
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNew 1,3,4-thiadiazoles as potential anticancer agents: pro-apoptotic, cell cycle arrest, molecular modelling, and ADMET profile†
Mohamed H. Hekal a,
Paula S. Faraga,
Magdy M. Hemdana,
Amira A. El-Sayeda,
Aya I. Hassaballaha and
Wael M. El-Sayed*b
a,
Paula S. Faraga,
Magdy M. Hemdana,
Amira A. El-Sayeda,
Aya I. Hassaballaha and
Wael M. El-Sayed*b
aDepartment of Chemistry, Faculty of Science, Ain Shams University, Abbassia, 11566 Cairo, Egypt
bDepartment of Zoology, Faculty of Science, Ain Shams University, Abbassia, 11566 Cairo, Egypt. E-mail: waelelhalawany@hotmail.com; wael_farag@sci.asu.edu.eg; Fax: +202 2684 2123; Tel: +202 2482 1633
First published on 25th May 2023
Abstract
A series of novel 1,3,4-thiadiazoles was synthesized via the reaction of N-(5-(2-cyanoacetamido)-1,3,4-thiadiazol-2-yl)benzamide (3) with different carbon electrophiles and evaluated as potential anticancer agents. The chemical structures of these derivatives were fully elucidated using various spectral and elemental analyses. Out of 24 new thiadiazoles, derivatives 4, 6b, 7a, 7d, and 19 have significant antiproliferative activity. However, derivatives 4, 7a, and 7d were toxic to the normal fibroblasts, and therefore were excluded from further investigations. Derivatives 6b and 19 with IC50 at less than 10 μM and with high selectivity were selected for further studies in breast cells (MCF-7). Derivative 19 arrested the breast cells at G2/M probably through inhibition of CDK1, while 6b significantly increased the sub-G1 percent of cells probably through induction of necrosis. These results were confirmed by the annexin V-PI assay where 6b did not induce apoptosis and increased the necrotic cells to 12.5%, and compound 19 significantly increased the early apoptosis to 15% and increased the necrotic cells to 15%. Molecular docking showed that compound 19 was like FB8, an inhibitor of CDK1, in binding the CDK1 pocket. Therefore, compound 19 could be a potential CDK1 inhibitor. Derivatives 6b and 19 did not violate Lipinski's rule of five. In silico studies showed that these derivatives have a low blood–brain barrier penetration capability and high intestinal absorption. Taken together, derivatives 6b and 19 could serve as potential anticancer agents and merit further investigations.
1. Introduction
Heterocyclic compounds are key structural moieties in several anticancer drugs. According to the FDA, most novel molecular anticancer agents contain heterocyclic rings within their structures.1 Due to their adaptability, heterocycle-based drugs can easily target various metabolic pathways and cellular processes in cancer pathology.2 Various heterocyclic compounds have been synthesized and reported for their potential applications as therapeutic agents. Among these compounds, the five-membered heterocyclic scaffolds, oxadiazoles, triazoles, and thiadiazoles are the most significant compounds. Thiadiazoles have been reported to exhibit several biological activities. Among the thiadiazole isomers, 1,3,4-derivatives showed preferable metabolic stability and availability.3 This is the most promising moiety with different therapeutic activities such as anticancer,4 antiviral,5 anti-inflammatory,6 antidiabetic activity,7 and antidepressant activity.8The interesting pharmacological properties of 1,3,4-thiadiazoles is attributed to their high aromaticity, in vivo stability, and lack of toxicity.9 Moreover, the capability of the hydrogen binding domain allows the use of 1,3,4-thiadiazole as one of the potential agents in many FDA-approved10 drugs such as desaglybuzole, acetazolamide, sulfamethizole, litronesib, filanesib, and methazolamide11 (Fig. 1).
 | ||
| Fig. 1 FDA-approved drugs of 1,3,4-thiadiazole derivatives showing their common pharmacophoric features. | ||
The present study aimed at synthesizing new thiadiazole derivatives and investigating the antiproliferative effect on various human cell lines as well as investigating the possible mechanism(s) of action through assessing the effect of these derivatives on cell cycle and apoptosis by flow cytometry. Different in silico and docking studies were performed to predict some of the pharmacological characteristics of the new thiadiazoles.
2. Experimental
2.1 Chemistry
All melting points were determined using uncorrected Griffin and George melting-point equipment (Griffin & George Ltd., Wembley, Middlesex, UK). The quality of the produced compounds was controlled using aluminum sheet silica gel F254 (Merck) thin layer chromatography (TLC). The KBr wafer approach was used to record IR-spectra using a Pye Unicam SP1200 spectrophotometer (Pye Unicam Ltd., Cambridge, UK). 1H-NMR spectra were obtained on a Bruker Avance (III) utilizing a Varian-Gemini 400 MHz and an internal standard of tetramethyl silane (chemical shifts in scale δppm), whereas 13C-NMR spectra were obtained at 100 MHz. In deuterated dimethyl sulfoxide, TMS was used as an internal standard (DMSO-d6). Chemical changes were described as elemental studies were performed using a PerkinElmer 2400 CHN elemental analyzer at the Microanalytical Unit, Faculty of Science, Ain Shams University, and satisfactory analytical data (±0.4) were obtained for all derivatives.![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1601 (CN). Anal. calcd for C9H8N4OS (220.25) C, 49.08; H, 3.66; N, 25.44; found: C, 49.18; H, 3.63; N, 25.60.
O), 1601 (CN). Anal. calcd for C9H8N4OS (220.25) C, 49.08; H, 3.66; N, 25.44; found: C, 49.18; H, 3.63; N, 25.60.Method (II): a mixture of N-(2-carbamothioylhydrazine-1-carbonothioyl)benzamide (1) (2.5 g, 10 mmol) and sulfuric acid 98% (10 mL) was stirred at room temperature for 1.5 h, kept in refrigerator overnight, and then poured onto cold water. The precipitated solid was separated by filtration, dried, and recrystallized from N,N-dimethyl formamide affording the pure compound 2 as yellow crystals; (1.1 g, 50%) (identity m.p., TLC, and IR).
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3] giving the pure compound 3 as yellow crystals; (2.1 g, 75%); m.p. 204–206 °C; IR (KBr) (υmax, cm−1): 3213 (NH), 3080 (CH aromatic), 2918, 2894 (CH aliphatic), 2257 (C
:3] giving the pure compound 3 as yellow crystals; (2.1 g, 75%); m.p. 204–206 °C; IR (KBr) (υmax, cm−1): 3213 (NH), 3080 (CH aromatic), 2918, 2894 (CH aliphatic), 2257 (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N), 1691, 1673 (CO), 1612 (CN). 1H-NMR (300 MHz, DMSO-d6) δppm: 3.88 (s, 2H, CH2), 7.46–7.95 (m, 5H, ArH), 11.69 (br s, 1H,
N), 1691, 1673 (CO), 1612 (CN). 1H-NMR (300 MHz, DMSO-d6) δppm: 3.88 (s, 2H, CH2), 7.46–7.95 (m, 5H, ArH), 11.69 (br s, 1H,  , exchangeable with D2O), 12.59 (br s, 1H,
, exchangeable with D2O), 12.59 (br s, 1H, 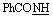 , exchangeable with D2O). 13C-NMR (100 MHz, DMSO-d6) δppm: 24.06, 115.46, 115.60, 128.53, 128.84, 131.83, 133.30, 160.11, 166.58, 167.40, 168.10, 178.14. Anal. calcd for C12H9N5O2S (287.30) C, 50.17; H, 3.16; N, 24.38; found: C, 50.27; H, 3.01; N, 24.30.O), 1636 (CN). 1H-NMR (300 MHz, DMSO-d6) δppm: 7.28–8.00 (m, 9H, ArH), 8.58 (s, 1H, CCH olefinic), 11.76 (br s, 1H,
, exchangeable with D2O). 13C-NMR (100 MHz, DMSO-d6) δppm: 24.06, 115.46, 115.60, 128.53, 128.84, 131.83, 133.30, 160.11, 166.58, 167.40, 168.10, 178.14. Anal. calcd for C12H9N5O2S (287.30) C, 50.17; H, 3.16; N, 24.38; found: C, 50.27; H, 3.01; N, 24.30.O), 1636 (CN). 1H-NMR (300 MHz, DMSO-d6) δppm: 7.28–8.00 (m, 9H, ArH), 8.58 (s, 1H, CCH olefinic), 11.76 (br s, 1H, 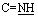 , exchangeable with D2O), 13.98 (br s, 2H,
, exchangeable with D2O), 13.98 (br s, 2H,  , exchangeable with D2O). 13C-NMR (100 MHz, DMSO-d6) δppm: 115.41, 118.12, 118.50, 124.71, 128.58 (2C), 128.70, 128.86, 129.40, 130.40, 131.94, 133.01, 133.27, 133.78, 142.42, 153.24, 156.25, 156.80, 168.39. Anal. calcd for C19H13N5O3S (391.41) %: C, 58.31; H, 3.35; N, 17.89; found: C, 58.28; H, 3.38; N, 17.70.
, exchangeable with D2O). 13C-NMR (100 MHz, DMSO-d6) δppm: 115.41, 118.12, 118.50, 124.71, 128.58 (2C), 128.70, 128.86, 129.40, 130.40, 131.94, 133.01, 133.27, 133.78, 142.42, 153.24, 156.25, 156.80, 168.39. Anal. calcd for C19H13N5O3S (391.41) %: C, 58.31; H, 3.35; N, 17.89; found: C, 58.28; H, 3.38; N, 17.70.The filtrate was acidified by cold dilute HCl. The deposited solid was filtered out, dried, and recrystallized from EtOH/dioxane [3:1] affording compound 5.
Olactone), 1679, 1655 (COamide), 1608 (CN). 1H-NMR (400 MHz, DMSO-d6) δppm: 7.48–7.57 (m, 5H, ArH), 7.66 (d, J = 7.24 Hz, 1H, ArH), 7.79 (d, J = 7.76 Hz, 1H, ArH), 8.00–8.08 (m, 2H, ArH), 9.04 (s, 1H, CCH olefinic), 12.20 (br s, 1H, NH, exchangeable with D2O), 13.74 (br s, 1H, NH, exchangeable with D2O). 13C-NMR (100 MHz, DMSO-d6) δppm: 104.51, 116.56, 117.96, 120.23, 123.32, 123.76, 126.28 (2C), 128.58, 130, 131.67, 134.63, 140.30 (2C), 152.43 (2C), 166.99 (2C), 169.65. Anal. calcd for C19H12N4O4S (392.39) %: C, 58.16; H, 3.08; N, 14.28; found: C, 58.20; H, 3.10; N, 14.25.N), 1660, 1641 (CO). 1H-NMR (400 MHz, DMSO-d6) δppm: 3.88 (s, 3H, OCH3), 5.59 (s, 2H, NH2, exchangeable with D2O), 7.18 (d, J = 8.80 Hz, 2H, ArH), 7.54 (t, J = 7.76 Hz, 2H, ArH), 7.67 (t, J = 7.4 Hz, 1H, ArH), 7.97–8.22 (m, 4H, ArH), 12.42 (br s, 1H,  , exchangeable with D2O). 13C-NMR (100 MHz, DMSO-d6) δppm: 66.13, 71.23, 116.39, 117.57, 121.90, 123.72, 124.02, 124.39, 126.28, 127.08 (2C), 127.74, 129.80, 130.83, 132.22, 135.78, 147.67, 152.57, 153.56, 153.81, 162.56, 170.13, 170.20. Anal. calcd for C23H15N7O3S (469.48) %: C, 58.84; H, 3.22; N, 20.88; found: C, 58.80; H, 3.28; N, 20.82.
, exchangeable with D2O). 13C-NMR (100 MHz, DMSO-d6) δppm: 66.13, 71.23, 116.39, 117.57, 121.90, 123.72, 124.02, 124.39, 126.28, 127.08 (2C), 127.74, 129.80, 130.83, 132.22, 135.78, 147.67, 152.57, 153.56, 153.81, 162.56, 170.13, 170.20. Anal. calcd for C23H15N7O3S (469.48) %: C, 58.84; H, 3.22; N, 20.88; found: C, 58.80; H, 3.28; N, 20.82.The reaction filtrate was poured onto ice cold dilute HCl with stirring. The crude material was filtered off and recrystallized from dioxane giving 6b.
N), 1681 (CO). 1H-NMR (400 MHz, DMSO-d6) δppm: 3.88 (s, 3H, OCH3), 7.18 (d, J = 8.80 Hz, 2H, ArH), 7.54 (t, J = 7.76 Hz, 2H, ArH), 7.67 (t, J = 7.4 Hz, 1H, ArH), 7.97–8.04 (m, 4H, ArH), 8.22 (s, 1H, CH olefinic), 11.11 (br s, 1H, NH, exchangeable with D2O), 11.81 (br s, 1H, NH, exchangeable with D2O). 13C-NMR (100 MHz, DMSO-d6) δppm: 20.58, 55.62, 112.29 (2C), 114.63 (2C), 118.80, 127.50 (2C), 128.99 (2C), 129.53 (2C), 137.06 (2C), 146.06 (2C), 159.78, 170.44, 178.66. Anal. calcd for C20H15N5O3S (405.43) %: C, 59.25; H, 3.73; N, 17.27; found: C, 59.30; H, 3.70; N, 17.30.N), 1660 (COamide). 1H-NMR (400 MHz, DMSO-d6) δppm: 7.57–8.17 (m, 8H, ArH), 8.29 (s, 1H, CHolefinic), 12.72 (br s, 1H, NH, exchangeable with D2O), 13.47 (br s, 1H, NH, exchangeable with D2O). 13C-NMR (100 MHz, DMSO-d6) δppm: 103.93, 112.94, 115.85, 117.93, 129.74 (2C), 131.93 (2C), 132.04, 136.85 (2C), 146.12, 146.39, 148.22, 156.91, 159.10, 160.49, 170.08, 170.69. Anal. calcd for C19H11Cl2N5O2S (444.29) %: C, 51.36; H, 2.50; Cl, 15.96; N, 15.76; found: C, 51.30; H, 2.43; Cl, 15.89; N, 15.80.N), 1686, 1665 (COamide). 1H-NMR (400 MHz, DMSO-d6) δppm: 7.53–8.02 (m, 9H, ArH), 8.29 (s, 1H, CHolefinic), 11.28 (br s, 1H,  , exchangeable with D2O), 12.40 (br s, 1H,
, exchangeable with D2O), 12.40 (br s, 1H, 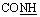 , exchangeable with D2O). 13C-NMR (100 MHz, DMSO-d6) δppm: 68.28, 70.01, 120.90, 123.02, 123.13, 125.59, 126.03, 126.71, 127, 128.11, 128.46, 128.55, 129.80, 129.89, 135.19, 146.74, 167.69, 170.62, 170.77. Anal. calcd for C19H12ClN5O2S (409.85) %: C, 55.68; H, 2.95; Cl, 8.65; N, 17.09; found: C, 55.70; H, 2.98; Cl, 8.72; N, 17.15.N), 1684, 1664 (COamide), 1614 (CN). 1H-NMR (400 MHz, DMSO-d6) δppm: 6.87–6.88 (m, 1H, ArH), 7.48–7.56 (m, 4H, ArH), 7.67 (t, J = 7.36 Hz, 2H, ArH), 7.97–8.08 (m, 1H, ArH), 8.20 (s, 1H, CHolefinic), 11.82 (br s, 1H,
, exchangeable with D2O). 13C-NMR (100 MHz, DMSO-d6) δppm: 68.28, 70.01, 120.90, 123.02, 123.13, 125.59, 126.03, 126.71, 127, 128.11, 128.46, 128.55, 129.80, 129.89, 135.19, 146.74, 167.69, 170.62, 170.77. Anal. calcd for C19H12ClN5O2S (409.85) %: C, 55.68; H, 2.95; Cl, 8.65; N, 17.09; found: C, 55.70; H, 2.98; Cl, 8.72; N, 17.15.N), 1684, 1664 (COamide), 1614 (CN). 1H-NMR (400 MHz, DMSO-d6) δppm: 6.87–6.88 (m, 1H, ArH), 7.48–7.56 (m, 4H, ArH), 7.67 (t, J = 7.36 Hz, 2H, ArH), 7.97–8.08 (m, 1H, ArH), 8.20 (s, 1H, CHolefinic), 11.82 (br s, 1H,  , exchangeable with D2O), 12.43 (br s, 1H,
, exchangeable with D2O), 12.43 (br s, 1H, 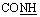 , exchangeable with D2O). 13C-NMR (100 MHz, DMSO-d6) δppm: 102.91, 117.04, 118.04, 121.51, 124.32, 124.76, 127.11 (2C), 129.58 (2C), 131.00, 132.67, 135.63, 141.39, 153.43, 167.98, 170.65. Anal. calcd for C17H11N5O3S (365.37) %: C, 55.89; H, 3.03; N, 19.17; found: C, 55.93; H, 3.10; N, 19.15.N), 1687, 1667 (COamide). 1H-NMR (400 MHz, DMSO-d6) δppm: 7.36 (t, J = 8.76 Hz, 1H, ArH), 7.54 (t, J = 7.84 Hz, 2H, ArH), 7.67 (t, J = 7.4 Hz, 1H, ArH), 7.97–7.99 (m, 3H, ArH), 8.18 (d, J = 4.96 Hz, 1H, ArH), 8.52 (s, 1H, CHolefinic), 11.82 (br s, 1H,
, exchangeable with D2O). 13C-NMR (100 MHz, DMSO-d6) δppm: 102.91, 117.04, 118.04, 121.51, 124.32, 124.76, 127.11 (2C), 129.58 (2C), 131.00, 132.67, 135.63, 141.39, 153.43, 167.98, 170.65. Anal. calcd for C17H11N5O3S (365.37) %: C, 55.89; H, 3.03; N, 19.17; found: C, 55.93; H, 3.10; N, 19.15.N), 1687, 1667 (COamide). 1H-NMR (400 MHz, DMSO-d6) δppm: 7.36 (t, J = 8.76 Hz, 1H, ArH), 7.54 (t, J = 7.84 Hz, 2H, ArH), 7.67 (t, J = 7.4 Hz, 1H, ArH), 7.97–7.99 (m, 3H, ArH), 8.18 (d, J = 4.96 Hz, 1H, ArH), 8.52 (s, 1H, CHolefinic), 11.82 (br s, 1H,  , exchangeable with D2O), 12.41 (br s, 1H,
, exchangeable with D2O), 12.41 (br s, 1H, 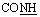 , exchangeable with D2O). 13C-NMR (100 MHz, DMSO-d6) δppm: 109.22, 119.18 (2C), 120.67, 123.35, 126.23, 126.67, 129.11, 131.85, 133.27, 134.76, 137.48, 143.22, 155.37 (2C), 169.99, 172.42. Anal. calcd for C17H11N5O2S2 (381.43) %: C, 53.53; H, 2.91; N, 18.36; found: C, 53.58; H, 2.80; N, 18.25.N), 1669 (CO). 1H-NMR (400 MHz, DMSO-d6) δppm: 7.59 (t, J = 7.32 Hz, 2H, ArH), 7.69 (t, J = 7.2 Hz, 1H, ArH), 8.15 (d, J = 7.48 Hz, 2H, ArH), 8.25 (d, J = 8.16 Hz, 2H, ArH), 8.40–8.45 (m, 2H, ArH + 1H, CHolefinic), 12.55 (br s, 1H, NH, exchangeable with D2O), 13.45 (br s, 1H, NH, exchangeable with D2O). 13C-NMR (100 MHz, DMSO-d6) δppm: 112.94, 113.29, 116.98, 117.83, 129.28, 129.52, 130.28 (2C), 131.05, 131.25, 134.62, 135.78, 148.16, 154.35, 160.65, 161.47, 170.82, 174.14, 174.73. Anal. calcd for C19H12N6O4S (420.40) %: C, 54.28; H, 2.88; N, 19.99; found: C, 54.30; H, 2.85; N, 19.90.N), 1667 (COamide). 1H-NMR (400 MHz, DMSO-d6) δppm: 3.85 (s, 3H, OCH3), 6.96 (d, J = 8.32 Hz, 1H, ArH), 7.56–7.78 (m, 5H, ArH), 8.11–8.15 (m, 2H, ArH + 1H, CHolefinic), 10.24 (br s, 1H, OH, exchangeable with D2O), 12.07 (br s, 1H, NH, exchangeable with D2O), 13.30 (br s, 1H, NH, exchangeable with D2O). 13C-NMR (100 MHz, DMSO-d6) δppm: 65.73, 66.81, 68.92, 116.04, 117.41, 120.44, 123.33, 123.69, 126.08, 128.56, 129.98, 131.66, 134.43, 140.38, 152.45, 158.87, 159.51, 166.93, 168.85, 169.66. Anal. calcd for C20H15N5O4S (421.43) %: C, 57.00; H, 3.59; N, 16.62; found: C, 57.09; H, 3.62; N, 16.58.N), 1648 (CO). 1H-NMR (400 MHz, DMSO-d6) δppm: 3.96 (s, 3H, CH3), 3.99 (s, 3H, CH3), 7.54–8.11 (m, 6H, ArH), 12.87 (br s, 1H, NH, exchangeable with D2O). 13C-NMR (100 MHz, DMSO-d6) δppm: 21.01, 21.13, 115.85, 116.66, 120.90, 123.13, 125.59, 126.71, 127.77, 129.80, 135.19, 152.70, 161.70, 167.67, 170.77, 180.28, 182.01. Anal. calcd for C17H13N5O2S (351.38) %: C, 58.11; H, 3.73; N, 19.93; found: C, 58.20; H, 3.65; N, 19.88.N), 1684 (CO), 1649 (CN). 1H-NMR (400 MHz, DMSO-d6) δppm: 2.15 (s, 3H, CH3), 7.57–8.30 (m, 11H, ArH), 13.37 (br s, 1H, NH, exchangeable with D2O). 13C-NMR (100 MHz, DMSO-d6) δppm: 22.83, 68.92, 116.04 (2C), 117.41, 120.44, 123.33, 123.69, 126.08 (2C), 128.56, 129.98, 131.66, 134.43, 140.38, 152.45, 158.87, 159.51, 166.93, 168.85 (2C), 169.66. Anal. calcd for C22H15N5O2S (413.46) %: C, 63.91; H, 3.66; N, 16.94; found: C, 63.88; H, 3.71; N, 16.81.N), 1734 (COester), br. 1673 (COamide), 1637 (CN). 1H-NMR (400 MHz, DMSO-d6) δppm: 1.19 (t, 3H,
, exchangeable with D2O). 13C-NMR (100 MHz, DMSO-d6) δppm: 109.22, 119.18 (2C), 120.67, 123.35, 126.23, 126.67, 129.11, 131.85, 133.27, 134.76, 137.48, 143.22, 155.37 (2C), 169.99, 172.42. Anal. calcd for C17H11N5O2S2 (381.43) %: C, 53.53; H, 2.91; N, 18.36; found: C, 53.58; H, 2.80; N, 18.25.N), 1669 (CO). 1H-NMR (400 MHz, DMSO-d6) δppm: 7.59 (t, J = 7.32 Hz, 2H, ArH), 7.69 (t, J = 7.2 Hz, 1H, ArH), 8.15 (d, J = 7.48 Hz, 2H, ArH), 8.25 (d, J = 8.16 Hz, 2H, ArH), 8.40–8.45 (m, 2H, ArH + 1H, CHolefinic), 12.55 (br s, 1H, NH, exchangeable with D2O), 13.45 (br s, 1H, NH, exchangeable with D2O). 13C-NMR (100 MHz, DMSO-d6) δppm: 112.94, 113.29, 116.98, 117.83, 129.28, 129.52, 130.28 (2C), 131.05, 131.25, 134.62, 135.78, 148.16, 154.35, 160.65, 161.47, 170.82, 174.14, 174.73. Anal. calcd for C19H12N6O4S (420.40) %: C, 54.28; H, 2.88; N, 19.99; found: C, 54.30; H, 2.85; N, 19.90.N), 1667 (COamide). 1H-NMR (400 MHz, DMSO-d6) δppm: 3.85 (s, 3H, OCH3), 6.96 (d, J = 8.32 Hz, 1H, ArH), 7.56–7.78 (m, 5H, ArH), 8.11–8.15 (m, 2H, ArH + 1H, CHolefinic), 10.24 (br s, 1H, OH, exchangeable with D2O), 12.07 (br s, 1H, NH, exchangeable with D2O), 13.30 (br s, 1H, NH, exchangeable with D2O). 13C-NMR (100 MHz, DMSO-d6) δppm: 65.73, 66.81, 68.92, 116.04, 117.41, 120.44, 123.33, 123.69, 126.08, 128.56, 129.98, 131.66, 134.43, 140.38, 152.45, 158.87, 159.51, 166.93, 168.85, 169.66. Anal. calcd for C20H15N5O4S (421.43) %: C, 57.00; H, 3.59; N, 16.62; found: C, 57.09; H, 3.62; N, 16.58.N), 1648 (CO). 1H-NMR (400 MHz, DMSO-d6) δppm: 3.96 (s, 3H, CH3), 3.99 (s, 3H, CH3), 7.54–8.11 (m, 6H, ArH), 12.87 (br s, 1H, NH, exchangeable with D2O). 13C-NMR (100 MHz, DMSO-d6) δppm: 21.01, 21.13, 115.85, 116.66, 120.90, 123.13, 125.59, 126.71, 127.77, 129.80, 135.19, 152.70, 161.70, 167.67, 170.77, 180.28, 182.01. Anal. calcd for C17H13N5O2S (351.38) %: C, 58.11; H, 3.73; N, 19.93; found: C, 58.20; H, 3.65; N, 19.88.N), 1684 (CO), 1649 (CN). 1H-NMR (400 MHz, DMSO-d6) δppm: 2.15 (s, 3H, CH3), 7.57–8.30 (m, 11H, ArH), 13.37 (br s, 1H, NH, exchangeable with D2O). 13C-NMR (100 MHz, DMSO-d6) δppm: 22.83, 68.92, 116.04 (2C), 117.41, 120.44, 123.33, 123.69, 126.08 (2C), 128.56, 129.98, 131.66, 134.43, 140.38, 152.45, 158.87, 159.51, 166.93, 168.85 (2C), 169.66. Anal. calcd for C22H15N5O2S (413.46) %: C, 63.91; H, 3.66; N, 16.94; found: C, 63.88; H, 3.71; N, 16.81.N), 1734 (COester), br. 1673 (COamide), 1637 (CN). 1H-NMR (400 MHz, DMSO-d6) δppm: 1.19 (t, 3H, 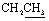 ), 4.06 (q, 2H,
), 4.06 (q, 2H, 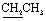 ), 4.59 (s, 2H,
), 4.59 (s, 2H, 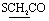 ), 6.75–6.77 (m, 5H, ArH), 7.66–8.07 (m, 5H, ArH), 11.96 (br s, 1H,
), 6.75–6.77 (m, 5H, ArH), 7.66–8.07 (m, 5H, ArH), 11.96 (br s, 1H, 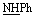 , exchangeable with D2O), 12.85–13.05 (br s, 2H,
, exchangeable with D2O), 12.85–13.05 (br s, 2H, 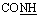 , exchangeable with D2O). Anal. calcd for C23H20N6O4S2 (508.57) %: C, 54.32; H, 3.96; N, 16.53; found: C, 54.25; H, 3.87; N, 16.59.N), 1741 (COKetone), br. 1670 (COamide). 1H-NMR (400 MHz, DMSO-d6) δppm: 4.10 (s, 2H,
, exchangeable with D2O). Anal. calcd for C23H20N6O4S2 (508.57) %: C, 54.32; H, 3.96; N, 16.53; found: C, 54.25; H, 3.87; N, 16.59.N), 1741 (COKetone), br. 1670 (COamide). 1H-NMR (400 MHz, DMSO-d6) δppm: 4.10 (s, 2H, 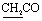 ), 6.72–6.77 (m, 5H, ArH), 7.64–8.05 (m, 5H, ArH), 12.95 (br s, 1H, NH, exchangeable with D2O), 14.69 (br s, H, NH, exchangeable with D2O). Anal. calcd for C21H14N6O3S2 (462.50) %: C, 54.54; H, 3.05; N, 18.17; found: C, 54.51; H, 3.13; N, 18.11.N), 1740 (COKetone), 1672 (COamide), 1638 (CN). 1H-NMR (400 MHz, DMSO-d6) δppm: 4.62 (s, 2H,
), 6.72–6.77 (m, 5H, ArH), 7.64–8.05 (m, 5H, ArH), 12.95 (br s, 1H, NH, exchangeable with D2O), 14.69 (br s, H, NH, exchangeable with D2O). Anal. calcd for C21H14N6O3S2 (462.50) %: C, 54.54; H, 3.05; N, 18.17; found: C, 54.51; H, 3.13; N, 18.11.N), 1740 (COKetone), 1672 (COamide), 1638 (CN). 1H-NMR (400 MHz, DMSO-d6) δppm: 4.62 (s, 2H, 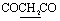 ), 7.49–7.59 (m, 5H, ArH), 7.84–8.04 (m, 5H, ArH), 11.89 (br s, 1H, NH, exchangeable with D2O), 12.50 (br s, 1H, NH, exchangeable with D2O). Anal. calcd for C22H14N6O4S2 (490.51) %: C, 53.87; H, 2.88; N, 17.13; found: C, 53.96; H, 2.94; N, 17.09.N), 1673 (COamide). 1H-NMR (400 MHz, DMSO-d6) δppm: 3.75–3.85 (m, 4H,
), 7.49–7.59 (m, 5H, ArH), 7.84–8.04 (m, 5H, ArH), 11.89 (br s, 1H, NH, exchangeable with D2O), 12.50 (br s, 1H, NH, exchangeable with D2O). Anal. calcd for C22H14N6O4S2 (490.51) %: C, 53.87; H, 2.88; N, 17.13; found: C, 53.96; H, 2.94; N, 17.09.N), 1673 (COamide). 1H-NMR (400 MHz, DMSO-d6) δppm: 3.75–3.85 (m, 4H, 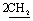 ), 6.72–6.77 (m, 5H, ArH), 7.55–8.05 (m, 5H, ArH), 9.21 (br s, 1H, NH, exchangeable with D2O), 13.01 (br s, 1H, NH, exchangeable with D2O). Anal. calcd for C21H16N6O2S2 (448.52) %: C, 56.24; H, 3.60; N, 18.74; found: C, 56.29; H, 3.55; N, 18.64.N), 1688 (COamide). 1H-NMR (400 MHz, DMSO-d6) δppm: 7.05–7.59 (m, 10H, ArH), 12.47 (br s, 1H,
), 6.72–6.77 (m, 5H, ArH), 7.55–8.05 (m, 5H, ArH), 9.21 (br s, 1H, NH, exchangeable with D2O), 13.01 (br s, 1H, NH, exchangeable with D2O). Anal. calcd for C21H16N6O2S2 (448.52) %: C, 56.24; H, 3.60; N, 18.74; found: C, 56.29; H, 3.55; N, 18.64.N), 1688 (COamide). 1H-NMR (400 MHz, DMSO-d6) δppm: 7.05–7.59 (m, 10H, ArH), 12.47 (br s, 1H, 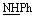 , exchangeable with D2O), 13.74 (br s, 1H, NH, exchangeable with D2O). 13C-NMR (100 MHz, DMSO-d6) δppm: 68, 116, 117, 120, 123.32, 123.76, 126, 128, 130, 131, 134, 140, 152, 166, 169, 180. Anal. calcd for C19H12N6O2S (388.41) %: C, 58.76; H, 3.11; N, 21.64; found: C, 58.65; H, 3.14; N, 21.59.N), 1669 (COamide). 1H-NMR (400 MHz, DMSO-d6) δppm: 3.5 (s, 1H,
, exchangeable with D2O), 13.74 (br s, 1H, NH, exchangeable with D2O). 13C-NMR (100 MHz, DMSO-d6) δppm: 68, 116, 117, 120, 123.32, 123.76, 126, 128, 130, 131, 134, 140, 152, 166, 169, 180. Anal. calcd for C19H12N6O2S (388.41) %: C, 58.76; H, 3.11; N, 21.64; found: C, 58.65; H, 3.14; N, 21.59.N), 1669 (COamide). 1H-NMR (400 MHz, DMSO-d6) δppm: 3.5 (s, 1H,  ), 7.46–7.67 (m, 5H, ArH), 8.12–8.13 (m, 5H, ArH), 9.24 (br s, 1H, NH, exchangeable with D2O), 13.10 (br s, 1H, NH, exchangeable with D2O), 13.15 (br s, 1H,
), 7.46–7.67 (m, 5H, ArH), 8.12–8.13 (m, 5H, ArH), 9.24 (br s, 1H, NH, exchangeable with D2O), 13.10 (br s, 1H, NH, exchangeable with D2O), 13.15 (br s, 1H, 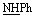 , exchangeable with D2O). Anal. calcd for C19H14N6O2S2 (422.48) %: C, 54.02; H, 3.34; N, 19.89; found: C, 54.05; H, 3.36; N, 19.85.N), 1664 (COamide). 1H-NMR (400 MHz, DMSO-d6) δppm: 2.67 (s, 3H,
, exchangeable with D2O). Anal. calcd for C19H14N6O2S2 (422.48) %: C, 54.02; H, 3.34; N, 19.89; found: C, 54.05; H, 3.36; N, 19.85.N), 1664 (COamide). 1H-NMR (400 MHz, DMSO-d6) δppm: 2.67 (s, 3H, 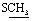 ), 2.70 (s, 3H,
), 2.70 (s, 3H, 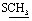 ), 7.51–8.26 (m, 5H, ArH), 13.25 (br s, 1H,
), 7.51–8.26 (m, 5H, ArH), 13.25 (br s, 1H,  , exchangeable with D2O), 14.74 (br s, 1H,
, exchangeable with D2O), 14.74 (br s, 1H, 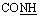 , exchangeable with D2O). 13C-NMR (100 MHz, DMSO-d6) δppm: 20.98 (2C), 69.90, 117.06, 118.04, 121.52, 127.10, 129.60, 131.00, 132.68, 133.65, 141.38, 153.43, 167.99, 170.64. Anal. calcd for C15H13N5O2S3 (391.48) %: C, 46.02; H, 3.35; N, 17.89; found: C, 46.15; H, 3.42; N, 17.93.:3] affording compounds 18–20, respectively.N), br. 1688 (COamide). 1H-NMR (400 MHz, DMSO-d6) δppm: 7.18–7.20 (m, 2H, ArH), 7.36–7.68 (m, 5H, ArH), 8.12 (d, J = 7.52 Hz, 2H, ArH), 11.24 (br s, 1H,
, exchangeable with D2O). 13C-NMR (100 MHz, DMSO-d6) δppm: 20.98 (2C), 69.90, 117.06, 118.04, 121.52, 127.10, 129.60, 131.00, 132.68, 133.65, 141.38, 153.43, 167.99, 170.64. Anal. calcd for C15H13N5O2S3 (391.48) %: C, 46.02; H, 3.35; N, 17.89; found: C, 46.15; H, 3.42; N, 17.93.:3] affording compounds 18–20, respectively.N), br. 1688 (COamide). 1H-NMR (400 MHz, DMSO-d6) δppm: 7.18–7.20 (m, 2H, ArH), 7.36–7.68 (m, 5H, ArH), 8.12 (d, J = 7.52 Hz, 2H, ArH), 11.24 (br s, 1H,  , exchangeable with D2O), 12.5 (br s, 2H,
, exchangeable with D2O), 12.5 (br s, 2H, 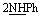 , exchangeable with D2O), 14.52 (br s, 1H,
, exchangeable with D2O), 14.52 (br s, 1H, 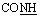 , exchangeable with D2O). Anal. calcd for C19H13N7O2S (403.42) %: C, 56.57; H, 3.25; N, 24.30; found: C, 56.47; H, 3.32; N, 24.41.N), br. 1665 (COamide), 1634 (CN). 1H-NMR (400 MHz, DMSO-d6) δppm: 6.73 (t, J = 7.24 Hz 1H, ArH), 6.96 (d, J = 8.72 Hz, 2H, ArH), 7.07 (d, J = 7.72 Hz, 1H, ArH), 7.21 (t, J = 8.24 Hz, 4H, ArH), 7.60 (d, J = 8.72 Hz, 1H, ArH), 10.15 (br s, 1H,
, exchangeable with D2O). Anal. calcd for C19H13N7O2S (403.42) %: C, 56.57; H, 3.25; N, 24.30; found: C, 56.47; H, 3.32; N, 24.41.N), br. 1665 (COamide), 1634 (CN). 1H-NMR (400 MHz, DMSO-d6) δppm: 6.73 (t, J = 7.24 Hz 1H, ArH), 6.96 (d, J = 8.72 Hz, 2H, ArH), 7.07 (d, J = 7.72 Hz, 1H, ArH), 7.21 (t, J = 8.24 Hz, 4H, ArH), 7.60 (d, J = 8.72 Hz, 1H, ArH), 10.15 (br s, 1H, 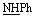 , exchangeable with D2O), 12.87 (br s, 1H, NH, exchangeable with D2O), 14.14 (br s, 1H, NH, exchangeable with D2O). 13C-NMR (100 MHz, DMSO-d6) δppm: 55.62, 112.29 (2C), 114.63 (2C), 118.80, 127.50, 128.99 (2C), 129.53 (2C), 137.06 (2C), 146.06, 154.68, 155.99 (2C), 159.78, 185.65. Anal. calcd for C19H12N6O3S (404.40) %: C, 56.43; H, 2.99; N, 20.78; found: C, 56.39; H, 2.85; N, 20.81.N), br. 1665 (COamide), 1633 (CN). 1H-NMR (400 MHz, DMSO-d6) δppm: 7.21 (d, J = 9.12 Hz, 1H, ArH), 7.36–7.39 (m, 2H, ArH), 7.59–7.63 (m, 2H, ArH), 8.01 (t, J = 6.8 Hz, 1H, ArH), 8.63 (d, J = 8.3 Hz, 2H, ArH), 11.9 (br s, 1H, NH, exchangeable with D2O), 12.34 (br s, 1H, NH, exchangeable with D2O). 13C-NMR (100 MHz, DMSO-d6) δppm: 66.81, 116.04, 117.41, 120.44, 123.33, 123.69, 126.08, 128.56, 129.98, 131.66, 134.43, 158.87, 159.51, 166.93, 168.85, 169.66, 185.16, 192.83. Anal. calcd for C18H10N6O4S (406.38) %: C, 53.20; H, 2.48; N, 20.68; found: C, 53.18H, 2.55; N, 20.74.
, exchangeable with D2O), 12.87 (br s, 1H, NH, exchangeable with D2O), 14.14 (br s, 1H, NH, exchangeable with D2O). 13C-NMR (100 MHz, DMSO-d6) δppm: 55.62, 112.29 (2C), 114.63 (2C), 118.80, 127.50, 128.99 (2C), 129.53 (2C), 137.06 (2C), 146.06, 154.68, 155.99 (2C), 159.78, 185.65. Anal. calcd for C19H12N6O3S (404.40) %: C, 56.43; H, 2.99; N, 20.78; found: C, 56.39; H, 2.85; N, 20.81.N), br. 1665 (COamide), 1633 (CN). 1H-NMR (400 MHz, DMSO-d6) δppm: 7.21 (d, J = 9.12 Hz, 1H, ArH), 7.36–7.39 (m, 2H, ArH), 7.59–7.63 (m, 2H, ArH), 8.01 (t, J = 6.8 Hz, 1H, ArH), 8.63 (d, J = 8.3 Hz, 2H, ArH), 11.9 (br s, 1H, NH, exchangeable with D2O), 12.34 (br s, 1H, NH, exchangeable with D2O). 13C-NMR (100 MHz, DMSO-d6) δppm: 66.81, 116.04, 117.41, 120.44, 123.33, 123.69, 126.08, 128.56, 129.98, 131.66, 134.43, 158.87, 159.51, 166.93, 168.85, 169.66, 185.16, 192.83. Anal. calcd for C18H10N6O4S (406.38) %: C, 53.20; H, 2.48; N, 20.68; found: C, 53.18H, 2.55; N, 20.74.2.2. Biological evaluation
2.3. In silico studies
3. Results and discussion
3.1 Chemistry
As a part of our ongoing research that aims at developing simple and efficient synthetic techniques for the synthesis of novel heterocyclic compounds with potential anticancer applications,21–31 the present study reported the synthesis of numerous new substituted 1,3,4-thiadiazole derivatives incorporating benzene moiety. N-(5-Amino-1,3,4-thiadiazol-2-yl)benzamide (2) was synthesized as a sole product through reaction of benzoylisothiocyanate with thiosemicarbzide in dry acetonitrile and then underwent cyclization according to the previously published methodology32–36 (Scheme 1). Treatment of compound 2 with ethyl cyanoacetate in the presence of a catalytic amount of triethylamine afforded N-(5-(2-cyanoacetamido)-1,3,4-thiadiazol-2-yl)benzamide (3) in a high yield, which was used as a key starting material (Scheme 1). The structures of compounds 2 and 3 were deduced from their elemental and spectral analyses.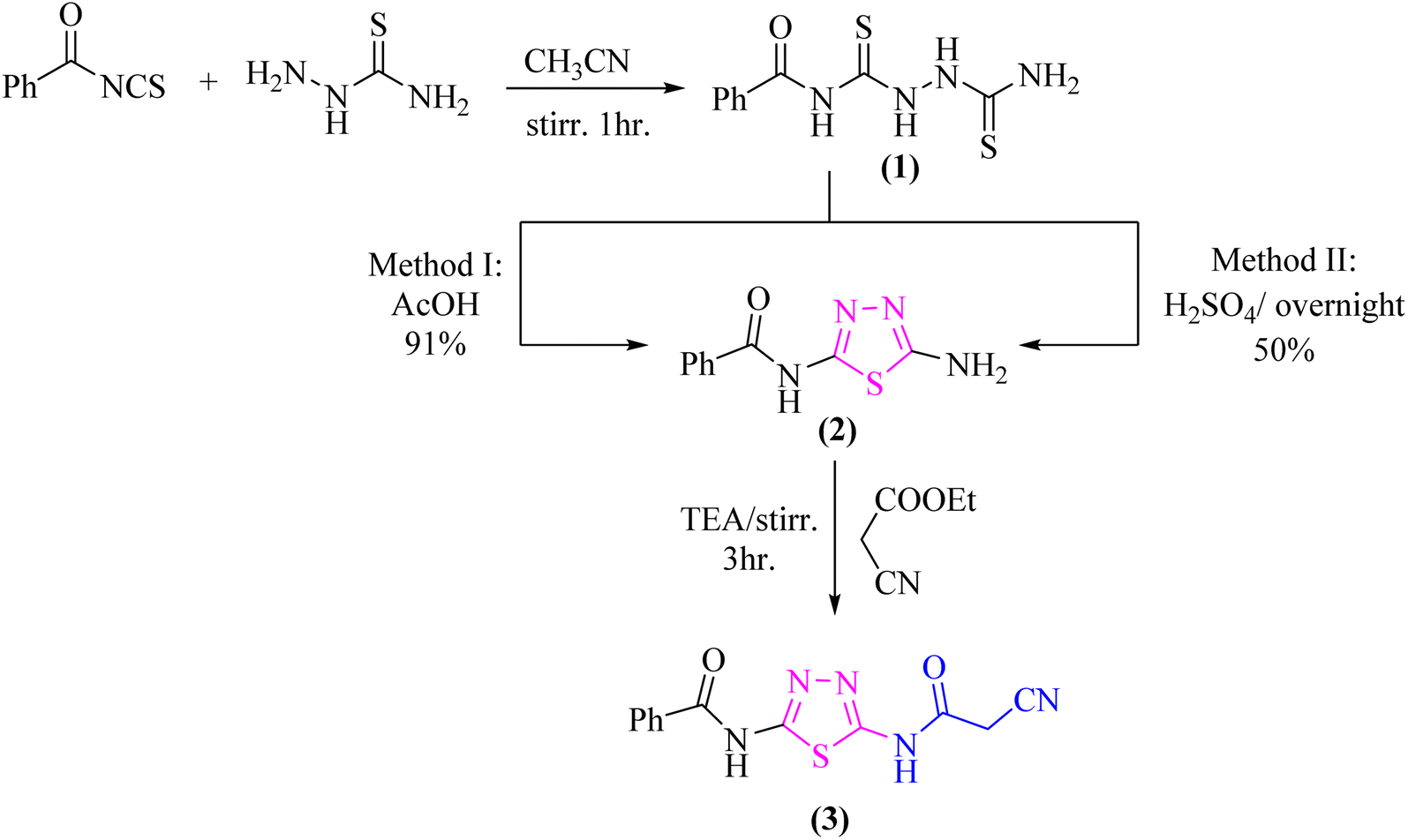 | ||
| Scheme 1 Synthesis of 1,3,4-thiadiazole derivative 3. | ||
The nucleophilic reactivity of the intermediate 3 towards various carbon electrophiles has been investigated. Refluxing compound 3 with salicylaldehyde in an ethanolic solution containing a catalytic amount of piperidine yielded 2-iminocoumarin derivative 4 (Scheme 2). The presence of absorption bands for –NH groups at 3409 and 3158 cm−1 along with the absence of absorption band for the nitrile group confirmed the assigned structure 4. In addition, acidification of the filtrate gave the expected coumarin derivative 5 (Scheme 2). Ample evidence for the assigned structure 5 was forthcoming from the correct elemental analysis and spectral data. The presence of a high frequency CO stretching at 1713 cm−1 was attributed to the coumarin oxo-group in the IR spectrum as well as the absence of NH proton of imino group at 11.76 ppm in the 1H-NMR spectrum of compound 5 agrees well with the proposed structure.
 | ||
| Scheme 2 Reaction of the β-ketonitrile 3 with some aromatic aldehydes and arylidene derivatives. | ||
Subjecting compound 3 to react with 2-(4-methoxybenzylidene)malononitrile gave a grey crystalline product upon reflux which was identified as 2-oxopyridine thiadiazole derivative 6a (major product, Scheme 2), while acidification of the reaction filtrate with cold dilute HCl furnished an orange precipitate that was detected as the arylidene derivative 6b (minor product). Furthermore, the Knoevenagel condensation of 1,3,4-thiadiazole derivative 3 with aromatic aldehydes namely; 2,4-dichlorobenzaldehde, p-chlorobenzaldehyde, furan-2-carbaldehyde, thiophene-2-carbaldehyde, p-nitro benzaldehyde, or 4-hydroxy-3-methoxy benzaldehyde for three hours in refluxing ethanol containing a catalytic amount of piperidine afforded the corresponding arylidene derivatives 7a–f, respectively (Scheme 2). Moreover, the reaction of compound 3 with ethyl 3-(4-chlorophenyl)-2-cyanoacrylate under the same reaction conditions afforded the arylidene derivative 7b rather than 2-oxopyridin derivative 8.
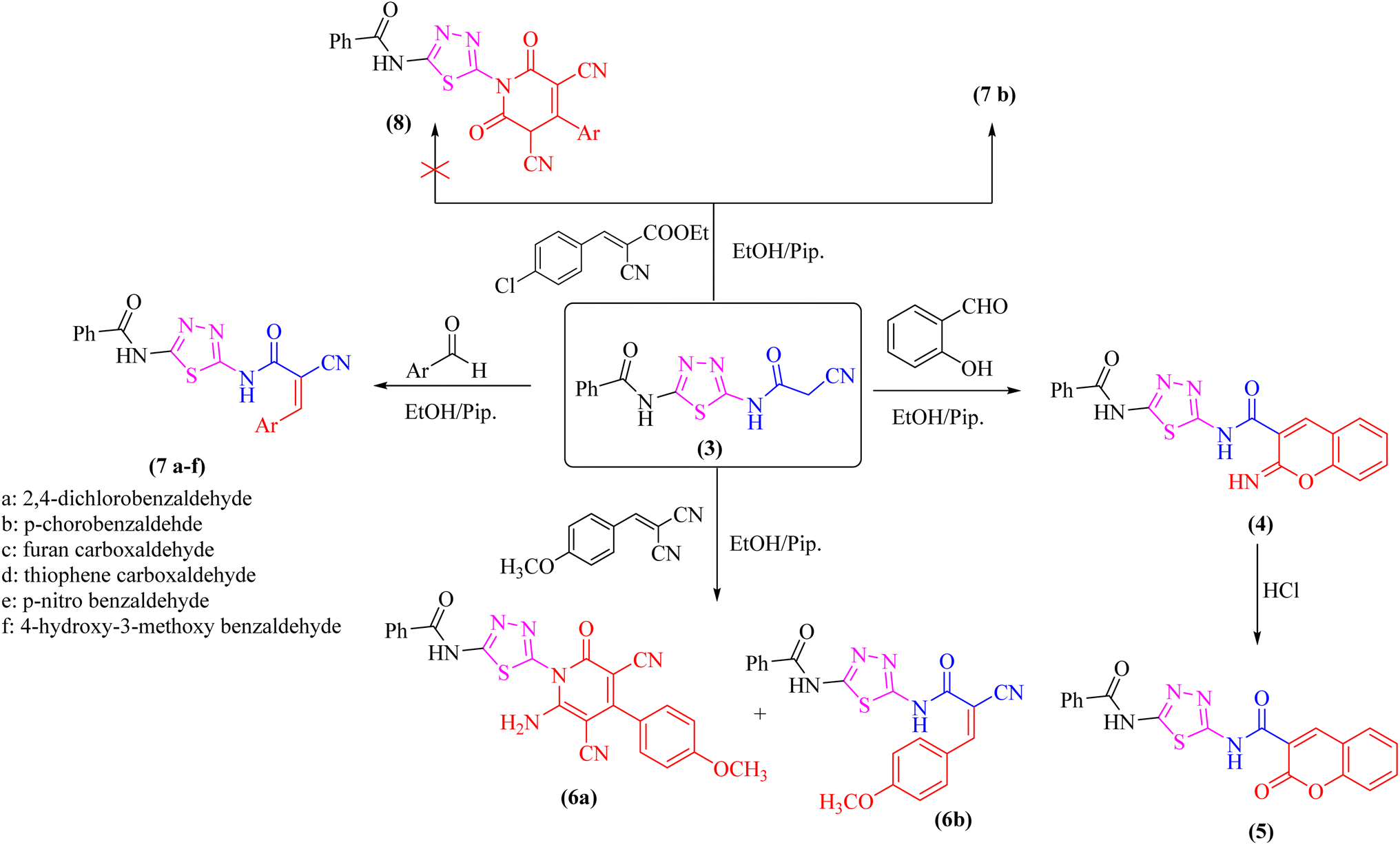
The proclivity of thiadiazole derivative 3 towards β-diketones was also investigated in this study. Thus, 2-pyridone derivatives 9 and 10 were obtained from the reaction of 3 with acetyl acetone and benzoyl acetone, respectively in ethanolic solution containing catalytic amount of fused sodium acetate (Scheme 3). 1H-NMR spectrum of compound 9 showed signals at 3.96 and 3.99 ppm which were attributed to protons of CH3 group, as well as the absence of a singlet signal for protons of CH2CN group which confirmed the proposed structure 9. Furthermore, the 13C-NMR spectrum revealed the presence of two methyl groups at δ 21.01 and 21.12 ppm (Scheme 3).
 | ||
| Scheme 3 Synthesis of some new 2-pyridone derivatives (9 and 10) from the cyanoacetamide 3. | ||
At room temperature, the active methylene moiety in the cyanoacetamide derivative 3 readily adds to one carbon donor such as phenyl isothiocyanate in DMF-containing potassium hydroxide forming a non-isolable intermediate potassium thiocarbamate salt (A), which in situ reacted with ethyl chloroacetate, chloroacetyl chloride, diethylmalonate, 1,2-dibromo ethane, methyliodide, or hydrochloric acid giving the corresponding ethyl thioacetate, 1,3-thiazolone, 4,6-dioxo-1,3-thiazinan, 1,3-thiazole, thiadiazolo[3,2-a]pyrimidin, and thioxopropanamide derivatives 11–16, respectively (Scheme 4). The structures of derivatives 11–16 were clarified using elemental analysis and spectral data (c.f. experimental). For instance, the appearance of strong absorption bands in the IR spectrum at 2195 and 1734 cm−1 for CN and COester groups, respectively was completely consistent with the suggested structure 11 and ruled out the cyclized compound 12.
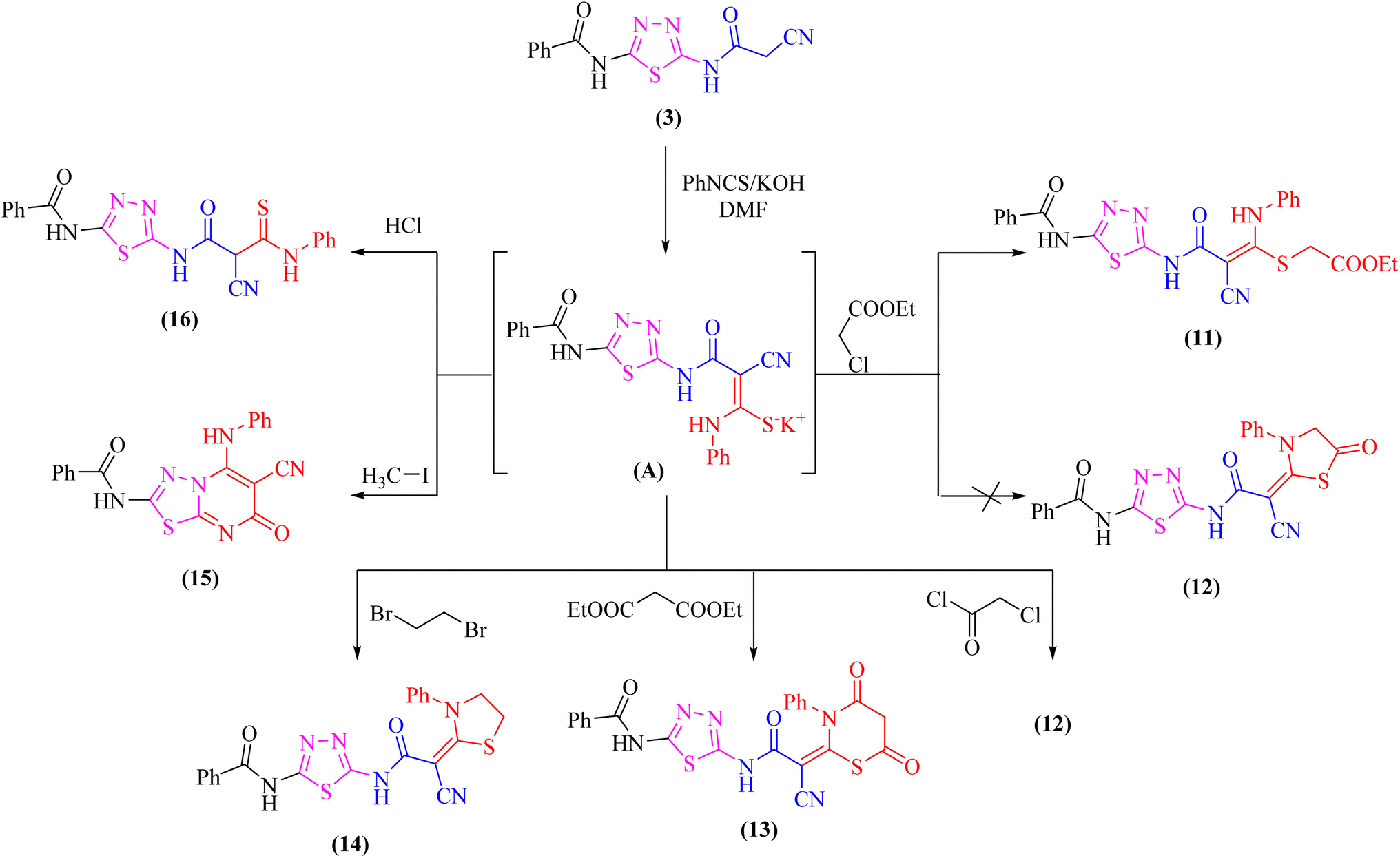 | ||
| Scheme 4 Reaction of potassium thiocarbamate (A) with some electrophiles. | ||
Likewise, addition of 1,3,4-thiadiazole derivative 3 to carbon disulfide in DMF-containing potassium hydroxide at room temperature gave the non-isolable dipotassium dithiocarbazate salt (B), which in situ reacted with methyl iodide affording the ketene S,S-dithioacetal 17 (Scheme 5). The elemental analysis and spectral data were used to determine the structure of compound 17.
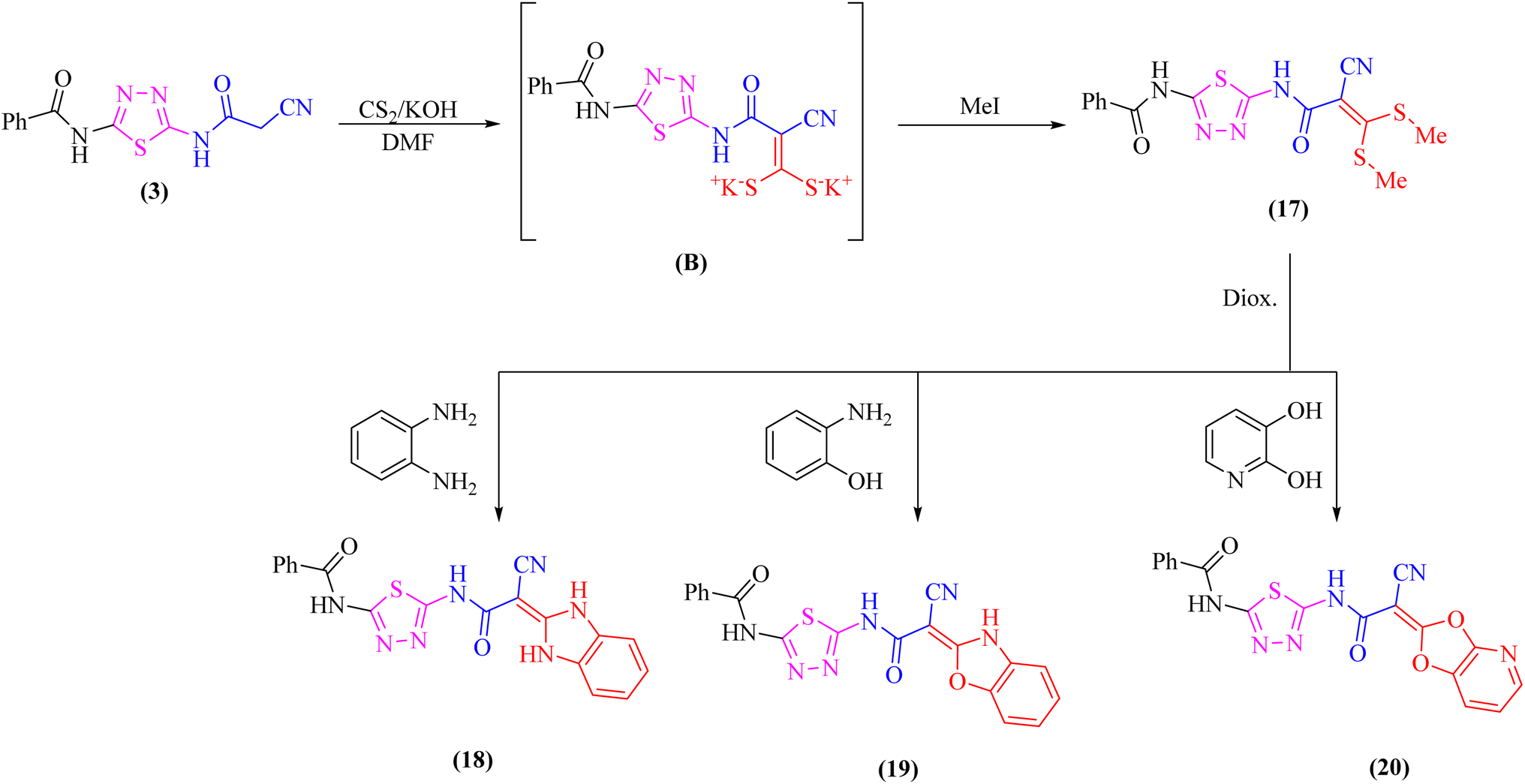 | ||
| Scheme 5 Treatment of the ketene S,S-dithioacetal 17 with some bidentate nucleophiles. | ||
Further, the reactivity of 17 towards some nitrogen and oxygen nucleophiles was investigated. Thus, compound 17 reacted under reflux with bidentate nucleophiles such as o-phenylenediamine, o-aminophenol, or 2,3-dihydroxy pyridine giving the corresponding benzimidazole derivative 18, benzoxazole derivative 19, and dioxolo[4,5-b]pyridine derivative 20, respectively (Scheme 5). The structures of compounds 18–20 were consistent with the 1H-NMR and 13C-NMR spectra which revealed the absence of two SCH3 groups.
3.2 Biological evaluation
| Cpds | MCF-7 | HCT-116 | PC-3 | HepG2 | WI-38 | SIc |
|---|---|---|---|---|---|---|
| a The IC50 was calculated from a dose–response curve. Doxorubicin (Dox) was used as a positive control. The data are expressed as mean ±SEM from three independent experiments.b Significant (less than 10 μM) antiproliferative activity as compared to the untreated cells.c SI: selectivity index = WI-38 IC50/IC50 against cancer cells. | ||||||
| DOX | 4.17 ± 0.2b | 5.23 ± 0.3b | 8.87 ± 0.6b | 4.50 ± 0.2b | 6.72 ± 0.5b | 1.2 |
| 2 | 12.36 ± 3.0 | 15.26 ± 2.8 | 30.01 ± 3.1 | 14.11 ± 1.5 | 17.56 ± 4.1 | 1.0 |
| 3 | 23.81 ± 1.9 | 25.72 ± 1.9 | 39.23 ± 2.8 | 31.26 ± 5.1 | 37.41 ± 2.5 | 1.2 |
| 4 | 8.25 ± 3.5b | 15.84 ± 3.2 | 20.97 ± 3.3 | 48.51 ± 2.9 | 29.35 ± 2.2 | 1.3 |
| 5 | 64.12 ± 3.8 | 47.22 ± 3.5 | 66.53 ± 3.9 | 63.93 ± 8.9 | 91.23 ± 4.6 | 1.5 |
| 6a | 41.57 ± 2.7 | 29.19 ± 2.3 | 35.76 ± 2.6 | 54.62 ± 6.8 | 18.64 ± 1.4 | 0.5 |
| 6b | 9.48 ± 0.8b | 8.15 ± 0.7b | 7.41 ± 0.6b | 15.88 ± 1.3 | 50.39 ± 0.9 | 4.9 |
| 7a | 5.51 ± 0.4b | 3.97 ± 0.2b | 9.10 ± 0.8b | 6.34 ± 0.9b | 9.18 ± 3.6b | 1.5 |
| 7b | 17.73 ± 0.6 | 16.18 ± 0.4 | 21.86 ± 1.1 | 14.68 ± 2.1 | 42.06 ± 2.7 | 2.4 |
| 7c | 28.13 ± 3.5 | 52.63 ± 3.7 | 13.30 ± 3.6 | 16.34 ± 1.8 | 22.57 ± 3.0 | 0.8 |
| 7d | 8.02 ± 1.4b | 34.14 ± 2.6 | 11.12 ± 4.7 | 66.10 ± 3.5 | 12.28 ± 1.2 | 0.4 |
| 7e | 45.11 ± 4.0 | 51.39 ± 4.1 | 75.22 ± 4.7 | 56.33 ± 7.8 | 23.39 ± 3.6 | 0.4 |
| 7f | 13.59 ± 1.1 | 10.92 ± 0.9 | 18.35 ± 1.5 | 21.26 ± 3.7 | 14.54 ± 0.3 | 0.9 |
| 9 | 14.13 ± 1.2 | 12.78 ± 1.1 | 15.38 ± 1.3 | 39.07 ± 2.7 | 50.39 ± 0.9 | 2.5 |
| 10 | 72.41 ± 4.1 | 68.01 ± 3.9 | 86.47 ± 4.8 | 19.40 ± 1.5 | >100 | 1.7 |
| 11 | 69.70 ± 3.9 | 63.12 ± 3.8 | 71.94 ± 4.0 | 52.87 ± 4.6 | 22.65 ± 3.8 | 0.4 |
| 12 | 84.81 ± 4.4 | 91.27 ± 4.6 | >100 | 69.54 ± 6.5 | >100 | 1.2 |
| 13 | 37.95 ± 3.2 | 46.14 ± 3.7 | 73.56 ± 4.3 | 45.21 ± 6.3 | 23.58 ± 3.2 | 0.5 |
| 14 | 36.23 ± 2.4 | 40.18 ± 3.1 | 92.28 ± 3.2 | >100 | 53.75 ± 3.4 | 0.8 |
| 15 | 12.74 ± 2.1 | 17.03 ± 1.4 | 10.49 ± 1.9 | 16.84 ± 1.7 | 19.38 ± 1.5 | 1.4 |
| 16 | 19.58 ± 1.6 | 21.62 ± 1.7 | 28.80 ± 2.3 | 14.68 ± 2.3 | 85.91 ± 1.9 | 4.1 |
| 17 | 15.10 ± 1.3 | 14.67 ± 0.9 | 12.17 ± 0.8 | 12.47 ± 1.0 | 81.20 ± 4.1 | 6.0 |
| 18 | 84.65 ± 4.6 | 56.85 ± 3.6 | 81.82 ± 4.5 | 47.33 ± 8.2 | >100 | 1.5 |
| 19 | 7.70 ± 0.5b | 5.98 ± 0.3b | 6.35 ± 0.5b | 19.40 ± 1.5 | 60.28 ± 3.7 | 6.1 |
| 20 | 36.78 ± 2.7 | 18.32 ± 1.7 | 31.39 ± 2.2 | 29.64 ± 3.1 | 42.74 ± 2.7 | 1.5 |
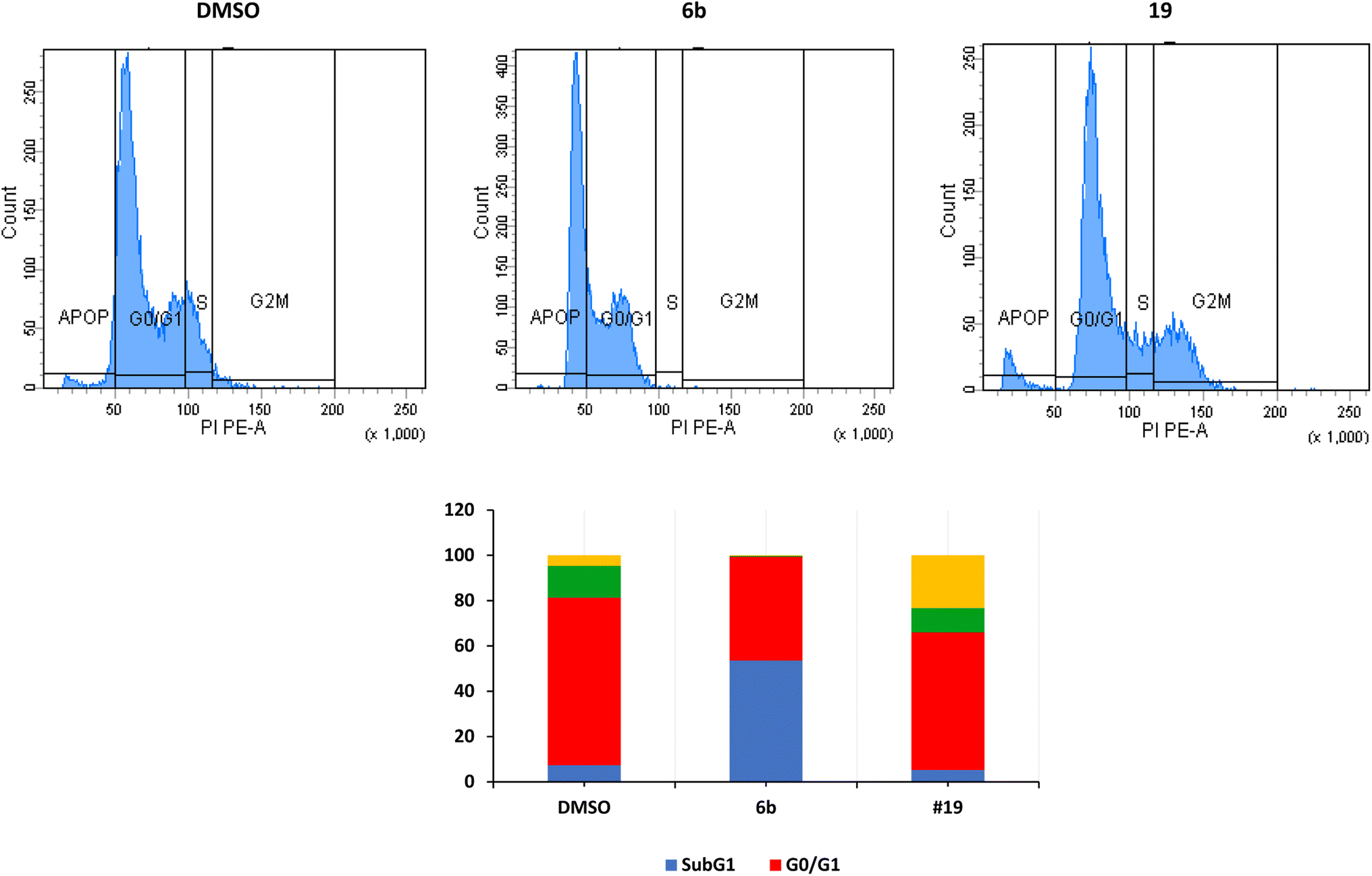 | ||
| Fig. 2 Cell cycle analysis of MCF7 after treatment with derivatives 6b and 19. Data are expressed as means ±SEM (n = 3). The top panel represents one sample, and the bottom figure represents the average of 3 independent experiments expressed as percentage of cells in every stage. | ||
The arrest of cells at the G2/M indicates a severe DNA damage that is beyond repair.38 The crucial event in cell-cycle progression through the G2/M checkpoint is the upregulation of expression of Chk2 protein and increase in the phosphorylation of Chk2 and CDC25C, leading to inhibition of the formation CDK1/cyclin B complex and cell cycle arrest at the G2/M phase.39,40 Many proteins such as p53, p21, and p38/MAPK could contribute to the arrest at the G2/M phase.41 p53 induces the transcription of p21 which inhibits CDK resulting in the arrest of cells at the G2/M.42,43 In the current study, we have not investigated these different pathways.
We have also measured apoptosis by annexin V-PI assay. The level of apoptotic cells either in the early stage (lower-right quadrant, LR) or late apoptotic stage (upper-right quadrant, UR), in addition to the necrotic cells in the upper left (UL) quadrant were all quantified by flow cytometry (Fig. 3). It is evident that compound 6b did not induce apoptosis and increased the necrotic cells by 12.5% compared to 0.1% when the cells were treated with DMSO. This confirms the data we have obtained from the cell cycle analysis. As expected, compound 19 significantly increased the early apoptosis to 15% and increased the necrotic cells to 15% (Fig. 3). Compound 19 evidently arrested the cells at the G2/M forcing them to apoptosis possibly through induction of p53 and different caspases.44–46 Derivatives 6b and 19 induced necrosis in addition to the apoptosis caused by derivative 19. Although the importance of necrosis in the cancer therapy is not as fully understood as apoptosis, recent studies suggest that necrosis or necroptosis have potential applications as anticancer target.47,48
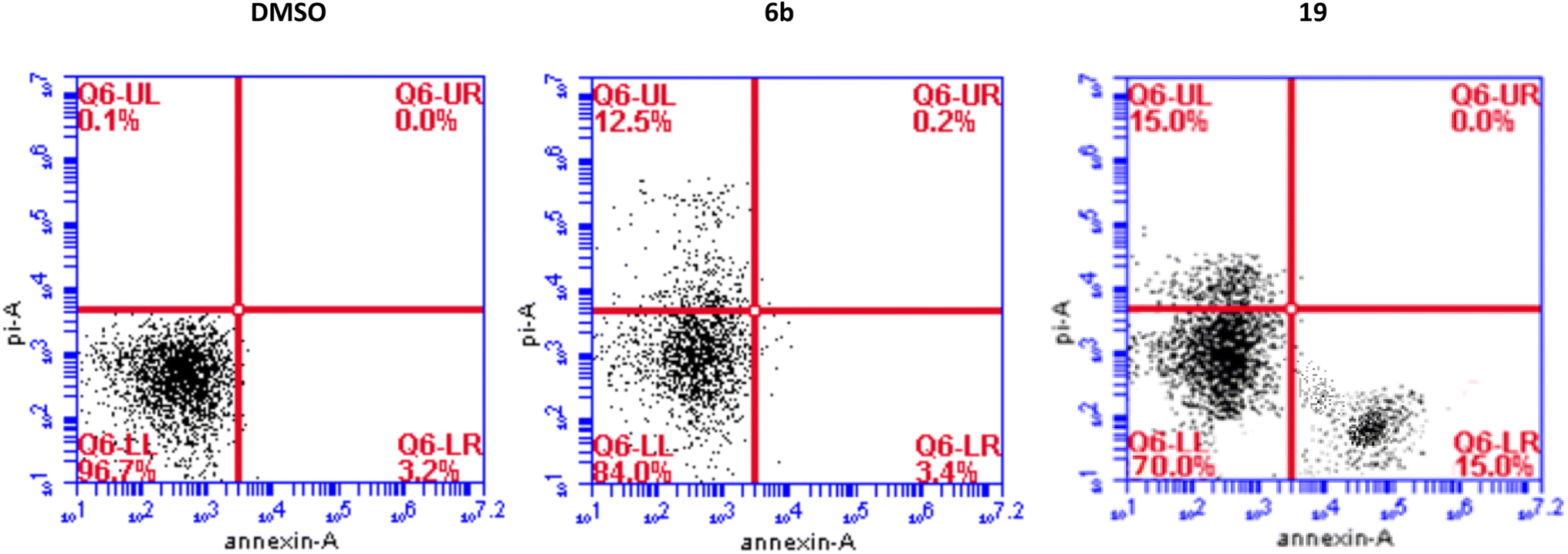 | ||
| Fig. 3 Detection of apoptosis by derivatives 6b and 19 in MCF7 cells by flow cytometry. | ||
3.3. In silico studies
It was shown that FB8, a native co-crystallized inhibitor of the CDK-1, forms four H-bonds with the amino acids Glu81, Leu83, Leu83 and Lys89, one ionic bond with Asp146, and one pi-bond with Phe80.49 More thorough research and comparisons with the docked FB8 of the CDK-1 binding pocket was conducted on the two most promising biologically active newly synthesized thiadiazol derivatives (6b and 19). After looking at the binding modes of the aforementioned compounds (Fig. 4), we concluded that:
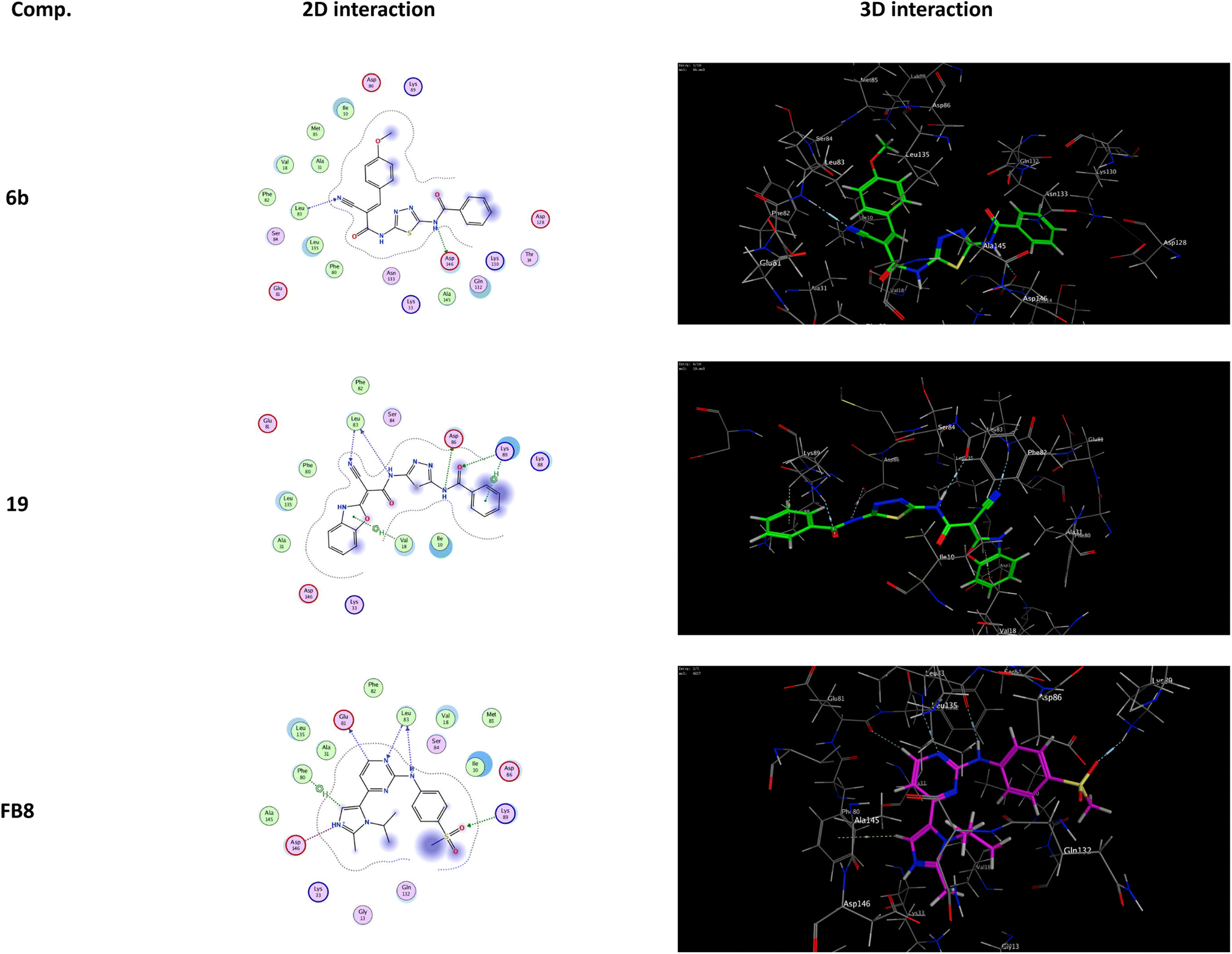 | ||
| Fig. 4 The interactions of the most promising derivatives (6b and 19) at CDK-1 binding pocket, alongside the docked co-crystallized inhibitor (FB8), are depicted in 2D and 3D interactions. H-bonds (dashed blue lines), ionic bond (red dashed lines) and H–pi bonds (green dashed lines). | ||
(a) Compound 6b formed a H-bond donor with the Asp146 amino acids at 2.97 Å and a H-bond acceptor with the Leu83 amino acid at 3.17 Å when positioned inside the largest binding pocket of the CDK-1 (S = −7.220 kcal mol−1 and RMSD = 1.713). It is not a good inhibitor of the enzyme.
(b) Compound 19 formed two H-bonds donor with Leu83 and Asp86 amino acids at 3.53 and 2.93 Å, respectively. Furthermore, it formed two H-bonds acceptor with Lys89 and Leu83 amino acid at 3.03 and 3.18 Å, respectively, as well as two pi–H bonds with Val18 and Lys89 amino acids at 3.78 and 3.66 Å, respectively inside the binding site of the CDK-1 (S = −6.671 kcal mol−1 and RMSD = 1.067). Therefore, compound 19 is a good CDK1 inhibitor candidate.
(c) Finally, it was shown that the docked co-crystallized inhibitor of the CDK-1 (FB8, S = −6.787 kcal mol−1 and RMSD = 1.124) formed two H-bonds donor with Glu81 and Leu83 amino acid at 3.39 and 2.99 Å, respectively. Furthermore, it formed two H-bonds acceptor with Leu83 and Lys89 amino acid at 3.42 and 3.13 Å, respectively. Also, formed one pi–H bond with Phe80 amino acids at 4.42 Å. In addition to one ionic bond with Asp146 amino acid at 4.42 Å.
P < 5. More than one of these requirements must be met for a compound to have a high level of bioavailability. Additionally, it was found that the number of rotatable bonds and the reduction in molecular flexibility were important indicators of excellent oral bioavailability.53 For substances with ten or fewer rotatable bonds and a molecular polar surface area of 140 Å or less, oral bioavailability is likely to be high.54Except for compound 11, which violates more than one of the Lipinski's rule of five, all tested, and reference compound (FB8) listed in Table 2 displayed drug-like molecular (DLM) nature, according to the study. Only one of Lipinski's rule of five was violated and only by compound 11. All other investigated molecules had logP, molecular weight, number of H-bond donors, and number of H-bond acceptors less than 5, 500, 5, and 10, respectively. Except for compound 11, which showed 13 rotatable bonds, all the examined molecules' topology polar surface areas (TPSA) and numbers of ratable bonds fall within the acceptable ranges of less than 10 and 140 Å, respectively.
| Comp | Lipinski's rule of five | Verber's rule | |||||
|---|---|---|---|---|---|---|---|
| logP |
Mol. wt | HBD | HBA | Violation of Lipinski's rule | Number of rotatable bonds | TPSA | |
| 2 | 1.13 | 220.25 | 2 | 3 | 0 | 3 | 109.14 |
| 3 | 0.95 | 287.30 | 2 | 5 | 0 | 6 | 136.01 |
| 4 | 2.66 | 391.40 | 3 | 6 | 0 | 6 | 149.21 |
| 5 | 2.57 | 392.39 | 2 | 6 | 0 | 6 | 142.43 |
| 6a | 2.42 | 469.48 | 2 | 7 | 0 | 6 | 187.95 |
| 6b | 2.58 | 405.43 | 2 | 6 | 0 | 8 | 145.24 |
| 7a | 3.59 | 444.29 | 2 | 5 | 0 | 7 | 136.01 |
| 7b | 3.07 | 409.85 | 2 | 5 | 0 | 7 | 136.01 |
| 7c | 1.86 | 365.37 | 2 | 6 | 0 | 7 | 149.15 |
| 7d | 2.53 | 381.43 | 2 | 5 | 0 | 7 | 164.25 |
| 7e | 1.91 | 420.40 | 2 | 7 | 0 | 8 | 181.83 |
| 7f | 2.25 | 421.43 | 3 | 7 | 0 | 8 | 165.47 |
| 9 | 2.39 | 351.38 | 1 | 5 | 0 | 4 | 128.91 |
| 10 | 3.30 | 413.45 | 1 | 5 | 0 | 5 | 128.91 |
| 11 | 3.00 | 508.57 | 3 | 7 | 2 | 13 | 199.64 |
| 12 | 2.32 | 462.50 | 2 | 6 | 0 | 7 | 181.62 |
| 13 | 1.88 | 490.51 | 2 | 7 | 0 | 7 | 198.69 |
| 14 | 2.62 | 448.52 | 2 | 5 | 0 | 7 | 164.55 |
| 15 | 2.57 | 388.40 | 2 | 5 | 0 | 5 | 140.42 |
| 16 | 2.45 | 422.48 | 3 | 5 | 0 | 9 | 180.13 |
| 17 | 2.16 | 391.49 | 2 | 5 | 0 | 8 | 186.61 |
| 18 | 1.78 | 403.42 | 4 | 5 | 0 | 6 | 167.59 |
| 19 | 2.11 | 404.40 | 3 | 6 | 0 | 6 | 164.94 |
| 20 | 1.60 | 406.37 | 2 | 8 | 0 | 6 | 167.36 |
| FB8 | 2.51 | 371.46 | 1 | 5 | 0 | 5 | 98.15 |
| Comp | BBB levela | Absorption levelb | Hepatotoxicity probabilityc | PPBd |
|---|---|---|---|---|
| a BBB level, blood–brain barrier level, 0 = very high, 1 = high, 2 = medium, 3 = low, 4 = very low.b Absorption level, 0 = good, 1 = moderate, 2 = poor, 3 = very poor.c Hepatotoxicity probability, value > 0.5 means toxic, value < 0.5 means non-toxic.d PPB, plasma protein binding, 0 means less than 90%, 1 means more than 90%, 2 means more than 95%. | ||||
| 2 | 0 | 0 | 0.857 | 0 |
| 3 | 1 | 0 | 0.901 | 0 |
| 4 | 0 | 0 | 0.834 | 2 |
| 5 | 4 | 2 | 0.518 | 2 |
| 6a | 1 | 0 | 0.909 | 2 |
| 6b | 1 | 0 | 0.878 | 2 |
| 7a | 4 | 1 | 0.89 | 2 |
| 7b | 4 | 1 | 0.530 | 2 |
| 7c | 4 | 1 | 0.887 | 2 |
| 7d | 4 | 2 | 0.87 | 2 |
| 7e | 4 | 2 | 0.516 | 2 |
| 7f | 4 | 2 | 0.894 | 2 |
| 9 | 0 | 0 | 0.86 | 2 |
| 10 | 1 | 0 | 0.974 | 1 |
| 11 | 0 | 0 | 0.907 | 2 |
| 12 | 2 | 0 | 0.859 | 2 |
| 13 | 4 | 2 | 0.905 | 2 |
| 14 | 4 | 2 | 0.955 | 2 |
| 15 | 1 | 0 | 0.905 | 2 |
| 16 | 1 | 0 | 0.865 | 2 |
| 17 | 1 | 0 | 0.794 | 2 |
| 18 | 0 | 0 | 0.92 | 2 |
| 19 | 1 | 0 | 0.973 | 1 |
| 20 | 0 | 0 | 0.912 | 2 |
| FB8 | 0 | 2 | 0.517 | 1 |
The results showed that BBB penetration levels varied from none (derivatives 2, 4, 9, 11, 18, and 20, in addition to FB8) to very high (derivatives 5, 7a–f, 13, and 14). Derivatives 3, 6a, 6b, 10, 12, 15–17, and 19 showed low penetration ability to the BBB. Most important that derivatives 6b and 19 are safe to the CNS.
Clinical trials for a number of pharmaceutical candidates have been documented as being unsuccessful because of absorption issues.56 Intestinal absorption is the proportion of a drug that is taken up by the intestinal mucosa. A drug is well-absorbed if at least 90% of it is absorbed into the bloodstream.57 All the tested compounds, except for 5, 7a–f, 13 and 14, which showed moderate to poor levels of absorption, have good absorption properties, according to the findings of these studies. All the novel compounds as well as the reference compound could have potential hepatotoxic effects according to predictions. All tested members and reference compound can bind plasma protein greater than 90% (highly bound), except for compounds 2 and 3, which showed plasma protein binding less than 90%.
3.4 Structure activity relationship (SAR) study
The SAR of the newly synthesized derivatives of the two series of thiadiazole and benzoxazole-based yields very promising results.(i) Concerning the antiproliferative activity of thiadiazole-based derivatives, compound 6b was found to be superior to compound 7a and then compound 7d on the four cell lines studied. This indicates that the 4-CH3 substituent on the side phenyl moiety is superior to the 2,4-diCl substituent, which is also superior to the thiophene moiety.
(ii) In contrast, we can conclude about the benzoxazole-based candidate compound 19 with benzo[d]oxazole ring together with the thiadiazole moiety achieved the best antiproliferative actions. The cyanoacetamido moiety in position-2 of the benzoxazole ring and benzamide side chain, demonstrated strong antiproliferative effects on all four cancer cell lines.
(iii) Derivative 19 shares some of the common characteristics of the known CDK1 inhibitors (Fig. 5). These include a hydrophobic head to occupy the hydrophobic region I, an H-bond donor in the spacer moiety, a flat heteroaromatic ring to occupy the ATP-binding pocket, and a hydrophobic tail to occupy the hydrophobic region I. In addition, derivative 19 has various H-bond donor and/or acceptor moieties such as cyanoacetamide. The presence of a linker (thiourea or cyanoacetamide) or rigidified to heteroaromatic ring facilitates the occupation to the hydrophobic region of the enzyme. A substituted phenyl ring and heteroaromatic ring with various electron-donating or withdrawing groups added to the aforementioned core probably improved the inhibiting potential of derivative 19.
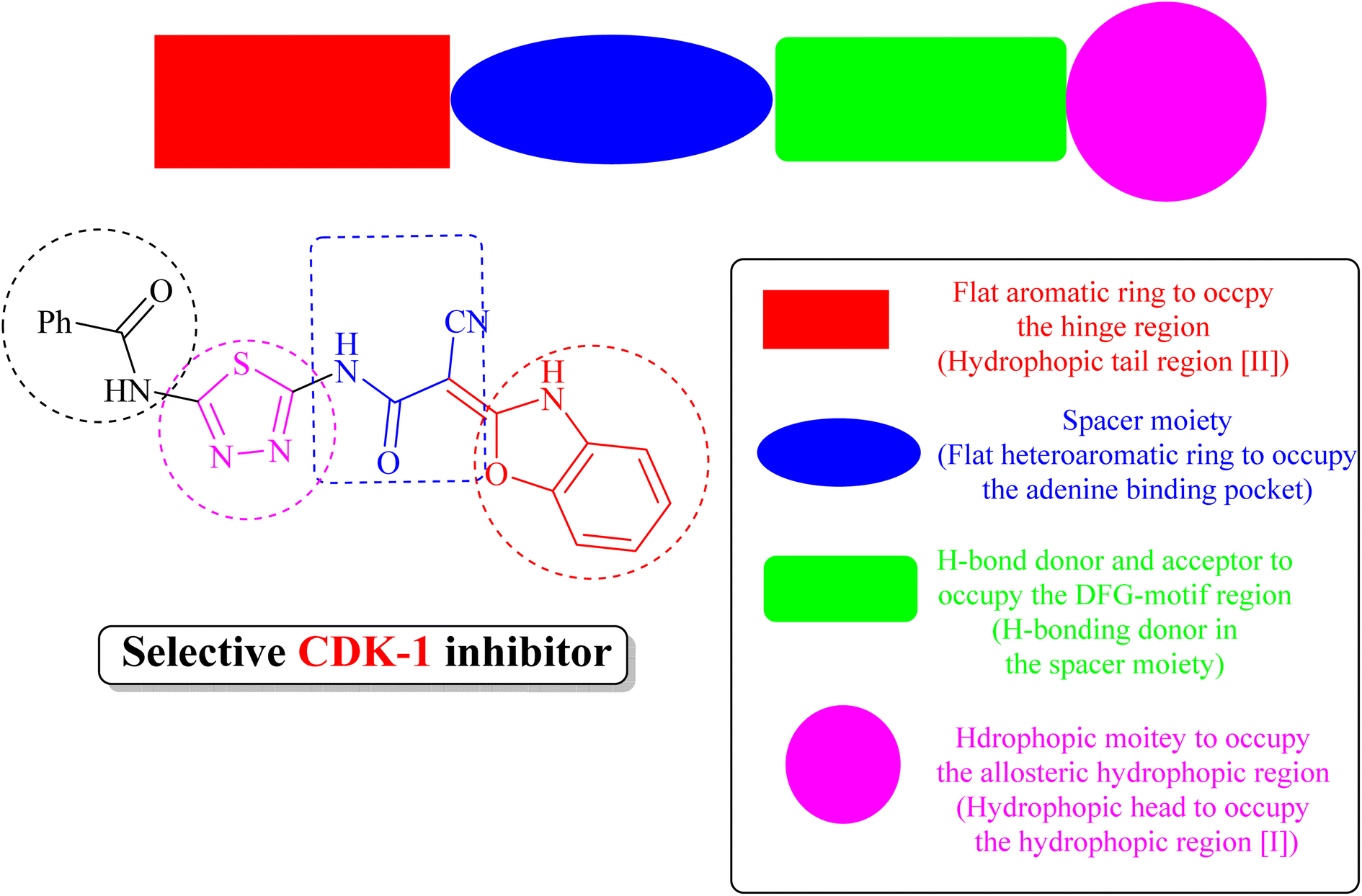 | ||
| Fig. 5 Derivative 19 as a potential inhibitor of CDK1. | ||
4. Conclusion
In this study, a group of 24 annulated 1,3,4-thiadiazole compounds were synthesized and investigated as anticancer agents. Only five derivatives had a significant antiproliferative activity against a panel of four cancer cell lines. However, three of these were not selective and caused toxicity to the normal cells. Two derivatives 6b and 19 were effective and selective. Compound 19 arrested the MCF-7 cells at the G2/M phase probably through inhibition of CDK1 and elicited apoptosis in these cells confirmed by the annexin V-PI assay. Compound 6b acted through a different pathway and significantly increased the sub-G1 percent of cells probably through induction of necrosis. Molecular docking showed that compound 19 is a potential CDK1 inhibitor candidate. Derivatives 6b and 19 did not violate Lipinski's rule of five and therefore, they are considered hits for further drug discovery research. In silico studies showed that these derivatives also have a low blood–brain barrier penetration capability and high intestinal absorption. Taken together, derivatives 6b and 19 could serve as potential anticancer agents and merit further investigations.Author contributions
Data curation: PSF and WME. Formal analysis: PSF and WME. Investigation: MHH, PSF, and WME. Writing – original draft: MHH, PSF, and WME. Conceptualization: MHH, MMH, WME. Supervision: MMH. Resources: AAE, AIH, WME. Software: PSF. Methodology: MHH, PSF, and WME. Validation: MHH, WME. Writing – review & editing: PSF, and WME. All authors read and approved the final version of the manuscript.Conflicts of interest
Authors declare that they have no conflict of interest.References
- A. Kumar, A. K. Singh, H. Singh, V. Vijayan, D. Kumar and J. Naik, et al., Pharmaceuticals, 2023, 16(2), 1–68 Search PubMed
.
- S. Pearce, Drug Discvery, 2017, 67, 66–70 Search PubMed
- J. Bostrom, A. Hogner, A. Llinas, E. Wellner and A. T. Plowright, J. Med. Chem., 2012, 55, 1817–1830 CrossRef PubMed
- C. D. Monte, S. Carradori, D. Secci, M. D'Ascenzio, P. Guglielmi, A. Mollica, S. Morrone, S. Scarpa, A. M. Aglianò, S. Giantulli and I. Silvestri, Eur. J. Med. Chem., 2015, 105, 245–262 CrossRef PubMed
- R. W. Sidwell, R. K. Robins and I. W. Hillyard, Pharmacol. Ther., 1979, 6, 123–146 CrossRef CAS PubMed
- M. D. Mullican, M. W. Wilson, D. T. Conner, C. R. Kostlan, D. J. Schrier and R. D. Dyer, J. Med. Chem., 1993, 36, 1090–1099 CrossRef CAS PubMed
- P. Datar and T. Deokule, Med. Chem., 2014, 4, 390–399 Search PubMed
- F. Clerici, D. Pocar, M. Guido, A. Loche, V. Perlini and M. Brufani, J. Med. Chem., 2001, 44, 931–936 CrossRef CAS PubMed
- H. Foks, D. P. Ksepko and K. Gobis, J. Heterocycl. Chem., 2014, 51, 507–512 CrossRef CAS
- G. Serban, Molecules, 2020, 25, 942 CrossRef CAS PubMed
- N. Shivakumara and P. M. Krishna, J. Mol. Struct., 2020, 1199, 126999 CrossRef
- P. Skehan, R. Storeng, D. Scudiero, A. Monks, J. McMahon, D. Vistica, J. T. Warren, H. Bokesch, S. Kenney and M. R. Boyd, J. Natl. Cancer Inst., 1990, 82, 1107–1112 CrossRef CAS PubMed
- A. Monks, D. Scudiero, P. Skehan, R. Shoemaker, K. Paull, D. Vistica, C. Hose, J. Langley, P. Cronise, A. Vaigro-Wolff, M. Gray-Goodrich, H. Campbell, J. Mayo and M. Boyd, J. Natl. Cancer Inst., 1991, 83, 757–766 CrossRef CAS PubMed
- W. M. El-Sayed, W. M. Hussin, A. A. Mahmoud and M. A. AlFredan, Biomed. Res. Int., 2013, 2013, 1–11 CrossRef PubMed
- M. A. Ismail, G. A. Abdelwahab, W. S. Hamama, E. Abdel-Latif, F. E. Fardous and W. M. El-Sayed, Arch. Pharm., 2022, e2100385 CrossRef PubMed
- A. A. Al-Karmalawy, M. A. Dahab, A. M. Metwaly, S. S. Elhady, E. B. Elkaeed, I. H. Eissa and K. M. Darwish, Front. Chem., 2021, 9, 24–44 Search PubMed
- H. Y. Aati, A. Ismail, M. E. Rateb, A. M. AboulMagd, H. M. Hassan and M. H. Hetta, Plants, 2022, 11, 2521 CrossRef CAS PubMed
- M. I. A. Hamed, K. M. Darwish, R. Soltane, A. Chrouda, A. Mostafa, N. M. Abo Shama, S. S. Elhady, H. S. Abulkhair, A. E. Khodir, A. A. Elmaaty and A. A. Al-karmalawy, RSC Adv., 2021, 11, 35536–35558 RSC
- K. El-Adl, M.-K. Ibrahim, M. S. Alesawy and I. H. Eissa, Bioorg. Med. Chem., 2021, 30, 115958 CrossRef CAS PubMed
- M. S. Alesawy, E. B. Elkaeed, A. A. Alsfouk, A. M. Metwaly and I. Eissa, Molecules, 2021, 26, 6593 CrossRef CAS PubMed
- M. H. Hekal, F. S. M. Abu El-Azm and S. R. Atta-Allah, Synth. Commun., 2019, 49, 2630–2641 CrossRef CAS
- M. H. Hekal, F. S. M. Abu El-Azm and H. A. Sallam, J. Heterocycl. Chem., 2019, 56, 795–803 CrossRef CAS
- M. A. Yousef, A. M. Ali, W. M. El-Sayed, W. Saber, H. H. Farag and T. Aboul-Fadl, Bioorg. Chem., 2020, 105, 104366 CrossRef CAS PubMed
- M. H. Hekal, A. M. El-Naggar, F. S. M. Abu El-Azm and W. M. El-Sayed, RSC Adv., 2020, 10, 3675–3688 RSC
- M. S. Salem, R. A. Hussein and W. M. El-Sayed, Med. Chem., 2019, 19, 620–626 CAS
- A. M. El-Naggar, A. K. Khalil, H. M. Zeidan and W. M. El-Sayed, Med. Chem., 2017, 17, 1644–1651 CAS
- W. A. Hussin, M. A. Ismail, A. M. Alzahrani and W. M. El-Sayed, Drug Des., Dev. Ther., 2014, 8, 963–972 CAS
- M. R. Franklin, P. J. Moos, W. M. El-Sayed, T. Aboul-Fadl and J. C. Roberts, Chem.–Biol. Interact., 2007, 168, 211–220 CrossRef CAS PubMed
- M. H. Hekal, S. S. Samir, Y. M. Ali and W. M. El-Sayed, Polycycl. Aromat. Compd., 2022, 42, 7644–7660 CrossRef CAS
- W. S. Qayed, M. A. Hassan, W. M. El-Sayed, J. R. A. Silva and T. Aboul-Fadl, Bioorg. Chem., 2022, 126, 105884 CrossRef CAS PubMed
- W. M. El-Sayed, T. Aboul-Fadl, J. C. Roberts, J. G. Lamb and M. R. Franklin, Toxicol. In Vitro, 2007, 21, 157–164 CrossRef CAS PubMed
- S. R. Atta-Allah, A. M. AboulMagd and P. S. Farag, Bioorg. Chem., 2021, 112, 104923 CrossRef CAS PubMed
- P. S. Farag, M. M. Hemdan and A. A. El-Sayed, J. Heterocycl. Chem., 2020, 57, 3428–3441 CAS
- P. S. Farag, A. M. AboulMagd, M. M. Hemdan and A. I. Hassaballah, Bioorg. Chem., 2023, 130, 106231 CrossRef CAS PubMed
- P. S. Farag, M. M. Hemdan and A. I Hassaballah, RSC Adv., 2022, 12, 10204–10208 RSC
- M. H. Hekal, P. S. Farag, M. M. Hemdan and W. M. El-Sayed, Bioorg. Chem., 2021, 115, 105176 CrossRef CAS PubMed
- S. L. Holbeck, J. M. Collins and J. H. Doroshow, Mol. Cancer Ther., 2010, 9(5), 1451–1460 CrossRef CAS PubMed
- A. Lezaja and M. Altmeyer, Cell Cycle, 2018, 17, 24–32 CrossRef CAS PubMed
- T. C. Tsou, F. Y. Tsai, S. C. Yeh and L. W. Chang, Arch. Toxicol., 2006, 80, 804–810 CrossRef CAS PubMed
- M. C. Chang, Y. J. Chen, E. J. Liou, W. Y. Tseng, C. P. Chan, H. J. Lin, W.-C. Liao, Y.-C. Chang, P.-Y. Jeng and J.-H. Jeng, Oncotarget, 2016, 7, 74473–74483 CrossRef PubMed
- M. Zhang, J. Qu, Z. Gao, Q. Qi, H. Yin, L. Zhu, Y. Wu, W. Liu, J. Yang and X. Huang, Front. Pharmacol., 2021, 11, 601468 CrossRef PubMed
- S. A. El-Metwally, A. K. Khalil and W. M. El-Sayed, Bioorg. Chem., 2020, 94, 103492 CrossRef CAS PubMed
- A. Arora and E. M. Scholar, J. Pharmacol. Exp. Ther., 2005, 315, 971–979 CrossRef CAS PubMed
- T. Ozaki and A. Nakagawara, J. Biomed. Biotechnol., 2011, 3, 994–1013 CAS
- M. Olsson and B. Zhivotovsky, Cell Death Differ., 2011, 18, 1441–1449 CrossRef CAS PubMed
- C. M. Pfeffer and A. T. K. Singh, Int. J. Mol. Sci., 2018, 19, 1–10 Search PubMed
- J. Yan, P. Wan, S. Choksi and Z. G. Liu, Trends Cancer, 2022, 8, 21–27 CrossRef CAS PubMed
- Z. G. Liu and D. Jiao, Cell Stress, 2019, 4, 1–8 CrossRef PubMed
- D. J. Wood, S. Korolchuk, N. J. Tatum, L. Wang, J. A. Endicott, M. E. M. Noble and M. P. Martin, Cell Chem. Biol., 2019, 26, 121–130 CrossRef CAS PubMed
- N. Malhotra, R. Gupta and P. KumarR, Neurochem. Int., 2021, 148, 105115 CrossRef CAS PubMed
- L. L. G. Ferreira and A. D. Andricopulo, Drug Discov. Today, 2019, 24, 1157–1165 CrossRef CAS PubMed
- T. Khan, M. Ali, A. Khan, P. Nisar, S. A. Jan and S. A. Z. K. Shinwari, Biomolecules, 2020, 10, 47 CrossRef CAS PubMed
- F. Wu, Y. Zhou, L. Li, X. Shen, G. Chen, X. Wang, X. Liang, M. Tan and Z. Huang, Front. Chem., 2020, 8, 726 CrossRef CAS PubMed
- D. F. Veber, S. R. Johnson, H.-Y. Cheng, B. R. Smith, K. W. Ward and K. D. Kopple, J. Med. Chem., 2002, 45, 2615–2623 CrossRef CAS PubMed
- H. Patel, K. Dhangar, Y. Sonawane, S. Surana, R. Karpoormath, N. Thapliyal, M. Shaikh, M. Noolvi and R. Jagtap, Arabian J. Chem., 2015, 11, 221–232 CrossRef
- A. K. Jain, S. Sharma, A. Vaidya, V. Ravichandran and R. K. Agrawal, Chem. Biol. Drug Des., 2013, 81, 557–576 CrossRef CAS PubMed
- A. Tahghighi and F. Babalouei, Iran. J. Basic Med. Sci., 2017, 20, 613–622 Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra02716c |
| This journal is © The Royal Society of Chemistry 2023 |