
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Back to the future of organolanthanide chemistry†
Nolwenn
Mahieu
 ,
Jakub
Piątkowski
,
Thomas
Simler
* and
Grégory
Nocton
*
,
Jakub
Piątkowski
,
Thomas
Simler
* and
Grégory
Nocton
*
LCM, CNRS, Ecole Polytechnique, Institut Polytechnique de Paris, Route de Saclay, 91120 Palaiseau, France. E-mail: gregory.nocton@polytechnique.edu; thomas.simler@polytechnique.edu
First published on 30th November 2022
Abstract
At the dawn of the development of structural organometallic chemistry, soon after the discovery of ferrocene, the description of the LnCp3 complexes, featuring large and mostly trivalent lanthanide ions, was rather original and sparked curiosity. Yet, the interest in these new architectures rapidly dwindled due to the electrostatic nature of the bonding between π-aromatic ligands and 4f-elements. Almost 70 years later, it is interesting to focus on how the discipline has evolved in various directions with the reports of multiple catalytic reactivities, remarkable potential in small molecule activation, and the development of rich redox chemistry. Aside from chemical reactivity, a better understanding of their singular electronic nature – not precisely as simplistic as anticipated – has been crucial for developing tailored compounds with adapted magnetic anisotropy or high fluorescence properties that have witnessed significant popularity in recent years. Future developments shall greatly benefit from the detailed reactivity, structural and physical chemistry studies, particularly in photochemistry, electro- or photoelectrocatalysis of inert small molecules, and manipulating the spins' coherence in quantum technology.
 Nolwenn Mahieu | Nolwenn Mahieu started her studies at the Ecole Normale Supérieure (ENS) Paris-Saclay in 2016. After a stay in Pr. Eric J. Schelter's group at the university of Pennsylvania working on imido–thorium complexes, she graduated in 2020 with a master in molecular chemistry from ENS Paris-Saclay. She then joined Dr G. Nocton's group at Ecole polytechnique for her PhD studies on the synthesis of organolanthanide complexes. |
 Jakub Piątkowski | Jakub Piątkowski was born in 1997 in Działdowo, Poland. In 2019, he received his B.S. degree in general chemistry from the University of Warsaw (Warsaw, Poland), under the supervision of Prof. Karol Grela, working on ruthenium-based olefin metathesis catalysts. After getting his M.S. degree in organic synthesis from the same alma mater within Grela's Group in 2021, he moved to France and joined Greg Nocton's Group at Ecole polytechnique (Palaiseau, France), where he carried out research on lanthanide complexes with large aromatic ligands and heterobimetallic species involving lanthanides and transition metals. After a short experience in Nocton's Group, he moved to the industrial sector, holding a position of medical chemist at CelonPharma (Warsaw, Poland), where he is currently conducting research in the development of new innovative pharmaceutical products. |
 Thomas Simler | Thomas Simler graduated from the Ecole Normale Supérieure in Lyon. He received his PhD in 2016 from the University of Strasbourg working on functionalised NHC and pincer complexes with Dr Pierre Braunstein and Dr Andreas A. Danopoulos. He then joined the group of Prof. Peter W. Roesky (KIT Karlsruhe) as an Alexander von Humboldt postdoctoral fellow and the group of Prof. Grégory Nocton at Ecole polytechnique for a second postdoctoral stay. In 2022, he was appointed CNRS researcher at Ecole polytechnique and focuses on the reactivity of low-valent lanthanide complexes for small molecule activation. |
 Grégory Nocton | Grégory Nocton was born in Reims, France in 1983. After a master of science at the Universities of Reims and Grenoble (2006), he obtained his PhD in 2009 in Grenoble with Prof. Marinella Mazzanti, working on the redox reactivity of uranium. He then joined UC Berkeley working with Prof. Richard A. Andersen and was appointed CNRS researcher in 2011 at Ecole polytechnique. He received the bronze medal from the CNRS (2016) and the Junior Prize of the Coordination Chemistry Division (French Chemical Society, 2021). He is Associate Professor at Ecole Polytechnique since 2017 and Vice-President of the chemistry department (2022). |
Introduction
During the winter of 1951, reading the journal Nature surprised many chemists.1 A new type of iron compound had been discovered (Fig. 1), and, with this discovery, a fantastic scientific adventure began. This story would culminate (but not end) with a Nobel Prize in 1973 for organometallic sandwich compounds. As several witnesses remember, the new structure at that time brought many questions related to the effects of π-coordination on the symmetry and physical properties of the complexes.2–4 A new field, similar to coordination chemistry, opened but with carbon-based ligands. The genesis of the metallocene success story lies in the chemical properties of the cyclopentadienyl (Cp) ligand, a small cyclic and mono-anionic ligand featuring 6π-electron Hückel aromaticity. Besides, this ligand framework is easy to modify and relatively robust.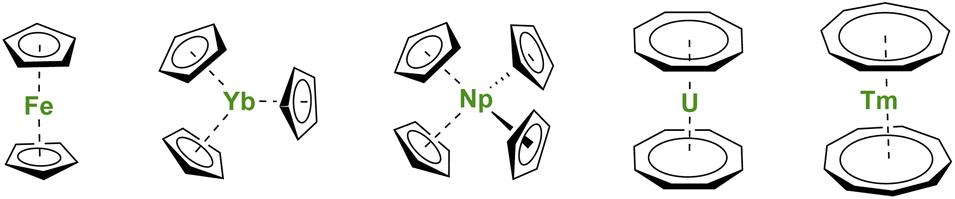 | ||
| Fig. 1 Molecular structures of ferrocene, Yb(Cp)3, Np(Cp)4, uranocene, and thulocene with unsubstituted CnHn aromatic ligands. | ||
Soon after the first report of unusual sandwich structures with the Cp ligand, several research groups became motivated to investigate its coordination behavior towards most metals of the periodic table, including, already at a very early stage, lanthanide ions.5–7 However, the result was not surprising, and several conclusions were drawn: (a) lanthanide ions are mostly trivalent with significantly larger ionic radii than most transition metal ions, and three Cp ligands easily fit around the metal center; (b) the metal–ligand bonding is principally ionic as demonstrated by the easy Cp ligand exchange from Ln(Cp)3 to iron halide to form ferrocene.5 The known divalent lanthanide analogs, Ln(Cp)2,8,9 are forming polymeric assemblies in the solid state to accommodate empty coordination sites with ligand electronic density, but these weak interactions can be broken by solvation.10–12 In all cases, the Cp ligands remain bent due to the bonding and respective sizes of the ligand and metal ion, as well as attractive dispersion or van der Waals interactions.13–15 The lanthanide series was completed with few of the actinides with similar findings,16 although the tetravalent states of Np or U allowed the wrapping of up to four Cp ligands around the metal centers (Fig. 1).17–20 In the original articles by Wilkinson, the magnetic moments of several Ln(Cp)3 complexes were measured at room temperature, and no significant deviations from the expected values were reported, except for the ytterbium complex Yb(Cp)3 (Fig. 1) exhibiting an unexpectedly low room temperature moment.6 This particular feature has been later explained by Denning et al.21 and relates to an unusual intermediate valent electronic structure for these, in appearance only, simple molecules.22
The size mismatch between the small cyclopentadienyl ligand and the large metallic f-elements has been easily corrected by increasing the size of the aromatic ligand used, either by substituting the hydrogens with bulkier groups23,24 or using larger aromatic rings.25,26 Both methods proved very efficient in accessing sandwich compounds of various charges and oxidation states with f-elements.
First, the use of large ligands such as the C8H8 ring, the dianionic cyclooctatetraenyl ligand (Cot), with uranium led to the formation of uranocene, U(Cot)2, a linear sandwich with a +4 metal oxidation state (Fig. 1). In the report from 1968, Streitwieser stated that the ligand size was nicely adapted to the f-orbitals, a situation similar to that found in between iron and Cp ligands.27 Lanthanide complexes supported by Cot ligands have been synthesized, but because of their predominant +3 oxidation state, the complexes are not neutral but anionic.28,29 The only exception to date is cerocene, Ce(Cot)2, the only neutral +4 complex of this family.30,31 The oxidation state in cerocene is also considered as intermediate valent,22,32,33 yet the overall complex remains neutral. The size of the aromatic ligand can be further increased to the C9H9 ring, corresponding to the monoanionic cyclononatetraenyl ligand (Cnt), which can be used with divalent Sm, Eu, Tm (Fig. 1) and Yb to form linear neutral sandwiches of 4f elements. Although the synthesis of the Cnt ligand was already reported in 1963,34,35 the first lanthanidocene Ln(Cnt)2 complexes were published in 2017 and 2018,36,37i.e. 50 years after the first report of uranocene. Note that the flexibility of the Cnt ligand also allows the formation of Ln(Cnt)3 complexes (vide infra).38
The neutral 4f-element sandwich complexes with unsubstituted aromatic ligands often suffer from poor solubility in organic solvents, which hinders both their characterization and the study of their reactivity. As such, derivatives of the versatile Cp and Cot ligands were used and constitute most of the sandwich complexes made in this area.23,25
Once the novelty of those arrangements wore off, the structural properties were primarily designed for the reactivity of the complexes. One key example is the development of the pentamethylcyclopentadienyl ligand (Cp*) and the corresponding SmII and YbII complexes as base-free or solvate adduct versions. Their vibrant chemistry prompted a generation of chemists to navigate through their stoichiometric reductive reactivity and catalytic properties, including in polymerization reactions.
The Ln(Cp*)2 complexes are bent sandwich compounds, which adducts of THF or diethyl ether were reported in the early 1980s (Fig. 2).39,40 The reduction potential of the SmII compound is lower than that of the YbII analog. It allows the reduction of CO,41 CO2,42 and even the non-polar and inert dinitrogen molecule: one N2 molecule can be reduced twice from two Sm(Cp*)2 fragments.43 The ytterbium complex is less reductive, yet it can easily reduce N-heteroaromatic fragments, such as bipyridine derivatives. The (Cp*)2Yb(bipy) complex was made originally in the early 1980s but only published in 2002.44 Indeed, during those years, its electronic structure remained difficult to rationalize until LIII-edge XANES measurements and CASSCF computations pointed towards intermediate valent states.45–48 Other examples of N-heteroaromatic cycles followed,48–52 contributing to straightening the rationalization of the single electron transfer in ytterbium complexes.22
 | ||
| Fig. 2 Molecular structures of Ln(Cp*)2 adducts, (Cp*)2LuMe, and (Cp*)2Yb(bipy). | ||
The reactivity of divalent organolanthanides extends to catalytic reactions, such as in ethylene polymerization,53,54 while the trivalent alkyl complex (Cp*)2LuMe (Fig. 2) reacts with methane,55,56 showing the vast scope of possible reactivity with one ligand set.
Larger substituents on the Cp ligands are also useful to stabilize kinetically “non-classical divalent lanthanides”57 as sandwich complexes of Tm (Fig. 3),58,59 Dy,60 and Nd,61 which still retain high reactivity for potential applications in the activation of small and inert molecules.61,62 Additionally, Lappert showed that using bulky substituents on the Cp rings from the original Ln(Cp)3 complexes allowed more straightforward reduction to form divalent lanthanide ions.63,64 This strategy has been more recently extended to almost all the lanthanide ions, even the most difficult to reduce, using multiple bulky Cp-derived ligands such as η5-C5H4(SiMe3) (Cp′) (Fig. 3), and showing the role of the ligand in the control of the redox properties of the lanthanide ion.65,66 Upon reduction, if the classical divalent lanthanide ions adopt a 4fn+1 configuration, in the other 4f-ions, the extra electron is promoted to the d-shell, opening a vast area of new applications for the redox chemistry of organolanthanides.
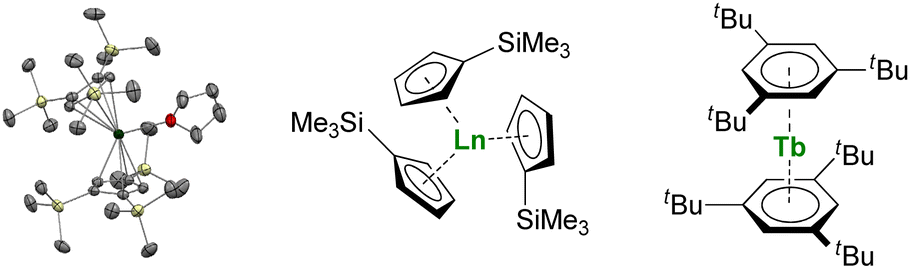 | ||
| Fig. 3 ORTEP plot of Tm(Cp′′′)2(thf) (Tm atom is in green, carbon in grey, silicon in yellow, and hydrogen are removed for clarity) and molecular structures of Ln(Cp′)3 and Tb(Bzttt)2. | ||
When the steric pressure induced by the ligands around the metal center reaches a certain level, the bulky ligands may enable an electron transfer to provide reductive chemistry, described as “sterically induced reduction”.67–69 A possible electronic contribution to these spontaneous reductions was also considered in the report of the Ln(Cnt)3 complexes. The corresponding synthesis was impossible for Sm or Yb but could be performed for the Tm and Y analogs.38 The original structures of tris-Cp arrangements made in 1954 found new horizons in the most recent studies.
Alongside reactivity studies, the fast development of molecules behaving as permanent magnets below a given temperature (Single Molecule Magnets, SMMs) became attractive.70 Rinehart and Long highlighted the relation between the ground mJ state of a given lanthanide metal ion and the coordination surroundings.71 Following this foundational perspective, the structural chemistry of 4f-organometallic sandwiches started to evolve for the formation of linear complexes with small and bulky Cp-based ligands,72–75 or large-size aromatic ligands,76 digging up the classic uranocene-like structures and building up the size of the Cp substituents' bulk from previous examples.58,77,78
Another great field evolution instance is the report of sandwich compounds supported by the neutral six-electron aromatic tris-tert-butylbenzene (Bzttt) ligands. Using the metal vapor technique, Cloke was able to synthesize a few so-called zero valent neutral compounds (Fig. 3).79–82 Yet, the spectroscopic oxidation state and magnetic properties remain to be fully explored (vide infra) and may re-open the case of C6 rings with lanthanides.
The structural chemistry of organolanthanide complexes moved forward and back multiple times following the timely active area of applications, but the molecules resisted time. It is likely that a few abandoned molecules of the past will serve as figureheads for future developments. Synthetic innovation and rigorous methods thus remain the critical contribution. Along this perspective, we will attempt to focus on several possible horizons for organolanthanides.
Reactivity and catalysis
Light-promoted redox reactions
Photoinduced transformations, which encompass photoredox catalysis, are becoming increasingly popular methods to promote chemical transformations by taking advantage of light irradiation to alter the redox properties of compounds. This area of chemistry has long been governed by using photosensitizers based on rare and precious transition metals such as Ru and Ir.83,84 For sustainable chemistry, the development of earth-abundant and cheaper photocatalysts is a significant objective.85 Lanthanide-based photosensitizers can be considered promising candidates based on their unique optical properties and higher abundance in the Earth's crust compared to platinum group metals. It is worth noting that Ce, the most readily available lanthanide, features an abundance similar to that of the 3d metals Cu, Ni, and Zn. In contrast, the rarest lanthanides, Tm and Lu, are still more abundant than Ru and Ir.86Upon light excitation, photosensitizers can behave as either potent reductants or oxidants, activating organic substrates through single electron transfer (SET) events, resulting in the formation of reactive radical intermediates. As a result of their partly filled 4f shell, most trivalent lanthanide ions absorb electromagnetic radiation in the visible region of the spectrum through parity-forbidden electronic transitions within the 4f shell. In addition to f–f transitions, electric-dipole allowed 4fn5d0 → 4fn−15d1 transitions are also accessible but usually occur at much higher energies, typically in the UV region.87 The application of lanthanide complexes in light-promoted transformations, especially reduction reactions, strongly relies on such 4f → 5d transitions. Upon photoirradiation, the 5d excited state features a stronger reducing character, i.e. a more negative redox potential than the ground state, potentially allowing the reduction of challenging substrates typically not reduced under standard conditions without light irradiation.88 Although 4f orbitals are strongly shielded and remain largely unperturbed by the surrounding donor ligands, the 5d orbitals are sensitive to the ligand environment. Thus, the energies of the 4f → 5d transitions can be tuned depending on the nature and geometry of the surrounding ligands.89 In this context, organometallic ligands are exciting candidates as they usually enforce rigid and well-defined geometric coordination environments, potentially allowing tuning of the photophysical properties.
A thermodynamically stable trivalent oxidation state characterizes all lanthanide ions. Only a few can easily shuttle between two oxidation states depending on their position in the lanthanide series and electronic configuration. The LnIV/LnIII couple is easily accessible for Ce, while the divalent oxidation state can be readily obtained in the case of Sm, Eu, and Yb complexes. These specific ions (CeIII, SmII, EuII, and YbII) are excellent candidates for photoinduced reduction reactions and applications in (photo)catalysis through light-promoted amplification of their reducing properties (Scheme 1).
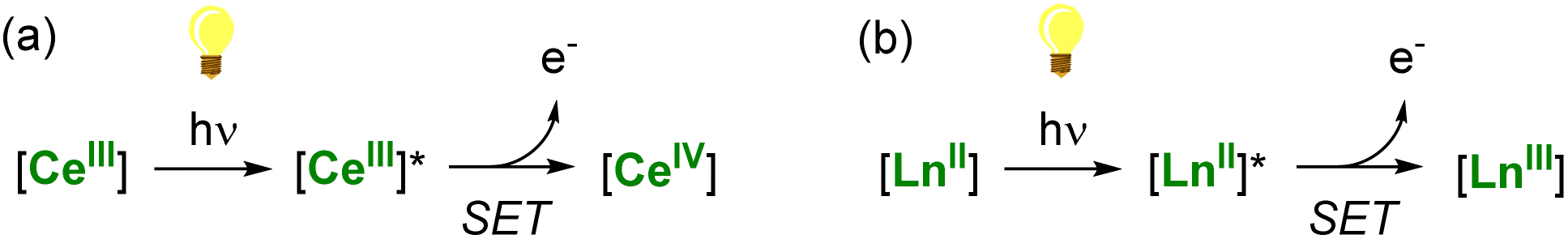 | ||
| Scheme 1 Light-promoted single-electron reduction reactions by trivalent (a) or divalent lanthanides (b). | ||
As a result of its 4f1 electronic configuration, the CeIII ion gives rise to a broad 4f1 → 4f05d1 transition in the near-UV/visible region and at the lowest energy compared to the other trivalent lanthanide ions. The energy of this transition involving 5d orbitals can be tuned by adjustment of the ligand environment.90 After photoexcitation, the redox-active Ce3+ ion acts as a highly reducing metalloradical that may participate in single-electron transfer (SET) reactions. The group of Schelter has primarily investigated the use of CeIII complexes as photosensitizers for light-induced reductive transformations of organic compounds,90 including challenging substrates such as benzyl or aryl chlorides.91–93 The hexachlorocerate(III) anion, [CeIIICl6]3−, was especially found to be a potent photosensitizer with an estimated excited-state reduction potential of −3.45 V vs. Fc+/Fc (ferrocenium/ferrocene couple).92
To design efficient CeIII photosensitizers, the relaxation of the excited state through nonradiative decay processes, such as ligand vibrational modes, should be minimized so that a maximum energy from light can be converted into chemical transformations.94 One strategy is to use rigid ligands and control C–H oscillators from proximity to the metal cation to minimize vibrational relaxation of the excited state. To this extent, developing new organocerium(III) complexes may be interesting. The recent development of the TbIV and PrIV chemistry may also have an increasing interest in light of the CeIV/CeIII photochemistry.95–100 However, organometallic Tb or Pr complexes in the +4 oxidation state have not yet been reported.
As single-electron reductants, divalent lanthanides, especially SmI2,101,102 have been extensively studied for their applications in organic chemistry.103–105 Compared to trivalent Ln ions, divalent lanthanides present 4fn5d0 → 4fn−15d1 transitions at lower energies, typically in the UV to IR region.106 Photoexcitation to promote the 4f → 5d transition has been reported to enhance the rate of several reduction reactions mediated by divalent lanthanide species (Scheme 2).88 For example, the photoexcitation of SmI2 was found to lead to a more potent SET reductant, capable of reducing substrates such as organic chlorides and nitriles typically not affected by SmI2 without light irradiation.107–110 Similarly, although YbI2 is a weaker SET reagent with a redox potential vs. NHE of −1.15 V, photoirradiation in the near UV (300–400 nm) led to a more potent reductant with a reducing power similar to that of SmI2 in THF (−1.55 V).111,112 With a half-filled 4f7 ground-state electronic configuration, EuII ions are remarkably stable towards oxidation and show only a weak reducing character (−0.35 V vs. NHE). Recently, the group of Allen has demonstrated that a EuII complex supported by an azacryptand macrocyclic ligand (see below) could reduce organic chlorides upon photoexcitation in the visible region and exhibited an estimated reduction potential of −2.8 V vs. NHE, much more negative than that of SmI2.113
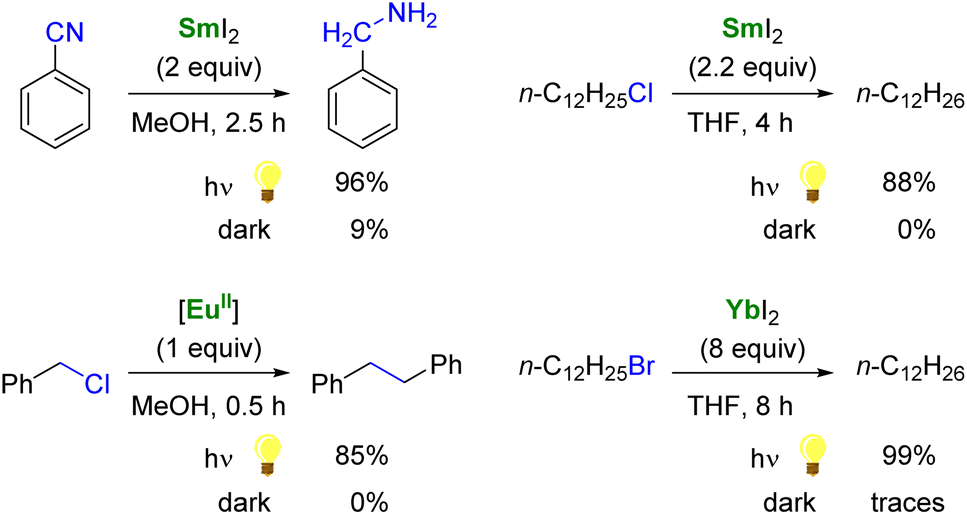 | ||
| Scheme 2 Examples of enhanced reductive reactivity of LnII (Ln = Sm, Eu, Yb) species upon photoirradiation.108,110–113 | ||
As discussed above, the organometallic chemistry of YbII and SmII complexes has been dominated by the Ln(Cp*)2(L)n (L = THF, Et2O; n = 0–2) complexes typically used for SET reactions. Although the group of Watson already reported in 1990 that the rate of C–F activation on fluorinated olefins and aromatics by Ln(Cp*)2(OEt2) (Ln = Yb, Eu) complexes could be enhanced upon visible-light irradiation,114 similar applications involving organolanthanide(II) complexes have remained relatively unexplored. The interesting photophysical properties of EuII complexes115 have led to the recent synthesis and study of divalent organoeuropium complexes with tunable luminescence properties depending on the ligand environment (Fig. 4).36,77,116–122 With the recent introduction of novel organometallic architectures as supporting ligands, such as large ring ligands,25,36,37 further development in this field is likely to be expected. The development of ligand structures inducing long luminescence lifetimes is highly desirable for light-promoted photoredox reactions to maximize the probability of an electron transfer from the organolanthanide complex to the substrate.
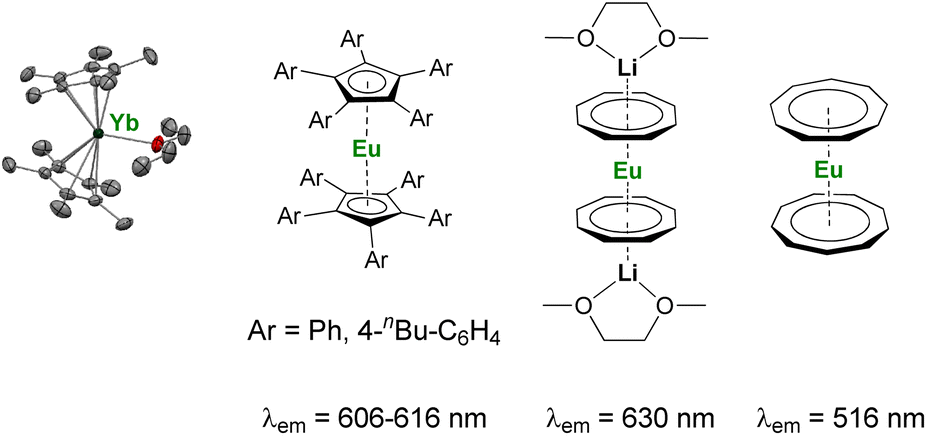 | ||
| Fig. 4 Example of organometallic divalent lanthanide complexes with potential applications in photoinduced reduction reactions.36,40,116,117,119,120 | ||
For application in organic transformations, new organometallic reducing agents based on EuII, SmII, and YbII may offer alternatives to “non-classical divalent lanthanide” species, such as TmI2, DyI2 and NdI2, which feature stronger reducing properties but are more challenging to synthesize and handle.57,123 The primary focus in the organometallic chemistry of reductive divalent lanthanides is prone to go back to the early years (before 2000) when EuII, SmII, and YbII were the major representatives. Detailed studies of the ligand effects on the 4f → 5d transitions may lead to photoredox catalysts with greater tunability and negative electrochemical potentials. The remaining challenge in using divalent lanthanide complexes for photoredox catalysis is the reduction of the oxidized trivalent complexes back to their divalent states. A challenge is also the sensitivity toward air and moisture of organolanthanide complexes.
Application in (photo)electrocatalysis
To use lanthanide complexes as SET reductants in catalytic transformations, different strategies can be used to regenerate the active, reducing species (CeIII, SmII, EuII, or YbII).In the field of divalent lanthanides, only very scarce examples have been reported, mainly focusing on the recycling of SmI2 when used in catalytic amounts.124 Three different strategies have been described to reduce the SmIII species back into SmII. The most common strategy corresponds to a chemical approach involving the addition of a sacrificial reductant. For example, elemental magnesium,125–129 mischmetal (a low-cost alloy of the light lanthanides)130,131 or Zn/Hg amalgam132 have been used for this purpose (Scheme 3). Typically, a silyl-based electrophilic reagent (Me3SiCl, Me3SiOTf) is added to trap the anionic organic product and favor its decoordination from the oxophilic metal center. This strategy involving an external reductant has been successfully applied in the first catalytic visible-light-promoted reductive coupling of benzyl chloride by a EuII complex coordinated by an azacryptand macrocyclic ligand (Scheme 4).113 In this reaction, Zn0 powder is used as a sacrificial reducing agent to reduce the EuIII complex formed after the SET step into its divalent EuII analog.
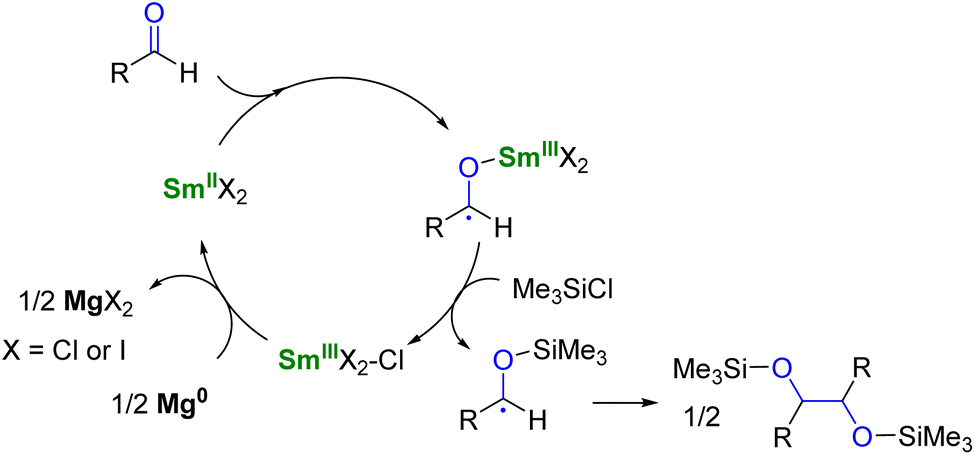 | ||
| Scheme 3 SmI2-catalyzed pinacol coupling reaction in the presence of Mg0 as co-reductant.125 | ||
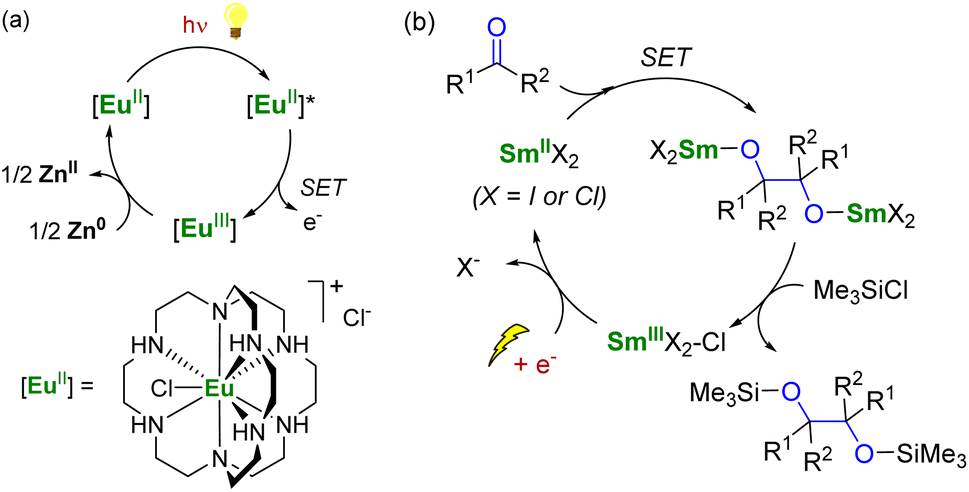 | ||
| Scheme 4 (a) (Photo)catalytic cycle with a EuII complex (a)113 and electrochemical approach for the recycling of SmII species (b).133 | ||
Although the chemical reduction approach is powerful, developing alternative methods that avoid using terminal reductants in stoichiometric quantities would be desirable. In this context, the group of Mellah has investigated electrochemical methods to reduce SmIII species back into SmII at the surface of an electrode (Scheme 4). The corresponding system has been applied in carbon–carbon coupling reactions such as pinacol formation and Barbier-type reactions,133 in the reduction of nitrobenzenes into azobenzenes,134 in the carboxylation of benzyl halides with CO2,135 and reductive alkoxylation of phthalimides into isoindolinone derivatives (using 10–20 mol% of SmII species).136 The best results were obtained using a Sm metal electrode, which, acting as a cathode, is not consumed during electrolysis. Other more conventional cathode materials such as platinum, carbon, nickel, lead, or stainless steel did not lead to the successful regeneration of the SmII active species. The extension of the electrochemical strategy to the recycling of divalent organometallic complexes, such as those depicted in Fig. 4, may be very promising for developing new electrocatalytic transformations with organolanthanide complexes.
Using another strategy, the group of Procter demonstrated that a radical relay approach could allow the use of SmI2 in catalytic amounts for organic transformations.137,138 In this strategy, no external reducing agent is necessary as the SmII species is regenerated through back electron transfer from a negatively charged organic intermediate (Scheme 5). However, this method requires the particular design of phenone substrates compatible with radical relay cyclization cascades and is, therefore, currently limited to very specific molecules. Despite its elegance, this strategy shows a heavy dependence on the nature of the substrate and the relative stability of the ketyl radical intermediates: tuning the ligand may be a key to developing this peculiar chemistry.138
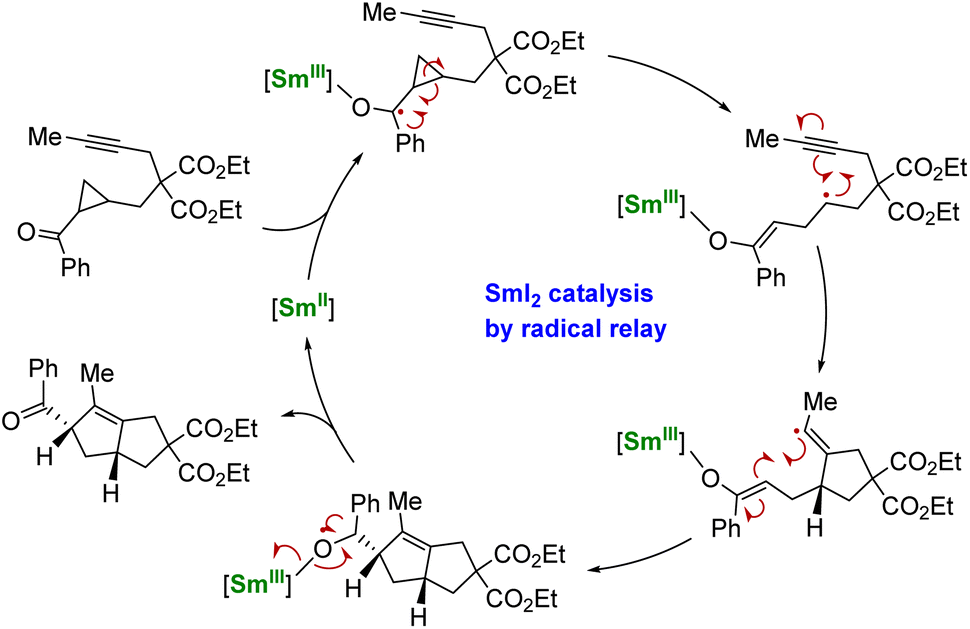 | ||
| Scheme 5 Schematic catalytic cycle of the SmII-induced electron transfer followed by cyclization and back electron transfer.137 | ||
Indeed, similar back electron transfer reactions between a radical anion intermediate and a SmIII metal center have been observed in several instances in organosamarium complexes but highly depend on the reaction conditions and nature of the substrates. For example, the reduction of diphenylacetylene,139 conjugated alkenes,140,141 and polycyclic aromatic hydrocarbons142 by Sm(Cp*)2(thf) was found to be reversible depending on the solvent used. While the different substrates are reduced in hydrocarbon solvents, leading to the formation of dinuclear SmIII complexes, these reactions can be reversed by adding THF, resulting in the regeneration of samarocene and the corresponding free alkyne/alkene. Similar observations involving YbII or SmII organometallic complexes have also been reported using different redox-active ligands.51,143,144
In the case of CeIV species obtained upon SET reductions induced by photoexcited CeIII complexes, the regeneration of the latter can also be achieved by different strategies. External reductants have been used to recycle the photoactive CeIII species, which allowed the use of a catalytic amount of cerium photosensitizer. For example, MN(SiMe3)2 (M = Na, K) has been used as a sacrificial reductant that effectively reduces the CeIV–Cl products to CeIII products through the formation of an aminyl ˙N(SiMe3)2 radical (Scheme 6a).91,94 The presence of an external reductant, such as Ce or Zn metal powder, was sometimes necessary to quench the aminyl radical and prevent the formation of by-products.91 Depending on the nature of the substrate, the organic radical generated after the SET reduction step may also be oxidized by the CeIV complex, which provides a way to regenerate the photoactive CeIII complexes for applications in catalytic transformations (Scheme 6b).94
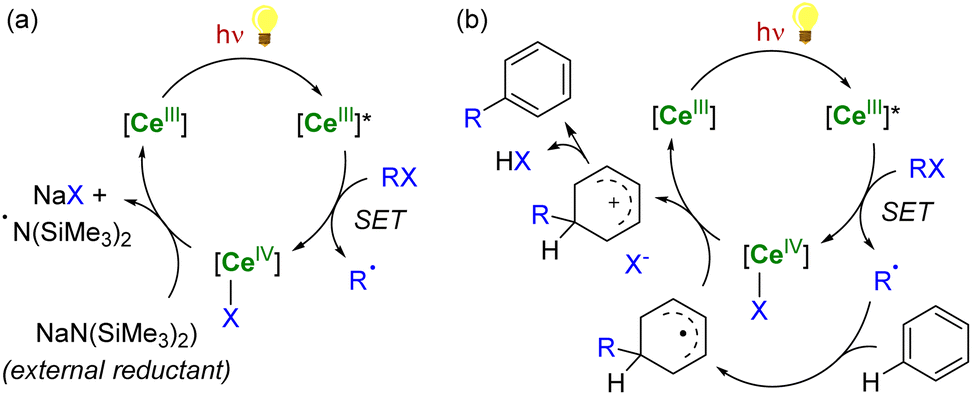 | ||
| Scheme 6 Schematic (photo)catalytic cycle involving the Ce(III/IV) couple with (a) or without (b) the use of an external sacrificial reductant.91,94 | ||
Another strategy for regenerating LnIII photosensitizers, which has been especially developed over the last decade, corresponds to the photoreduction of the corresponding LnIV complexes with the concomitant formation of organic radicals. This strategy may be used for lanthanide compounds featuring a relatively stable +IV oxidation state, such as CeIV and TbIV species, with 4f0 and 4f7 electron configurations, respectively. These oxidizing ions display low-energy metal-to-ligand charge transfer (LMCT) absorptions, typically in the near UV and visible region. The corresponding excited states are usually not emissive but reactive.87 Upon irradiation, CeIV complexes undergo LMCT transitions which promote homolysis of the metal–ligand bond, leading to the photoreduction of the lanthanide center together with the generation of reactive ligand-centered radicals (Scheme 7). It should also be mentioned that this photochemical strategy had already been reported more than 30 years ago for recycling LnII (Ln = Sm, Eu, Yb) species for catalytic applications,145–147 but has only recently seen a renaissance in Ce chemistry.90,148,149

Using this procedure, reactive heteroatom-centered radical species such as carboxyl,150 alkoxy151 and chlorine92,152 radicals can be formed and used to activate other substrates under mild conditions. This strategy has recently been employed by Zuo and Schelter in several photocatalytic transformations induced by soluble cerium complexes and relying on the +IV/+III redox couple of the cerium center.90,148,149 For example, cerium photoredox catalysis has been applied in photocatalytic dehydrogenation of amines, C–C bond cleavage and functionalization of alcohols,153–157 C–H activation of alkanes152,158,159 and functionalization of aryl substrates.93 Although commercially available CeIII salts (chlorides or triflates) are typically used in these photocatalytic reactions, the development of new stable LnIV complexes may be attractive for photocatalytic applications and to control the homolysis of the LnIV–ligand bonds.
In organometallic chemistry, synthesizing organocerium(IV) complexes is challenging, which can be traced back to the strong oxidizing character of the CeIV ion and the reducing properties of carbon-based anionic ligands.160 Therefore, only limited examples of organometallic cerium(IV) complexes have been reported in the literature, and the development of an organometallic scaffold allowing the stabilization of CeIV species is highly desirable. To date, organometallic CeIV complexes have been mostly limited to metallocene structures with examples of CeIV complexes bearing monoanionic cyclopentadienyl (Cp) ligands,161–167 dianionic cyclooctatetraenyl rings,30,31,168,169 and substituted pentalene dianionic ligands170,171 (Fig. 5). Improvement of the kinetic stability of CeIV organometallic complexes, especially by tuning the steric properties of the ligands, may be a direction of future work to go toward catalysis by organocerium complexes.
 | ||
| Fig. 5 Examples of CeIV metallocene complexes. | ||
Single-ion magnets and quantum technologies
This point was demonstrated in 2003 by Ishikawa et al. with a single-metal ion complex of Tb bis-phthalocyanine Tb(Pc)2− (Fig. 6),70,175 which presented a remanent field under zero applied magnetic field. This critical study also underlined that the relaxation paths were multiple and that the surrounding of the central metal ion was crucial for maximizing the anisotropy and preventing the under-barrier relaxation sources from excited mJ states. The single-ion-magnet property was extended to 5f-ions with the neutral diphenyl bis(pyrazolyl borate) complex U(Ph2BPz2)3.176,177 However, a key challenge remained: to maximize the metal–metal magnetic exchange in f-elements combining anisotropy and high ground mJ value. This strategy particularly advanced by using bridging radical ligands, such as N23− (Fig. 6), for which the strong magnetic exchange with f-elements provides strong metal–metal alignment and, inevitably, single-magnet properties.178–180 This powerful strategy is currently still used for the design of f-element single-molecule magnets, particularly with organometallic fragments (vide infra).181
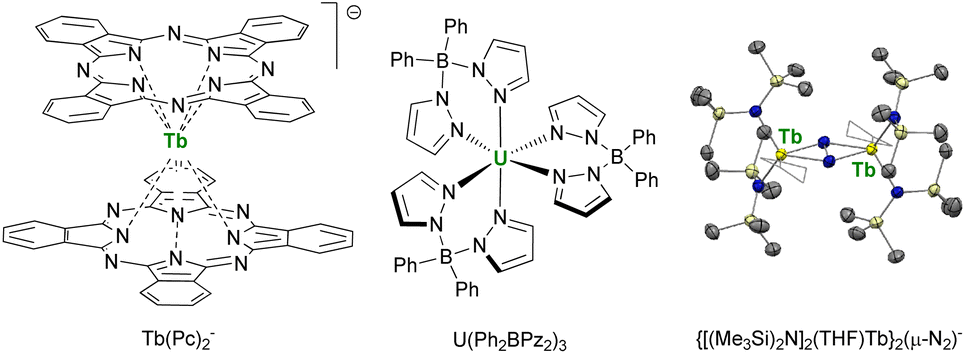 | ||
| Fig. 6 Molecular structures of Tb(Pc)2−,70U(Ph2BPz2)3,176,177 and ORTEP plot of {[(Me3Si)2N]2(THF)Tb}2(μ-N2)−,180 (terbium atoms are in yellow, nitrogen atoms in blue, silicon atoms in pale yellow, carbon atoms in grey, hydrogen atoms are not represented, and coordinated thf molecules are represented in wireframe). | ||
These early studies strongly emphasized two points: the control of the ground mJ value is critical for maximizing the anisotropy. In contrast, the nature and energy of the excited mJ states are critical for controlling the relaxation paths. From these starting points, Rinehart and Long elaborated a comprehensive description of the relationship between the coordination environment of the 4f-elements and the relative energies of the mJ states using a simple electrostatic model.71 This article was followed by a series of similar important notions,182 which moved the field to the rational design of SMMs. From then on, the unique structural arrangements accessible using organometallic complexes started to play a more significant role.
A short review of the compounds known from the organolanthanide chemistry that would have an approaching adapted structure established that the (Cpttt)2Dy(X) (X = Br, I, BH4) complexes made in 2007 by Nief and co-workers (Fig. 7),60 and the ErIII representative of the K[Ln(Cot)2] family, pioneered by Streitwieser,29 would meet most criteria. In the latter, the chelation of the potassium ion led to the isolated Er(Cot)2− anion (Fig. 7) that was reported with record SMMs properties at the time.183 The formation of heteroleptic neutral π-sandwich complexes was also investigated and led to good SMMs properties,184,185 particularly with the large monoanionic Cnt ligand.186,187 The Cot–Er pair is highly relevant as an excellent example of the perfect adequation between the metal and the ligand coordination for maximal anisotropy: this is referred to as ligand–metal pair anisotropy.188,189 The concept has been extended to divalent thulium, an ion with prolate-shaped density. However, the SMM properties of the corresponding complex (Fig. 7) remain very modest due to the decrease of the ground mJ states to ±7/2.76
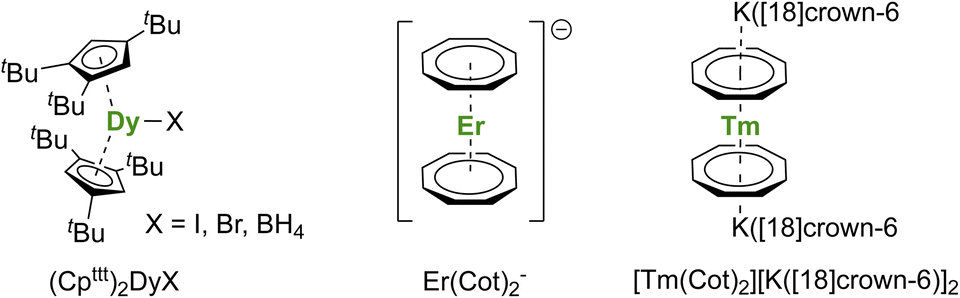 | ||
| Fig. 7 Molecular structures of (Cpttt)2Dy(μ-I)K([18]crown-6),60Er(Cot)2−,183 and [Tm(Cot)2][K([18]crown-6)]2.76 | ||
For the Dy(III) case, building on the previous observations that steric bulk on the Cp substituents could provide linearity to the corresponding sandwich complexes,77,120,190 the Cpttt was found a suitable ligand that enforces axial ligand field. However, a problem remained owing to the presence of the halide counteranion coordinated in the equatorial sphere, even upon reduction.60 The key solution consisted of using [H(SiEt3)2]+[B(C6F5)4]− as a halide abstraction reagent, leading to the separated [Dy(Cpttt)2]+[B(C6F5)4]− ion-pair (Fig. 8).72,73 The SMM properties of this molecule hammered the previous record with a 60 K blocking temperature and a 1541(11) cm−1 energy barrier. A few months later, this landmark was increased to 80 K, i.e. above the liquid nitrogen temperature, by increasing the bulk of the ligand with the use of the CpiPr5 ligand (Fig. 8),74 a ligand previously used for the formation of Ln(CpiPr5)2 (Ln = Sm, Eu, Yb).77,191,192 The modulation of the Cp substituents leads to variations in the complexes' geometry, directly impacting the magnetic properties. Additionally, the strategy was extended to non-classical divalent lanthanide complexes featuring a linear geometry (Fig. 8), which is remarkable, considering the larger size of the LnII ions.193 However, reducing the lanthanide ions does not necessarily fill the f-shell, resulting in complications in the magnetic analysis because of 5d and 6s contributions.
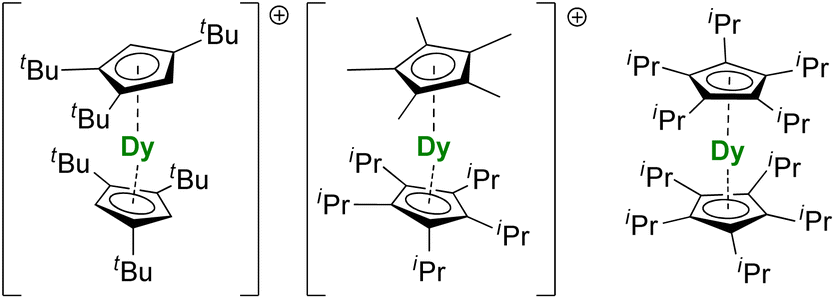
In addition, a few lanthanide complexes bearing aromatic rings with sizes other than C5 or C8 also showed interesting SMM properties.194 One intriguing question remains in the case of the Ln(Bzttt)2 family of complexes (Fig. 3) reported by Cloke and co-workers, since the ligand is intermediate between a large ring and a strongly localized aromatic anion.79–82 In these compounds, the electron count and charge on the ligand will depend on the actual oxidation state of the lanthanide ion.
Radical bridge for strong metal–metal enhancement
After the description of radical-induced metal alignment with reduced dinitrogen radicals, a series of compounds was reported with the objective of increasing the energetic barrier. For such a purpose, the target of choice corresponds to soluble and versatile lanthanide fragments associated with redox-active bridging ligands. For example, the N23− bridging ligand can be easily replaced by another π-accepting ligand such as N-aromatic heterocycles. The known {(Cp*)2Ln}2(μ-bipym) (bipym = bipyrimidine) complex was an obvious choice because of the symmetrical and bridging nature of the ligand,195 which would compare well with the terminal bipyridine ligand.46,47 However, this complex, obtained upon reaction of two equivalents of divalent lanthanide precursor with free bipym, features a doubly reduced dianion bridge. Therefore, no radical is present on the ligand, which renders the corresponding complex of limited interest in terms of magnetic exchange. The more appealing radical bridged [{(Cp*)2Ln}2(μ-bipym)]+ cations (Fig. 9) could be obtained by first coordination of bipym to cationic {(Cp*)2Ln}+ fragments, followed by single-electron reduction. As a result, both lanthanide ions are trivalent, and the bipym is in a radical mono-anionic form.196 These compounds exhibit SMM behavior, with barriers essentially smaller than those in the N23−-radical-bridged analogs,179,180 but can be compared to the values obtained in complexes with larger ligands such as the 2,3,5,6-tetra(2-pyridyl)pyrazine (tppz)197 or in related trimetallic arrangements using the μ3-hexaazatrinaphthylene (HAN)198 ligand (Fig. 9). The great advantage of such systems is that both the organometallic fragment and the bridging ligand can be easily and independently tuned: the influence of the coordination sphere around the lanthanide ion,199 as well as that of the bridging ligand electronic properties, through the use of electron-donating and withdrawing substituents, could be evaluated.200 The two conclusions of this remarkable series of works by Demir and Long are that the best SMMs should possess maximal magnetic exchange while retaining as much as possible an axial field. The ligand's size and electronics (nature of the bonding) are both critical in this matter.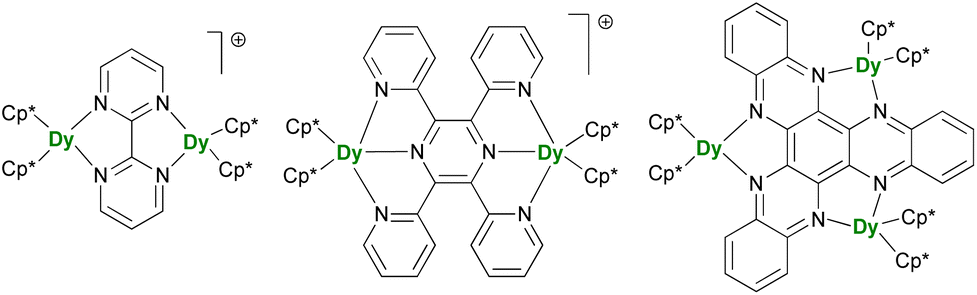 | ||
| Fig. 9 Molecular structures of [{(Cp*)2Dy}2(μ-bipym)]+,196[{(Cp*)2Dy}2(μ-tppz)]+,197 and (Cp*)6Dy3(μ3-HAN).198 | ||
An ideal way to embrace both conditions would be to have one single electron placed between two lanthanide ions, each possessing a strong and highly localized axial field. This strategy was recently addressed by Long and coworkers, who designed the {CpiPr5LnI}2(μ-I)2 dimeric structures in which the CpiPr5 ligands are each capping one lanthanide ion. The rest of the metal coordination sphere was completed by iodide ligands.201 The single electron that would favor the magnetic exchange was placed by simply reducing one metal center to afford a mixed-valent organometallic complex, {CpiPr5Ln}2(μ-I)3, in which both metal ions can magnetically communicate. When the extra electron is placed in the f-shell, the coupling is not strong; however, when it is placed in the d-shell, the coupling is maximum, and the Hund's rules are verified. As a result, all metallic spins align, leading to a giant coercive field and ultrahard magnetism at high temperatures (60 K).201
Beyond these remarkable results, there are a few take-home points: the blocking temperature and effective barrier can reach very high values so that feasible quantum applications may be considered in the future. The ligand design is a crucial parameter to find adapted metal–ligand anisotropy, and it seems that π-coordinating ligands are particularly well-suited for designing high-performance SMMs and providing the correct geometry. These characteristics rely on how the relaxation paths are selected, an essential key for designing future quantum devices.
Relaxation paths analysis
As mentioned above, the thermally activated barrier, corresponding to the Orbach model, is not satisfactory as a sole criterion for describing the SMM properties.202 This is mainly due to possible under-the-barrier magnetic relaxation paths via Raman (spin phonon) and quantum tunneling, which need to be unraveled. It is essential to rationalize how these processes can be avoided using an appropriate molecular design.203Several approaches were proposed, such as using rigid ligands to increase the energy of the phonons and avoid them from meeting with the energies of the mJ excited states.202,204 A second approach, which is appealing, consists in preventing resonance phonon transitions from the magnetic energy window. For this purpose, it is necessary to relate the structural features to the energetic ladder, which has been attempted on several occasions and is an important current objective in the theoretical chemistry community.205 As such, in the dysprosocenium series, the reasons for recording SMM behavior can be understood by two distinct phenomena. First, the near linear arrangement of the ligands allows maximal splitting of the energy states. In their article, Chilton et al. argued that the larger crystal field splitting is the primary driver of the slower relaxation rate for [Dy(Cp*)(CpiPr5)]+ compared to [Dy(Cpttt)2]+.205 Then, the first complex also has the advantage of possessing electronic states that are off-resonance with vibronic states. The investigation of such an interplay between the structural features and the electronic structure is of great importance for bringing the rational design of SMM even further. Combining theory and luminescence spectroscopy could be instrumental in rationalizing a potential correlation.206,207
Additionally, the development of heterocyclic ligands such as phospholyl or new aminoborolide ligands could bring new data to enable broader comprehension of the magnetic relaxation pathways.208–210
Quantum devices
A very promising application of the structure–physical property relationship made over the most recent years lies in the development of quantum devices. Molecular qubits are increasingly cited as potential tools for tailoring quantum algorithms211 since they are highly tunable and would be easily excited by external physical stimuli, such as microwaves or light.212 Organometallic compounds have recently been highlighted as potent molecules to pursue in this direction.213Aromi and Sessoli proposed lanthanide coordination compounds in this direction,214–216 and the report of the electronic relaxation of Lu(Cp′)3− (Fig. 10) further increased the interest.217,218 Indeed, the sizeable hyperfine coupling constant, AISO = 428.5 G, can be associated with a significant s-character to the magnetic orbital carrying the spin. In turn, spin–orbit coupling is minimized and spin–lattice relaxation is reduced in systems with J > 1/2. Maximizing AISO is this key, which can be done by modifying the ligand set around the metal center. With this in mind, high-field EPR measurements were performed on the bulky tris(aryloxide) Lu(OAr*)3− (OAr* = 2,6-Ad2-4tBu-C6H2O, Ad is for adamantyl, Fig. 10), which displayed a massive AISO value of 3467 MHz, similar to that in a BiII radical,219 with giant clock transition.218 The control of the bonding is primordial for manipulating the electronic properties and systematic structure/properties correlations will be necessary for the design of future molecules or reinvestigating ghosts from the past.

Materials, such as Er3+:CaWO4 and Yb3+:YSO (YSO = yttrium orthosilicate, Y2SiO5),220,221 have shown interesting coherence properties, and the molecular extension to these molecules shall bring much information to the field. The use of carbon-based ligands, which possess 99% 12C (I = 0) and only 1% 13C (I = 1/2) and are relatively easy to deuterate at specific positions, should bring valuable information. In contrast, access to multimetallic compounds with close-but-different ligand fields would be appealing. These requirements are reminiscent of the lanthanide multiple-decker chemistry developed with large aromatic ligands such as Cot and its substituted analogs.25,222 Monometallic sandwich compounds have already shown promising results, as in the case of titanium for example, which also demonstrated that the presence of protons could not be as critical as anticipated with such ligands (Fig. 10).213 The use of multiple oxidation states, such as in divalent thulium, a simple f13 (I = 1/2) ion which ground mJ state and hyperfine coupling constants can be easily tuned,223,224 is also a future topic of interest while the population of the 5d-shell (often hybridized with the 6s-orbital) in non-classical divalent lanthanides will provide many future research directions.76,225
Conclusions
What started as a search for peculiar geometry promptly developed into a vast array of original chemistry, from single electron reductants to polymerization catalysts. The interplay between the aromatic ligand and the lanthanide center was better understood through these different applications. This allowed, in turn, a rational tuning of the ligand to harness the intrinsic magnetic and electronic properties of the metallic center, leading to the recent breakthrough in these fields. Given the constant back-and-forth shuttle between old structures and new outlooks, highlighted in this perspective, a modern look at classic lanthanide organometallic compounds from the past will likely lead to a bright future.Author contributions
GN designed the structure of the perspective. All authors contributed to the writing of the article.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding from the European Research Council (ERC) under the European Union's Horizon H2020 research program (grant agreement no. 716314) and from ANR (French National Research Agency) under project number ANR-19-CE07-0019-1. CNRS and Ecole Polytechnique are thanked for funding.References
- T. J. Kealy and P. L. Pauson, Nature, 1951, 168, 1039–1040 CrossRef CAS.
- P. Laszlo and R. Hoffmann, Angew. Chem., Int. Ed., 2000, 39, 123–124 CrossRef CAS.
- H. Werner, Angew. Chem., Int. Ed., 2012, 51, 6052–6058 CrossRef CAS PubMed.
- J. Okuda, Eur. J. Inorg. Chem., 2017, 2017, 217–219 CrossRef CAS.
- G. Wilkinson and J. M. Birmingham, J. Am. Chem. Soc., 1954, 76, 6210 CrossRef CAS.
- J. M. Birmingham and G. Wilkinson, J. Am. Chem. Soc., 1956, 78, 42–44 CrossRef.
- P. Štěpnička, Dalton Trans., 2022, 51, 8085–8102 RSC.
- E. O. Fischer and H. Fischer, Angew. Chem., Int. Ed. Engl., 1964, 3, 132–133 CrossRef.
- E. O. Fischer and H. Fischer, J. Organomet. Chem., 1965, 3, 181–187 CrossRef.
- G. Deacon, A. Koplick and T. Tuong, Aust. J. Chem., 1984, 37, 517–525 CrossRef.
- C. Apostolidis, G. B. Deacon, E. Dornberger, F. T. Edelmann, B. Kanellakopulos, P. MacKinnon and D. Stalke, Chem. Commun., 1997, 1047–1048 RSC.
- M. Schultz, C. J. Burns, D. J. Schwartz and R. A. Andersen, Organometallics, 2000, 19, 781–789 CrossRef.
- R. A. Andersen, J. M. Boncella, C. J. Burns, J. C. Green, D. Hohl and N. Rösch, J. Chem. Soc., Chem. Commun., 1986, 405–407 RSC.
- L. Perrin, L. Maron, O. Eisenstein, D. J. Schwartz, C. J. Burns and R. A. Andersen, Organometallics, 2003, 22, 5447–5453 CrossRef.
- S. Labouille, C. Clavaguéra and F. Nief, Organometallics, 2013, 32, 1265–1271 CrossRef.
- D. Seyferth, Organometallics, 2004, 23, 3562–3583 CrossRef.
- E. O. Fischer and Y. Hristidu, Z. Naturforsch., B, 1962, 17, 275–276 CrossRef.
- F. Baumgärtner, E. O. Fischer, B. Kanellakopulos and P. Laubereau, Angew. Chem., Int. Ed. Engl., 1968, 7, 634 CrossRef.
- J. H. Burns, J. Am. Chem. Soc., 1973, 95, 3815–3817 CrossRef.
- M. S. Dutkiewicz, C. Apostolidis, O. Walter and P. L. Arnold, Chem. Sci., 2017, 8, 2553–2561 RSC.
- R. G. Denning, J. Harmer, J. C. Green and M. Irwin, J. Am. Chem. Soc., 2011, 133, 20644–20660 CrossRef PubMed.
- M. Tricoire, N. Mahieu, T. Simler and G. Nocton, Chem.–Eur. J., 2021, 27, 6860–6879 CrossRef PubMed.
- F. Benner, F. Delano, E. R Pugliese and S. Demir, in Comprehensive Organometallic Chemistry IV, ed. G. Parkin, K. Meyer and D. O'hare, Elsevier, Oxford, 2022, pp. 98–184 Search PubMed.
- A. J. Gremillion and J. R. Walensky, in Comprehensive Organometallic Chemistry IV, ed. G. Parkin, K. Meyer and D. O'hare, Elsevier, Oxford, 2022, pp. 185–247 Search PubMed.
- O. Stetsiuk, V. Cemortan, T. Simler and G. Nocton, in Comprehensive Organometallic Chemistry IV, ed. G. Parkin, K. Meyer and D. O'hare, Elsevier, Oxford, 2022, pp. 550–581 Search PubMed.
- O. Walter, in Comprehensive Organometallic Chemistry IV, ed. G. Parkin, K. Meyer and D. O'hare, Elsevier, Oxford, 2022, pp. 582–606 Search PubMed.
- A. Streitwieser and U. Mueller-Westerhoff, J. Am. Chem. Soc., 1968, 90, 7364 CrossRef.
- F. Mares, K. Hodgson and A. Streitwieser, J. Organomet. Chem., 1970, 24, C68–C70 CrossRef.
- K. O. Hodgson, F. Mares, D. F. Starks and A. Streitwieser, J. Am. Chem. Soc., 1973, 95, 8650–8658 CrossRef.
- A. Greco, S. Cesca and W. Bertolini, J. Organomet. Chem., 1976, 113, 321–330 CrossRef.
- M. D. Walter, C. H. Booth, W. W. Lukens and R. A. Andersen, Organometallics, 2009, 28, 698–707 CrossRef.
- N. M. Edelstein, P. G. Allen, J. J. Bucher, D. K. Shuh, C. D. Sofield, N. Kaltsoyannis, G. H. Maunder, M. R. Russo and A. Sella, J. Am. Chem. Soc., 1996, 118, 13115–13116 CrossRef.
- O. Mooßen and M. Dolg, Chem. Phys. Lett., 2014, 594, 47–50 CrossRef.
- T. J. Katz and P. J. Garratt, J. Am. Chem. Soc., 1963, 85, 2852–2853 CrossRef.
- T. J. Katz and P. J. Garratt, J. Am. Chem. Soc., 1964, 86, 5194–5202 CrossRef.
- K. Kawasaki, R. Sugiyama, T. Tsuji, T. Iwasa, H. Tsunoyama, Y. Mizuhata, N. Tokitoh and A. Nakajima, Chem. Commun., 2017, 53, 6557–6560 RSC.
- M. Xémard, S. Zimmer, M. Cordier, V. Goudy, L. Ricard, C. Clavaguéra and G. Nocton, J. Am. Chem. Soc., 2018, 140, 14433–14439 CrossRef.
- O. Stetsiuk, L. La Droitte, V. Goudy, B. Le Guennic, O. Cador and G. Nocton, Organometallics, 2022, 41, 133–140 CrossRef.
- T. D. Tilley, R. A. Andersen, B. Spencer, H. Ruben, A. Zalkin and D. H. Templeton, Inorg. Chem., 1980, 19, 2999–3003 CrossRef.
- W. J. Evans, I. Bloom, W. E. Hunter and J. L. Atwood, J. Am. Chem. Soc., 1981, 103, 6507–6508 CrossRef.
- W. J. Evans, J. W. Grate, L. A. Hughes, H. Zhang and J. L. Atwood, J. Am. Chem. Soc., 1985, 107, 3728–3730 CrossRef.
- W. J. Evans, C. A. Seibel and J. W. Ziller, Inorg. Chem., 1998, 37, 770–776 CrossRef.
- W. J. Evans, T. A. Ulibarri and J. W. Ziller, J. Am. Chem. Soc., 1988, 110, 6877–6879 CrossRef.
- M. Schultz, J. M. Boncella, D. J. Berg, T. D. Tilley and R. A. Andersen, Organometallics, 2002, 21, 460–472 CrossRef CAS.
- C. H. Booth, M. D. Walter, M. Daniel, W. W. Lukens and R. A. Andersen, Phys. Rev. Lett., 2005, 95, 267202 CrossRef CAS.
- C. H. Booth, M. D. Walter, D. Kazhdan, Y.-J. Hu, W. W. Lukens, E. D. Bauer, L. Maron, O. Eisenstein and R. A. Andersen, J. Am. Chem. Soc., 2009, 131, 6480–6491 CrossRef CAS PubMed.
- C. H. Booth, D. Kazhdan, E. L. Werkema, M. D. Walter, W. W. Lukens, E. D. Bauer, Y.-J. Hu, L. Maron, O. Eisenstein, M. Head-Gordon and R. A. Andersen, J. Am. Chem. Soc., 2010, 132, 17537–17549 CrossRef CAS PubMed.
- G. Nocton, C. H. Booth, L. Maron and R. A. Andersen, Organometallics, 2013, 32, 5305–5312 CrossRef CAS.
- M. D. Walter, M. Schultz and R. A. Andersen, New J. Chem., 2006, 30, 238–246 RSC.
- M. D. Walter, D. J. Berg and R. A. Andersen, Organometallics, 2006, 25, 3228–3237 CrossRef CAS.
- G. Nocton, C. H. Booth, L. Maron and R. A. Andersen, Organometallics, 2013, 32, 1150–1158 CrossRef CAS.
- G. Nocton, W. W. Lukens, C. H. Booth, S. S. Rozenel, S. A. Medling, L. Maron and R. A. Andersen, J. Am. Chem. Soc., 2014, 136, 8626–8641 CrossRef CAS PubMed.
- Z. Hou and Y. Wakatsuki, Coord. Chem. Rev., 2002, 231, 1–22 CrossRef CAS.
- F. Allouche, K. W. Chan, A. Fedorov, R. A. Andersen and C. Copéret, Angew. Chem., Int. Ed., 2018, 57, 3431–3434 CrossRef CAS PubMed.
- P. L. Watson, J. Am. Chem. Soc., 1983, 105, 6491–6493 CrossRef CAS.
- P. L. Watson and G. W. Parshall, Acc. Chem. Res., 1985, 18, 51–56 CrossRef CAS.
- F. Nief, Dalton Trans., 2010, 39, 6589–6598 RSC.
- F. Jaroschik, F. Nief and L. Ricard, Chem. Commun., 2006, 426–428 RSC.
- F. Jaroschik, F. Nief, X.-F. Le Goff and L. Ricard, Organometallics, 2007, 26, 3552–3558 CrossRef CAS.
- F. Jaroschik, F. Nief, X.-F. Le Goff and L. Ricard, Organometallics, 2007, 26, 1123–1125 CrossRef CAS.
- F. Jaroschik, A. Momin, F. Nief, X.-F. Le Goff, G. B. Deacon and P. C. Junk, Angew. Chem., Int. Ed., 2009, 48, 1117–1121 CrossRef CAS PubMed.
- T. Simler, K. N. McCabe, L. Maron and G. Nocton, Chem. Sci., 2022, 13, 7449–7461 RSC.
- M. C. Cassani, D. J. Duncalf and M. F. Lappert, J. Am. Chem. Soc., 1998, 120, 12958–12959 CrossRef.
- P. B. Hitchcock, M. F. Lappert, L. Maron and A. V. Protchenko, Angew. Chem., Int. Ed., 2008, 47, 1488–1491 CrossRef.
- W. J. Evans, Organometallics, 2016, 35, 3088–3100 CrossRef.
- J. C. Wedal and W. J. Evans, J. Am. Chem. Soc., 2021, 143, 18354–18367 CrossRef PubMed.
- W. J. Evans, J. Organomet. Chem., 2002, 647, 2–11 CrossRef.
- W. J. Evans, J. Alloys Compd., 2009, 488, 493–510 CrossRef.
- C. Ruspic, J. R. Moss, M. Schürmann and S. Harder, Angew. Chem., Int. Ed., 2008, 47, 2121–2126 CrossRef CAS PubMed.
- N. Ishikawa, M. Sugita, T. Ishikawa, S.-y. Koshihara and Y. Kaizu, J. Am. Chem. Soc., 2003, 125, 8694–8695 CrossRef CAS PubMed.
- J. D. Rinehart and J. R. Long, Chem. Sci., 2011, 2, 2078–2085 RSC.
- C. A. P. Goodwin, F. Ortu, D. Reta, N. F. Chilton and D. P. Mills, Nature, 2017, 548, 439–442 CrossRef CAS.
- F.-S. Guo, B. M. Day, Y.-C. Chen, M.-L. Tong, A. Mansikkamäki and R. A. Layfield, Angew. Chem., Int. Ed., 2017, 56, 11445–11449 CrossRef CAS PubMed.
- F.-S. Guo, B. M. Day, Y.-C. Chen, M.-L. Tong, A. Mansikkamäki and R. A. Layfield, Science, 2018, 362, 1400–1403 CrossRef CAS.
- B. M. Day, F.-S. Guo and R. A. Layfield, Acc. Chem. Res., 2018, 51, 1880–1889 CrossRef CAS.
- J. Moutet, J. Schleinitz, L. La Droitte, M. Tricoire, F. Pointillart, F. Gendron, T. Simler, C. Clavaguéra, B. Le Guennic, O. Cador and G. Nocton, Angew. Chem., Int. Ed., 2021, 60, 6042–6046 CrossRef CAS.
- H. Sitzmann, T. Dezember, O. Schmitt, F. Weber, G. Wolmershäuser and M. Ruck, Z. Anorg. Allg. Chem., 2000, 626, 2241–2244 CrossRef CAS.
- F. Nief, B. T. de Borms, L. Ricard and D. Carmichael, Eur. J. Inorg. Chem., 2005, 637–643 CrossRef CAS.
- J. G. Brennan, F. G. N. Cloke, A. A. Sameh and A. Zalkin, J. Chem. Soc., Chem. Commun., 1987, 1668–1669 RSC.
- D. M. Anderson, F. G. N. Cloke, P. A. Cox, N. Edelstein, J. C. Green, T. Pang, A. A. Sameh and G. Shalimoff, J. Chem. Soc., Chem. Commun., 1989, 53–55 RSC.
- F. G. N. Cloke, Chem. Soc. Rev., 1993, 22, 17–24 RSC.
- P. L. Arnold, M. A. Petrukhina, V. E. Bochenkov, T. I. Shabatina, V. V. Zagorskii, G. B. Sergeev and F. G. N. Cloke, J. Organomet. Chem., 2003, 688, 49–55 CrossRef CAS.
- J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102–113 RSC.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- C. B. Larsen and O. S. Wenger, Chem.–Eur. J., 2018, 24, 2039–2058 CrossRef.
- S. Cotton, in Lanthanide and Actinide Chemistry, John Wiley & Sons, Ltd., Chichester (U.K.), 2006, pp. 9–22 Search PubMed.
- A. Vogler and H. Kunkely, Inorg. Chim. Acta, 2006, 359, 4130–4138 CrossRef.
- R. Barraza and M. J. Allen, Molecules, 2020, 25, 3892 CrossRef.
- S. Cotton, in Lanthanide and Actinide Chemistry, John Wiley & Sons, Ltd., Chichester (U.K.), 2006, pp. 61–87 Search PubMed.
- Y. Qiao and E. J. Schelter, Acc. Chem. Res., 2018, 51, 2926–2936 CrossRef PubMed.
- H. Yin, P. J. Carroll, J. M. Anna and E. J. Schelter, J. Am. Chem. Soc., 2015, 137, 9234–9237 CrossRef PubMed.
- H. Yin, Y. Jin, J. E. Hertzog, K. C. Mullane, P. J. Carroll, B. C. Manor, J. M. Anna and E. J. Schelter, J. Am. Chem. Soc., 2016, 138, 16266–16273 CrossRef.
- Y. Qiao, Q. Yang and E. J. Schelter, Angew. Chem., Int. Ed., 2018, 57, 10999–11003 CrossRef CAS PubMed.
- H. Yin, P. J. Carroll, B. C. Manor, J. M. Anna and E. J. Schelter, J. Am. Chem. Soc., 2016, 138, 5984–5993 CrossRef CAS PubMed.
- N. T. Rice, I. A. Popov, D. R. Russo, J. Bacsa, E. R. Batista, P. Yang, J. Telser and H. S. La Pierre, J. Am. Chem. Soc., 2019, 141, 13222–13233 CrossRef CAS.
- C. T. Palumbo, I. Zivkovic, R. Scopelliti and M. Mazzanti, J. Am. Chem. Soc., 2019, 141, 9827–9831 CrossRef CAS.
- A. R. Willauer, C. T. Palumbo, R. Scopelliti, I. Zivkovic, I. Douair, L. Maron and M. Mazzanti, Angew. Chem., Int. Ed., 2020, 59, 3549–3553 CrossRef CAS PubMed.
- A. R. Willauer, C. T. Palumbo, F. Fadaei-Tirani, I. Zivkovic, I. Douair, L. Maron and M. Mazzanti, J. Am. Chem. Soc., 2020, 142, 5538–5542 CrossRef CAS.
- A. R. Willauer, I. Douair, A.-S. Chauvin, F. Fadaei-Tirani, J.-C. G. Bünzli, L. Maron and M. Mazzanti, Chem. Sci., 2022, 13, 681–691 RSC.
- T. P. Gompa, A. Ramanathan, N. T. Rice and H. S. La Pierre, Dalton Trans., 2020, 49, 15945–15987 RSC.
- P. Girard, J. L. Namy and H. B. Kagan, J. Am. Chem. Soc., 1980, 102, 2693–2698 CrossRef CAS.
- H. B. Kagan, Inorg. Chim. Acta, 1987, 140, 3–6 CrossRef CAS.
- H. B. Kagan, Tetrahedron, 2003, 59, 10351–10372 CrossRef CAS.
- K. C. Nicolaou, S. P. Ellery and J. S. Chen, Angew. Chem., Int. Ed., 2009, 48, 7140–7165 CrossRef CAS PubMed.
- M. Szostak, N. J. Fazakerley, D. Parmar and D. J. Procter, Chem. Rev., 2014, 114, 5959–6039 CrossRef CAS PubMed.
- M. Suta and C. Wickleder, J. Lumin., 2019, 210, 210–238 CrossRef CAS.
- W. G. Skene, J. C. Scaiano and F. L. Cozens, J. Org. Chem., 1996, 61, 7918–7921 CrossRef CAS.
- A. Ogawa, Y. Sumino, T. Nanke, S. Ohya, N. Sonoda and T. Hirao, J. Am. Chem. Soc., 1997, 119, 2745–2746 CrossRef CAS.
- E. Prasad, B. W. Knettle and R. A. Flowers II, Chem.–Eur. J., 2005, 11, 3105–3112 CrossRef.
- C. N. Rao and S. Hoz, J. Org. Chem., 2012, 77, 4029–4034 CrossRef PubMed.
- A. Ogawa, S. Ohya, Y. Sumino, N. Sonoda and T. Hirao, Tetrahedron Lett., 1997, 38, 9017–9018 CrossRef.
- Y. Sumino, N. Harato, Y. Tomisaka and A. Ogawa, Tetrahedron, 2003, 59, 10499–10508 CrossRef.
- T. C. Jenks, M. D. Bailey, J. L. Hovey, S. Fernando, G. Basnayake, M. E. Cross, W. Li and M. J. Allen, Chem. Sci., 2018, 9, 1273–1278 RSC.
- P. L. Watson, T. H. Tulip and I. Williams, Organometallics, 1990, 9, 1999–2009 CrossRef.
- J. Garcia and M. J. Allen, Eur. J. Inorg. Chem., 2012, 2012, 4550–4563 CrossRef CAS PubMed.
- A. C. Thomas and A. B. Ellis, Organometallics, 1985, 4, 2223–2225 CrossRef CAS.
- S. Harder, D. Naglav, C. Ruspic, C. Wickleder, M. Adlung, W. Hermes, M. Eul, R. Pöttgen, D. B. Rego, F. Poineau, K. R. Czerwinski, R. H. Herber and I. Nowik, Chem.–Eur. J., 2013, 19, 12272–12280 CrossRef PubMed.
- T. Tsuji, N. Hosoya, S. Fukazawa, R. Sugiyama, T. Iwasa, H. Tsunoyama, H. Hamaki, N. Tokitoh and A. Nakajima, J. Phys. Chem. C, 2014, 118, 5896–5907 CrossRef.
- T. Tsuji, S. Fukazawa, R. Sugiyama, K. Kawasaki, T. Iwasa, H. Tsunoyama, N. Tokitoh and A. Nakajima, Chem. Phys. Lett., 2014, 595–596, 144–150 CrossRef.
- R. P. Kelly, T. D. M. Bell, R. P. Cox, D. P. Daniels, G. B. Deacon, F. Jaroschik, P. C. Junk, X. F. Le Goff, G. Lemercier, A. Martinez, J. Wang and D. Werner, Organometallics, 2015, 34, 5624–5636 CrossRef.
- T. Simler, T. J. Feuerstein, R. Yadav, M. T. Gamer and P. W. Roesky, Chem. Commun., 2019, 55, 222–225 RSC.
- H. Ramanantoanina, L. Merzoud, J. T. Muya, H. Chermette and C. Daul, J. Phys. Chem. A, 2020, 124, 152–164 CrossRef PubMed.
- M. Szostak and D. J. Procter, Angew. Chem., Int. Ed., 2012, 51, 9238–9256 CrossRef CAS PubMed.
- S. Maity, Eur. J. Org. Chem., 2021, 2021, 5312–5319 CrossRef CAS.
- R. Nomura, T. Matsuno and T. Endo, J. Am. Chem. Soc., 1996, 118, 11666–11667 CrossRef CAS.
- H. C. Aspinall, N. Greeves and C. Valla, Org. Lett., 2005, 7, 1919–1922 CrossRef CAS PubMed.
- T. Ueda, N. Kanomata and H. Machida, Org. Lett., 2005, 7, 2365–2368 CrossRef CAS PubMed.
- F. Orsini and E. M. Lucci, Tetrahedron Lett., 2005, 46, 1909–1911 CrossRef CAS.
- S. Maity and R. A. Flowers, J. Am. Chem. Soc., 2019, 141, 3207–3216 CrossRef CAS PubMed.
- F. Hélion and J.-L. Namy, J. Org. Chem., 1999, 64, 2944–2946 CrossRef.
- M.-I. Lannou, F. Hélion and J.-L. Namy, Tetrahedron, 2003, 59, 10551–10565 CrossRef CAS.
- E. J. Corey and G. Z. Zheng, Tetrahedron Lett., 1997, 38, 2045–2048 CrossRef CAS.
- L. Sun, K. Sahloul and M. Mellah, ACS Catal., 2013, 3, 2568–2573 CrossRef CAS.
- Y.-F. Zhang and M. Mellah, ACS Catal., 2017, 7, 8480–8486 CrossRef CAS.
- S. Bazzi, E. Schulz and M. Mellah, Org. Lett., 2019, 21, 10033–10037 CrossRef CAS PubMed.
- Y.-F. Zhang and M. Mellah, Org. Chem. Front., 2022, 9, 1308–1314 RSC.
- H.-M. Huang, J. J. W. McDouall and D. J. Procter, Nat. Catal., 2019, 2, 211–218 CrossRef CAS.
- S. Agasti, N. A. Beattie, J. J. W. McDouall and D. J. Procter, J. Am. Chem. Soc., 2021, 143, 3655–3661 CrossRef CAS.
- W. J. Evans, I. Bloom, W. E. Hunter and J. L. Atwood, J. Am. Chem. Soc., 1983, 105, 1401–1403 CrossRef CAS.
- W. J. Evans, T. A. Ulibarri and J. W. Ziller, J. Am. Chem. Soc., 1990, 112, 219–223 CrossRef CAS.
- W. J. Evans, D. G. Giarikos, C. B. Robledo, V. S. Leong and J. W. Ziller, Organometallics, 2001, 20, 5648–5652 CrossRef.
- W. J. Evans, S. L. Gonzales and J. W. Ziller, J. Am. Chem. Soc., 1994, 116, 2600–2608 CrossRef.
- A. A. Trifonov, Y. A. Kurskii, M. N. Bochkarev, S. Muehle, S. Dechert and H. Schumann, Russ. Chem. Bull., 2003, 52, 601–606 CrossRef.
- J. Wang, R. I. J. Amos, A. S. P. Frey, M. G. Gardiner, M. L. Cole and P. C. Junk, Organometallics, 2005, 24, 2259–2261 CrossRef.
- A. Ishida, S. Toki and S. Takamuku, Chem. Lett., 1985, 14, 893–896 CrossRef.
- A. Ishida, S. Toki and S. Takamuku, J. Chem. Soc., Chem. Commun., 1985, 1481–1483 RSC.
- T. Kondo, M. Akazome and Y. Watanabe, J. Chem. Soc., Chem. Commun., 1991, 757–758 RSC.
- H. Tsurugi and K. Mashima, J. Am. Chem. Soc., 2021, 143, 7879–7890 CrossRef.
- A. Prieto and F. Jaroschik, Curr. Org. Chem., 2022, 26, 6–41 CrossRef.
- R. A. Sheldon and J. K. Kochi, J. Am. Chem. Soc., 1968, 90, 6688–6698 CrossRef.
- L. Chang, Q. An, L. Duan, K. Feng and Z. Zuo, Chem. Rev., 2022, 122, 2429–2486 CrossRef PubMed.
- Q. Yang, Y.-H. Wang, Y. Qiao, M. Gau, P. J. Carroll, P. J. Walsh and E. J. Schelter, Science, 2021, 372, 847–852 CrossRef.
- J.-J. Guo, A. Hu, Y. Chen, J. Sun, H. Tang and Z. Zuo, Angew. Chem., Int. Ed., 2016, 55, 15319–15322 CrossRef.
- A. Hu, Y. Chen, J.-J. Guo, N. Yu, Q. An and Z. Zuo, J. Am. Chem. Soc., 2018, 140, 13580–13585 CrossRef.
- A. Hu, J.-J. Guo, H. Pan, H. Tang, Z. Gao and Z. Zuo, J. Am. Chem. Soc., 2018, 140, 1612–1616 CrossRef.
- J. Schwarz and B. König, Chem. Commun., 2019, 55, 486–488 RSC.
- K. Zhang, L. Chang, Q. An, X. Wang and Z. Zuo, J. Am. Chem. Soc., 2019, 141, 10556–10564 CrossRef PubMed.
- A. Hu, J.-J. Guo, H. Pan and Z. Zuo, Science, 2018, 361, 668–672 CrossRef PubMed.
- Q. An, Z. Wang, Y. Chen, X. Wang, K. Zhang, H. Pan, W. Liu and Z. Zuo, J. Am. Chem. Soc., 2020, 142, 6216–6226 CrossRef.
- R. Anwander, M. Dolg and F. T. Edelmann, Chem. Soc. Rev., 2017, 46, 6697–6709 RSC.
- A. Gulino, M. Casarin, V. P. Conticello, J. G. Gaudiello, H. Mauermann, I. Fragala and T. J. Marks, Organometallics, 1988, 7, 2360–2364 CrossRef.
- W. J. Evans, T. J. Deming and J. W. Ziller, Organometallics, 1989, 8, 1581–1583 CrossRef.
- P. Dröse, A. R. Crozier, S. Lashkari, J. Gottfriedsen, S. Blaurock, C. G. Hrib, C. Maichle-Mössmer, C. Schädle, R. Anwander and F. T. Edelmann, J. Am. Chem. Soc., 2010, 132, 14046–14047 CrossRef PubMed.
- A. D. Sutton, D. L. Clark, B. L. Scott and J. C. Gordon, Inorganics, 2015, 3, 589–596 CrossRef.
- D. Schneider, N. Harmgarth, F. T. Edelmann and R. Anwander, Chem.–Eur. J., 2017, 23, 12243–12252 CrossRef.
- L. Hirneise, C. Maichle-Mössmer and R. Anwander, Inorg. Chem., 2021, 60, 18211–18224 CrossRef PubMed.
- L. Hirneise, J. Langmann, G. Zitzer, L. Ude, C. Maichle-Mössmer, W. Scherer, B. Speiser and R. Anwander, Organometallics, 2021, 40, 1786–1800 CrossRef.
- T. R. Boussie, D. C. Eisenberg, J. Rigsbee, A. Streitwieser and A. Zalkin, Organometallics, 1991, 10, 1922–1928 CrossRef.
- U. Kilimann, R. Herbst-Irmer, D. Stalke and F. T. Edelmann, Angew. Chem., Int. Ed. Engl., 1994, 33, 1618–1621 CrossRef.
- A. Ashley, G. Balazs, A. Cowley, J. Green, C. H. Booth and D. O'Hare, Chem. Commun., 2007, 1515–1517 RSC.
- G. Balazs, F. G. N. Cloke, J. C. Green, R. M. Harker, A. Harrison, P. B. Hitchcock, C. N. Jardine and R. Walton, Organometallics, 2007, 26, 3111–3119 CrossRef CAS.
- R. Sessoli, D. Gatteschi, A. Caneschi and M. A. Novak, Nature, 1993, 365, 141–143 CrossRef CAS.
- A. M. Ako, I. J. Hewitt, V. Mereacre, R. Clérac, W. Wernsdorfer, C. E. Anson and A. K. Powell, Angew. Chem., Int. Ed., 2006, 45, 4926–4929 CrossRef CAS.
- O. Kahn, Molecular Magnetism, VCH, New York, N.Y., 1993 Search PubMed.
- N. Ishikawa, M. Sugita, T. Okubo, N. Tanaka, T. Iino and Y. Kaizu, Inorg. Chem., 2003, 42, 2440–2446 CrossRef CAS.
- L. Maria, M. Paula Campello, Â. Domingos, I. Santos and R. Andersen, J. Chem. Soc., Dalton Trans., 1999, 2015–2020 RSC.
- J. D. Rinehart and J. R. Long, J. Am. Chem. Soc., 2009, 131, 12558–12559 CrossRef CAS.
- W. W. Lukens, N. Magnani and C. H. Booth, Inorg. Chem., 2012, 51, 10105–10110 CrossRef CAS.
- J. D. Rinehart, M. Fang, W. J. Evans and J. R. Long, Nat. Chem., 2011, 3, 538–542 CrossRef CAS PubMed.
- J. D. Rinehart, M. Fang, W. J. Evans and J. R. Long, J. Am. Chem. Soc., 2011, 133, 14236–14239 CrossRef CAS.
- D. N. Woodruff, R. E. P. Winpenny and R. A. Layfield, Chem. Rev., 2013, 113, 5110–5148 CrossRef CAS.
- J.-L. Liu, Y.-C. Chen and M.-L. Tong, Chem. Soc. Rev., 2018, 47, 2431–2453 RSC.
- K. R. Meihaus and J. R. Long, J. Am. Chem. Soc., 2013, 135, 17952–17957 CrossRef CAS.
- S.-D. Jiang, B.-W. Wang, H.-L. Sun, Z.-M. Wang and S. Gao, J. Am. Chem. Soc., 2011, 133, 4730–4733 CrossRef CAS PubMed.
- M. D. Korzyński, M. Bernhardt, V. Romankov, J. Dreiser, G. Matmon, F. Pointillart, B. Le Guennic, O. Cador and C. Copéret, Chem. Sci., 2022, 13, 10574–10580 RSC.
- L. Münzfeld, C. Schoo, S. Bestgen, E. Moreno-Pineda, R. Köppe, M. Ruben and P. W. Roesky, Nat. Commun., 2019, 10, 3135 CrossRef.
- M. Tricoire, L. Münzfeld, J. Moutet, N. Mahieu, L. La Droitte, E. Moreno-Pineda, F. Gendron, J. D. Hilgar, J. D. Rinehart, M. Ruben, B. Le Guennic, O. Cador, P. W. Roesky and G. Nocton, Chem.–Eur. J., 2021, 27, 13558–13567 CrossRef CAS.
- J. D. Hilgar, M. G. Bernbeck, B. S. Flores and J. D. Rinehart, Chem. Sci., 2018, 9, 7204–7209 RSC.
- J. D. Hilgar, M. G. Bernbeck and J. D. Rinehart, J. Am. Chem. Soc., 2019, 141, 1913–1917 CrossRef CAS PubMed.
- G. Nocton and L. Ricard, Dalton Trans., 2014, 43, 4380–4387 RSC.
- M. D. Walter, G. Wolmershäuser and H. Sitzmann, J. Am. Chem. Soc., 2005, 127, 17494–17503 CrossRef CAS.
- S. Lauk and A. Schäfer, Eur. J. Inorg. Chem., 2021, 2021, 5026–5036 CrossRef CAS.
- C. A. Gould, K. R. McClain, J. M. Yu, T. J. Groshens, F. Furche, B. G. Harvey and J. R. Long, J. Am. Chem. Soc., 2019, 141, 12967–12973 CrossRef CAS PubMed.
- K. L. M. Harriman, J. J. Le Roy, L. Ungur, R. J. Holmberg, I. Korobkov and M. Murugesu, Chem. Sci., 2017, 8, 231–240 RSC.
- D. J. Berg, J. M. Boncella and R. A. Andersen, Organometallics, 2002, 21, 4622–4631 CrossRef CAS.
- S. Demir, J. M. Zadrozny, M. Nippe and J. R. Long, J. Am. Chem. Soc., 2012, 134, 18546–18549 CrossRef CAS PubMed.
- S. Demir, M. Nippe, M. I. Gonzalez and J. R. Long, Chem. Sci., 2014, 5, 4701–4711 RSC.
- C. A. Gould, L. E. Darago, M. I. Gonzalez, S. Demir and J. R. Long, Angew. Chem., Int. Ed., 2017, 56, 10103–10107 CrossRef CAS.
- S. Demir, M. I. Gonzalez, L. E. Darago, W. J. Evans and J. R. Long, Nat. Commun., 2017, 8, 2144 CrossRef PubMed.
- C. A. Gould, E. Mu, V. Vieru, L. E. Darago, K. Chakarawet, M. I. Gonzalez, S. Demir and J. R. Long, J. Am. Chem. Soc., 2020, 142, 21197–21209 CrossRef CAS.
- C. A. Gould, K. R. McClain, D. Reta, J. G. C. Kragskow, D. A. Marchiori, E. Lachman, E.-S. Choi, J. G. Analytis, R. D. Britt, N. F. Chilton, B. G. Harvey and J. R. Long, Science, 2022, 375, 198–202 CrossRef CAS.
- K. S. Pedersen, J. Dreiser, H. Weihe, R. Sibille, H. V. Johannesen, M. A. Sørensen, B. E. Nielsen, M. Sigrist, H. Mutka, S. Rols, J. Bendix and S. Piligkos, Inorg. Chem., 2015, 54, 7600–7606 CrossRef CAS.
- L. Escalera-Moreno, J. J. Baldoví, A. Gaita-Ariño and E. Coronado, Chem. Sci., 2018, 9, 3265–3275 RSC.
- A. Lunghi, F. Totti, R. Sessoli and S. Sanvito, Nat. Commun., 2017, 8, 14620 CrossRef.
- D. Reta, J. G. C. Kragskow and N. F. Chilton, J. Am. Chem. Soc., 2021, 143, 5943–5950 CrossRef CAS.
- H.-D. Amberger, H. Reddmann and F. T. Edelmann, J. Organomet. Chem., 2005, 690, 2238–2242 CrossRef CAS.
- F. Guégan, J. Jung, B. Le Guennic, F. Riobé, O. Maury, B. Gillon, J.-F. Jacquot, Y. Guyot, C. Morell and D. Luneau, Inorg. Chem. Front., 2019, 6, 3152–3157 RSC.
- P. Evans, D. Reta, G. F. S. Whitehead, N. F. Chilton and D. P. Mills, J. Am. Chem. Soc., 2019, 141, 19935–19940 CrossRef CAS PubMed.
- D. P. Mills and P. Evans, Chem.–Eur. J., 2021, 27, 6645–6665 CrossRef CAS PubMed.
- J. C. Vanjak, B. O. Wilkins, V. Vieru, N. S. Bhuvanesh, J. H. Reibenspies, C. D. Martin, L. F. Chibotaru and M. Nippe, J. Am. Chem. Soc., 2022, 144, 17743–17747 CrossRef CAS.
- A. Gaita-Ariño, F. Luis, S. Hill and E. Coronado, Nat. Chem., 2019, 11, 301–309 CrossRef PubMed.
- S. L. Bayliss, D. W. Laorenza, P. J. Mintun, B. D. Kovos, D. E. Freedman and D. D. Awschalom, Science, 2020, 370, 1309–1312 CrossRef CAS.
- L. C. de Camargo, M. Briganti, F. S. Santana, D. Stinghen, R. R. Ribeiro, G. G. Nunes, J. F. Soares, E. Salvadori, M. Chiesa, S. Benci, R. Torre, L. Sorace, F. Totti and R. Sessoli, Angew. Chem., Int. Ed., 2021, 60, 2588–2593 CrossRef CAS PubMed.
- D. Aguilà, L. A. Barrios, V. Velasco, O. Roubeau, A. Repollés, P. J. Alonso, J. Sesé, S. J. Teat, F. Luis and G. Aromí, J. Am. Chem. Soc., 2014, 136, 14215–14222 CrossRef.
- G. Aromí, F. Luis and O. Roubeau, in Lanthanides and Actinides in Molecular Magnetism, 2015, pp. 185–222 Search PubMed.
- M. Atzori and R. Sessoli, J. Am. Chem. Soc., 2019, 141, 11339–11352 CrossRef CAS PubMed.
- M. R. MacDonald, J. E. Bates, J. W. Ziller, F. Furche and W. J. Evans, J. Am. Chem. Soc., 2013, 135, 9857–9868 CrossRef CAS PubMed.
- K. Kundu, J. R. K. White, S. A. Moehring, J. M. Yu, J. W. Ziller, F. Furche, W. J. Evans and S. Hill, Nat. Chem., 2022, 14, 392–397 CrossRef CAS PubMed.
- R. J. Schwamm, J. R. Harmer, M. Lein, C. M. Fitchett, S. Granville and M. P. Coles, Angew. Chem., Int. Ed., 2015, 54, 10630–10633 CrossRef CAS PubMed.
- H.-J. Lim, S. Welinski, A. Ferrier, P. Goldner and J. J. L. Morton, Phys. Rev. B, 2018, 97, 064409 CrossRef CAS.
- M. Le Dantec, M. Rančić, S. Lin, E. Billaud, V. Ranjan, D. Flanigan, S. Bertaina, T. Chanelière, P. Goldner, A. Erb, R. B. Liu, D. Estève, D. Vion, E. Flurin and P. Bertet, Sci. Adv., 2021, 7, eabj9786 CrossRef CAS PubMed.
- L. Münzfeld, A. Hauser, P. Hädinger, F. Weigend and P. W. Roesky, Angew. Chem., Int. Ed., 2021, 60, 24493–24499 CrossRef PubMed.
- C. A. P. Goodwin, N. F. Chilton, G. F. Vettese, E. Moreno Pineda, I. F. Crowe, J. W. Ziller, R. E. P. Winpenny, W. J. Evans and D. P. Mills, Inorg. Chem., 2016, 55, 10057–10067 CrossRef CAS PubMed.
- M. Xémard, M. Cordier, F. Molton, C. Duboc, B. Le Guennic, O. Maury, O. Cador and G. Nocton, Inorg. Chem., 2019, 58, 2872–2880 CrossRef PubMed.
- A.-M. Ariciu, D. H. Woen, D. N. Huh, L. E. Nodaraki, A. K. Kostopoulos, C. A. P. Goodwin, N. F. Chilton, E. J. L. McInnes, R. E. P. Winpenny, W. J. Evans and F. Tuna, Nat. Commun., 2019, 10, 3330 CrossRef PubMed.
Footnote |
| † This perspective is dedicated to Prof. Peter Junk on the occasion of his 60th birthday. |
| This journal is © The Royal Society of Chemistry 2023 |