
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanochemical protocol facilitates the generation of arylmanganese nucleophiles from unactivated manganese metal†
Rina
Takahashi
a,
Pan
Gao
b,
Koji
Kubota
 *ab and
Hajime
Ito
*ab
*ab and
Hajime
Ito
*ab
aDivision of Applied Chemistry and Frontier Chemistry Center, Faculty of Engineering, Hokkaido University, Sapporo, Hokkaido, Japan. E-mail: kbt@eng.hokudai.ac.jp; hajito@eng.hokudai.ac.jp
bInstitute for Chemical Reaction Design and Discovery (WPI-ICReDD), Hokkaido University, Sapporo, Hokkaido, Japan
First published on 28th November 2022
Abstract
The direct synthesis of organomanganese reagents from organic halides and manganese metal remains a challenge. Current solution-based approaches require the preparation of activated manganese (Rieke manganese) or the use of multiple metal additives to promote the insertion of manganese metal into a carbon–halogen bond. Here, we show that a mechanochemical ball-milling protocol facilitates the generation of various arylmanganese nucleophiles from aryl halides and commercially available, unactivated manganese metal without the need for complicated pre-activation processes and metal additives. These manganese-based carbon nucleophiles can be used directly for one-pot addition reactions with various electrophiles and palladium-catalyzed cross-coupling reactions under bulk-solvent-free mechanochemical conditions. Importantly, all experimental operations can be conducted under atmospheric conditions.
Introduction
Organomanganese reagents play a unique role in organic synthetic chemistry because of their mild reactivity, distinctive chemoselectivity, and low toxicity.1 Most organomanganese reagents are typically prepared via the following methods: (1) the insertion of manganese metal into a carbon–halogen bond (direct synthesis);2–4 (2) metathesis between an organolithium or organomagnesium reagent and a manganese halide;5 or (3) a directed deprotonation using TMP2Mn·2LiCl.6 Given the ready availability of manganese metal, and the mild reaction conditions and high atom efficiency associated with direct synthesis, this method can be expected to afford convenient access to versatile organomanganese reagents. Although manganese metal has a stronger reduction potential than zinc, direct synthesis is only applicable to highly reactive organic halides, such as allylic halides or α-haloesters when commercial manganese metal is used.2,3 One of the main reasons for this low reactivity is the presence of a usually tightly bound oxide layer on its surface.3a Therefore, the preparation of activated manganese (Rieke manganese) using strong reducing reagents, such as alkali metals, is often required when aryl halides are used as substrates (Scheme 1a).4 Unfortunately, this greatly reduces the practical utility of arylmanganese nucleophiles. Recently, Knochel et al. have reported that the direct synthesis of arylmanganese reagents from unactivated manganese metal can be achieved using LiCl, PbCl2, and InCl3 additives (Scheme 1b).3b Although this achievement is remarkable, this solution-based approach still requires multiple metal additives, long reaction times (24 h), the use of dry organic solvents, and inert-gas-line techniques. All of which represent considerable drawbacks from both an environmental and an economic perspective. Therefore, the development of an operationally simple protocol that does not require pre-activation of the reagents would be highly desirable as it can be expected to advance the widespread application of arylmanganese nucleophiles in organic synthesis.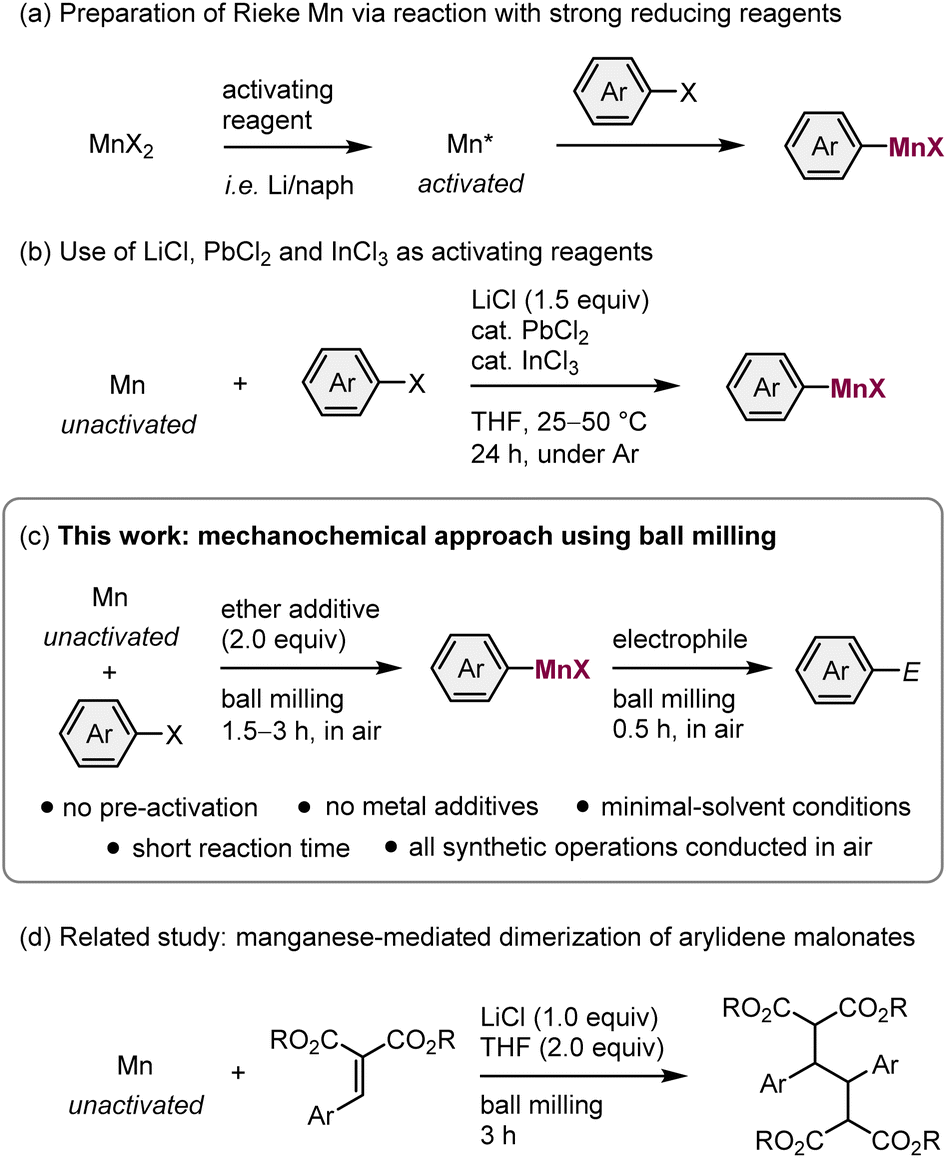 | ||
| Scheme 1 Direct insertion of manganese metal into carbon–halogen bonds. (a) Use of Rieke manganese metal. (b) Use of multiple metal additives. (c) Ball-milling approach (this work). (d) Related study on mechanochemical activation of manganese metal. | ||
Recently, a mechanochemical synthetic technique that uses ball milling has attracted significant attention as a new tool for organic reactions.7 The advantages of mechanochemical synthesis include avoiding the use of potentially harmful organic solvents, shorter reaction times, and operational simplicity. Moreover, recent studies have revealed that strong mechanical agitation during the ball-milling process can activate zero-valent metals in the bulk state, facilitating surface reactions with organic halides.8 For example, Browne et al. have reported the direct synthesis of organozinc reagents from metallic zinc facilitated by ball milling.9 The groups of Harrowfield, Birke, Hanusa, Bolm, and our own group have independently reported the mechanochemical synthesis of Grignard reagents via the direct insertion of magnesium metal into organic halides.10,11 More recently, we have demonstrated that a ball-milling approach allows the generation of heavy Grignard reagents based on calcium from aryl halides and unactivated calcium metal.12 Notably, these protocols do not involve the pre-activation of the metals and are applicable to a broad range of substrates.
These achievements led us to consider the mechanochemical generation of organomanganese reagents via the direct synthesis from unactivated manganese metal, which is barely reactive in solution (Scheme 1c). We envisaged that the mechanical impact of ball milling could remove the unreactive oxide layer covering the surface of commercially available manganese metal, thus facilitating the direct insertion of manganese metal into organic halide bonds without the need for a complicated pre-activation process.4 Whilst our study was already underway, Browne et al. reported that commercial manganese metal can be activated using ball-milling to realize the reductive dimerization of arylidene malonates (Scheme 1d).13 However, in that report, the direct synthesis of organomanganese nucleophiles from alkyl halides and the subsequent nucleophilic addition to electrophiles was unsuccessful whilst other organic halides were not explored.13 Herein, we report the first successful example of the direct synthesis of arylmanganese nucleophiles from unactivated manganese metal and aryl halides using mechanochemistry. These reactions are highly efficient and went to completion within 3 hours to form the desired arylmanganese nucleophiles. These nucleophiles were then reacted with various electrophiles under mechanochemical conditions. The direct synthesis of alkylmanganese nucleophiles was also achieved using this protocol, although there is room to improve the efficiency. Notably, the entire synthetic procedure can be conducted under atmospheric conditions without using any special precautions or synthetic techniques.
Results and discussion
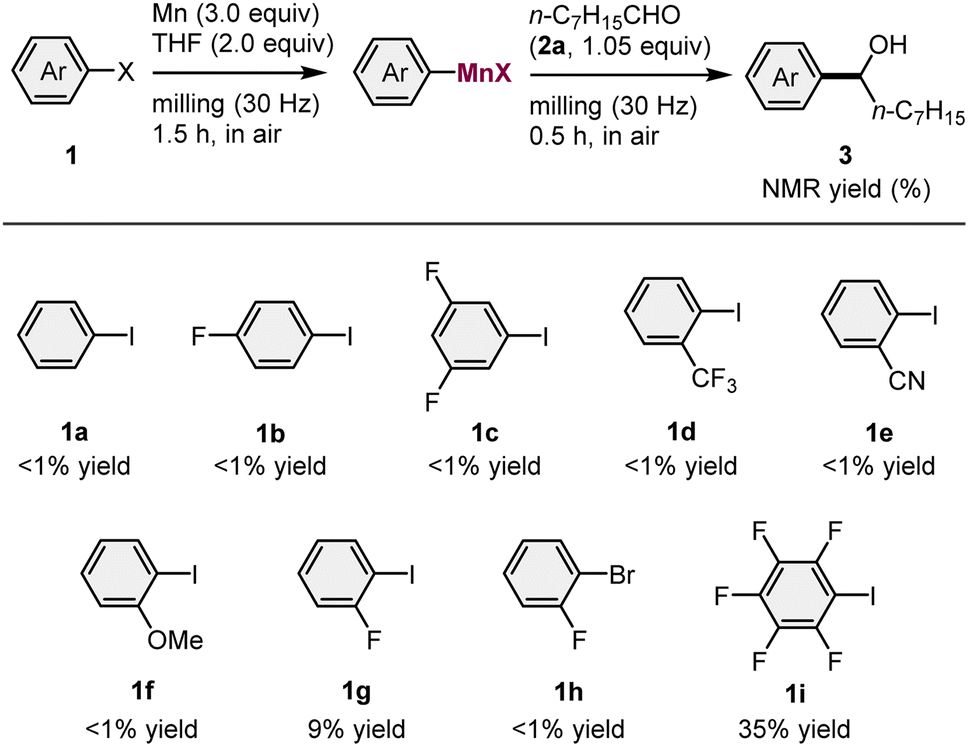
We began by attempting the preparation of various arylmanganese species from different aryl iodides (1) (1.0 equiv.) and commercial manganese metal (Wako, powder, >98% purity) (3.0 equiv.), using a Retch MM400 mixer mill (5 mL stainless-steel milling jar with a 10 mm-diameter stainless-steel ball), followed by the subsequent nucleophilic addition to an aldehyde 2a (1.05 equiv.) (Table 1). Based on a previous study on the isolation of THF-coordinated arylmanganese species by Westerhausen et al.,14 2.0 equivalents of tetrahydrofuran (THF) were used to facilitate the formation of the organomanganese nucleophile. Unfortunately, aryl iodides 1a–1f, which bear electron-donating and -withdrawing groups, did not react under the applied conditions. However, the substrate bearing a fluorine atom at the ortho position (1g) afforded the desired product 3g in 9% yield. No reaction occurred when the corresponding aryl bromide (1h) was used. We found that the reaction of pentafluorophenyl iodide (1i) underwent direct metalation to form the corresponding addition product (3i) in 35% yield. These results suggest that only ortho-halogenated aryl iodides are reactive in the direct manganese metalation under mechanochemical conditions, a finding which is consistent with the results of Knochel's previously reported solution-based method.3b The use of activated organic halides such as benzyl bromide only provided the homocoupling products. This is probably due to a facile radical–radical coupling or the nucleophilic substitution of the substrate by an in situ-generated benzylmanganese nucleophile.| a Conditions: 1 (1.0 mmol), Mn powder (3.0 mmol), THF (2.0 mmol), and 2a (1.05 mmol) in a stainless-steel milling jar (5 mL) with a stainless-steel ball (10 mm). |
|---|
|
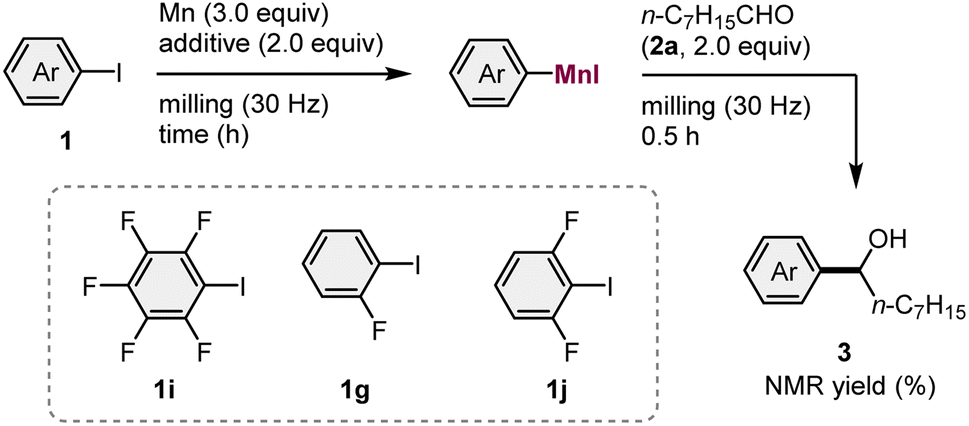
Next, we proceeded to optimize the reaction conditions using pentafluorophenyl iodide (1i) as the substrate and 2.0 equivalent of 2a as a trapping reagent (Table 2). The reaction without the addition of an ether additive provided a trace amount of 3i (2% yield; entry 1). The addition of THF significantly promoted the generation of the arylmanganese nucleophile and 3i was obtained in 54% yield (entry 2). After screening further ether additives (entries 3–8), we found that the reaction with methyl tert-butyl ether (MTBE) gave the highest yield (75% yield; entry 3). Next, the mechanochemical conditions for the synthesis of 3g from 1g were optimized (entries 9–11). While 1g showed much lower reactivity toward the metalation than 1i, the reaction in the presence of THF gave the corresponding addition product (3g) in 25% yield (entry 9). Prolonging the reaction time for the metalation step led to a slightly improved yield (30% yield; entry 10). Interestingly, the addition of MTBE, which was effective for the reaction of 1i, did not produce 3g (<1%; entry 11). Next, 1,3-difluoro-2-iodobenzene (1j) was used as a substrate (entries 12–14). While 1j also showed lower reactivity than 1i, the reaction with manganese metal in the presence of THF for 3 hours afforded the desired product (3j) in 69% yield (entry 13). Again, the use of MTBE did not lead to the formation of 3j, indicating that MTBE is specifically effective for the highly electron-deficient pentafluorophenyl iodide 1i.
|
|
||||
|---|---|---|---|---|
| Entry | Substrate | Additive | Time (h) | Yieldb (%) |
| a Conditions: 1(1.0 mmol), Mn powder (3.0 mmol), additive (2.0 mmol), and 2a (2.0 mmol) in a stainless-steel milling jar (5 mL) with a stainless-steel ball (10 mm). b Determined via19F NMR analysis with an internal standard. Isolated yields are shown in parenthesis. | ||||
| 1 | 1i | — | 1.5 | 2 |
| 2 | 1i | THF | 1.5 | 54 |
| 3 | 1i | MTBE | 1.5 | 75 (54) |
| 4 | 1i | Anisole | 1.5 | 66 |
| 5 | 1i | Et2O | 1.5 | 64 |
| 6 | 1i | CPME | 1.5 | 57 |
| 7 | 1i | 1,2-DME | 1.5 | 40 |
| 8 | 1i | 1,4-Dioxane | 1.5 | 10 |
| 9 | 1g | THF | 1.5 | 25 |
| 10 | 1g | THF | 3 | 30 (25) |
| 11 | 1g | MTBE | 3 | <1 |
| 12 | 1j | THF | 1.5 | 25 |
| 13 | 1j | THF | 3 | 69 (67) |
| 14 | 1j | MTBE | 1.5 | <1 |
With the optimized conditions for the generation of the arylmanganese reagents in hand, a series of one-pot mechanochemical reactions with various electrophiles were evaluated (Scheme 2). Following the reaction between 1i and the unactivated manganese metal, benzaldehyde (2b) was added to the mixture and subsequent ball milling afforded the desired addition product (3k) in 60% yield. The reaction with cyclohexanone (2c) also produced the corresponding product (4) in 45% yield. In addition, we found that phenyl isocyanate (5a) efficiently reacted with the mechanochemically generated arylmanganese nucleophile to give the desired amide (6a) in 69% yield. Furthermore, a mechanochemical palladium-catalyzed cross-coupling reaction between aryl iodide and the arylmanganese nucleophile in the presence of a Pd-PEPPSI-iPr catalyst15,16 furnished the corresponding biaryl compound (7) in 71% yield under high-temperature ball-milling conditions using a heat gun (for details, see the ESI†).17
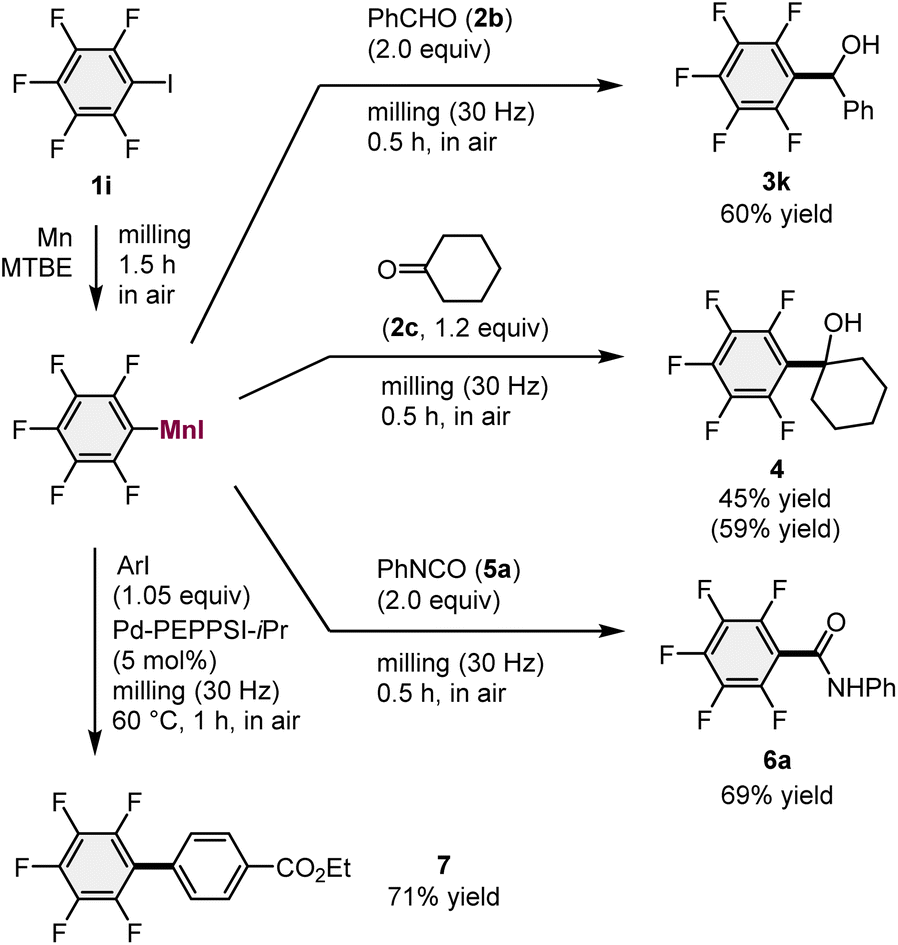 | ||
| Scheme 2 The use of mechanochemically generated arylmanganese nucleophiles for reactions with various electrophiles. aFor the reaction conditions, please see the ESI.†bIsolated yields are shown. NMR yields are shown in parenthesis. | ||
Next, the scope of aryl halides that can be used for the mechanochemical generation of arylmanganese nucleophiles was investigated (Table 3). Cyclohexyl isocyanate (5b) was used as a trapping reagent and the reactions of several fluorinated aryl iodides (1g, 1i–1k) under the optimized conditions proceeded smoothly to give the corresponding amides (6b–6e) in moderate to good yields (38–79%). Polyfluorinated substrates bearing methoxy, dimethylamino, or siloxy groups (1l–1n) were successfully converted into the desired products (6f–6h) in 28–70% yield. The iodide moiety of 5-bromo-1,3-difluoro-2-iodobenzene (1o) selectively reacted with manganese metal to furnish the desired product (6i) in 75% yield. We found that 1,3-dichloro-2-iodobenzene (1p) also reacted under the applied conditions to furnish 6j in 55% yield. The reaction of 1-bromo-3-fluoro-2-iodobenzene (1q) produced the desired amide (6k) in 70% yield. In the case of 1,3-dibromo-2-iodobenzene (1r), the desired product (6m) was obtained, albeit that the bromide moiety also reacted to form side product 6n. In addition to aryl iodides, electron-deficient polyfluorinated aryl bromides (1s–1v) could be used as substrates to afford the corresponding amides 39–69% yield. Even though the aryl-halide substrate scope of our reaction is limited compared to that of the Rieke method,4 it is comparable to that of Knochel's conditions.3b
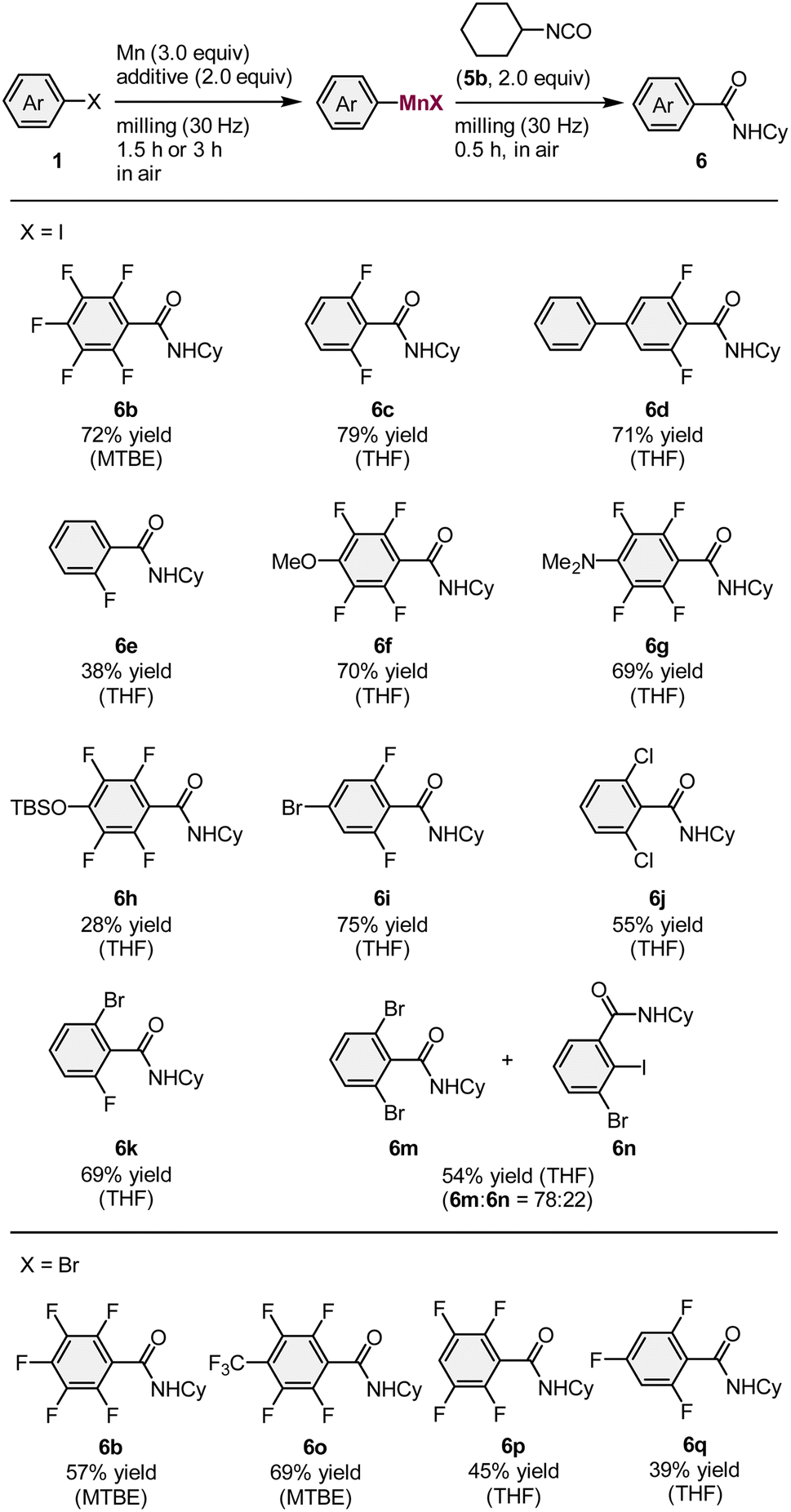
The direct formation of alkylmanganese nucleophiles from unactivated alkyl halides can be accomplished using Rieke manganese.4d We found that the mechanochemical reaction of alkyl iodide 8 with commercial manganese metal, followed by reaction with 5b formed the desired product (9) in 17% yield (Scheme 3). Although the yield needs to be improved, this is the first example of the direct generation of alkylmanganese nucleophiles from unactivated alkyl halides and commercial, unactivated manganese metal.
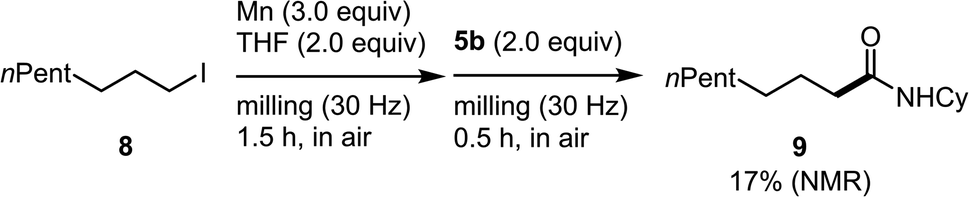 | ||
| Scheme 3 Reaction of alkyl iodide 8 and unactivated manganese metal under mechanochemical conditions. | ||
We conducted a number of nucleophilic addition reactions with 5b after exposing the mechanochemically generated arylmanganese nucleophile 1i to air for different lengths of time (5–60 min; Fig. 1). In the optimized procedure, 5b was added immediately after opening the jar, and the jar was closed as rapidly as possible. The time of exposure to air was less than one minute, and the desired product 6b was obtained in 91% NMR yield (Fig. 1). We found that, even though there were no obvious changes in its appearance, the yield of 6b decreased when the generated arylmanganese species were exposed to air for 5 min or more (Fig. 1). Although pentafluorobenzene was detected in the 1H NMR spectrum after the exposure of the manganese nucleophile to air, byproducts derived from oxygen or carbon dioxide, such as phenol and benzoic acid, were not detected. These results suggest that the decreased yield is most likely caused by protonation resulting from exposure to moisture in the air.
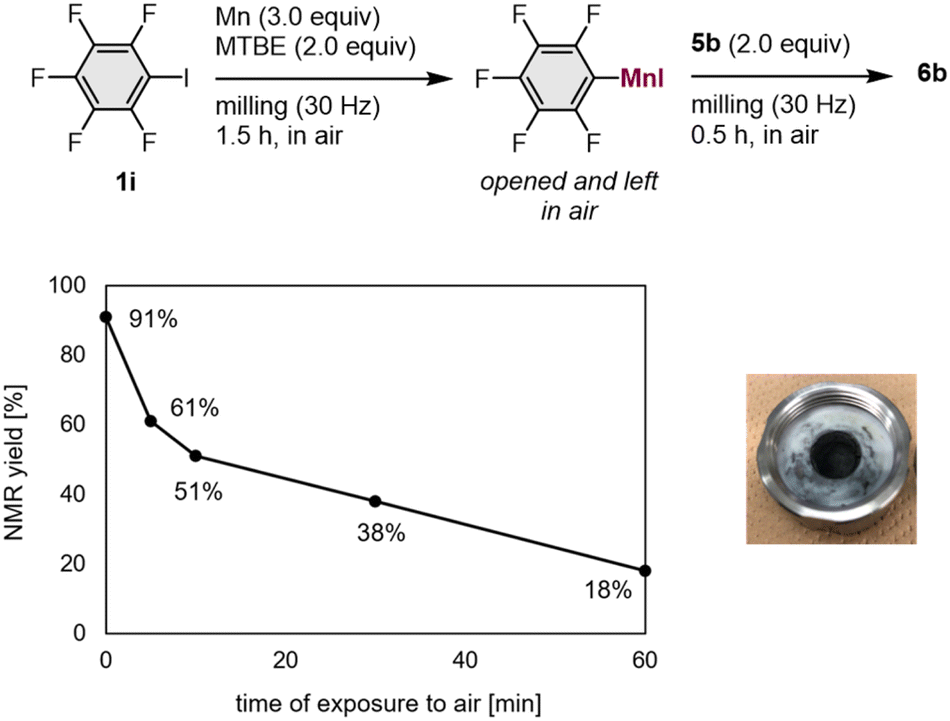 | ||
| Fig. 1 Stability of organomanganese reagents in air and the appearance of the crude mixture. | ||
In order to confirm that the arylmanganese nucleophiles are indeed generated under our mechanochemical conditions, preliminary mechanistic studies were performed (Scheme 4). First, we conducted a deuteration experiment (Scheme 4a) wherein the mechanochemically formed arylmanganese nucleophile was quenched by the addition of CD3CO2D (10 equiv.) immediately after opening the jar in order to prevent undesired protonation by atmospheric moisture. The corresponding deuterated product (10) was obtained in a high deuteration ratio (95% D), suggesting that an arylmanganese nucleophile was most likely generated under mechanochemical conditions. Next, in order to test the hypothesis that transition-metals, such as iron and chromium, which could potentially leach from the stainless-steel milling jar and balls, catalyse the direct metalation or subsequent nucleophilic addition, we investigated the reaction using jars made of different materials such as tungsten carbide (WC) or zirconium oxide (Scheme 4b). Although the use of a 5 mL jar is optimal for this scale, we compared the reactions with 10 mL jars and 10 mm balls without changing the reaction scale because 10 mL is the minimum volume for the commercially available jars made of these materials. The results confirmed that the direct metalation and subsequent nucleophilic addition to 5b proceeded regardless of the material of the milling jars and balls. Thus, we excluded the possibility that the reaction is catalysed by metals leached from the ball milling equipment (Scheme 4b). Finally, we assessed the use of unactivated manganese metal in solution (Scheme 4c). When we conducted the reaction between unactivated manganese metal and 1i in THF (1.0 M) at room temperature, the corresponding amide (6b) was not detected and a large amount of unreacted 1i was recovered (93%). This result indicates that mechanical activation of the manganese metal via ball milling is essential for the generation of arylmanganese nucleophiles. When Knochel's method was applied to the direct metalation of 1i, 6b was obtained in 83% yield.3b This result suggests that manganese-based carbon nucleophiles (Ar–MnX) similar to those formed under Knochel's conditions are also most likely formed under the mechanochemical conditions.
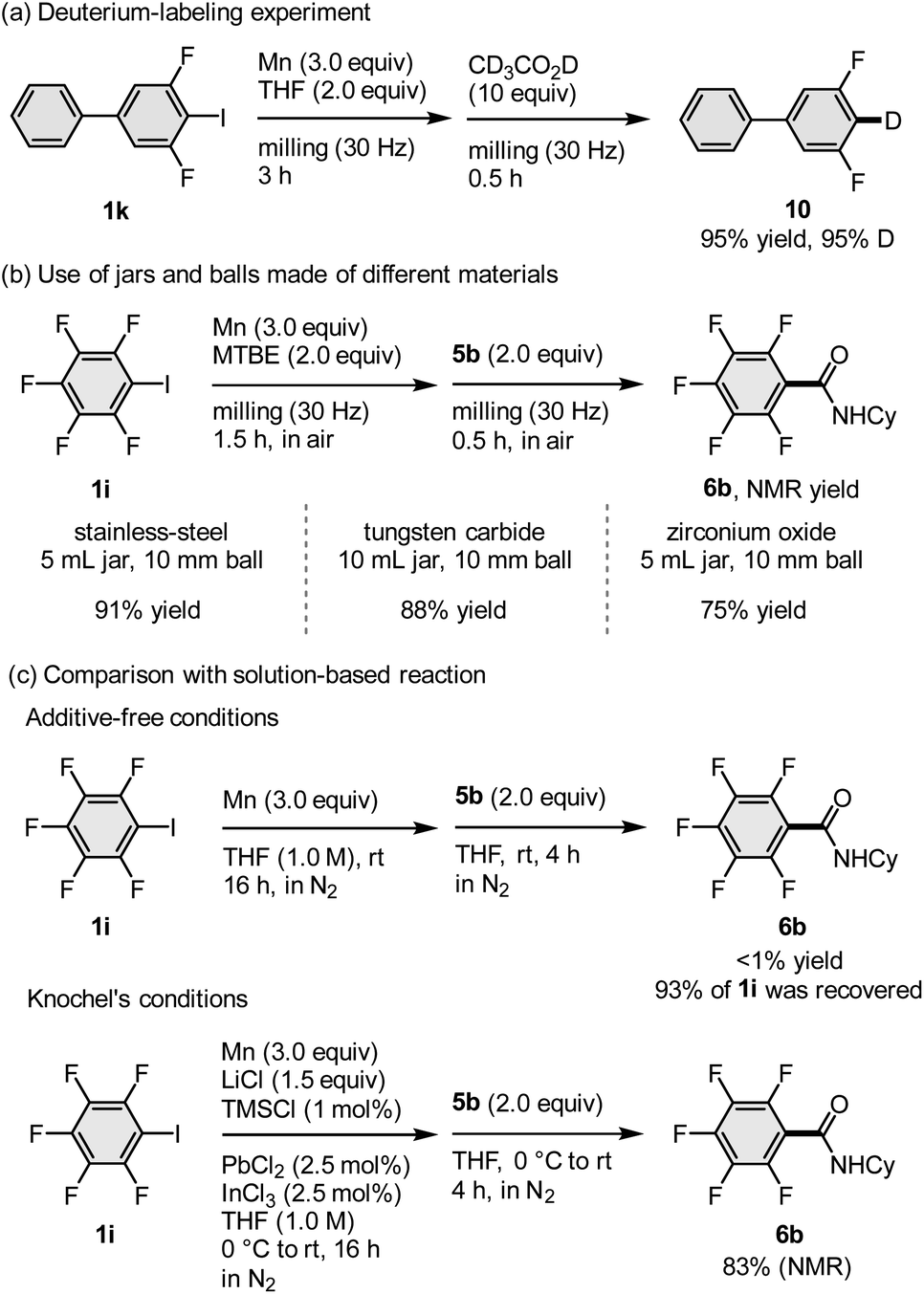 | ||
| Scheme 4 Mechanistic studies. (a) Deuterium-labeling study. (b) Effect of milling jars and balls made of different materials. (c) Comparison with solution-based conditions. aFor the reaction conditions, please see the ESI.† | ||
To validate our hypothesis that the mechanical impact provided by ball milling can reduce or remove the unreactive oxide layer covering the surface of the commercial manganese metal, we analyzed its surface using X-ray photoelectron spectroscopy (XPS) (Fig. 2). When commercial manganese metal used in this study was subjected to XPS analysis, we found that a signal derived from the Mn–O bonds was detected on its surface (Fig. 2a). The manganese oxide content can be estimated based on the Mn/O atom ratio.3a The XPS analysis of the ball-milled sample revealed that the Mn ratio on the surface was increased after ball milling (Fig. 2b). This result suggests that ball milling can increase the reactive surface area of manganese metal via mechanical removal of the oxide layer, thus supporting our hypothesis.
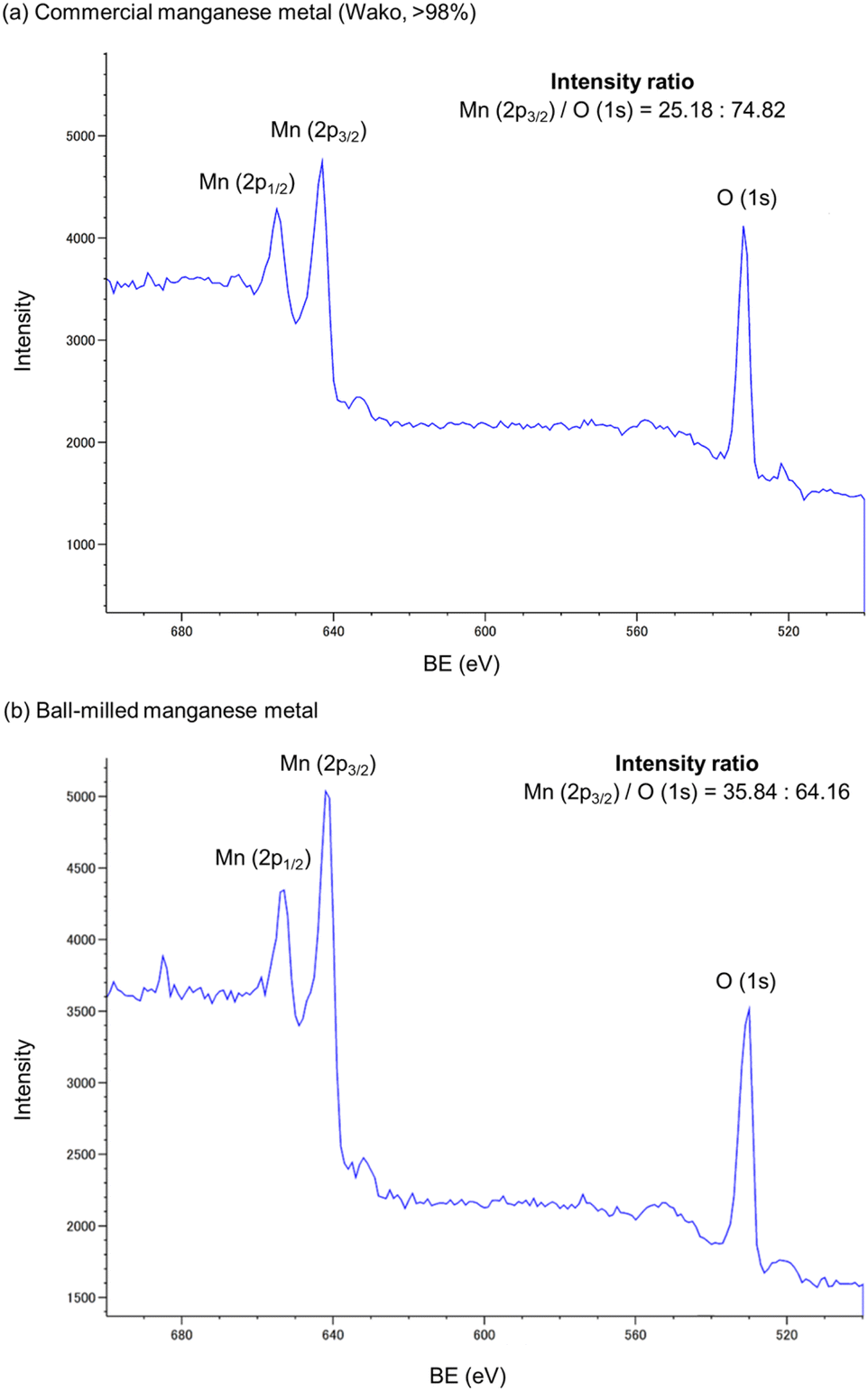 | ||
| Fig. 2 X-Ray photoelectron spectra of commercial manganese metal (a) before ball milling and (b) after ball milling. | ||
Conclusions
In summary, we have demonstrated that a mechanochemical protocol using ball milling, that does not require metal additives and complicated pre-activation processes, allows the generation of arylmanganese nucleophiles via the reaction between an aryl halide and commercial, unactivated manganese metal. The mechanochemically generated arylmanganese reagents can be used as carbon nucleophiles for the one-pot addition to various electrophiles and palladium-catalyzed cross-coupling reactions under bulk-solvent-free ball-milling conditions. Notably, all synthetic operations can be conducted under atmospheric conditions. We therefore anticipate that this work will provide a new platform for the rapid discovery of novel organic transformations mediated by organomanganese species.Data availability
All experimental data is available in the ESI.†Author contributions
K. K. and H. I. conceived and designed the study. R. T., K. K. and H. I. co-wrote the paper. R. T. and P. G. performed the chemical experiments and analyzed the data. K. K. performed the X-ray photoelectron spectroscopy analysis. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Japan Society for the Promotion of Science (JSPS) via KAKENHI grants 22H00318, 21H01926, 22K18333 and 22H05328; the JST via CREST grant JPMJCR19R1; FOREST grant JPMJFR201I; as well as by the Institute for Chemical Reaction Design and Discovery (ICReDD), which was established by the World Premier International Research Initiative (WPI), MEXT, Japan. We thank Ms. Tamae Seo for her help in cross-checking experiments.Notes and references
- For selected reviews and examples on the use of organomanganese reagents in organic synthesis, see: (a) G. Cahiez, C. Duplais and J. Buendia, Chem. Rev., 2009, 109, 1434–1476 CrossRef CAS PubMed; (b) G. Cahiez and E. Métais, Tetrahedron Asymmetry, 1997, 8, 1373–1376 CrossRef CAS; (c) G. Cahiez and E. Metais, Tetrahedron Lett., 1995, 36, 6449–6452 CrossRef CAS.
- For the direct synthesis of organomanganese reagents from commercial manganese metal and reactive organic halides without pre-activation, see: G. Cahiez and P.-Y. Chavant, Tetrahedron Lett., 1989, 30, 7373–7376 CrossRef CAS.
- For examples on the direct synthesis of organomanganese reagents from commercial manganese metal with activating reagents, see: (a) K. Takai, T. U. T. Hayashi and T. Moriwake, Tetrahedron Lett., 1996, 37, 7049–7052 CrossRef CAS; (b) Z. Peng and P. Knochel, Org. Lett., 2011, 13, 3198–3201 CrossRef CAS PubMed.
- For examples of the direct synthesis of organomanganese reagents from activated manganese metal, see: (a) T. Hiyama, M. Obayashi and A. Nakamura, Organometallics, 1982, 1, 1249–1251 CrossRef CAS; (b) R. D. Rieke, S.-H. Kim and X. Wu, J. Org. Chem., 1997, 62, 6921–6927 CrossRef CAS; (c) A. Fürstner and H. Brunner, Tetrahedron Lett., 1996, 37, 7009–7012 CrossRef; (d) S.-H. Kim, M. V. Hanson and R. D. Rieke, Tetrahedron Lett., 1996, 37, 2197–2200 CrossRef CAS; (e) H. Kakiya, S. Nishimae, H. Shinokubo and K. Oshima, Tetrahedron, 2001, 57, 8807–8815 CrossRef CAS; (f) J. Tang, H. Shinokubo and K. Oshima, Synlett, 1998, 1075–1076 CrossRef CAS.
- For examples of the synthesis of organomanganese reagents via metathesis with carbon nucleophiles, see: (a) G. Cahiez, D. Bernard and J. F. Normant, Synthesis, 1977, 130–133 CrossRef CAS; (b) G. Cahiez and B. Laboue, Tetrahedron Lett., 1989, 30, 3545–3546 CrossRef CAS; (c) G. Friour, G. Cahiez and J. F. Normant, Synthesis, 1984, 37–40 CrossRef CAS; (d) G. Cahiez, D. Luart and F. Lecomte, Org. Lett., 2004, 6, 4395–4398 CrossRef CAS PubMed; (e) Y. Ahn, W. W. Doubleday and T. Cohen, Synth. Commun., 1995, 25, 33–41 CrossRef CAS; (f) I. Klement, H. Stadtmüller, P. Knochel and G. Cahiez, Tetrahedron Lett., 1997, 38, 1927–1930 CrossRef CAS; (g) K. Ritter and M. Hanack, Tetrahedron Lett., 1985, 26, 1285–1288 CrossRef CAS; (h) G. Cahiez and B. Figadere, Tetrahedron Lett., 1986, 26, 4445–4448 CrossRef; (i) M. T. Reetz, K. Rölfing and N. Griebenow, Tetrahedron Lett., 1994, 35, 1969–1972 CrossRef CAS; (j) G. Cahiez, L. Razafintsalama, B. Laboue and F. Chau, Tetrahedron Lett., 1998, 39, 849–852 CrossRef CAS; (k) G. Cahiez and B. Laboue, Tetrahedron Lett., 1989, 30, 7369–7372 CrossRef CAS; (l) C. Boucley, G. Cahiez, S. Carini, V. Cerè, M. Comes-Franchini, P. Knochel, S. Pollicino and A. Ricci, J. Organomet. Chem., 2001, 624, 223–228 CrossRef CAS.
- S. H. Wunderlich, M. Kienle and P. Knochel, Angew. Chem., Int. Ed., 2009, 48, 7256–7260 CrossRef CAS PubMed.
- For selected reviews on the use of ball milling for organic synthesis, see: (a) S. L. James, C. J. Adams, C. Bolm, D. Braga, P. Collier, T. Friščić, F. Grepioni, K. D. M. Harris, G. Hyett, W. Jones, A. Krebs, J. Mack, L. Maini, A. G. Orpen, I. P. Parkin, W. C. Shearouse, J. W. Steed and D. C. Waddell, Chem. Soc. Rev., 2012, 41, 413–447 RSC; (b) G.-W. Wang, Chem. Soc. Rev., 2013, 42, 7668–7700 RSC; (c) J.-L. Do and T. Friščić, ACS Cent. Sci., 2017, 3, 13–19 CrossRef CAS PubMed; (d) J. G. Hernández and C. Bolm, J. Org. Chem., 2017, 82, 4007–4019 CrossRef PubMed; (e) T.-X. Métro, J. Martinez and F. Lamaty, ACS Sustainable Chem. Eng., 2017, 5, 9599–9602 CrossRef; (f) T. K. Achar, A. Bose and P. Mal, Beilstein J. Org. Chem., 2017, 13, 1907–1931 CrossRef CAS PubMed; (g) O. Eguaogie, J. S. Vyle, P. F. Conlon, M. A. Gîlea and Y. Liang, Beilstein J. Org. Chem., 2018, 14, 955–970 CrossRef CAS PubMed; (h) J. L. Howard, Q. Cao and D. L. Browne, Chem. Sci., 2018, 9, 3080–3094 RSC; (i) J. Andersen and J. Mack, Green Chem., 2018, 20, 1435–1443 RSC; (j) C. Bolm and J. G. Hernández, Angew. Chem., Int. Ed., 2019, 58, 3285–3299 CrossRef CAS PubMed; (k) T. Friščić, C. Mottillo and H. M. Titi, Angew. Chem., Int. Ed., 2020, 59, 1018–1029 CrossRef PubMed; (l) K. Kubota and H. Ito, Trends Chem., 2020, 2, 1066–1081 CrossRef CAS; (m) A. Porcheddu, E. Colacino, L. De Luca and F. Delogu, ACS Catal., 2020, 10, 8344–8394 CrossRef CAS; (n) J. A. Leitch and D. L. Browne, Chem.–Eur. J., 2021, 27, 9721–9726 CrossRef CAS PubMed.
- A. C. Jones, J. A. Leitch, S. E. Raby-Buck and D. L. Browne, Nat. Synth., 2022, 1, 763–775 CrossRef.
- (a) Q. Cao, J. L. Howard, E. Wheatley and D. L. Browne, Angew. Chem., Int. Ed., 2018, 57, 11339–11343 CrossRef CAS PubMed; (b) Q. Cao, R. T. Stark, I. A. Fallis and D. L. Browne, ChemSusChem, 2019, 12, 2554–2557 CrossRef CAS PubMed; (c) J. Yin, R. T. Stark, I. A. Fallis and D. L. Browne, J. Org. Chem., 2020, 85, 2347–2354 CrossRef CAS PubMed.
- (a) J. M. Harrowfield, R. J. Hart and C. R. Whitaker, Aust. J. Chem., 2001, 54, 423–425 CrossRef CAS; (b) V. Birke, C. Schütt, H. Burmeier and W. K. L. Ruck, Fresenius Environ. Bull., 2011, 20, 2794–2805 CAS; (c) I. R. Speight and T. P. Hanusa, Molecules, 2020, 25, 570–578 CrossRef CAS PubMed; (d) R. Takahashi, A. Hu, P. Gao, Y. Gao, Y. Pang, T. Seo, J. Jiang, S. Maeda, H. Takaya, K. Kubota and H. Ito, Nat. Commun., 2021, 12, 6691 CrossRef CAS PubMed; (e) V. S. Pfennig, R. C. Villella, J. Nikodemus and C. Bolm, Angew. Chem., Int. Ed., 2022, 61, e202116514 CrossRef CAS PubMed.
- Yang, Dai, and co-workers have reported magnesium-mediated reductive radical homo-coupling reactions of polyhaloarenes using mechanochemistry; for details, see: H. Chen, J. Fan, Y. Fu, C.-L. Do-Thanh, X. Suo, T. Wang, I. Popovs, D.-E. Jiang, Y. Yuan, Z. Yang and S. Dai, Adv. Mater., 2021, 33, 2008685 CrossRef CAS PubMed.
- P. Gao, J. Jiang, S. Maeda, K. Kubota and H. Ito, Angew. Chem., Int. Ed., 2022, e202207118 CAS.
- W. I. Nicholson, J. L. Howard, G. Magri, A. C. Seastram, A. Khan, R. R. A. Bolt, L. C. Morrill, E. Richards and D. L. Browne, Angew. Chem., Int. Ed., 2021, 60, 23128–23133 CrossRef CAS PubMed.
- R. Fischer, H. Görls, M. Friedrich and M. Westerhausen, J. Organomet. Chem., 2009, 694, 1107–1111 CrossRef CAS.
- C. J. O'Brien, E. A. B. Kantchev, C. Valente, N. Hadei, G. A. Chass, A. Lough, A. C. Hopkinson and M. G. Organ, Chem.–Eur. J., 2006, 12, 4743–4748 CrossRef PubMed.
- (a) G. Cahiez and S. Marquais, Tetrahedron Lett., 1996, 37, 1773–1776 CrossRef CAS; (b) A. Leleu, Y. Fort and R. Schneider, Adv. Synth. Catal., 2006, 348, 1086–1092 CrossRef CAS.
- T. Seo, N. Toyoshima, K. Kubota and H. Ito, J. Am. Chem. Soc., 2021, 143, 6165–6175 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sc05468j |
| This journal is © The Royal Society of Chemistry 2023 |