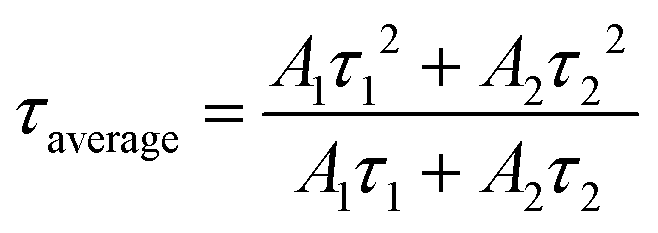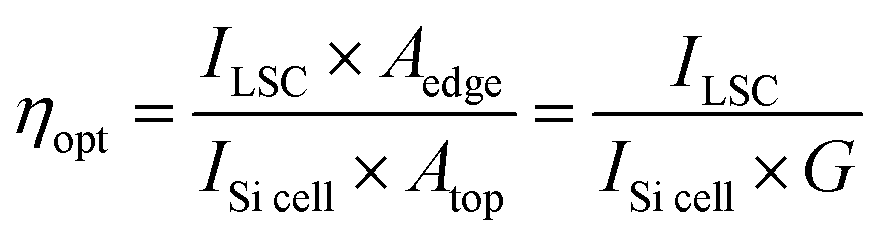DOI:
10.1039/D2NH00360K
(Communication)
Nanoscale Horiz., 2023,
8, 83-94
Boosting efficiency of luminescent solar concentrators using ultra-bright carbon dots with large Stokes shift†
Received
1st August 2022
, Accepted 18th October 2022
First published on 19th October 2022
Abstract
Luminescent solar concentrators (LSCs) are able to collect sunlight from a large-area to generate electric power with a low cost, showing great potential in building-integrated photovoltaics. However, the low efficiency of large-area LSCs caused by the reabsorption losses is a critical issue that hampers their practical applications. In this work, we synthesized novel yellow emissive carbon dots (CDs) with a large Stokes shift of 193 nm, which exhibit nearly zero reabsorption. The quantum yield (QY) of the yellow emitting CDs is up to 61%. The yellow emitting CDs can be employed to fabricate high-performance large-area LSCs due to successful suppression of the reabsorption losses. The as-prepared LSCs are able to absorb 14% of the sunlight as the absorption of the CDs matches well with the sun's spectrum. The large-area LSC (10 × 10 cm2) with a laminated structure based on the yellow emitting CDs achieves an optical conversion efficiency (ηopt) of 4.56% and power conversion efficiency (ηPCE) of 4.1% under natural sunlight (45 mW cm−2), which are significantly higher than other previously reported works with similar sizes. Furthermore, the prepared high-performance LSCs show good stability. This method of synthesizing novel CDs for high-efficiency LSCs provides a useful platform for future study and practical application of LSCs.
New concepts
Luminescent solar concentrators (LSCs) have potential in building-integrated photovoltaics. Unfortunately, the reabsorption losses of luminescent materials dramatically lead to the low efficiency of large-area LSCs. Herein, we report novel yellow emissive carbon dots (CDs) with a large Stokes shift of 193 nm, which exhibit nearly zero reabsorption. The quantum yield (QY) of the yellow CDs is up to 61%. The large-area LSC (10 × 10 cm2) using the yellow CDs that suppress reabsorption losses can obtain an optical conversion efficiency (ηopt) of 4.56% and power conversion efficiency (ηPCE) of 4.1% under natural sunlight (45 mW cm−2), which are significantly higher than other previously reported works with similar sizes.
|
1. Introduction
Solar photovoltaic (PV) technology has become the fastest growing renewable energy technology due to increased energy demand, the imperative reduction of carbon emissions, and environmental concerns.1–4 In addition, with the requirements of people's daily life, high performance batteries and modules are continuously developed, and the continuous renewal and iteration of products force the cost competition in the manufacturing industry to become increasingly fierce. Due to the limited spectral response of PV, it can only effectively collect part of the solar spectrum, which seriously limits the PV efficiency. Thus, it is an urgent problem that needs to be solved to realize the efficient PV conversion of solar energy.5 Luminescent solar concentrators (LSCs) can solve this problem well since they provide a straightforward approach to harvest and concentrate sunlight.6,7 LSCs were first reported in 1976 by Weber and Lambe.8 An LSC commonly consists of a plate of a transparent material with a high refractive index containing luminescent species (luminophores).7,9–16 These luminophores can re-emit the absorbed sunlight in a large fraction of the solar spectrum into a waveguide mode via total internal reflection at longer wavelengths through photoluminescence (PL) processes, and the re-emitted light will propagate toward the slab edges where small-area PVs are placed.
Luminophores play a major role in an aspect strongly marked by improving the power conversion efficiency (ηPCE) of the LSCs. To date, many types of luminescent species have been used in the study of LSCs, including organic dyes, polymers, semiconducting polymers, metal complexes, metal oxides, upconversion nanocrystals, quantum dots (QDs), perovskite nanocrystals and fluorescent protein complexes.10–12,17–19 Among them, QDs are the most emerging member in the family of LSC luminophores due to their excellent optical properties, such as high fluorescent quantum yield (QY) and good photostability compared to traditional fluorescent materials like organic dyes.1–3,11,16 To date, a variety of QDs have been used in LSCs.10,11,20–26 Carbon dots (CDs) in particular show promise in LSCs because of their unique optical properties which have been applied in many fields such as biological imaging, medical diagnosis, light emitting devices and energy converting devices.3,11 CDs are nanoparticles with a typical size of less than 10 nm. Most CDs contain abundant non-toxic elements (C, N and O) on the earth, and large amounts of synthesis can be carried out through simple and green methods by using rich and low-cost carbon sources, and sustainable synthetic processes.27 Since CDs were first reported for LSC fabrication in 2017,11 many efforts have been devoted to prepare CD-based LSCs.3,13,21,22,27–35 Unfortunately, the currently reported ηPCE of CD-based LSCs is still low, which dramatically hinders their potential application in building-integrated photovoltaics (BIPVs).
In this work, we designed and synthesized novel ultra-bright yellow emissive CDs with a large Stokes shift and long fluorescent lifetime using a one-step solvothermal method. The yellow emitting CDs exhibited strong concentration-dependent QYs. By optimizing the CD concentration, the QY of the yellow emitting CDs reached ∼61%. Besides, the yellow emitting CDs with a Stokes shift of 193 nm enable reabsorption of nearly zero which greatly suppresses the reabsorption loss. We also demonstrated a high-performance large-area LSC using the yellow emitting CDs as luminophores. The optical conversion efficiency (ηopt) and ηPCE of the yellow emitting CDs integrated large-area LSC (10 × 10 cm2) reached 4.56% and 4.1% under natural sunlight (45 mW cm−2), respectively. These efficiencies are higher than other previously reported works with similar sizes. The high-efficiency LSCs using eco-friendly yellow emitting CDs as fluorophores provide a useful platform for future studies and practical application of LSCs.
2. Results and discussion
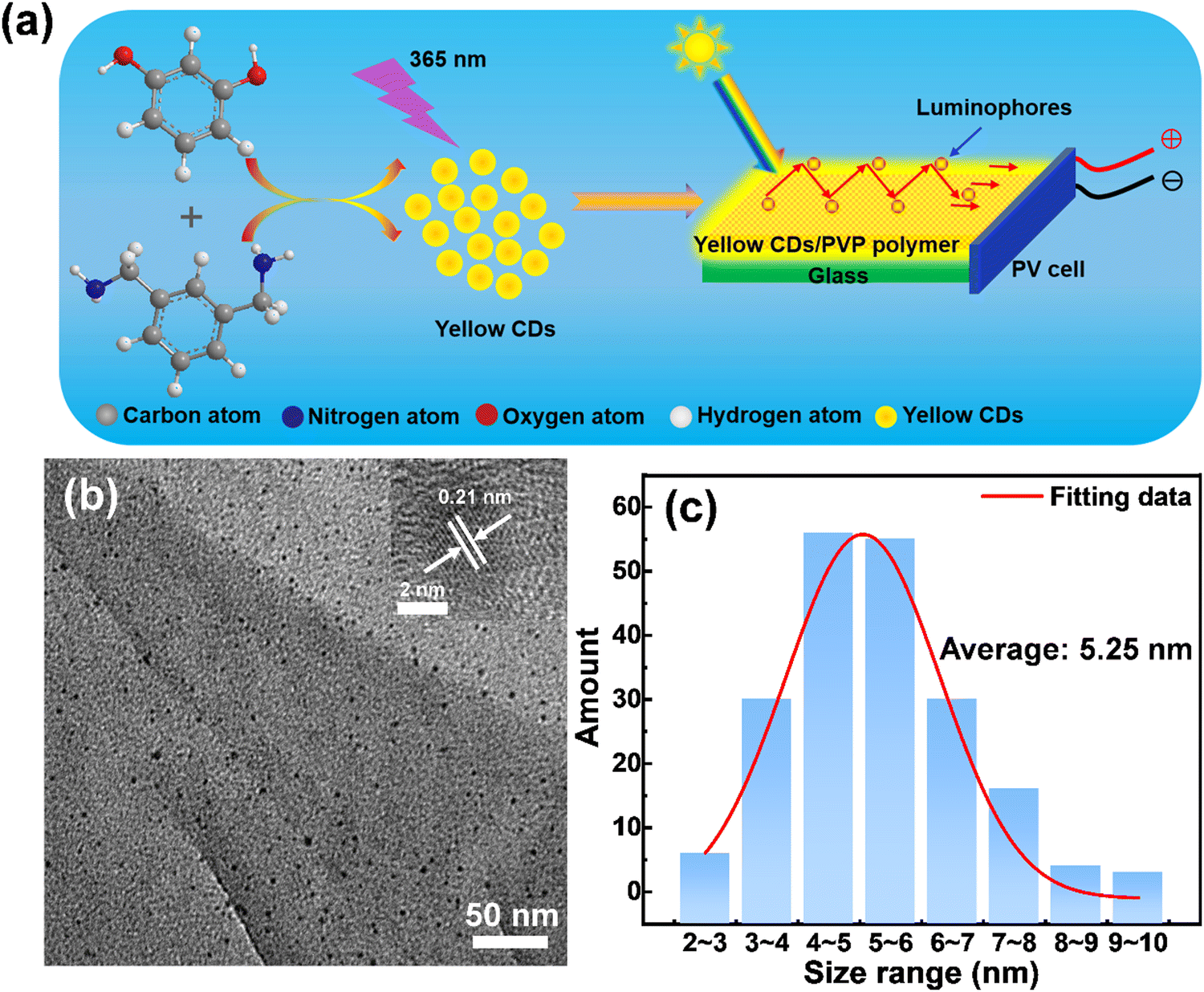
Yellow emissive CDs were synthesized through a solvothermal approach. Details of the synthetic process are shown in Fig. 1a. The reaction was carried out at 180 °C for 6 hours with Resorcinol (Rc) as the carbon source, m-phenyldimethylamine (mPDA) as the nitrogen source and absolute ethanol as the solvent. Transmission electron microscopy (TEM) was used to characterize the size and morphology of the prepared yellow emitting CDs. A typical low-magnification TEM image of yellow emitting CDs is presented in Fig. 1b. The as-prepared yellow emitting CDs have an average size of 5.25 nm (Fig. 1c), which was measured according to more than 200 particles by TEM images. The inset of Fig. 1b is the high-resolution TEM (HRTEM) image of the as-synthesized yellow emitting CDs, showing an obvious lattice spacing of 0.21 nm. The surface chemical groups of the yellow emitting CDs were tested by FT-IR spectroscopy. As shown in the FT-IR spectrum (Fig. S1, ESI†), the absorption peak range at 3300–3500 cm−1 results from the stretching vibrations of N–H or O–H. The absorption peak range at 2800–3000 cm−1 indicates the existence of stretching vibrations of C–H. A clearly observed peak at 1645 cm−1 can be attributed to the stretching vibration of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C or CN. Another sharp peak at 1381 cm−1 originated from stretching vibrations of C–C or C–N. One absorption peak between 1028 cm−1 and 1199 cm−1 belongs to the bending vibration of C–H or C–O. Therefore, the FT-IR results show that the core structure of the obtained yellow emitting CDs could contain conjugated bonds and primary amino groups of precursor molecules. At the same time, the FT-IR result also confirms that the structure of the yellow emitting CDs is similar to those previously reported results.36–38 This is further corroborated by following XPS measurements.
C or CN. Another sharp peak at 1381 cm−1 originated from stretching vibrations of C–C or C–N. One absorption peak between 1028 cm−1 and 1199 cm−1 belongs to the bending vibration of C–H or C–O. Therefore, the FT-IR results show that the core structure of the obtained yellow emitting CDs could contain conjugated bonds and primary amino groups of precursor molecules. At the same time, the FT-IR result also confirms that the structure of the yellow emitting CDs is similar to those previously reported results.36–38 This is further corroborated by following XPS measurements.
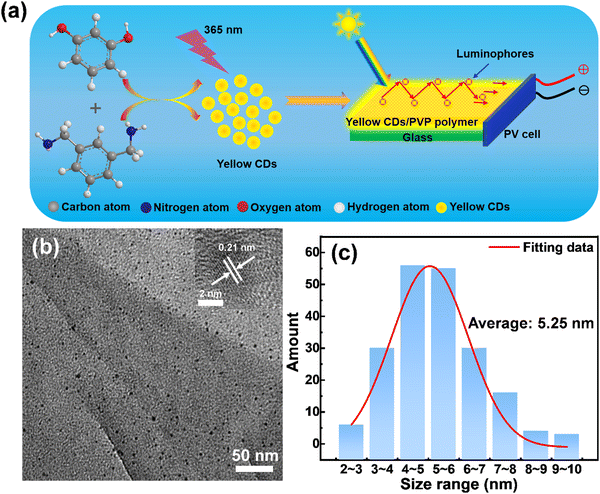 |
| | Fig. 1 (a) Chemical reactions used for the synthesis of CDs and schematic illustration of the LSCs based on CDs. (b) Typical TEM image of the yellow emitting CDs and HRTEM image of the yellow emitting CDs in the upper right corner of (b). (c) Size distribution of the yellow emitting CDs. | |
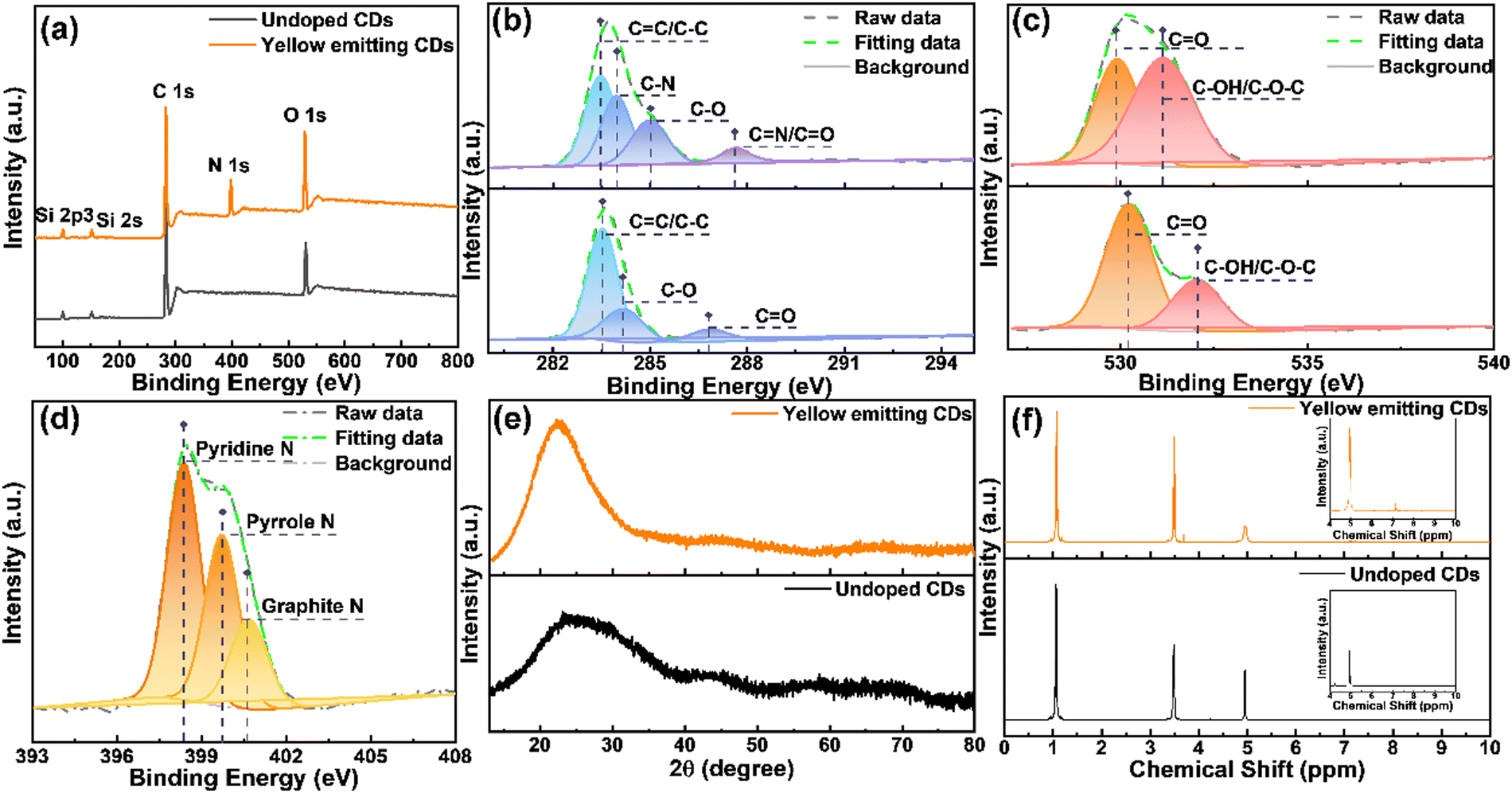
X-Ray photoelectron spectroscopy (XPS) and X-ray powder diffraction (XRD) are two of the best techniques to characterize the structure and composition of materials. In order to better compare the advantages of the as-synthesized yellow emitting CDs, we compared them with CDs without nitrogen (N) doping. As shown in Fig. 2a, the XPS spectrum of yellow emitting CDs only shows three peaks which are located at 532.2, 400.2 and 284.8 eV, belonging to oxygen (O 1s), nitrogen (N 1s) and carbon (C 1s) elements, respectively. It shows that these prepared yellow emitting CDs are comprised of C, N, and O elements with corresponding contents of 67.27%, 14.62%, and 18.11%. Compared with the yellow emitting CDs, the CDs without N doping obviously have no N peak. Detailed XPS analysis was performed to study the bonding structure and the differences between the yellow emitting CDs and the CDs without N doping. The upper part corresponds to the yellow emitting CDs. Fig. 2b displays the high-resolution XPS spectrum of C 1s, which is able to be fitted to four peaks located at 287.6 eV (CN/CO), 285.1 eV (C–O), 284.0 eV (C–N) and 283.6 eV (CC/C–C).37–39 It is worth noting that, for CDs without N-doping, there is obviously no C–N in the high-resolution spectrum of C 1s. Moreover, the high-resolution XPS spectra of O 1s and N 1s are also depicted in Fig. 2c and d. The high-resolution XPS spectrum of O 1s is able to be divided into two peaks located at 530 eV (CO) and 531 eV (C–OH/C–O–C).38–40 The N 1s spectrum is able to be assigned into three peaks located at 398.4, 399.7 and 400.8 eV due to the pyridinic-N, pyrrolic-N and graphitic-N, respectively. These reveal the formation of polyaromatic structures containing C–N and CN in yellow-emitting CDs. The XRD pattern shows a broad peak centered at approximately 22° (Fig. 2e), which confirms the (100) crystal plane of graphite carbon,38,41 but XRD cannot identify the amorphous composition. To further identify the structure and composition of the synthetic CDs containing N-doped and undoped CDs, they were characterized by nuclear magnetic resonance (1H NMR and 13C NMR, DMSO-d6, ppm) spectroscopy and mass spectrometry (MS). The 1H NMR spectra of yellow emitting CDs and undoped CDs are illustrated in Fig. 2f. The signal between 1.0 and 2.0 ppm is assigned to protons in aliphatic chains, such as alkane CH; while the signal peaks between 2 and 4 ppm correspond to protons of hydroxyl and/or carbonyl a-hydrogen.42–45 In addition, the signal near 3.5 ppm corresponds to α-H (amino) and the signal around 5.0 ppm corresponds to amide H.46 These non-aromatic hydrogens should be located on the CD surface. The signal peaks between 6.0 and 9.0 ppm correspond to aromatic protons.42,43–45,47–49 Obviously, with the introduction of N doping, both five- and six-membered ring forms may be present in the CD structure due to the possible structural changes of the CDs caused by N doping. Therefore, compared with the CDs without N doping, the N-doped CDs have significantly more signal peaks between 6.0 and 9.0. Specifically, in the 1H NMR spectrum of the N-doped yellow-emitting CDs, the 1H NMR proton shifts on the five-membered rings in the CDs were in the range of 6.0–7.0 ppm, while those on the six-membered rings were in the range of 7.0–7.5.46 In addition, as shown in Fig. S2a and b (ESI†), the 13C-NMR spectra (DMSO-d6, ppm) of yellow-emitting CDs and undoped CDs were further used to characterize the CDs. Both spectra showed sharp signal peaks about 18.0 ppm and around 58 ppm, and this is the methyl carbon and methylene carbon of ethanol, respectively, which can be attributed to the residual ethanol during the ethanol treatment.50 Similarly, there is also a signal peak at approximately 40 ppm for both, which can be attributed to the resident carbon peak of the DMSO solvent. Compared to the CDs without N-doping, some extra signal peaks are noticeable between 100 ppm and 200 ppm after N-doping. The large emerging signal observed in the 13C-NMR spectra of the N-doped yellow-emitting CDs in the range of 100–140 ppm compared to the undoped CDs may originate from the signal of the sp2 carbon atom, which is further evidence of the formation of the intact sp2 structural domain during the synthesis of the yellow emitting CDs.44,48 The resonance signal also clearly observed in the range of 155 to 180 ppm is attributed to the binding of sp2 carbon atoms to hydroxyl groups.45 Besides, mass spectrometric (MS) analysis of yellow emitting CDs and CDs without N-doping was also applied as an assistant analysis (Fig. S3, ESI†). As shown in Fig. S3a (ESI†), we estimated the properties of CHCA, undoped CDs, and yellow-emitting CDs as matrices for MALDI-TOF MS analysis, and the green stars indicate fragment peaks originating from the matrix. When CHCA is used as an assisted matrix, extra fragment ion peaks are clearly observed in the spectra of yellow emission CDs according to these spectra (m/z 136.19, m/z 210.27, m/z 228.29, m/z 276.28, m/z 346.46, m/z 379.18, m/z 438.56, m/z 481.35, m/z 572.35 and m/z 598.37), which can be attributed to the formation of some small fragments during the carbonized dehydration condensation of the reaction precursors. Besides, some other slight fragment ion peaks are also observed, which indicate the presence of residual polymer structures in the CDs.41,51 Similar results were obtained using the DHB matrix, see Fig. S3b (ESI†). Both NMR spectra and MS spectra can imply that CDs contain polyaromatic ring systems, –OH and amide groups, i.e., the reverse proves the successful introduction of N doping.
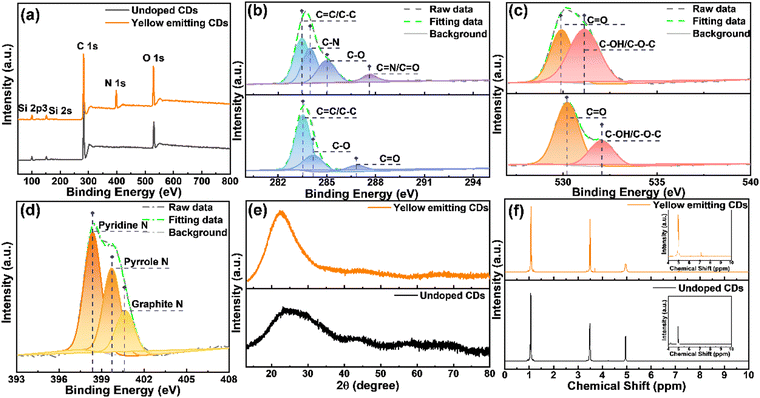 |
| | Fig. 2 (a) XPS survey spectrum of the synthesized yellow emitting CDs and undoped CDs. High-resolution XPS spectra of C 1s (b) and O 1s (c) for yellow emitting CDs (up) and undoped CDs (down). High-resolution XPS spectra of N 1s for yellow emitting CDs (d). (e) The XRD spectra of yellow emitting CDs (up) and undoped CDs (down). (f) 1H NMR spectra of yellow emitting CDs (up) and undoped CDs (down). | |
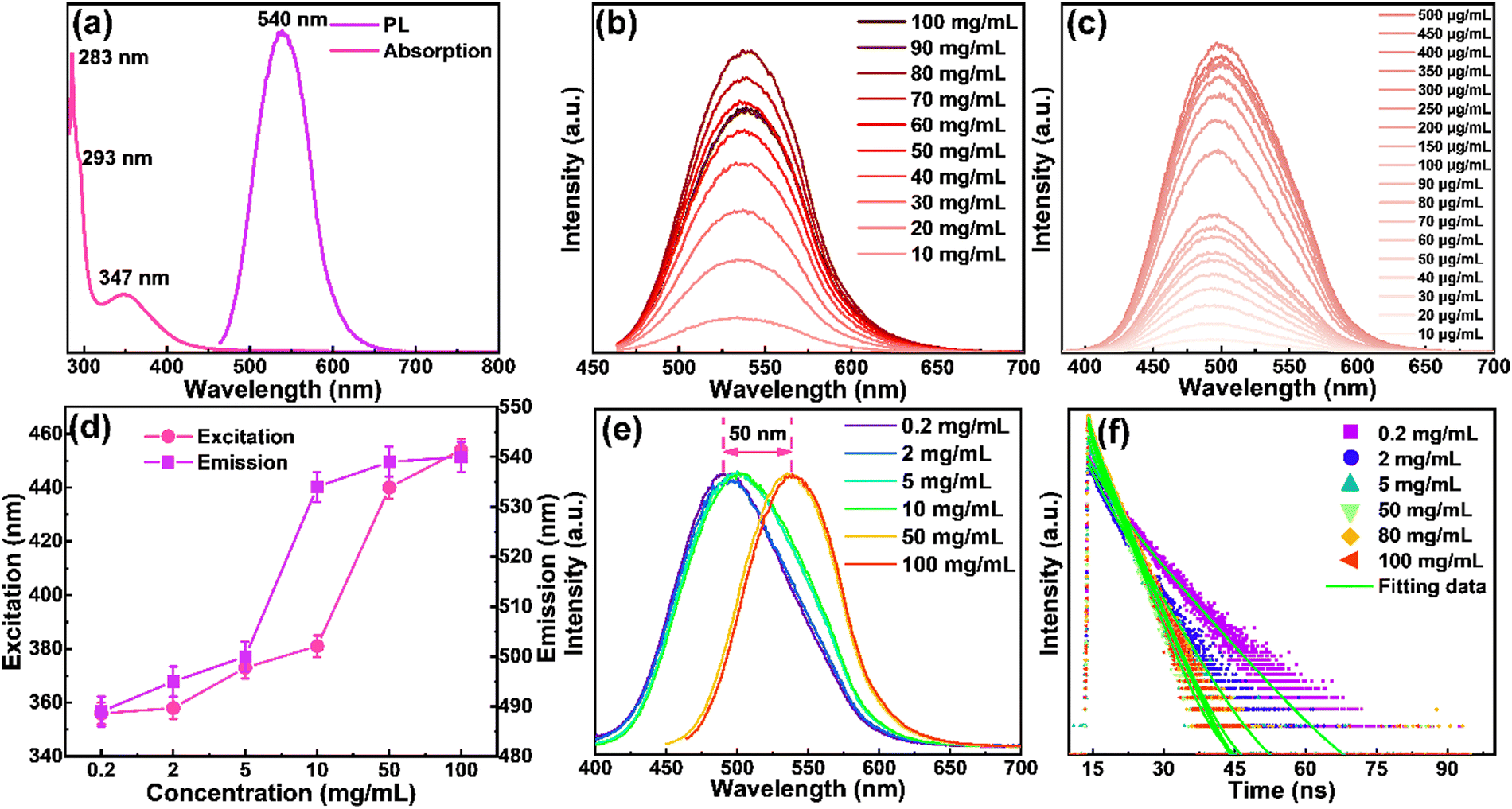
The optical properties of the yellow emitting CDs in ethanol solutions were investigated by UV-vis absorption and fluorescence spectra. As shown in Fig. 3a, the strongest peak centered at 283 nm, and a shoulder peak centered at 293 nm originated from π–π* transition of aromatic sp2 domains. The peak centered at 347 nm may be derived from the surface energy states.30,52 The yellow emitting CDs show absorption covering from 280 nm to ∼500 nm. The emission spectrum of the yellow emitting CDs ranges from 450 to 650 nm with a maximum peak at 540 nm when they were excited at 454 nm. The as-prepared CDs have a QY of ∼61% when they are dispersed in solution (Fig. S4a, ESI†). Meanwhile, the photophysical properties and device performance of the N-doped CDs are dramatically improved compared to those of non-N-doped CDs (Fig. S4, ESI†). The difference between the maximum of the absorption and emission peaks is usually defined as the Stokes shift, which is considered as one of the important factors in regulating the performance of LSCs. A small Stokes shift always leads to the loss of reabsorbed energy.30 According to Fig. 3a, the prepared yellow emitting CDs have a large Stokes shift of 193 nm and there is little spectral overlapping between the absorption and emission, revealing that the prepared yellow emitting CDs can successfully suppress the reabsorption loss. Since the concentration of luminophores plays an important role in the performance of LSCs, we investigated the concentration effect to better understand the optical properties of the CDs. We first analyze the overall high concentration, and find that while increasing the yellow emitting CD concentration from 10 to 100 mg mL−1 (Fig. 3b) with a fixed excitation wavelength (454 nm), a slight redshift (∼5 nm) of the PL spectra is observed (Fig. 3b), but the intensity of the peaks increases and decreases obviously. Interestingly, the emission intensity of the yellow emitting CDs increased nearly linear when the concentration changed from 10 to 80 mg mL−1 (Fig. S5, ESI†). However, further increasing the concentration induces the decrease of emission intensity (Fig. 3b). The collisional quenching phenomena can explain the change of decreased luminescence intensity of yellow emitting CDs at higher concentration.30,41 The PL peak position of the CDs during the concentration variation process hardly changed, indicating that the PL behavior of the yellow emitting CDs is not concentration-dependent at this concentration range (10–80 mg mL−1). In order to prove this phenomenon, we continued to reduce the concentration of yellow emitting CDs (0.2–10 mg mL−1) and found that the results were the same as above, and the obtained result is illustrated in Fig. 3c. Then, in order to study the different excitation-dependence of yellow emitting CDs, different concentrations including 0.2 mg mL−1, 2 mg mL−1, 5 mg mL−1, 10 mg mL−1, 50 mg mL−1 and 100 mg mL−1 were selected. At the same time, for different concentrations of the yellow emitting CDs, their optimal excitation and emission were also different. Compared with the high concentration, with the decrease of concentration, both excitation and emission have an obvious blueshift (Fig. 3d). Between low to high concentrations, the overall peak position shifted about 50 nm (Fig. 3e). In order to study the excitation-dependence of the yellow emitting CDs at different concentrations in more detail, we carried out a systematic excitation-dependence study for the above six concentrations (Fig. S6, ESI†). With further increasing the excitation wavelength, the fluorescence intensity decreased, revealing that yellow emitting CDs are more sensitive in response to the UV and blue ranges. In addition, the redshift of the emission peaks appeared with the increase of excitation wavelengths, and the emission peak induced fluctuation of about 20 nm under different excitations (Fig. S6, ESI†). Therefore, the results clearly show that these yellow emitting CDs absorb UV light and then re-emit at higher wavelengths e.g., in the blue or green light range.
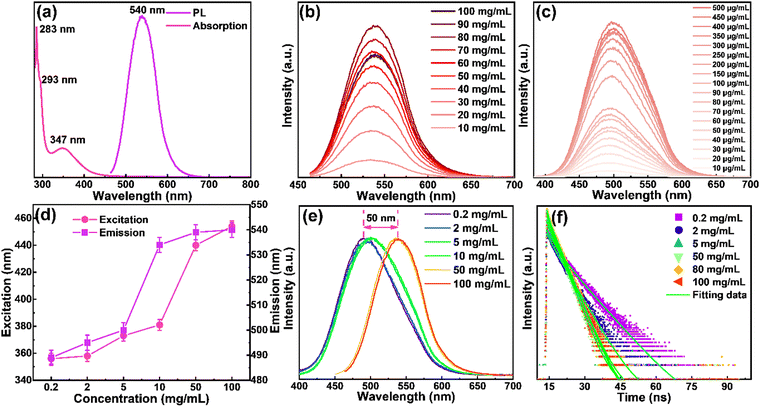 |
| | Fig. 3 (a) The absorption and emission spectra of the yellow emitting CDs dispersed in ethanol solution. (b) The PL spectra of the yellow emitting CDs under different concentrations at an excitation wavelength of 454 nm. (c) The PL spectra of the yellow emitting CDs under lower concentrations at an excitation wavelength of 454 nm. (d) The relationship between excitation and emission at different concentrations. (e) Optimum PL spectra of the CDs with different concentrations from 0.2 to 100 mg mL−1. (f) The corresponding time-resolved PL decay curves. | |
As mentioned above, under different yellow emitting CD concentrations, the optimal emission of yellow emitting CDs was also different as shown in Fig. 3e. In addition, when increasing the concentration, the optimal emission peak position had a distinct redshift, which can be attributed to the reabsorption or reemission processes.53 When reabsorption occurs, high-energy photons will be reabsorbed, and then re-emitted as low-energy photons, resulting in a reduced luminescence intensity and redshift of the PL peak position.53 We assume that this PL red-shifting with increasing the concentrations at different excitation wavelengths could be ascribed to two possible mechanisms, namely, concentration-dependent nonradiative energy transfer and reabsorption processes.13,19,53,54 Due to the introduction of extra nonradiative pathways, the energy transfer will not only lead to a spectral redshift, but also reduce the PL lifetime.19,53 To prove this hypothesis, time-resolved PL decay curves of yellow emitting CDs were investigated (excitation wavelength at 454 nm). According to Fig. 3f, the PL decay curves exhibited double exponential lifetimes. The fluorescence behavior was on a nanosecond timescale for yellow emitting CDs, with the longest lifetime of 6.7 ns. Detailed data are presented in Table 1 and the average lifetime (τaverage) was calculated through eqn (1):22,52
| | 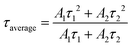 | (1) |
where
A1 and
A2 are the pre-exponential factors; and
τ1 and
τ2 are the fitted lifetimes, respectively. The decay curves of the yellow emitting CDs include two components: the fast component (
τ1) and slow component (
τ2).
τ1 is ascribed to the radiative recombination of eigenstates and
τ2 is attributed to recombination processes of surface states.
52,55 And also, it can be seen that the PL lifetimes were slightly reduced with increasing the loadings from 0.2 to 80 mg mL
−1. Further increasing the concentration of CDs, there is no significant variation in the lifetimes. Based on
eqn (2) and (3), the non-radiative decay (
Knrad) increases from 0.10 (10 mg mL
−1) to 0.15 (50 mg mL
−1) and 0.16 (80 mg mL
−1) with the increase of the concentration, indicating that the higher concentration would contribute to an energy transfer process because (i) the spatial distance between CDs favors a higher concentration in this process; (ii) there is still a small overlap between the absorption and emission spectrum.
| | | τmeasured = 1/(Krad + Knrad) | (2) |
| | | QY = Krad/(Krad + Knrad) | (3) |
where
Krad and
Knrad are the radiative decay and non-radiative decay respectively. As shown in Fig. S7 and S8 (ESI
†), the fluorescence lifetimes were also studied under different excitation. The variation trend of the whole fluorescence lifetime is similar to that in
Fig. 3f. However, when exciting the CDs with a high energy (short wavelength), the fluorescence lifetime is higher than that with a low energy (long wavelength), and the detailed data are provided in Tables S1 and S2 (ESI
†). This result can be explained by the fact that high energy excitation involves multiple bands and multiple relaxation process; on the contrary, low energy excitation is more surface structure related excitation, which may lead to a shorter fluorescence lifetime due to defects and other non-radiative recombination processes. In order to better verify this explanation, we fixed the low concentration (0.2 mg mL
−1) and high concentration (80 mg mL
−1) of the CDs respectively and adjusted different excitation, and the results are completely consistent with the previous results. In Fig. S9 and S10 (ESI
†), once the excitation wavelength was increased, and the corresponding fluorescence lifetime decreased slightly. In addition, the fluorescence lifetime at a low concentration was significantly higher than that at a high concentration because there is no or small energy transfer at a low concentration (see the data in Tables S3 and S4, ESI
†).
Table 1 The fluorescence lifetime for double exponential lifetimes of yellow emitting CDs in different concentrations
| Concentration (mg mL−1) |
τ
1 (ns) |
τ
2 (ns) |
τ
average (ns) |
R
2
|
| 0.2 |
3.02 ± 0.11 |
7.75 ± 0.17 |
∼6.70 |
0.995 |
| 2 |
2.02 ± 0.19 |
5.13 ± 0.07 |
∼4.89 |
0.997 |
| 5 |
0.67 ± 0.02 |
3.91 ± 0.02 |
∼3.79 |
0.998 |
| 50 |
0.75 ± 0.02 |
3.46 ± 0.01 |
∼3.30 |
0.999 |
| 80 |
0.77 ± 0.02 |
3.59 ± 0.01 |
∼3.39 |
0.999 |
| 100 |
0.85 ± 0.02 |
3.64 ± 0.02 |
∼3.41 |
0.999 |
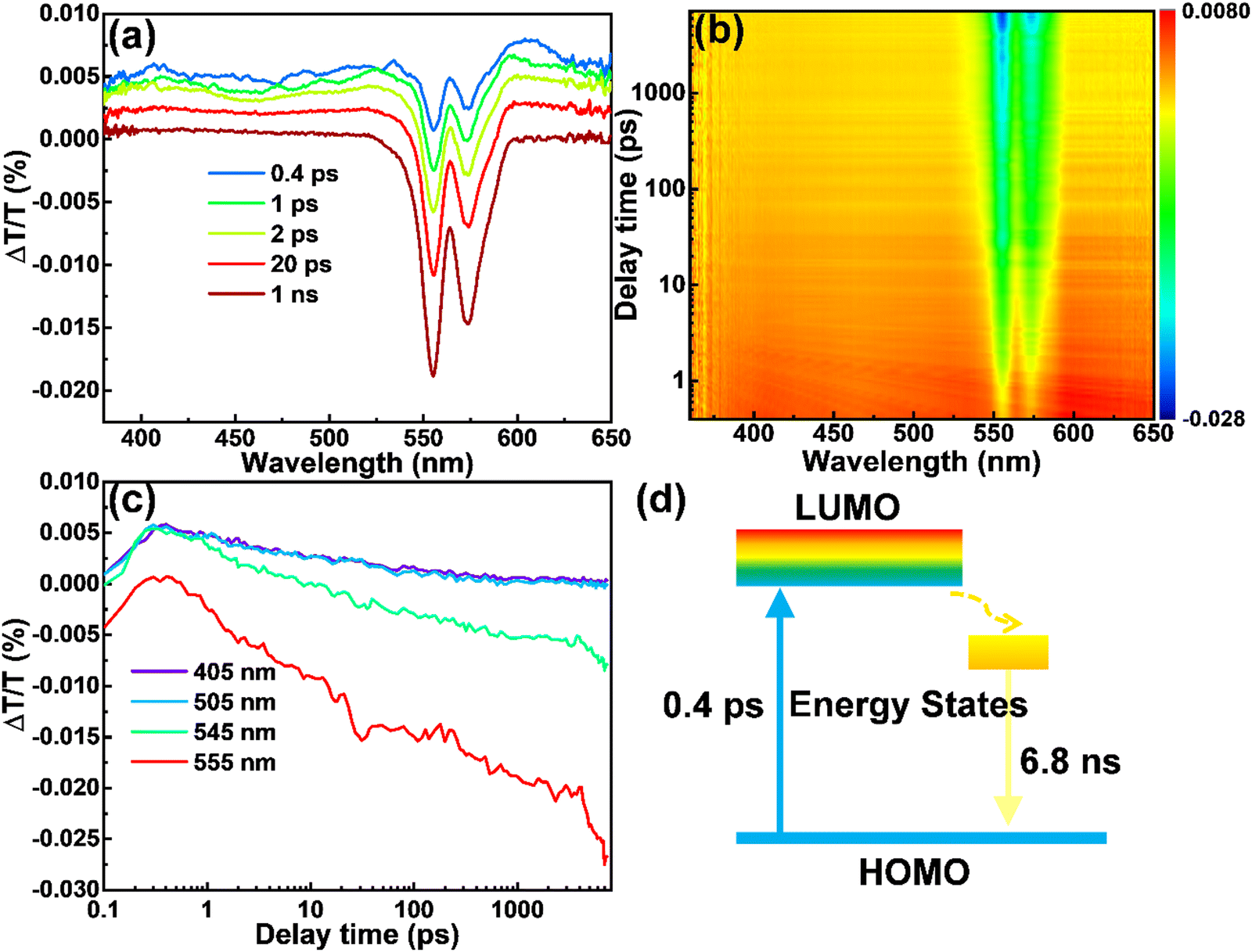
In order to explain the origination of the large Stokes shift and high QY of the CDs produced in this work, we performed ultra-fast transient absorption spectroscopy (TA). The TA spectra of the CDs dispersed in methanol were shown in Fig. 4a and b, with the wavelength range of 360–650 nm, scan delay time from 0.1 ps to 7 ns and pump of 280 nm. The negative peak centered at 555 nm corresponds to simulated emission (SE), consistent with their PL spectra. The negative peak centered at 574 nm is due to the scattering effect of the pump (280 nm). The relatively weaker positive features from 360 to 530 nm correspond to the excited state absorption (ESA). As in the detection range, we did not find any of the photo-bleaching caused by ground state absorption (GSA), so we deduce that GSA may be lower than 400 nm, consistent with the steady state absorption peak (347 nm) of the CDs. This TA measurement confirms the large Stokes shift in our CDs. It is obvious that at 0.4 ps (blue in Fig. 4a), the SE has already shown the spectral characteristics observed in steady-state emission. This phenomenon indicates that the timescale of Stokes shift of the yellow emitting CDs takes place on a much shorter timescale than 0.4 ps. Even in the temporal window of 1 ns, no obvious intensity loss of SE is observed (maroon in Fig. 4a). Meanwhile, we found that the amplitude of the emissive component is much higher than that of the nonradiative decay component, which explains the high QY of the as-prepared CDs. Further decay measurement indicates that the SE has a decay time of 6.8 ns, in agreement with the time-resolved PL result (Fig. 4c). Based on the TA data, the energy structure and exciton dynamics are shown in Fig. 4d. With the excitation, the electron jumps to the down-shifted lowest unoccupied molecular orbital (LUMO), further decays to the internal energy state between the LUMO and the highest occupied molecular orbital (HOMO), and recombines with a hole by releasing a photon. The large Stokes shift originates from the internal energy state, which may be induced by the N dopants.
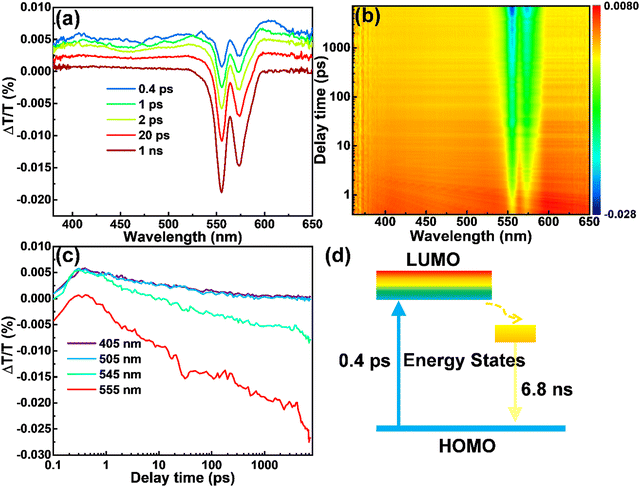 |
| | Fig. 4 The ΔT/T spectra of the yellow emitting CDs. (a) The spectra for selected time delays. (b) The mapping of ΔT/T. (c) The dynamics for selected wavelengths. (d) Schematic illustration for the proposed energy diagram and exciton dynamics. | |
Based on the above optical performance study, the as-prepared yellow emitting CDs with different concentrations (20 mg mL−1, 40 mg mL−1, 60 mg mL−1, 80 mg mL−1, and 100 mg mL−1) were dispersed into PVP ethanol solutions and the single-layer LSCs were fabricated by drop-casting. The yellow emitting CD-integrated single-layer LSCs (5 × 5 cm2) are semi-transparent (Fig. S11a, ESI†). A concentrated light can be obviously seen on the edges of the LSC and “LSC” words on the bottom of LSC can be clearly detected. As shown in Fig. S12 (ESI†), the yellow emitting CDs inside the LSCs exhibited a relatively independent PL spectrum at different concentrations. With the increase of yellow emitting CD concentration, the PL intensity also increased and reached the strongest at 80 mg mL−1. However, the intensity of PL decreased after further increasing the yellow emitting CDs concentration (Fig. S12a, ESI†). At the same time, the peak position was accompanied by a slight redshift, which is consistent with the PL behavior of the CDs in ethanol solution. If the amount of yellow emitting CDs/PVP mixture is changed, the PL phenomenon is similar to that of different concentrations (Fig. S12b, ESI†). When the amount of the yellow emitting CDs/PVP mixture was 2.5 mL, the PL intensity reached the maximum. Once the amount continued to increase the PL intensity decreased inversely. Through the above PL detection for LSCs, the optimal concentration and dosage are 80 mg mL−1 and 2.5 mL. It is found that there was little change in fluorescence intensity after five weeks’ storage of the LSCs (Fig. S12c and d, ESI†). Moreover, the different excitation and emission phenomena of yellow emitting CDs/PVP composite films were almost the same as those of the yellow emitting CD solution (Fig. S6 and S13, ESI†). This shows that there was little influence on the fluorescence intensity when yellow emitting CDs were embedded in PVP films.
To evaluate the performance of the LSC devices, PCE measurements were carried out. The photocurrent density and photovoltage (J–V) response of the commercial Si solar cell is shown in Fig. S14 (ESI†). As shown in Table S5 (ESI†), the Si solar cell has a Jsc of 36.88 mA cm−2, Voc of 0.54 V, fill factor (FF) of 75.76% and PCE of 15.19% under one sun illumination (AM 1.5G). The J–V curves were measured for different LSC devices (Fig. S15, ESI†), and the detailed parameters are shown in Tables S5 and S6 (ESI†). Under illumination of 100 mW cm−2, ηopt (power coming out from LSC edge/power coming in from the top surface) could be obtained for the device (5 × 5 cm2, detailed calculation is shown in the ESI†).56 We further measured and calculated the ηopt value of LSCs by the power meter coupled at the edge of the LSCs, which is defined as the number of photons emitted from the edges of the LSC divided by the number of photons incident on its upper surface. ηopt is a key index to evaluate the performance of LSCs. ηopt can be calculated through eqn (4):3,30,57
| | 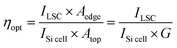 | (4) |
where
ILSC is the measured short-circuit current when the solar cell is attached on the LSC device edge, and
ISi![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) cell
cell is the measured short circuit current which directly irradiates the same solar cell with the same light source. While
Aedge is the edge area of the LSCs where the solar cell is attached,
Atop is the top area of the LSC. The ratio of
Atop/
Aedge is generally termed as geometric gain (
G). In our case, the
G was calculated to be 6.25 for the 5 × 5 cm
2 and 2.5 for the 10 × 10 cm
2 LSC.
In order to study the influence of the G factor on the performance of the LSCs, LSCs with different sizes, but with identical film thickness were fabricated (Fig. S15a, ESI†). The as-prepared LSCs have G factors of 2.5, 6.25 and 10, respectively. The J–V curves were measured for the different sizes of LSC devices (Fig. S15b, ESI†). With the increase of the size of LSCs, there is no significant variation of the measured PCEs (Fig. S15c, ESI†). We are still able to obtain an optical efficiency of 4.1% for the LSC with a G factor of 10 (dimension: 8 × 8 × 0.2 cm3). Therefore, it can be concluded that the impact of the G factor on large-size LSCs is relatively small, and this is mainly because of the large Stokes shift of the yellow emitting CDs. As shown in Fig. S15d (ESI†), the LSC showed an obvious concentration-dependent J–V behavior by optimizing the concentration of the yellow emitting CDs. When the concentration was 80 mg mL−1, the PCE performance of the device reached the best as shown in Fig. S15e (ESI†). The ηopt and ηPCE of the yellow emitting CD-integrated LSC reached up to 4.26% and 3.72% under illumination of 100 mW cm−2, respectively. To investigate the performance stability of the LSCs (concentration of 80 mg mL−1), we measured the PCE each week for five weeks (Fig. S15f, ESI†). In addition to the obvious increase in PCE in the first week, this might be mainly caused by the fact that the yellow emitting CDs/PVP composite film is not completely dried on the first day. After that, the PCE of the LSC devices hardly changed, indicating that the LSCs have good stability. Furthermore, the optical properties of the yellow emitting CDs/PVP film LSCs with various dosages were investigated (Fig. S12b and S12d, ESI†). For different yellow emitting CDs/PVP mixtures of dosage in LSCs, the result of J–V behavior was almost the same as the different concentrations. Due to the fact that more yellow emitting CDs are contained in the higher dosage film, the PL intensity of the yellow emitting CDs/PVP film LSCs increased first and then gradually decreased with the increase of the dosage. As shown in Fig. S15g (ESI†), the change of photocurrent density was similar to that of the PL intensity with the dosage increases. This shows that the LSC displayed a clear dosage-dependent J–V behavior by optimizing the dosage of the yellow emitting CDs. When the dosage was 2.5 mL, the PCE performance of the LSC device reached the best value (Fig. S15h, ESI†). The ηopt and ηPCE of the yellow emitting CDs integrated LSC could reach 4.78% and 4.14% under illumination of 100 mW cm−2, respectively. Even after the LSC devices were placed under natural sunlight irradiation for five weeks, the PCE hardly changed, indicating that the LSCs have good stability (Fig. S15i, ESI†). The stability of other parameters is shown in Fig. S16 and Tables S5, S6 (ESI†).
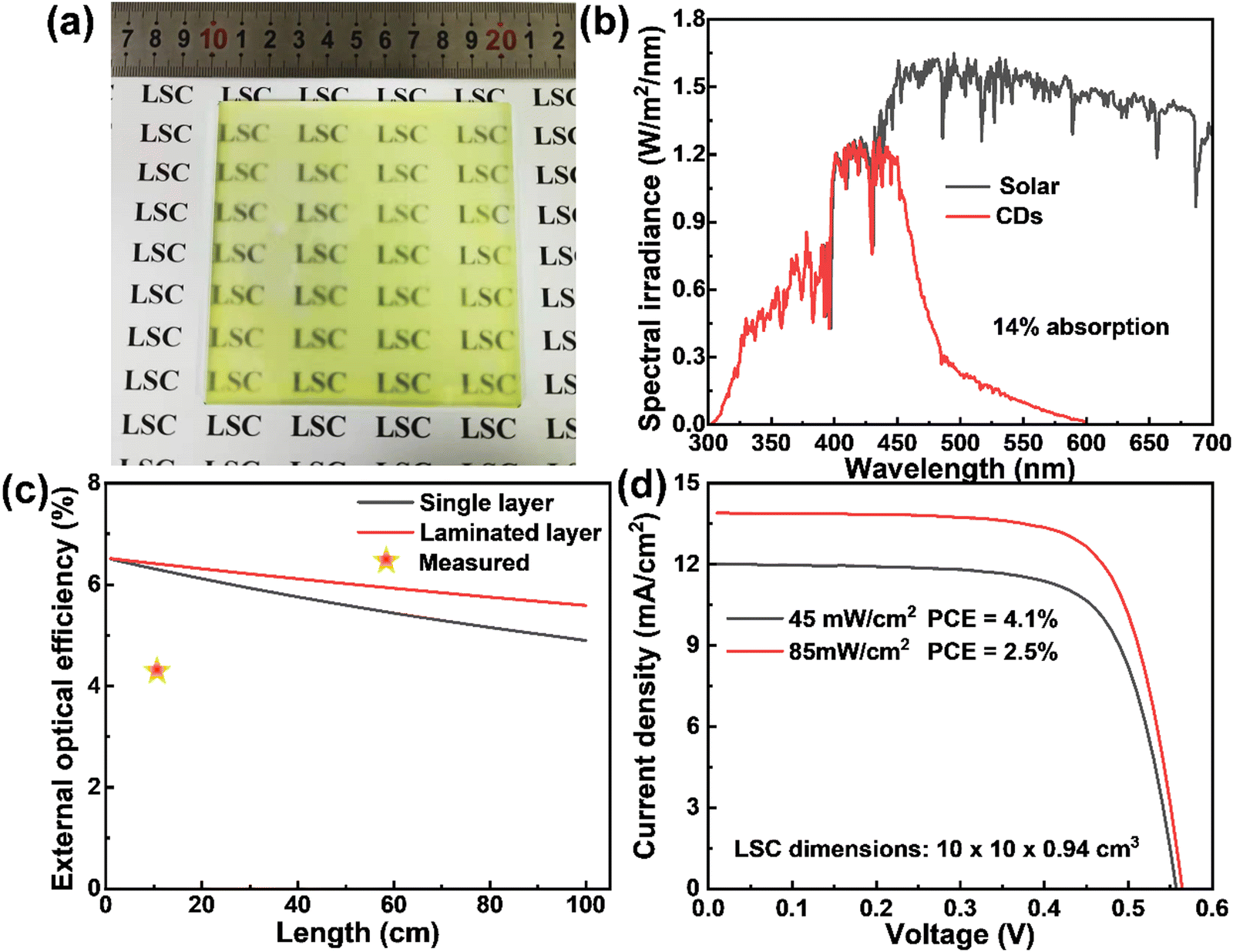
In order to get close to life and realize commercial large-scale applications, we optimized the CD concentration, dosage and preparation conditions, and prepared a large-are LSC (10 × 10 cm2) with a laminated structure as shown in Fig. 5a. Typically, the CDs/PVP composite film was formed on the glass surface by drop casting. Another glass slide was then put on the top of the CDs/PVP film to construct a laminated structure. The as-prepared LSC based on CDs can absorb 14% of the sunlight in the UV-vis range as the absorption of the CDs matches well with the sun's spectrum (Fig. 5b). We theoretically calculated the ηopt of the LSCs using the reported method (details are in the ESI†).36 As shown in Fig. 5c, ηopt of the yellow emitting CD-based LSCs with a concentration of 80 mg mL−1 in the PVP film decreased with increasing the size of the LSC. Benefiting from the laminated structure compared to the single-layer structure, the laminated LSCs exhibited an ηopt of 5.6% and even the lateral size of the LSCs is 1 m2, which shows promising potential for realizing BIPV. In addition, the ηopt of the laminated LSC maintains 86% of the initial value when the lateral size is 1 m2, indicating the reabsorption energy loss can be largely suppressed since the Stokes shift of yellow emitting CDs is very large. We measured the PCE of the LSCs by coupling a calibrated Si solar cell. The LSC had a PCE of 2.5% and 4.1% under the natural environment of 85 mW cm−2 and 45 mW cm−2, respectively (Fig. 5d). The ηopt of the yellow emitting CDs integrated large-area LSCs reached 2.78% and 4.56% under 85 mW cm−2 and 45 mW cm−2, respectively. The obtained ηopt of the large-area LSC is higher than other previously reported works with similar sizes (Table 2). The high efficiency achieved in this work might be attributed to the two following reasons: (i) efficient absorption of the sunlight; (ii) less reabsorption energy loss benefiting from the high QY, large Stokes shift and laminated LSC structure. It should be noted that this laminated structure is fit for the real structure of warm-keeping windows, in which two pieces of glass are usually used.
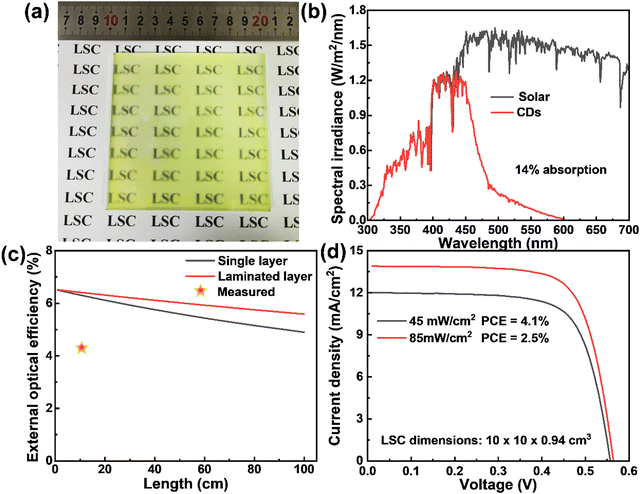 |
| | Fig. 5 (a) Photographs of the laminated LSC based on yellow emitting CDs. (b) Calculated solar absorption of the LSCs based on yellow emitting CDs and solar spectrum (AM 1.5G). (c) The calculated external optical efficiency of LSCs based on yellow emitting CDs with a QY of 61% and a concentration of 80 mg mL−1. The red star marker is the external optical efficiency of the LSCs measured by a power meter for the LSCs with lateral sizes of 10 × 10 cm2. (d) J–V response of the LSC with a Si cell attached on the edge under natural sunlight of 85 mW cm−2 and 45 mW cm−2, respectively. | |
Table 2 External optical efficiencies of the LSCs based on different fluorophores
| Fluorophores |
Type |
Absorption range |
η
opt (%) |
LSC area (cm2) |
Ref. |
| CDs |
Single layer |
280–500 |
4.56 |
10 × 10 |
This work |
| CDs |
Single layer |
300–550 |
2.6 |
10 × 10 |
22
|
| CDs |
Single layer |
300–650 |
3.0 |
10 × 10 |
22
|
| CDs |
Two layer tandem |
300–660 |
4.3 |
10 × 10 |
22
|
| CDs |
Single layer |
300–600 |
3.94 |
2 × 2 |
11
|
| CDs |
Two layer tandem |
300–700 |
1.1 |
10 × 10 |
28
|
| CDs and perovskite QDs |
Three layer tandem |
300–400 |
3 |
10 × 10 |
13
|
| CDs |
Two layer tandem |
300–650 |
— |
10 × 10 |
31
|
| DDAB@AuNCs/PLMA |
Single layer |
300–500 |
1.85 |
10 × 10 |
58
|
| CdSe/CdS QDs |
Sandwich |
300–580 |
2.25 |
10 × 10 |
59
|
| Perovskite nanoplatelets |
Single layer |
300–550 |
0.87 |
10 × 10 |
60
|
| Perovskite nanocrystals |
Single layer |
300–520 |
2.4 |
10 × 10 |
17
|
| CDs and AIE molecules |
Two layer tandem |
30–550 |
3.55 |
6.5 × 2.5 |
33
|
| CDs and Ag NPs |
Single layer |
— |
0.9 |
5 × 5 |
32
|
| CDs |
Three layer tandem |
300–610 |
2.3 |
8 × 8 |
35
|
| CDs |
Three layer tandem |
300–600 |
4.03 |
5 × 5 |
61
|
| CDs |
Single layer |
300–500 |
2.7 |
10 × 10 |
3
|
| Copper-doped InP/ZnSe QDs |
Single layer |
300–620 |
3.4 |
10 × 10 |
26
|
| Silica encapsulation of CdSe/CdS QRs |
Single layer |
300–650 |
2.06 |
10 × 10 |
62
|
| Iodine-based perovskite nanoplatelets |
Single layer |
300–750 |
1.1 |
10 × 10 |
63
|
| CdSe/CdS QDs and CDs |
Two layer tandem |
300–520 |
1.4 |
10 × 10 |
64
|
3. Conclusions
In summary, highly emissive yellow emitting CDs with a large Stokes shift of 193 nm, which exhibit nearly zero reabsorption, were reported. The QY of the yellow emitting CDs reached as high as 61%. The fluorescence properties of different concentrations were systematically studied, and the results showed that the corresponding excitation and emission of the yellow emitting CDs were also different with a concentration change. In addition, the PL emission was red-shifted along with a reduced PL intensity when increasing the excitation wavelengths. At the same time, the fluorescence lifetime corresponding to different concentrations and different excitation were also different. The longest fluorescence lifetime of yellow emitting CDs could reach 6.70 ns. By optimizing the concentration and dosage, LSCs with a size of 5 × 5 cm2 were prepared, and the ηopt could reach 4.78%. For better commercialization, a high-efficiency large-area LSC was successfully fabricated using these yellow emissive CDs. The ηopt and ηPCE of the CD-integrated large-area LSC (10 × 10 cm2) reached up to 4.56% and 4.1% under natural sunlight (45 mW cm−2), respectively. The efficiencies are higher than other previously reported works with similar sizes. The high-performance large-area LSCs based on eco-friendly CDs with nearly zero reabsorption offers a useful platform for future study and practical application of LSCs.
Conflicts of interest
The authors declare no competing interests.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21774098), and the 111 project (No. B18038). H. Zhao acknowledges support from the Shandong Natural Science Funds for Distinguished Young Scholar (ZR2020JQ20).
References
- J. Roncali, Luminescent Solar Collectors: Quo Vadis?, Adv. Energy Mater., 2020, 10(36), 2001907 CrossRef CAS.
- Y. Zhou, H. Zhao, D. Ma and F. Rosei, Harnessing the properties of colloidal quantum dots in luminescent solar concentrators, Chem. Soc. Rev., 2018, 47(15), 5866–5890 RSC.
- H. Zhao, G. Liu, S. You, F. V. A. Camargo, M. Zavelani-Rossi, X. Wang, C. Sun, B. Liu, Y. Zhang, G. Han, A. Vomiero and X. Gong, Gram-scale synthesis of carbon quantum dots with a large Stokes shift for the fabrication of eco-friendly and high-efficiency luminescent solar concentrators, Energy Environ. Sci., 2021, 14(1), 396–406 RSC.
- M. G. Debije and P. P. C. Verbunt, Thirty Years of Luminescent Solar Concentrator Research: Solar Energy for the Built Environment, Adv. Energy Mater., 2012, 2(1), 12–35 CrossRef CAS.
- D. Cambie, J. Dobbelaar, P. Riente, J. Vanderspikken, C. Shen, P. H. Seeberger, K. Gilmore, M. G. Debije and T. Noel, Energy-Efficient Solar Photochemistry with Luminescent Solar Concentrator Based Photomicroreactors, Angew. Chem., Int. Ed., 2019, 58(40), 14374–14378 CrossRef CAS PubMed.
- B. R. Sutherland, Cost Competitive Luminescent Solar Concentrators, Joule, 2018, 2(2), 203–204 CrossRef CAS.
- M. G. Debije, R. C. Evans and G. Griffini, Laboratory protocols for measuring and reporting the performance of luminescent solar concentrators, Energy Environ. Sci., 2021, 14(1), 293–301 RSC.
- W. H. Weber and J. Lambe, Luminescent greenhouse collector for solar radiation, Appl. Opt., 1976, 15(10), 2299–2300 CrossRef CAS.
- F. Meinardi, A. Colombo, K. A. Velizhanin, R. Simonutti, M. Lorenzon, L. Beverina, R. Viswanatha, V. I. Klimov and S. Brovelli, Large-area luminescent solar concentrators based on ‘Stokes shift-engineered’ nanocrystals in a mass-polymerized PMMA matrix, Nat. Photonics, 2014, 8(5), 392–399 CrossRef CAS.
- H. Zhao, Y. Zhou, D. Benetti, D. Ma and F. Rosei, Perovskite quantum dots integrated in large-area luminescent solar concentrators, Nano Energy, 2017, 37, 214–223 CrossRef CAS.
- Y. Li, P. Miao, W. Zhou, X. Gong and X. Zhao, N-doped carbon-dots for luminescent solar concentrators, J. Mater. Chem. A, 2017, 5(40), 21452–21459 RSC.
- A. Marinins, R. Zandi Shafagh, W. van der Wijngaart, T. Haraldsson, J. Linnros, J. G. C. Veinot, S. Popov and I. Sychugov, Light-Converting Polymer/Si Nanocrystal Composites with Stable 60-70% Quantum Efficiency and Their Glass Laminates, ACS Appl. Mater. Interfaces, 2017, 9(36), 30267–30272 CrossRef CAS PubMed.
- H. Zhao, D. Benetti, X. Tong, H. Zhang, Y. Zhou, G. Liu, D. Ma, S. Sun, Z. M. Wang, Y. Wang and F. Rosei, Efficient and stable tandem luminescent solar concentrators based on carbon dots and perovskite quantum dots, Nano Energy, 2018, 50, 756–765 CrossRef CAS.
- J. A. H. P. Sol, G. H. Timmermans, A. J. Breugel, A. P. H. J. Schenning and M. G. Debije, Multistate Luminescent Solar Concentrator “Smart” Windows, Adv. Energy Mater., 2018, 8(12), 1702922 CrossRef.
- K. Wu, H. Li and V. I. Klimov, Tandem luminescent solar concentrators based on engineered quantum dots, Nat. Photonics, 2018, 12(2), 105–110 CrossRef CAS.
- R. Mazzaro and A. Vomiero, The Renaissance of Luminescent Solar Concentrators: The Role of Inorganic Nanomaterials, Adv. Energy Mater., 2018, 8(33), 1801903 CrossRef.
- H. Zhao, R. Sun, Z. Wang, K. Fu, X. Hu and Y. Zhang, Zero-Dimensional Perovskite Nanocrystals for Efficient Luminescent Solar Concentrators, Adv. Funct. Mater., 2019, 29(30), 1902262 CrossRef.
- Z. Li, A. Johnston, M. Wei, M. I. Saidaminov, J. Martins de Pina, X. Zheng, J. Liu, Y. Liu, O. M. Bakr and E. H. Sargent, Solvent-Solute Coordination Engineering for Efficient Perovskite Luminescent Solar Concentrators, Joule, 2020, 4(3), 631–643 CrossRef CAS.
- H. Y. Huang, M. J. Talite, K. B. Cai, R. J. Soebroto, S. H. Chang, W. R. Liu, W. C. Chou and C. T. Yuan, Utilizing host-guest interaction enables the simultaneous enhancement of the quantum yield and Stokes shift in organosilane-functionalized, nitrogen-containing carbon dots for laminated luminescent solar concentrators, Nanoscale, 2020, 12(46), 23537–23545 RSC.
- S. Han, G. Chen, C. Shou, H. Peng, S. Jin and C. C. Tu, Visibly Transparent Solar Windows Based on Colloidal Silicon Quantum Dots and Front-Facing Silicon Photovoltaic Cells, ACS Appl. Mater. Interfaces, 2020, 12(39), 43771–43777 CrossRef CAS.
- Z. Li, X. Zhao, C. Huang and X. Gong, Recent advances in green fabrication of luminescent solar concentrators using nontoxic quantum dots as fluorophores, J. Mater. Chem. C, 2019, 7(40), 12373–12387 RSC.
- J. Li, H. Zhao, X. Zhao and X. Gong, Red and yellow emissive carbon dots integrated tandem luminescent solar concentrators with significantly improved efficiency, Nanoscale, 2021, 13(21), 9561–9569 RSC.
- J. t Schiphorst, M. L. M. K. H. Y. K. Cheng, M. van der Heijden, R. L. Hageman, E. L. Bugg, T. J. L. Wagenaar and M. G. Debije, Printed luminescent solar concentrators: Artistic renewable energy, Energy Build., 2020, 207, 109625 CrossRef.
- F. Meinardi, F. Bruni and S. Brovelli, Luminescent solar concentrators for building-integrated photovoltaics, Nat. Rev. Mater., 2017, 2(12), 17072 CrossRef CAS.
- Y. You, X. Tong, W. Wang, J. Sun, P. Yu, H. Ji, X. Niu and Z. M. Wang, Eco-Friendly Colloidal Quantum Dot-Based Luminescent Solar Concentrators, Adv. Sci., 2019, 6(9), 1801967 CrossRef.
- S. Sadeghi, H. Bahmani Jalali, S. B. Srivastava, R. Melikov, I. Baylam, A. Sennaroglu and S. Nizamoglu, High-Performance, Large-Area, and Ecofriendly Luminescent Solar Concentrators Using Copper-Doped InP Quantum Dots, iScience, 2020, 23(7), 101272 CrossRef CAS.
- G. Liu, X. Wang, G. Han, J. Yu and H. Zhao, Earth abundant colloidal carbon quantum dots for luminescent solar concentrators, Mater. Adv., 2020, 1(2), 119–138 RSC.
- Y. Zhou, D. Benetti, X. Tong, L. Jin, Z. M. Wang, D. Ma, H. Zhao and F. Rosei, Colloidal carbon dots based highly stable luminescent solar concentrators, Nano Energy, 2018, 44, 378–387 CrossRef CAS.
- H. Zhao, G. Liu and G. Han, High-performance laminated luminescent solar concentrators based on colloidal carbon quantum dots, Nanoscale Adv., 2019, 1(12), 4888–4894 RSC.
- F. Mateen, M. Ali, H. Oh and S.-K. Hong, Nitrogen-doped carbon quantum dot based luminescent solar concentrator coupled with polymer dispersed liquid crystal device for smart management of solar spectrum, Sol. Energy, 2019, 178, 48–55 CrossRef CAS.
- Y. Han, X. Zhao, A. Vomiero, X. Gong and H. Zhao, Red and green-emitting biocompatible carbon quantum dots for efficient tandem luminescent solar concentrators, J. Mater. Chem. C, 2021, 9(36), 12255–12262 RSC.
- X. Liu, D. Benetti and F. Rosei, Semi-transparent luminescent solar concentrators based on plasmon-enhanced carbon dots, J. Mater. Chem. A, 2021, 9(41), 23345–23352 RSC.
- W. Ma, W. Li, R. Liu, M. Cao, X. Zhao and X. Gong, Carbon dots and AIE molecules for highly efficient tandem luminescent solar concentrators, Chem. Commun., 2019, 55(52), 7486–7489 RSC.
- X. Gong, W. Ma, Y. Li, L. Zhong, W. Li and X. Zhao, Fabrication of high-performance luminescent solar concentrators using N-doped carbon dots/PMMA mixed matrix slab, Org. Electron., 2018, 63, 237–243 CrossRef CAS.
- L. ZdraZil, S. Kalytchuk, K. Hola, M. Petr, O. Zmeskal, S. Kment, A. L. Rogach and R. Zboril, A carbon dot-based tandem luminescent solar concentrator, Nanoscale, 2020, 12(12), 6664–6672 RSC.
- V. I. Klimov, T. A. Baker, J. Lim, K. A. Velizhanin and H. McDaniel, Quality Factor of Luminescent Solar Concentrators and Practical Concentration Limits Attainable with Semiconductor Quantum Dots, ACS Photonics, 2016, 3(6), 1138–1148 CrossRef CAS.
- X. Xu, L. Mo, Y. Li, X. Pan, G. Hu, B. Lei, X. Zhang, M. Zheng, J. Zhuang, Y. Liu and C. Hu, Construction of Carbon Dots with Color-Tunable Aggregation-Induced Emission by Nitrogen-Induced Intramolecular Charge Transfer, Adv. Mater., 2021, 2104872 CrossRef CAS.
- X. Wang, Y. Ma, Q. Wu, Z. Wang, Y. Tao, Y. Zhao, B. Wang, J. Cao, H. Wang, X. Gu, H. Huang, S. Li, X. Wang, F. Hu, M. Shao, L. Liao, T. K. Sham, Y. Liu and Z. Kang, Ultra-Bright and Stable Pure Blue Light-Emitting Diode from O, N Co-Doped Carbon Dots, Laser Photonics Rev., 2021, 15(3), 2000412 CrossRef CAS.
- B. Wang, Z. Wei, L. Sui, J. Yu, B. Zhang, X. Wang, S. Feng, H. Song, X. Yong, Y. Tian, B. Yang and S. Lu, Electron-phonon coupling-assisted universal red luminescence of o-phenylenediamine-based carbon dots, Light: Sci. Appl., 2022, 11(1), 172 CrossRef CAS.
- B. Wang, J. Yu, L. Sui, S. Zhu, Z. Tang, B. Yang and S. Lu, Rational Design of Multi-Color-Emissive Carbon Dots in a Single Reaction System by Hydrothermal, Adv. Sci., 2020, 8(1), 2001453 CrossRef.
- B. Wang, H. Song, Z. Tang, B. Yang and S. Lu, Ethanol-derived white emissive carbon dots: the formation process investigation and multi-color/white LEDs preparation, Nano Res., 2021, 15(2), 942–949 CrossRef.
- B. Zhi, X. Yao, M. Wu, A. Mensch, Y. Cui, J. Deng, J. J. Duchimaza-Heredia, K. J. Trerayapiwat, T. Niehaus, Y. Nishimoto, B. P. Frank, Y. Zhang, R. E. Lewis, E. A. Kappel, R. J. Hamers, H. D. Fairbrother, G. Orr, C. J. Murphy, Q. Cui and C. L. Haynes, Multicolor polymeric carbon dots: synthesis, separation and polyamide-supported molecular fluorescence, Chem. Sci., 2020, 12(7), 2441–2455 RSC.
- B. Bartolomei, A. Bogo, F. Amato, G. Ragazzon and M. Prato, Nuclear Magnetic Resonance Reveals Molecular Species in Carbon Nanodot Samples Disclosing Flaws, Angew. Chem., Int. Ed., 2022, 61(20), 202200038 CrossRef PubMed.
- Q. Zhang, R. Wang, B. Feng, X. Zhong and K. K. Ostrikov, Photoluminescence mechanism of carbon dots: triggering high-color-purity red fluorescence emission through edge amino protonation, Nat. Commun., 2021, 12(1), 6856 CrossRef CAS.
- F. Yuan, T. Yuan, L. Sui, Z. Wang, Z. Xi, Y. Li, X. Li, L. Fan, Z. Tan, A. Chen, M. Jin and S. Yang, Engineering triangular carbon quantum dots with unprecedented narrow bandwidth emission for multicolored LEDs, Nat. Commun., 2018, 9(1), 2249 CrossRef PubMed.
- X. Yang, L. Ai, J. Yu, G. I. N. Waterhouse, L. Sui, J. Ding, B. Zhang, X. Yong and S. Lu, Photoluminescence mechanisms of red-emissive carbon dots derived from non-conjugated molecules, Sci. Bull., 2022, 67(14), 1450–1457 CrossRef CAS.
- Z. Wang, F. Yuan, X. Li, Y. Li, H. Zhong, L. Fan and S. Yang, 53% Efficient Red Emissive Carbon Quantum Dots for High Color Rendering and Stable Warm White-Light-Emitting Diodes, Adv. Mater., 2017, 29(37), 1702910 CrossRef PubMed.
- F. Yuan, P. He, Z. Xi, X. Li, Y. Li, H. Zhong, L. Fan and S. Yang, Highly efficient and stable white LEDs based on pure red narrow bandwidth emission triangular carbon quantum dots for wide-color gamut backlight displays, Nano Res., 2019, 12(7), 1669–1674 CrossRef CAS.
- J. Xu, Q. Liang, Z. Li, V. Y. Osipov, Y. Lin, B. Ge, Q. Xu, J. Zhu and H. Bi, Rational Synthesis of Solid-State Ultraviolet B Emitting Carbon Dots via Acetic Acid-Promoted Fractions of sp(3) Bonding Strategy, Adv. Mater., 2022, 34(17), 2200011 CrossRef CAS PubMed.
- S. Ren, B. Liu, M. Wang, G. Han, H. Zhao and Y. Zhang, Highly bright carbon quantum dots for flexible anti-counterfeiting, J. Mater. Chem. C, 2022, 10(31), 11338–11346 RSC.
- K. De Bruycker, A. Welle, S. Hirth, S. J. Blanksby and C. Barner-Kowollik, Mass spectrometry as a tool to advance polymer science, Nat. Rev. Chem., 2020, 4(5), 257–268 CrossRef.
- S. Lu, L. Sui, J. Liu, S. Zhu, A. Chen, M. Jin and B. Yang, Near-Infrared Photoluminescent Polymer-Carbon Nanodots with Two-Photon Fluorescence, Adv. Mater., 2017, 29(15), 1603443 CrossRef PubMed.
- Y. Song, S. Zhu, S. Xiang, X. Zhao, J. Zhang, H. Zhang, Y. Fu and B. Yang, Investigation into the fluorescence quenching behaviors and applications of carbon dots, Nanoscale, 2014, 6(9), 4676–4682 RSC.
- M. J. Talite, H. Y. Huang, Y. H. Wu, P. G. Sena, K. B. Cai, T. N. Lin, J. L. Shen, W. C. Chou and C. T. Yuan, Greener Luminescent Solar Concentrators with High Loading Contents Based on in Situ Cross-Linked Carbon Nanodots for Enhancing Solar Energy Harvesting and Resisting Concentration-Induced Quenching, ACS Appl. Mater. Interfaces, 2018, 10(40), 34184–34192 CrossRef CAS PubMed.
- M. J. Talite, H. Y. Huang, K. B. Cai, K. C. Capinig Co, P. A. Cynthia Santoso, S. H. Chang, W. C. Chou and C. T. Yuan, Visible-Transparent Luminescent Solar Concentrators Based on Carbon Nanodots in the Siloxane Matrix with Ultrahigh Quantum Yields and Optical Transparency at High-Loading Contents, J. Phys. Chem. Lett., 2020, 11(2), 567–573 CrossRef CAS PubMed.
- M. Zhang, Y. Ma, H. Wang, B. Wang, Y. Zhou, Y. Liu, M. Shao, H. Huang, F. Lu and Z. Kang, Chiral Control of Carbon Dots via Surface Modification for Tuning the Enzymatic Activity of Glucose Oxidase, ACS Appl. Mater. Interfaces, 2021, 13(4), 5877–5886 CrossRef CAS PubMed.
- Y. Zhou, D. Benetti, Z. Fan, H. Zhao, D. Ma, A. O. Govorov, A. Vomiero and F. Rosei, Near Infrared, Highly Efficient Luminescent Solar Concentrators, Adv. Energy Mater., 2016, 6(11), 1501913 CrossRef.
- H. Y. Huang, K. B. Cai, Y. R. Sie, K. Li, J. M. Yeh and C. T. Yuan, Eco-Friendly, High-Loading Luminescent Solar Concentrators with Concurrently Enhanced Optical Density and Quantum Yields While Without Sacrificing Edge-Emission Efficiency, Sol. RRL, 2019, 3(5), 1800347 CrossRef.
- G. Liu, R. Mazzaro, Y. Wang, H. Zhao and A. Vomiero, High efficiency sandwich structure luminescent solar concentrators based on colloidal quantum dots, Nano Energy, 2019, 60, 119–126 CrossRef CAS.
- M. Wei, F. P. G. de Arquer, G. Walters, Z. Yang, L. N. Quan, Y. Kim, R. Sabatini, R. Quintero-Bermudez, L. Gao, J. Z. Fan, F. Fan, A. Gold-Parker, M. F. Toney and E. H. Sargent, Ultrafast narrowband exciton routing within layered perovskite nanoplatelets enables low-loss luminescent solar concentrators, Nat. Energy, 2019, 4(3), 197–205 CrossRef CAS.
- J. Wang, J. Wang, Y. Xu, J. Jin, W. Xiao, D. Tan, J. Li, T. Mei, L. Xue and X. Wang, Controlled Synthesis of Long-Wavelength Multicolor-Emitting Carbon Dots for Highly Efficient Tandem Luminescent Solar Concentrators, ACS Appl. Energy Mater., 2020, 3(12), 12230–12237 CrossRef CAS.
- S. Li, H. Liu, W. Chen, Z. Zhou, D. Wu, R. Lu, B. Zhao, J. Hao, L. Yang, H. Yang, R. Cai, B. Xu, K. Wang and X. W. Sun, Low reabsorption and stability enhanced luminescent solar concentrators based on silica encapsulated quantum rods, Sol. Energy Mater. Sol. Cells, 2020, 206, 110321 CrossRef CAS.
- G. Liu, R. Mazzaro, C. Sun, Y. Zhang, Y. Wang, H. Zhao, G. Han and A. Vomiero, Role of refractive index in highly efficient laminated luminescent solar concentrators, Nano Energy, 2020, 70, 104470 CrossRef CAS.
- G. Liu, H. Zhao, F. Diao, Z. Ling and Y. Wang, Stable tandem luminescent solar concentrators based on CdSe/CdS quantum dots and carbon dots, J. Mater. Chem. C, 2018, 6(37), 10059–10066 RSC.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *b,
Xiujian
Zhao
*b,
Xiujian
Zhao