
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An ionic liquid synthesis route for mixed-phase sodium titanate (Na2Ti3O7 and Na2Ti6O13) rods as an anode for sodium-ion batteries
Pooja
Kumari
,
Yining
Li
and
Rebecca
Boston
 *
*
Department of Materials Science and Engineering, University of Sheffield, S1 3JD, Sheffield, UK. E-mail: R.boston@sheffield.ac.uk
First published on 13th June 2023
Abstract
Sodium ion batteries represent a sustainable alternative to Li-ion technologies. Challenges with material properties remain, however, particularly with regards the performance of anodes. We report a rapid, energy-efficient ionic liquid synthesis method for mixed phase Na2Ti3O7 and Na2Ti6O13 rods. This method is based on a novel phase-transfer route which produces pure functional materials via a dehydrated IL. The structure of the synthesised materials was characterised using powder X-ray diffraction, which confirms the formation of a mixed Na2Ti3O7 and Na2Ti6O13 phase, with majority Na2Ti3O7 phase, in contrast to previous synthesis methods. Scanning and transmission electron microscopy analysis reveals a rod morphology, with an average diameter and length of 87 nm ± 3 nm and 1.37 μm ± 0.07 μm, respectively. The initial discharge and charge capacity of Na2Ti3O7 nanorods were measured as 325.20 mA h g−1 and 149.07 mA h g−1, respectively, at 10 mA g−1 between 0.01–2.5 V. We attribute the enhanced performance to the higher weight fraction of Na2Ti3O7 phase vs. previous reports, demonstrating the potential of the ionic liquid method when applied to sodium titanate materials.
Introduction
Lithium-ion batteries (LIBs) are likely to be unable to meet the large-scale demands for global energy storage, due to limited Li reserves.1,2 Sodium-ion batteries (SIBs) are regarded as a potential alternative to LIBs for a range of large-scale energy storage applications, due to a higher abundance of constituent materials,3 price4 and safety.5 Some of the key challenges which currently restrict implementation of Na-ion technologies stem from the larger radius of sodium ions (1.02 Å) vs. Li (0.76 Å), alongside the need for reliable synthesis methods which produce nanostructured materials through energy-efficient means. In order to optimise electrochemical performance and make Na-ion a genuine competitor for Li-ion, the choice of electrode material and structural optimisation are key areas for focus.6 Nanostructured materials could provide shorter electron and ion transport path7 and provide a larger surface area to enhance the number of reaction sites,8 and therefore represents a viable pathway to improve battery performance.9 Cathode materials have been an area of intense research,10–13 however the materials for anodes have had less attention, particularly with respect to synthesis method.Of the many materials under consideration for anodes, the sodium titanates in the Na2TinO2n+1 (2 ≤ n ≤ 9) family have been highlighted as a promising candidates.14–16 Recently the Na2Ti6O13 and Na2Ti3O7 compositions have attracted attention due to their abundance, low toxicity, low cost, and low voltage plateaux.4,14,17 Na2Ti6O13 has a high ionic conductivity and excellent cycling stability (c. 78% after 3000 cycles15) at high rate because of its tunnel structure and large interlayer spacing, which is able to accommodate the volume expansion (c. 1%) upon Na+ (de)insertion.17–19 Na2Ti6O13, however, has a low theoretical capacity of 49.5 mA h g−1 (for one Na+ ion (de)insertion, at a current density of 1C), which restricts the practical application of Na2Ti6O13 as an anode.15 In contrast, Na2Ti3O7 can uptake 2 Na+ ions per formula unit (Na2Ti3O7 + 2Na+ + 2e− → Na4Ti3O7) at a low average voltage of 0.3 V vs. Na+/Na,20 leading to a high theoretical capacity of 178 mA h g−1 (at a current density of 1C).17,18 This is attributed to the zigzag layered structure consisting of edge-linked TiO6 octahedra, with Na+ ions able to insert in the interlayer space (as large as ca. 8 Å).21 The low (de)insertion potential decreases the likelihood of Na dendrites/mossy structures forming during cycling and offers a high energy density with a high and safe operating potential window.15 This composition, however, suffers from poor cycling stability caused by structural distortion during cycling and poor electronic conductivity resulting from a large bandgap (3.7 eV).22 Neither Na2Ti3O7 nor Na2Ti6O13, therefore, exhibit properties to make them viable anode candidates in isolation.
To improve the electrochemical performance of Na2Ti3O7 in terms of rate capability and cycling stability, various approaches have been undertaken, such as nanostructured electrode fabrication (to reduce the diffusion length of Na+ ions) with different morphologies.14,17,23,24 Also, carbon coating has been used as conducting additive and/or as an elastic buffer.25,26 Previous research has demonstrated that a mixed phase of nanocomposite Na2Ti3O7 and Na2Ti6O13 can provide excellent electrochemical performance, by combining the advantages of both the polymorphs (high capacity of Na2Ti3O7 and the long-term cyclability and high rate performance of Na2Ti6O13).18,19,23,27 This synergistic effect has been previously attributed to the two different types of reactions occurring during cycling. Na2Ti3O7 was shown to undergo a phase transition, concurrent with the formation of a solid solution in the Na2Ti6O13 phase, acting to offset the disadvantageous behaviour of each of the phases independently.28 These previous studies still suffer with low reversible specific capacity and cycling stability. There are a number of possible causes for this, including morphology (linked to synthesis method), or different phase fractions of the polymorphs. For example, Wang et al. reported that a hydrothermally synthesised mixture Na2Ti3O7 and Na2Ti6O13 nanotube array (chemically engraved on metallic Ti foil) offers enhanced stability,27 although the specific capacity was only 80 mA h g−1 for 200 cycles at 400 mA g−1, and these nanotubes were poorly crystalline and contained impurities. Similarly, Cech et al. reported that a mixed sodium titanate phase of Na2Ti3O7 (42%) and Na2Ti6O13 (58%) nanorods synthesised using a microwave-assisted hydrothermal method, resulted in better cycling stability than the single phase of Na2Ti3O7 and Na2Ti6O13. The measured initial discharge capacity was quite low (90 mA h g−1 at C/50),29 however, layered Na2Ti3O7 (26.22%) and tunnel Na2Ti6O13 (73.78%) microrods, synthesised by a hydrothermal method, showed excellent cycling performance and rate capability in addition to high capacities (initial discharge and charge capacity of 212.52 and 122.23 mA h g−1, respectively) at 20 mA g−1.23 Furthermore, Wu et al. mentioned that the electrochemical behaviour of mixed NTO anode in terms of reversible capacity and cycling stability could be related to the layered percent.23 Interestingly, in all of these studies, Na2Ti6O13 phase is present as the majority phase, and is known to exhibit lower capacity vs. Na2Ti3O7. Hence, increasing the fraction of Na2Ti3O7 may therefore be a viable method to further increase capacity.
Previous studies into the preparation of the mixed phases have examined a multitude of synthesis processes. In one such study, solid state synthesis has been used to produce NTO nanorods via the addition of Vulcan carbon (0–90 wt%), and these nanorods exhibited a high specific capacity (0–0.2 V range) and excellent rate capability depending on added wt% of Vulcan carbon and quantity of Na2Ti6O13 in the sample.18 Therein, Ho et al. reported the role of carbon in synthesising the mix phase of NTO (a higher wt% of carbon resulted in higher wt% of Na2Ti6O13) on morphology and electrochemical performance (143 mA h g−1 (0 wt%) to 170 mA h g−1 (90 wt%) at 0.2C). Similarly, in a solvothermal synthesis method, Chandel et al. also used Cetyltrimethylammonium Bromide (CTAB) as a carbon source to fabricate the Na2Ti3O7/Na2Ti6O13 (Na2Ti3O7: 18%, Na2Ti6O13: 82%) nanocomposite, where the involved reactions between CO2 (produced due to the presence of CTAB) and Na2Ti3O7 help to form another polymorph of Na2Ti6O13.17 The synthesised NTO nanocomposite with carbon delivered a high reversible capacity (161 mA h g−1) and excellent rate capability.17 Also, Hwang et al. reported a carbon coated (sucrose octaacetate as carbon precursor) mixed phase of NTO microrods, prepared using supercritical methanol followed by a subsequent heat treatment also demonstrated excellent cycling stability and high rate performance.14 Whilst relatively successful, these composite approaches generally require a long (24–48 h) and/or sophisticated synthesis processes such as hydrothermal,23,27 or solid state reaction18 methods, and in all of these, the fraction of Na2Ti3O7 is low. There is a need, therefore, to develop simple reaction methods to produce NTO composite materials, which also generate the opportunity to increase the proportion of Na2Ti3O7 phase.
One such method is an ionic liquid-based synthesis, which has been demonstrated as a non-aqueous route to produce nanoscale oxide materials. Green et al.30 showed that ionic liquids such as 1-ethyl 3-methylimidazolium acetate could be used as a solvent for cellulose, thereby accessing the morphological directing properties of this as a biological template. They successfully synthesised a range of nanoscale complex oxides using this technique, including sodium potassium niobate, and importantly, demonstrated that aqueous solutions of precursor metal salts could be combined with alkoxide precursors (with an intermediate heating step to drive off water for the aqueous salts), without the detrimental and often violent formation of binary oxide materials. More recently, Mottram et al.31 showed that the addition of cellulose to this reaction was not needed, by showing that nanoscale templating in La-doped strontium titanate could be achieved using the ionic liquid alone, further simplifying the reaction mixture. Thus this non-aqueous synthesis route shows substantial promise for use in the synthesis of nanoscale titanate materials.
Herein, we synthesise mixed phase NTO via the simple ionic liquid method as set out by Green et al., producing materials using less energy vs. solid state and without using an additional carbon source (during the synthesis process, as has been often used previously) whilst maintaining good battery performance as discharge and charge-specific capacities of 325.20 mA h g−1 and 149.07 mA h g−1, respectively, in the voltage window of 0.01–2.5 V at 10 mA g−1 (1C = 178 mA g−1).
Experimental
Synthesis
All chemicals were obtained from Sigma-Aldrich (U.K.) and used without further purification. Initially, 5 mL of a 0.4 M aqueous solution of sodium acetate was prepared and added to 5 mL of the ionic liquid 1-ethyl-3-methylimidazolium acetate. The mixture was heated to 90 °C under stirring for 2 h until all of the water had evaporated, forming a thick gel. A stoichiometric volume of titanium isopropoxide (C12H28O4Ti) was then added under vigorous stirring. This produced a pale yellow gel which was calcined in a chamber furnace in air at 800 °C for 12 h, heated at a rate of 5 °C min−1 to fabricate the NTO powder. The resulting white powder was ground by hand ready for further characterisation.X-ray diffraction (XRD) was performed using a PANalytical Aeris powder diffractometer with Ni-filtered Cu Kα radiation (λ = 1.5406), to observe the crystal structure and purity of the calcined sample. Phase analysis for the samples was performed using High Score (PANalytical) software, and Rietveld refinements were conducted using GSAS. A shifted Chebyshev function with 15 terms was required to fit the background.
The morphology of the as-synthesised powders was observed using scanning electron microscopy using an Inspect F50 SEM (FEI/Thermoscientific). Samples were prepared by using affixing powder on carbon tape and sputter-coated with a thin layer of gold for imaging. Energy dispersive X-ray analysis (spectra and mapping) was also collected on the Inspect F50 SEM, using an Oxford Instruments EDS detector (at 15 kV accelerating voltage) samples were coated with carbon for these measurements. Image J software was used to perform SEM analysis and with rod sizes calculated from a minimum of 500 rods, measured on both the long and short axis. An average on each dimension was taken, and the errors quoted in the manuscript are one standard deviation calculated from these datasets.
Powders were further examined by transmission electron microscopy (JEOL JEM-F200 TEM) at 200 kV, for both imaging and selected area electron diffraction. Samples were prepared by suspension in ethanol and then drop-cast onto a holey carbon-coated Cu grid.
Raman spectroscopy was collected using a Renishaw inVia Raman microscope. The measurement was carried out with a 514 nm Ar laser (green laser), and the laser power used was 10%. Three regions were selected for each sample, to avoid non-representative data caused by the uneven distribution of NTOs.
Electrochemistry
The negative electrode (anode) slurry was prepared by mixing 70 wt% active material (NTO powder), 20 wt% carbon black (Timical Super C65, MTI), as a conductive agent and 10 wt% binder (Polyvinylidene fluoride PVDF, MTI). A Thinky ARE-250 Mixer was used to prepare the slurry. A sufficient volume of N-methyl-2-pyrrolidone, NMP (anhydrous 99.5%, Sigma-Aldrich) was used as a solvent to dissolve the PVDF and form the slurry. This was then uniformly coated (thickness c. 250 μm) onto carbon-coated aluminium foil using a micrometre adaptable doctor blade followed by drying under vacuum at 80 °C for 24 h. The dried slurry was then pressed using a rolling mill (MTI, MSK-HRP-MR100DC), to ensure close contact between the current collector and the anode material. The coated foil was cut into disks with a diameter of 12 mm (with an area of 1.13 cm2) before being used as an electrode in the battery. All the battery testing was performed on 2032-type coin cells. The half-cell was assembled in an Ar-filled glove box (MBraun, O2 & H2O < 0.5 ppm) using the as-prepared anode as the working electrode, GF/F Whatman glass microfiber filter as a separator, and sodium metal (sodium ingot, 99.8%, Alfa Aesar) as the counter electrode. 1 M NaPF6, dissolved in ethylene carbonate (EC) and diethyl carbonate (DEC) solvent (volume ratio of EC![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DEC = 1:1) was used as an electrolyte. All the electrochemical measurements were performed at different current densities with a cut-off voltage window of 0.01–2.5 V (vs. Na+/Na) in a Maccor Series 4000 battery cycler at room temperature. A Maccor series 4300 battery cycler was used to conduct Cyclic Voltammetry (CV) measurements between cut-off voltages of 0.0 V and 3.0 V (vs. Na+/Na) at a scan rate of 0.08 mV s−1. No deduction of the contribution of carbon black (conductive agent) was made when calculating the specific capacity of sodium titanate anode material.
:DEC = 1:1) was used as an electrolyte. All the electrochemical measurements were performed at different current densities with a cut-off voltage window of 0.01–2.5 V (vs. Na+/Na) in a Maccor Series 4000 battery cycler at room temperature. A Maccor series 4300 battery cycler was used to conduct Cyclic Voltammetry (CV) measurements between cut-off voltages of 0.0 V and 3.0 V (vs. Na+/Na) at a scan rate of 0.08 mV s−1. No deduction of the contribution of carbon black (conductive agent) was made when calculating the specific capacity of sodium titanate anode material.
Results and discussion
The crystal structure and phase mixture of the synthesised NTO was characterised by powder XRD. Fig. 1(a) shows that the product is highly crystalline, and the phases can be indexed to a mixed phase composition of monoclinic Na2Ti3O7 (P1 21/m 1; JCPDS no. 96-231-0332), monoclinic Na2Ti6O13 (C1 2/m 1; JCPDS no. 96-400-0749), and a cubic phase, likely to be NaCl, likely present as a contaminant in the ionic liquid. | ||
| Fig. 1 (a) XRD pattern of as-prepared Na2Ti3O7 (represented with a star symbol). The secondary phase is indicated using a filled diamond (Na2Ti6O13) with the NaCl impurity represented by a filled circle symbol. (b) Rietveld refinement of the sample, (black symbols experimental pattern; red line calculated pattern; blue line difference curve, with a reduced χ2 of 3.77. | ||
A preliminary Rietveld refinement (Fig. 1(b)) was used to determine lattice parameters and phase fraction. The Na2Ti3O7 lattice parameters were found to be a = 8.5637(3) Å, b = 3.8014(8) Å, c = 9.1283(3) Å, with α = 90°, β = 101.598°, γ = 90°, agreeing well with previously published results.32 The Na2Ti6O13 phase lattice parameters were also in agreement with previous research14,17 of a = 15.1546(4) Å, b = 3.7517(7) Å, c = 9.1420(8) Å, with α = 90°, β = 99.263°, γ = 90°. The lattice parameter of the cubic phase was found to be 5.660(2) Å which is in good agreement with the lattice parameter of NaCl, confirming the presence of this phase. Weight fractions were found to be 58% Na2Ti3O7, 28% Na2Ti6O13 and 14% NaCl. Previous research17,18 into composite NTO materials have predominantly focused on high Na2Ti6O13 fractions, and this demonstrates an advantage of using the ionic liquid method.
Raman spectra of both Na2Ti3O7 and Na2Ti6O13 are reported in the literature.7,33,34Fig. 2 shows the Raman spectrum of the NTO powder. The observed peaks (red labels in Fig. 2) agree well with reported Na2Ti3O7 data.33 XRD analysis revealed that Na2Ti6O13 was also present, and this is corroborated by the Raman, with the blue arrows matching with the typical wavenumbers for Na2Ti6O13.33
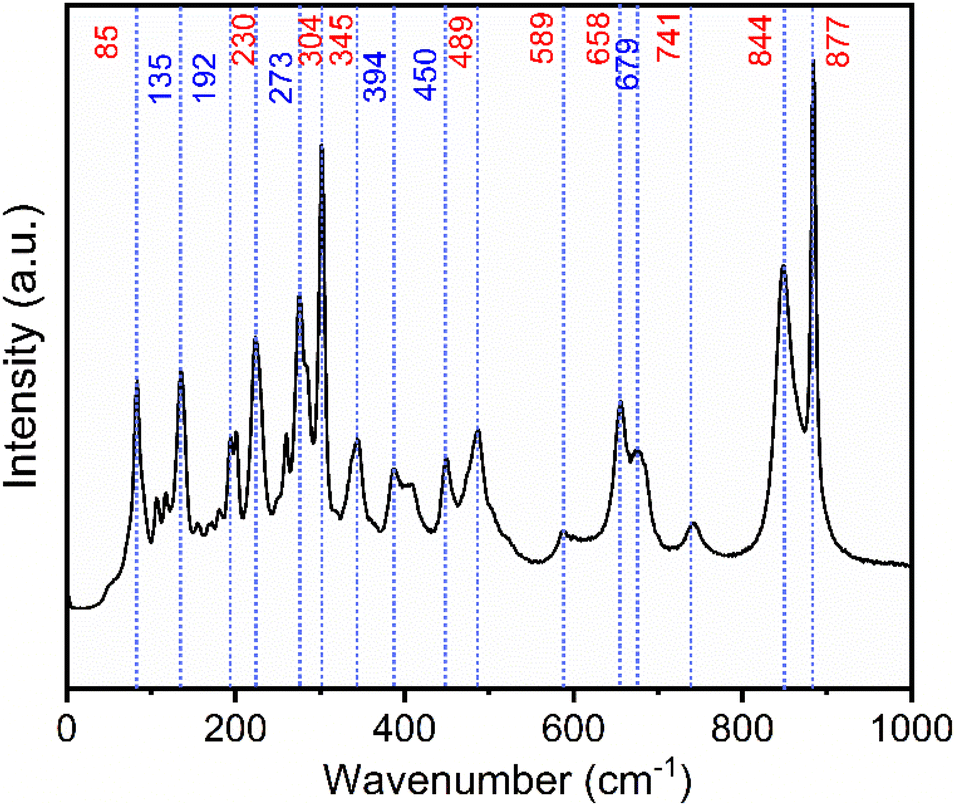 | ||
| Fig. 2 Raman spectrum of NTO sample, the red arrows correspond to the Na2Ti3O7 phase and blue arrows Na2Ti6O13. | ||
The electrochemical performance of the nanostructured electrodes is highly dependent on their surface area, particle size, crystal structure and morphology.35 Hence, the morphology of the synthesised samples was investigated by scanning electron microscopy (SEM) and transmission electron microscopy (TEM). As shown in Fig. 3(a), the SEM image demonstrates that the microstructure consists of rods, with an average diameter and length of 87 nm ± 3 nm and 1.37 μm ± 0.07 μm, respectively. There are some larger outliers on these values however. These microstructures were further investigated using transmission electron microscopy (TEM). The TEM image Fig. 3(b) shows a typical collection of rods. High-resolution TEM (HRTEM) and selected area electron diffraction (SAED) were used to confirm that the rods were single crystalline and exist in both Na2Ti3O7 and Na2Ti6O13 phases. Fig. 3(c) shows the HRTEM of a representative Na2Ti3O7 rod, along with the corresponding SAED pattern (Fig. 3(d)). The lattice fringes and SAED spot spacing are in good agreement with the (00l) direction along the length of the rod. This can be observed in the HRTEM as periodicity in the lattice fringes, with the (003) spacing also resolved (Fig. 3(c)). Similarly, the Na2Ti6O13 phase can also be observed in the rod morphology. Fig. 3(e) confirms this through the observable lattice fringes along the (020) direction, and corresponding SAED (Fig. 3(f)).
 | ||
| Fig. 3 (a) SEM image of the mixed phase rods, (b) low-resolution TEM image of the rods, (c) HRTEM image of a Na2Ti3O7 rod with inset low-resolution image and one to resolve lattice fringes, (d) corresponding SAED pattern, (e) HRTEM image of a Na2Ti6O13 rod with low-resolution inset image to resolve lattice fringes, (f) corresponding SAED pattern. | ||
The XRD in Fig. 1 indicates the presence of NaCl; this is supported by energy-dispersive X-ray analysis shown in Fig. 4, where EDX spectrum (Fig. 4b with corresponding image in (a)) and mapping (Fig. 4(c)–(g)) proves the presence of Cl in the sample. In Fig. 4(c)–(e), Na, Ti, and O are observed as major phases in the EDX maps. Cl and K are also present as in homogeneously distributed regions (Fig. 4(f) and (g)). The bright spot in Fig. 4(e), Na, is also present in the Cl map (Fig. 4f) indicating NaCl exists as a discrete phase, rather than in the rods themselves. The presence of carbon (Fig. 4(h) in the EDX spectrum is due to carbon coating. A small peak of K is also observed in the EDX spectrum that also could be present as a contaminant in the ionic liquid, again present as KCl (corroborated by the maps in Fig. 4(f), Cl, and Fig. 4(g), K), although this is present in too small a quantity to be observed using the X-ray diffraction. Whilst these impurity phases might represent a disadvantage, their removal should be possible, either through the use of a higher purity ionic liquid.
 | ||
| Fig. 4 (a) SEM image of the mixed phase rods, and ((b)) corresponding EDX spectrum. EDX elemental maps of (c) Ti, (d) O, (e) Na, (f) Cl, (g) K, and (h) C. According to the EDX mapping Na, Ti, and O are distributed uniformly and taking a major role in the synthesized product. However, Cl and K are also present in the sample as contaminant product that are coming from IL. | ||
From the chemical and physical characterisation, the ionic liquid method has a number of notable advantages vs. e.g. solid state and hydrothermal methods. The material processing time and number of processing steps are significantly reduced, and the synthesis itself is safe and straightforward to perform. The products are highly crystalline, in contrast to many of those prepared by e.g. hydrothermal methods, and show a higher weight fraction of Na2Ti3O7 phase than is generally reported in mixed phase samples, which should lead to higher specific capacity.
Electrochemical measurements
To study the electrochemical properties of the redox couples, cyclic voltammetry (CV) measurements were performed at 0.08 mV s−1 scan rate in a voltage range of 0 to 3.0 V. Fig. 5(a) shows the CV curves of the prepared mixed phase NTO electrode for the initial five cycles. For the first cathodic scan (Na+ ion insertion), the electrolyte reductive reaction started from c. 1.2 V, with a further strong reductive peak located at 0.35 V23,32 (only present in the first scan). This is assigned to the solid electrolyte interphase (SEI) formation on the surface of the electrode36 through electrolyte decomposition or side reactions of the electrolyte reduction.32,37 A reversible reductive peak at around 0.04 V which was maintained over up to five cycles is attributed to the Na+ ion insertion into the layered Na2Ti3O7 (Ti4+ reduced to Ti3+).32,37 | ||
| Fig. 5 Electrochemical performance of NTO electrodes (a) cyclic voltammogram curves in a voltage range of 0.0–3.0 V at a scanning rate of 0.08 mV s−1, (b) charge–discharge profiles in a voltage range of 0.01–2.5 V at current density of 10 mA g−1, (c) cycling performance of NTO electrode at current density of 10 mA g−1 (d) rate capability of NTO electrode. | ||
During the anodic scan, an oxidation peak at 0.33 V was observed with a shoulder peak at 0.44 V which corresponds to the extraction of Na+ from Na2Ti3O7.29,37 Furthermore, a redox pair at the high potentials of 0.8 V (cathodic scan) and 0.9 V (ref. 23) (anodic scan) was observed, which corresponds to Na+ ion insertion/extraction for Na2Ti6O13 phase.14,29 During subsequent cycles, the large anodic peak at 0.33 V gradually fades and the other peak at 0.44 V becomes more prominent. These two oxidation peaks have also been observed by Rudola et al. for pure Na2Ti3O7 electrodes when the CV measurements were performed between 0.1 and 2.5 V.38 The dominating second peak is assigned to the structural changes in the layered Na2Ti3O7 structure upon cycling.38 However, Pan et al. suggested that this could be associated with the intermediate phase formation during charging and discharging.32 According to Hwang et al., the reason behind two oxidative peaks may be the presence of two distinct Na+ ion storage sites in the Na2Ti3O7 structure, where an ion extraction site at 0.33 V could be kinetically more favourable, however, energetically less favourable. On the other hand, the Na+ ion extraction site at 0.44 V could be energetically more favourable and kinetically less favourable.14 Thus, as soon as the Na+ ion storage site at 0.44 V is activated during the subsequent cycles, Na+ ions tend to diffuse out of the 0.44 V site.14 From the reports available to date, the origin of these two oxidation peaks is not known.
The electrochemical performance was investigated by galvanostatic discharge–charge measurements (Fig. 5(b)), over a voltage window of 0.01 to 2.5 V at a current density of 10 mA g−1 (1C = 178 mA g−1). The initial discharge and charge capacity of NTO (with a low average Na insertion voltage of around 0.2 V) has been observed as 325.20 mA h g−1 and 149.07 mA h g−1, respectively, which is higher than the available reports of mixed-phase of NTO14,17,23,29 and may be attributed to the higher proportion of Na2Ti3O7 phase than these reports. The first discharge (Na+ ion insertion) profile exhibited multiple voltage plateau that are in good agreement with the redox pairs observed in the CV measurements. Specifically, the first discharge curve shows three slopping regions at about 0.60 V, 0.20 V, and 0.12 V, where the plateau at 0.60 V may be originated from the SEI formation or side-reactions of the electrolyte reduction and disappeared in subsequent cycles.39 The other discharge voltage plateaus at 0.2 V and 0.12 V might have been caused by the Na+ ion insertion in the NTO structure and in carbon black (used as a conducting agent in the prepared anode composite material), respectively, in good agreement with the previous research.14 Furthermore, for the initial charge process (Na+ ion deinsertion), two voltage plateaux have been observed at around 0.07 V and 0.38 V, which are associated with Na+ ion deinsertion from carbon black and NTO. Even after 50 cycles, the discharge and charge capacities were still maintained at 91.47 mA h g−1 and 89.81 mA h g−1, respectively, which is higher than the available report on single phase monoclinic Na2Ti3O7 electrode,37 mixed phase of Na2Ti3O7 and Na2Ti6O13,29 and carbon coated NTO (C-NTO-1100), where a carbon content (0.7 wt%) was used to synthesise mixed phase of Na2Ti3O7 (54 wt%), and Na2Ti6O13 (46 wt%).14 NTO discharge/charge profile exhibited the lowest potential plateau (about 0.20 V), which is crucial to improving the cell voltage and the power density of NIBs.23 The coulombic efficiency for initial cycles has been observed at only 45.83%, however, it reaches about 98.18% after subsequent cycles (Fig. 5(c)), which is commonly observed in transition metal oxide as electrodes for rechargeable batteries.37 The low initial coulombic efficiency is a known feature but is still under investigation and may originate from the irreversible formation of solid electrolyte interphase (SEI) and/or side-reactions of the electrolyte decomposition which is also supported by the CV profile (Fig. 5(a)).32 The improved coulombic efficiency (up to 98%) and cycling stability, can be attributed to the Na2Ti6O13 phase, due to the lower volume expansion of Na2Ti6O13 (below 1%) than Na2Ti3O7 (ca. 130%).18,23,30 The amplified electrochemical performance of the mixed phase of NTO nanorods can be attributed to the synergistic effect of high stability from Na2Ti6O13 and the higher theoretical capacity of Na2Ti3O7 that also observed in previous report.17,18,28 The shorter diffusion length and higher surface to volume ratio of nanorods is also playing a vital role here, further improving on previous work. Fig. 5(c) shows the cycling performance of the NTO electrode at a current density of 10 mA g−1 for initial 50 cycles. After 50 cycles, the capacity retention from the second cycle (2nd discharge capacity = 170.73 mA h g−1) of NTO was observed 52.50%. After investigating the battery performance, the rate performance of the prepared NTO electrode was tested by increasing the current rate from 10 mA g−1 to 1000 mA g−1 (or 0.05C to 5C). Fig. 5(d) demonstrates the cyclic performance of NTO electrode at different C-rates as the current density increases, the specific capacity decreases gradually. It has been shown that the surface of the active material only takes part in the electrochemical reaction at high current densities due to the kinetic restrictions, which causes the capacity to fade at a high C rate.17 The result shows that reversible capacities are 135.5 mA h g−1 after the first two cycles at 10 mA g−1, 111.05 mA h g−1 at 20 mA g−1, 95.86 mA h g−1 at 40 mA g−1, 61.12 mA h g−1 at 200 mA g−1, 46.33 mA h g−1 at 400 mA g−1, 31.26 mA h g−1 at 1000 mA g−1. Even after the high C-rate (5C in this work), the initial specific capacity at 10 mA g−1 was almost recovered, demonstrating the excellent cell integrity, however, couldn't retain the 1st cycle capacity, which may be due to the irreversible SEI formation/side-reactions of the electrolyte reduction in initial cycles and could be associated with the crystal structure of Na2Ti3O7 and Na2Ti6O13.14 Furthermore, even at a high C rate (1 A g−1), the reversible capacity is still measured to be 31.26 mA h g−1 suggesting the excellent rate capability of the prepared anode material as compared to previous reports.14
Conclusions
In summary, we have demonstrated that using a simple and rapid ionic liquid synthesis method a mixed phase of sodium titanates (Na2Ti6O13 & Na2Ti3O7) is easy to fabricate and this is achieved without the addition of a carbon source during synthesis, in contrast to previous similar work. This provides proof of concept for the use of 1-ethyl-3-methylimidazolium acetate ionic liquid for the synthesis of mixed phase sodium titanate rods. XRD, EDX and TEM confirm the co-existence of the Na2Ti3O7 and Na2Ti6O13 phases in the rod morphology observed by SEM. Some impurity phases were also observed as discrete materials in the sample, arising from impurities in the ionic liquid. Rietveld refinement of the obtained XRD pattern suggested the higher Na2Ti3O7/Na2Ti6O13 ratio that leads to the high specific capacity of the prepared nanocomposite. The synthesised NTO nanorods manifest high specific discharge and charge capacity (325.20 mA h g−1 and 149.07 mA h g−1) with excellent cycling retention (98% capacity is retained after 50 cycles at 10 mA g−1) at room temperature for Na+ ion storage. We attribute the higher capacity and increased cyclability arises due to a synergistic effect between the Na2Ti3O7 and Na2Ti6O13, in excess of previous reports due to the high fraction of Na2Ti3O7 phase. It is clear even at this early stage of optimisation, the decrease in temperature, energy costs and synthesis time (calcination time) provided by the ionic synthesis method is very encouraging and deserves further investigation.Author contributions
PK designed and synthesised samples, performed experiments, and conducted data analysis. YL performed Raman experiment on the prepared material and conducted data analysis. RB designed the project, conducted Rietveld refinement and helped to analyse TEM data. All authors wrote the manuscript and contributed to general discussion.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Faraday Institution “Nexgenna” project (grant number. FIRG018). The authors wish to acknowledge the Henry Royce Institute for Advanced Materials, funded through EPSRC grants EP/R00661X/1, EP/S019367/1, EP/P02470X/1 and EP/P025285/1, and EP/V034804/1, for access to the X-ray diffraction facility, and the Royce Small Equipment Fund for the battery assembly facilities. They also thank the Sorby Centre for Electron Microscopy for use of the SEM and TEM. RB thanks Dr Nik Reeves-McLaren for the helpful discussions regarding Rietveld refinement. Authors also wish to thank the battery lab at Kroto Research Institute and the Henry Royce Institute for running battery measurements in a Maccor Series 4000 battery cycler and in a Maccor Series 4300 battery cycler.References
- M. Sawicki and L. L. Shaw, RSC Adv., 2015, 5, 53129–53154 RSC.
- D. Larcher and J. M. Tarascon, Nat. Chem., 2015, 7, 19–29 CrossRef CAS PubMed.
- D. M. De Carolis, D. Vrankovic and S. A. Kiefer, et al. , Energy Technol., 2021, 9, 2000856 CrossRef CAS PubMed.
- M. M. Doeff, J. Cabana and M. Shirpour, J. Inorg. Organomet. Polym. Mater., 2013, 24, 5–14 CrossRef.
- A. Rudola, C. J. Wright and J. Barker, Energy Mater. Adv., 2021, 1–12 Search PubMed.
- N. T. Ruiz, A. R. Armstrong and H. Alptekin, et al. , JPhys Energy, 2021, 3, 031503 CrossRef.
- S. Anwer, Y. Huang and J. Liu, et al. , ACS Appl. Mater. Interfaces, 2017, 9, 11669–11677 CrossRef CAS PubMed.
- P. Kumari, K. Awasthi and S. Agarwal, et al. , RSC Adv., 2019, 9, 29549–29555 RSC.
- M. R. Panda, A. R. Kathribail and B. Modak, et al. , Electrochim. Acta, 2021, 392, 139026 CrossRef CAS.
- Q. Liu, Z. Hu, M. Chen and C. Zou, et al. , Small, 2019, 15, 1805381 CrossRef PubMed.
- Y. Lyu, Y. Liu and Z. E. Yu, et al. , Sustainable Mater. Technol., 2019, 21, 00098 Search PubMed.
- L. Zhao, T. Zhang and H. Zhao, et al. , Mater. Today Nano, 2020, 10, 100072 CrossRef.
- X. Zhang, X. Rui and D. Chen, et al. , Nanoscale, 2019, 11, 2556–2576 RSC.
- J. Hwang, H. S. Cahyadi and W. Chang, et al. , J. Supercrit. Fluids, 2019, 148, 116–129 CrossRef CAS.
- A. Rudola, K. Saravanan and S. Devaraj, et al. , Chem. Commun., 2013, 49, 7451–7453 RSC.
- W. Zou, J. Li and Q. Deng, et al. , Solid State Ionics, 2014, 262, 192–196 CrossRef CAS.
- S. Chandel, S. Lee and S. Lee, et al. , J. Electroanal. Chem., 2020, 877, 114747 CrossRef CAS.
- C.-K. Ho, C.-Y. V. Li and K.-Y. Chan, et al. , Ind. Eng. Chem. Res., 2016, 55, 10065–10072 CrossRef CAS.
- T. Song, S. Ye, H. Liu and Y. G. Wang, J. Alloys Compd., 2018, 767, 820–828 CrossRef CAS.
- W. Wang, C. Yu and Z. Lin, et al. , Nanoscale, 2013, 5, 594–599 RSC.
- C. Piffet, B. Vertruyen and F. Hatert, et al. , J. Energy Chem., 2022, 65, 210–218 CrossRef CAS.
- J. Xu, C. Ma and M. Balasubramanian, et al. , Chem. Commun., 2014, 50, 12564–12567 RSC.
- C. Wu, W. Hua and Z. Zhang, et al. , Adv. Sci., 2018, 5, 1800519 CrossRef PubMed.
- P. Senguttuvan, G. Rousse and V. Seznec, et al. , Chem. Mater., 2011, 23, 4109–4111 CrossRef CAS.
- F. Xie, L. Zhang and D. Suniec, et al. , Adv. Mater., 2017, 29, 1700989 CrossRef PubMed.
- Z. Zhou, H. Xiao and F. Zhang, et al. , Electrochim. Acta, 2016, 211, 430–436 CrossRef CAS.
- X. Wang, Y. Li, Y. Gao, Z. Wang and L. Chen, Nano Energy, 2015, 13, 687–692 CrossRef CAS.
- C. Wu, W. Hua and Z. Zhang, et al. , Adv. Sci., 2018, 5, 1800519 CrossRef PubMed.
- O. Cech, P. Vanysek and L. Chladil, et al. , ECS Trans., 2016, 74, 331–337 CrossRef CAS.
- D. C. Green, S. Glatzel and A. M. Collins, et al. , Adv. Mater., 2012, 24, 5767–5772 CrossRef CAS PubMed.
- L. Mottram, D. Z. C. Martin, N. Reeves-McLaren and R. Boston, J. Am. Ceram. Soc., 2018, 101, 4468–4471 CrossRef CAS.
- H. Pan, X. Lu and X. Yu, et al. , Adv. Energy Mater., 2013, 3, 1186–1194 CrossRef CAS.
- C. E. Bamberger and G. M. Begun, J. Am. Ceram. Soc., 1987, 70, 48–51 CrossRef.
- Y. V. Kolenko, A. I. Gavrilov and A. V. Garshev, et al. , J. Phys. Chem. B, 2006, 110, 4030–4038 CrossRef CAS PubMed.
- S. Y. Lai, J. P. Maehlen and T. J. Preston, et al. , Nanoscale Adv., 2020, 2, 5335–5342 RSC.
- S. Chandel, Zulkifli, J. Kim and A. K. Rai, Dalton Trans., 2022, 51, 11797 RSC.
- Y. Cao, Q. Ye and F. Wang, et al. , Adv. Funct. Mater., 2020, 30, 2003733 CrossRef.
- A. Rudola, K. Saravanan and C. W. Masona, et al. , J. Mater. Chem. A, 2013, 1, 2653–2662 RSC.
- W. Wang, C. Yu and Y. Liu, et al. , RSC Adv., 2013, 3, 1041–1044 RSC.
| This journal is © The Royal Society of Chemistry 2023 |