
Adaptive particle patterning in the presence of active synthetic nanomotors†
Dazhi
Xie
a,
Shaoming
Fu
a,
Dongmei
Fu
a,
Bin
Chen
b,
Weidong
He
a,
Haiying
Liang
 b,
Yingfeng
Tu
b,
Daniela A.
Wilson
c and
Fei
Peng
*a
b,
Yingfeng
Tu
b,
Daniela A.
Wilson
c and
Fei
Peng
*a
aSchool of Materials Science and Engineering, Sun Yat-Sen University, Guangzhou 510275, China
bGuangdong Provincial Key Laboratory of New Drug Screening, School of Pharmaceutical Sciences, Southern Medical University, Guangzhou 510515, China
cInstitute for Molecules and Materials, Radboud University, Nijmegen, 6525 AJ, The Netherlands
First published on 8th March 2023
Abstract
For the maintenance of a biological system, spatial organization of material condensates within the cell through the dissipation of energy is crucial. Besides directed transport via microtubules, material arrangement can be achieved via motor protein facilitated adaptive active diffusiophoresis. For example, the distribution of membrane proteins during the cell division of Escherichia coli is affected by the MinD system. Synthetic active motors exhibit the ability to simulate natural motors. Here we propose an active Au–Zn nanomotor driven by water and discovered an interesting adaptive interaction mode of the diffusiophoretic Au–Zn nanomotors with passive condensate particles in different environments. It is found that the attraction/repulsion between the nanomotor and passive particles is adaptive, while an interesting hollow pattern is formed with a negatively charged substrate and a cluster pattern is favored with a positively charged substrate.
Introduction
In nature, the transportation and distribution of materials lead to spatiotemporal organization within cells. Besides the directional cargo transport via microtubules, active diffusiophoresis driven by natural motor proteins is considered to constitute a novel mechanism for coupling energy dissipation with pattern formation and spatiotemporal organization, including positioning nucleoid and material condensates, for example, the protein self-organizing MinD system in Escherichia coli.1 The distribution of materials to create a complex structure and a large-scale pattern is the basis for a complicated and delicate function. The as-formed material condensates pattern can adapt according to changes in the environment, allowing the system to remain flexible and stable at the same time.2–4 Exploring the mechanism behind this feature has attracted booming research interest.5–8 In recent years, self-powered active micro-nanomotors have emerged as promising candidates for synthetic natural motor counterparts.9 These synthetic nanomotors, first fabricated in 2004,10 can be propelled via bubble, self-electrophoresis, diffusiophoresis, and an external physical field.11–16 For all mechanisms, a locally established chemical or physical gradient is required for propulsion and autonomous movement. These gradients can also constitute the communication bridge between nanomotor individuals and the environment to simulate real-life collective behaviour.Sen et al. pioneered communication and collective behaviour among active nanomotor individuals.17 Yet in nature, the interaction with passive particles is more universal. Guan et al. reported TiO2 and ZnO, demonstrating the phenomenon of a prey–predator interaction with passive colloids.18 Recently, Tang et al. found that ion exchange between predator and prey can further enhance the interaction.19 Yet different from real-life behaviour, the behaviour of the above active–passive systems is simple, and their interaction in different environments is largely overlooked. The flexibility of the active system behaviour patterns remains to be uncovered.20 For further exploration, a new platform is highly desirable.
Here we report an attractive Au–Zn nanotube motor, capable of autonomously moving in DI water (with pure water as the energy input, no acid needed and no bubbles generated during the propulsion). The interference of bubbles in the flow field was eliminated. During the propulsion, a clear and strong concentration gradient was observed to build up, which was considered to impact and interact with the PS bead sharing the same space via the resulting electroosmotic field. Interesting phenomena such as chasing, attraction–repulsion, attraction–no repulsion, and adaptive movement changing were observed. In addition, a collective pattern of the Au–Zn nanomotor–PS cluster was discovered. Depending on the environment, i.e. the position and substrate potential, two organizing patterns (hollow or cluster) were found, which resulted from the individual communication influenced by the environment, reflecting the adaptivity and complexity of the interaction between the active nanomotor cluster and the environment. Away from the substrate (environment wall), pattern formation is unfavorable, while a hollow pattern is formed with a negatively charged substrate and a cluster pattern is formed with a positively charged substrate. This is very consistent with the working mode of the protein self-organizing MinD system in Escherichia coli, that is, the dimer MinD is adsorbed onto the membrane to recruit proteins in the solution, and MinDE repels non-dissociable membrane proteins to form a large-scale gradient.21 This switchable interaction with the environment is expected to be used to explain the intracellular self-organization protein pattern and indicates that the Au–Zn nanomotor system emerges as a ready platform for exploring non-equilibrium dynamics. This non-specific transport and distinct pattern-forming system discovery is not a coincidence, but highlights that such occurrences are presumably masked in cells with more sophisticated interactions.
Experimental
Materials and methods
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 = DI water:DMSO) was added to a 10 μl Au–Zn nanomotor dispersion, followed by imaging using an inverted fluorescence nanoscope. For observing the interaction between the Au–Zn nanomotor and PS beads, 10 μL of the solution of the Au–Zn nanomotor in DI water and 10 μL of the PS bead solution (amine 1 μm PS or carboxyl 1 μm PS or 10 μm PS, purchased from Sigma) were placed on a glass slide (microscope slides and adhesion microscope slides, purchased from CITOTEST) and imaged with an inverted microscope (Nikon Ti2-A). It was observed with a 40× microscope and videos were recorded using NIS-Element Viewer.
:1 = DI water:DMSO) was added to a 10 μl Au–Zn nanomotor dispersion, followed by imaging using an inverted fluorescence nanoscope. For observing the interaction between the Au–Zn nanomotor and PS beads, 10 μL of the solution of the Au–Zn nanomotor in DI water and 10 μL of the PS bead solution (amine 1 μm PS or carboxyl 1 μm PS or 10 μm PS, purchased from Sigma) were placed on a glass slide (microscope slides and adhesion microscope slides, purchased from CITOTEST) and imaged with an inverted microscope (Nikon Ti2-A). It was observed with a 40× microscope and videos were recorded using NIS-Element Viewer.
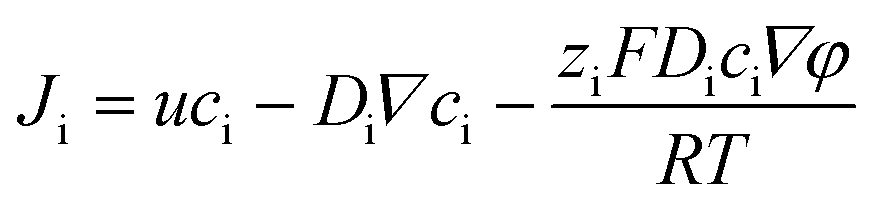 | (1) |
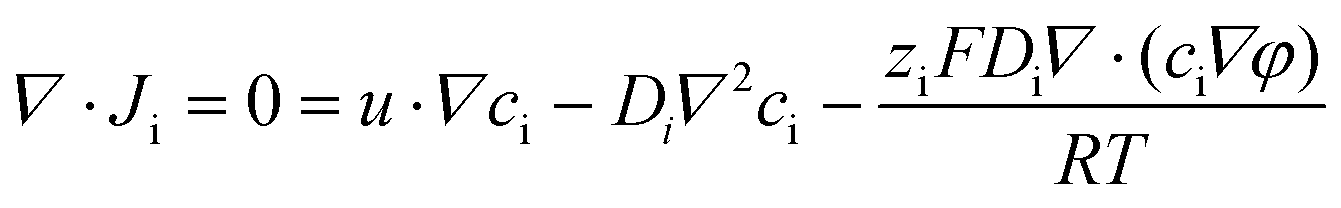 | (2) |
where u is the fluid velocity, F is the Faraday constant, φ is the electrostatic potential, R is the gas constant, T is the absolute temperature, and ci, Di, and zi are the concentration, diffusion coefficient, and charge of species i, respectively.
The electric potential (φ) in eqn (1) was calculated using the Poisson equation:
| −ε0εr∇2 = ρe = F(z+c+ + z−c−) | (3) |
The zinc ion flux was determined using the following equations:

The value of Δt takes 4 days to reach 345600 s; the value of ΔC was determined using the pore number of the polycarbonate membrane and the zinc volume of a single nanomotor; A represents the area, and the value of A was determined using the pore number of the polycarbonate film and the zinc area of a single nanomotor. The pore number of the polycarbonate membrane was 3.98 × 10−5. The area and volume of a single nanomotor were 8.9 μm2 and 0.37 μm3, respectively. Finally, the flux of Zn2+ on the zinc surface was 1.6 × 10−6 mol (m2 s)−1 (Table 1).
| Name | Description | Value |
|---|---|---|
| Flux_in | Zn2+ flux on the zinc surface of the nanomotor | 1.6 × 10−6 mol (m2 s)−1 |
| D_Zn | Zn2+ diffusion coefficient | 0.7 × 10−9 m2 s−1 |
| C_Zn0 | Zn2+ initial concentration | 0 mol m−3 |
| U m | Electric field mobility | 3.7 × 10−12 s mol kg−1 |
Results and discussion
Mechanism of the Au–Zn nanomotor
Au–Zn tubular nanomotors were synthetized via a facile approach (Fig. 1A). With the polycarbonate film as the template, nanotubes were electrochemically deposited with one end of gold and one end of zinc. Fig. 1B shows the SEM image of the surface of the polycarbonate film after depositing Au–Zn nanomotors. Through scanning electron microscopy (SEM) (Fig. 1C), the average diameter of the nanomotor was determined to be 414 ± 70 nm, the average length of the entire tube was 4.76 ± 1.3 μm, and the average length of the gold segment was 1.57 ± 0.3 μm (Fig. 1D). From the SEM image, the gold segment appears to be brighter than the zinc segment due to higher conductivity. With energy dispersive spectroscopy (EDS) (Fig. 1E and Fig. S1†), the copresence and distribution of Au and Zn could be clearly seen, with the Au atomic quantification being 91.8% and the Zn atomic quantification being 8.1%. The as-obtained Au–Zn nanomotor can therefore move spontaneously in pure DI water (Fig. 1F and S2, ESI Video S1†), with an average speed of 6.4 ± 1.4 μm s−1 (Table S1†). For the propulsion, the driving force comes from the reaction of the zinc segment with water, as shown in eqn (4).| Zn + 2H+ ↔ Zn2+ + H2 | (4) |
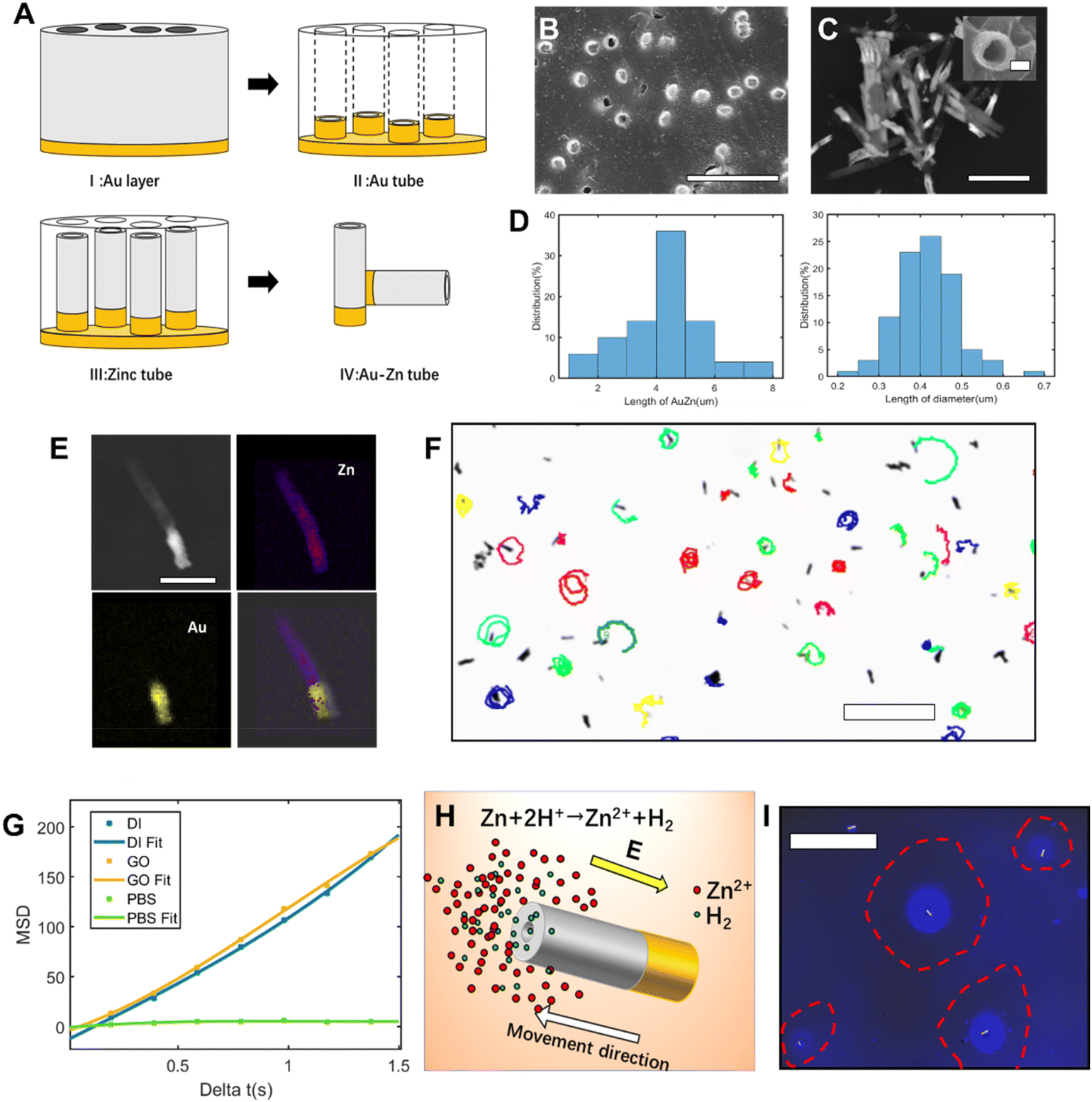 | ||
| Fig. 1 Morphology and motion evaluation of Au–Zn nanomotors. (A) Schematic for the preparation of the tubular Au–Zn nanomotor. I: A layer of 100 nm gold evaporated on the polycarbonate film with 400 nm pore. II: The gold tube electrically deposited on the conductive film. III: The electrolyte was replaced to continue the electrodeposition of the zinc tube segment. IV: The Au–Zn nanomotor was released by removing the gold layer via physical friction and dissolution of the polycarbonate. (B) SEM image of the Au–Zn nanomotor array after removing the gold layer. Scale bars: 2 μm. (C) The SEM side-view image of the Au–Zn nanomotors. (Inset: front view of the Au nozzle of the nanomotor.) Scale bars: 0.2 μm (c inset), 3 μm (c). (D) Length and diameter distribution of the nanomotor. (E) EDS elemental mapping of the nanomotor. Scale bars: 2 μm. (F) The trajectory of the Au–Zn nanomotor in DI water in 10 seconds. Scale bars: 50 μm. (G) MSD analyses of the Au–Zn nanomotor in DI water, 5% glucose solution, and PBS. (H) Schematic illustration of the diffusion of Zn2+ and H2; the nanomotor moves to the high concentration Zn2+ region. (I) Blue fluorescence images of the inverted microscope. Blue fluorescence indicates the distribution of Zn2+. Scale bars: 100 μm. | ||
The hydrogen ions in water are reduced in presence of the zinc segment to produce hydrogen and zinc ions. But no bubbles were observed under the microscope. Different from previous water-consuming metal motors, our bubble-free system ensures a system without interference from bubbles.22–24 With the accumulated Zn2+ concentration gradient along the zinc (active)–gold (inert) nanomotor, a self-built electric field is expected to form, propelling the nanomotor to the Zn direction (Fig. 1H).25 Since there is only a diffusing zinc cation, the propulsion mechanism of the Au–Zn nanomotor is attributed to ionic self-diffusiophoresis, in which the motor moves in the direction of the higher ion level region.26,27
On fitting the mean square displacement (MSD) value against time (Fig. 1G), the MSD was found to increase linearly with time. There was no difference in the performance of the nanomotor in deionized water and 5% glucose solution, indicating that the presence of glucose molecules did not affect the propulsive ability of the motor. The moving ability of the nanomotor was hindered in the PBS solution, mainly because pH buffering by PBS modifies the rate of the zinc oxidation reaction, which relies on protons, and modifies the charge on the nanomotor surface.25 For confirmation of the zinc gradient, the zinc ion fluorescent probe Zinquin was used to detect the zinc ions produced by nanomotors. The fluorescent probe can polymerize into fluorescent polymer particles in the presence of zinc ions. From the Zn2+ fluorescence (blue) image (Fig. 1I), the blue fluorescent region highlighted by a red circle indicates the accumulated fluorescent product from the polymerization of the probe and Zn2+ from the nanomotor. The dark area inside the red circle is a forbidden area generated by the nanomotor, and a portion of the fluorescent products is repelled outside. Since the nanomotor continuously reacts with water to generate Zn2+, and the Zn2+ continuously polymerizes with the fluorescent probe, the nanomotor keeps spraying out fluorescent beads (ESI Video S2,† see the spraying out of luminous polymer beads from the polymerization of Zn2+ and fluorescent probes). As the reaction between zinc and water is mild enough, the nanomotor can maintain the movement ability for a sustained time. The propulsion ability of the Au–Zn nanomotor can be maintained in DI water for a prolonged period of 4–5 days. To the best of our knowledge, this is the longest life span for self-consuming nanomotors powered by chemical energy.
Self-built field around the nanomotor
After investigating the moving mechanism of the individual nanomotor, the interaction between the nanomotor and passive particles was explored. According to the previous literature, electroosmotic flow is considered as a main medium of interaction between nanomotors and the environment. As the Au–Zn nanomotor continuously reacts with water, a decreasing gradient field of Zn2+ which points away from the nanomotor surface is formed around it (Fig. 2A). This leads to a net electric field which points away from the nanomotor surface. For the negatively charged glass substrate (CITOTEST 1A5101, ζW = −85 mV (ref. 28)), positively charged protons were adsorbed on the substrate. The Zn2+ electric field acts on the proton and creates a repelling and outward electroosmotic flow from the nanomotor towards the substrate. Since the fluid flow is continuous, an inward fluid is generated in the upper liquid layer.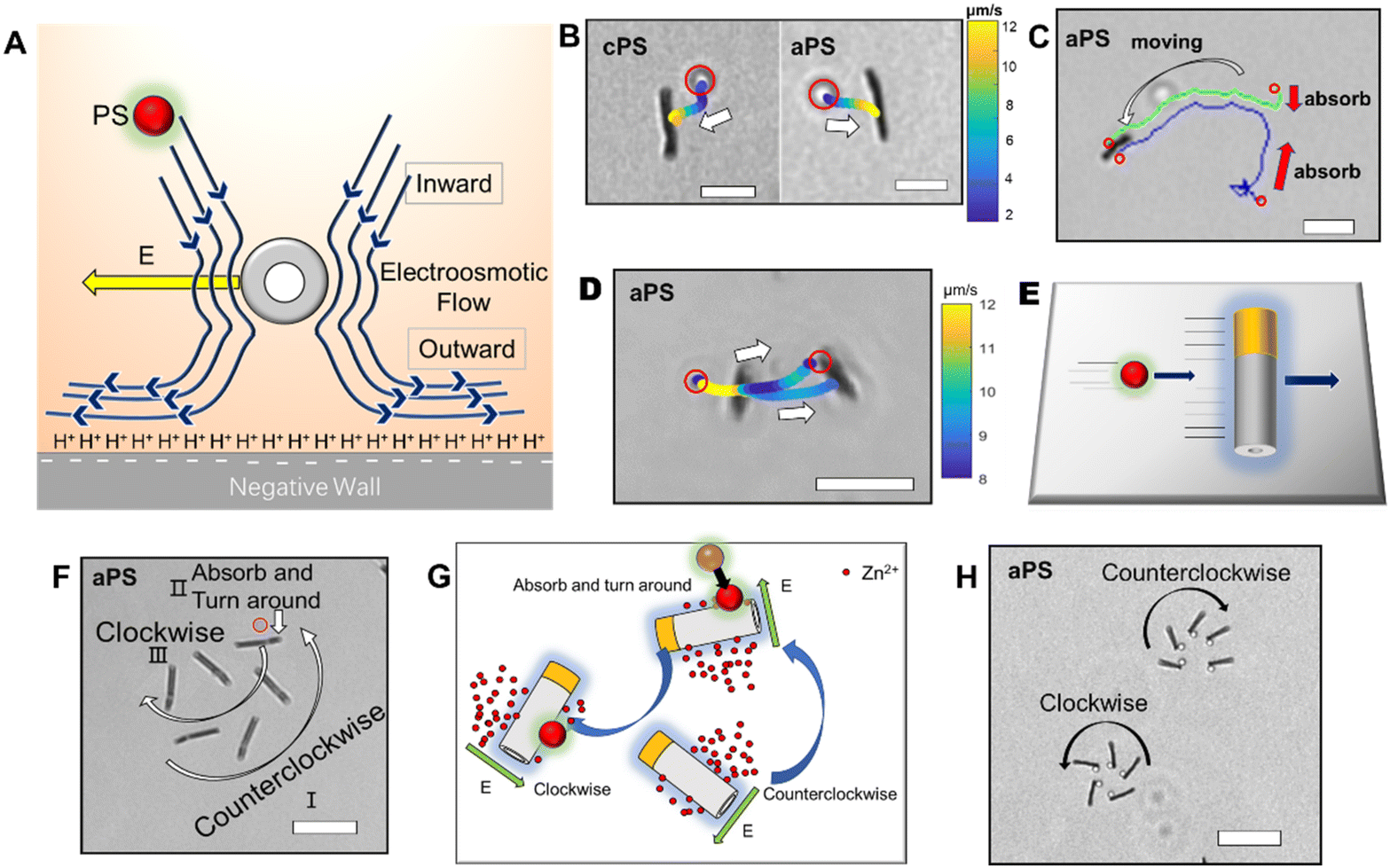 | ||
| Fig. 2 Interactions between the Au–Zn nanomotor and PS beads. (A) A cross-section scheme of the self-built electric field and the electroosmotic flow generated by the tubular nanomotor. (B) The trajectory of amine PS beads and carboxyl PS beads; the speed correlated with the PS bead position is shown. Scale bars: 5 μm. (C) The Au–Zn nanomotor was loaded with two amine PS beads during the propulsion. The red circle represents amine PS beads. Scale bars: 20 μm. (D) The trajectory diagrams of amine PS tracer particles chasing the Au–Zn nanomotor. Scale bars: 10 μm. (E) Schematic diagram of an amine PS bead particle chasing the nanomotor. (F) The trajectory diagram of the Au–Zn nanomotor resteering after the adsorption of amine PS beads. Scale bars: 10 μm. (G) Schematic diagram of the interaction between the nanomotor and amine PS tracer particles. (H) The trajectory diagram of the circular motion of the Au–Zn nanomotor due to the interaction of the amine PS beads. Scale bars: 10 μm. All results are obtained with negatively charged substrates. | ||
As shown in Fig. 2B, the nanomotor was suspended in DI water,29 with the carboxyl PS bead (hereinafter referred to as cPS, ζcPS = −34 mV, diameter 1 μm) as the passive particle added to the solution. In the vicinity (2 μm away) of a single Au–Zn nanomotor, the cPS bead falling to the height of the nanomotor can be quickly adsorbed towards the nanomotor surface (ESI Video S3†), which could be the equilibrium position of the surface slip flow.18 A quasi-stable nanomotor–cPS complex was then formed. Since the nanomotor was far from the substrate, the cPS beads were not repelled by the electroosmotic flow below. The zeta potential of the nanomotor was measured as −10.2 mV, which did not seem to affect its adsorption of cPS beads that were also negatively charged. The motion of 15 cPSs was recorded and the velocity was averaged to obtain the curve shown in Fig. S3a.† The speed of cPSs reached 12.5 ± 4 μm s−1 when it was adsorbed, which indicated there was an attraction field around the nanomotor. Then, amine PS with a positive charge (hereinafter referred to as aPS, ζaPS = +38 mV, diameter 1 μm) was selected as the passive particle, and a similar phenomenon was observed (Fig. 2B and ESI Video S4†). The aPS was also adsorbed to the surface of the Au–Zn nanomotor and formed a quasi-stable complex. Speeds of 15 such aPSs were recorded and averaged (Fig. S3a†). It was found that the speed of aPS during adsorption was 10.6 ± 2.0 μm s−1; speeds were not significantly different with sPS, which indicated electrostatic forces did not play an important role in the interaction process.27 Here Zn2+ from the reaction of the Au–Zn nanomotor with water forms self-built electric field points away from the nanomotor and the positive electric field force hinders the movement of the positively charged aPS and on the other hand accelerates the negatively charged aPS. Since the value of the aPS zeta potential is less than that of the glass slide, the osmotic flow dominates, and is enough to overcome the phoretic motion of the particles. As illustrated in Fig. 2C, the nanomotor adsorbed two aPS beads on its way forward. Such attraction–no repulsion interactions are not common in conventional active–passive particle systems, and it could be that the continuous motion of the nanomotors hindered the electroosmotic flow to interact with the PS beads. This indicates that when the nanomotor is in motion and far from the substrate, it interacts with the passive PS beads in a way that favors adsorbing and forming a quasi-stable complex.
The trajectories of aPS and cPS passive particles are demonstrated respectively in Fig. 2B, D, and colors are used to indicate their moving speed: yellow indicates high speed and blue indicates low speed. It can be observed that the velocity of the PS particle decreases with increasing distance from the nanomotor, which means that the closer the Au–Zn nanomotor is, the higher the electroosmotic velocity. Since only a slight difference was observed for the adsorption of the amine PS and carboxyl PS, for the following experiments we used amine PS as the passive particles. If aPS beads can be attracted behind a moving nanomotor, a chase phenomenon will occur, and the passive particle should have a long-distance high-speed motion. As shown in Fig. 2D, an interesting chase phenomenon was observed for the nanomotor and aPS beads. With the nanomotor moving to the right, the aPS beads were attracted by the nanomotor's adsorption field. The aPS bead was observed to accelerate and therefore catch up with the nanomotor (Fig. 2E), and was combined into a quasi-stable aPS–nanomotor complex. The speed of aPS beads can reach 17 μm s−1 during the chasing process (Fig. S3b and ESI Video S5†). As observed from the video, when the motion of the nanomotor is paused (bound on substrate, no more displacement), the aPS beads fall off, probably because the equilibrium position of the surface slip flow of the nanomotor changes.
In the above chase phenomenon, the loading of PS beads does not change the moving direction of the nanomotor. An interesting phenomenon will appear when the PS beads confront the moving nanomotor. As shown in Fig. 2F, when the nanomotor rotates counterclockwise to adsorb the aPS beads in the front, the rotation direction of the nanomotor will be reversed to be clockwise and aPS beads will be adsorbed in the middle of the nanomotor (ESI Video S6†). This phenomenon could result from the fact that the nanomotor moved towards the high zinc ion region (zinc segment) at the beginning. Upon adsorption of the aPS tracer particle, the local chemical reaction was hindered, causing the zinc ion concentration on both sides of the nanomotor to reverse, thereby reversing the rotation direction of the nanomotor (Fig. 2G). As observed in Fig. 2H, the adsorption position of the PS beads indeed influenced the moving direction of the nanomotor. Judging from the moving direction of the motor, aPS beads were adsorbed on the Au side of the nanomotor (ESI Video S7†). With aPS beads being loaded at the Au end, the nanomotor moves in a circular way toward the Au/aPS side. With the other side being unaffected, the trajectory is biased towards the Au/aPS side. The response of the nanomotor to the adsorbed particles reflects the flexibility of movement in mimicking intelligent organisms.
To investigate the impact of the nanomotor adsorption field on larger passive particles, we used 10 μm PS (diameter 10 μm, no surface modification of groups, ζ10PS = −18.5 mV). Fig. S4† shows the collision of the nanomotor with 10 μm PS beads. A quasi-stable aPS–nanomotor complex was still formed and 10 μm PS beads were pushed to move forward. Edge chemotaxis in which nanomotors exhibit motion along geometric boundaries occurs, for the nanomotor and the 10 μm PS. The nanomotor rotates around the edge of the 10 μm PS bead (Fig. S5†). Due to the large size of the 10 μm PS relative to the nanomotor, there is no stable equilibrium point for surface slip flow of the 10 μm PS on the surface of the nanomotor.30
From quasi-stable complex to self-organizing patterns
The interaction between the nanomotor and the passive particles is not only limited to individual interaction and quasi-stable complex formation. Upon retention of the active nanomotor, the direction of the fluid flow is influenced by the direction of the electric field formed by the nanomotor.28–31 The direction of the electric field formed by the Zn2+ diffusion of the Au–Zn nanomotor points away from the nanomotor. When the Au–Zn nanomotor approaches a negatively charged substrate, the Zn2+ electric field acts on the protons absorbed on the negatively charged substrate electric double layer (EDL), creating a repelling and outward electroosmotic flow from the nanomotor to the substrate. Again, since the fluid is a continuous circuit, an inward electroosmotic flow is generated in the upper layer. As shown in Fig. 3A, the nanomotor pauses on a negatively charged substrate. The aPS beads in the upper layer are first adsorbed by the inward electroosmotic flow above the nanomotor, and then are repelled by the strong outward electroosmotic flow near the nanomotor. Close to the substrate, the vicinity of the nanomotor is dominated by the strong outward electroosmotic flow. The attraction and combination of aPS beads and the nanomotor into a quasi-stable complex are not favorable. At this time, in response to changes in the environment, the interaction mode with the passive particles becomes repulsive. In the process of ejection, the speed of the aPS tracer particles can reach 10 μm s−1 (Fig. 3B and ESI Video S8†). This attractive–repulsive process can also be seen in Fig. S6.† This attraction–repulsion interaction can lead to a forbidden zone effect in the system of active and passive particles. Therefore, a circular forbidden zone is formed around the nanomotor, and the aPS beads are excluded from the forbidden zone. 10 μm PS tracer particles were used to clearly show the forbidden zone phenomenon (Fig. 3C). During this process, the nanomotors can agglomerate. The more nanomotors produce a higher concentration of zinc ions, the larger the area of the zinc ion gradient field, and the larger the forbidden zone area. The size of the forbidden zone is related to the zinc ion distribution as the zinc ion gradient providing the electric field drives the electroosmotic flow and thus the particle motion. Therefore, the size of the forbidden zone is proportional to the size of the zinc ion distributed area (ESI Video S9†).32 Fig. S7† shows the complete process of the nanomotor's first attractive and then repulsive interactions with external passive particles. Firstly, the nanomotor is in motion, and an aPS bead is adsorbed on the nanomotor surface. Then the nanomotor approaches and stops on the negatively charged substrate. The aPS beads fall off and are quickly repelled from the nanomotor. The key determining the state of the Au–Zn nanomotor is the distance from the substrate, and the direction of the electroosmotic flow around the nanomotor to change from inward to outward, resulting in a repulsive mode of interaction with the passive particles.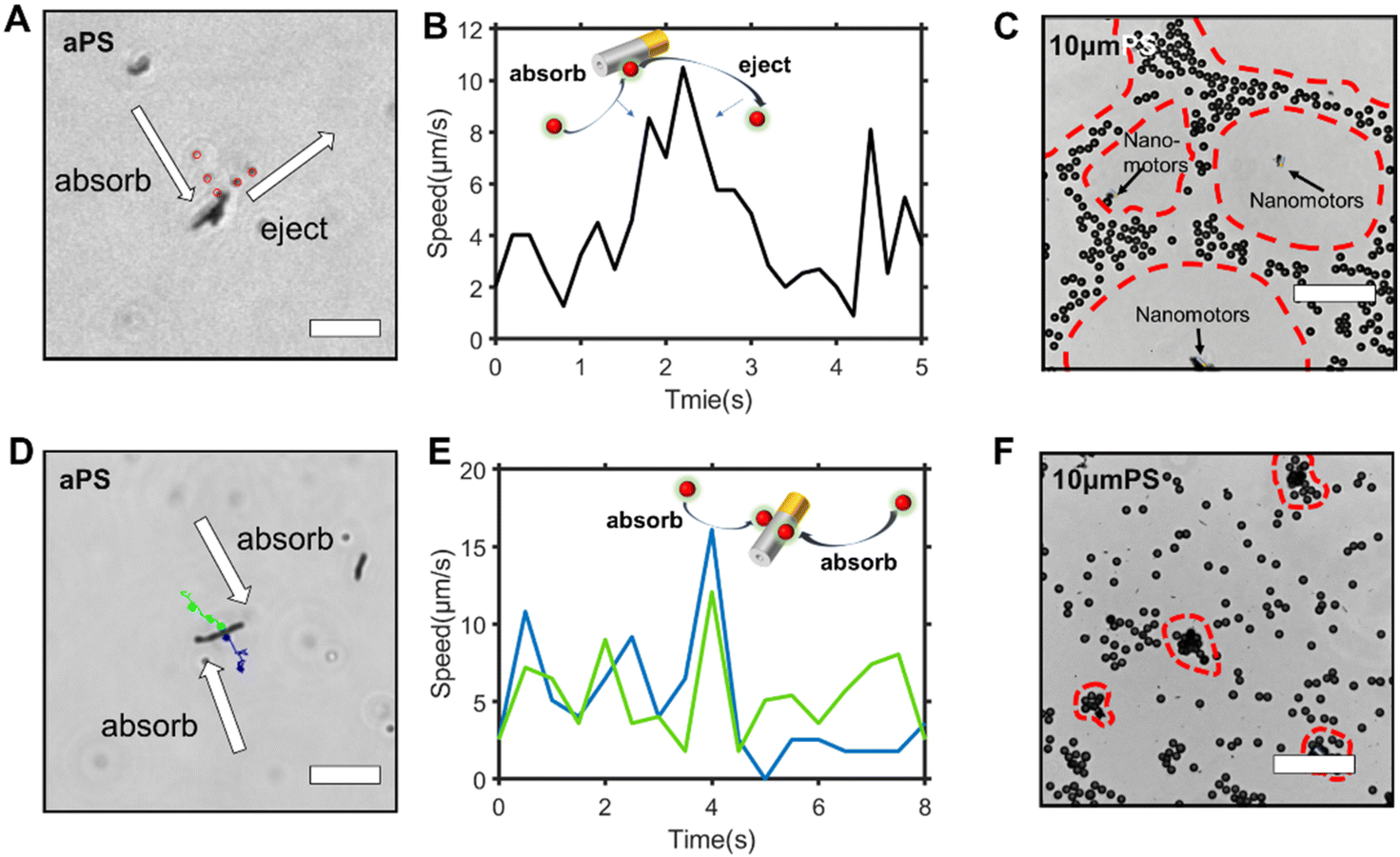 | ||
| Fig. 3 Adaptive repulsive–attractive interactions between the Au–Zn nanomotor and PS beads. (A) The Au–Zn nanomotor stopped on the negatively charged substrate; the amine PS beads were first adsorbed and then ejected out. Scale bars: 10 μm. (B) The speed diagram of amine PS tracer particles given in (A). (C) The emergence of collective patterns of 10 μm PS in the presence of Au–Zn nanomotors on the negative substrate. Scale bars: 100 μm. (D) The trajectory diagram of amine PS beads on positively charged substrates adsorbed by Au–Zn nanomotors. Scale bars: 10 μm. (E) The migration speed of amine PS beads adsorbed by Au–Zn nanomotors on positively charged substrates. (F) The emergence of a swarm cluster of the Au–Zn nanomotor and 10 μm PS on the positive substrate. Scale bars: 100 μm. | ||
To verify that the attraction/repulsion of the nanomotor is determined by the substrate, we replaced the negatively charged substrate with a positively charged glass substrate (CITOTEST 188105W) with OH− adsorbed on surface. With the self-built Zn2+ electric field of the nanomotor constant, the reversal of the zeta potential of the substrate led to the electroosmotic flow direction changing from outward repulsion to inward attraction between the nanomotor and the substrate. As observed in Fig. 3D, the Au–Zn nanomotor was attracted to the glass slide due to the electrostatic force and two aPS beads were adsorbed at the middle of the nanomotor (ESI Video S10†). With 10 μm PS beads, the forbidden area did not appear. 10 μm PS beads and nanomotors formed clusters or scattered around. These results are in line with our expectations. When the inward electroosmotic flow becomes dominant, the interaction between the nanomotor and the passive particles becomes attractive (ESI Video S11†). With differently charged substrates, the interaction mode with the passive particles is adaptive. Intracellular components aggregate into condensates at the right time as well as in space to perform their corresponding functions. The transport of substances within the cells can be achieved by active processes based on non-specific, purely physical mechanisms, a phenomenon that is still poorly studied, and the formation of controlled patterning driven by Au–Zn nanomotors is expected to shed new light on how membrane-free condensates are formed and their physicochemical nature.
Conclusions
For our Au–Zn nanomotor, the inert Au segments do not undergo a chemical reaction during the propulsion, therefore the nanomotor is driven via diffusiophoresis instead of self-electrophoresis. The propulsion force for the nanomotor motion is obtained using eqn (5). The macroscopic zinc does not react with DI water. With the size of zinc being reduced to the nanoscale, the specific surface area of the zinc increases, and the reaction of zinc with hydrogen can occur. This reaction is mild and can allow our nanomotor to work continuously for 4–5 days.The Au–Zn nanomotor adsorbs hydrogen ions on the surface of the zinc segment and releases zinc ions. The diffusion rate of hydrogen ions is much larger than that of Zn2+ (DH+ = 9.31 × 10−9, DZn2+ = 0.7 × 10−9). When the reaction is carried out in air-perfused water and H+ continuously consumed, the following equilibrium reaction (eqn (6)) will occur to maintain constant hydrogen ion concentration:
| H2O + CO2 ↔ H+ + HCO3− | (5) |
| H2O ↔ H+ + OH− | (6) |
In addition, the reaction of the nanomotor with DI water is very slow, so the hydrogen ion concentration around the nanomotor is assumed to remain constant. The initial concentration of zinc ions in DI water is 0, the nanomotor continuously releases zinc ions, and an outwardly decreasing Zn2+ concentration area is formed around the nanomotor, thus generating a net outward electric field. The zinc ion flux on the surface of the nanomotor is calculated using a simple chemical reaction rate formula, and the result is 1.6 × 10−6 mol m−2 s−1 (ESI†). COMSOL was used to simulate the self-built electric field formed by the outer diffusion of zinc ions (Fig. 4A). The simulated electric field diagram shows the electric field formed by zinc ions points toward the outward direction.
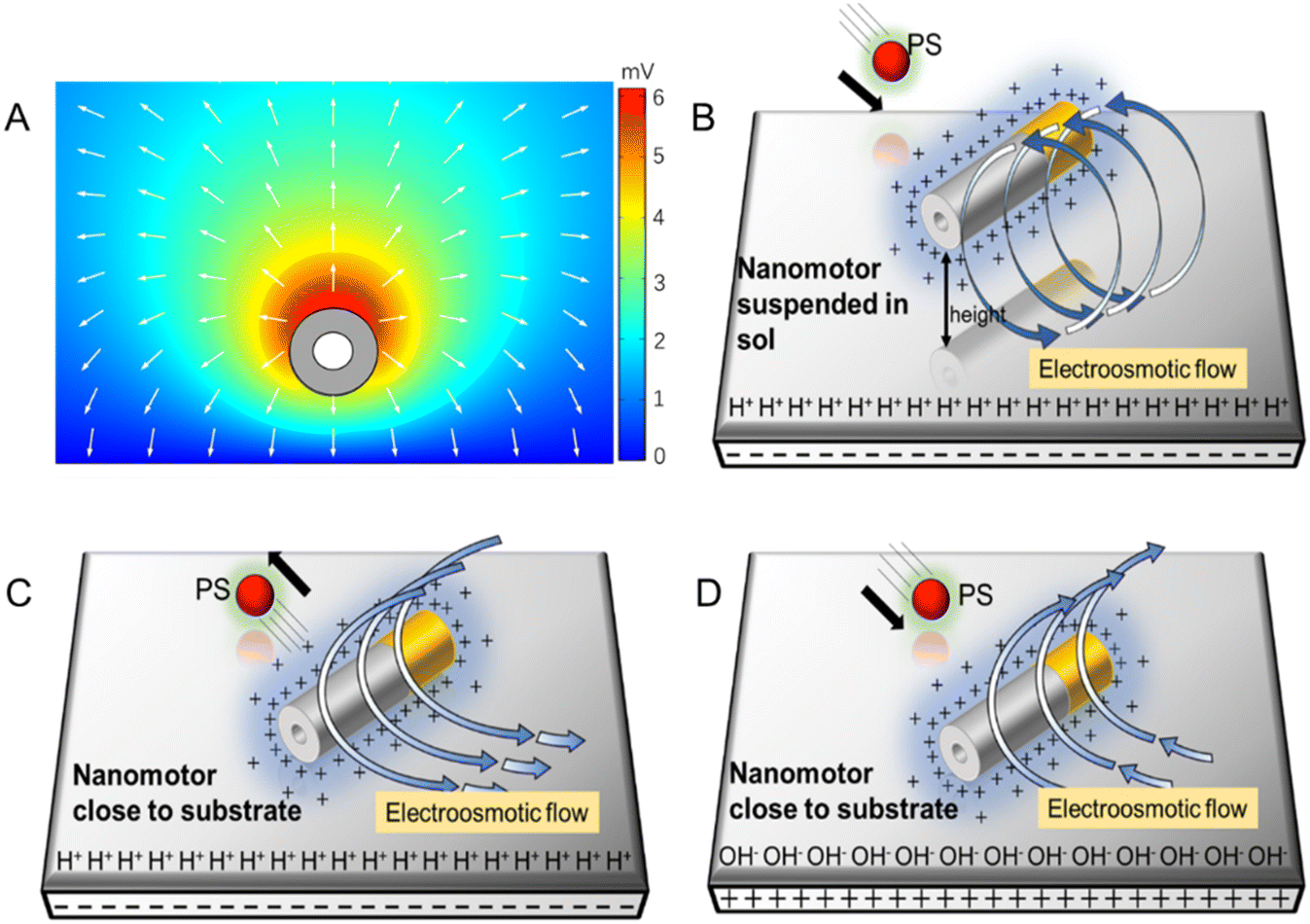 | ||
| Fig. 4 Schematic demonstration of adaptive repulsive–attractive interactions between the Au–Zn nanomotor and PS beads. (A) Electric potential diagram simulated by COMSOL (V stands for the voltage unit). (B) Schematic diagram of the electroosmotic flow formed by a nanomotor suspended in solution, away from a negatively charged substrate. The Zn2+-induced electric field repels the positively charged protons adsorbed on the negatively charged substrate, thus creating an outward electroosmotic flow from the nanomotor to the substrate. (C) Schematic diagram of the electroosmotic flow formed by a nanomotor; the Zn2+ electric field repels the protons adsorbed on the negatively charged substrate, creating an outward flow from the nanomotor to the substrate. (D) Schematic diagram of the electroosmotic flow formed by a nanomotor; the Zn2+-induced electric field attracts the negatively charged hydroxide adsorbed on the positively charged substrate, thus creating an inward electroosmotic flow from the substrate to the nanomotor. The symbol (+) near the Au–Zn nanomotor represents the zinc ion. | ||
The propulsion mechanism of diffusiophoresis can be expressed using eqn (7):33
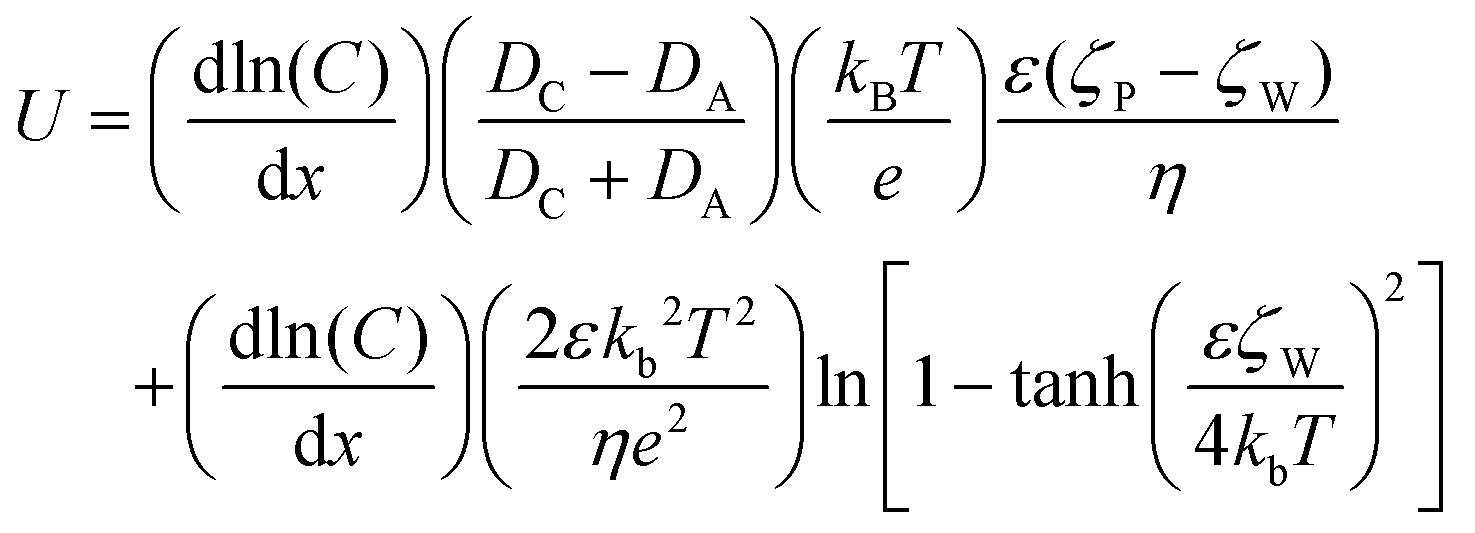 | (7) |
The formula expresses the velocity of the diffusiophoresis particles close to the wall. U is the particle speed, C is the electrolyte concentration, DCDA is the diffusion coefficient of cations and anions, kB is Boltzmann's constant, T is the temperature, e is the elementary charge, ε is the dielectric constant of the solution, η is the viscosity of the solution, ζW is the zeta potential of the wall, and ζP is the zeta potential of the particle. The first term of eqn (3) represents part of the electric field. In our Au–Zn nanomotor only Zn2+ are produced on the motor surface, so DA = 0. The second part is the thickness of the EDL, because the thickness of the EDL on the wall is related to the electrolyte concentration, which causes the fluid to flow from the area with a higher electrolyte concentration to the area with a lower electrolyte concentration.
Next, we analyzed the reason why the nanomotor has different interaction modes with passive particles in different positions and under different glass substrates. The zeta potential of the glass slide substrate where the nanomotor is located is negative (ζW = −85 mV), so there are protons inside the electrical double layer of the slider. The electric field of the nanomotor acts on the protons, thereby generating an electroosmotic flow outward along the substrate. Since the fluid is continuous, an inward electroosmotic flow is generated from the top. When the nanomotor is suspended on the substrate, the inward flow field dominates near the nanomotor (Fig. 4B). The adsorbed PS beads are located at the equilibrium position of the slip flow on the surface of the Au–Zn nanomotor and are unaffected by the electroosmotic flow below. Therefore, the nanomotor has the function of capturing passive particles when moving. When the nanomotor pauses on the substrate, the outward flow field dominates (Fig. 4C). The nanomotor becomes capable of repelling nearby substances, that is, it serves a nanopump. After continuous attraction–repulsion interactions, a forbidden zone is formed around the Au–Zn nanomotor.
When the substrate is changed into a positively charged glass slide, the EDL of the glass slide adsorbs the negatively charged OH−, and the electric field of the nanomotor does not change, so it will attract OH− and form an electroosmotic flow inward along the glass slide (Fig. 4D). In addition, because the nanomotor is negatively charged, the nanomotor and the substrate are electrostatically attracted, so if a positively charged substrate is used, there is no such situation that the nanomotor is suspended on the substrate.34
Protein space self-organization is the key to cell metabolism and self-replication. The most representative system is E. coli MinCDE, which has become a model for studying reaction–diffusion systems. However, there is still no reliable explanation for the pattern of protein self-organization in the model. We think the above-mentioned experimental observations can be used to explain the molecular mechanism of action. After the MinD dimer in the system is attached to the cell membrane, it has the ability to recruitproteins in the solution. This is similar to this experiment, i.e., when the nanomotor is suspended on a positively charged substrate, it will attract PS beads to form clusters. The change in the environment allows the nanomotor to acquire the ability to adsorb the attached PS beads. In addition, MinDE can reject membrane proteins to form large-scale protein gradients. This is similar to this experiment, where the motor is suspended on the negatively charged substrate to repel PS beads to form a hole pattern. We believe that our system will explain the basic interaction principle of protein self-organization systems such as MinCDE to form self-organized protein patterns.
Life is known to be based on entities joining together to create a large-scale structure. If self-propulsion or active diffusiophoresis did not occur, organisms would be limited to slow passive processes to move deoxyribonucleic acids and proteins within cells or tissues, and many of life's complex structures and functions might never emerge.35 Au–Zn nanomotors that are self-propelled and can switch between two modes of interaction, attraction and repulsion, with particles in the environment are similar to the diffusion–antagonism concept that formed the Turing patch. The interaction is not limited to passive particles, but can also be with another active material or even a biological entity. The Au–Zn nanomotor platform is expected to shed new light on the organizing behavior of the active material in biological evolution, the formation of condensates and cyto component clusters.
In summary, we have demonstrated an interesting Au–Zn nanomotor system driven via diffusiophoresis: in addition to accomplishing quasi-stable complex formation with passive particles, the system forms patterns in response to the position and environment zeta potential. Away from the substrate (environment wall), pattern formation is not favored, while with the positively charged substrate a cytoskeleton-like pattern is formed and with the negatively charged substrate a cluster pattern is formed. Such nanomotor-driven diffusiophoresis and the ability to pattern matter in a controllable manner (or phase separation) could be associated with living systems. The spatiotemporal organization of material condensates within the cell is crucial for function maintenance. Besides cargo transport via specific protein interactions, condensate arrangement and general active transport within the cell can be achieved via adaptive active diffusiophoresis. Previous studies in a biologically relevant context suggested that a self-organizing protein pattern occurs in the process of MinD-mediated protein transport in bacteria. During condensate assembly via liquid–liquid phase separation, membranes modulate the biogenesis and dynamics of phase-separated condensates by serving as assembly platforms or by forming direct contacts. For our system, the diffusiophoresis involving particle fluxes and self-organization in response to the substrate/environment wall suggests that a flux-centered viewpoint will be appropriate for general transport and multicomponent mixtures out of equilibrium. Thus, it could be particularly important to investigate self-driven organization and macroscopic patterns. Simple as it is, this mechanism could be interpreted as an alternative, rudimentary chem–physical interaction mode and as such might be present and prevalent in early forms of life.
Author contributions
D. Xie is responsible for experiments and article writing; D. Fu and S. Fu provided theoretical support; W. He and B. Chen provided experimental equipment; D. A. Wilson and F. Peng are instructors who provided theoretical support and article writing suggestions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (grant no. 21805318, 51973241 and 22175083) and the Guangdong Provincial Science Foundation for Distinguished Young Scholars (grant no. 2018B030306007).References
- B. Ramm, P. Glock, J. Mücksch, P. Blumhardt, D. A. García-Soriano, M. Heymann and P. Schwille, Nat. Commun., 2018, 9, 3942 CrossRef PubMed.
- W. Bialek, A. Cavagna, I. Giardina, T. Mora, E. Silvestri, M. Viale and A. M. Walczak, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 4786–4791 CrossRef CAS PubMed.
- J. K. Parrish and L. Edelstein-Keshet, Science, 1999, 284, 99–101 CrossRef CAS PubMed.
- C. P. Brangwynne, C. R. Eckmann, D. S. Courson, A. Rybarska, C. Hoege, J. Gharakhani, F. Julicher and A. A. Hyman, Science, 2009, 324, 1729–1732 CrossRef CAS PubMed.
- A. Bricard, J.-B. Caussin, N. Desreumaux, O. Dauchot and D. Bartolo, Nature, 2013, 503, 95–98 CrossRef CAS PubMed.
- T. Sanchez, D. T. N. Chen, S. J. DeCamp, M. Heymann and Z. Dogic, Nature, 2012, 491, 431–434 CrossRef CAS PubMed.
- J. Yu, B. Wang, X. Du, Q. Wang and L. Zhang, Nat. Commun., 2018, 9, 3260 CrossRef PubMed.
- M. Rubenstein, A. Cornejo and R. Nagpal, Science, 2014, 345, 795–799 CrossRef CAS PubMed.
- D. P. Singh, U. Choudhury, P. Fischer and A. G. Mark, Adv. Mater., 2017, 29, 7 Search PubMed.
- W. F. Paxton, K. C. Kistler, C. C. Olmeda, A. Sen, S. K. St Angelo, Y. Y. Cao, T. E. Mallouk, P. E. Lammert and V. H. Crespi, J. Am. Chem. Soc., 2004, 126, 13424–13431 CrossRef CAS PubMed.
- B. Chen, L. Liu, K. Liu, F. Tong, S. H. Wang, D. M. Fu, J. B. Gao, J. M. Jiang, J. F. Ou, Y. C. Ye, D. A. Wilson, Y. F. Tu and F. Peng, Adv. Funct. Mater., 2021, 31, 11 Search PubMed.
- F. Peng, Y. Tu and D. A. Wilson, Chem. Soc. Rev., 2017, 46, 5289–5310 RSC.
- Z. Wang, S. H. Wang, K. Liu, D. M. Fu, Y. C. Ye, J. B. Gao, L. Liu, D. A. Wilson, Y. F. Tu and F. Peng, Appl. Mater. Today, 2020, 21, 7 Search PubMed.
- J. Orozco, V. Garcia-Gradilla, M. D'Agostino, W. Gao, A. Cortes and J. Wang, ACS Nano, 2013, 7, 818–824 CrossRef CAS PubMed.
- C. R. Chen, F. Z. Mou, L. L. Xu, S. F. Wang, J. G. Guan, Z. P. Feng, Q. W. Wang, L. Kong, W. Li, J. Wang and Q. J. Zhang, Adv. Mater., 2017, 29, 1603374 CrossRef PubMed.
- W. Gao, S. Sattayasamitsathit, J. Orozco and J. Wang, J. Am. Chem. Soc., 2011, 133, 11862–11864 CrossRef CAS PubMed.
- W. Wang, W. T. Duan, S. Ahmed, A. Sen and T. E. Mallouk, Acc. Chem. Res., 2015, 48, 1938–1946 CrossRef CAS PubMed.
- F. Z. Mou, X. F. Li, Q. Xie, J. H. Zhang, K. Xiong, L. L. Xu and J. G. Guan, ACS Nano, 2020, 14, 406–414 CrossRef CAS PubMed.
- C. Wu, J. Dai, X. Li, L. Gao, J. Wang, J. Liu, J. Zheng, X. Zhan, J. Chen, X. Cheng, M. Yang and J. Tang, Nat. Nanotechnol., 2021, 16, 288–295 CrossRef CAS PubMed.
- J. Deseigne, O. Dauchot and H. Chate, Phys. Rev. Lett., 2010, 105, 098001 CrossRef PubMed.
- A. Merino-Salomon, L. Babl and P. Schwille, Curr. Opin. Cell Biol., 2021, 72, 106–115 CrossRef CAS PubMed.
- Q. H. Cui, T. H. Le, Y. J. Lin, Y. B. Miao, I. T. Sung, W. B. Tsai, H. Y. Chan, Z. H. Lin and H. W. Sung, Nano Energy, 2019, 66, 104120 CrossRef CAS.
- R. Mundaca-Uribe, B. E. F. de Avila, M. Holay, P. L. Venugopalan, B. Nguyen, J. R. Zhou, A. Abbas, R. H. Fang, L. F. Zhang and J. Wang, Adv. Healthcare Mater., 2020, 9, 2000900 CrossRef CAS PubMed.
- Z. H. Lin, C. Y. Gao, D. L. Wang and Q. He, Angew. Chem., Int. Ed., 2021, 60, 8750–8754 CrossRef CAS PubMed.
- D. Velegol, A. Garg, R. Guha, A. Kar and M. Kumar, Soft Matter, 2016, 12, 4686–4703 RSC.
- X. Chen, C. Zhou and W. Wang, Chem. – Asian J., 2019, 14, 2388–2405 CrossRef CAS PubMed.
- J. L. Moran and J. D. Posner, Annu. Rev. Fluid Mech., 2017, 49, 511–540 CrossRef.
- Y. Gu and D. Li, J. Colloid Interface Sci., 2000, 226, 328–339 CrossRef CAS.
- D. Takagi, J. Palacci, A. B. Braunschweig, M. J. Shelley and J. Zhang, Soft Matter, 2014, 10, 1784–1789 RSC.
- C. Zhou, H. Zhang, Z. H. Li and W. Wang, Lab Chip, 2016, 16, 1797–1811 RSC.
- W. T. Duan, R. Liu and A. Sen, J. Am. Chem. Soc., 2013, 135, 1280–1283 CrossRef CAS PubMed.
- X. Wang, L. Baraban, V. R. Misko, F. Nori, T. Huang, G. Cuniberti, J. Fassbender and D. Makarov, Small, 2018, 14, 1802537 CrossRef PubMed.
- J. L. Anderson, Annu. Rev. Fluid Mech., 1989, 21, 61–99 CrossRef.
- T. Huang, B. Ibarlucea, A. Caspari, A. Synytska, G. Cuniberti, J. de Graaf and L. Baraban, Eur. Phys. J. E, 2021, 44, 39 CrossRef CAS PubMed.
- G. Popkin, Nature, 2016, 529, 16–18 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3nr00462g |
| This journal is © The Royal Society of Chemistry 2023 |