
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNanozymes with versatile redox capabilities inspired in metalloenzymes†
Rocío
López-Domene
 ab,
Krishan
Kumar
a,
Jose Eduardo
Barcelon
c,
Gabriela
Guedes
b,
Ana
Beloqui
*ad and
Aitziber L.
Cortajarena
*bd
ab,
Krishan
Kumar
a,
Jose Eduardo
Barcelon
c,
Gabriela
Guedes
b,
Ana
Beloqui
*ad and
Aitziber L.
Cortajarena
*bd
aPOLYMAT and Department of Applied Chemistry, Faculty of Chemistry, University of the Basque Country UPV/EHU, Paseo Manuel Lardizabal 3, Donostia-San Sebastián, 20018, Spain. E-mail: ana.beloquie@ehu.eus
bCenter for Cooperative Research in Biomaterials (CIC biomaGUNE), Basque Research and Technology Alliance (BRTA), Paseo de Miramón 194, Donostia-San Sebastián, 20014, Spain. E-mail: alcortajarena@cicbiomagune.es
cDonostia International Physics Center, Paseo Manuel de Lardizabal 4, 20018 Donostia-San Sebastian, Spain
dIKERBASQUE, Basque Foundation for Science, Plaza Euskadi 5, Bilbao, 48009, Spain
First published on 2nd October 2023
Abstract
Metalloenzymes represent exemplary systems in which an organic scaffold combines with a functional inorganic entity, resulting in excellent redox catalysts. Inspired by these natural hybrid biomolecules, biomolecular templates have garnered significant attention for the controlled synthesis of inorganic nanostructures. These nanostructures ultimately benefit from the protection and colloidal stabilization provided by the biomacromolecule. In this study, we have employed this strategy to prepare nanozymes with redox capabilities, utilizing the versatile catalytic profile of Pt-loaded nanomaterials. Thus, we have investigated protein-templated Pt-based nanoclusters of different sizes and compositions, which exhibit remarkable oxidase, catalase, and reductase-like activities. The interplay between the composition and catalytic activity highlighted the size of the nanocluster as the most prominent factor in determining the performance of the nanozymes. Additionally, we have demonstrated the use of protein-templated nanozymes as potential co-catalysts in combination with enzymes for coupled reactions, under both sequential and concurrent one-pot conditions. This study provides valuable insights into nanozyme design and its wide range of applications in the design of complex catalytic systems.
1. Introduction
Enzymes represent excellent biomaterials capable of facilitating chemical reactions under mild conditions. However, these biocatalysts often suffer from limited stability and robustness, as well as narrow operating ranges concerning pH, temperature, and chemical environments. Consequently, the development of alternative catalysts to enzymes has gained considerable attention, leading to the emergence of nanozymes—synthetic nanomaterials that exhibit enzyme-like catalytic properties. To date, more than 540 distinct nanomaterials have been reported as nanozymes, with a majority incorporating metal components such as gold (Au), silver (Ag), palladium (Pd), copper (Cu), or platinum (Pt).1–6 These metals possess unique electronic and surface properties that confer high catalytic efficiency for various chemical conversions. Among the breadth of nanomaterials exhibiting enzyme-like activities, such as nanoparticles, graphene, carbon nanotubes, metal–organic frameworks, and quantum dots, metal nanoclusters have emerged as particularly promising nanozymes. However, the catalytic potential of metal nanoclusters has not been fully explored, and their remarkable catalytic features remain largely unexploited in the field.7–10Metal nanoclusters (NCs) are small assemblies of a few to hundreds of metal atoms, known for their distinctive electronic and geometric characteristics. Consequently, they have attracted considerable interest in the fields of bioimaging, catalysis, lighting, and sensing.11–17 The reactivity and substrate interaction of metal clusters are strongly influenced by factors such as surface structure, size, and composition. However, it has been observed that as the size of the particles decreases, the surface free energy increases significantly. This phenomenon triggers the agglomeration of clusters into larger particles during the fabrication process, leading to significant alterations in their key properties. For this reason, strategies that pursue the colloidal stabilization of these nanomaterials are demanded.
Inspired by the composition and structure of metalloenzymes, protein-templated nanoclusters have emerged as a remarkable strategy for enhancing the stability, activity, and selectivity of the inorganic nanomaterial. This biomimetic approach involves utilizing a protein backbone to embrace the inorganic entity, creating a favorable environment that prevents the aggregation of nanoclusters. The protein template plays a major role in guiding the formation of metal clusters with distinct sizes, shapes, and configurations, resulting in metal-based catalytic sites that emulate the active sites found in natural enzymes. As a result, the protein-nanocluster hybrid provides both stability and a conducive microenvironment that protects the catalyst from deactivation.
Furthermore, the protein-nanocluster hybrid can be customized to address specific reactions. For example, Wang et al. synthesized bovine serum albumin (BSA)-protected gold nanoclusters (Au NCs) by the in situ reduction of gold ions using the tyrosine residues of the protein.18 This biohybrid showed peroxidase enzyme-like activity over a wide pH and temperature range. The redox capabilities of protein-NC hybrids have been demonstrated using BSA-AuNCs or lysozyme-PtNCs for the oxidation of 3,3′,5,5′-tetramethylbenzidine (TMB), or ferritin-PtNCs for the disproportionation of hydrogen peroxide.19–21 However, most of the reported protein-templated NCs are prepared using proteins that have not been previously engineered. As a result, the allocation of the nanoclusters on the protein is not controlled, which can result in the aggregation of the biohybrids and/or limited control over the properties of the NC. In our recent works, we employed an engineered protein with a well-defined structure to direct the allocation of the nanoclusters and serve as a shield, protecting the metal clusters against aggregation, oxidation, and other detrimental effects.15,22,23 This approach permits the fabrication of an artificial catalytic pocket in which the nanoclusters act as the catalytic entity. By using a consensus tetratricopeptide repeat (CTPR) engineered protein template, we demonstrated the precise modulation of the catalytic properties, which depended not only on the nanocluster composition, but also on the chemical environment, including the coordinating residues to which the nanomaterial is exposed.15 Moreover, our approach offers several advantages, such as being environmentally friendly, simple, efficient, scalable, and avoids the need for hazardous reagents.
In this study, we have investigated the catalytic versatility of CTPR-templated nanozymes, with a specific focus on platinum (Pt)-loaded nanoclusters. These biohybrids, inspired by the structure and configuration of natural metalloenzymes, exhibit various redox performances, including oxidase, catalase, and reductase-like activities. We have examined the influence of NC size and composition on the catalytic oxidation of small molecules, the disproportionation of hydrogen peroxide, and the reduction of p-nitrophenol. Through this investigation, we have unveiled major connections between the features of the NCs and their catalytic performance in those reactions. Furthermore, we aimed to explore the potential interfacing of protein-templated nanozymes with natural enzymes in two-step reactions. By combining the best-performing biohybrid with Candida antarctica Lipase B (CALB) enzyme, we sought to broaden the application scope of the biohybrids while enhancing the overall process efficiency and streamlining.21,24 Specifically, we tested the reduction of p-nitrophenylbutyrate (PNB) to p-aminophenol (PAP), using both a two-step sequential approach and a one-pot concurrent method, making use of the joint action of the engineered biohybrid and the CALB enzyme. This investigation highlights the compatibility and synergistic effects of protein-templated nanozymes with natural enzymes in complex reaction systems.
2. Results and discussion
2.1. Synthesis and characterization of protein-templated nanozymes
The synthesis of the biohybrids was carried out by leveraging the capability of engineered consensus tetratricopeptide repeat (CTPR) proteins to coordinate metals, accommodate, and eventually stabilize the nanoclusters within their designed binding pocket. Specifically, the CTPR mutant utilized in this work has been engineered with a metal-coordination module designed to accommodate the artificial catalytic site of the hybrid.17 This binding module consists of 16 histidines oriented towards the inner cavity with its concave face (Fig. 1A). The hybrid nanozyme is formed throughout the in situ assembly of the nanocluster in the proximity of the metal-binding module, as previously described.15 To achieve this, the desired amount of metal salt precursor, e.g., K2PtCl4 for Pt nanozymes, was added to the CTPR solution at pH 10, followed by a 72 h incubation at 50 °C. We hypothesize that the functional groups, particularly the binding residues, of the protein can play an important role as “carriers” of the seeded metal cations under these conditions, facilitating the formation of hot spots that eventually lead to the synthesis of the nanoclusters.15 Subsequently, the metal cations were reduced in situ by the addition of sodium ascorbate as a reducing agent. Complete details on the nanocluster synthesis and purification procedures can be found in the ESI section,† providing a comprehensive overview of the methodology employed in this study. | ||
Fig. 1 (A) Proposed configuration of CTPR-templated Pt nanoclusters based on the CTPR8-WT structure (PDB ID: 2AVP) and depiction of the nanoclusters fabricated in this work. In grey, the 16 histidines comprising the engineered binding site are highlighted, with the accommodated Pt nanocluster. The inset graph displays the size of each nanozyme measured by TEM. (B) MALDI-ToF spectra of CTPR and protein-templated nanozymes. Free protein is detected with a well-defined peak at m/z 26![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 200. Only Pt11, Pt14, and Pt46 hybrids could be detected (for further details see ESI†). (C) ICP-MS measured Pt loads per protein for the nanozymes synthesized in this work, with the corresponding Pt equivalents. (D) XPS spectra of the nanozymes focused in the Pt4f region. Grey and blue arrows highlight the maximum of the Pt4f bands for Au7Pt74 (72.6 eV) and Pt11 nanozymes (73.2 eV), respectively. 200. Only Pt11, Pt14, and Pt46 hybrids could be detected (for further details see ESI†). (C) ICP-MS measured Pt loads per protein for the nanozymes synthesized in this work, with the corresponding Pt equivalents. (D) XPS spectra of the nanozymes focused in the Pt4f region. Grey and blue arrows highlight the maximum of the Pt4f bands for Au7Pt74 (72.6 eV) and Pt11 nanozymes (73.2 eV), respectively. | ||
In this study, our objective was to investigate the interplay between the size, composition, and redox capability of the biohybrids. To achieve this, we designed a series of Pt nanozymes with an increasing number of atoms, as well as a biohybrid which composition was doped with a second metal cation, namely Au. The synthesis process involved the seeding of CTPR proteins with increasing amounts of PtII, specifically 32, 64, 160, and 320 equivalents per protein. In parallel, Au/Pt bimetallic nanozymes were achieved by the coaddition of 320 and 32 equivalents of Pt and Au, respectively. Overall, a total of five Pt-based protein-templated nanozymes were successfully synthesized and characterized.
After the synthesis protocol, the protein-templated nanozymes were efficiently recovered with yields of 72 and 90% of the initial protein amount, for the lowest to the highest metal cation loads, respectively (Table S1†). The synthesis of the nanoclusters was validated by Transmission Electron Microscopy (TEM) imaging, and the size of the nanomaterials ranged from 1.29 to 1.95 nm, achieving larger nanomaterials for the largest Pt loads (Fig. 1A, Fig. S1 and Table S2†). Matrix-assisted Laser Desorption/Ionization Time-of-Flight Mass Spectrometry (MALDI-ToF MS) analysis confirmed the absence of unmodified protein, indicating a high conversion efficiency (Fig. 1B). The total metal content per unit of protein, which correlates with the size of the nanocluster, was determined by Inductively Coupled Plasma Mass Spectrometry (ICP-MS) (Table S3†). Hence, the biohybrids were coded as Ptx and PtxAuy, where x and y are the number of atoms per protein, as measured by ICP-MS. For example, Pt34 refers to a CTPR-protected Pt nanocluster with an average of 34 atoms of Pt. Fig. 1C illustrates that nanozymes with the highest Pt content could only be achieved by the coaddition of Au atoms to the synthesis reaction. The seeding of 32 equivalents of Au resulted in nanoclusters with an increased Pt content of 38%—from 46 to 74 atoms of Pt—for the same seeded equivalents of Pt, i.e., 320.
X-ray Photoelectron Spectroscopy (XPS) measurements were conducted on the protein-templated nanozymes (Fig. 1D, Fig. S2 and S3†), revealing compelling findings. The Pt4f bands exhibited a notable shift to lower binding energies as the nanoclusters increased in size, ranging from 73.2 to 72.6 eV. The highest energies found in smaller nanoclusters are indicative of PtII species. With the enlargement of the nanoclusters, a second component, likely Pt0, emerged, leading to a shift of the band to lower binding energies. The presence of Pt0 exclusively in larger nanoclusters may be attributed to the oxidation of Pt in the presence of air. The larger three-dimensional structure of the material in Au7Pt74 and Pt46 nanoclusters might maintain the reduced state of the Pt, whereas the higher exposure of the few Pt atoms in Pt11 and Pt14 could dictate the prevalence of the PtII species.
2.2. Redox capability of the nanozymes
We recently explored the peroxidase-like activity of similar protein-templated nanozymes, which surpassed many of the Pt-based hybrid nanozymes reported so far. In this study, we explore the oxidation potential of a set of CTPR-templated nanozymes, loaded with different Pt contents in the absence of hydrogen peroxide, focusing first on oxidation reactions. In the first experiment, the oxygen consumption rate of Au7Pt74 nanozyme (from 1 to 10 μM) in the presence of pyrogallol (PG), as the substrate, was monitored in situ using an oxygen dipping probe (Fig. S4A†). Concentration-dependent oxygen consumption was observed. To analyze these results, we defined the oxygen consumption 50 (OC50) index as the time required for the nanozyme to reduce the oxygen content in the solution by half. Accordingly, OC50 values ranged from 40 to 140 s for nanozyme concentrations of 10 to 1 μM, respectively (Fig. S4B†). Fig. 2A clearly shows that the largest nanoclusters exhibited the fastest oxygen consumption rates under the same oxidation conditions when comparing the entire set of nanozymes. As a general trend, the oxidation efficiency, as measured by OC50, could be correlated with the size of the nanocluster following an exponential trend, ranking the nanozymes from most to least active as follows: Au7Pt74 > Pt46 > Pt34 > Pt16 > Pt11 (Table S4 and Fig. S5†). Thus, while Au7Pt74 and Pt46 nanozymes achieved a 50% reduction in oxygen concentration within 50 and 83 seconds, respectively, the Pt11 nanozymes required more than 1400 seconds to reach similar oxidation yields. Furthermore, nanozymes with low Pt content, i.e., Pt11 and Pt16, were unable to completely deplete the oxygen from the solution, indicating the limited catalytic potential of these hybrids. Conversely, the catalytic performance of Pt46 and Au7Pt74 nanozymes did not exhibit significant differences despite the increase in the number of Pt atoms. This effect might be attributed to the exposed surface area, which might be, in this size range, the dominating factor in understanding the catalytic performance of the nanoclusters.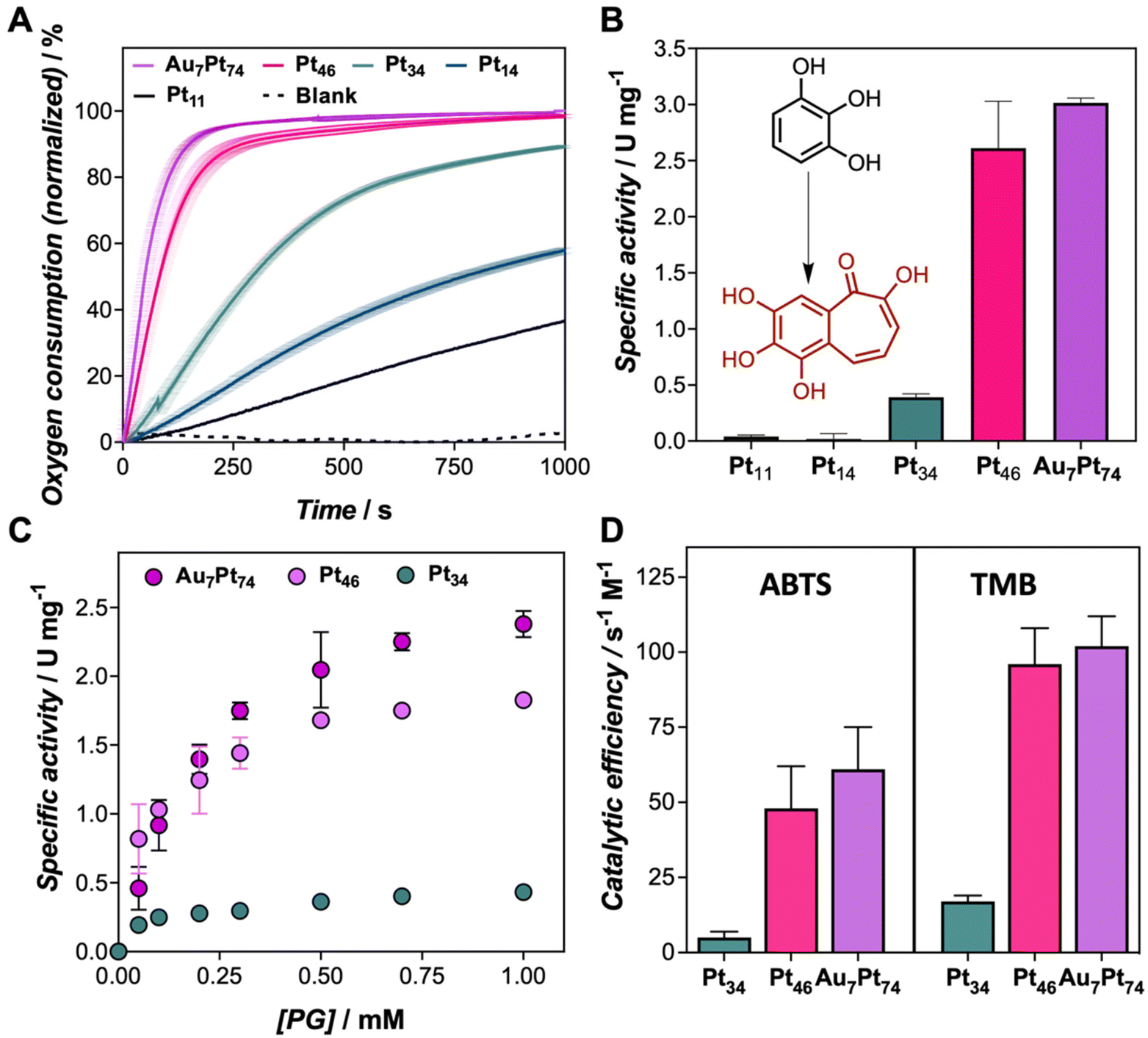 | ||
| Fig. 2 (A) Oxygen consumption measured for the set of nanozymes (reaction conditions: 5 μM of nanozymes, 3 mM of pyrogallol in 50 mM PB buffer at pH 7 and 25 °C). (B) Specific activity, in U mg−1, measured for the set of nanozymes for the oxidation of pyrogallol (reaction conditions: 1 μM of nanozymes, 1 mM of pyrogallol in 50 mM PB buffer at pH 7 and 25 °C). The chemical structure of PG and the proposed structure for the oxidized PG are inserted. (C) Michaelis–Menten curves for Au7Pt74, Pt46, and Pt34 using 1 μM of nanozymes and 0–1 mM of pyrogallol in 50 mM PB buffer at pH 7. (D) Catalytic efficiency measured for the oxidation of TMB and ABTS reagents by Au7Pt74, Pt46, and Pt34. | ||
The catalytic performance of the nanozymes was ascertained by UV-Vis spectroscopy (Fig. S6†). The oxidation of PG could be traced by the increase of absorbance at 420 nm. The specific activity (Fig. 2B and Fig. S7†) of the nanozymes followed a similar profile to that determined by the monitoring of oxygen in the reaction milieu. Apparent kinetic parameters (appKM, appVmax, appkcat, and appkcat/KM) were determined for PG and two more chromogenic substrates, 3,3′,5,5′-Tetramethylbenzidine (TMB), and 3,3-TMB, and 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) (ABTS) to assess the oxidation potential of the nanozyme set (Fig. S8, S9 and Tables S5 to S7†). The smallest nanozymes, Pt11 and Pt16, failed to oxidize these compounds under assayed conditions. For this reason, a detailed analysis of the kinetic parameters was exclusively performed for Au7Pt74, Pt46, and Pt34. As depicted in Fig. 2C, the nanozymes followed the Michaelis–Menten profile. There was an evident disparity in the catalytic rates of Au7Pt74 and Pt46 compared to Pt34, which may be attributed to the size of the nanocluster. Since the catalytic mechanism of these nanozymes involves the interaction of the organic compound with the material's surface, smaller nanoclusters reach surface saturation more rapidly (Fig. 2C). Furthermore, larger nanoclusters showed lower appKm values, indicating higher substrate affinity. An increase of 12 atoms of Pt resulted in a decrease from 1.22 to 0.79 mM for ABTS, as well as a significant increase of the turnover numbers from 0.006 to 0.038 s−1 for ABTS and from 0.003 to 0.022 s−1 for TMB. It is noteworthy that our developed nanozymes displayed significantly lower appKM values compared to previously reported nanozymes. For instance, Pt nanoclusters stabilized by a lysozyme showed appKM of 0.63 mM, while the three engineered nanozymes (Au7Pt74, Pt46, and Pt34) displayed tighter binding with appKM values, ranging from 0.18 to 0.22 mM for TMB substrate.20 This analysis is consistent with the oxygen consumption measurements and reveals a clear rise in terms of catalytic efficiency for Pt46 and Au7Pt47 compared to Pt34 hybrids (Fig. 2D).
Along with the oxidation capability of the protein-templated nanozymes, we examined their catalase-like performance. Reported Pt-based nanomaterials have shown catalase-like activity due to the ability of Pt atoms to promote the dismutation of hydrogen peroxide. Interestingly, the addition of our Pt nanozymes into a hydrogen peroxide solution resulted in rapid bubble formation. The dismutation reaction was more prominent at higher pH levels (Fig. S10†). Notably, the nanozymes demonstrated catalase activity within the pH range of 7–8 and even at pH 5 and 6, which is considered challenging for artificial enzymes.25,26 All the nanozymes in the set displayed some degree of catalase-like performance, with the biohybrids loaded with higher amounts of Pt showing the highest activity (Fig. 3). These nanozymes were able to remove more than 90% of a 50 μM hydrogen peroxide solution in 20 min under diluted conditions (1 μM).
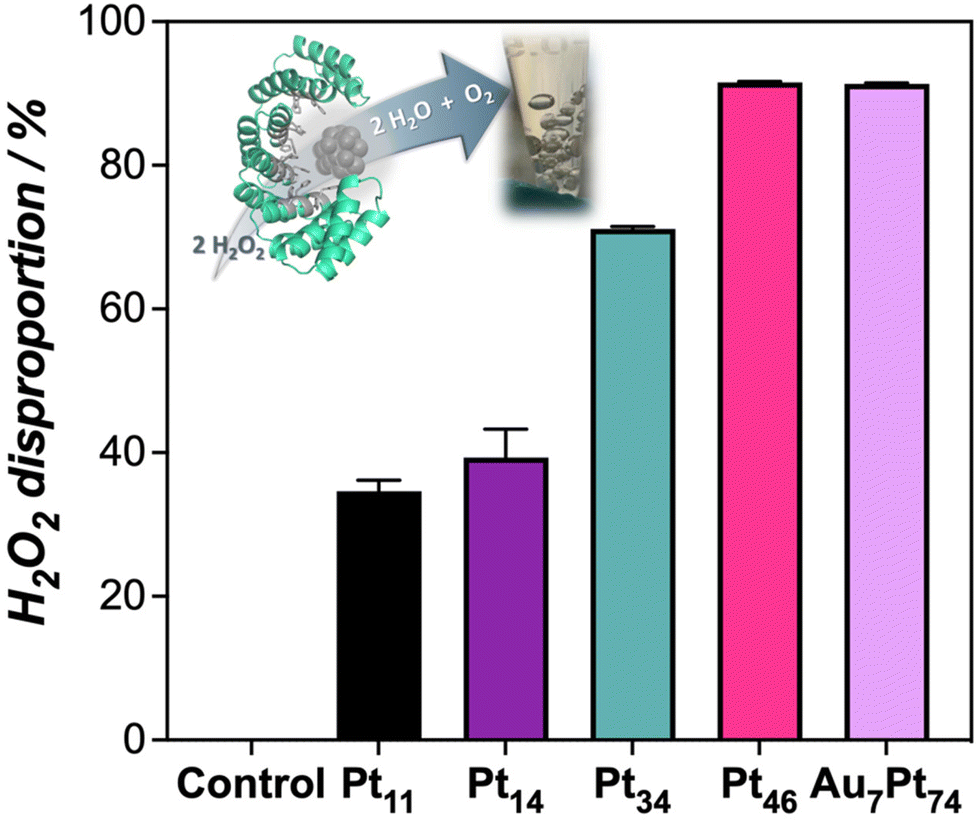 | ||
| Fig. 3 Catalase-like activity of the protein-templated nanozymes measured as the % of H2O2 removed from solution. Inset image shows the formation of bubbles upon the addition of the nanozymes to the H2O2 solution. | ||
2.3. Use of nanozymes in combination with enzymes in coupled reactions
Finally, to demonstrate the catalytic versatility of the engineered biohybrids, the catalytic reduction of p-nitrophenol (PNP) to p-aminophenol (PAP) using borohydride ions (BH4−) was used as a model reaction. This reaction can be monitored by UV-Vis spectroscopy, tracking the disappearance of the PNP band at 400 nm and the subsequent emergence of a band at 300 nm corresponding to PAP.27,28 It is important to note that in the absence of the nanozyme, there were no significant changes observed in the absorption spectra of the reaction mixture (Fig. S11†). As denoted in Fig. 4A, the reduction reaction became evident only upon the addition of the nanozymes. While all the tested nanozymes triggered the reduction of the PNP (Fig. S12†), only the most active biohybrids, namely Au7Pt74, Pt46, and Pt34, evidenced the formation of PAP at 300 nm. The top-performing nanozymes (1 μM) catalyzed the reduction of the 4-nitrophenolate ion (0.22 mM) in approximately 10 minutes.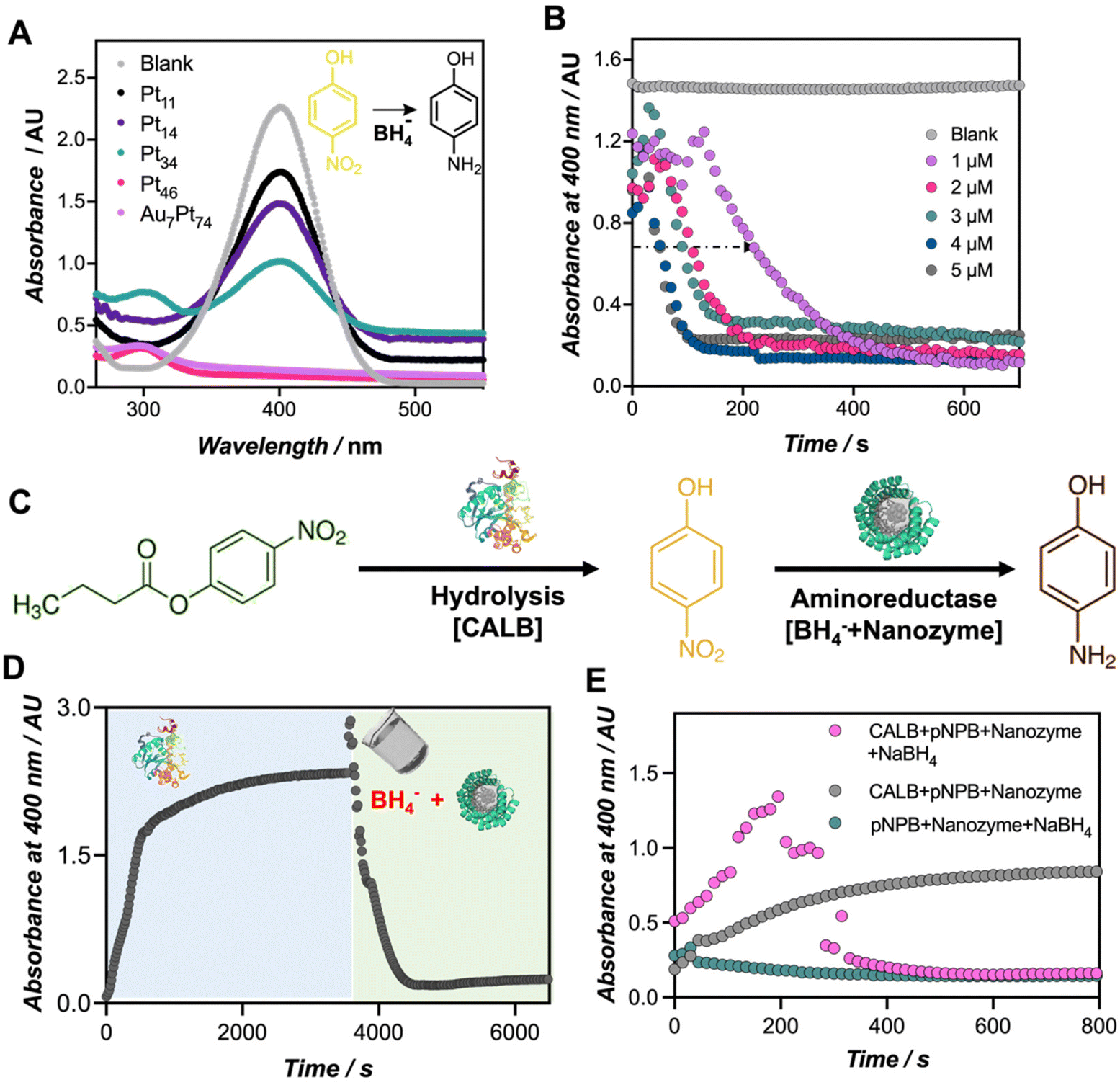 | ||
| Fig. 4 Catalytic performance of nanozymes in the reduction of PNP to PAP and enzyme-coupled reactions. (A) Monitoring the reduction of PNP to PAP by UV-Vis for each of the nanozymes. (B) Reduction kinetics monitored at 400 nm at different concentrations of Au7Pt74 nanozyme. (C) Scheme of the sequential reaction carried out in this work. First, the PNB is hydrolyzed by CALB lipase and, in a second reaction, the released PNP is reduced by the joint action of NaBH4 and the nanozyme. (D) Monitoring of the PNP absorbance after the sequential addition of the catalysts (CALB first—in blue —, and then the nanozymes—in green); (E) monitoring of the PNP absorbance along the one-pot concurrent reduction from PNB to PAP. | ||
To the best of our knowledge, this is the first example in which Pt-based nanoclusters are applied for the reduction of PNP using borohydride ions. Other examples include the use of stabilized Pt nanoparticles such as graphene@CALB-PdNPs-CuNPs based catalysts,29 or Pt NPs stabilized by guar gum.30 In this work, we specifically investigated the influence of the number of platinum atoms in facilitating efficient catalysis. It is known that in the first stage, the reactants are adsorbed on the surface of the nanocluster, which mediates electron transfer between BH4− and PNP.31 The induction time required for the activation of the intermediates was herein observed at different nanozyme concentrations (Fig. 4B and Fig. S13†). Notably, increasing the nanozyme concentration, from 1 to 5 μM, significantly reduced the induction time from 140 to 40 seconds, respectively, for Au7Pt74 nanozyme. These results align with previous reports that have elucidated that varying the concentration of catalyst, temperature, and pH influences the induction time.32
Finally, we explored the diverse applications of these nanozymes in different scenarios. The use of protein-templated nanozymes holds great potential for sequential, as well as concurrent chemoenzymatic reactions. As a proof-of-concept, we assayed our best-performing biohybrid, Au7Pt74, in a sequential cascade reaction with CALB enzyme to convert p-nitrophenyl butyrate (PNB) to PAP (Fig. 4C). In the first step, we added the CALB enzyme to the PNB solution. Subsequently, the nanozyme hybrid was added together with the NaBH4 hydride donor. We performed the reaction in a 96-well plate, and the formation and conversion of PNP were monitored at 400 nm (Fig. 4D). Under the applied conditions, CALB catalyzed the complete hydrolysis of PNB in less than 30 min, as indicated by the saturation of the signal at 400 nm. In the second step, the co-addition of the nanozyme and NaBH4 triggered a sudden increase in pH, leading to a rise in the extinction coefficient of the p-nitrophenolate chromophore (Fig. S13†). Subsequently, this co-addition of the nanozyme and NaBH4 resulted in the disappearance of the signal corresponding to PNP and the emergence of a band at 300 nm, likely indicating the formation of PAP, and confirming the occurrence of this second reduction reaction (Fig. S14†).33
Motivated by these results, we proceeded to perform the same reaction under one-pot conditions, which would provide insights into the compatibility of the biohybrids with functional proteins such as enzymes. For this purpose, we prepared a cocktail comprising both natural (CALB) and artificial (Au7Pt74) enzymes along with the hydride donor, NaBH4. To monitor this reaction by UV-Vis spectroscopy, two reaction controls were tested. As depicted in Fig. 4E, in the absence of the CALB enzyme, no PNP conversion was detected (green dots in Fig. 4E), emphasizing the role of the p-nitrophenolate species in the reduction mechanism to PAP.34,35 Additionally, we observed that the presence of a hydride donor, NaBH4, is necessary to initiate the reduction reaction (grey dots in Fig. 4E), which was also accompanied by the rise of the band at 300 nm corresponding to PAP (Fig. S15†). Finally, the co-addition of all the reactants led to the release of PNP, which, at a given concentration, is likely further converted in situ to PAP, evidenced by a sudden drop of the absorbance at 400 nm. Therefore, we have successfully demonstrated the efficient concurrence of two catalytic transformations driven by a natural enzyme and a nanozyme within the same reaction environment.
3. Conclusions
This study reveals compelling conclusions on the design and versatility of Pt nanoclusters stabilized by CTPR protein. It has been demonstrated that the in situ synthesis procedure can be controlled for the achievement of nanoclusters comprising 11 to 74 Pt atoms. Interestingly, the addition of a small fraction of gold atoms into the synthesis, i.e., 10% (n/n), triggers up to a 38% increase in the incorporation of Pt into the nanomaterial. However, the role of the gold atoms appears to be merely structural. The kinetic profile of the metal nanoclusters fits well with the Michaelis–Menten model, indicating the successful development of robust nanozymes with redox capabilities. Moreover, there is an exponential relationship between the dimensions and the catalytic performance of the hybrids. While the smallest hybrids, i.e., Pt11, Pt16, and Pt34, exhibit limited redox catalysis, Pt46, and Au7Pt74, with similar catalytic performance, exhibit oxygen consumption rates up to 200 times higher. Moreover, an insightful structural and functional characterization of the hybrids reveals that the catalytic performance is not solely influenced by the size; rather, parameters such as surface area and the oxidation state of the Pt atoms must also be taken into consideration.We have explored the application of protein-templated nanozymes in sequential and concurrent chemoenzymatic reactions. Our focus was centered on assessing the performance of the most effective biohybrid in catalyzing a sequential cascade reaction coupled to CALB enzyme, resulting in the successful conversion of PNB into PAP. This demonstrates the capability of interfacing protein-templated nanozymes with natural enzymes, which broadens the applicability of these nanozymes. Thus, the demonstrated catalytic versatility, together with the possibility to use them together with other enzymes, underline the role of CTPR-templated Pt nanoclusters as promising catalysts in various technological fields, such as environmental remediation, biosensing, biomedicine, and energy conversion among others.
Conflicts of interest
There are not conflicts to declare.Acknowledgements
A. L. C. acknowledges support by the Agencia Estatal de Investigación Grant PID2019-111649RB-I00 and PID2022-137977OB-100 funded by MCIN/AEI/10.13039/501100011033 and Grant PDC2021-120957-I00 funded by MCIN/AEI/10.13039/501100011033 and by the “European Union NextGenerationEU/PRTR”. This work was performed under the Maria de Maeztu Units of Excellence Program from Q5 the Spanish State Research Agency grant no. MDM-2017-0720. A. B. gratefully acknowledges the financial support from the Spanish Research Agency (AEI) for the financial support (PID2022-142128NB-I00 funded by MCIN/AEI/10.13039/501100011033 and by the “European Union NextGenerationEU/PRTR”; RYC2018-025923-I from RyC program – MCIN/AEI/10.13039/501100011033 and FSE “invierte en tu futuro”), BBVA Foundation – IN[21]_CBB_QUI_0086, and UPV/EHU – GIU21-033. K.K. thanks JdC Grant FJC2021-047607-I funded by MCIN/AEI/ 10.13039/501100011033. We thank Fernando López-Gallego for support with the oxygen consumption measurements.References
- N. Pajooheshpour, M. Rezaei, A. Hajian, A. Afkhami, M. Sillanpää, F. Arduini and H. Bagheri, Sens. Actuators, B, 2018, 275, 180–189 CrossRef CAS.
- J. Xie, Y. Zheng and J. Y. Ying, J. Am. Chem. Soc., 2009, 131, 888–889 CrossRef CAS PubMed.
- B. Jiang and M. Liang, Chin. J. Chem., 2021, 39, 174–180 CrossRef CAS.
- Y. Jiang, Z. Ding, M. Gao, C. Chen, P. Ni, C. Zhang, B. Wang, G. Duan and Y. Lu, Chin. J. Chem., 2021, 39, 3369–3374 CrossRef CAS.
- H. Wei and E. Wang, Chem. Soc. Rev., 2013, 42, 6060–6093 RSC.
- J. Wu, X. Wang, Q. Wang, Z. Lou, S. Li, Y. Zhu, L. Qin and H. Wei, Chem. Soc. Rev., 2019, 48, 1004–1076 RSC.
- L. Chen, T. Hou, Y. Tan, C. Guo, B. Wang, L. Ge and F. Li, ACS Sustainable Chem. Eng., 2022, 10, 2750–2760 CrossRef CAS.
- H. Wang, P. Li, D. Yu, Y. Zhang, Z. Wang, C. Liu, H. Qiu, Z. Liu, J. Ren and X. Qu, Nano Lett., 2018, 18, 3344–3351 CrossRef CAS PubMed.
- X. Niu, X. Li, Z. Lyu, J. Pan, S. Ding, X. Ruan, W. Zhu, D. Du and Y. Lin, Chem. Commun., 2020, 56, 11338–11353 RSC.
- L. Zhao, Z. Wu, G. Liu, H. Lu, Y. Gao, F. Liu, C. Wang, J. Cui and G. Lu, J. Mater. Chem. B, 2019, 7, 7042–7051 RSC.
- W. Huang, X. Ma, O. Sato and D. Wu, Chem. Soc. Rev., 2021, 50, 3176–3191 Search PubMed.
- S. I. Tanaka, J. Miyazaki, D. K. Tiwari, T. Jin and Y. Inouye, Angew. Chem., Int. Ed., 2011, 50, 431–435 CrossRef CAS PubMed.
- F. Molaabasi, M. Sarparast, M. Shamsipur, L. Irannejad, A. A. M. Movahedi, A. Ravandi, B. H. Verdom and R. Ghazfar, Sci. Rep., 2018, 8, 14507 CrossRef PubMed.
- X. Meng, I. Zare, X. Yan and K. Fan, Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol., 2020, 12, e1602 CrossRef PubMed.
- R. L. Domene, S. V. Díaz, E. Modin, A. Beloqui and A. L. Cortajarena, Adv. Funct. Mater., 2023, 2301131 CrossRef.
- N. Goswami, K. Zhenga and J. Xie, Nanoscale, 2014, 6, 13328 RSC.
- A. Aires, V. F. Luna, J. F. Cestau, R. D. Costa and A. L. Cortajarena, Nano Lett., 2020, 20, 2710–2716 CrossRef CAS PubMed.
- X. X. Wang, Q. Wu, Z. Shan and Q. M. Huang, Biosens. Bioelectron., 2011, 26, 3614–3619 CrossRef CAS PubMed.
- G. L. Wang, L. Y. Jin, Y. M. Dong, X. M. Wu and Z. J. Li, Biosens. Bioelectron., 2015, 64, 523–529 CrossRef CAS PubMed.
- C. J. Yu, T. H. Chen, J. Y. Jiang and W. L. Tseng, Nanoscale, 2014, 6, 9618–9624 RSC.
- J. Fan, J. J. Yin, B. Ning, X. Wu, Y. Hu, M. Ferrari, G. J. Anderson, J. Wei, Y. Zhao and G. Nie, Biomaterials, 2011, 32, 1611–1618 CrossRef CAS PubMed.
- A. Aires, A. Sousaraei, M. Möller, J. C. Gonzalez and A. L. Cortajarena, Nano Lett., 2021, 21, 9347–9353 CrossRef CAS PubMed.
- A. Aires, I. Llarena, M. Moller, J. C. Smirnov, J. C. Gonzalez and A. L. Cortajarena, Angew. Chem., Int. Ed., 2019, 58, 6214–6219 CrossRef CAS PubMed.
- N. L. Garcia, A. J. Alesanco, A. V. Campoy, O. Abian and J. M. Palomo, ACS Appl. Mater. Interfaces, 2021, 13, 5111–5124 CrossRef PubMed.
- D. Xu, L. Wu, H. Yao and L. Zhao, Small, 2022, 37, 2203400 CrossRef PubMed.
- J. Li, W. Liu, X. Wu and X. Gao, Biomaterials, 2015, 48, 37–44 CrossRef CAS PubMed.
- S. Shirin, S. Roy, A. Rao and P. P. Pillai, J. Phys. Chem. C, 2020, 124, 19157–19165 CrossRef CAS.
- S. Roy, A. Rao, G. Devatha and P. P. Pillai, ACS Catal., 2017, 7, 7141–7145 CrossRef CAS.
- N. L. Garcia, E. P. Urriolabeitia and J. M. Palomo, ACS Appl. Nano Mater., 2023, 6, 704–713 CrossRef.
- S. Pandey and S. B. Mishra, Carbohydr. Polym., 2014, 113, 525–531 CrossRef CAS PubMed.
- T. A. Thiel, X. Zhang, B. Radhakrishnan, R. van de Krol, F. F. Abdi, M. Schroeter, R. Schomäcker and M. Schwarze, RSC Adv., 2022, 12, 30860–30870 RSC.
- R. D. Neal, R. A. Hughes, P. Sapkota, S. Ptasinska and S. Neretina, ACS Catal., 2020, 10, 10040–10050 CrossRef CAS.
- J. Strachan, C. Barnett, A. F. Masters and T. Maschmeyer, ACS Catal., 2020, 10, 5516–5521 CrossRef CAS.
- R. Grzeschik, D. Schäfer, T. Holtum, S. Küpper, A. Hoffmann and S. Schlücker, J. Phys. Chem. C, 2020, 124, 2939–2944 CrossRef CAS.
- C. Kästner and A. F. Thünemann, Langmuir, 2016, 32, 7383–7391 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3nr03443g |
| This journal is © The Royal Society of Chemistry 2023 |