
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEstablishing carbon–carbon double bond position and configuration in unsaturated fatty acids by gas-phase infrared spectroscopy†
Carla
Kirschbaum
 ab,
Reuben S. E.
Young‡
cd,
Kim
Greis
ab,
Jan Philipp
Menzel
ce,
Sandy
Gewinner
b,
Wieland
Schöllkopf
b,
Gerard
Meijer
b,
Gert
von Helden
b,
Tim
Causon
f,
Venkateswara R.
Narreddula§
ce,
Berwyck L. J.
Poad
cde,
Stephen J.
Blanksby
*cde and
Kevin
Pagel
*ab
ab,
Reuben S. E.
Young‡
cd,
Kim
Greis
ab,
Jan Philipp
Menzel
ce,
Sandy
Gewinner
b,
Wieland
Schöllkopf
b,
Gerard
Meijer
b,
Gert
von Helden
b,
Tim
Causon
f,
Venkateswara R.
Narreddula§
ce,
Berwyck L. J.
Poad
cde,
Stephen J.
Blanksby
*cde and
Kevin
Pagel
*ab
aInstitut für Chemie und Biochemie, Freie Universität Berlin, Altensteinstraße 23a, 14195 Berlin, Germany. E-mail: kevin.pagel@fu-berlin.de
bFritz-Haber-Institut der Max-Planck-Gesellschaft, Faradayweg 4-6, 14195 Berlin, Germany
cSchool of Chemistry and Physics, Queensland University of Technology, Brisbane, QLD 4000, Australia. E-mail: stephen.blanksby@qut.edu.au
dCentral Analytical Research Facility, Queensland University of Technology, Brisbane, QLD 4000, Australia
eCentre for Materials Science, Queensland University of Technology, Brisbane, QLD 4000, Australia
fInstitute of Analytical Chemistry, University of Natural Resources and Life Sciences Vienna, 1190 Vienna, Austria
First published on 25th January 2023
Abstract
Fatty acids are an abundant class of lipids that are characterised by wide structural variation including isomeric diversity arising from the position and configuration of functional groups. Traditional approaches to fatty acid characterisation have combined chromatography and mass spectrometry for a description of the composition of individual fatty acids while infrared (IR) spectroscopy has provided insights into the functional groups and bond configurations at the bulk level. Here we exploit universal 3-pyridylcarbinol ester derivatization of fatty acids to acquire IR spectra of individual lipids as mass-selected gas-phase ions. Intramolecular interactions between the protonated pyridine moiety and carbon–carbon double bonds present highly sensitive probes for regiochemistry and configuration through promotion of strong and predictable shifts in IR resonances. Gas-phase IR spectra obtained from unsaturated fatty acids are shown to discriminate between isomers and enable the first unambiguous structural assignment of 6Z-octadecenoic acid in human-derived cell lines. Compatibility of 3-pyridylcarbinol ester derivatization with conventional chromatography-mass spectrometry and now gas-phase IR spectroscopy paves the way for comprehensive structure elucidation of fatty acids that is sensitive to regio- and stereochemical variations and with the potential to uncover new pathways in lipid metabolism.
Introduction
The full extent of molecular diversity within the lipidome remains an open question that presents significant challenges to contemporary methods for lipid structure determination.1 Much of the potential for structural diversity in lipid biology arises from wide variation in the molecular structures of fatty acids (FA) that represent major building blocks of more complex lipids and are subject to substantial modification through changes in supply and metabolism.2 Currently the LIPID MAPS database lists over 8600 distinct lipids in the fatty acyl category that includes over 3800 lipids classified as unsaturated fatty acids.3 Much of this structural variation arises from differences in the degree of unsaturation and the position and configuration of the carbon–carbon bonds within the fatty acids resulting, in many instances, in isomeric structures. Recent investigations have revealed that changes in the position or configuration of carbon–carbon double bonds in fatty acids can be key markers for changes in diet or metabolism but are difficult to visualize against a complex background of hundreds (or even thousands) of other lipids that include double bond or stereoisomers of the marker compound.4–7 The unambiguous assignment of the molecular structure of individual fatty acids with explicit assignment of double bond (DB) position(s) and configuration (i.e., cis/trans or E/Z) is thus critical to a complete understanding of the lipidome and for mapping the full potential of lipid metabolism.Identifying individual FAs within a complex mixture of isomers is a significant challenge to even the most advanced analytical methods. Gas chromatography-mass spectrometry (GC-MS) of fatty acid methyl esters (FAMEs) has been the most widely used method for FA analysis.8 For monounsaturated FAs, shifts in retention time can be indicative of DB position and stereochemistry.9 Recent GC-MS investigations of FAs in cancer have, however, identified poor resolution or even co-elution of some isomers.4,10 The challenge of FA identification is further confounded by the identical electron ionization mass spectra of isomeric FAMEs.11,12 Utilization of 3-pyridylcarbinol esters11,13 and other derivatization14–21 or ion activation22,23 strategies can instead yield diagnostic mass spectral fragments for DB position assignment in FAs. However, apart from rare exceptions,24–28 these tandem MS approaches are agnostic to DB geometry. Conversely, techniques such as nuclear magnetic resonance (NMR)29 or vibrational spectroscopy30,31 are capable of quantifying the relative amount of cis versus trans unsaturation in a bulk lipid extract. These stereochemical assignments, however, cannot typically be ascribed to DB position(s) or indeed to individual fatty acids except where sufficient quantities of individual lipids can be purified from the bulk.29–31
Rapid advances in the spectroscopy of gaseous ions present an exciting possibility to establish a universal approach to fatty acid structure elucidation that combines mass spectrometric and spectroscopic insights on mass-selected populations of ionized lipids.
Recently, we demonstrated that both the position and geometry of DBs in 1-deoxysphingosine can be characterized simultaneously using cryogenic gas-phase infrared (IR) spectroscopy.32 Here the shifts in the N–H vibrational frequencies were found to be sensitive to the position and stereochemistry of carbon–carbon double bonds within the lipid due to a through-space charge–olefin interaction in the gas-phase [M + H]+ cations. While the power of combining mass spectrometry and gas-phase IR spectroscopy was clearly demonstrated for this system, the reliance on the N–H resonance meant that direct spectroscopic analysis of other lipid classes would require modification to establish a more universal experimental approach. A promising example of such modifications are 3-pyridylcarbinol ester derivatives of fatty acids. These esters have been widely deployed for GC-MS analysis of isomers where retention of charge at the pyridine-nitrogen facilitates radical-driven dissociation or carbon–carbon bonds and promotes isomer-selective mass spectra.8,9,11–13
Herein we demonstrate the potential of 3-pyridylcarbinol esters as universal derivatives to enable gas-phase IR spectroscopy of ionized fatty acids. This approach exploits the protonation of the pyridine-nitrogen during electrospray ionization to promote the favoured intramolecular charge–olefin interaction within the gaseous lipid ions. Subjecting these mass-selected ions populations to cryogenic gas-phase IR spectroscopy provides a rich fingerprint infrared spectrum that can be deployed to assign DB-position and -stereochemistry within FAs even in mixtures without the need for prior chromatographic separation. Application of the developed workflow to two human prostate cancer cell lines (PC-3 and LNCaP) identifies the presence of four isomers of FA 18:1, including the unusual FA 18:1 (Δ6) isomer where the DB is in position-6 with respect to the carboxylate moiety. While this isomer has been identified in prostate cell lines previously,5 IR spectroscopy explicitly assigns the cis-configuration to this unsaturated FA despite it representing <10 mol% of the isomer population.
Results and discussion
For this study, a set of FA 18:1 double bond regio and geometric isomers (6Z, 6E, 8Z, 9Z, 9E, 11Z, 11E) along with a FA 18:0 reference sample were derivatized with 3-pyridylcarbinol. 3-Pyridylcarbinol esters of FAs enhance the ionization in positive ion mode via protonation of the pyridyl-nitrogen. In addition, the protonated label serves as a sensitive double bond reporter in gas-phase IR spectroscopy, similar to protonated amines reported before.32 Herein, the protonated 3-pyridylcarbinol esters of FA 18:1 isomers were investigated by cryogenic gas-phase IR spectroscopy in superfluid helium droplets.33 Briefly, derivatized FAs are ionized, mass-selected, and then captured in helium droplets. The absorption of IR radiation is indirectly measured by mass detection of lipid ions released from the droplets. Previously, the technique was used to study ionic complexes of FAs with ammonium derivatives; however, differences in the IR spectra of DB isomers were subtle and mostly indistinguishable.34 In contrast, covalently bound 3-pyridylcarbinol enables a clear distinction between FA 18:1 DB isomers in the 1000–1800 cm−1 range (Fig. 1a). In particular, the fingerprint region (1050–1250 cm−1) offers unique features for each isomer. In addition, the carbonyl (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) stretching frequency (1700–1800 cm−1) is shifted depending on the DB configuration and proximity to the ester. Analogous shifts of the CO stretching frequency were observed for 3-picolylamides, which were investigated as an alternative modification strategy but not further considered because they were largely outperformed by 3-pyridylcarbinol esters (Fig. S1–2†). Computation of gas-phase structures using density functional theory (DFT) at the PBE0+D3/6-311+G(d,p) level of theory reveals charge–olefin interactions between the protonated 3-pyridylcarbinol moiety and the DB, as previously reported for sphingolipids.32,35 For comparison, an IR spectrum of the 3-pyridylcarbinol ester of FA 18:0 was recorded, in which the absence of charge–olefin interactions translates into a reduced signal intensity in the N–H bending region between 1350–1500 cm−1 (Fig. S11†). According to the computed IR spectra, the N–H stretching vibrations are also diagnostic for charge–olefin interactions (Fig. S12†).
O) stretching frequency (1700–1800 cm−1) is shifted depending on the DB configuration and proximity to the ester. Analogous shifts of the CO stretching frequency were observed for 3-picolylamides, which were investigated as an alternative modification strategy but not further considered because they were largely outperformed by 3-pyridylcarbinol esters (Fig. S1–2†). Computation of gas-phase structures using density functional theory (DFT) at the PBE0+D3/6-311+G(d,p) level of theory reveals charge–olefin interactions between the protonated 3-pyridylcarbinol moiety and the DB, as previously reported for sphingolipids.32,35 For comparison, an IR spectrum of the 3-pyridylcarbinol ester of FA 18:0 was recorded, in which the absence of charge–olefin interactions translates into a reduced signal intensity in the N–H bending region between 1350–1500 cm−1 (Fig. S11†). According to the computed IR spectra, the N–H stretching vibrations are also diagnostic for charge–olefin interactions (Fig. S12†).
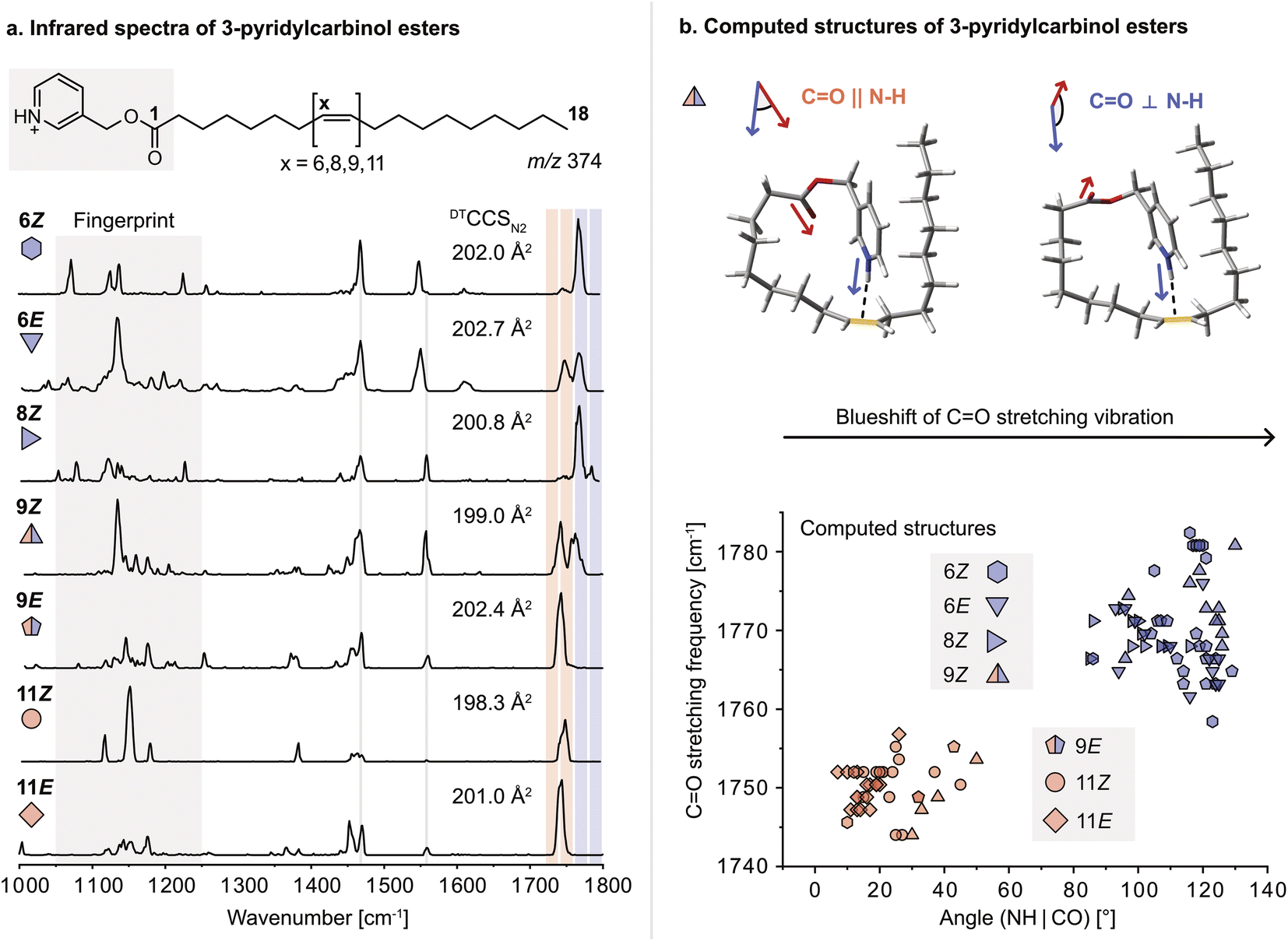 | ||
| Fig. 1 Gas-phase infrared spectra, collision cross sections and structures of FA 18:1 double bond isomers derivatized with 3-pyridylcarbinol. (a) The double bond position and configuration influence the frequency of the carbonyl stretching vibration and the diagnostic fingerprint region (1050–1250 cm−1). (b) The carbonyl stretching frequencies correlate with the relative orientation of the carbonyl bond and the N–H bond, as illustrated by two computed structures of the 9Z isomer. Large angles (>80°) in the computed structures induce a blueshift of the predicted carbonyl stretching frequency, which is indicated by blue filling of the respective symbol. Mixed-colour symbols show that a split CO band is predicted by computation. | ||
Upon further investigation of the computed conformers featuring charge–olefin interactions, a correlation was found between the CO stretching frequency and the angle between the N–H– and the CO vectors (Fig. 1b and S3†). If the two vectors have cosine similarity (<60°), CO stretching frequencies below 1760 cm−1 are predicted, whereas computed structures with angles larger than 80° display a significant blueshift of the carbonyl stretching vibration. The predicted frequencies match exceptionally well with the experimental spectra. The only exceptions are the 6E and 9E isomers, which are predicted to feature a single blueshifted and a split CO band, respectively.
As charge–olefin interactions determine the conformation of 3-pyridylcarbinol esters, they also influence the ion mobility of DB isomers. The collision cross sections (CCS) determined by drift tube ion mobility-mass spectrometry (DTIM-MS) increase with decreasing distance of the DB from the charge group and are larger for E than for Z isomers, in agreement with previous reports.36 The CCSs of the computed structures are consistently underestimated but their trends are in general agreement with the measured values (Fig. S19†). The relative CCS differences are, however, too small to allow for the analysis of isomeric mixtures solely by DTIM-MS. On the contrary, IR spectroscopy yields sufficiently diagnostic absorption bands for each regio- and geometric isomer to enable mixture analysis.
In order to benchmark the ability to analyse isomeric mixtures of FA 18:1 3-pyridylcarbinol esters by IR spectroscopy, synthetic binary and ternary mixtures of the 8Z, 9Z and 11Z isomers were prepared. FA 18:1 (9Z) and (11Z) are the two most abundant isomers in mammalian cells, whereas the 8Z isomer has only recently been discovered to be elevated in cancer cells.4 IR spectra of isomeric mixtures were recorded in the fingerprint region and CO stretching region and were deconvolved by composite spectrum regression implemented in OriginPro 2021 using the three reference spectra (Fig. 2). As all isomers are modified with the same charge carrier, ionization efficiencies are expected to be comparable, and the isomer distribution in the gas phase should hence reflect the distribution in solution. Previous reports from other compound classes have revealed that relative isomer abundances of 3–5% can be determined in isomeric mixtures,37,38 while the present work demonstrated measured isomeric ratios to be within ±8% of the known FA isomer composition. The average relative error is ±10% of the isomer abundance obtained by spectral deconvolution. Overall, the results demonstrate that the deconvolution of composite IR spectra allows for the identification and accurate relative quantification of DB isomers in mixtures. In the case of the synthetic binary and ternary mixtures, a spectral range of only 300 cm−1 is sufficient for isomer assignment and relative quantification.
 | ||
| Fig. 2 Gas-phase infrared spectra of synthetic FA 18:1 (8Z), (9Z) and (11Z) 3-pyridylcarbinol ester standards and synthetic binary and ternary mixtures of known composition. Composite spectrum regression of the infrared spectra obtained from synthetic mixtures in the fingerprint and carbonyl stretching region yields the expected isomer contributions. | ||
After the successful validation of the spectral deconvolution approach for synthetic mixtures of biologically relevant isomers, the derivatization procedure was optimized for biological lipid extracts to eliminate isobaric impurities. The technique was tested on a commercially available reference mixture (Restek 35077 Food Industry FAME mix), which contains FA 18:1 (9Z) and (9E) isomers. The IR spectrum of the FA 18:1 mixture after derivatization is shown in Fig. 3a. The composite spectrum regression yielded a ratio of approximately 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 9Z:9E. As a control experiment, liquid chromatography-ozone-induced dissociation (LC-OzID-MS)39 tandem mass spectrometry was used to confirm isomer assignment and relative abundance by integration of the chromatogram (Fig. 3b). Both techniques yield similar isomer ratios (Fig. 3c).
:1 9Z:9E. As a control experiment, liquid chromatography-ozone-induced dissociation (LC-OzID-MS)39 tandem mass spectrometry was used to confirm isomer assignment and relative abundance by integration of the chromatogram (Fig. 3b). Both techniques yield similar isomer ratios (Fig. 3c).
 | ||
| Fig. 3 Gas-phase infrared spectra and chromatograms of FA 18:1 from a synthetic standard mix (37 Mix Restek) and two lipid extracts from cancer cell lines (1 = LNCaP (TOFA), 2 = PC-3) derivatized with 3-pyridylcarbinol. (a) Infrared spectra were deconvolved by composite spectrum regression using reference spectra. (b) Relative quantification of isomers by liquid chromatography-ozone-induced dissociation was performed by summing the integrals of OzID product chromatograms (aldehyde and Criegee; colored and dotted lines) and the associated deconvolved precursor chromatogram (shaded Gaussian peak area) for each isomer. Heptadeuterated oleic acid (9Z d7) is derived from the SPLASH LipidoMix analytical standard that was added to the cancer cell samples prior to lipid extraction. (c) Gas-phase infrared spectroscopy and LC-OzID-MS yield similar isomeric ratios. | ||
Subsequently, hydrolysed lipid extracts from two human cancer cell lines, LNCaP (treated with TOFA, a dual SCD-1 and ACC1 inhibitor) (1) and PC-3 (2), were derivatized with 3-pyridylcarbinol and measured by IR spectroscopy and confirmed using LC-OzID-MS. Linear combination of the component spectra of FA 18:1 (6Z), (8Z), (9Z) and (11Z) using the isomer ratios determined by composite spectrum regression (grey trace) shows a reasonable fit of the predicted model to the experimental spectrum. The reference spectra and mixture spectra were measured with a delay of several months, and the discrepancy in band intensities between the simulated and experimental spectra in the region between 1350 and 1600 cm−1 can be explained by fluctuations of the laser pulse energy and focus over time. Due to the multiple photon absorption process, the relative band intensities are influenced by the photon fluence. On the contrary, the band positions, i.e., the absorption frequencies, which are crucial for the structural assignment, are reliable and not affected by the experimental conditions.
As is observed in Fig. 3c, the two cancer cell lines display significant variation in their FA 18:1 isomer populations. Most importantly, the presence of FA 18:1 (Δ6) DB – previously reported with unknown geometry in PC-3 cells5 – was confirmed for the first time as being in the cis configuration by IR spectroscopy. In the IR spectrum of PC-3 (Fig. 3a, cell line 2), the redshifted peak at 1550 cm−1 is diagnostic for position Δ6 (cf.Fig. 1a), and the Z configuration was verified by composite spectrum regression, yielding a contribution of 0% from the 6E standard. Furthermore, a significant amount of the 8Z isomer was found in the PC-3 lipid extract, in accordance with previous findings.10 The increased amount of FA 18:1 (8Z) in PC-3 cells can be seen by the increased intensity of the blueshifted CO band. It should be noted that the Δ6 isomer observed in the LC chromatograms of LNCaP (TOFA) at a lower ratio than in the PC-3 cells (Fig. 3b, cell line 1) is below the detection limit of the spectroscopic approach. The characteristic redshifted peak at 1550 cm−1 is not captured by the spectral deconvolution because its intensity is too low compared to the background noise. However, it is difficult to define a universal detection limit for the identification of individual FAs in the presence of other isomers because it varies with the absorption intensity, the probed wavenumber range and the similarity of the component spectra in that particular range. All FA isomers that have been detected in this work by IR spectroscopy have a relative abundance of at least 10% according to the LC-OzID-MS reference method. While likely also below the limit of detection, the Δ5 isomer (observed in Fig. 3b) was not included as a standard in the linear regression model for IR analysis as it was not commercially available.
The isomer abundances determined by IR spectroscopy deviate from the values determined by the LC-OzID-MS reference method up to ±12%, which corresponds to a relative error of ±30% of the isomer abundance. The estimated relative error of relative quantities of octadecenoic acids determined by LC-OzID-MS is ±20% (based on the triplicate measurement of a pooled human plasma – NIST 1950 – standard reference material, total hydrolysed fatty acid content, average coefficient of variation of relative quantities of all detected octadecenoic acids).39 A determined 10% relative quantity, for example, should thus be read as 8–12%. Overall, the analysis of two hydrolysed lipid extracts of biological origin showcases the power of gas-phase IR spectroscopy for the analysis of isomeric mixtures, even in complex matrices.
Conclusions
In conclusion, we here present an innovative approach to the characterization of FA DB-position and -geometry by 3-pyridylcarbinol ester derivatization and subsequent gas-phase IR spectroscopy. The 3-pyridylcarbinol modification, commonly used for FA analysis within GC-MS platforms,8,9,11–13 promotes a charge–olefin interaction between the pyridyl group and the FA DB. This interaction affects the molecular conformation and thereby influences the CO stretching and fingerprint region vibrations. The shifts of the CO stretching frequency are predictable by computational chemistry as a function of the proximity of the DB to the ester. Using spectral deconvolution, individual FAs can be identified even in the presence of multiple isomeric species. Given the sensitivity of the vibrational spectra to the regio- and stereo-chemistry of carbon–carbon double bonds in unsaturated lipids, it is reasonable to speculate that the structures of other functionalized FAs, such as hydroxylated and nitrated FAs, could also be determined using this approach.
Lastly, the technique was employed to detect and identify the isomers of mass-selected FA 18:1 in cancer cell lines by deconvolution of the mixture spectra. We were able to identify the presence of four FA 18:1 isomers and establish the DB geometry in FA 18:1 (Δ6) from PC-3 cancer cells as being cis despite it representing less than 10 mol% of the isomer population. Gas-phase IR spectroscopy enables explicit assignment of the position and configuration of double bonds for individual unsaturated FAs in the presence of multiple isomeric variants. Such detailed structure elucidation of mass-selected lipids is a powerful and complementary approach to LC-MS technologies, which thus empowers lipid discovery and reveals otherwise-invisible changes in the lipidome.
Materials and methods
Synthesis of 3-pyridylcarbinol esters and 3-picolylamides
C18:1 fatty acid standards were purchased as follows: 11Z-octadecenoic acid ≥97%, Sigma Aldrich; 11E-octadecenoic acid 99%, Matreya; 9Z-octadecenoic acid 99%, Sigma Aldrich; 9E-octadecenoic acid ≥97%, TCI; 6Z-octadecenoic acid ≥98%, TCI; 6E-octadecenoic acid ≥99%, Nu-Chek Prep; 8Z-octadecenoic acid ≥99%, Larodan; Food Industry FAME Mix of 37 fatty acid methyl esters including 9E- and 9Z-octadecanoic acid, Restek.Lipids were extracted from PC-3 and LNCaP (TOFA) cells using methods similar to those described by Matyash et al.40 as documented in detail in Young et al.5 with no amendments. Approximately 350000 cells from the lipid extract suspensions were added to clean 2 mL glass vials and were dried under N2 gas. Fatty acids were hydrolysed from intact lipids using 200 μL of MeOH (LC-MS grade, Fisher Scientific, Scorseby, Australia) and 200 μL of aqueous KOH [1.75 M] added to each sample vial. Vials were capped (PTFE cap insert) and lightly agitated for 30 s without the contents touching the cap insert. Samples were heated at 75 °C for 2 hours using a dry block heater (Ratek, Saratoga, United States). Samples were removed from heat and allowed to cool to RT. The reaction was quenched using 800 μL H2O and acidified to pH 1.5 using 120 μL HCl [5 M]. 500 μL of n-hexane was added and vials were vortexed for 30 s before allowing the two phases to settle. The supernatant was collected in a new 4 mL glass vial via pipette before the biphasic extraction process was repeated for an additional 500 μL of n-hexane. Collection vials containing fatty acids in n-hexane were dried under N2 gas.
C18:1 fatty acids were derivatized with 3-pyridinemethanol to yield 3-pyridylcarbinol esters. For lipid extract samples, 250 μL ACN, 50 μL 3-pyridinemethanol in ACN [110 mM], 50 μL DIPEA in ACN [350 mM] and 50 μL of HBTU in ACN [110 mM] were added to each vial before 30 s vortex. For fatty acid standard samples, 50 μL C18:1 in ACN [35 mM], 200 μL ACN, 50 μL 3-pyridinemethanol in ACN [110 mM], 50 μL DIPEA in ACN [350 mM] and 50 μL of HBTU in ACN [110 mM] were added to each vial before 30 s vortex. Samples were heated at 50 °C for 17 hours using a dry heat block before being removed from heat and allowed to cool to RT. 1 mL of H2O was added along with 200 μL saturated aqueous NaCl solution and 2 mL of MTBE. Samples were vortexed for 30 s and the supernatant was collected in a 2 mL glass vial via pipette. Samples in 2 mL glass vials were dried under N2 gas and washed with an additional 200 μL H2O, 200 μL saturated aqueous NaCl solution and 750 μL MTBE. Samples were mixed by inversion before the supernatant was transferred to a new 2 mL glass vial via pipette. Samples were dried under N2 gas and capped with N2 gas in the head space prior to inter-laboratory shipping.
18:1 fatty acids were derivatized with 3-picolylamine to yield 3-picolylamides. 133 μL of 3-picolylamine (2.67 μmol, 3 equiv.) in DMF (HPLC grade) [20 mM] was added to the C18:1 fatty acid (0.889 μmol, 1 equiv.) in a 4 mL vial before 30 s vortex. 26 μL HATU (2.67 μmol, 3 equiv.) in DMF [0.1 M] and 44 μL DIPEA (4.44 μmol, 5 equiv.) in DMF [0.1 M] were added before 1 min vortex. The resulting mixture was heated at 65 °C for 30 min before cooling to RT. The mixture was diluted with 1.5 mL H2O (optima grade) and 1.5 mL MTBE (HPLC grade). 100 μL of saturated NH4Cl solution was added before 30 s vortex. The layers were separated, and the aqueous layer was again extracted with 1.5 mL MTBE. The organic layers were combined and MTBE removed under N2 gas.
3-Pyridylcarbinol ester and 3-picolylamide derivatives of fatty acids were resuspended in MeOH to a concentration of 100 μM for infrared spectroscopy.
Gas-phase infrared spectroscopy in superfluid helium droplets
Gas-phase IR spectra of ionized lipids were recorded on a custom-built instrument described previously.33,41 Ions are generated by nano-electrospray ionization using Pd/Pt-coated needles and applying a voltage of 0.7–1.0 kV. The ions of interest are m/z-selected in a quadrupole and bent by 90° by a quadrupole bender into a hexapole ion trap. The ion trap is filled with helium buffer gas, which is additionally cooled to 90 K by liquid nitrogen. Ions are thus decelerated and thermalized by buffer gas cooling and trapped by DC and RF potentials. Superfluid helium droplets are generated by expanding pressurized helium (60 bar) through the cooled nozzle (21 K) of a pulsed Even–Lavie valve (10 Hz). After the buffer gas is pumped out of the trap, the trapped ions can be picked up by the helium droplets, which traverse the trap coaxially. Once inside a droplet, the ion is cooled to the intrinsic droplet temperature of 0.4 K.42 The doped droplets travel towards the interaction region, where the droplet beam overlaps in space and time with the pulsed beam of the Fritz Haber Institute free-electron laser (FHI FEL).43 The macropulse repetition rate of the FHI FEL is 10 Hz, and each macropulse contains 104 micropulses, which allow for the rapid sequential absorption of multiple photons by the analyte ion inside the helium droplet. Upon the absorption of one IR photon, the vibrational energy is dissipated by the evaporation of helium atoms and the ion is cooled to the initial droplet temperature of 0.4 K. The helium shell around the ion can thus be regarded as an IR-transparent cryostat preventing ion heating during photon absorption while not disturbing vibrations of the analyte because superfluid helium lacks viscosity. After the absorption of multiple photons, the bare ion is released from the droplet and detected on a time-of-flight mass analyser. IR spectra are generated by scanning the tuneable FHI FEL in steps of 2 cm−1 while monitoring the ion count on the detector. Spectra are averaged from two individual scans.Composite spectrum regression
IR spectra of synthetic and biological mixtures of FA 18:1 3-pyridylcarbinol esters were deconvolved using reference spectra of standard substances. Prior to the deconvolution, mixture spectra and reference spectra were binned in 2 cm−1 steps in OriginPro 2021 using the 1D binning application. The binned data are represented in Table S15 and S16.† The binned mixture spectra were then deconvolved using the composite spectrum regression application implemented in OriginPro 2021 to yield the relative abundance of each isomer contained in the mixture. The model minimizes the error between the measured composite spectrum and the sum spectrum of the component spectra by optimizing the weighting factors, i.e., the isomer abundances. The experimental mixture spectra can be simulated by linear combination of the reference spectra using the weighting factors obtained from the composite spectrum regression (Fig. S21† and 3). The R2 values are >0.91 for the synthetic mixtures in a reduced wavenumber range, and >0.83 for the biological samples in the full wavenumber range, as indicated in the ESI (Table S15 and S16).† All isomer abundances reported in this work were determined based on IR spectra averaged from two individual scans. The maximum variation of isomer abundances obtained from individual measurements of the same sample is ±12%.Computational details
Gas-phase structures and harmonic IR spectra of protonated FA 18:1 3-pyridylcarbinol esters and 3-picolylamides were computed by an initial conformational sampling and subsequent optimization of selected structures by density functional theory (DFT). The conformational sampling of each isomer was performed using CREST44 with the semiempirical method GFN2-xTB45 and default settings. Several conformers were selected for further optimization at the PBE0+D3/6-311+G(d,p) level of theory in Gaussian 16.46 Harmonic IR spectra of the DFT-optimized conformers were computed at the same level of theory and scaled by an empirical factor of 0.965, in accordance with previous publications.32,34,47 Free energies (ΔF) were computed at the ion trap temperature of 90 K because the conformational ensemble at 90 K is shock-frozen in the helium droplets. The relative energy values refer to the lowest-energy conformer. Collision cross sections (CCSs) of all computed conformers were calculated with DFT-computed Merz–Singh–Kollman charges48 using the software HPCCS49 which is based on the trajectory method.50 The CCSs were computed at 298.15 K (25 °C) in nitrogen, and the average CCS of all conformers below a threshold of ΔF = 10 kJ mol−1 was determined for each isomer.Ion mobility-mass spectrometry
For determination of DTCCSN2 values, an Agilent 6560 IM-QTOFMS (Agilent Technologies, Santa Clara, CA) equipped with a Dual JetStream Electrospray ion source was used. Each isomer was injected separately and eluted from a ZORBAX Eclipse Plus C18 column (50 mm × 2.1 mm i.d. and 1.8 μm dp) using a linear gradient (solvent A: 0.1% formic acid; solvent B: 0.1% formic acid: 99.9% acetonitrile) and a flow rate of 200 μL min−1. The instrument was operated in the 1700 extended dynamic range mode with positive polarity. The drying gas flow was set to 13 L min−1 (275 °C), sheath gas flow was 12 L min−1 (350 °C) with a nebuliser pressure of 30 psi. The trapping funnel fill time was set to 1000 μs and the 78 cm drift tube was operated using a field strength of 10.9 V cm−1 (entrance 1074 V, exit 224 V). The maximum drift time was set to 50 ms and the trap release time to 150 μs. An acquisition rate of 1.1 frames per second was realised by summing 14 IM transients per frame.For secondary (single-field) DTCCSN2 calibration, an infusion of ESI Tune Mix ESI-L (G1969-85000, Agilent Technologies) prepared according to the manufacturer's instructions was used to determine a linear calibration function using the method and DTCCSN2 values reported in Stow et al.51 The calibration parameters were subsequently applied to all measurement files in a single sequence. All data processing for DTCCSN2 determination was performed using Agilent MassHunter IM-MS Browser (10.0, Build 10.0.100027.0).
Photoreactions
E/Z isomerization reactions were carried out in a custom-made photoreactor consisting of a Styrofoam box containing an aluminium lining on the inside, a UV lamp, emitting broadband 254 nm light and a quartz glass vial (crimped, airtight). 0.15 mL of a solution of 35 mM (5.3 mmol) FA 18:1 (11Z), 35 mM (5.3 mmol) FA 18:1 (9Z), 35 mM (5.3 mmol) FA 18:1 (9E), 35 mM (5.3 mmol) FA 18:1 (8Z), 35 mM (5.3 mmol) FA 18:1 (6Z), (in the following referred to as 18:1 mix) in n-hexane was prepared, transferred to a quartz glass vial, crimped airtight and deoxygenated by a stream of argon for 5 minutes. The vial was placed inside the photoreactor at 3 cm from the lamp and irradiated for 12 h. The temperature inside the photo reactor was monitored and maintained between 20–35 °C throughout. The photochemically isomerized fatty acid standards were employed for the assignment of E/Z isomers, as described further below.Ultra-high performance liquid chromatography-ozone-induced dissociation-mass spectrometry (UPLC-OzID-MS)
For LC-MS analysis, extracted lipids were derivatized with 1-(4-(aminomethyl)phenyl)pyridinium (AMPP) as described previously in Young et al.5 5 μL of the Restek 37 Mix of fatty acid methyl esters (FAME) and blanks were derivatized using the same method. The lipids were stored in sealed vials at −20 °C.Analysis was performed using a Waters Acquity (UPLC i-Class; CSH, C18 reverse phase column, length 100 mm, inner diameter 2.1 mm, particle size 1.7 μm) liquid chromatography system coupled with a Waters SYNAPT G2-Si (Z-Spray, T-Wave Ion Mobility; TOF) mass spectrometer previously modified to allow introduction of ozone into the IMS cell.52 Samples were prepared in 1.5 mL LC-MS vials with PTFE septum and kept in the autosampler of the LC system at 10 °C prior to analysis. The column was kept at 60 °C during analysis.
Liquid chromatography was performed with a linear gradient at a flow rate of 0.4 mL min−1. Mobile phase A was water with 0.1% formic acid; mobile phase B was acetonitrile with 0.1% formic acid. The injection volume varied depending on the sample from 1 μL to 10 μL. Initially the mobile phase consisted of 10% B (90% A), and was increased to 20% B at 0.5 min, then linearly increased from 0.5 min to 18 min (100% B). The mobile phase was then kept isocratic until 20 min at 100% B and was reduced to 10% B at 22 min to 25 min to recondition the column for the next sample injection.
Using positive polarity, the mass spectrometer was operated in “sensitivity mode” allowing for a mass resolution of ∼15000. A capillary voltage of 3.0 kV was applied, with a sampling cone of 40 V and source offset set to 80 V. The source temperature was set to 120 °C and the desolvation temperature to 550 °C. Cone gas was set to 100 L h−1, desolvation gas 900 L h−1 and nebulizer 6.5 bar. IMS gas controls were set to: Trap 2.0 mL min−1, He-Cell 180 mL min−1, IMS 10 mL min−1. IMS travelling wave velocity and height were 650 ms−1 and 28 V, while the transfer travelling wave velocity and height were 1000 ms−1 and 2 V, respectively.
For OzID, the ion mobility cell is filled with a mixture of nitrogen, ozone and oxygen, in place of just nitrogen in normal operation. Ozone was produced from high purity oxygen using a corona discharge based high-concentration ozone generator (Ozone Solutions TG-40), producing a flow of 400 mL min−1 with 200–250 g m−3 ozone in oxygen. Ozone in oxygen was introduced to the IMS gas flow through a needle valve, that was adjusted to yield an ultimate pressure of ∼3 mbar in the IMS cell. Ozone was catalytically destroyed using a manganese dioxide-based catalyst before being exhausted from the laboratory.
A solution of 200 pg mL−1 Leucine Enkephalin (LeuEnk, YGGFL, H-Tyr-Gly-Gly-Phe-Leu-OH, m/z 556.2771) in a mixture of 50% acetonitrile and 50% water with 0.1% formic acid was used as a Lockmass solution. Lockmass correction was performed upon loading of the raw LC-MS dataset into Skyline. Data was acquired from 100 to 750 m/z at a scan time of 0.1 seconds. Data analysis was performed with Skyline MS and chromatograms and results were visualized with Origin 2018 64 bit and Excel.
Analysis of UPLC-OzID-MS data
The presence of characteristic fragments associated with cleavage of double bonds during ozonolysis (leading to aldehyde and Criegee fragments) provides evidence of the position of the double bond. The associated extracted ion chromatograms (XICs) provide further evidence for correct identification (coelution of associated aldehyde and Criegee fragments). Relative quantification is performed by integration of the extracted ion chromatograms of both precursor and associated fragment chromatograms. The precursor XIC is deconvolved as a sum of Gaussian expressions to determine the contribution of each isomer. The retention time of the characteristic fragments is used to inform the retention time position of the Gaussian expressions used for deconvolution. Apart from the aldehyde and Criegee fragmentation pathways, no other fragmentation pathways are detectable. Thus, relative quantification of isomers is performed as described. The E/Z configuration was determined as described in the next section, employing LC-OzID-MS data of photochemically isomerized fatty acid standards.Assignment of E/Z configuration based on relative retention times and comparison with photochemically isomerized fatty acid standards
Analysis of the mixture of fatty acids prior to irradiation reveals that no FA 18:1 (11E) or FA 18:1 (6E) can be detected, refer to Fig. S26 and S27.† Note, that the apparent aldehyde neutral loss from FA 18:1 (8Z) at 8.20 min is due to overoxidation of the FA 18:1 (9E) eluting at the same retention time. For this reason, the confidence of the assignment of a relative retention time of FA 18:1 (8E) is low. After irradiation, small amounts of the respective E fatty acids are detectable, revealing their relative retention times, refer to Fig. S28 and S29.† A retention time comparison allows determination of the configuration of the double bond in respective fatty acids as detected in prostate cancer cell lines (PC-3 and LNCaP-TOFA), which were previously subjected to the same derivatization and LC-OzID-MS analysis procedure, refer to Fig. S32.† The analysis reveals that for all eight fatty acids investigated here, features are identified at the expected relative retention time (PC-3 and LNCaP-TOFA). Nevertheless, relative amounts of each E fatty acid are far lower than the respective Z fatty acid.Data availability
All authors confirm that the data supporting the studies are available within the article and its ESI.†Author contributions
CK: data curation, formal analysis, investigation, project administration, visualization, and writing – original draft. RSEY: formal analysis, investigation, visualization, and writing – original draft. KG: formal analysis, investigation, and writing – review & editing. JPM: formal analysis, investigation, visualization, and writing – original draft. SG: investigation. WS: investigation and writing – review & editing. GM: resources and writing – review & editing. GvH: methodology and resources. TC: formal analysis, investigation, and writing – review & editing. VRN: investigation. BLJP: funding acquisition, investigation, and writing – review & editing. SJB: conceptualization, funding acquisition, methodology, project administration, resources, supervision, and writing – original draft. KP: conceptualization, funding acquisition, project administration, resources, supervision, and writing – original draft.Conflicts of interest
SJB holds patents on ozone-induced dissociation technology (A method for the determination of the position of unsaturation in a compound, US8242439 and US7771943). The work presented in this publication was supported in-part through funding from Waters Corporation and the Australian Research Council through the Linkage Partnership Scheme (LP18010023).Acknowledgements
CK is grateful for financial support by the Fonds der Chemischen Industrie in the form of a Kekulé fellowship. KG thanks the Fonds National de la Recherche, Luxembourg, for funding the project GlycoCat (13549747). KP acknowledges generous funding by the European Research Council, ERC-2019-CoG-863934-GlycoSpec. This work was financially supported by the Australian Government through award of an RTP scholarship (to RSEY) and the Australian Research Council through the Discovery Program (DP190101486) and the Linkage Program (LP180100238, partnered with Waters Corporation). The project team acknowledge the Central Analytical Research Facility operated by the Queensland University of Technology for provision of instrumentation and training in support of this project. TC thanks EQ VIBT GmbH and the BOKU Core Facility Mass Spectrometry for providing mass spectrometry instrumentation.References
- A. Shevchenko and K. Simons, Nat. Rev. Mol. Cell Biol., 2010, 11, 593–598 CrossRef CAS PubMed.
- B. Brügger, Annu. Rev. Biochem., 2014, 83, 79–98 CrossRef PubMed.
- M. Sud, E. Fahy, D. Cotter, A. Brown, E. A. Dennis, C. K. Glass, A. H. Merrill, R. C. Murphy, C. R. Raetz, D. W. Russell and S. Subramaniam, Nucleic Acids Res., 2007, 35, D527–D532 CrossRef CAS PubMed.
- K. Vriens, S. Christen, S. Parik, D. Broekaert, K. Yoshinaga, A. Talebi, J. Dehairs, C. Escalona-Noguero, R. Schmieder, T. Cornfield, C. Charlton, L. Romero-Perez, M. Rossi, G. Rinaldi, M. F. Orth, R. Boon, A. Kerstens, S. Y. Kwan, B. Faubert, A. Mendez-Lucas, C. C. Kopitz, T. Chen, J. Fernandez-Garcia, J. A. G. Duarte, A. A. Schmitz, P. Steigemann, M. Najimi, A. Hagebarth, J. A. Van Ginderachter, E. Sokal, N. Gotoh, K. K. Wong, C. Verfaillie, R. Derua, S. Munck, M. Yuneva, L. Beretta, R. J. DeBerardinis, J. V. Swinnen, L. Hodson, D. Cassiman, C. Verslype, S. Christian, S. Grunewald, T. G. P. Grunewald and S. M. Fendt, Nature, 2019, 566, 403–406 CrossRef PubMed.
- R. S. E. Young, A. P. Bowman, E. D. Williams, K. D. Tousignant, C. L. Bidgood, V. R. Narreddula, R. Gupta, D. L. Marshall, B. L. J. Poad, C. C. Nelson, S. R. Ellis, R. M. A. Heeren, M. C. Sadowski and S. J. Blanksby, Cell Rep., 2021, 34, 108738 CrossRef CAS PubMed.
- C. Guijas, C. Meana, A. M. Astudillo, M. A. Balboa and J. Balsinde, Cell Chem. Biol., 2016, 23, 689–699 CrossRef CAS PubMed.
- A. B. Oteng and S. Kersten, Adv Nutr., 2020, 11, 697–708 CrossRef PubMed.
- W. W. Christie, Lipids, 1998, 33, 343–353 CrossRef CAS PubMed.
- W. W. Christie, J. Chromatogr. A, 1988, 447, 305–314 CrossRef CAS.
- C. Ferreri, A. Sansone, S. Buratta, L. Urbanelli, E. Costanzi, C. Emiliani and C. Chatgilialoglu, Cancers, 2020, 12, 900 CrossRef CAS PubMed.
- D. J. Harvey, Biol. Mass Spectrom., 1982, 9, 33–38 CrossRef CAS.
- J. Y. Zhang, Q. T. Yu, B. N. Liu and Z. H. Huang, Biol. Mass Spectrom., 1988, 15, 33–44 Search PubMed.
- W. W. Christie, E. Y. Brechany and R. T. Holman, Lipids, 1987, 22, 224–228 CrossRef CAS PubMed.
- C. L. Feider, L. A. Macias, J. S. Brodbelt and L. S. Eberlin, Anal. Chem., 2020, 92, 8386–8395 CrossRef CAS PubMed.
- C. E. Randolph, D. J. Foreman, S. K. Betancourt, S. J. Blanksby and S. A. McLuckey, Anal. Chem., 2018, 90, 12861–12869 CrossRef CAS PubMed.
- H. T. Pham, A. J. Trevitt, T. W. Mitchell and S. J. Blanksby, Rapid Commun. Mass Spectrom., 2013, 27, 805–815 CrossRef CAS PubMed.
- V. R. Narreddula, N. R. Boase, R. Ailuri, D. L. Marshall, B. L. J. Poad, M. J. Kelso, A. J. Trevitt, T. W. Mitchell and S. J. Blanksby, Anal. Chem., 2019, 91, 9901–9909 CrossRef CAS PubMed.
- X. Ma and Y. Xia, Angew. Chem., Int. Ed., 2014, 53, 2592–2596 CrossRef CAS PubMed.
- M. C. Thomas, T. W. Mitchell, D. G. Harman, J. M. Deeley, J. R. Nealon and S. J. Blanksby, Anal. Chem., 2008, 80, 303–311 CrossRef CAS PubMed.
- Y. Zhao, H. Zhao, X. Zhao, J. Jia, Q. Ma, S. Zhang, X. Zhang, H. Chiba, S. P. Hui and X. Ma, Anal. Chem., 2017, 89, 10270–10278 CrossRef CAS PubMed.
- M. Koktavá, J. Valášek, D. Bezdeková, V. Prysiazhnyi, B. Adamová, P. Beneš, J. Navrátilová, M. Hendrych, P. Vlček, J. Preisler and A. Bednařík, Anal. Chem., 2022, 94, 8928–8936 CrossRef PubMed.
- M. Fang, Y. Rustam, M. Palmieri, O. M. Sieber and G. E. Reid, Anal. Bioanal. Chem., 2020, 412, 2339–2351 CrossRef CAS PubMed.
- H. J. Yoo and K. Håkansson, Anal. Chem., 2010, 82, 6940–6946 CrossRef CAS PubMed.
- H. T. Pham, M. B. Prendergast, C. W. Dunstan, A. J. Trevitt, T. W. Mitchell, R. R. Julian and S. J. Blanksby, Int. J. Mass Spectrom., 2015, 390, 170–177 CrossRef CAS.
- T. Baba, J. L. Campbell, J. C. Y. Le Blanc and P. R. S. Baker, Anal. Chem., 2017, 89, 7307–7315 CrossRef CAS PubMed.
- S.-T. Kuo, S. Tang, D. H. Russell and X. Yan, Int. J. Mass Spectrom., 2022, 479, 116889 CrossRef CAS.
- G. Feng, M. Gao, L. Wang, J. Chen, M. Hou, Q. Wan, Y. Lin, G. Xu, X. Qi and S. Chen, Nat. Commun., 2022, 13, 2652 CrossRef CAS PubMed.
- R. S. E. Young, C. L. Flakelar, V. R. Narreddula, L. J. Jekimovs, J. P. Menzel, B. L. J. Poad and S. J. Blanksby, Anal. Chem., 2022, 94, 16180–16188 CrossRef CAS PubMed.
- Y. Miyake and K. Yokomizo, J. Am. Oil Chem. Soc., 1998, 75, 801–805 CrossRef CAS.
- H. Azizian and J. K. Kramer, Lipids, 2005, 40, 855–867 CrossRef CAS PubMed.
- S. Du, M. Su, C. Wang, Z. Ding, Y. Jiang and H. Liu, Anal. Chem., 2022, 94, 2891–2900 CrossRef CAS PubMed.
- C. Kirschbaum, E. M. Saied, K. Greis, E. Mucha, S. Gewinner, W. Schöllkopf, G. Meijer, G. von Helden, B. L. J. Poad, S. J. Blanksby, C. Arenz and K. Pagel, Angew. Chem., Int. Ed., 2020, 59, 13638–13642 CrossRef CAS PubMed.
- A. I. González Flórez, E. Mucha, D. S. Ahn, S. Gewinner, W. Schöllkopf, K. Pagel and G. von Helden, Angew. Chem., Int. Ed., 2016, 55, 3295–3299 CrossRef PubMed.
- C. Kirschbaum, K. Greis, M. Lettow, S. Gewinner, W. Schöllkopf, G. Meijer, G. von Helden and K. Pagel, Anal. Bioanal. Chem., 2021, 413, 3643–3653 CrossRef CAS PubMed.
- B. L. J. Poad, A. T. Maccarone, H. Yu, T. W. Mitchell, E. M. Saied, C. Arenz, T. Hornemann, J. N. Bull, E. J. Bieske and S. J. Blanksby, Anal. Chem., 2018, 90, 5343–5351 CrossRef CAS PubMed.
- R. Wojcik, I. K. Webb, L. Deng, S. V. Garimella, S. A. Prost, Y. M. Ibrahim, E. S. Baker and R. D. Smith, Int. J. Mol. Sci., 2017, 18, 183 CrossRef PubMed.
- C. Kirschbaum, K. Greis, E. Mucha, L. Kain, S. Deng, A. Zappe, S. Gewinner, W. Schöllkopf, G. von Helden, G. Meijer, P. B. Savage, M. Marianski, L. Teyton and K. Pagel, Nat. Commun., 2021, 12, 1201 CrossRef CAS PubMed.
- A. H. Abikhodr, V. Yatsyna, A. Ben Faleh, S. Warnke and T. R. Rizzo, Anal. Chem., 2021, 93, 14730–14736 CrossRef CAS PubMed.
- J. P. Menzel, R. S. E. Young, A. H. Benfield, J. Scott, L. M. Butler, S. T. Henriques, B. L. J. Poad and S. J. Blanksby, bioRxiv, 2022, preprint, DOI:10.1101/2022.10.24.513604.
- V. Matyash, G. Liebisch, T. V. Kurzchalia, A. Shevchenko and D. Schwudke, J. Lipid Res., 2008, 49, 1137–1146 CrossRef CAS PubMed.
- E. Mucha, A. I. González Flórez, M. Marianski, D. A. Thomas, W. Hoffmann, W. B. Struwe, H. S. Hahm, S. Gewinner, W. Schöllkopf, P. H. Seeberger, G. von Helden and K. Pagel, Angew. Chem., Int. Ed., 2017, 56, 11248–11251 CrossRef CAS PubMed.
- J. P. Toennies and A. F. Vilesov, Angew. Chem., Int. Ed., 2004, 43, 2622–2648 CrossRef CAS PubMed.
- W. Schöllkopf, S. Gewinner, H. Junkes, A. Paarmann, G. von Helden, H. Bluem and A. M. M. Todd, Proc. SPIE, 2015, 9512, 95121L CrossRef.
- P. Pracht, F. Bohle and S. Grimme, Phys. Chem. Chem. Phys., 2020, 22, 7169–7192 RSC.
- C. Bannwarth, S. Ehlert and S. Grimme, J. Chem. Theory Comput., 2019, 15, 1652–1671 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman, D. J. Fox, Gaussian 16, Rev. A.03, 2016 Search PubMed.
- C. Kirschbaum, K. Greis, L. Polewski, S. Gewinner, W. Schöllkopf, G. Meijer, G. von Helden and K. Pagel, J. Am. Chem. Soc., 2021, 143, 14827–14834 CrossRef CAS PubMed.
- U. C. Singh and P. A. Kollman, J. Comput. Chem., 1984, 5, 129–145 CrossRef CAS.
- L. Zanotto, G. Heerdt, P. C. T. Souza, G. Araujo and M. S. Skaf, J. Comput. Chem., 2018, 39, 1675–1681 CrossRef CAS PubMed.
- M. F. Mesleh, J. M. Hunter, A. A. Shvartsburg, G. C. Schatz and M. F. Jarrold, J. Phys. Chem., 1996, 100, 16082–16086 CrossRef CAS.
- S. M. Stow, T. J. Causon, X. Zheng, R. T. Kurulugama, T. Mairinger, J. C. May, E. E. Rennie, E. S. Baker, R. D. Smith, J. A. McLean, S. Hann and J. C. Fjeldsted, Anal. Chem., 2017, 89, 9048–9055 CrossRef CAS PubMed.
- B. L. J. Poad, M. R. Green, J. M. Kirk, N. Tomczyk, T. W. Mitchell and S. J. Blanksby, Anal. Chem., 2017, 89, 4223–4229 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sc06487a |
| ‡ Present address: Molecular Horizons, University of Wollongong, Wollongong, NSW 2522, Australia. |
| § Present address: QML Pathology Queensland, PO Box 2280, Mansfield QLD 4122, Australia. |
| This journal is © The Royal Society of Chemistry 2023 |