DOI:
10.1039/D2SC05858H
(Edge Article)
Chem. Sci., 2023,
14, 2353-2360
Saddle-shaped aza-nanographene with multiple odd-membered rings†
Received
23rd October 2022
, Accepted 3rd January 2023
First published on 3rd January 2023
Abstract
A saddle-shaped aza-nanographene containing a central 1,4-dihydropyrrolo[3,2-b]pyrrole (DHPP) has been prepared via a rationally designed four-step synthetic pathway encompassing intramolecular direct arylation, the Scholl reaction, and finally photo-induced radical cyclization. The target non-alternant, nitrogen-embedded polycyclic aromatic hydrocarbon (PAH) incorporates two abutting pentagons between four adjacent heptagons forming unique 7−7−5−5−7−7 topology. Such a combination of odd-membered-ring defects entails a negative Gaussian curvature within its surface with a significant distortion from planarity (saddle height ≈ 4.3 Å). Its absorption and fluorescence maxima are located in the orange-red region, with weak emission originating from the intramolecular charge-transfer character of a low-energy absorption band. Cyclic voltammetry measurements revealed that this stable under ambient conditions aza-nanographene underwent three fully reversible oxidation steps (two one-electron followed by one two-electron) with an exceptionally low first oxidation potential of Eox1 = −0.38 V (vs. Fc/Fc+).
Introduction
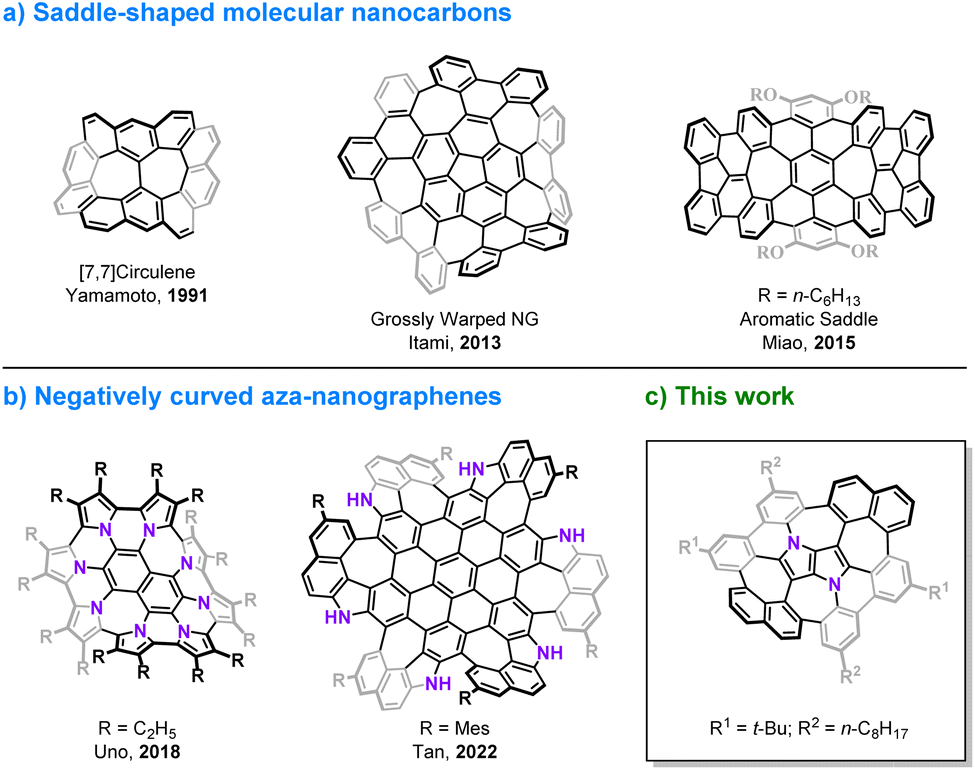
The discovery of curved carbon allotropes, i.e. fullerenes1 and cylindrical carbon nanotubes2 has resulted in a paradigm shift in the chemistry of polyarenes and has given rise to modern materials science. Aromatic compounds, which in the early days of organic chemistry were considered to have a tendency to be planar, can actually adopt various nonplanar molecular shapes.3,4 The development of contemporary synthetic methods in combination with researchers' imagination and creativity has enabled the preparation of elusive, three-dimensional structures including circulenes,5–9 cycloparaphenylenes (CPPs),10–13 carbon nanobelts (CNBs),14–18 large phenine frameworks19,20 and impressive, all-benzene interlocked catenanes and trefoil knots.21 Moreover, embedding non-hexagonal rings in a graphenic lattice causes warping of the π-system, due to geometric mismatch of adjacent rings. The presence of a five-membered ring introduces a positive Gaussian curvature as in the case of the smallest curved cutouts of C60, i.e. corannulene,22–26 and sumanene27,28 and their π-extended analogs,29–32 while the incorporation of rings larger than hexagons e.g. heptagons33 or octagons34 leads to a negative curvature and a saddle-shaped geometry (Fig. 1a).30,35–39 The chemistry of distorted molecular nanocarbons continues to evolve, not only as a consequence of curiosity or aesthetic appeal, but also due to their concrete contribution to understanding the inherent nature of aromaticity,40–44 by delivering experimental verification for the distribution of electrons in their π-extended scaffolds.45,46 Furthermore, history has shown that the emergence of various carbon allotropes has indeed led to the discovery of previously unforeseen and unpredicted functions and applications.47,48 Doping heteroatoms into a carbonaceous backbone of the π-systems is an elegant and efficient method of tuning their basic physicochemical and electronic characteristics.49,50 In particular, interest in nitrogen-embedded, nonplanar polyarenes has gained significant momentum recently, resulting in the construction of some intriguing, mainly positively curved molecules.51–56 Examples of negatively curved aza-nanographenes, however, remain rather scarce (Fig. 1b).57–60 Here, we present the synthesis and properties of a saddle-shaped polyarene containing the most electron-rich small aromatic molecule, 1,4-dihydropyrrolo[3,2-b]pyrrole.61 This 16-ring nitrogen-containing molecular nanocarbon comprises two central pentagons confined between four adjacent heptagons, a lattice defect, which to the best of our knowledge, has not yet been identified in graphene.62–64
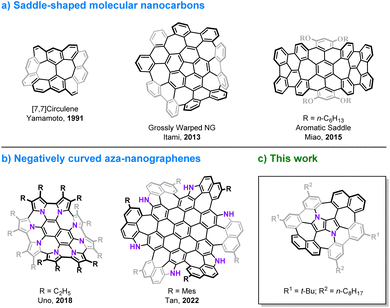 |
| | Fig. 1 Representative examples of negatively curved polyarenes containing multiple odd-membered rings. | |
Results and discussion
Design and synthesis
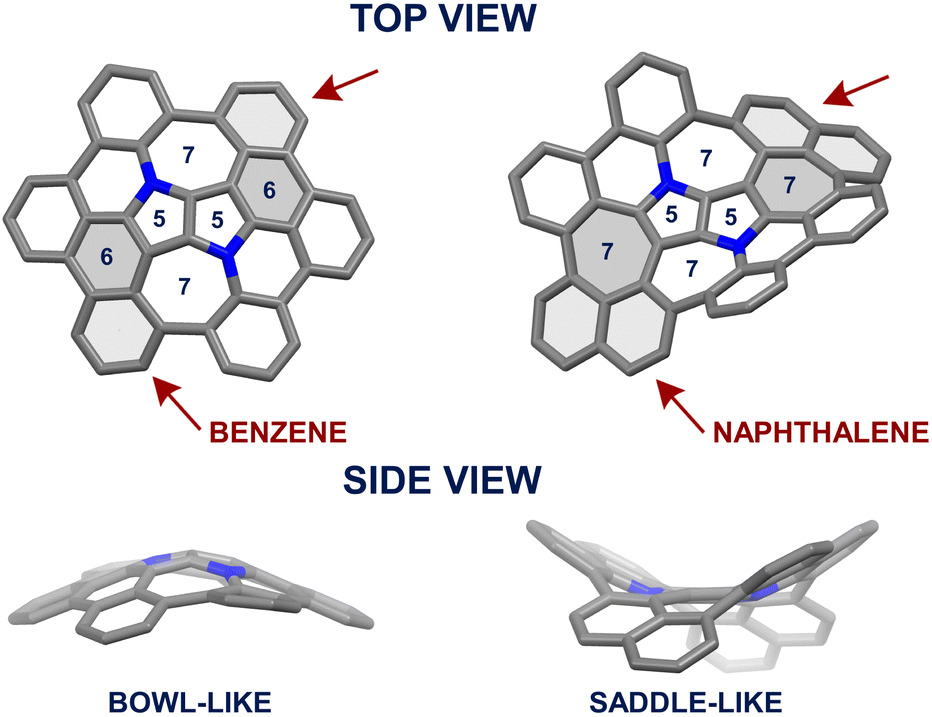
Recently, we reported the on-surface,65 followed by an in-solution66 syntheses of an aza-buckybowl composed of a 1,4-dihydropyrrolo[3,2-b]pyrrole (DHPP) core encircled by six benzene rings bonded with each other, possessing the inverse Stone–Thrower–Wales (7–5–5–7) defect. In the latter endeavor, the implemented synthetic pathway encompassed DHPP formation, the Scholl reaction,67–70 and subsequent sequential intramolecular direct arylation.71 The success of the applied synthetic strategy leading to a series of consecutive couplings of peripheral rings around the central DHPP core prompted us to design and prepare a saddle-shaped DHPP-based nanographene. We envisaged that a seemingly small modification in the structure, i.e., replacing two hexagons with two heptagons next to the central unit (Fig. 2) realized by an imaginary ‘insertion’ of additional benzene rings (naphthalene vs. benzene) would entail a drastic change in the molecular geometry and thus in the overall physicochemical features.
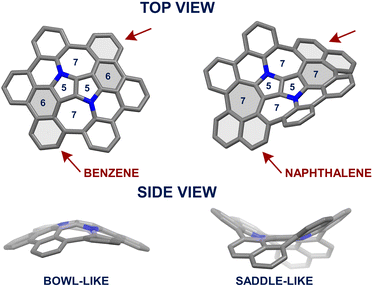 |
| | Fig. 2 Left-hand side: crystal structure (CCDC 2064960†)66 of bowl-shaped DHPP-based nanographene. Right-hand side: the computed model of its π-extended counterpart adopting a saddle-shaped geometry. | |
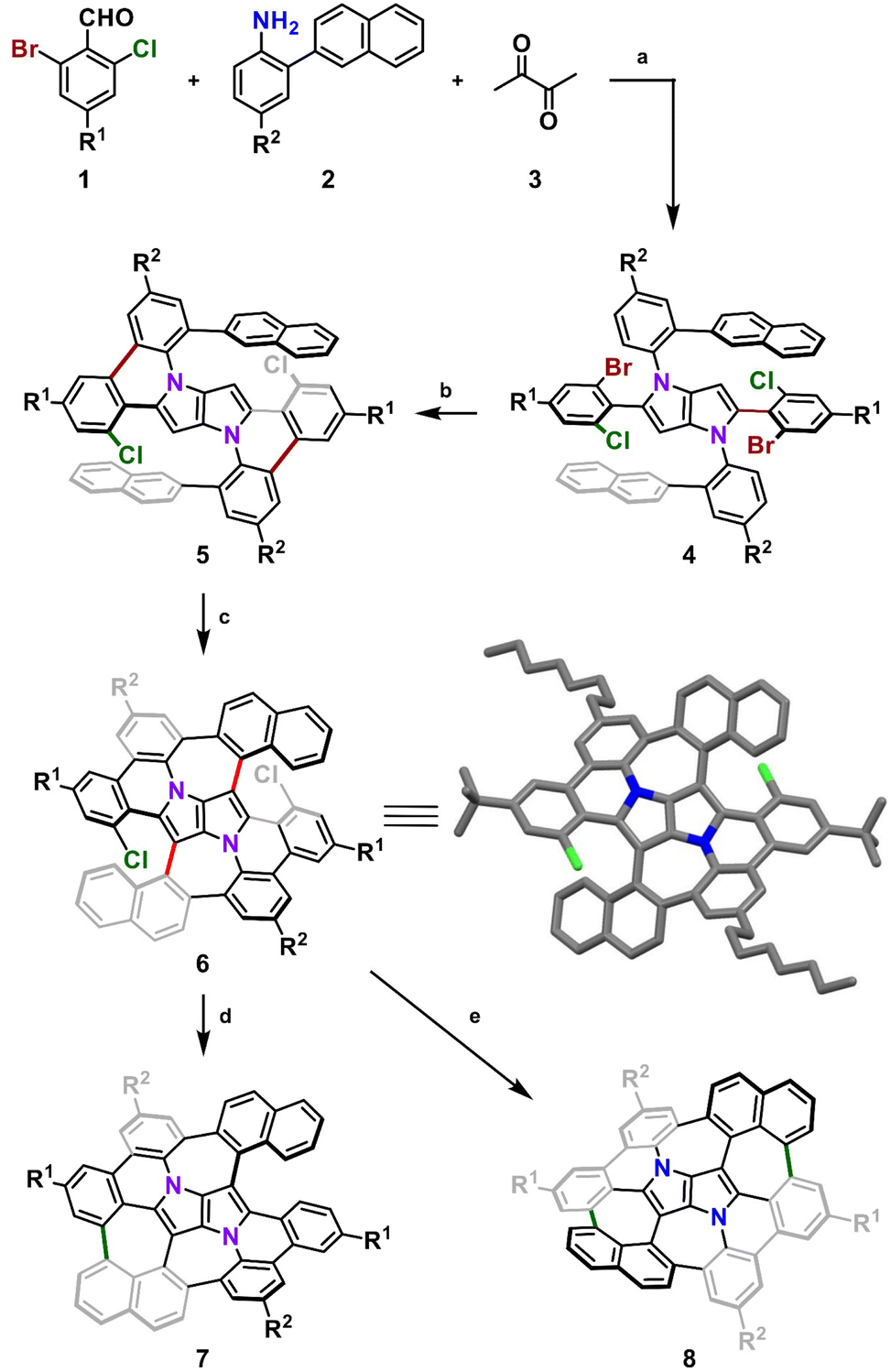
We began with designing a suitable DHPP which would be a platform for further intramolecular transformations. Our analysis resulted in a tetra-aryl pyrrolo[3,2-b]pyrrole (TAPP) substituted with two 2-(naphthalen-2-yl)phenyl groups on the pyrrolic nitrogen atoms and two phenyl residues at positions 2 and 5 of the pyrrolo[3,2-b]pyrrole unit, both carrying a bromine and a chlorine atom at their ortho positions. Finally, to ensure sufficient solubility of the final compound and its parent precursors in common organic solvents, we decided to introduce two tert-butyl groups and two n-octyl chains on the periphery.
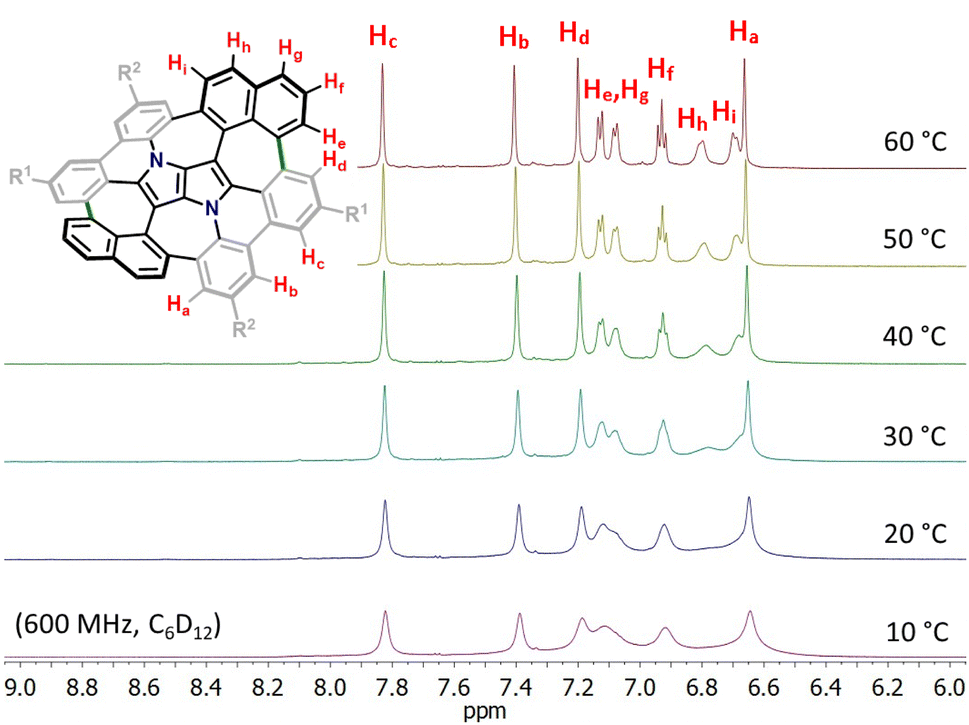
Consequently, suitable starting materials were prepared, that is, 2-bromo-4-(tert-butyl)-6-chlorobenzaldehyde (1) and 2-(naphthalen-2-yl)-4-octylaniline (2) (see the ESI† for details). The one-pot multicomponent condensation, catalyzed by iron(III) perchlorate,72 between 1, 2 and butane-2,3-dione (3) gave TAPP 4 in 49% yield (Scheme 1). Compound 4 was then subjected to selective intramolecular direct arylation through exclusive double C–Br activation, while the two C–Cl bonds remained intact. The reaction was carried out with Pd(OAc)2, Cs2CO3 and PPh3 in toluene at 120 °C for 4 h, giving compound 5 in excellent yield (91%). Then, this product was subjected to the double Scholl reaction mediated by iron(III) triflate. Importantly, as we have already demonstrated, the specific order of consecutive steps is crucial to avoid the unwanted 1,2-aryl shift triggered by the oxidative aromatic coupling.73,74 Also, as previously reported, FeCl3 proved to be the most efficient oxidizing agent with respect to interactions with DHPPs; however, in this particular case, side products were formed. As shown before, the excessive amount of FeCl3 could lead to undesired chlorination of the PP core.66 Therefore, we used iron(III) triflate instead to exclude the possibility of any uncontrolled chlorination processes.75 The reaction was carried out in a dichloroethane/nitromethane mixture at 80 °C for 6 h. The desired compound 6 containing two newly formed seven-membered rings was isolated with a yield of 24%, which is only slightly lower (24% vs. 29%) than the yield of the analogous Scholl reaction carried out on the synthesis of the aza-buckybowl reported earlier.66 It is worth mentioning that this reaction is highly regioselective, and double oxidation takes place predominantly at the α-positions of both naphthalene subunits, which was unequivocally confirmed by X-ray diffraction (Scheme 1, inset). Then, the rationally designed compound 6 was subjected to the final double C–Cl bond activation. The reaction was carried out with Pd(OAc)2 and HPt-Bu2MeBF4 in a mixture of DBU/DMA in a pressure tube at 180 °C for 24 h. However, it turned out that the desired coupling took place only on one side of the molecule, whereas dehalogenation occurred on the other side, giving a saddle–helicene hybrid 7 isolated in 52% yield. Therefore, we resorted to a different strategy, namely photo-induced radical cyclization.60 To our delight, subjecting 6 to a reaction with potassium tert-butoxide in DMSO irradiated with 455 nm blue LEDs at ambient temperature for 20 h gave the desired saddle-shaped nanographene 8 with a yield of 75%. Its structure was confirmed through nuclear magnetic resonance (NMR) spectroscopy, and high-resolution mass spectrometry (HRMS). Its superb solubility allowed for NMR measurements in cyclohexane-d12. The variable temperature 1H NMR spectrum of nanographene 8 measured in cyclohexane-d12 at 10–60 °C (Fig. 3) reveals the dynamic behavior of the molecule in solution and rapid equilibration of naphthalene protons on the perimeter (Fig. 3). Further 2D-NMR studies allowed each signal to be assigned to the respective proton (see the ESI† for details).
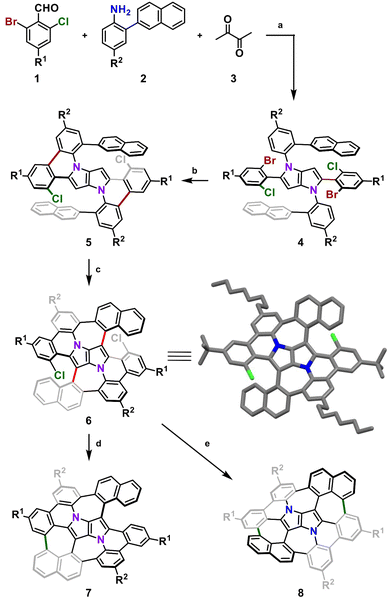 |
| | Scheme 1 Synthetic route to molecular saddle 8. Reagents and conditions: (a) Fe(ClO4)3·xH2O (6 mol%), AcOH/toluene, 50 °C, 16 h, 49%; (b) Pd(OAc)2 (20 mol%), Cs2CO3 (2.4 equiv.), PPh3 (48 mol%), toluene, 120 °C, 4 h, 91%; (c) Fe(OTf)3 (6.0 equiv.), DCE/MeNO2, 80 °C, 6 h, 24%; (d) Pd(OAc)2 (2.0 equiv.), HPt-Bu2MeBF4 (6.0 equiv.), DBU/DMA, 180 °C, 24 h, 52%; (e) t-BuOK (10.0 equiv.), DMSO, blue LEDs, rt, 20 h, 75%. DMA – dimethylacetamide, DCE – 1,2-dichloroethane; inset: X-ray crystal structure of 6. R1 = t-Bu, R2 = n-C8H17. | |
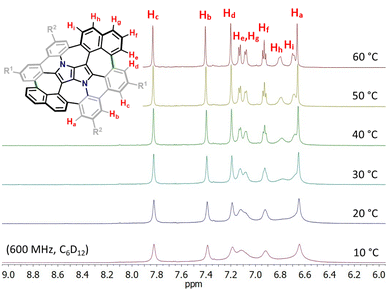 |
| | Fig. 3 Variable temperature 1H-NMR spectrum of the molecular N-doped saddle 8 in the aromatic region (from 6.0 to 9.0 ppm) recorded in cyclohexane-d12 (600 MHz, 10–60 °C). | |
X-ray crystallography analysis
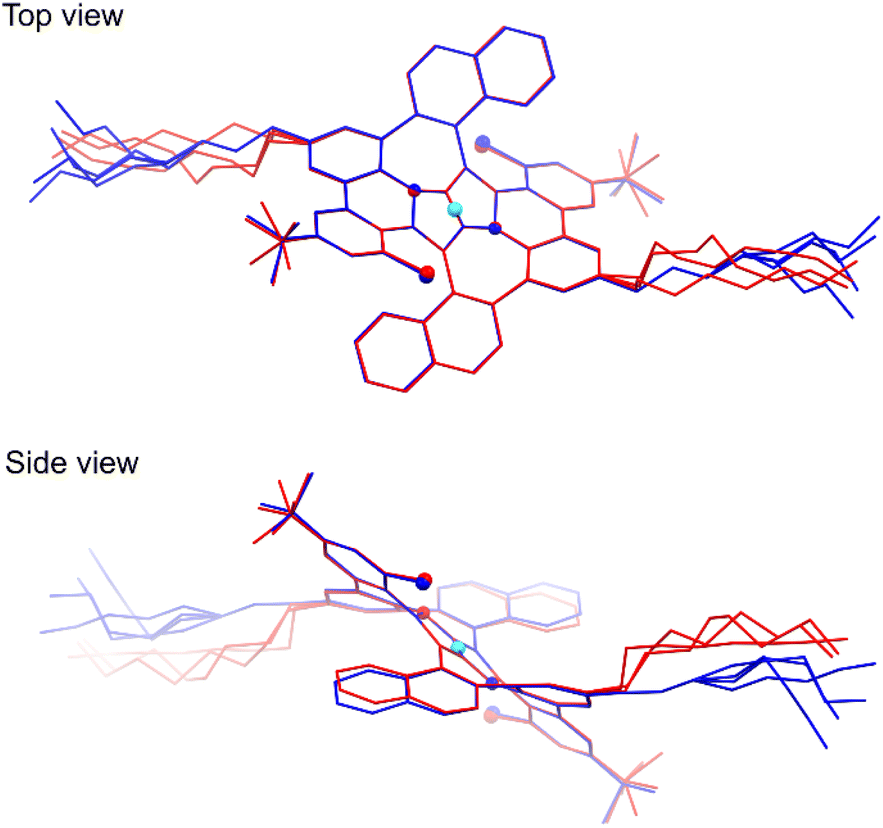
In spite of many attempts it was not possible to crystallize the final product 8, only an amorphous solid was obtained instead. However, during slow evaporation of THF/MeOH/acetone solution X-ray quality crystals of the intermediate compound 6 were obtained confirming its anticipated structure. All the details concerning the crystal data and the structure refinement are located in Table S1 of the ESI.† The crystal lattice contains two centrosymmetric conformers of 6 with slightly different geometry. Hence, the symmetry of the molecule from the X-ray studies fully agrees with the NMR data. The structure is severely disordered especially in the n-octyl chains. The overlay of the two molecules with adjacent five-membered rings atoms fitted is presented in Fig. 4. As it can be seen the core of the molecules fits almost ideally, even seven-membered rings are distorted to the same extent. Whereas the biggest geometry difference occurs in the alkyl chains, the naphthalene moieties deviate as well.
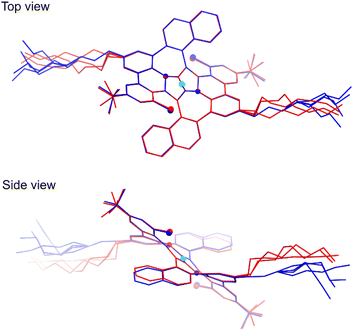 |
| | Fig. 4 Top and side views of the overlay of two differently colored molecules form the crystal structure of 6 (CCDC 2210403†). The cyan sphere represents the center of symmetry; smaller and bigger globes denote the N and Cl atoms, respectively. Overlay was performed for all eight atoms of two adjacent five-membered rings. The THF molecule and all H atoms have been omitted for clarity. | |
Theoretical investigation of conformational flexibility
Quantum chemical calculations were performed for the aza-nanographene 8 structure with the tert-butyl groups and octyl-chains replaced by hydrogen atoms (8H). The equilibrium geometries of the possible conformers of 8H and the saddle-points between them in the closed-shell ground state (S0) were determined with the MP2 method. Local minima and saddle-points were verified by computation of the Hessian of the DFT-optimized structures (see the ESI† for details). The dense accumulation of non-hexagonal rings around the central point of 8H leads to its double-concave molecular structure (Fig. 5). The central DHPP core is nearly planar, while the peripheral phenanthridine and naphthalene components are heavily twisted out of plane to give a saddle height of 4.3 Å (Fig. 5b). These moieties arrange themselves in such a way that both naphthalene subunits are facing downwards and simultaneously both phenanthridine subunits are facing upwards (or conversely, in the case of its enantiomer) (Fig. 5a). The racemization mechanism of (M,P,M,P)-8H to (P,M,P,M)-8H follows a complex quintuple-well potential model through two transition states TS1 and TS2 and the meso form (M,M,P,P)-8H. The calculated energy barrier of 19.6 kcal mol−1 is very similar to that of Itami's grossly warped nanographene (theoretical value: 18.9 kcal mol−1).30 Although the racemization energy barrier is markedly higher than that of the DHPP-based buckybowl (theoretical value: 5.4 kcal mol−1),66 it is still not high enough to ensure conformational stability at ambient temperature, which is supported by the variable temperature 1H NMR experiments (vide supra). The overall potential-energy (PE) landscape of the racemization process of 8H is shown in Fig. 5d.
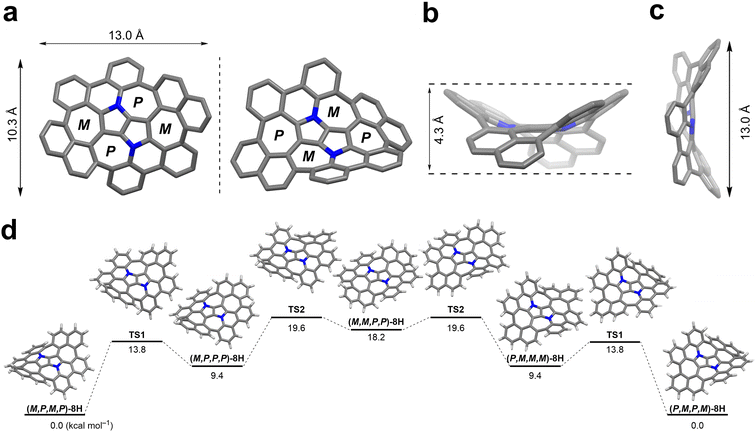 |
| | Fig. 5 Images showing the warped structure of the truncated molecular saddle 8H. (a) Top view of the 8H enantiomer pair. (b) Side view along the a axis. (c) Side view along the b axis. (d) The potential energy landscape of 8H. All geometries were optimized at the MP2/cc-pVDZ level of theory (zero-point energy corrected). | |
Photophysical and electrochemical properties
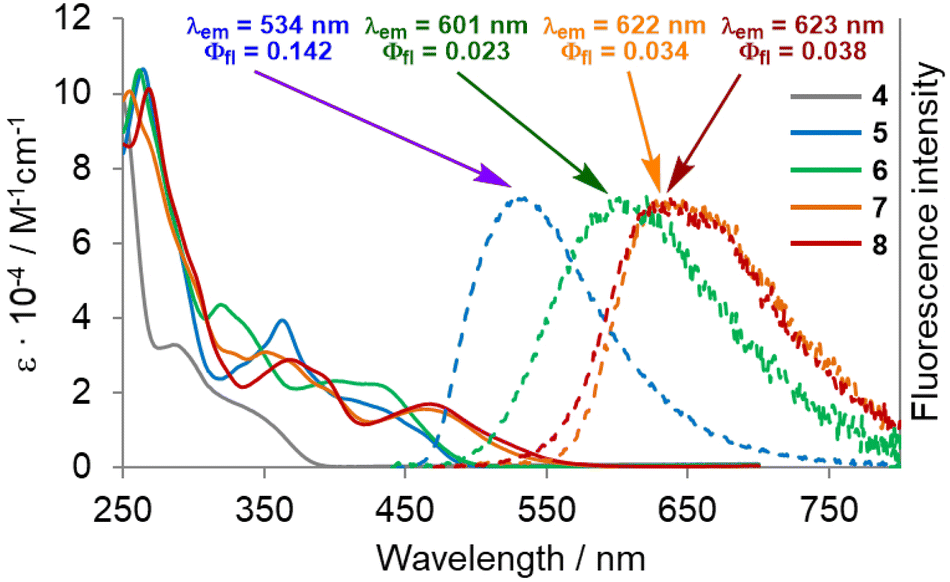
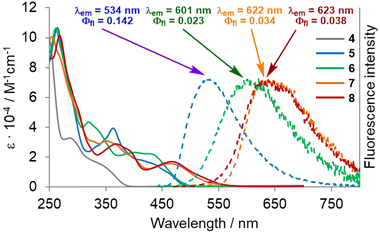
In order to unravel the impact of the molecular geometry and the consecutive extensions of the π-system on the overall photophysical characteristics, we measured the UV-vis absorption and fluorescence spectra of compounds 4–8. The results are shown in Table 1 and Fig. 6. The tetraaryl-substituted DHPP 4 shows a main absorption band comprising two peaks located in the ultraviolet region of the spectrum at λabs = 285 and 334 nm with molar extinction coefficients (ε) reaching 33![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1 and 16000 M−1 cm−1, respectively. Dye 5 obtained from 4 through intramolecular direct arylation possesses a higher degree of conjugation, which is clearly manifested in its optical features. A large bathochromic shift in absorption is observed, accompanied by a hyperchromic shift. Moreover, unlike 4, compound 5 exhibits an emission. In its absorption spectrum, there are two broad bands, the first located between 320 and 400 nm and the second between 400 and 450 nm with a shoulder extending to ca. 480 nm. Higher-energy transitions show a peak located at λabs = 362 nm with ε = 39000 M−1 cm−1. Lower-energy transitions exhibit faint maxima at ca. λabs = 410 with shoulders at 430 and 460 nm (ε = 18000, 16000 and 10000 M−1 cm−1). Dye 5 is a weak green light emitter (λem = 534 nm) with a fluorescence quantum yield (Φfl) of 0.14. The fluorescence maximum of 6, possessing two seven-membered rings, is strongly bathochromically shifted (67 nm) with respect to 5 (λem = 601 vs. 534 nm) with an accompanying evident decrease in emission intensity to Φfl = 0.023. The unusually high Stokes' shift value determined for 6, namely 6500 cm−1 (169 nm), stems from the profound differences in its S1 excited state and S0 ground state geometries.
000 M−1 cm−1 and 16000 M−1 cm−1, respectively. Dye 5 obtained from 4 through intramolecular direct arylation possesses a higher degree of conjugation, which is clearly manifested in its optical features. A large bathochromic shift in absorption is observed, accompanied by a hyperchromic shift. Moreover, unlike 4, compound 5 exhibits an emission. In its absorption spectrum, there are two broad bands, the first located between 320 and 400 nm and the second between 400 and 450 nm with a shoulder extending to ca. 480 nm. Higher-energy transitions show a peak located at λabs = 362 nm with ε = 39000 M−1 cm−1. Lower-energy transitions exhibit faint maxima at ca. λabs = 410 with shoulders at 430 and 460 nm (ε = 18000, 16000 and 10000 M−1 cm−1). Dye 5 is a weak green light emitter (λem = 534 nm) with a fluorescence quantum yield (Φfl) of 0.14. The fluorescence maximum of 6, possessing two seven-membered rings, is strongly bathochromically shifted (67 nm) with respect to 5 (λem = 601 vs. 534 nm) with an accompanying evident decrease in emission intensity to Φfl = 0.023. The unusually high Stokes' shift value determined for 6, namely 6500 cm−1 (169 nm), stems from the profound differences in its S1 excited state and S0 ground state geometries.
Table 1 Spectroscopic and electrochemical properties of compounds 4–8a
| Cmpd. |
λ
abs [nm] |
ε × 103 [M−1 cm−1] |
λ
em [nm] |
Φ
fl
|
E
ox1
1/2
[V] |
E
HOMO [eV] |
|
Measured in CH2Cl2.
Determined with coumarin 153 in EtOH as a standard.
Cyclic voltammograms were measured in a deoxygenated solution of 0.1 M tetrabutylammonium perchlorate in anhydrous CH2Cl2, with a glassy-carbon working electrode, a Ag/AgCl/NaClsat reference electrode, and a platinum-foil auxiliary electrode (scan rate v = 100 mV s−1, Ar, rt). Oxidation potentials are given relative to Fc/Fc+.
|
|
4
|
346 |
14 |
— |
— |
0.35 |
−5.15 |
|
5
|
410 |
20 |
534 |
0.14 |
0.05 |
−4.85 |
|
6
|
432 |
22 |
601 |
0.023 |
0.18 |
−4.98 |
|
7
|
461 |
16 |
622 |
0.034 |
−0.12 |
−4.68 |
|
8
|
468 |
17 |
623 |
0.038 |
−0.38 |
−4.42 |
 |
| | Fig. 6 UV-vis absorption spectra (solid lines) and fluorescence spectra (dashed lines) of dyes 4–8 measured in dichloromethane. | |
In both cases of π-extension, leading to compounds 7 and 8, a red-shift of the absorption and emission spectra can be observed. The final saddle-shaped aza-nanographene 8 exhibits three prominent bands in the absorption spectrum. The first band is located between 280 and 330 nm with a distinctive shoulder at 300 nm with ε = 52000 M−1 cm−1. The second band corresponding to lower-energy transitions, found between 330 and 420 nm, shows a peak at λabs = 367 nm (ε = 29000 M−1 cm−1) with a shoulder at 390 nm (ε = 25000 M−1 cm−1). Finally the third band corresponds to the longest wavelength absorption at 468 nm, ε = 17000 M−1 cm−1 tailing up to 580 nm. The maximum absorption is red-shifted by 7 nm compared to 7. Interestingly, both compounds 7 and 8 have nearly identical emission maxima in the orange/red region of the spectrum with λem = 622 and 623 nm, and exhibit similar fluorescence quantum yields of Φfl = 0.034 and 0.038, respectively. Unlike the DHPP-buckybowl,66 molecular saddle 8 does not show any dependence of the emission features on solvent polarity (see the ESI† for details).
A direct comparison of the photophysics of dye 8 with the previously reported aza-buckybowl66 reveals that they behave in analogous manner. In both cases absorption peaks at approx. 470 nm with long tail reaching beyond 550 nm. Both the aza-buckybowl and 8 possess a broad, weak red emission. A more pronounced deformation of the molecule 8 is reflected by a small bathochromic shift of the emission maximum by ca. 8 nm compared to the respective buckybowl.
To further elucidate the nature of electronic transitions, the absorption spectrum of the lowest-energy conformer (M,P,M,P)-8H was computed with the aid of two different theoretical methods: TD-DFT and ADC(2). The resulting spectrum is shown in Fig. S13 of the ESI.† Apart from the expected energy shift, both computed spectra are consistent with each other and are in good agreement with the low-energy part of the observed absorption spectrum of 8.
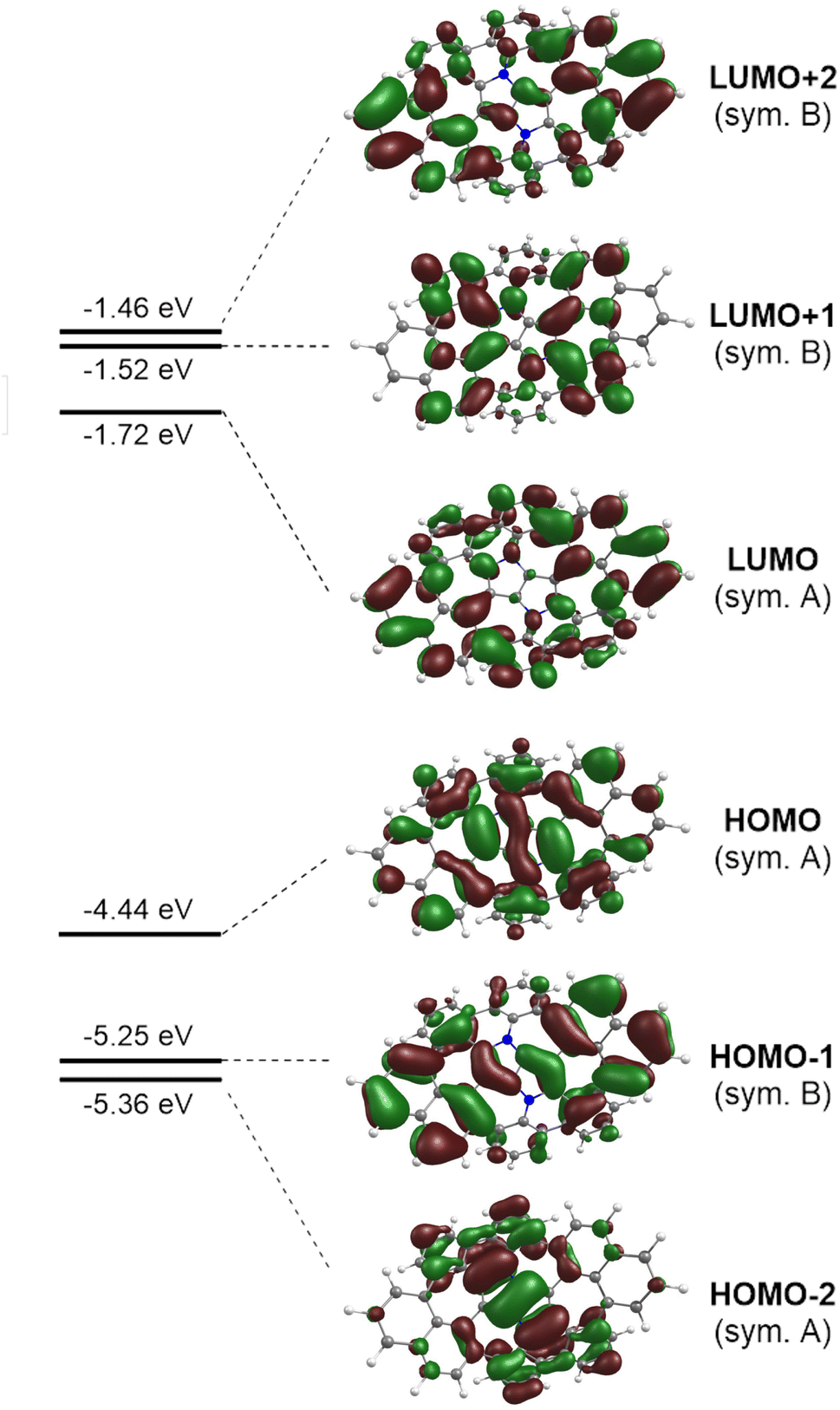
The photophysical parameters of the lowest excited states of 8H computed with the aid of the ADC(2)/cc-pVDZ ab initio method are summarized in Table S3 of the ESI.† On inspection of the results it can be noticed that the series of excited singlet states that comprise the low-energy part of the absorption spectrum involve electronic excitations from the two highest occupied molecular orbitals (HOMO−2 and HOMO) in the C2 symmetry point group of the (M,P,M,P)-8H conformer. Both HOMOs are localized largely on the DHPP core. Optical excitation of the system expels an electron from the central core into the (naphthalene and benzene) molecular edges (Fig. 7).
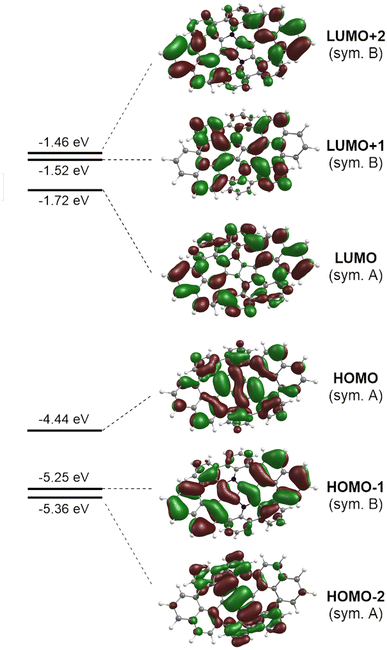 |
| | Fig. 7 Selected Kohn–Sham molecular orbitals of the truncated molecular saddle 8H and their energy levels calculated with the aid of DFT at the B3LYP/cc-pVDZ level of theory. An isovalue of 0.02 was used for the depiction of each MO, the view corresponds to the top face of the molecule. | |
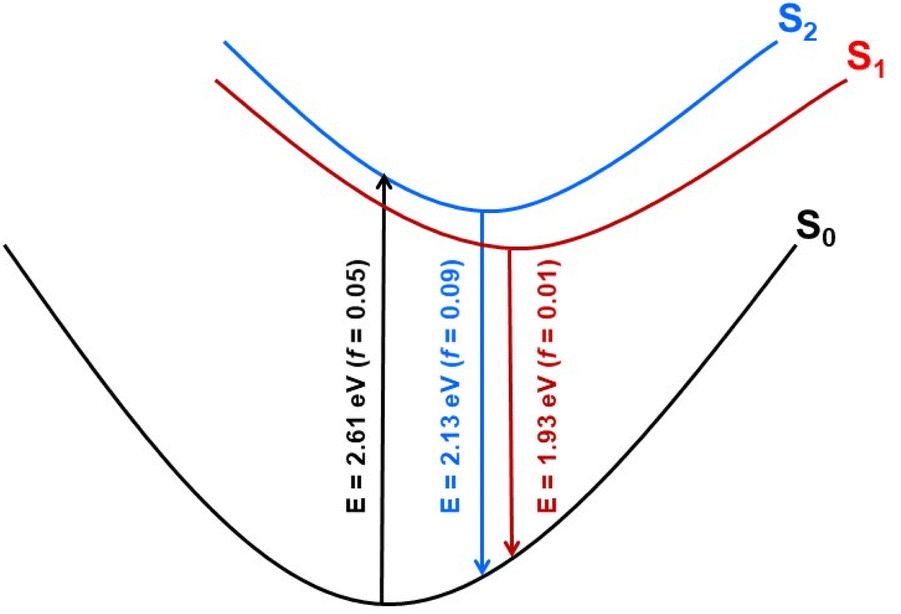
Geometry optimization of the two lowest excited singlet states (1A(S1) and 1B(S2)) with the aid of the ADC(2) method resulted in the determination of their adiabatic energy and energy of the vertical fluorescence expected from these states (Fig. 8). Additionally, the natural transition orbitals (NTOs)76 of the hole and the electron created by electronic excitation of these states were determined. These data are shown in Fig. S14 of the ESI.† From this it can be seen that both states are stabilized by a similar energy loss due to geometry optimization, and the state of the A symmetry is by about 0.22 eV (5.1 kcal mol−1) more stable than the state of the B symmetry, both ‘vertically’ (Table S3†) and adiabatically (Fig. S14†). The singlet state of the B symmetry lies above the 1A even at the optimized geometry of the former state. We therefore conclude that the latter state represents the lowest (emitting) singlet state (S1) of the 8H (and 8) compound, although its oscillator strength for emission is one order of magnitude smaller than for emission from the 1B state (Fig. S14†). The NTOs inspection of the two excited states indicates that the fluorescence from these states is approximately perpendicularly polarized to each other.
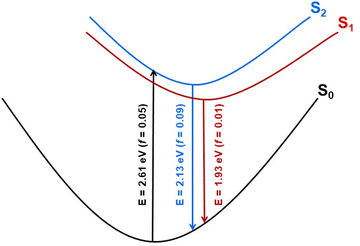 |
| | Fig. 8 Schematic potential energy profiles of the lowest excited states of 8H constructed on the basis of theoretical results. Vertical arrows illustrate vertical absorption from the ground state (black) and fluorescence (blue and red) from the lowest excited singlet states with values of transition energy (E) and oscillator strength (f) computed at the ADC(2)/cc-pVDZ theoretical level. | |
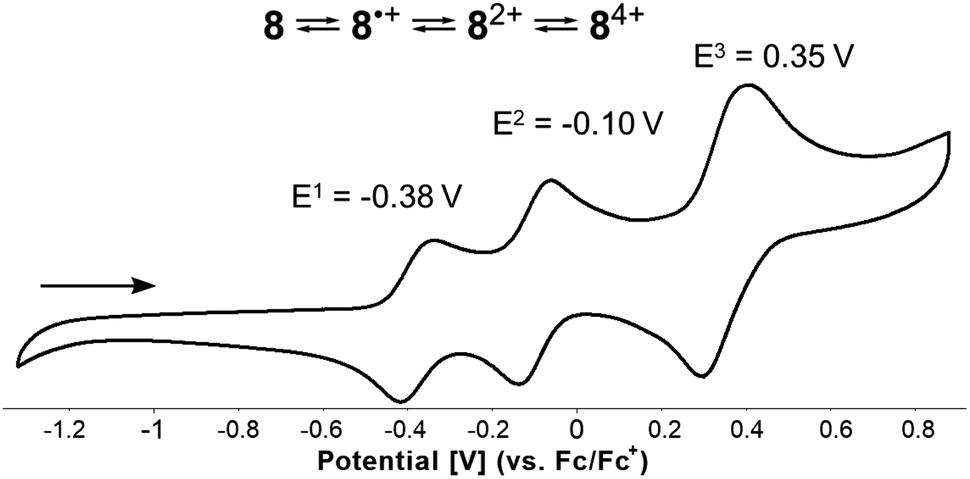
Finally, the well-known, electron-rich nature of 1,4-dihydropyrrolo[3,2-b]pyrroles renders them potent candidates for application in the field of organic optoelectronics. Therefore, voltammetric experiments were performed to investigate the electrochemical properties of all synthesized DHPP-based compounds 4–8. Cyclic voltammetry measurements revealed that all obtained DHPPs are easily oxidizable with the obvious trend that as the degree of conjugation is increased, the lower the first oxidation potential (Tables 1 and S2†). The parent TAPP 4, underwent two one-electron oxidation steps, the first being reversible, followed by an irreversible process. Concurrently compounds 5–7 underwent two one-electron reversible oxidation steps (see the ESI†). Intriguingly, target molecular saddle 8 underwent as many as three fully reversible oxidation steps. The first two were one-electron processes, whereas the third was two-electron process. The half-wave potentials measured for 8 referenced to the ferrocene/ferrocenium ion couple were Eox1 = −0.38 V, Eox2 = −0.10 V, and Eox3 = +0.35 V (Fig. 9). HOMO energy levels (EHOMO) were estimated from the first oxidation potentials (Table 1). The calculated energy of the HOMO level of 8H (EHOMO = −4.44 eV, Fig. 7) was in excellent agreement with the experimental data for 8i.e., EHOMO = −4.42 eV. Thus, HOMO of dye 8 is located ca. 0.3 eV above HOMO of previously described buckybowl (EHOMO = −4.71 eV).66 The exceptionally low value of the first oxidation potential of 8, nearly as low as the potential measured for the core-expanded azacoronene that has up to eight nitrogen atoms (Eox1 = −0.52 V)57 indicates its remarkable tendency to oxidize. To date, this is a record-breaking oxidation potential value in the chemistry of dihydropyrrolopyrroles.77
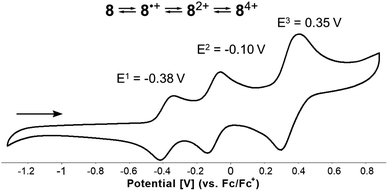 |
| | Fig. 9 Cyclic voltammogram of the N-doped molecular saddle 8. | |
Conclusions
By employing a rationally designed synthetic strategy, we have demonstrated the successful solution phase synthesis of a saddle-shaped aza-nanographene containing a pyrrolopyrrole core. This 16-ring nitrogen-doped, highly twisted framework was easily and efficiently formed in four steps with an overall yield of 8%. Our approach undoubtedly shows that the constant growth of the synthetic toolbox allows scientists to fine-tune the structure and properties of previously unimaginable organic materials. This heavily distorted π-system provides valuable information regarding the impact of closely concentrated odd-membered-ring defects on the overall physicochemical properties of the otherwise planar graphenic sheet. To the best of our knowledge this is the first nanographene displaying a unique 7–7–5–5–7–7 topology. Furthermore, it is the first example of a π-extended 1,4-dihydropyrrolo[3,2-b]pyrrole that exhibits three fully reversible oxidation steps, with a record low for its first oxidation potential value. We have proven that replacing the previously described 7–5–5–7 ring system with 7–7–5–5–7–7 one leads to three dramatic changes: (1) the new aza-nanographene is markedly (0.3 eV) easier to oxidize; (2) distortion from planarity doubles (4.3 Å vs. 2.1 Å) and (3) negative curvature replaces positive curvature. The warped double-concave structure holds potential for supramolecular research and its use in the construction of future outstanding nitrogen-enriched nanocarbon materials is envisioned.
Data availability
The data supporting this article have been uploaded as part of the ESI.†
Author contributions
M. K. conceived the idea and wrote the manuscript. M. K. performed all synthetic experiments. Ł. D. performed crystallographic studies and M. K. C. supervised them. A. L. S. performed computational studies, analyzed data, wrote and reviewed the manuscript. D. T. G. supervised the project, performed formal analysis, wrote and reviewed the manuscript. All the authors discussed the results and commented on the manuscript. All authors have given approval to the final version of the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The work was financially supported by the Polish National Science Centre, Poland (OPUS 2020/37/B/ST4/00017), the Foundation for Polish Science (TEAM POIR.04.04.00-00-3CF4/16-00 and START scholarship no. 039.2017 to M. K.). M. K. is a recipient of a scholarship awarded by the Polish Ministry of Education and Science to outstanding young scientists. The authors thank Dr David C. Young for proofreading the manuscript.
Notes and references
- H. W. Kroto, J. R. Heath, S. C. O'Brien, R. F. Curl and R. E. Smalley, Nature, 1985, 318, 162–163 CrossRef CAS
 .
.
- S. Iijima, Nature, 1991, 354, 56–58 CrossRef CAS .
- M. A. Majewski and M. Stępień, Angew. Chem., 2019, 131, 90–122 CrossRef .
- Y. Segawa, D. R. Levine and K. Itami, Acc. Chem. Res., 2019, 52, 2760–2767 CrossRef CAS PubMed .
-
(a) S. K. Pedersen, K. Eriksen, H. Ågren, B. F. Minaev, N. N. Karaush-Karmazin, O. Hammerich, G. V. Baryshnikov and M. Pittelkow, J. Am. Chem. Soc., 2020, 33, 14058–14063 CrossRef PubMed ;
(b) K. Yamamoto, T. Harada, M. Nakazaki, T. Naka, Y. Kai, S. Harada and N. Kasai, J. Am. Chem. Soc., 1983, 105, 7171–7172 CrossRef CAS .
- K. Yamamoto, Y. Saitho, D. Iwaki and T. Ooka, Angew. Chem., Int. Ed., 1991, 30, 1173–1174 CrossRef .
- C. N. Feng, M. Y. Kuo and Y. T. Wu, Angew. Chem., Int. Ed., 2013, 52, 7791–7794 CrossRef CAS PubMed .
- Y. Sakamoto and T. Suzuki, J. Am. Chem. Soc., 2013, 135, 14074–14077 CrossRef CAS PubMed .
- M. Krzeszewski, H. Ito and K. Itami, J. Am. Chem. Soc., 2022, 144, 862–871 CrossRef CAS PubMed .
- R. Jasti, J. Bhattacharjee, J. B. Neaton and C. R. Bertozzi, J. Am. Chem. Soc., 2008, 130, 17646–17647 CrossRef CAS PubMed .
- H. Takaba, H. Omachi, Y. Yamamoto, J. Bouffard and K. Itami, Angew. Chem., Int. Ed., 2009, 48, 6112–6116 CrossRef CAS PubMed .
- S. Yamago, Y. Watanabe and T. Iwamoto, Angew. Chem., Int. Ed., 2010, 49, 757–759 CrossRef CAS PubMed .
- F. E. Golling, S. Osella, M. Quernheim, M. Wagner, D. Beljonne and K. Müllen, Chem. Sci., 2015, 6, 7072–7078 RSC .
- G. Povie, Y. Segawa, T. Nishihara, Y. Miyauchi and K. Itami, Science, 2017, 356, 172–175 CrossRef CAS PubMed .
- G. Povie, Y. Segawa, T. Nishihara, Y. Miyauchi and K. Itami, J. Am. Chem. Soc., 2018, 140, 10054–10059 CrossRef CAS PubMed .
- K. Y. Cheung, S. Gui, C. Deng, H. Liang, Z. Xia, Z. Liu, L. Chi and Q. Miao, Chem, 2019, 5, 838–847 CAS .
- K. Y. Cheung, K. Watanabe, Y. Segawa and K. Itami, Nat. Chem., 2021, 13, 255–259 CrossRef CAS PubMed .
- Y. Segawa, T. Watanabe, K. Yamanoue, M. Kuwayama, K. Watanabe, J. Pirillo, Y. Hijikata and K. Itami, Nat. Synth., 2022, 1, 535–541 CrossRef .
- K. Ikemoto, J. Lin, R. Kobayashi, S. Sato and H. Isobe, Angew. Chem., Int. Ed., 2018, 57, 8555–8559 CrossRef CAS PubMed .
- K. Ikemoto, M. Akiyoshi, T. Mio, K. Nishioka, S. Sato and H. Isobe, Angew. Chem., Int. Ed., 2022, 61, e202204035 CAS .
- Y. Segawa, M. Kuwayama, Y. Hijikata, M. Fushimi, T. Nishihara, J. Pirillo, J. Shirasaki, N. Kubota and K. Itami, Science, 2019, 365, 272–276 CrossRef CAS PubMed .
- W. E. Barth and R. G. Lawton, J. Am. Chem. Soc., 1971, 93, 1730–1745 CrossRef .
- M. M. Hashemi, M. S. Bratcher and L. T. Scott, J. Am. Chem. Soc., 1992, 114, 1920–1921 CrossRef .
- L. T. Scott, M. M. Hashemi, D. T. Meyer and H. B. Warren, J. Am. Chem. Soc., 1991, 113, 7082–7084 CrossRef CAS .
- E. Nestoros and M. C. Stuparu, Chem. Commun., 2018, 54, 6503–6519 RSC .
- L. T. Scott, P. C. Cheng, M. M. Hashemi, M. S. Bratcher, D. T. Meyer and H. B. Warren, J. Am. Chem. Soc., 1997, 119, 10963–10968 CrossRef CAS .
- H. Sakurai, T. Daiko and T. Hirao, Science, 2003, 301, 1878 CrossRef CAS PubMed .
- S. Higashibayashi and H. Sakurai, Chem. Lett., 2011, 40, 122–128 CrossRef CAS .
- R. B. M. Ansems and L. T. Scott, J. Am. Chem. Soc., 2000, 122, 2719–2724 CrossRef CAS .
- K. Kawasumi, Q. Zhang, Y. Segawa, L. T. Scott and K. Itami, Nat. Chem., 2013, 5, 739–744 CrossRef CAS PubMed .
- K. Shoyama and F. Würthner, J. Am. Chem. Soc., 2019, 141, 13008–13012 CrossRef CAS PubMed .
- Z.-Z. Zhu, Z.-C. Chen, Y.-R. Yao, C.-H. Cui, S.-H. Li, X.-J. Zhao, Q. Zhang, H.-R. Tian, P.-Y. Xu, F.-F. Xie, X.-M. Xie, Y.-Z. Tan, S.-L. Deng, J. M. Quimby, L. T. Scott, S.-Y. Xie, R.-B. Huang and L.-S. Zheng, Sci. Adv., 2019, 5, eaaw0982 CrossRef CAS PubMed .
- Chaolumen, I. A. Stepek, K. E. Yamada, H. Ito and K. Itami, Angew. Chem., Int. Ed., 2021, 60, 23508–23532 CrossRef CAS PubMed .
- G. González Miera, S. Matsubara, H. Kono, K. Murakami and K. Itami, Chem. Sci., 2022, 13, 1848–1868 RSC .
- S. H. Pun and Q. Miao, Acc. Chem. Res., 2018, 51, 1630–1642 CrossRef CAS PubMed .
- K. Y. Cheung, X. Xu and Q. Miao, J. Am. Chem. Soc., 2015, 137, 3910–3914 CrossRef CAS PubMed .
- K. Y. Cheung, C. K. Chan, Z. Liu and Q. Miao, Angew. Chem., 2017, 56, 9003–9007 CrossRef CAS PubMed .
- I. R. Márquez, S. Castro-Fernández, A. Millán and A. G. Campaña, Chem. Commun., 2018, 54, 6705–6718 RSC .
- S. H. Pun, C. K. Chan, J. Luo, Z. Liu and Q. Miao, Angew. Chem., Int. Ed., 2018, 57, 1581–1586 CrossRef CAS PubMed .
- M. Randić, Chem. Rev., 2003, 103, 3449–3605 CrossRef PubMed .
- M. A. Dobrowolski, A. Ciesielski and M. K. Cyrański, Phys. Chem. Chem. Phys., 2011, 13, 20557–20563 RSC .
- O. El Bakouri, D. W. Szczepanik, K. Jorner, R. Ayub, P. Bultinck, M. Solà and H. Ottosson, J. Am. Chem. Soc., 2022, 144, 8560–8575 CrossRef CAS PubMed .
- Bharat, R. Bhola, T. Bally, A. Valente, M. K. Cyrański, Ł. Dobrzycki, S. M. Spain, P. Rempała, M. R. Chin and B. T. King, Angew. Chem., Int. Ed., 2010, 49, 399–402 CrossRef CAS PubMed .
- Y. Tanaka, N. Fukui and H. Shinokubo, Nat. Commun., 2020, 11, 3873 CrossRef CAS PubMed .
- C. Liu, Y. Ni, X. Lu, G. Li and J. Wu, Acc. Chem. Res., 2019, 52, 2309–2321 CrossRef CAS PubMed .
- J. C. Buttrick and B. T. King, Chem. Soc. Rev., 2017, 46, 7–20 RSC .
- M. F. L. De Volder, S. H. Tawfick, R. H. Baughman and A. J. Hart, Science, 2013, 339, 535–539 CrossRef CAS PubMed .
- W.-S. Wong and M. Stępień, Trends Chem., 2022, 7, 573–576 CrossRef .
- M. Stępień, E. Gońka, M. Żyła and N. Sprutta, Chem. Rev., 2017, 117, 3479–3716 CrossRef PubMed .
- A. Borissov, Y. K. Maurya, L. Moshniaha, W.-S. Wong, M. Żyła-Karwowska and M. Stępień, Chem. Rev., 2022, 122, 565–788 CrossRef CAS PubMed .
-
(a) Q. Tan, S. Higashibayashi, S. Karanjit and H. Sakurai, Nat. Commun., 2012, 3, 10–15 Search PubMed ;
(b) S. K. Pedersen, K. Eriksen and M. Pittelkow, Angew. Chem., Int. Ed., 2019, 58, 18419 CrossRef CAS PubMed .
-
(a) C. G. Claessens, D. González-Rodríguez and T. Torres, Chem. Rev., 2002, 102, 835–854 CrossRef CAS PubMed ;
(b) D. Myśliwiec, M. Stępień and M. Stȩpień, Angew. Chem., Int. Ed., 2013, 52, 1713–1717 CrossRef PubMed .
- S. Ito, Y. Tokimaru and K. Nozaki, Angew. Chem., Int. Ed., 2015, 54, 7256–7260 CrossRef CAS PubMed .
- H. Yokoi, Y. Hiraoka, S. Hiroto, D. Sakamaki, S. Seki and H. Shinokubo, Nat. Commun., 2015, 6, 8215 CrossRef PubMed .
- S. Higashibayashi, P. Pandit, R. Haruki, S. I. Adachi and R. Kumai, Angew. Chem., Int. Ed., 2016, 55, 10830–10834 CrossRef CAS PubMed .
- J. Wagner, P. Zimmermann Crocomo, M. A. Kochman, A. Kubas, P. Data and M. Lindner, Angew. Chem., Int. Ed., 2022, 61, e202202232 CrossRef CAS PubMed .
- K. Oki, M. Takase, S. Mori, A. Shiotari, Y. Sugimoto, K. Ohara, T. Okujima and H. Uno, J. Am. Chem. Soc., 2018, 140, 10430–10434 CrossRef CAS PubMed .
- T. Kirschbaum, F. Rominger and M. Mastalerz, Chem.–Eur. J., 2020, 26, 14560–14564 CrossRef CAS PubMed .
- P. An, R. Li, B. Ma, R. He, Y. Zhang, M. Xiao and B. Zhang, Angew. Chem., Int. Ed., 2021, 60, 24478–24483 CrossRef CAS PubMed .
- Z. Qiu, X. Chen, Y. Huang, R. Wei, K. Chu, X. Zhao and Y. Tan, Angew. Chem., Int. Ed., 2022, 61, e202116955 CAS .
-
(a) M. Krzeszewski, D. Gryko and D. T. Gryko, Acc. Chem. Res., 2017, 50, 2334–2345 CrossRef CAS PubMed ;
(b) G. Sanil, B. Koszarna, Y. M. Poronik, O. Vakuliuk, B. Szymański, D. Kusy and D. T. Gryko, The Chemistry of 1,4-Dihydropyrrolo[3,2-b]pyrroles, Adv. Heterocyclic Chem., 2022, 138, 335–409 CrossRef .
- F. Banhart, J. Kotakoski and A. V. Krasheninnikov, ACS Nano, 2011, 5, 26–41 CrossRef CAS PubMed .
-
T. Xu and L. Sun, in Defects in Advanced Electronic Materials and Novel Low Dimensional Structures, Elsevier, 2018, pp. 137–160 Search PubMed .
- Y. Fei, Y. Fu, X. Bai, L. Du, Z. Li, H. Komber, K. H. Low, S. Zhou, D. L. Phillips, X. Feng and J. Liu, J. Am. Chem. Soc., 2021, 143, 2353–2360 CrossRef CAS PubMed .
- S. Mishra, M. Krzeszewski, C. A. Pignedoli, P. Ruffieux, R. Fasel and D. T. Gryko, Nat. Commun., 2018, 9, 1714 CrossRef PubMed .
- M. Krzeszewski, Ł. Dobrzycki, A. L. Sobolewski, M. K. Cyrański and D. T. Gryko, Angew. Chem., Int. Ed., 2021, 60, 14998–15005 CrossRef CAS PubMed .
- M. Grzybowski, K. Skonieczny, H. Butenschön and D. T. Gryko, Angew. Chem., Int. Ed., 2013, 52, 9900–9930 CrossRef CAS PubMed .
- M. Grzybowski, B. Sadowski, H. Butenschön and D. T. Gryko, Angew. Chem., Int. Ed., 2020, 59, 2998–3027 CrossRef CAS PubMed .
- P. Rempala, J. Kroulík and B. T. King, J. Am. Chem. Soc., 2004, 126, 15002–15003 CrossRef CAS PubMed .
- Y. Zhang, S. H. Pun and Q. Miao, Chem. Rev., 2022, 122, 14554–14593 CrossRef CAS PubMed .
- W. Hagui, H. Doucet and J.-F. Soulé, Chem, 2019, 5, 2006–2078 CAS .
- M. Tasior, O. Vakuliuk, D. Koga, B. Koszarna, K. Górski, M. Grzybowski, Ł. Kielesiński, M. Krzeszewski and D. T. Gryko, J. Org. Chem., 2020, 85, 13529–13543 CrossRef CAS PubMed .
- M. Krzeszewski, K. Sahara, Y. M. Poronik, T. Kubo and D. T. Gryko, Org. Lett., 2018, 20, 1517–1520 CrossRef CAS PubMed .
- J. Liu, A. Narita, S. Osella, W. Zhang, D. Schollmeyer, D. Beljonne, X. Feng and K. Müllen, J. Am. Chem. Soc., 2016, 138, 2602–2608 CrossRef CAS PubMed .
- J. P. Lewtak, D. Gryko, D. Bao, E. Sebai, O. Vakuliuk, M. Ścigaj and D. T. Gryko, Org. Biomol. Chem., 2011, 9, 8178–8181 RSC .
- R. L. Martin, J. Chem. Phys., 2003, 118, 4775–4777 CrossRef CAS .
- A. Janiga, M. Krzeszewski and D. T. Gryko, Chem.–Asian J., 2015, 10, 212–218 CrossRef CAS PubMed .
Footnote |
| † Electronic supplementary information (ESI) available: Details of the synthesis and spectroscopic characterization of new compounds, crystallographic and electrochemical data. CCDC 2210403. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2sc05858h |
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence b,
Andrzej L.
Sobolewski
b,
Andrzej L.
Sobolewski