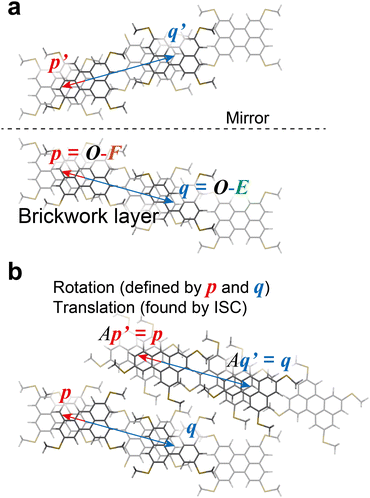 |
| | Fig. 4 Construction of the neighboring layer in the iBW structure of MT-perylene from the BW structure. The mirror plane can be chosen arbitrarily and is y = 8.5 Å in the present case (a). Rotation matrix A in (b) is defined in eqn (1) to ensure Aq′ = q (and Ap′ = p). | |
2.3 Simulation of the iBW structure by the ISC protocol
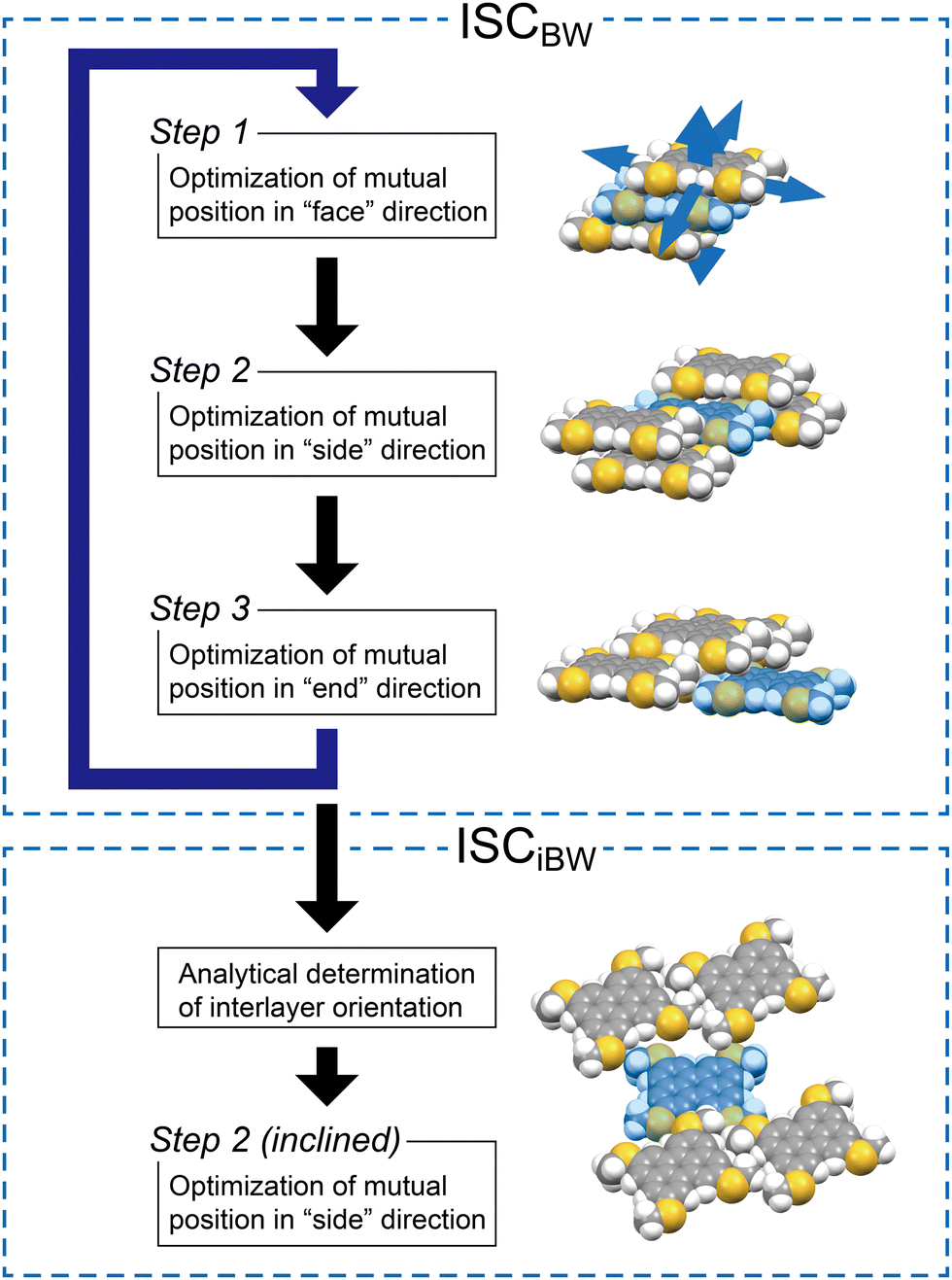
The original ISC protocol for the BW structures is as follows (Fig. 5): the molecules F, E, and FE are placed temporarily at van der Waals contacts with O. In step 1, the relative position of two stacking molecules to O, F and FE, is searched by minimizing the sum of intermolecular interaction energies (Eint)36 between O–F and O–FE while moving F and FE as a cluster with respect to O and by using the Gaussian16 program.37 This two-molecular (2M) stacking optimization defines a BW layer consisting of six molecules surrounding O: F, FE, and their symmetrically translated molecules, E, –F, –F–E, and –E. Next, in step 2, the BW layers are piled up in the positive side-to-side direction by optimizing the positions of S, SF, and SFE and in the negative side-to-side direction by −S, −S−F, and −S−F−E. Note that there are two ways to pile the neighboring layers up in step 2, which corresponds to polymorph candidates of the BW structures (vide infra). Then, molecule E and molecules related to E (FE, SE, SFE, etc.), which were temporarily defined in step 1, are finely optimized in step 3. These steps are repeated typically three times to determine the final BW structure with high quality (Fig. 5, ISCBW). In the case of MT-perylene, the simulated BW structure nicely reproduced the experimental BW structure as in the case of MT-pyrene and MT-peropyrene (Table 2).
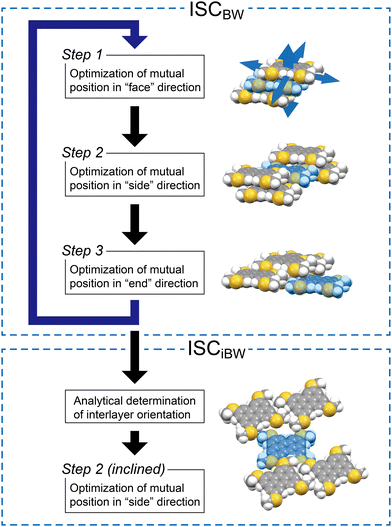 |
| | Fig. 5 Schematic representation of the ISC protocol for BW structures (ISCBW) and renewed ISC covering iBW structures (ISCiBW). | |
Table 2 Comparison of cell parameters between the predicted and experimental crystal structures of MT-perylene
| MT-perylene |
Brickwork (BW) |
Inclined brickwork (iBW) |
| Simulated |
Experimentala |
Simulated |
Experimentala |
|
Data obtained at T = 100 K.
Space group was not explicitly determined.
E
int(15): sum of intermolecular interaction energies (Eint) between O and its 14 neighboring molecules.
|
| Crystal system |
(Triclinic)a |
Triclinic |
(Triclinic)a |
Monoclinic |
| Space group |
(P1)b |
P |
(P1)b |
P21/n |
|
a/Å |
4.98 |
4.8621(3) |
4.98 |
4.8336(2) |
|
b/Å |
9.17 |
9.3471(5) |
21.93 |
21.9293(8) |
|
c/Å |
11.17 |
11.0114(8) |
9.17 |
9.3596(3) |
|
α/° |
89.6 |
90.500(5) |
89.1 |
90 |
|
β/° |
101.8 |
100.650(5) |
92.4 |
91.596(3) |
|
γ/° |
92.4 |
91.424(4) |
91.2 |
90 |
|
V/Å3 |
499.4 |
491.61(5) |
1001.2 |
991.71(6) |
E
int(15)/kcal mol−1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) c c |
−100.7 |
−107.9 |
−101.8 |
−110.5 |
| Ref. |
34
|
35
|
This work |
35
|
For the simulation of the iBW structure of MT-perylene, the final BW structure by the ISCBW protocol was used; two vectors, p and q, extracted from the simulated BW structure, are used to define the rotation matrix A according to eqn (1). Then, three molecules in the neighboring layers, –S, SF, and SFE, are mirrored by the xz plane and rotated by matrix A, and then, as described above for step 2, the positions of these molecules optimized by searching the minimum of the sum of Eints (∑Eint) between O–S, O–SF, and O–SFE are calculated, and molecule –S is placed at the position of energy minimum (Fig. 5, step 2 (inclined) in ISCiBW). Note that the translation operation after the mirror and rotation operations define the initial position of the molecules for searching for the energy minimum, and the glide symmetry is not assumed in the simulation.
Table S3 (ESI†) summarizes the centers of mass of molecules of the simulated iBW structure of MT-perylene in comparison to the experimental iBW structure. It should be emphasized that the molecular positions are virtually identical, and the root-mean-square deviation of the 15 molecules (evaluated atom-wise, RMSD15) is as small as 0.27 Å, which is comparable to the search step size in the simulation.38 The triclinic cell parameters extracted from the optimized molecular clusters are summarized in Table 2 (see also the crystallographic information files (CIFs)‡ ESI†). Note that although the glide symmetry of the P21/n space group was not assumed, the crystallographic α and γ angles of the simulated iBW structure of MT-perylene are almost 90°, which is a characteristic of a monoclinic P21/n space group. The other crystallographic parameters including the cell axes (a, b, and c) and the angle (β) were also successfully reproduced. The relative stability of the polymorphs can be evaluated by lattice energy, and in the present case, we calculated the sum of intermolecular interaction energies (Eint(15)) between O and its 14 neighboring molecules, which is a similar parameter to the lattice energy.39 The experimental iBW structure of MT-perylene is energetically slightly more stable than the BW structure, which was also reproduced in the present ISC simulations (Table 2).
2.4 iBW structure of MS-pyrene
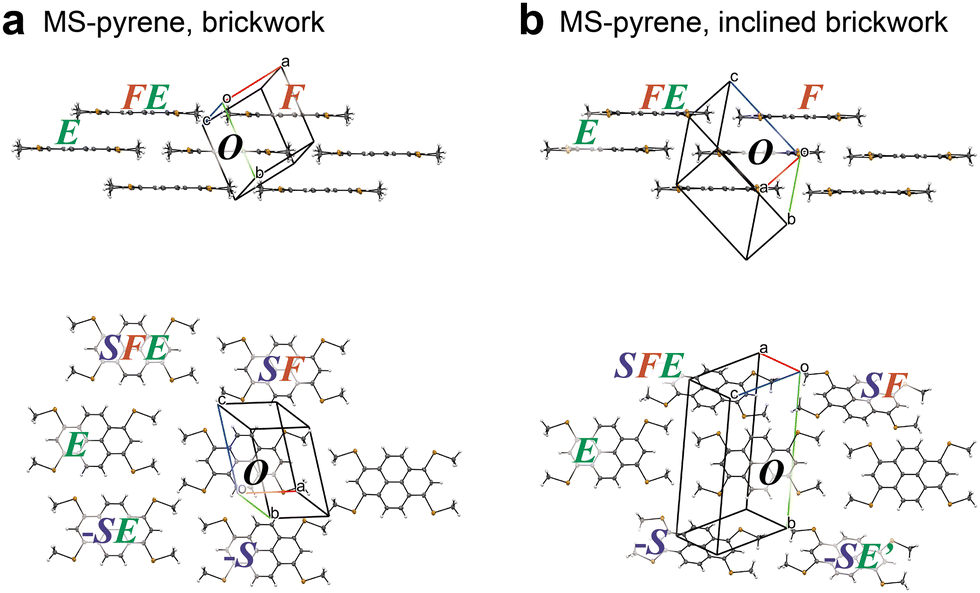
As shown in Fig. 6, MS-pyrene also has an iBW structure polymorph like MT-perylene. However, the BW layers in the BW and iBW polymorphs are noticeably different. The p and q vectors in the experimental BW structure, pBW = (5.510, 0.254, 3.581) and qBW = (−14.316, 1.748, −0.045), do not match the p and q vectors in the iBW structure, piBW = (3.835, −1.653, 3.534) and qiBW = (−14.400, 1.241, 0.183). The pBW and qBW in the simulated structure of MS-pyrene by the ISCBW protocol (Fig. 5) match the pBW and qBW, not the piBW and qiBW of the experimental structures. This strongly implies that there are other ways to construct the BW layers. Close inspection of two BW layers of MS-pyrene revealed that the intermolecular interaction energy between FE and O is much smaller than that between F and O in the iBW structure. This means that the iBW structure of MS-pyrene is more one-dimensional (1D) than the BW structure. To simulate such a 1D-like stacking structure, we tested the positional search in step 1 by just using one stacking molecular pair of O and F. This resulted in an optimal position of F = (3.50, −1.50, 3.50), which afforded a p vector roughly matching piBW. Although such a BW structure with a 1D-like stacking structure was so far not found in the actual crystals, performing the ISC with such a one-molecular (1M) stacking in step 1 results in the determination of the finalized p = (3.70, −1.60, 3.49) and q = (−14.59, 1.20, 0.22), which matches the piBW and qiBW of the experimental structure with good accuracy. We then proceeded to step 2 for iBW structures. The RMSD15 of the simulated structure with respect to the experimental one was as low as 0.27 Å, indicating that the iBW structure of MS-pyrene could also be simulated. Note that for MS-pyrene the simulated crystallographic α and γ angles (see Table 3) were very close to 90°, reproducing the monoclinic cell, similar to the case of MT-perylene. As already mentioned, the ISCiBW protocol (as well as the ISCBW protocol) does not assume the monoclinic cell, yet the resulting simulated structures are close to the monoclinic cells, indicating the utility of the protocols.
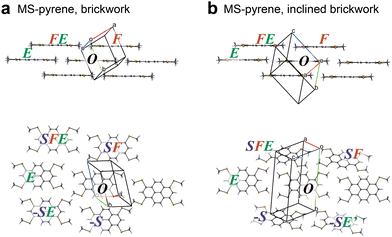 |
| | Fig. 6 Crystal structures of MS-pyrene featuring BW (a) and iBW (b) structures. The structures are shown from the side (top) and top (bottom) of the molecular plane. | |
Table 3 Comparison of cell parameters between the predicted and experimental inclined brickwork structures of MS-pyrene (1M-ZL-iBW) and MT-peropyrene (1M-ZL-iBW)
|
|
MS-pyrene (1M-ZL-iBW) |
MT-peropyrene (1M-CL-iBW) |
| Simulated |
Experimentala |
Simulated |
Experimentala |
|
Data obtained at T = 100 K.
Space group was not explicitly determined.
E
int(15): sum of intermolecular interaction energies (Eint) between O and its 14 neighboring molecules.
|
| Crystal system |
(Triclinic)b |
Monoclinic |
(Triclinic)b |
Triclinic |
| Space group |
(P1)b |
P21/n |
(P1)b |
P |
|
a/Å |
5.33 |
5.4709(2) |
5.15 |
5.3250(2) |
|
b/Å |
16.52 |
16.2641(8) |
13.99 |
13.4178(6) |
|
c/Å |
10.37 |
10.1056(5) |
16.78 |
16.3151(4) |
|
α/° |
90.3 |
90 |
109.4 |
108.191(3) |
|
β/° |
91.8 |
93.349(4) |
90.1 |
91.312(3) |
|
γ/° |
90.1 |
90 |
99.9 |
99.886(4) |
|
V/Å3 |
913.2 |
897.65(7) |
1120.9 |
1087.36(8) |
|
E
int(15)/kcal mol−1c |
−103.6 |
−112.8 |
−123.7 |
−132.4 |
| Ref. |
This work |
31
|
This work |
This work |
2.5 Conditional branching in the simulation of BW-related crystal structures
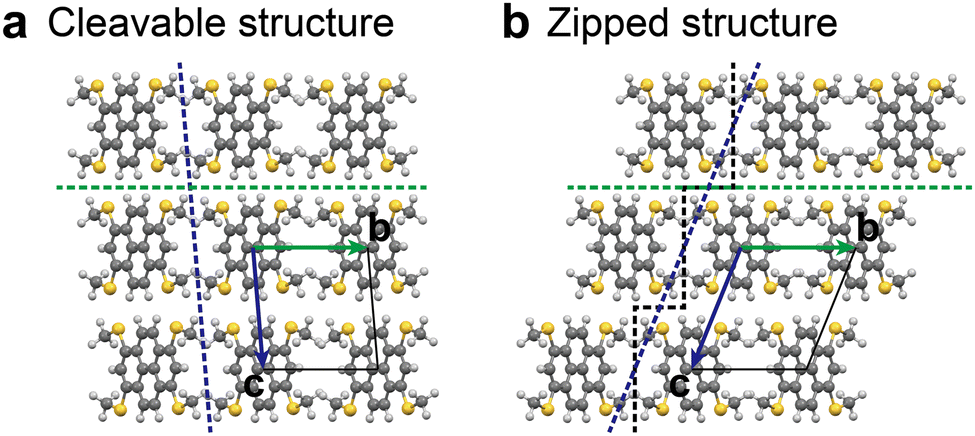
In step 2 of the ISCBW protocol (Fig. 5), there generally exist two ways to pile the BW layers up, as already reported in the previous paper.34Fig. 7 shows the case of MT-pyrene as an example. These two structures can be distinguished by the molecular arrangement in the crystallographic bc plane: a polymorph with a molecular array that is “cleavable” along the crystallographic ac-plane, and another polymorph with a not cleavable molecular array, as the molecules are “zipped” by the methylchalcogeno groups.31 Thus, we call the first one “cleavable layer (CL)” and the other “zipped layer (ZL)” in the interlayer structures.
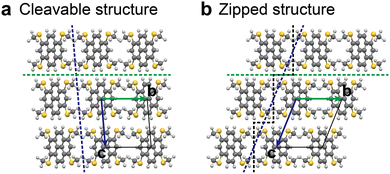 |
| | Fig. 7 Two polymorph candidates of MT-pyrene that arise in step 2 in the original ISC protocol: (a) “cleavable” structure along the crystallographic ac-plane; (b) another structure, where the molecular array is not cleavable as the molecules are “zipped” by the methylthio groups. | |
In most cases, the CL polymorph is energetically more favorable than the ZL polymorph in the ISC simulations. However, the experimentally elucidated structures of MT-pyrene and 1,3,6,8-tetramethoxypyrene (MO-pyrene) are classified into the ZL polymorph. Therefore, it is important to simulate both polymorphs by the ISC protocols. Out of MO-, MT-, MS-pyrene, MT-perylene, and MT-peropyrene, the CL (ZL) polymorph was noticeably more stable for MT-perylene and MT-peropyrene (MO-pyrene). For MT- and MS-pyrene, the Eint(15) of the CL and ZL polymorphs were competing. With these results in mind, we tried to find the CL polymorph of MT-pyrene and the ZL polymorph of MS-pyrene (vide infra).
To summarize the updated ISC protocols, it is now possible to simulate BW structures with one-/two-molecular (1M/2M) stacking configurations in step 1, two different interlayer configurations of the BW layers (CL/ZL) in step 2, and two different interlayer orientations of the BW layers (BW/iBW) in the final step. These three branching points can potentially result in eight BW-related crystal structures (polymorph candidates): e.g., 1M-CL-iBW structure, 2M-ZL-BW structure, and so on. As examples, the eight simulated structures of MT-perylene are summarized in Fig. 8 (see also ESI† for MT- and MS-pyrene, and MT-peropyrene). For MT-perylene, the two polymorph candidates, which were simulated to be the most energetically favorable, 2M-CL-BW and 2M-CL-iBW structures, were the experimentally confirmed ones.35
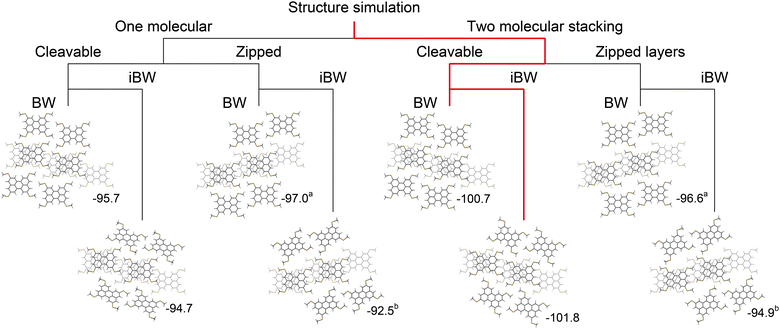 |
| | Fig. 8 Eight polymorph candidates of MT-perylene crystal structures simulated through three conditional branching. The energetically favourable two polymorphs, 2M-CL-BW and 2M-CL-iBW, were experimentally confirmed. The values are Eint(15) (kcal mol−1), and the alphabets in the superscript form denote almost identical structures, e.g., the 1M-ZL-BW and 2D-ZL-BW polymorphs are virtually identical. | |
2.6 Search for new polymorphs that were predicted to be energetically favorable
With the above-simulated crystal structures in hand, we have tried to find other polymorphs of these materials that correspond to the simulated polymorph candidates. For all materials, the crystals were grown using the physical vapor transport (PVT) method under a flow of nitrogen or argon gas.40 For MT-peropyrene with quite high vaporizing pressure, the pressure was reduced to <20 Pa, while for other materials, PVT was performed under atmospheric pressure. By extensive search, we managed to find the 2M-CL-BW polymorph for MT-pyrene and 1M-CL-iBW polymorph for MT-peropyrene (RMSD15 of corresponding simulated structures were 0.26 and 0.48 Å, respectively).§ Interestingly, the MT-peropyrene iBW structure, unlike those of MS-pyrene and MT-perylene, was of triclinic P space group with two half-molecules in the asymmetric unit. The 1M-CL-iBW polymorph of MT-peropyrene experimentally found is the first example of an iBW structure without glide symmetry in the present methylchalcogenolated PAH series.
The polymorphs experimentally confirmed so far, the 2M-ZL-BW and 2M-CL-BW polymorphs of MT-pyrene, the 2M-CL-BW and 2M-CL-iBW polymorphs of MT-perylene, and the 2M-CL-BW and 1M-CL-iBW polymorphs of MT-peropyrene, are energetically favorable polymorphs among the eight polymorph candidates simulated by the ISC protocols. On the contrary, the 2M-ZL-BW polymorph of MS-pyrene was not found, although it is estimated to be the most energetically stable out of the simulated structures. If it exists, the 2M-CL-BW polymorph of MS-pyrene is expected to show better two-dimensionality than the known 2M-CL-BW structure and thus is a more promising structure compared to the existing two polymorphs (2M-CL-BW and 1M-ZL-iBW polymorphs). Nevertheless, it can be said that the renewed ISC protocols can successfully simulate various BW-related structures and even predict the energetical stability (i.e., the likelihood of the appearance) of each polymorph to some extent.
3. Conclusions
Close inspection and analyses of the BW and iBW structures of MT-perylene experimentally found encouraged us to expand our recently developed ISC protocol for the simulation of methylchalcogenated PAH-based molecular semiconductors. Thus, renewed ISC protocols enable us to simulate a variety of BW-related polymorph candidates through three conditional branching based on the stacking mode in the BW layer, and configuration and orientation of the interlayer structure.
It was found that MT-, MS-pyrene, MT-perylene, and MT-peropyrene all afforded two polymorphs out of (and no polymorphs outside of) the eight simulated ones. These structures include the new polymorphs of MT-pyrene and MT-peropyrene found in this work. The monoclinic nature of the MS-pyrene and MT-perylene iBW structures and the triclinic nature of the MT-peropyrene iBW structures were successfully simulated without assuming a particular symmetry in the simulations. Therefore, the method, “in silico crystallization”, is now a powerful tool to simulate the possible crystal structures and understand their relationships in the methylchalcogenolated PAH-based molecular semiconductors. It is also expected that this approach can be expanded to other crystal structure types such as pitched π-stack and herringbone crystal structures of molecules with planar molecular structures.
Author contributions
Kirill Bulgarevich: experiment and computational design, methodology, experiment and calculation, validation, investigation, data curation, writing – original draft. Kazuo Takimiya: validation, investigation, writing – review & editing, funding acquisition, supervision.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was financially supported by JSPS KAKENHI Grant Numbers JP19H00906, JP20H05865, JP22K14293 and JP23H00307, and the Mitsubishi Foundation. We gratefully acknowledge the Advanced Center for Computing and Communication (ACCC) of RIKEN for the use of the Supercomputer System in theoretical calculations and the Center for Computational Materials Science, Institute for Materials Research, Tohoku University for the use of MASAMUNE-IMR (MAterials science Supercomputing system for Advanced MUlti-scale simulations towards NExt-generation-Institute for Materials Research).
Notes and references
- V. Coropceanu, J. Cornil, D. A. da Silva Filho, Y. Olivier, R. Silbey and J.-L. Brédas, Charge Transport in Organic Semiconductors, Chem. Rev., 2007, 107, 926–952 CrossRef CAS PubMed.
- G. Schweicher, Y. Olivier, V. Lemaur and Y. H. Geerts, What Currently Limits Charge Carrier Mobility in Crystals of Molecular Semiconductors?, Isr. J. Chem., 2014, 54, 595–620 CrossRef CAS.
- K. Takimiya, M. Nakano, H. Sugino and I. Osaka, Design and elaboration of organic molecules for high field-effect-mobility semiconductors, Synth. Met., 2016, 217, 68–78 CrossRef CAS.
- A. N. Sokolov, S. Atahan-Evrenk, R. Mondal, H. B. Akkerman, R. S. Sánchez-Carrera, S. Granados-Focil, J. Schrier, S. C. B. Mannsfeld, A. P. Zoombelt, Z. Bao and A. Aspuru-Guzik, From computational discovery to experimental characterization of a high hole mobility organic crystal, Nat. Commun., 2011, 2, 437 CrossRef PubMed.
- S. Obata, T. Miura and Y. Shimoi, Theoretical prediction of crystal structures of rubrene, Jpn. J. Appl. Phys., 2013, 53, 01AD02 CrossRef.
- S. L. Price, Predicting crystal structures of organic compounds, Chem. Soc. Rev., 2014, 43, 2098–2111 RSC.
- H. Ishii, S. Obata, N. Niitsu, S. Watanabe, H. Goto, K. Hirose, N. Kobayashi, T. Okamoto and J. Takeya, Charge mobility calculation of organic semiconductors without use of experimental single-crystal data, Sci. Rep., 2020, 10, 2524 CrossRef CAS PubMed.
- J. Yang, S. De, J. E. Campbell, S. Li, M. Ceriotti and G. M. Day, Large-Scale Computational Screening of Molecular Organic Semiconductors Using Crystal Structure Prediction, Chem. Mater., 2018, 30, 4361–4371 CrossRef CAS.
- K. Takimiya, S. Shinamura, I. Osaka and E. Miyazaki, Thienoacene-Based Organic Semiconductors, Adv. Mater., 2011, 23, 4347–4370 CrossRef CAS PubMed.
- C. Wang, H. Dong, W. Hu, Y. Liu and D. Zhu, Semiconducting π-Conjugated Systems in Field-Effect Transistors: A Material Odyssey of Organic Electronics, Chem. Rev., 2012, 112, 2208–2267 CrossRef CAS PubMed.
- H. Sirringhaus, 25th Anniversary Article: Organic Field-Effect Transistors: The Path Beyond Amorphous Silicon, Adv. Mater., 2014, 26, 1319–1335 CrossRef CAS PubMed.
- G. Schweicher, G. Garbay, R. Jouclas, F. Vibert, F. Devaux and Y. H. Geerts, Molecular Semiconductors for Logic Operations: Dead-End or Bright Future?, Adv. Mater., 2020, 32, 1905909 CrossRef CAS PubMed.
- D. J. Gundlach, Y. Y. Lin, T. N. Jackson, S. F. Nelson and D. G. Schlom, Pentacene organic thin-film transistors-molecular ordering and mobility, IEEE Electron Device Lett., 1997, 18, 87–89 CAS.
- H. Ebata, T. Izawa, E. Miyazaki, K. Takimiya, M. Ikeda, H. Kuwabara and T. Yui, Highly Soluble [1]Benzothieno[3,2-b]benzothiophene (BTBT) Derivatives for High-Performance, Solution-Processed Organic Field-Effect Transistors, J. Am. Chem. Soc., 2007, 129, 15732–15733 CrossRef CAS PubMed.
- H. Iino, T. Usui and J.-I. Hanna, Liquid crystals for organic thin-film transistors, Nat. Commun., 2015, 6, 6828 CrossRef CAS PubMed.
- T. Yamamoto and K. Takimiya, Facile Synthesis of Highly π-Extended Heteroarenes, Dinaphtho[2,3-b:2′,3′-f]chalcogenopheno[3,2-b]chalcogenophenes, and Their Application to Field-Effect Transistors, J. Am. Chem. Soc., 2007, 129, 2224–2225 CrossRef CAS PubMed.
- T. Okamoto, C. Mitsui, M. Yamagishi, K. Nakahara, J. Soeda, Y. Hirose, K. Miwa, H. Sato, A. Yamano, T. Matsushita, T. Uemura and J. Takeya, V-Shaped Organic Semiconductors With Solution Processability, High Mobility, and High Thermal Durability, Adv. Mater., 2013, 25, 6392–6397 CrossRef CAS PubMed.
- C. Mitsui, T. Okamoto, M. Yamagishi, J. Tsurumi, K. Yoshimoto, K. Nakahara, J. Soeda, Y. Hirose, H. Sato, A. Yamano, T. Uemura and J. Takeya, High-Performance Solution-Processable N-Shaped Organic Semiconducting Materials with Stabilized Crystal Phase, Adv. Mater., 2014, 26, 4546–4551 CrossRef CAS PubMed.
- J. Takeya, M. Yamagishi, Y. Tominari, R. Hirahara, Y. Nakazawa, T. Nishikawa, T. Kawase, T. Shimoda and S. Ogawa, Very high-mobility organic single-crystal transistors with in-crystal conduction channels, Appl. Phys. Lett., 2007, 90, 102120 CrossRef.
- H. Takenaka, T. Ogaki, C. Wang, K. Kawabata and K. Takimiya, Selenium-Substituted β-Methylthiobenzo[1,2-b:4,5-b′]dithiophenes: Synthesis, Packing Structure, and Transport Properties, Chem. Mater., 2019, 31, 6696–6705 CrossRef CAS.
- C. Wang, D. Hashizume, M. Nakano, T. Ogaki, H. Takenaka, K. Kawabata and K. Takimiya, “Disrupt and induce” intermolecular interactions to rationally design organic semiconductor crystals: from herringbone to rubrene-like pitched π-stacking, Chem. Sci., 2020, 11, 1573–1580 RSC.
- K. Takimiya, T. Ogaki, C. Wang and K. Kawabata, Crystal Structures of Dimethoxyanthracens: A Clue to a Rational Design of Packing Structures of π-Conjugated Molecules, Chem. – Asian J., 2020, 15, 915–919 CrossRef CAS PubMed.
- K. Takimiya, K. Bulgarevich, K. Sahara, K. Kanazawa, H. Takenaka and K. Kawabata, What Defines a Crystal Structure? Effects of Chalcogen Atoms in 3,7-Bis(methylchalcogeno)benzo[1,2-b:4,5-b′]dichalcogenophene-Based Organic Semiconductors, Chin. J. Chem., 2022, 40, 2546–2558 CrossRef CAS.
- K. Kanazawa, K. Bulgarevich, K. Kawabata and K. Takimiya, Uncovered Effects of thieno[2,3-b]thiophene Substructure in a Tetrathienoacene Backbone: Reorganization Energy and Intermolecular Interaction, Chem. Mater., 2023, 35, 280–288 CrossRef CAS.
- K. Kanazawa, K. Bulgarevich, K. Kawabata and K. Takimiya, Methylthiolation of Acenes: Change of Crystal Structure from Herringbone to Rubrene-like Pitched π-Stacking Structure, Cryst. Growth Des., 2023, 23, 5941–5949 CrossRef CAS.
- J. E. Anthony, The Larger Acenes: Versatile Organic Semiconductors, Angew. Chem., Int. Ed., 2008, 47, 452–483 CrossRef CAS PubMed.
- J. E. Anthony, D. L. Eaton and S. R. Parkin, A Road Map to Stable, Soluble, Easily Crystallized Pentacene Derivatives, Org. Lett., 2002, 4, 15–18 CrossRef CAS PubMed.
- M. M. Payne, S. R. Parkin, J. E. Anthony, C.-C. Kuo and T. N. Jackson, Organic Field-Effect Transistors from Solution-Deposited Functionalized Acenes with Mobilities as High as 1 cm2/V·s, J. Am. Chem. Soc., 2005, 127, 4986–4987 CrossRef CAS PubMed.
- S. Miao, A. L. Appleton, N. Berger, S. Barlow, S. R. Marder, K. I. Hardcastle and U. H. F. Bunz, 6,13-Diethynyl-5,7,12,14-tetraazapentacene, Chem. – Eur. J., 2009, 15, 4990–4993 CrossRef CAS PubMed.
- Z. Liang, Q. Tang, J. Xu and Q. Miao, Soluble and Stable N-Heteropentacenes with High Field-Effect Mobility, Adv. Mater., 2011, 23, 1535–1539 CrossRef CAS PubMed.
- K. Bulgarevich, S. Horiuchi, T. Ogaki and K. Takimiya, 1,3,6,8-Tetrakis(methylchalcogeno)pyrenes: Effects of Chalcogen Atoms on the Crystal Structure and Transport Properties, Chem. Mater., 2022, 34, 6606–6616 CrossRef CAS.
- G. Heywang and S. Roth, Radical Cation Salts of 1,3,6,8-Tetrakis(methylthio)-pyrene—New Easily Accessible Compounds with High Electrical Conductivity and Excellent Stability, Angew. Chem., Int. Ed. Engl., 1991, 30, 176–177 CrossRef.
- K. Takimiya, K. Bulgarevich, M. Abbas, S. Horiuchi, T. Ogaki, K. Kawabata and A. Ablat, “Manipulation” of Crystal Structure by Methylthiolation Enabling Ultrahigh Mobility in a Pyrene-Based Molecular Semiconductor, Adv. Mater., 2021, 33, 2102914 CrossRef CAS PubMed.
- K. Bulgarevich, S. Horiuchi and K. Takimiya, Crystal-Structure Simulation of Methylthiolated Peri-Condensed Polycyclic Aromatic Hydrocarbons for Identifying Promising Molecular Semiconductors: Discovery of 1,3,8,10-tetrakis(methylthio)peropyrene Showing Ultrahigh Mobility, Adv. Mater., 2023, 35, 2305548 CrossRef PubMed.
- K. Takimiya, K. Bulgarevich and S. Horiuchi, Contrasted behaviours of methylthiolated perylene and pyrene as organic semiconductors: implications of molecular electronic structure and crystal structure, J. Mater. Chem. C, 2023, 11, 10809–10815 RSC.
- S. F. Boys and F. Bernardi, The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors, Mol. Phys., 1970, 19, 553–566 CrossRef CAS.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 Rev. C.01, 2016 Search PubMed.
- M. K. Dudek and K. Drużbicki, Along the road to crystal structure prediction (CSP) of pharmaceutical-like molecules, CrystEngComm, 2022, 24, 1665–1678 RSC.
- S. P. Thomas, P. R. Spackman, D. Jayatilaka and M. A. Spackman, Accurate Lattice Energies for Molecular Crystals from Experimental Crystal Structures, J. Chem. Theory Comput., 2018, 14, 1614–1623 CrossRef CAS PubMed.
- R. A. Laudise, C. Kloc, P. G. Simpkins and T. Siegrist, Physical vapor growth of organic semiconductors, J. Cryst. Growth, 1998, 187, 449–454 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental and computational details, simulated crystal structures. CCDC 2256594–2256596 and 2280111. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3mh01055d |
| ‡ Simulated crystal structures are available as crystallographic information format (see ESI†) |
| § Experimental crystallographic data for new polymorphs of MT-pyrene (2M-CL-BW, CCDC 2256594–2256596) and MT-peropyrene (1M-CL-iBW, CCDC 2280111).† |
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a and
Kazuo
Takimiya
a and
Kazuo
Takimiya