
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Advances in Cu nanocluster catalyst design: recent progress and promising applications
Sourav
Biswas
 a,
Saikat
Das
*b and
Yuichi
Negishi
*ab
a,
Saikat
Das
*b and
Yuichi
Negishi
*ab
aDepartment of Applied Chemistry, Faculty of Science, Tokyo University of Science, Kagurazaka, Shinjuku-ku, Tokyo 162-8601, Japan. E-mail: negishi@rs.tus.ac.jp
bResearch Institute for Science & Technology, Tokyo University of Science, Tokyo 162-8601, Japan. E-mail: saikatdas@rs.tus.ac.jp
First published on 26th September 2023
Abstract
The quest for cleaner pathways to the production of fuels and chemicals from non-fossil feedstock, efficient transformation of raw materials to value-added chemicals under mild conditions, and control over the activity and selectivity of chemical processes are driving the state-of-the-art approaches to the construction and precise chemical modification of sustainable nanocatalysts. As a burgeoning category of atomically precise noble metal nanoclusters, copper nanoclusters (Cu NCs) benefitting from their exclusive structural architecture, ingenious designability of active sites and high surface-to-volume ratio qualify as potential rationally-designed catalysts. In this Minireview, we present a detailed coverage of the optimal design strategies and controlled synthesis of Cu NC catalysts with a focus on tuning of active sites at the atomic level, the implications of cluster size, shape and structure, the ligands and heteroatom doping on catalytic activity, and reaction scope ranging from chemical catalysis to emerging photocatalysis and electrocatalysis.
 Sourav Biswas | Sourav Biswas is currently a postdoctoral research fellow of Professor Yuichi Negishi laboratory in the Department of Applied Chemistry at Tokyo University of Science (TUS). He received his PhD degree in Chemistry (2020) from National Institute of Technology Durgapur, India under the supervision of Prof. Sujit S. Panja. His current research interests focus on the synthesis and finding potential applications of new copper nanoclusters and silver cluster-assembled materials. |
 Saikat Das | Saikat Das is an Assistant Professor of the Research Institute for Science and Technology at TUS. He received his PhD degree in Chemistry and Physics of Polymers (2018) from Jilin University, China under the supervision of Prof. Teng Ben. His research interests encompass the designed synthesis of metal–organic frameworks and covalent organic frameworks for energy and environmental applications. |
 Yuichi Negishi | Yuichi Negishi is a Professor of the Department of Applied Chemistry at TUS. He received his PhD degree in Chemistry (2001) from Keio University under the supervision of Prof. Atsushi Nakajima. Prior to joining TUS in 2008, he was employed as an Assistant Professor at Keio University and the Institute for Molecular Science. His research interests include the structural and functional exploration of atomically precise metal nanoclusters, metal nanocluster-assembled materials, and covalent organic frameworks. |
1. Introduction
Metal nanoclusters (NCs), composed of a few to a few hundred metal atoms with metal–metal connectivity and falling within the size range of 1–3 nm, have recently gained significant attention because of their unique size-dependent properties.1–5 These properties encompass electronic behavior, optical characteristics, and magnetic attributes, distinguishing them from both bulk materials and isolated atoms. Consequently, metal NCs have been subject to extensive investigation for their potential utilization in diverse fields such as catalysis, sensing, energy storage, and bioimaging.6–11 Moreover, the synthesis of metal NCs has emerged as a rapidly advancing area of research, as scientists strive to develop methods for producing clusters with precise sizes, shapes, and compositions.2,12,13 This pursuit has led to a deeper comprehension of the fundamental properties exhibited by metal NCs and their promising prospects for practical applications. Over the past few decades, the study of metal NCs has made significant strides, fostering increased knowledge and opening doors for new applications and synthetic techniques.9,14–21 As researchers continue to explore novel applications and refine synthetic methods, the field of metal NCs is expected to experience continued growth and further advancements.The study of metal NCs has a rich history dating back to the early 20th century, when metal nanoparticles (NPs) were first observed in the colloidal state.22 However, it was not until the 1980s that the field of metal NCs research began to rapidly develop with the development of advanced analytical techniques in X-ray diffraction, advanced microscopy and surface spectroscopy.1,2 These techniques enabled researchers to study the size-dependent properties of metal NPs, which were found to exhibit unique electronic, optical, and magnetic properties that differed from those of bulk materials or individual atoms.
Gold (Au) and silver (Ag) NCs can be synthesized using chemical reduction, template-assisted synthesis, and electrochemical methods.2,23–33 These methods have resulted in significant production of these NCs. On the other hand, the synthesis of copper (Cu) NCs, which belong to the most abundant and lightest congener in the group 11, poses challenges due to the low half-cell reduction potential and high chemical reactivity.34 As a result, Cu NCs are still considered new in this field. In accordance with the other NC structures, Cu NCs are also formed by core–shell structural architecture where the metallic cores are protected by the ligands or metal–ligand staple motifs.35–37 Typically, the core of Cu NCs adopts polyhedral geometries, while the presence of additional ligands, alongside thiolate or phosphine or alkynyls ligands, contributes to a diverse range of polyhedral crystal structures and conformational variations.38–54 The precise structural architecture and high surface-to-volume ratio of Cu NCs offer significant advantages that open up unique opportunities for chemical catalysis. These unique attributes, coupled with the redox potential of the Cu atom, make them positioned as highly desirable photocatalyst and electrocatalysts for various photocatalytic and electrochemical reactions, with a particular focus on energy conversion and storage technologies.55 The catalytic performance of these NCs is directly influenced by changes in their structural architecture, making the tuning of structural characteristics a crucial aspect for future progress in catalysis. Over the course of Cu NC research numerous new Cu NCs have been discovered and assessed for their catalytic potential, with classification based on structural architecture into superatom-based Cu(0)/Cu(I) NCs and Cu(I) NCs. Therefore, in this concise review article, our primary objective was to conduct a thorough investigation and comparison of the diverse structural architectures of Cu NCs and their respective catalytic activities. We aspired to provide valuable insights and guidelines for future research endeavors in this budding field. Our approach went beyond a mere performance comparison of Cu NCs. Instead, we delved deep into their intricate structural compositions to unravel the fundamental factors influencing their catalytic properties. Through meticulous examination and rigorous analysis, we sought to identify the key determinants driving their efficiency in various catalytic reactions. By understanding the subtle relationships between specific structural attributes and catalytic performances, we aimed to distinguish the most efficient Cu NCs for specific applications, equipping researchers with valuable knowledge to drive advancements in this exciting domain. In this context, we discussed the application of Cu NCs on the basis of three main catalytic reaction pathways, namely: chemical catalysis, photocatalysis and electrocatalysis as presented in Fig. 1.
 | ||
| Fig. 1 Recent advancement of Cu NCs as catalyst. | ||
2. Effect of Cu NCs in chemical catalysis
2.1. Hydrogenation reaction
Since the discovery of CuH complexes in early 1800, it has been used in various catalytic reactions for the reduction of the intermediates; however, it took another few decades to be applied in organic medium.56–58 The discovery of the organocopper species named as Stryker's reagent, efficiently broadened up the utilities of this material by constructing some useful bonds rather than its solely reductive nature.59,60 Since then, organocopper species has drawn a tremendous attention as a superior catalyst as the utility of Cu appeared most economic. Researchers have been actively engaged in exploring alternative approaches to achieve more effective catalysis by focusing on precise structural architectures. Within this context, the intriguing potential of sulfhydryl groups has emerged, as they exhibit remarkable reducing abilities in alkaline environments and can form stable covalent bonds with metal ions. Moreover, the formation of strong metal–sulfur bonds plays a crucial role in safeguarding the metallic architecture against aggregation, thereby enhancing overall stability. By harnessing these properties, scientists aim to develop innovative catalytic systems capable of delivering enhanced performance and efficiency. Sun et al. synthesized [Cu25H10(SPhCl2)18]3− NC and investigated its effectiveness in the hydrogenation of ketones to alcohols.61 The NC was found to consist of a Cu@Cu12 core, which exhibited a centered twinned cuboctahedral geometric architecture, unlike the cuboctahedral building block observed in bulk Cu metal with a face-centered cubic structure (Fig. 2). The core was shielded by a shell of metal atoms forming a truncated v3 tetrahedral shape, where each distorted hexagonal face of the shell aligned with a trigonal face of the Cu@Cu12 core. The Cu12(SPhCl2)18 shell was connected via thiolate bridging, and the core was connected to the shell through six hydride atoms, with four additional hydrides solely bound to the core via connecting Cu atoms (Fig. 2). The researchers found that the hydrides solely attached to the core were more exposed and accessible for catalytic hydrogenation reactions. Through theoretical calculations, they identified the most active catalytic sites, where the carbon in the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group selectively interacted with a specific H atom, while the oxygen atom bound to three Cu atoms, occupies the position of the missing hydride to form the intermediate. The structural stability of the Cu NC ensures a consistent reaction rate throughout the entire catalytic process. An intriguing characteristic of this nanocatalyst lies in the presence of ten hydrogen atoms that exist in a hydride state. These hydride species serve a dual purpose by not only improving the structural stability but also serving as a H source during the catalytic reaction. This distinctive attribute significantly enhances the overall efficiency and effectiveness of the catalytic process, offering a promising pathway for sustainable and efficient hydrogenation reactions.
O group selectively interacted with a specific H atom, while the oxygen atom bound to three Cu atoms, occupies the position of the missing hydride to form the intermediate. The structural stability of the Cu NC ensures a consistent reaction rate throughout the entire catalytic process. An intriguing characteristic of this nanocatalyst lies in the presence of ten hydrogen atoms that exist in a hydride state. These hydride species serve a dual purpose by not only improving the structural stability but also serving as a H source during the catalytic reaction. This distinctive attribute significantly enhances the overall efficiency and effectiveness of the catalytic process, offering a promising pathway for sustainable and efficient hydrogenation reactions.
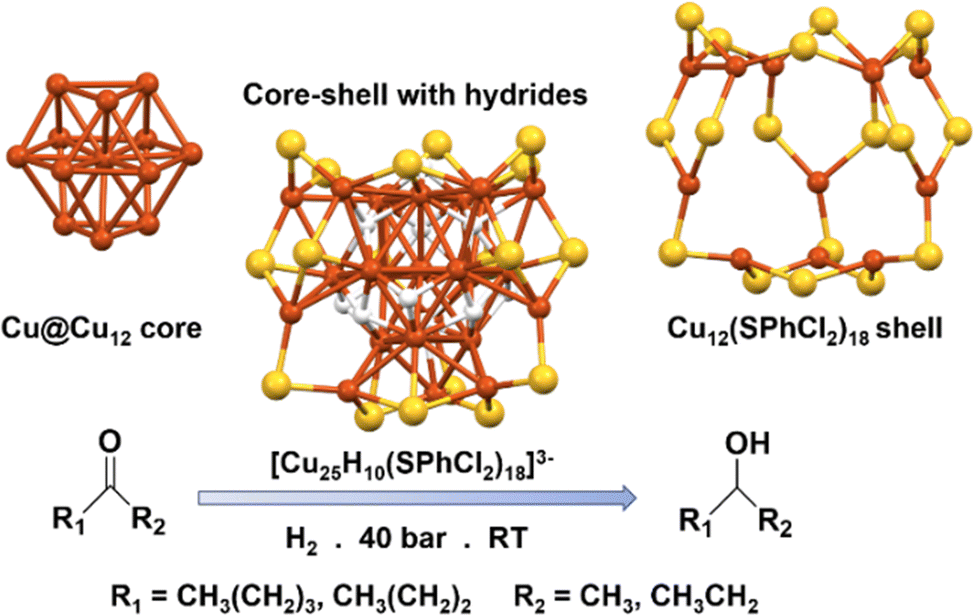 | ||
| Fig. 2 Structural architecture of [Cu25H10(SPhCl2)18]3− NC and its effectivity towards hydrogenation reaction. Cu displayed in brown color; S displayed in yellow color and H displayed in white color. Other ligands were removed for clarity. Figure created based on the cif file deposition of ref. 61. | ||
Recently Luo et al. introduced a novel metal-deficient chiral Cu NC, denoted as [Cu57H20(PET)36(PPh3)4]+ (PET: phenylethanethiolate) NC, composed of a unique architecture featuring a Cu8 cubic kernel and a Cu49(PET)36(PPh3)4 shell.62 The outermost shell exhibits a distinct Cu atom deficiency, as evidenced by the total symmetric structure of reported [Cu58H20(S-CH2CH2CH3)36(PPh3)8]2+ NC.63 Despite the presence of eight PPh3 units capping the outermost cubic shell in the reported nanocluster, a single copper atom vacancy defect emerges at one corner of this shell in the [Cu57H20(PET)36(PPh3)4]+ NC, leading to alterations in the PPh3 ligand attachments. Interestingly, while a vacancy exists in the outermost shell, it does not impact the arrangement of hydrides and instead serves to establish a connection between the core and the inner shells. Employing theoretical calculations, the researchers unravelled a fascinating aspect of this metal-deficient Cu NC—the presence of a more exposed linked hydride position due to the vacancy of a metal atom within the shell. This exposed position was identified as the active catalytic site responsible for the hydrogenation of nitroaromatics. To validate the catalytic potential of the Cu NC, the researchers monitored the reduction process by measuring the absorbance of nitroaromatics. Strikingly, the results demonstrated that in the presence of BH4−, the Cu NC achieved full conversion of p-nitrophenol to p-aminophenol in just 20 minutes. In stark contrast, in the absence of the catalyst, the absorbance peak intensity remained unchanged after the same time duration, underscoring the crucial role of the Cu NC as a highly effective catalyst in the hydrogenation reaction. This study not only sheds light on the significance of metal-deficient Cu NCs in catalytic applications but also highlights the promising potential of defect-engineered NCs for enhancing reactivity and selectivity in chemical transformations, particularly in the context of nitroaromatic hydrogenation reactions.
2.2. C–N bond formation reaction
Lee et al. presented a novel Cu NC, denoted as [Cu32(PET)24H8Cl2]2−, designed for the carbonylation of aniline to synthesize carbamates.64 This NC showcases a unique core–shell architecture, comprising a Cu14 core and a Cu18(PET)24Cl2 shell (Fig. 3). The Cu14 core is formed through the assembly of two Cu8 square antiprisms, sharing their edges. This core–shell connection is upheld by the presence of eight hydride atoms. On the other hand, the metal–ligand shell of the NC consists of two distinct components: the simpler triangular Cu2PET motifs and the more intricate Cu7(PET)11Cl motifs. The core is connected to the triangular Cu2PET motifs on opposing sides, aligning two Cu atoms from each motif parallel to the shared edge of the two square antiprisms. This arrangement establishes a cohesive structure, ensuring the integration of the core and the metal–ligand shell in the NC. They anticipated equilibrium interactions between amines, such as anilines, and solution-phase NCs due to the use of tetramethylethylenediamine as an assisting agent during the synthesis of the NCs. Taking advantage of this concept, they utilized this NC for carbamate formation, eliminating the need for toxic gases, which had been crucial factors in previous methods.65 In the conducted experiment, Cu NCs dissolved in CDCl3 were subjected to a reaction with aniline and aniline derivatives, which were substituted with methyl and halide groups. The reaction was performed using diisopropyl azodicarboxylate under inert conditions. To evaluate the influence of Cu NCs, control experiments were also conducted, wherein the Cu NC was substituted with a CuI and a CuPET complex, while maintaining the same reaction conditions. The yields obtained from these control experiments did not surpass 6.1%, demonstrating the superior catalytic performance of this NC in comparison to the alternative Cu sources. This significant difference highlights the effectiveness of this NC in carbamate formation reaction. The mechanism proposed for aniline carbonylation focuses on the creation of highly reactive oxyacyl radicals through the breakdown of diisopropyl azodicarboxylate on the catalyst. The outer surface of the NC plays a critical role in facilitating selective interactions between aniline or aniline radicals and itself, thus promoting the targeted formation of carbamate products. The interactions observed between tetramethylethylenediamine and the Cu cluster during the synthesis process bear resemblance to those seen in the aniline carbonylation reaction. However, when CuI or CuPET is used instead, which lack or minimize such specific interactions, the aniline radicals undergo uncontrolled combination, resulting in reduced selectivity for the carbamate product. Consequently, the absence of these tailored interactions hinders the desired outcome. So, the precise structural architecture appeared here to stabilize the intermediates which can promote the reaction pathways. | ||
| Fig. 3 Structural architecture of [Cu32(PET)24H8Cl2]2− NC and its effectivity towards C–N bond formation reaction. Cu displayed in brown color; S displayed in yellow color; Cl displayed in green color and H displayed in white color. Other ligands were removed for clarity. Figure created based on the cif file deposition of ref. 64. | ||
2.3. Click reaction
Cook et al. successfully synthesized [Cu20(CCPh)12(OAc)6] NC, representing a rare 2e− Cu superatom.66 This NC exhibits a unique tetrahedral [Cu4]2+ core, surrounded by a [Cu16(CCPh)12(OAc)6]2− shell (Fig. 4). They utilized this NC as both a homogeneous and heterogeneous catalyst for the [3+2] cycloaddition reaction. In the case of the homogeneous reaction, they directly employed this NC in the catalytic reaction. For the heterogeneous catalyst design, they immobilized this NC on silica support. Interestingly, they achieved high yields for the cycloaddition of alkyne with benzyl azide in both the homogeneous and heterogeneous catalyst mediums. Based on their findings, they proposed a mechanistic pathway for this catalytic reaction. The initial step involves the reaction between the bound phenylacetylide ligand with benzyl azide, leading to the generation of an NC bound triazolate fragment. This fragment then undergoes subsequent protonolysis with the incoming alkyne, generating 1-benzyl-4-phenyl-1H-1,2,3-triazole and a new Cu-bound acetylide ligand. It is noteworthy that both the catalysts showed similar effectiveness during the catalytic reaction. However, they observed that the Cu NC in the homogeneous catalytic reaction was less stable under those reaction conditions. In contrast, the extended X-ray absorption fine structure confirmed the stability of the heterogeneous catalyst even after the catalytic reaction had taken place. Notably, this study highlighted the significance of the exposed catalytic sites on the surface of the Cu NCs in stabilizing reaction intermediates during the click reaction. This observation is of paramount importance, as it underscores the pivotal role played by these exposed catalytic sites in facilitating and driving the catalytic process effectively. | ||
| Fig. 4 Structural architecture of [Cu20(CCPh)12(OAc)6] NC and its effectivity towards [3+2] cycloaddition reaction. Cu displayed in brown color; O displayed in red color and C displayed in light dark color. Other ligands were removed for clarity. Figure created based on the cif file deposition of ref. 66. | ||
2.4. Reduction of ferricyanide to ferrocyanide
Dong et al. recently reported Cu18H(PET)14(PPh3)6(isothiocyanate)3 NC and its effectivity toward the reduction of ferricyanide.67 The structure is visualized as a conventional core–shell structure which consists of a triple-helical Cu15 core and [(Cu isothiocyanate)3(PET)14(PPh3)6] shell. They observed that the clustered NCs display photoluminescence emission in the deep-red spectrum and, in addition, exhibit impressive efficacy in catalyzing electron transfer reactions. However, due to their insolubility in water, these NCs are considered heterogeneous catalysts for the reduction of Fe(CN)63− in the presence of NaBH4. To assess its catalytic power, the reduction process using this NC was closely monitored using absorbance spectrum. Specifically, they tracked the intensity change of the [Fe(CN)6]3− peak at 420 nm. Upon adding the NCs and NaBH4, they observed a rapid decrease in absorption intensity at 420 nm, which completely vanished within 14 seconds. This observation indicated the complete conversion of Fe(III) to Fe(II), accompanied by a color change to colorless in the solution. To verify the role of the Cu NC, they further conducted a control experiment without the NCs. In this case, the reduction of Fe(III) took significantly longer, approximately 16 minutes. The notable disparity in reaction times strongly suggests that the Cu NCs exhibit exceptional catalytic reactivity for the reduction of [Fe(CN)6]3− through electron transfer reactions.2.5. Styrene oxidation
Wang et al. studied the effectivity of bovine serum albumin (BSA) capped Cu NC in styrene oxidation reaction.68 BSA was chosen as the model protein to stabilize the clusters and provide steric protection to synthesize a water-soluble Cu NC. The process of styrene oxidation led to the formation of multiple reaction products with a remarkable selectivity of 70% towards the production of benzaldehyde, which emerged as the predominant product. Furthermore, approximately 70% of the initial styrene was successfully converted during the course of the oxidation reaction. However, when the oxidation reaction was conducted without a catalyst, the progress was considerably sluggish. The conversion of styrene under these conditions was merely 7%, indicating that the catalyst played a vital role. Significantly, the high selectivity towards benzaldehyde and its remarkable conversion of styrene highlights the importance in facilitating the desired reaction pathway and promoting the formation of the primary product. However, they have not yet provided a detailed mechanistic pathway, it can be inferred that the exceptional selectivity arises from the ability of catalyst to stabilize the reaction intermediates. This stabilization likely occurs through interactions between the intermediate species and the Cu NC surface. So, the exposed interactable surface area on the Cu NCs is likely to play a crucial role in accommodating and supporting reaction intermediates, ultimately influencing the selectivity towards benzaldehyde formation.2.6. Synthesis of indole derivatives
Wang et al. introduced a new Cu NC with bis-pyrazolate ligands, showcasing remarkable chemical stability and its utilization in synthesis of indolizines from 2-pyridinecarboxaldehyde derivatives (Fig. 5).69 The synthesized Cu NC comprises a highly twisted [Cu8]8+ ring, capped by eight ligands, each coordinated by two Cu+ cations. Under optimized conditions, utilizing the Cu NC as a catalyst with a low loading of 0.5 mol% in specific solvents and at a specific temperature, led to high yields of indolizines. The remarkable chemical stability of the Cu NC played a pivotal role in this catalytic transformation, as compared to other Cu salts, which exhibited significantly lower yields. The reaction kinetics exhibited a pseudo-first-order rate dependence on phenylacetylene concentration. Moreover, this Cu NC demonstrated exceptional reusability, maintaining its catalytic performance over ten cycles without any degradation. Through further exploration of different substrates, the NC-catalyzed indolizine synthesis demonstrated good tolerance and versatility. So, the broad substrate scope observed in this catalytic system is of significant importance, as it opens up numerous possibilities for the synthesis of diverse indolizine derivatives. Indolizine derivatives are known for their high medicinal chemistry applications, making them crucial building blocks in drug discovery and development. Therefore, the successful application of the Cu NC catalyst in this context holds promise for advancing the field of medicinal chemistry and facilitating the synthesis of potentially valuable pharmaceutical compounds. | ||
| Fig. 5 Structural architecture of Cu8(bis-pyrazolate)8 NC and its effectivity towards the synthesis of indole derivatives. Cu displayed in brown color; N displayed in blue color and C displayed in light dark color. Other ligands were removed for clarity. Figure created based on the cif file deposition of ref. 69. | ||
2.7. Synthesis of silanol
Precisely controlling the synthesis of metal NCs doped with heteroatoms is crucial for obtaining a deeper understanding of how doping affects electronic structure and catalytic properties; however, this control presents considerable challenges when dealing with metals of significantly different atomic sizes, as exemplified by Cu and platinum (Pt). Lee et al. designed Pt-doped Cu NC, [Pt2Cu34(PET)22Cl4]2− NC, by utilizing the unique reactivity of [Cu32(PET)24Cl2H8]2− NC with Pt4+ ions.70 The structural analysis of the single crystalline architecture unveils a fascinating arrangement: two platinum (Pt) atoms form direct bonds at the centers of an intricately interlocking and partially complete biicosahedron core (designated as Pt2Cu18). This distinctive core configuration gains added stability from an enveloping shell composed of sixteen Cu atoms coordinated with twenty-two PET ligands and four Cl atoms. Utilizing density functional theory (DFT) for electronic structure analysis, it has been discerned that this alloy nanocluster exhibits superatomic characteristics, featuring 10 electrons. They employed this alloy NC in silanol formation and observed its effectiveness when supported on a TiO2 surface. A comparison with the parent Cu32 NC revealed significantly lower catalytic activity. Furthermore, the study identified the favorable influence of temperature on the catalytic efficiency in this reaction up to 50 °C.3. Effect of Cu NCs in photocatalysis
3.1. C–C cross coupling reaction
Nematulloev et al. conducted a study to explore the catalytic properties of [Cu28H10(C7H7S)18(PPh3)3] NC in selective C–C cross-coupling reactions (Fig. 6).71 This NC features a core–shell structure, comprising a Cu13 anti-cuboctahedron core and a Cu15(C7H7S)18(PPh3)3 shell. Notably, all the hydrides are located around the core of the NCs. An intriguing observation made by the researchers was that the NCs exhibit a tetrahedral shape, apart from a vacancy defect in one of the vertices. This defect results in the absence of a Cu atom and a phosphine ligand. Consequently, the framework experiences a distortion, leading to a reduction in symmetry and an alteration in the packing mode of the NCs. Theoretical calculations suggested that the surface vacancy created by this defect serves as an effective catalytic site. In order to examine the impact of a particular defect on catalytic activity, the defective NC was utilized as a catalyst in the Sonogashira C–C coupling reaction. The NC demonstrated a remarkable catalytic efficiency, resulting in an impressive yield of 82% for the desired C–C coupling product, while successfully preventing the formation of undesired homocoupling byproducts. In contrast, when conventional Cu salts such as CuCl or Cu(CH3CN)4 BF4 were employed as catalysts under the same conditions, the desired Sonogashira product was obtained in lower yields (61% and 34%, respectively), accompanied by significant amounts of the undesired homocoupling byproduct (34% and 18%, respectively). This control experiment highlighted the superior performance of the NC over conventional Cu salts. Once optimized reaction conditions were established, the researchers proceeded to investigate the scope of the reaction using different aryl iodides. In general, aryl iodides possessing neutral, electron-rich, and electron-withdrawing groups exhibited high reactivity, leading to the desired products with yields of up to 84%. The absorbance spectrum of this NC exhibited a broad range of absorbance between 400 and 500 nm, suggesting its potential for photochemical activity under blue-light irradiation. Upon interaction with iodides, the NC generated highly reactive Ar-radicals. These Ar-radicals subsequently initiated an attack on the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C bond, resulting in the formation of vinylic-type C-radical intermediates. Therefore, the NC played a crucial role in activating the CC bond by forming a π-alkynyl complex, facilitating the facile attack by the Ar-radical. Ultimately, the desired Sonogashira C–C coupled product was achieved through the base-induced deprotonation of the vinyl cation. It is worth mentioning that they compared the yield of Sonogashira C–C coupled product formation with another Cu NC. That Cu NC is formulated as [Cu61(StBu)26S6Cl6H14]+, and it is currently the largest structurally-solved core–shell Cu cluster containing Cu(0) ever reported.72 This particular NC possesses a distinctive structure consisting of a quasi-elongated triangular gyrobicupola Cu19 core and Cu42(StBu)26S6Cl6 shell. Despite its unique and intricate structure, the researchers found that this NC did not yield higher C–C coupled product compared to previous experiments. This could be attributed to its larger cluster size, suggesting that other factors, such as the reaction conditions or the specific catalytic properties of the NC, may play a significant role in determining the yield of the Sonogashira C–C coupled product.
C bond, resulting in the formation of vinylic-type C-radical intermediates. Therefore, the NC played a crucial role in activating the CC bond by forming a π-alkynyl complex, facilitating the facile attack by the Ar-radical. Ultimately, the desired Sonogashira C–C coupled product was achieved through the base-induced deprotonation of the vinyl cation. It is worth mentioning that they compared the yield of Sonogashira C–C coupled product formation with another Cu NC. That Cu NC is formulated as [Cu61(StBu)26S6Cl6H14]+, and it is currently the largest structurally-solved core–shell Cu cluster containing Cu(0) ever reported.72 This particular NC possesses a distinctive structure consisting of a quasi-elongated triangular gyrobicupola Cu19 core and Cu42(StBu)26S6Cl6 shell. Despite its unique and intricate structure, the researchers found that this NC did not yield higher C–C coupled product compared to previous experiments. This could be attributed to its larger cluster size, suggesting that other factors, such as the reaction conditions or the specific catalytic properties of the NC, may play a significant role in determining the yield of the Sonogashira C–C coupled product.
 | ||
| Fig. 6 Structural architecture of [Cu28H10(C7H7S)18(PPh3)3] NC and its effectivity towards photocatalytic C–C cross coupling reaction. Cu displayed in brown color; S displayed in yellow color and P displayed in pink color. Other ligands were removed for clarity. Hydrides were not detected by SCXRD. Figure created based on the cif file deposition of ref. 71. | ||
3.2. C–N coupling reaction
Sagadevan et al. made a significant discovery of Cu NC research to find its capability of activating aryl chlorides in cross-coupling reactions.73 They introduced [Cu61H14(StBu)26S6Cl6] NC, which proved highly effective in facilitating C–N bond-forming reactions under visible-light irradiation at mild condition (Fig. 7). As we discussed earlier, this specific structural architecture of NCs played a crucial role in its catalytic properties. In this study also they have cited the similar thoughts of dependence of the structural architecture on catalytic properties. They investigated the catalytic activity of this NC in the amination of carbazole with p-bromobenzonitrile under blue-light irradiation. Remarkably, the desired C–N-arylation product was obtained with an impressive yield of 88% when this Cu NC was employed as the catalyst. In contrast, the use of CuCl as the catalyst resulted in a significantly lower yield of only 18%, highlighting the superior effectiveness of the Cu NC with its precise structural architecture. Control experiments were conducted to gain further insights into the reaction mechanism. These experiments confirmed that the presence of both blue-light irradiation and a base, in addition to the Cu NC catalyst, was essential for the successful occurrence of the amination reaction. This suggests that the catalytic activity of the Cu NC is triggered and facilitated by the combination of light and the presence of a base, which likely play critical roles in the activation of reactants and the stabilization of reaction intermediates. Interestingly, during the study, cyanobenzene was detected as a side product, likely resulting from the in situ formation of an arene radical and a hydrogen atom from the solvent. Then they applied optimized conditions to perform the amination of p-chlorobenzonitrile, and the desired product was obtained with an 85% yield, even with low reaction temperatures and a small amount of catalyst loading. However, when p-chlorobenzonitrile and p-chloroanisole were subjected to the reaction using only 5 mol% of CuCl as the catalyst, the product was obtained in trace amounts. So, they proposed a mechanism based on the facts, which involves the formation of a NC-amine complex to initiate the reaction upon exposure to blue light, leading to the production of a photoexcited state. This photoexcited state then undergoes a single-electron transfer process, donating an electron to the aryl halide. This results in the formation of an intermediate complex, along with the generation of a halide anion and an arene radical. In the subsequent step, the aryl radical interacts with the oxidized intermediate, following either a radical-radical bound pathway or a reductive elimination pathway. Consequently, the desired C–N coupled product is formed, while simultaneously regenerating the original Cu NC catalyst, allowing it to initiate a new catalytic cycle. The results underscore the potential for harnessing NC catalysts in synthetic transformations, providing valuable insights into the role of structural architecture and external factors such as light and base in influencing catalytic efficiency. | ||
| Fig. 7 Structural architecture of [Cu61H14(StBu)26S6Cl6] NC and its effectivity towards photocatalytic C–N coupling reaction. Cu displayed in brown color; S displayed in yellow color and Cl displayed in green color. Other ligands were removed for clarity. Hydrides were not detected by SCXRD. Figure created based on the cif file deposition of ref. 73. | ||
3.3. Click reaction
Dong et al. made an intriguing discovery involving two Cu NCs: [Cu58H20PET36(PPh3)4]2+ (Cu58) NC and its surface-defective counterpart [Cu57H20(PET)36(PPh3)4]+ (Cu57) NC.74 These NCs share a remarkably similar structure, comprising five concentric metal shells which is similar to the previous discussed [Cu57H20(PET)36(PPh3)4]+ and [Cu58H20(S-CH2CH2CH3)36(PPh3)8]2+ NCs. The crucial difference lies in Cu57, where a single surface Cu atom is absent just like the previously discussed [Cu57H20(PET)36(PPh3)4]+ NC. Surprisingly, this seemingly minor modification has a profound impact on their reactivity. It has been observed that Cu57 exhibited exceptional activity in photoinduced [3+2] azide–alkyne cycloaddition. The superior catalytic performance of Cu57 can be attributed to the presence of an active catalytic site resulting from the removal of one surface Cu atom. This subtle alteration between Cu58 and Cu57 emphasizes the significance of the local structure surrounding the Cu vacancy in dictating catalytic activity. To delve deeper into the phenomenon, they calculated electrostatic potential (ESP) maps on the molecular van der Waals surface of both Cu58 and Cu57. The elimination of the surface Cu atom in Cu57 led to a significant reduction in ESP around the Cu vacancy. This observation led them to propose that the π-electron nature of the alkyne could substitute the PET ligands, which have a low ESP. Based on this value, they confirmed that the Cu atoms surrounding the vacant site become active catalytic centers in Cu57, contributing to the heightened catalytic activity observed. Overall, Dong et al.'s findings shed light on the remarkable influence of a single surface Cu atom on the reactivity of these Cu NCs.3.4. Sulfoxidation of sulfides
Zhang and colleagues reported two quasi-structurally isomeric Cu NCs and compared their effectivity in photocatalytic oxidation of sulfides.75 Both the clusters composed of a similar Cu13 metallic architecture and protected by CZ-PrAH (9-(prop-2-yn-1-yl)-9H-carbazole) and H4TC4A (p-tert-butylthiacalix[4]arene) ligands. However, the main structural difference is the attachment of Cl− to the metallic architecture due to the induces solvent effect during the crystallization process. So, the formula of these NCs are [Cu13Na(CZ-PrA)6(TC4A)2(CH3OH)]·CH3OH·CH2Cl2·CH3COCH3 and [Cu13Na2(CZ-PrA)6(TC4A)2Cl(CH3OH)2]. Upon conducting a comparative analysis of the two isomers, notable disparities in their charge transfer mechanisms were observed. Particularly, the attachment of chloride to the Cu NCs caused a significant blue shift in the absorption spectrum, distinguishing it from the other Cu NCs. This alteration in the charge transfer process had an impact on the energy band structure, leading to higher 1O2 and lower O2˙− yields compared to the other Cu NCs when activating molecular oxygen. As a result of its unique chloride attachment, the Cu NCs demonstrated an outstanding level of selectivity in the realm of photocatalytic reactions. Specifically, it showcased remarkable efficiency in the selective oxidation of dimethyl sulfide. The mechanism behind this exceptional selectivity lies in the specific chemical interactions between the Cu NCs and the chloride ions. The attachment of chloride ions onto the Cu NCs surface creates an enhanced active site, boosting its capability to interact specifically with dimethyl sulfide molecules. Consequently, the Cu NCs with chloride attachment becomes a highly efficient catalyst for the targeted oxidation reaction. The implications of this discovery are substantial, as it opens up new possibilities for designing and engineering highly selective photocatalysts tailored for various chemical reactions.4. Effect of Cu NCs in electrocatalysis
4.1. CO2 reduction reaction (CO2 RR)
The electrochemical method for CO2 reduction holds great promise as a viable approach to convert CO2 into valuable products.76 Among the various catalysts, Cu-based catalysts have emerged as the most effective and highly selective options for this purpose.77,78 As a result, atomically precise Cu NCs with well-structured architectures have garnered significant attention for research in this field, showcasing their immense potential. Tang et al. have demonstrated the exceptional selectivity of the structurally precise Cu32H20(S2P(OiPr)2)12 NC in electrocatalytic CO2 reduction, even at low overpotentials (Fig. 8).79 The overall architecture of this NC comprises a distorted hexacapped rhombohedral Cu14 core, which is nestled between two nest-like triangular cupola fragments composed of meta-ligand motifs. Each motif contains nine Cu atoms, while the core is encircled by twenty hydrides that establish connections with the motifs. These hydrides can be categorized through their coordination as twelve μ3-H, six μ4-H, and two μ5-H. Theoretical calculations have uncovered that the involvement of negatively charged hydrides within this NC is pivotal in determining the selectivity of the reduction process. Specifically, it favors the production of HCOOH over CO at lower overpotentials. The NC reveals that the lattice hydride channel exhibits a higher energy preference for CO2 reduction compared to the proton reduction channel. Out of the hydrides, the μ3-H hydrides exhibit the most favorable binding to CO2 molecules, effectively enhancing the CO2 reduction process. The primary product of CO2 reduction on this NC is predicted to be HCOOH, achieved through a two-step lattice hydrogen reduction pathway. The initial step in the process involves the direct reaction of CO2 with a capped hydride, resulting in the formation of an intermediate. This intermediate then undergoes further reaction with another interstitial hydride, ultimately yielding HCOOH. The hydride participating in the reaction can be readily regenerated through proton reduction. Theoretical calculations indicate that, at low overpotentials, the competitive nature of hydrogen evolution is reduced compared to HCOOH formation. As a result, HCOOH becomes the predominant product at these potentials, achieving a remarkable faradaic efficiency of up to 89% at −0.5 V (vs. RHE). Remarkably, this NC displays unique selectivity when compared to its nanoparticle counterparts, offering a much lower overpotential for HCOOH production than conventional Sn- or Bi-based catalysts. | ||
| Fig. 8 Effect of the structural architecture of Cu32H20(S2P(OiPr)2)12 NC in CO2RR. Cu displayed in brown color; S displayed in yellow color; P displayed in pink color; O displayed in red color and H displayed in white color. Other ligands were removed for clarity. Figure created based on the cif file deposition of ref. 79. | ||
Li and co-workers also investigated the significance of hydride in Cu NCs during CO2RR.80 They studied a [Cu25H22(PPh3)12]Cl NC as model, featuring a Cu13 icosahedral core surrounded by a metal–ligand shell containing four Cu3 triangles at the tetrahedral sites. This cluster exhibited the highest metal-to-hydride ratio with 22 hydrides at the surface capping sites. The electrochemical CO2RR on this catalyst followed a two-electron pathway, resulting in the predominant formation of HCOOH (formic acid) or CO. The presence of abundant surface-capped hydrides in the Cu NC enhances the accessibility of hydrogen for CO2 reduction, enabling it to originate from two potential sources: either protons in the solution (H+ + e−) or directly from the capping hydrides (H*). Furthermore, the researchers observed that the NC's surface played a crucial role in stabilizing the intermediate HCOO*, leading to the formation of formic acid. Thus, the product was generated through two distinct mechanisms: the proton-reduction mechanism or the hydride-proton mechanism, both exhibiting comparable thermodynamic and kinetic feasibility. Interestingly, the DFT analysis indicated that the HCOOH product did not undergo further reduction to H2COOH, suggesting a specific selectivity towards HCOOH formation. This investigation underscores the structural dependence of CO2RR by Cu NCs, where the presence of hydride ligands determines the optimal reaction pathway.
In a recent study by Liu et al., the selectivity of Cu NCs (Cu NCs) in CO2RR was compared by investigating a series of Cu NCs (Fig. 9).81 The researchers synthesized three Cu8 isomers: Cu8(H)-(9H-carbazole-9-carbodithioate)6PF6 (referred to as Cu8-1), Cu8(tBuS)4(9H-carbazole-9-carbodithioate)4 (Cu8-2), and Cu8(tBuS)4(O-ethylcarbonodithiolate)4 (Cu8-3). While these structures share a similar Cu8 metallic architecture, the distribution of ligands varies among each case, resulting in distinct geometries of the Cu8 core. In the Cu8-1 NC, a slightly twisted cubic geometry of Cu8 core is observed. On the other hand, Cu8-2 exhibits a splitting of the Cu8 core geometry into ditetrahedral configurations. In the case of Cu8-3, although the Cu8 geometry is similar to the ditetrahedral configuration, it is attached to a different ligand. The researchers identified that all three Cu NCs are active in electrochemical CO2RR. However, the Cu8 NCs with the ditetrahedral core configuration displayed the highest current density and better efficiency in formic acid formation compared to the cubic geometry. However, the cubic geometry provides the highest amount of hydrogen evolution compared to the other two NCs. Theoretical studies have shed light on the competitive nature of the hydrogen evolution reaction (HER) on cubic core-shaped Cu8 clusters compared to the ditetrahedron-shaped Cu8 clusters. The investigations also uncovered lower free energy values for the adsorbed intermediate on the ditetrahedron-shaped Cu8 clusters. As a result, the combination of suppressed HER and reduced free energy barrier for CO2RR intermediates works synergistically to enhance the reactivity and selectivity of the catalytic CO2 reduction process in these Cu NCs.
 | ||
| Fig. 9 Effect of the structural architecture of Cu8 NC in selectivity of CO2RR product formation. Cu displayed in brown color; S displayed in yellow color; N displayed in blue color and C displayed in light dark color. Other ligands were removed for clarity. Figure created based on the cif file deposition of ref. 81. | ||
Chen et al. reported the effect of N and S anchored carbon matrix (Cu-S1N3) and atomically dispersed Cu clusters in electrochemical CO2 RR.82 The Cu-S1N3/Cux composite exhibits an exceptional 100% FECO when operated at −0.65 V vs. RHE. Notably, it maintains a consistently high FECO of over 90% within a range of −0.55 to −0.75 V, surpassing the performance of analogous materials with Cu–N4 configuration (FECO only 54% at −0.7 V) and Cu-S1N3 configuration (FECO 70% at −0.7 V). One of the key factors contributing to this remarkable performance lies in the unsymmetrical Cu-S1N3 atomic interface integrated into the carbon basal plane. This unique structure optimizes the binding energy for the crucial intermediate *COOH, surpassing the performance of the Cu–N4 site. Furthermore, the adjacent Cu NC play a significant role by facilitating the protonation of *CO2−. This is achieved through two mechanisms: first, by accelerating water dissociation, and second, by providing *H species to the Cu-S1N3 active sites. In summary, the exceptional performance of the Cu-S1N3/Cux composite in achieving high FECO can be attributed to its unique atomic configuration, specifically the unsymmetrical Cu-S1N3 interface, which enhances the binding energy for the intermediate *COOH, while the presence of adjacent Cux species further aids in promoting protonation through accelerated water dissociation and providing additional *H species to the active sites of Cu-S1N3.
In their thorough investigation, Wu et al. explored the catalytic properties of Cu6(2-mercaptobenzimidazole)6 in CO2 RR to produce hydrocarbons (Fig. 10).83 In particular, it was observed that each Cu atom displayed a μ3 coordination attachment, leading to a unique Cu-S2N1 geometry, distinct from the previous example. Theoretical calculations provided valuable insights into the significance of this geometry. Notably, the specific coordination between the C atom of CO2 and the Cu-S2N1 site significantly lowered the formation energy barrier of *COOH, which is a critical step in the CO2RR process. Consequently, this promoted the preferential formation of *CO while impeding the undesired formation of HCOOH. Another noteworthy discovery was that the Cu-S2N1 sites effectively stabilized the *CO intermediate, facilitating its further hydrogenation and enabling C–C coupling, which ultimately led to the generation of hydrocarbons. The experimental results were highly remarkable. This NC exhibited an optimal FE of 65.5% for hydrocarbons, with 42.5% CH4 and 23% C2H4 at a potential of −1.4 V versus RHE. Furthermore, the corresponding industrial-level hydrocarbons' partial current density reached an impressive −183.4 mA cm−2. Of particular significance was the complete suppression of HCOOH formation at all detected potentials, distinguishing this NC from other C-based nanocatalysts. So, the distinctive structural features of this NC, particularly the symmetry-broken distorted trigonal geometry, enabled specific binding of CO2 and facilitated hydrocarbon formation while inhibiting HCOOH formation. These findings hold exciting prospects for the development of highly efficient and selective catalysts for CO2 conversion and utilization in future applications.
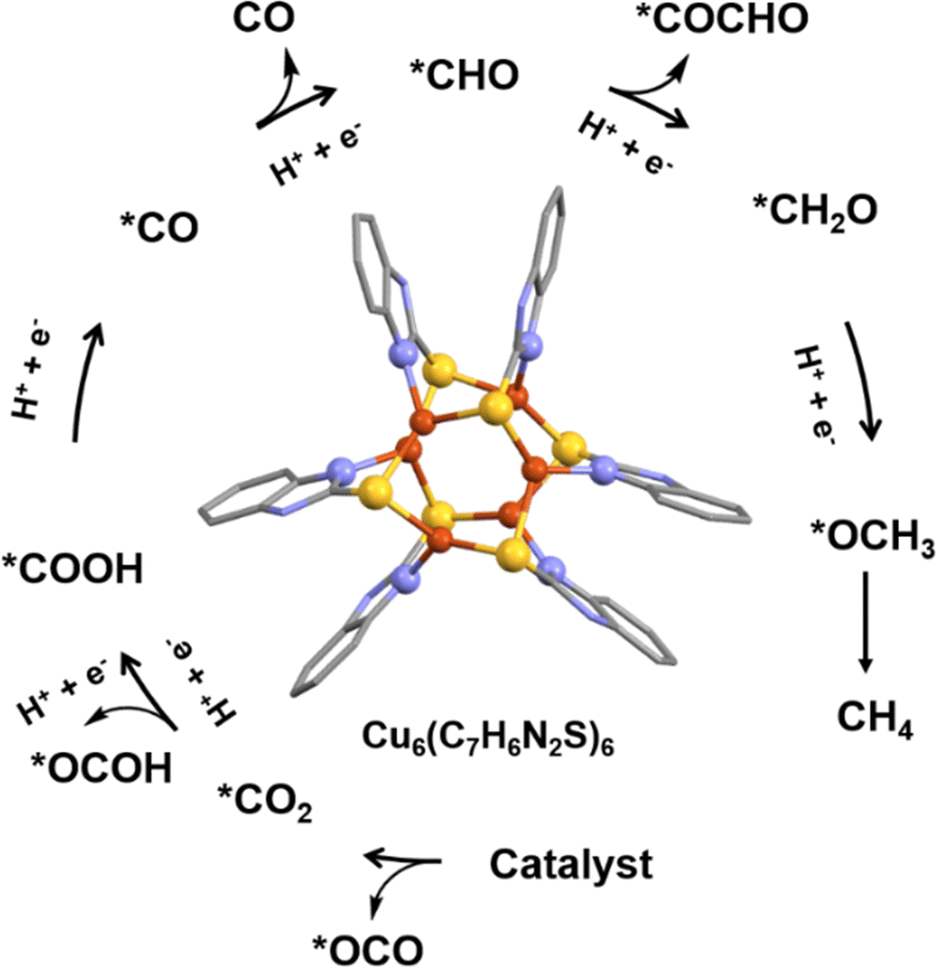 | ||
| Fig. 10 Effect of the structural architecture and the specific combinatorial active sites for the selective CO2RR product formation. Cu displayed in brown color; S displayed in yellow color; N displayed in blue color and C displayed in light dark color. Other ligands were removed for clarity. Figure created based on the cif file deposition of ref. 83. | ||
4.2. Oxygen reduction reaction (ORR)
The ORR is a vital step in energy conversion technologies, such as in fuel cells and metal–air batteries, involving the reduction of oxygen to water.84 While platinum (Pt)-based catalysts exhibit excellent activity in ORR, their scarcity and high cost hinder widespread adoption.85 As a promising alternative, researchers are exploring non-precious metals for high-performance catalysts. In this regard, Cu NCs are gradually emerging as a potential solution. In an early study conducted by Wei et al., they introduced subnanometer-sized Cu NCs protected with 2-mercapto-5-n-propylpyrimidine as a novel electrocatalyst for the ORR.86 Through extensive ESI-MS measurement, they confirmed the presence of dominant fragments consisting of Cu8 clusters within the protected Cu NCs. The electrocatalytic activity of these Cu NCs was evaluated as cathode catalysts for ORR. Remarkably, the onset potential of ORR (−0.07 V vs. Ag/AgCl) was found to be comparable to that of Au11 NCs (−0.08 V vs. Ag/AgCl) and certain commercial Pt catalysts. This discovery highlights the significant potential of these low-cost Cu NCs as efficient cathode materials in alkaline fuel cells. The findings of this study demonstrate the promise of Cu NCs as a viable alternative to expensive Pt-based catalysts for ORR. The comparable onset potential to Au11 NCs and commercial Pt catalysts suggests that these Cu NCs could serve as cost-effective and sustainable electrocatalysts for ORR, paving the way for further exploration and development in the field of alkaline fuel cells.In a subsequent study, Han et al. successfully synthesized a chiral Cu NC with S-bridging ligands.87 The cluster consisted of thirteen Cu atoms and twelve mercaptobenzoxazole ligands, forming a distinct structure with cap and core molecules. The interaction between C–H and π orbitals played a significant role in connecting these components. While the cluster demonstrated stability in air, it exhibited different oxidation states of Cu atoms, making it intriguing for redox and catalytic activities. The researchers observed that the synthesized Cu NC maintained both stability and activity when used as a cathode catalyst for the ORR. In contrast to the N2-saturated KOH electrolyte, which displayed a featureless voltammetric profile, the introduction of O2 led to a significant increase in the cathodic current for the Cu cluster. The onset potential for the ORR was measured at −0.3 V (vs. Ag/AgCl). The electrochemical behavior of the S-bridged Cu cluster showed distinct differences between N2 and O2 environments, as evidenced by the double-layer response. At −0.7 V, each O2 molecule underwent the acquisition of 2.3 electrons, indicating that the primary product formed was H2O2, accompanied by a small amount of H2O resulting from O2 reduction. As the potential became more negative, the number of electrons increased to 3.3 at −1.1 V, indicating a shift towards H2O as the predominant product, while H2O2 remained a minor component. Additionally, the Cu clusters exhibited remarkable electrocatalytic activity in the reduction of CO2, leading to the production of 20% CO and 78% H2 as the primary products at −1.05 V, with a minor quantity of CH4 also being generated. These findings highlight the versatile electrocatalytic capabilities of the S-bridged Cu NC, not only as an efficient cathode catalyst for ORR but also for the reduction of CO2. The ability to selectively produce H2 and CO from CO2 reduction showcases the potential of these Cu clusters in sustainable energy conversion and storage applications.
4.3. Oxygen evolution reaction (OER)
The OER is a fundamental electrochemical process that plays a pivotal role in water splitting, a crucial component of renewable energy technologies like water electrolysis and photoelectrochemical cells.88 It involves the oxidation of water to produce O2 gas, H+, and e−. However, the OER faces challenges due to its high energy barrier and slow kinetics, which hinder efficient and sustainable energy conversion. Conventional catalysts employed for the OER encompass materials such as Ir, Ru, Ni, and their respective oxides.89 These catalysts have been extensively studied and utilized to enhance the performance of OER systems. Cu NCs are now being started to be utilized as promising contenders in the field of OER catalysts. In a study by Hu et al., a block-like [Cu12(μ4-SCH3)6(NH3)12][Cu12(μ4-SCH3)6Cl12] NC was synthesized and evaluated for its electrocatalytic properties in alkaline media.90 The synthesized Cu NC displayed a distinctive structural feature, consisting of two separate 0D Cu12S6 cores, maintaining a remarkably high level of symmetry. This NC was characterized by a cationic [Cu12(μ4-SCH3)6(NH3)12]6+ part and an anionic [Cu12(μ4-SCH3)6Cl12]6− part. Within the spherical unit of the Cu NC, each Cu+ center was interconnected by two μ4-SCH3 ligands and one N or Cl atom, leading to a planar trigonal coordination geometry. Concerning its electrocatalytic performance, the Cu NC exhibited a low overpotential of 377 mV at 10 mA cm−2 and an onset potential of 1.59 V (vs. RHE) during the OER. The overpotential values of the Cu NC were notably superior to those of well-established sulfur-based catalysts for the OER, and it even outperformed noble catalysts like RuO2. This enhancement in OER activity was attributed to the presence of sulfur in the catalyst, suggesting that the sulfur sites acted as active sites, significantly improving the overall OER efficiency. These findings highlight the promising potential of Cu NCs, specifically the [Cu12(μ4-SCH3)6(NH3)12][Cu12(μ4-SCH3)6Cl12] NC, as efficient catalysts for the OER. The unique structure and sulfur-doping contribute to the improved OER activity, offering valuable insights for the development of sustainable and cost-effective OER catalysts.4.4. Nitrate reduction reaction (NO3 RR)
The NO3RR is a significant process involving the reduction of nitrate (NO3−) ions.91 Metallic Cu has been widely recognized as a classical catalyst for this reaction due to its versatility in transforming nitrate into various nitrogen-containing species.92 The products of NO3RR include nitrogen monoxide (NO), nitrogen gas (N2), ammonia (NH3), and hydrazine (N2H4). These products have important applications in environmental processes and various industries. However, the reaction rate of NO3RR is often sluggish, particularly during the hydrogenation step, presenting a challenge for efficient catalysis. Recently, there has been great interest in exploring the potential of atom-precise Cu NCs to enhance the reaction rate. These Cu NCs offer advantageous capabilities that can improve the efficiency of the process. To investigate the effect of Cu NCs in electrocatalytic nitrate conversion to ammonia, Luo et al. demonstrated the effectiveness of utilizing single-stranded deoxyribonucleic acid as a template for their newly synthesized Cu NCs.93 Notably, they observed an impressive NH3 yield rate of 2.62 mg h−1 cm−2 and a remarkable faradaic efficiency (FE) of 96.8% at −0.6 V (vs. RHE) during NO3RR. Even more noteworthy is that this high efficiency was consistently maintained over an extended period. The researchers attributed this success to the role of the DNA template in facilitating the distribution of water towards the interfacial region through a hydrogen bond network. This process effectively enhanced proton generation on the electrode surface, promoting smooth NO3RR kinetics, and ultimately leading to improved ammonia production through enhanced hydrogenation. These results highlight the significance of not only the structural architecture of Cu NCs but also the surface engineering of the catalysts. The surface modifications offer promising opportunities for developing efficient and sustainable catalytic processes, making a significant contribution to the field of catalysis.5. Conclusions
This article provides an in-depth exploration of the remarkable advancements in Cu NC synthesis and their efficacy in various catalytic applications. The synthesis of Cu NCs has posed challenges due to their lower half reduction potential in comparison to noble metals, raising concerns about their stability for direct catalytic applications. To address this, researchers have concentrated on achieving precise control during synthesis and carefully selecting ligands to enhance NC stability by controlling their structural architectures. Despite these efforts, the specific structural architecture of Cu NCs can sometimes hinder their ability to achieve desired products, mainly due to low selectivity and limited accessibility of active catalytic sites during reactions. Steric hindrance of ligands and surface defect sites also significantly influence catalyst design. As a result, catalyst design is a complex process, highly dependent on the particular application objective. So, we summarize the key factors here that play a crucial role in achieving the optimal catalyst design for different catalytic scenarios.1. The presence and accessibility of hydrides within the molecular structure of Cu NC play a crucial role in facilitating hydrogenation reactions and electrochemical CO2 reduction. These hydrides serve as active sites where hydrogen atoms can bind during the hydrogenation process or where CO2 molecules can undergo reduction to form valuable carbon-based products. When hydrides are readily accessible, the reactants can easily interact with the active sites, promoting faster and more controlled reactions. Furthermore, the proper design and engineering of materials with accessible hydrides can significantly enhance the overall performance of the catalyst.
2. The accessibility of Cu atoms in Cu NC catalyst is a key factor influencing the effectiveness in various chemical reactions, particularly those involving C–N bond formation, C–C coupling, click reactions, electron transfer processes, and photocatalytic reactions. Cu atoms can act as highly versatile reaction centers, facilitating the formation of crucial chemical bonds and stabilizing reaction intermediates. The accessibility of these Cu atoms allows reactants to approach the active sites efficiently, leading to enhanced catalytic activity. Moreover, controlling the size of NCs is crucial for optimizing their reactivity and selectivity in specific reactions. The optimum size ensures a balance between a high surface area, providing more active sites, and avoiding excessive agglomeration, which could limit accessibility and hinder the reaction.
3. The role of defect sites in Cu NC catalyst design is a pivotal factor for enhanced reactivity and selectivity of Cu NC based catalytic reaction. These defects, which manifest on the metal or ligand components, play a crucial role by exposing active sites to reactants, thereby facilitating more efficient catalytic processes and promoting selectivity in product formation. Remarkably, this superior performance extends across both chemical reactions and electrocatalytic processes, highlighting the paramount influence of defect engineering in unlocking the full potential of Cu NC catalysts.
4. The attachment of specific auxiliary ligands to catalysts plays a pivotal role in various catalytic reactions by ensuring optimum interactions with reactants, intermediates, or electron transfer processes. These ligands can modify the electronic and steric properties of the catalyst, influencing its selectivity and efficiency. By tailoring the ligands to the specific requirements of the reaction, catalysts can be fine-tuned to promote desired pathways and suppress unwanted side reactions. Additionally, these auxiliary ligands can facilitate electron transfer processes, enabling the catalyst to participate in redox reactions more effectively. As a result, the choice and design of auxiliary ligands are critical considerations in the development of efficient catalytic systems for diverse chemical transformations, from organic synthesis to industrial processes.
5. While the precise mechanisms underlying the impact of doping on Cu NCs are not yet comprehensively elucidated, specific instances provide compelling evidence of the enhanced catalytic efficacy of doped Cu NCs. Typically, the alterations in electronic structures resulting from doping play a pivotal role in actively modulating the catalytic activities of these reactions. These changes in electronic structure can influence the reactivity of Cu NCs and their suitability as catalysts for various chemical processes.
In conclusion, our exploration of the interplay between structural features and catalytic properties of Cu NCs aims to provide valuable guidance to researchers and industries in selecting the most suitable Cu NCs for their specific catalytic applications. By understanding the intricate relationship between structural design and catalytic behavior, we envision the development of efficient catalysts tailored for diverse chemical transformations. This summary opens up exciting possibilities in industrial and environmental applications, paving the way for more sustainable and environmentally friendly processes. As we continue to advance in NC catalysis, we are poised to witness transformative breakthroughs that will shape the future of catalytic science and its impact on the world.
6. Outlook
The continuous emergence of breakthroughs in NC synthesis and catalyst design heralds a transformative era for catalytic science, with a profound impact on society. Each new discovery contributes to a deeper understanding of the intricate interplay between NC structures, ligands, and catalytic properties. This growing knowledge empowers researchers to fine-tune catalysts with unprecedented precision, unlocking the potential for enhanced efficiency, selectivity, and stability in catalytic processes. As we look to the future, several key factors emerge as enticing avenues of exploration for our co-researchers, shaping the direction of this research area:1. Synthesis of diverse Cu NCs with different geometries and electronic structures: this approach enhances our understanding of how catalytic properties correlate with their structural variations. It empowers us to craft catalysts more precisely for specific reactions. Furthermore, knowledge of their electronic structures simplifies the assessment of their suitability for various applications. Therefore, the synthesis methods should encompass various approaches, including the manipulation of available free valence electrons, to achieve this diversity.
2. Defect-induced Cu NC and doped Cu NC synthesis: focusing on the synthesis of defect-induced Cu NCs and doped Cu with exposed sites will provide invaluable information about the active catalytic sites, offering precise insights into their role in promoting catalytic efficiency.
3. Identification of ligand effects on catalysis: investigating Cu NCs with different ligands and comparing their catalytic properties will help identify the impact of ligand interactions on catalytic activity. This knowledge will be essential for fine-tuning catalysts for desired reactions.
4. Role of hydrides in catalytic efficiency: conducting a comparative study of Cu NCs with and without hydrides will shed light on the specific role of hydrides in enhancing catalytic efficiency and product selectivity.
5. Exploring less ligand-attached Cu NCs: by synthesizing Cu NCs with fewer surface ligands, we can investigate the influence of ligand density on active catalytic sites and charge stabilization, providing insights into their overall catalytic performance.
6. Identifying wider catalytic applications: exploring and expanding the range of catalytic applications for existing Cu NCs based on their unique properties will unlock new possibilities in industrial processes, environmental remediation, and energy conversion technologies.
By pursuing these avenues, we can revolutionize catalytic science and harness the full potential of Cu NCs for sustainable and efficient catalysis. Collaborative efforts among researchers will be pivotal in advancing this field, fostering knowledge exchange and driving the translation of discoveries into practical applications. In conclusion, the future of catalytic science holds tremendous promise as we continue to delve into NC synthesis and catalyst design. With a focus on innovation, interdisciplinary collaboration and sustainability, we are poised to usher in a new era of catalytic advancements, benefitting society and the environment alike. Through the tireless pursuit of knowledge and breakthroughs, we can drive positive changes and contribute to a greener, more prosperous world.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Japan Society for the Promotion of Science (JSPS) KAKENHI (grant no. 20H02698, 22K19012, and 23H00289), Scientific Research on Innovative Areas “Aquatic Functional Materials” (grant no. 22H04562), the Yazaki Memorial Foundation for Science and Technology, and the Ogasawara Foundation for the Promotion of Science and Engineering.Notes and references
- R. Jin, C. Zeng, M. Zhou and Y. Chen, Chem. Rev., 2016, 116, 10346–10413 CrossRef CAS PubMed.
- I. Chakraborty and T. Pradeep, Chem. Rev., 2017, 117, 8208–8271 CrossRef CAS PubMed.
- W. Kurashige, Y. Niihori, S. Sharma and Y. Negishi, Coord. Chem. Rev., 2016, 320, 238–250 CrossRef.
- Y. Niihori, S. Miyajima, A. Ikeda, T. Kosaka and Y. Negishi, Small Sci., 2023, 2300024 CrossRef CAS.
- S. Hossain, Y. Niihori, L. V. Nair, B. Kumar, W. Kurashige and Y. Negishi, Acc. Chem. Res., 2018, 51, 3114–3124 CrossRef CAS PubMed.
- Y. Du, H. Sheng, D. Astruc and M. Zhu, Chem. Rev., 2019, 120, 526–622 CrossRef PubMed.
- H. Hirai, S. Ito, S. Takano, K. Koyasu and T. Tsukuda, Chem. Sci., 2020, 11, 12233–12248 RSC.
- X. Kang and M. Zhu, Chem. Soc. Rev., 2019, 48, 2422–2457 RSC.
- S. Biswas, S. Das and Y. Negishi, Coord. Chem. Rev., 2023, 492, 215255 CrossRef CAS.
- L. Shang, S. Dong and G. U. Nienhaus, Nano Today, 2011, 6, 401–418 CrossRef CAS.
- L. Liu and A. Corma, Chem. Rev., 2018, 118, 4981–5079 CrossRef CAS PubMed.
- Y. Jin, C. Zhang, X.-Y. Dong, S.-Q. Zang and T. C. W. Mak, Chem. Soc. Rev., 2021, 50, 2297–2319 RSC.
- S. Biswas, A. K. Das and S. Mandal, Acc. Chem. Res., 2023, 56, 1838–1849 CrossRef CAS PubMed.
- H. Yu, B. Rao, W. Jiang, S. Yang and M. Zhu, Coord. Chem. Rev., 2019, 378, 595–617 CrossRef CAS.
- Y.-P. Xie, Y.-L. Shen, G.-X. Duan, J. Han, L.-P. Zhang and X. Lu, Mater. Chem. Front., 2020, 4, 2205–2222 RSC.
- Y. Tao, M. Li, J. Ren and X. Qu, Chem. Soc. Rev., 2015, 44, 8636–8663 RSC.
- D. Yazaki, T. Kawawaki, T. Tanaka, D. Hirayama, Y. Shingyouchi and Y. Negishi, Energy Adv., 2023, 2, 1148–1154 RSC.
- T. Kawawaki, Y. Mitomi, N. Nishi, R. Kurosaki, K. Oiwa, T. Tanaka, H. Hirase, S. Miyajima, Y. Niihori and D. Osborn, Nanoscale, 2023, 15, 7272–7279 RSC.
- V. Truttmann, H. Drexler, M. Stöger-Pollach, T. Kawawaki, Y. Negishi, N. Barrabés and G. Rupprechter, ChemCatChem, 2022, 14, e202200322 CrossRef CAS PubMed.
- H. Tsunoyama, H. Sakurai, Y. Negishi and T. Tsukuda, J. Am. Chem. Soc., 2005, 127, 9374–9375 CrossRef CAS PubMed.
- B. Kumar, T. Kawawaki, N. Shimizu, Y. Imai, D. Suzuki, S. Hossain, L. V. Nair and Y. Negishi, Nanoscale, 2020, 12, 9969–9979 RSC.
- S. Bayda, M. Adeel, T. Tuccinardi, M. Cordani and F. Rizzolio, Molecules, 2019, 25, 112 CrossRef PubMed.
- R. Jin, Nanoscale, 2010, 2, 343–362 RSC.
- A. K. Das, S. Biswas, S. S. Manna, B. Pathak and S. Mandal, Chem. Sci., 2022, 13, 8355–8364 RSC.
- S. Biswas, A. K. Das, S. S. Manna, B. Pathak and S. Mandal, Chem. Sci., 2022, 13, 11394–11404 RSC.
- H. Qian, M. Zhu, Z. Wu and R. Jin, Acc. Chem. Res., 2012, 45, 1470–1479 CrossRef CAS PubMed.
- Q.-M. Wang, Y.-M. Lin and K.-G. Liu, Acc. Chem. Res., 2015, 48, 1570–1579 CrossRef CAS PubMed.
- B. Zhang, J. Chen, Y. Cao, O. J. H. Chai and J. Xie, Small, 2021, 17, 2004381 CrossRef CAS PubMed.
- X. Du and R. Jin, ACS Nano, 2019, 13, 7383–7387 CrossRef CAS PubMed.
- Y. Negishi, K. Nobusada and T. Tsukuda, J. Am. Chem. Soc., 2005, 127, 5261–5270 CrossRef CAS PubMed.
- T. Kawawaki, A. Ebina, Y. Hosokawa, S. Ozaki, D. Suzuki, S. Hossain and Y. Negishi, Small, 2021, 17, 2005328 CrossRef CAS PubMed.
- S. Hossain, Y. Imai, Y. Motohashi, Z. Chen, D. Suzuki, T. Suzuki, Y. Kataoka, M. Hirata, T. Ono and W. Kurashige, Mater. Horiz., 2020, 7, 796–803 RSC.
- S. Hossain, Y. Imai, D. Suzuki, W. Choi, Z. Chen, T. Suzuki, M. Yoshioka, T. Kawawaki, D. Lee and Y. Negishi, Nanoscale, 2019, 11, 22089–22098 RSC.
- X. Liu and D. Astruc, Coord. Chem. Rev., 2018, 359, 112–126 CrossRef CAS.
- Z. Wang, B. Chen and A. L. Rogach, Nanoscale Horiz., 2017, 2, 135–146 RSC.
- A. Baghdasaryan and T. Bürgi, Nanoscale, 2021, 13, 6283–6340 RSC.
- S. Shahsavari, S. Hadian-Ghazvini, F. H. Saboor, I. M. Oskouie, M. Hasany, A. Simchi and A. L. Rogach, Mater. Chem. Front., 2019, 3, 2326–2356 RSC.
- A. K. Das, S. Biswas, V. S. Wani, A. S. Nair, B. Pathak and S. Mandal, Chem. Sci., 2022, 13, 7616–7625 RSC.
- S. Sharma, K. K. Chakrahari, J.-Y. Saillard and C. W. Liu, Acc. Chem. Res., 2018, 51, 2475–2483 CrossRef CAS PubMed.
- K. K. Chakrahari, J. H. Liao, S. Kahlal, Y. C. Liu, M. H. Chiang, J. Y. Saillard and C. Liu, Angew. Chem., Int. Ed., 2016, 55, 14704–14708 CrossRef CAS PubMed.
- R. S. Dhayal, J.-H. Liao, Y.-R. Lin, P.-K. Liao, S. Kahlal, J.-Y. Saillard and C. W. Liu, J. Am. Chem. Soc., 2013, 135, 4704–4707 CrossRef CAS PubMed.
- B.-L. Han, Z. Liu, L. Feng, Z. Wang, R. K. Gupta, C. M. Aikens, C.-H. Tung and D. Sun, J. Am. Chem. Soc., 2020, 142, 5834–5841 CrossRef CAS PubMed.
- S. Nematulloev, R. W. Huang, J. Yin, A. Shkurenko, C. Dong, A. Ghosh, B. Alamer, R. Naphade, M. N. Hedhili, P. Maity, M. Eddaudi, O. F. Mohammed and O. M. Bakr, Small, 2021, 17, 2006839 CrossRef CAS PubMed.
- R.-W. Huang, J. Yin, C. Dong, A. Ghosh, M. J. Alhilaly, X. Dong, M. N. Hedhili, E. Abou-Hamad, B. Alamer, S. Nematulloev, Y. Han, O. F. Mohammed and O. M. Bakr, J. Am. Chem. Soc., 2020, 142, 8696–8705 CrossRef PubMed.
- P. P. Sun, B. L. Han, H. G. Li, C. K. Zhang, X. Xin, J. M. Dou, Z. Y. Gao and D. Sun, Angew. Chem., Int. Ed., 2022, 61, e202200180 CrossRef CAS PubMed.
- H. Li, H. Zhai, C. Zhou, Y. Song, F. Ke, W. W. Xu and M. Zhu, J. Phys. Chem. Lett., 2020, 11, 4891–4896 CrossRef CAS PubMed.
- F. Ke, Y. Song, H. Li, C. Zhou, Y. Du and M. Zhu, Dalton Trans., 2019, 48, 13921–13924 RSC.
- X. Lin, J. Tang, C. Zhu, L. Wang, Y. Yang, R. A. Wu, H. Fan, C. Liu and J. Huang, Chem. Sci., 2023, 14, 994–1002 RSC.
- J. Tang, C. Liu, C. Zhu, K. Sun, H. Wang, W. Yin, C. Xu, Y. Li, W. Wang, L. Wang, R. Wu, C. Liu and J. Huang, Nanoscale, 2023, 15, 2843–2848 RSC.
- C. Dong, R.-W. Huang, C. Chen, J. Chen, S. Nematulloev, X. Guo, A. Ghosh, B. Alamer, M. N. Hedhili, T. T. Isimjan, Y. Han, O. F. Mohammed and O. M. Bakr, J. Am. Chem. Soc., 2021, 143, 11026–11035 CrossRef CAS PubMed.
- S. Zeng, X. Ge, H. Deng, S. Hao, Z. Zhang, B. K. Teo and C. Sun, J. Cluster Sci., 2023, 1–5 Search PubMed.
- X. Lin, J. Tang, C. Zhu, L. Wang, Y. Yang, H. Fan, C. Liu and J. Huang, Chem. Sci., 2023, 14, 994–1002 RSC.
- T. Jia, Z.-J. Guan, C. Zhang, X.-Z. Zhu, Y.-X. Chen, Q. Zhang, Y. Yang and D. Sun, J. Am. Chem. Soc., 2023, 145, 10355–10363 CrossRef CAS PubMed.
- S. K. Barik, S. C. Huo, C. Y. Wu, T. H. Chiu, J. H. Liao, X. Wang, S. Kahlal, J. Y. Saillard and C.-W. Liu, Chem. – Eur. J., 2020, 26, 10471–10479 CrossRef CAS PubMed.
- Y. Liu, J. Yu, Y. Lun, Y. Wang, Y. Wang and S. Song, Adv. Funct. Mater., 2023, 2304184 CrossRef.
- G. V. Goeden and K. G. Caulton, J. Am. Chem. Soc., 1981, 103, 7354–7355 CrossRef CAS.
- M. T. Pirnot, Y. M. Wang and S. L. Buchwald, Angew. Chem., Int. Ed., 2016, 55, 48–57 CrossRef CAS PubMed.
- S. A. Bezman, M. R. Churchill, J. A. Osborn and J. Wormald, J. Am. Chem. Soc., 1971, 93, 2063–2065 CrossRef.
- J.-X. Chen, J. F. Daeuble, D. M. Brestensky and J. M. Stryker, Tetrahedron, 2000, 56, 2153–2166 CrossRef CAS.
- D. M. Brestensky, D. E. Huseland, C. McGettigan and J. M. Stryker, Tetrahedron Lett., 1988, 29, 3749–3752 CrossRef CAS.
- C. Sun, N. Mammen, S. Kaappa, P. Yuan, G. Deng, C. Zhao, J. Yan, S. Malola, K. Honkala and H. Häkkinen, ACS Nano, 2019, 13, 5975–5986 CrossRef CAS PubMed.
- G.-G. Luo, Z.-H. Pan, B.-L. Han, G.-L. Dong, C.-L. Deng, M. Azam, Y.-W. Tao, J. He, C.-F. Sun and D. Sun, Angew. Chem., Int. Ed., 2023, 62, e202306849 CrossRef CAS PubMed.
- S. Biswas, S. Hossian, T. Kosaka, J. Sakai, D. Arima, Y. Niihori, M. Mitsui, D.-E. Jiang, S. Das, S. Wang and Y. Negishi, Chem. Commun., 2023, 59, 9336–9339 RSC.
- S. Lee, M. S. Bootharaju, G. Deng, S. Malola, W. Baek, H. Hakkinen, N. Zheng and T. Hyeon, J. Am. Chem. Soc., 2020, 142, 13974–13981 CrossRef CAS PubMed.
- Z. H. Guan, H. Lei, M. Chen, Z. H. Ren, Y. Bai and Y. Y. Wang, Adv. Synth. Catal., 2012, 354, 489–496 CrossRef CAS.
- A. W. Cook, Z. R. Jones, G. Wu, S. L. Scott and T. W. Hayton, J. Am. Chem. Soc., 2018, 140, 394–400 CrossRef CAS PubMed.
- G. Dong, Z. Pan, B. Han, Y. Tao, X. Chen, G.-G. Luo, P. Sun, C. Sun and D. Sun, Angew. Chem., Int. Ed., 2023, 62, e202302595 CrossRef CAS PubMed.
- C. Wang, C. Wang, L. Xu, H. Cheng, Q. Lin and C. Zhang, Nanoscale, 2014, 6, 1775–1781 RSC.
- Y.-M. Wang, X.-C. Lin, K.-M. Mo, M. Xie, Y.-L. Huang, G.-H. Ning and D. Li, Angew. Chem., Int. Ed., 2023, 62, e202218369 CrossRef CAS PubMed.
- S. Lee, M. S. Bootharaju, G. Deng, S. Malola, H. Häkkinen, N. Zheng and T. Hyeon, J. Am. Chem. Soc., 2021, 143, 12100–12107 CrossRef CAS PubMed.
- S. Nematulloev, A. Sagadevan, B. Alamer, A. Shkurenko, R. Huang, J. Yin, C. Dong, P. Yuan, K. E. Yorov, A. A. Karluk, W. J. Mir, B. E. Hasanov, M. N. Hedhili, N. M. Halappa, M. Eddaoudi, O. F. Mohammed, M. Rueping and O. M. Bakr, Angew. Chem., Int. Ed., 2023, 62, e202303572 CrossRef CAS PubMed.
- A. Ghosh, R.-W. Huang, B. Alamer, E. Abou-Hamad, M. N. Hedhili, O. F. Mohammed and O. M. Bakr, ACS Mater. Lett., 2019, 1, 297–302 CrossRef CAS.
- A. Sagadevan, A. Ghosh, P. Maity, O. F. Mohammed, O. M. Bakr and M. Rueping, J. Am. Chem. Soc., 2022, 144, 12052–12061 CrossRef CAS PubMed.
- C. Dong, R.-W. Huang, A. Sagadevan, P. Yuan, L. Gutiérrez-Arzaluz, A. Ghosh, S. Nematulloev, B. Alamer, O. F. Mohammed, I. Hussain, M. Rueping and O. M. Bakr, Angew. Chem., Int. Ed., 2023, 62, e202307140 CrossRef CAS PubMed.
- C. Zhang, Z. Wang, W.-D. Si, L. Wang, J.-M. Dou, Z.-Y. Gao, C.-H. Tung and D. Sun, ACS Nano, 2022, 16, 9598–9607 CrossRef CAS PubMed.
- S. J. Davis, K. Caldeira and H. D. Matthews, Science, 2010, 329, 1330–1333 CrossRef CAS PubMed.
- D. W. DeWulf, T. Jin and A. J. Bard, J. Electrochem. Soc., 1989, 136, 1686 CrossRef CAS.
- Y. Hori, K. Kikuchi, A. Murata and S. Suzuki, Chem. Lett., 1986, 897–898 CrossRef CAS.
- Q. Tang, Y. Lee, D.-Y. Li, W. Choi, C. W. Liu, D. Lee and D.-E. Jiang, J. Am. Chem. Soc., 2017, 139, 9728–9736 CrossRef CAS PubMed.
- F. Li and Q. Tang, J. Catal., 2020, 387, 95–101 CrossRef CAS.
- L. J. Liu, Z. Y. Wang, Z. Y. Wang, R. Wang, S. Q. Zang and T. C. Mak, Angew. Chem., Int. Ed., 2022, 61, e202205626 CrossRef CAS PubMed.
- D. Chen, L. H. Zhang, J. Du, H. Wang, J. Guo, J. Zhan, F. Li and F. Yu, Angew. Chem., Int. Ed., 2021, 60, 24022–24027 CrossRef CAS PubMed.
- Q.-J. Wu, D.-H. Si, P.-P. Sun, Y.-L. Dong, S. Zheng, Q. Chen, S.-H. Ye, D. Sun, R. Cao and Y.-B. Huang, Angew. Chem., Int. Ed., 2023, 62, e202306822 CrossRef CAS PubMed.
- K. Gong, F. Du, Z. Xia, M. Durstock and L. Dai, Science, 2009, 323, 760–764 CrossRef CAS PubMed.
- H. Tsunoyama, A. Ohnuma, K. Takahashi, A. Velloth, M. Ehara, N. Ichikuni, M. Tabuchi and A. Nakajima, Chem. Commun., 2019, 55, 12603–12606 RSC.
- W. Wei, Y. Lu, W. Chen and S. Chen, J. Am. Chem. Soc., 2011, 133, 2060–2063 CrossRef CAS PubMed.
- H. Han, Y. Yao, A. Bhargava, Z. Wei, Z. Tang, J. Suntivich, O. Voznyy and R. D. Robinson, J. Am. Chem. Soc., 2020, 142, 14495–14503 CrossRef CAS PubMed.
- N.-T. Suen, S.-F. Hung, Q. Quan, N. Zhang, Y.-J. Xu and H. M. Chen, Chem. Soc. Rev., 2017, 46, 337–365 RSC.
- X. Xie, L. Du, L. Yan, S. Park, Y. Qiu, J. Sokolowski, W. Wang and Y. Shao, Adv. Funct. Mater., 2022, 32, 2110036 CrossRef CAS.
- L. Hu, A. Zheng, Y. Kang, T. Wen and J. Zhang, Chem. Commun., 2020, 56, 3967–3970 RSC.
- H. Wang, J. Huang, J. Cai, Y. Wei, A. Cao, B. Liu and S. Lu, Small Meth., 2023, 2300169 CrossRef CAS PubMed.
- W. Jung and Y. J. Hwang, Mater. Chem. Front., 2021, 5, 6803–6823 RSC.
- W. Luo, S. Wu, Y. Jiang, P. Xu, J. Zou, J. Qian, X. Zhou, Y. Ge, H. Nie and Z. Yang, ACS Appl. Mater. Interfaces, 2023, 15, 18928–18939 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |