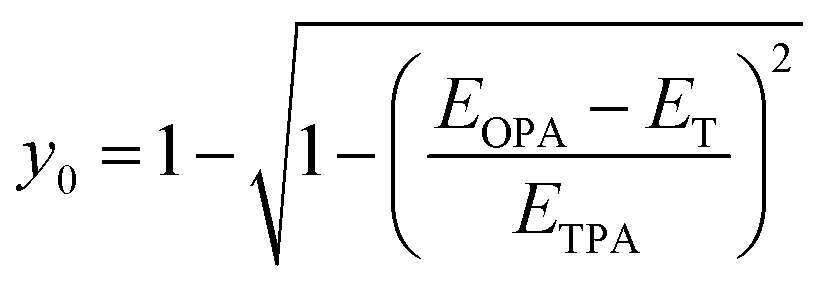DOI:
10.1039/D2SC06612B
(Edge Article)
Chem. Sci., 2023,
14, 1978-1985
Unveiling a new aspect of oxocarbons: open-shell character of 4- and 5-membered oxocarbon derivatives showing near-infrared absorption†
Received
1st December 2022
, Accepted 14th January 2023
First published on 16th January 2023
Abstract
Oxocarbon derivatives consisting of 4- and 5-membered rings, referred to as croconaine and squaraine dyes and regarded as closed-shell molecules, are found to have an intermediate open-shell character from the experimental results of 1H-NMR, ESR spectroscopy, SQUID magnetometric analysis, and X-ray crystallography. We employed two chalcogenopyrylium moieties with O and S chalcogen atoms as substitutions on oxocarbons. The singlet–triplet energy gaps (ΔES–T) associated with the degree of diradical nature are smaller for croconaines than for squaraines and smaller for thiopyrylium than for pyrylium groups. The diradical nature impacts the electronic transition energy that decreased with a decreasing degree of diradical contribution. They exhibit substantial two-photon absorption in the region over 1000 nm. The diradical character y of the dye was determined experimentally from the observed one- and two-photon absorption peaks and the triplet energy level. The present finding provides new insight into diradicaloids with the contribution of non-Kekulé oxocarbon and also showcases the correlation between the electronic transition energy and their diradical character.
Introduction
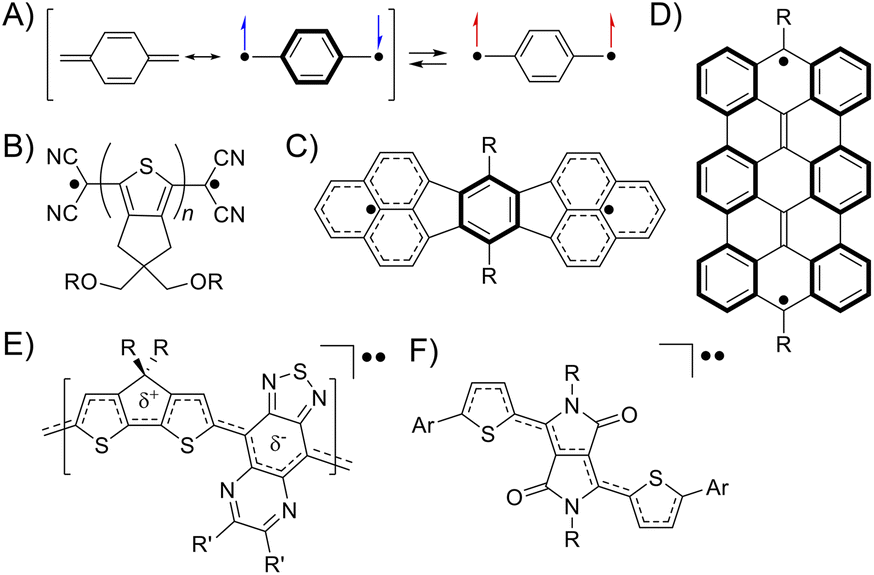
A singlet diradicaloid is a molecule in which the two antiparallel electrons are partially coupled but not fully coupled in the ground state.1 The degree can be expressed using the diradical character y of an intermediate value between 0 and 1, where y = 0 and 1 represent closed-shell and open-shell electronic structures, respectively.2 Then, the bond order represented by 1 − y is between 1 and 0, meaning partial dissociation of the chemical bond.3 The electrons in such partially dissociated bonds are weakly bound to nuclei and easily fluctuate. Because of this, singlet diradicaloids exhibit intriguing functionalities such as electric conductivity, optical nonlinearity including two-photon absorption (TPA), and singlet fission.4–6 Over the last two decades, diverse classes of stable diradicaloids have been demonstrated including quinoidal polycyclic aromatic hydrocarbons, quinoidal oligothiophenes, and graphene fragments.4,7 The open-shell character in this class of diradicaloids is known to arise from the recovery of Clar's aromatic sextet rings (Fig. 1A–D). Furthermore, singlet diradicaloids have become an important concept recently in many fields of chemistry. For example, acenes were once recognized as closed-shell molecules described with a Kekulé-type structure; however, singlet diradicals formed in the resonance structures can explain the instability of oligoacenes (pentacene or longer) and reactive sites of short acenes (such as anthracene).8 More recently, some groups demonstrated that donor–accepter conjugated polymers and diketopyrrolopyrrole derivatives, generally known as closed-shell species, exhibited the intermediate open-shell character that impacted their optoelectronic properties (Fig. 1E and F).9,10 Thus, reviewing closed-shell molecules from the viewpoint of singlet diradicaloids can provide us with a new aspect in understanding their molecular properties.
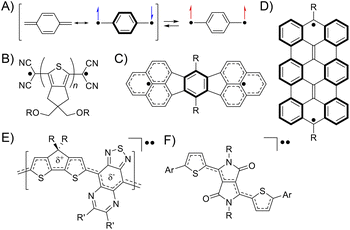 |
| | Fig. 1 The electronic structure of quinodimethane drawn with the resonance of closed-shell and open-shell structures (A). Typical examples of diradicaloids, quinoidal oligothiophenes (B),7a quinoidal polycyclic aromatic hydrocarbons (C),4 and graphene fragments (D)7b illustrated in their open-shell resonance forms. A donor–accepter conjugated polymer (E)9 and diketopyrrolopyrrole derivatives (F)10 showing the intermediate open-shell character (A). | |
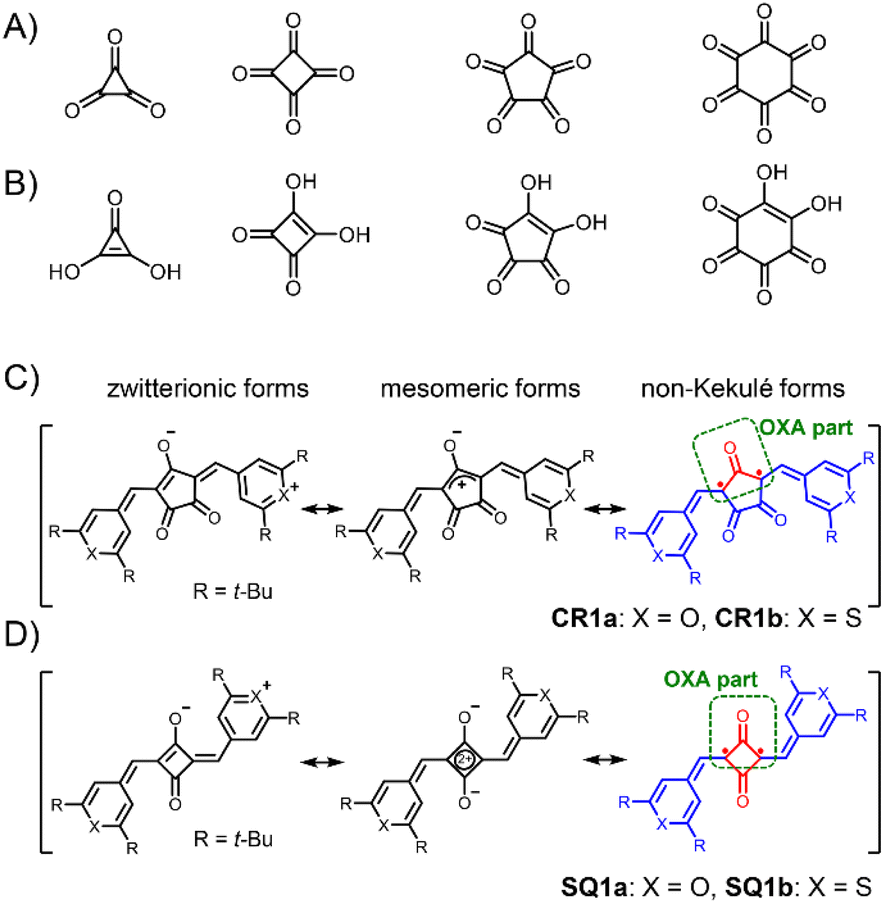
Oxocarbons (CO)n, cyclic polycarbonyls or oxa derivatives of radialenes, as shown in Fig. 2A, have attracted chemists' interest for more than a century because not only of their highly symmetric structure but also of their unique properties.11 The derivatives with four- and five-membered rings, squaric acid (n = 4) and croconic acid (n = 5), in which all of the carbon atoms are bonded to carbonyl and enol oxygen atoms (Fig. 2B),12 undergo dehydration reactions following conjugated addition with nucleophiles to give squaraine and croconaine dyes with π-conjugation chains in their two arms.13 They have been extensively studied and applied as functional dyes in various optoelectronic applications such as photoconductors, nonlinear optics, solar cells, and photodetectors.14–18 Some of the oxocarbon derivatives were known to show strong absorption at long wavelength as in the deep red or near infrared (NIR) region.19 Fabian et al. pointed out in the late 80s that the diradical nature of the compounds shifts the absorption to a longer wavelength by increasing diradical character and suggested that squaraines and croconaines have such a diradical nature represented with non-Kekulé structures (in red, Fig. 2C and D).20 Their diradical forms can also be viewed as oxyallyl (OXA) radicals, known to be the reaction intermediate of enduring interest.21 In spite of the pioneering work and the following contributions based on theoretical calculations,22 these dye molecules have been considered and treated as closed-shell species because no experimental evidence has been presented, to the best of our knowledge. In this paper, we shed light on the diradical character of squaraine and croconaine dyes to obtain a deeper understanding of strong one-photon absorption (OPA) and TPA of squaraine and croconaine dyes, especially in the context of the relationship between NIR-absorption and the diradical nature. We synthesized squaraine and croconaine dyes with chalcogenopyrylium moieties for two variations of chalcogen atoms of X = O and S with different electron affinities. Resonance structures of these four dyes formally can be drawn as zwitterionic, mesomeric, and non-Kekulé forms (Fig. 2C and D). The singlet diradical nature is expected from the contribution of the non-Kekulé forms. From the results of 1H-NMR, ESR, X-ray crystallography, and DFT calculations, we clarified that these dyes can be regarded as singlet diradicaloids, which supports the theoretical prediction. The influences of the number of the ring member (n = 4, 5) and chalcogen atoms (X = O, S) in the terminal groups on the absorption and diradical character are also investigated. The characteristic strong NIR OPA peaks with different wavelengths were observed. The TPA spectra showed a systematic correspondence with the OPA peaks depending on the symmetry selection rule and its break by vibronic coupling. With the spectroscopic information of OPA, TPA, and ESR, diradical character y0 of the dye molecules is experimentally determined, supporting the intermediate diradical nature of these molecules.
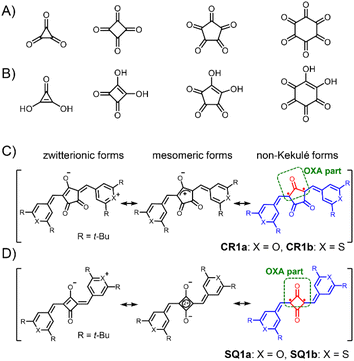 |
| | Fig. 2 Neutral oxocarbons (CO)n (A). Conjugated acids of oxocarbon bianions (CO)n2− (B). General resonance structures including zwitterionic, mesomeric, and non-Kekulé forms for the present croconaine dyes (CR1a–b, C) and squaraine dyes (SQ1a–b, D). Non-Kekulé diradical parts are shown in red. Oxyallyl (OXA) parts are shown in a green round box. | |
Results and discussion
Synthesis and NMR measurements
Croconaine dyes with pyrylium (X = O, CR1a) and thiopyrylium components (X = S, CR1b) were synthesized by condensation of the corresponding chalcogenopyrylium salts with croconic acid (see ESI†). Squaraine dyes with pyrylium (SQ1a) and thiopyrylium (SQ1b) were also prepared by the same method.23 They are stable in the solid state even at high temperature, and also stable in solution under dark conditions (Fig. S1†), and therefore allowed us to measure reliable 1H-NMR, ESR, and electronic absorption spectra as well as magnetometry using a superconducting quantum interference device (SQUID). However, CR1a–b and SQ1a gradually decomposed in solution in ambient air and light with half-life values in the range from 9 to 61 h, although SQ1b did not decompose at all (Fig. S2†).
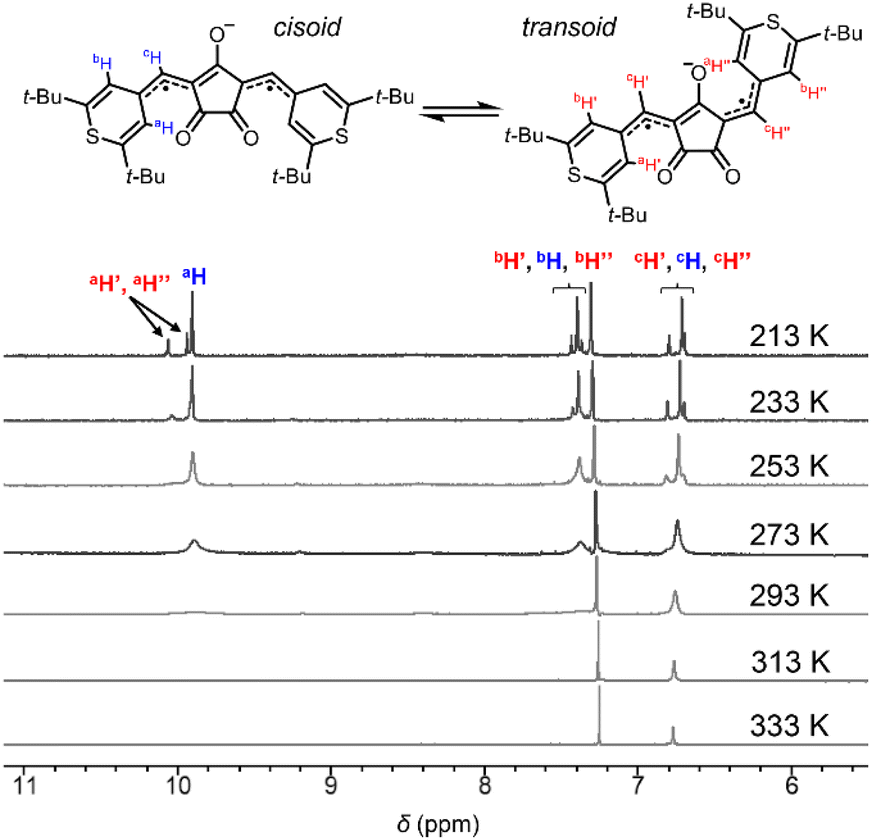
Singlet diradicaloids have smaller excitation energy;20 thus singlet–triplet energy gaps (ΔES–T) between the singlet ground state and the triplet state become smaller and may allow thermal excitation to the triplet state from the singlet ground state. Due to the thermally populated triplet species, singlet diradicaloids generally exhibited unique temperature dependence in the 1H-NMR spectrum.7b–e It is interesting to note that at 293 K or higher, no proton signals were observed for CR1a–b in the aromatic region (7.0–11.0 ppm) while broad signals attributed to a methine proton (6.4–6.8 ppm) were observed (Fig. 3, S3 and S4†). The broad signals that appeared in the aromatic region became more intense and sharper with decreasing temperature. At 213 K, only sharp signals attributed to cisoid and transoid conformers were observed for CR1a–b. In the same manner, SQ1a–b showed distinct proton signals attributed to cisoid and transoid conformers below 253 K. The signals in the aromatic region (6.0–10.0 ppm) became broader at 273 K and higher in CDCl3 (Fig. S5 and S7†), and then disappeared at a temperature over 403 K in DMSO-d6 (Fig. S6 and S8†). We confirmed that the disappeared proton signals were recovered with a decrease in temperature. The temperature dependence in 1H-NMR was similar to that in diradicaloids with thermally accessible triplet states.7 The temperature at which the signal broadening starts is 383 K for SQ1a, 293 K for SQ1b, 293 K for CR1a, and 253 K for CR1b (Fig. S3–S8†), so ΔES–T is considered to become smaller in this order.
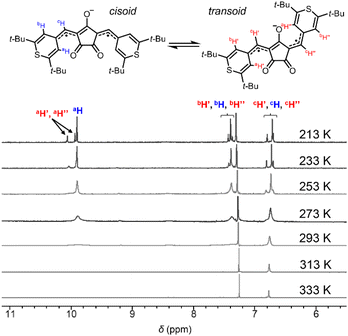 |
| | Fig. 3 Variable temperature 1H-NMR spectra (CDCl3, 213–333 K) of CR1b. | |
ESR and SQUID measurements
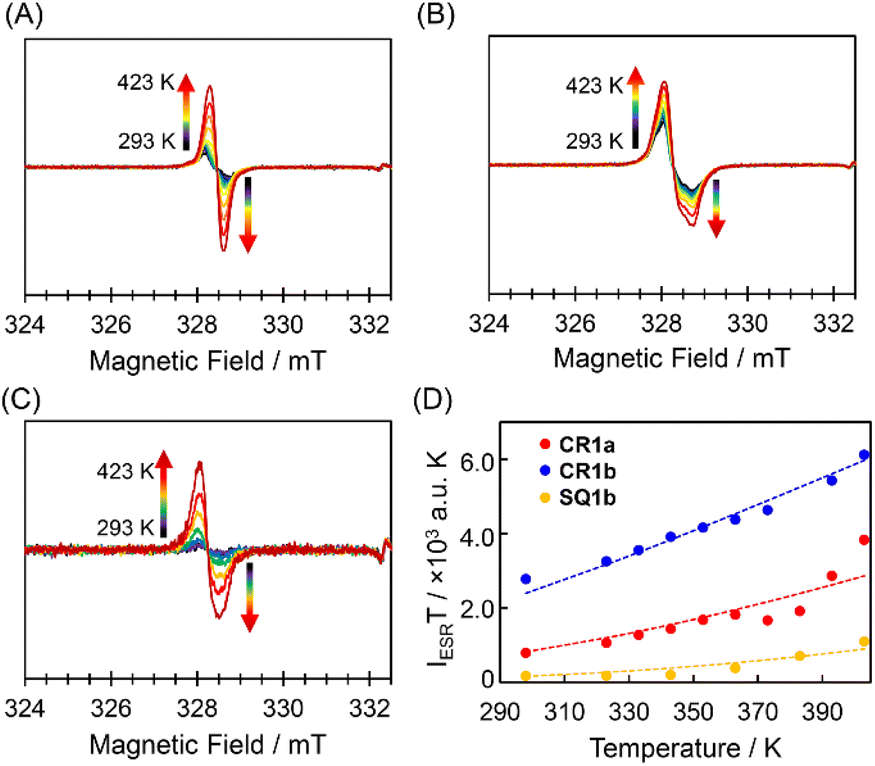
The triplet species of these dyes were also observed by ESR spectroscopy (Fig. 4). Broad ESR signals were observed at g = 2.003 for CR1a, g = 2.006 for CR1b, and g = 2.005 for SQ1b. The product of ESR intensity (IESR) and temperature (T) decreased with a decrease in temperature. The careful curve fitting of the temperature-dependent IESRT values by using the Bleaney–Bowers equation (see Section 4 in ESI†)24 gave ΔES–T of 0.170, 0.129, and 0.100 eV for SQ1b, CR1a, and CR1b, respectively (Fig. 4D). Thus, ESR studies can be considered experimental indications of the presence of thermally populated triplet species CR1b, CR1a, and SQ1b. For SQ1a, the Bleaney–Bowers analysis could not be adapted because of its very weak ESR signals. The signal appeared above 383 K, showing that the thermally excited triplet state was generated at this temperature or higher (Fig. S9†). The signal intensity was much weaker than those of the other three dyes, suggesting that SQ1a has larger ΔES–T than the others.
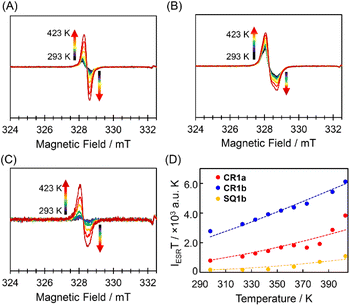 |
| | Fig. 4 ESR spectra of the powder samples CR1a (A, g = 2.003), CR1b (B, g = 2.006), and SQ1b (C, g = 2.005) and temperature–IESRT plots based on ESR data of CR1a (red), CR1b (blue), and SQ1b (orange) (D); the dotted lines are the fitted plots by using the Bleaney–Bowers equation. | |
Furthermore, SQUID measurements were carried out for the solid sample of the present dyes (Fig. S10†). Similar to the variable-temperature (VT)-ESR data, the product of the molar magnetic susceptibility (χM) and temperature (T) for CR1a, CR1b, SQ1a, and SQ1b increased with an increase in temperature. From the fitting of the χMT plots with the Bleaney–Bowers equation, the ΔES–T was given as 0.179 eV (2080 K) for SQ1a, followed by 0.163 eV (1890 K) for SQ1b, 0.13 eV (1500 K) for CR1a, and 0.107 eV (1240 K) for CR1b (Fig. S10†). The temperature dependence observed in 1H-NMR and ESR, and SQUID magnetometry proved that the present dyes have sufficiently small ΔES–T enabling thermal excitation from the singlet ground state to the triplet state. Thus the small ΔES–T value is associated with the diminishment of the covalency of π-bonds and hence suggests an increasing degree of the diradical character for the present dyes.
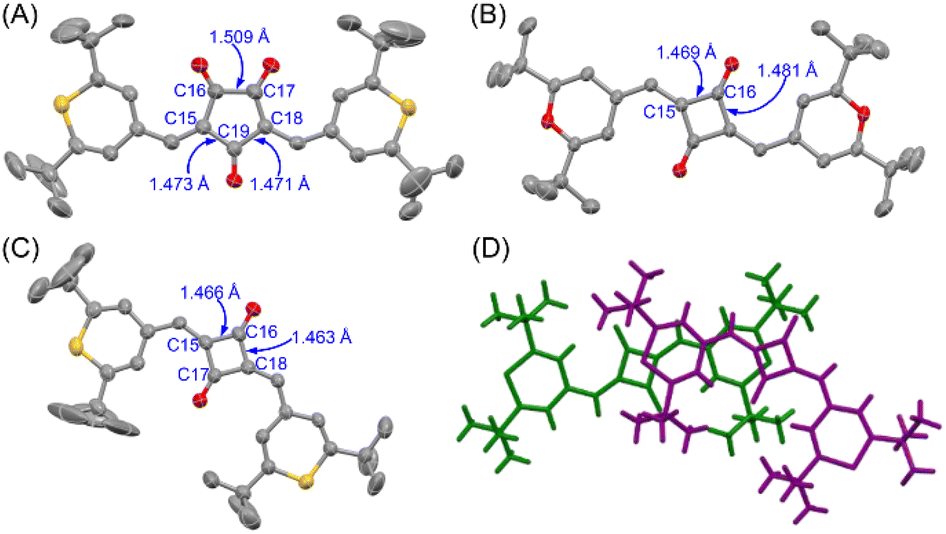
X-ray structures
To obtain structural information, X-ray crystallographic analysis was carried out for the single crystals of CR1b, SQ1a, and SQ1b recrystallized from CHCl3/hexane solution (Fig. 5, S11, Tables S1 and S2†). CR1b adopts a highly planar cis conformation. The C–C bond distances of the underside of the oxyallyl ring shown in Fig. 5A were close to the C(sp2)–C(sp2) bond length (C15–C19, C18–C19: 1.47 Å), whereas the C–C bond distance of the upper side of the oxyallyl ring was close to the C(sp3)–C(sp3) bond length (C16–C17; 1.51 Å). This indicates that the C–C bonds on the underside comprise the π-conjugated system. SQ1a adopts a trans conformation in the crystalline state. The C–C bond lengths at the cyclobutenedione component ranged from 1.46 to 1.49 Å, showing that the entire cyclobutenedione skeleton incorporates the π-conjugated system. Interestingly, the crystal of SQ1b with thiopyrylium components contained both the trans conformer and C2v-symmetric cis conformer. This substantiates the assignment of 1H-NMR spectra where two series of signals attributable to transoid and cisoid conformations were observed at low temperature.
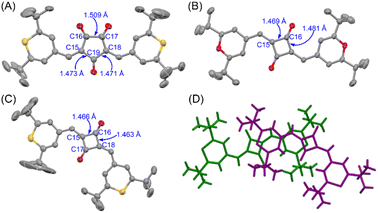 |
| | Fig. 5 Single crystal X-ray structures at 293 K and C–C bond lengths (in Å) of CR1b (A), SQ1a (B), and cisSQ1b (C) with thermal ellipsoids at 50% probability. Solvent molecules and hydrogen atoms are omitted for clarity. The packing structure of the transoid (green) and cisoid (purple) isomers of SQ1b (D). | |
Bond length alternation analysis
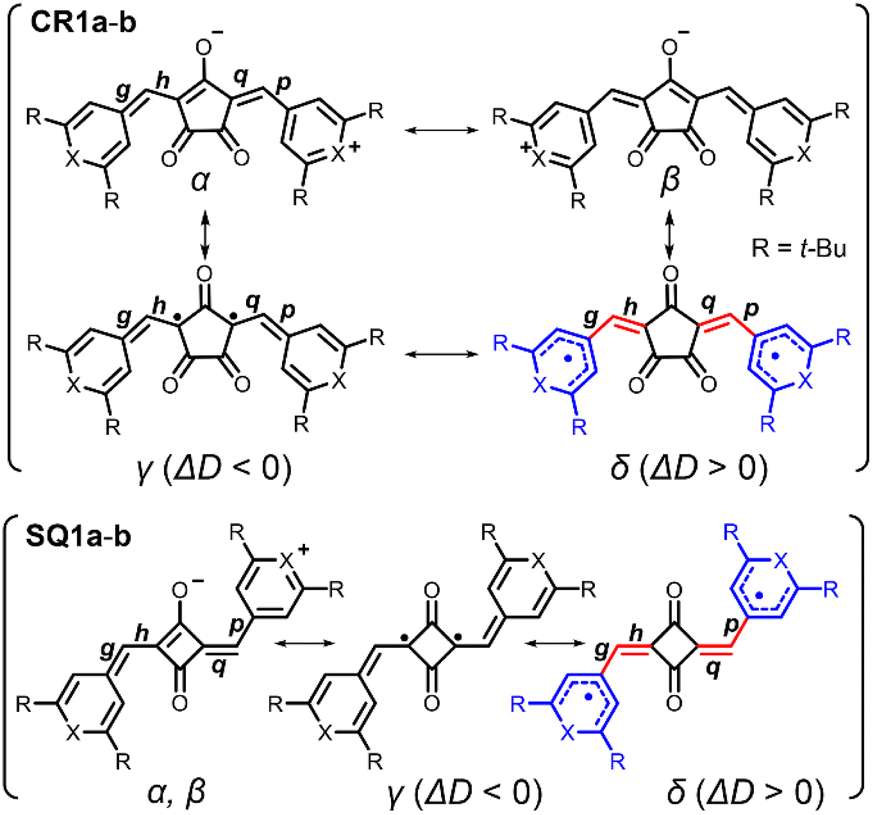
Bond-length alternation (BLA) analysis was employed to obtain deeper insights into the diradical character of the present dyes. Here we focus on the difference in the averaged bond length along the oxyallyl part, ΔD = ave(g, p) − ave(h, q), where ave(a, b) means the arithmetic average of the lengths of bonds a and b. Bonds g, p (outer) and h, q (inner) are shown in Fig. 6. For closed-shell structures (zwitterionic, α, and β forms in Fig. 6), ΔD = 0 because (g, p) and (h, q) are the pairs of double (short) and single (long) bonds and give the same value. For diradical structures (γ and δ forms), ΔD ≠ 0 since the outer (g, p) and inner (h, q) bonds are not equivalent. More precisely, ΔD < 0 for γ form and ΔD > 0 for δ form. This is because for the δ form, g and p (outer) are single bonds, h and q (inner) are double bonds (red-highlighted), and unpaired electrons were located on chalcogenopyrylium components (blue-highlighted). Opposite things occur for the γ form. We calculated ΔD from the bond lengths of the X-ray structures of CR1b, SQ1a, and SQ1b and found that ΔD > 0 for all dyes (Table 1). This suggests that the δ form makes a more significant contribution to their resonance structure than the γ form which we initially envisioned according to the prediction in the literature.20 This idea was supported by the full analysis of the resonance of diradical structures of CR1a–b, where the resonance is composed of nine δ forms with ΔD > 0, three γ forms with ΔD < 0, and twelve forms with ΔD = 0 (Fig. S12†). For SQ1a–b, the number of resonance forms with ΔD > 0 (form δ) is also larger than that with ΔD < 0 (Fig. S13†). From the perspective of the number of resonance forms, the present dyes have large contribution from the diradical δ form with ΔD > 0, that is, the outer bonds (g, p) are longer than the inner bonds (h, q).
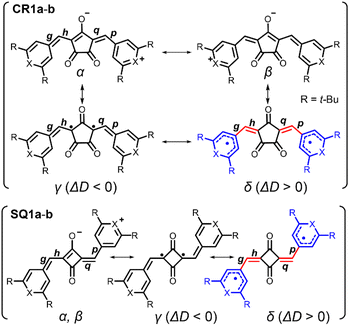 |
| | Fig. 6 Resonance structures of CR1a–b and SQ1a–b. Red-highlighted parts indicate methine parts (bonds g, h, p, and q) with bond length alternation. Blue-highlighted parts are chalcogenopyrylium rings in which unpaired electrons are delocalized. The difference between bond lengths at methine parts ΔD is obtained as follows; ΔD = (averaged length of g, p) − (averaged length of h, q). | |
Table 1 Differences between the average length of bonds g, p and average length of h, q, i.e., ΔD = ave(g, p) − ave(h, q), obtained from X-ray structures, optimized closed-shell singlet structures, and optimized open-shell singlet/triplet structures of CR1b and SQ1a–b. Here, ave(a, b) is defined as the averaged bond length of bonds a and b
|
|
ΔD/Å |
| X-raya |
Closed-shell singletb |
Open-shell singletc |
Tripletc |
|
Obtained from X-ray structures measured at 293 K.
Obtained from optimized closed-shell singlet structures calculated at CAM-RB3LYP/6-31G(d,p).
Obtained from optimized open-shell singlet and triplet structures calculated at CAM-UB3LYP/6-31G(d,p).
|
|
CR1b
|
0.025 |
0.0032 |
0.0241 |
0.0353 |
|
SQ1a
|
0.0029 |
−0.0123 |
−0.0027 |
0.0448 |
|
SQ1b (transoid) |
0.019 |
−0.007 |
0.0135 |
0.0577 |
| (Cisoid) |
0.025 |
−0.0074 |
0.0134 |
0.0576 |
The BLA analysis was also supported by the DFT calculations carried out at the CAM-(U)B3LYP/6-31G(d,p) level. For CR1b, the experimental ΔD value from the X-ray structure (0.0250 Å) was found to be between those for the DFT-optimized structures of the closed-shell singlet states (0.0032 Å) and the open-shell triplet states (0.0353 Å) (Tables 1 and S3†). This also supported the substantial contribution of the diradical resonance forms δ of these dyes. ΔD from X-ray structures is increased in the following order: SQ1a < SQ1b < CR1b.
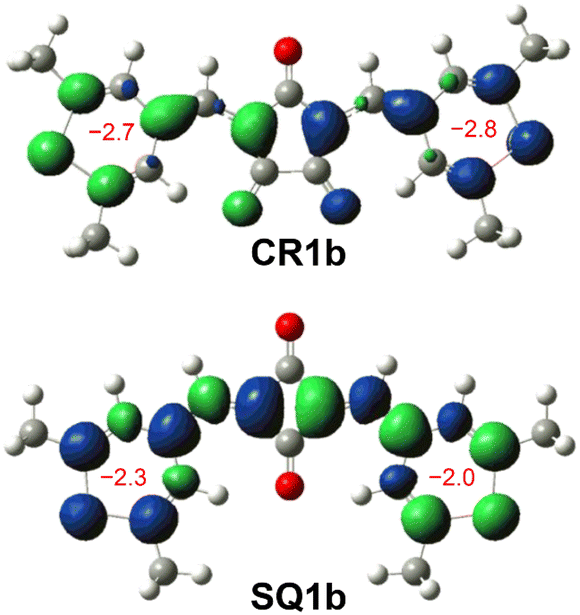
The BLA analysis of the chalcogenopyrylium components shows the contribution of the δ-form open-shell diradical in which unpaired electrons were delocalized over chalcogenopyrylium skeletons (Tables S3–S6†). Indeed, nucleus independent chemical shift (NICS) calculations (UB3LYP/6-31G*) predicted a slightly negative value (−2.8 to −2.0) for the chalcogenopyrylium rings (Fig. 7 and S14†), suggesting that they exhibit a non-aromatic nature and have pyran-like and thiopyran-like structures.25 Their spin densities at the open-shell singlet states calculated at the CAM-UB3LYP/6-31G(d,p) level are mainly distributed to the methine carbons as well as chalcogenopyrylium skeletons (Fig. S15†). This also supports the contribution of diradical δ-form.
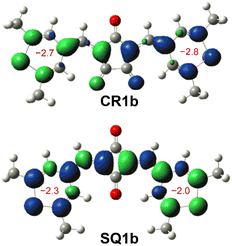 |
| | Fig. 7 Mulliken spin density maps of open-shell singlet states for CR1b (top) and cisoid SQ1b (bottom) calculated at the CAM-UBLYP/6-31G(d,p) level of theory with an isovalue of 0.002 (green: up spin; blue: down spin). NICS(1) values of thiopyrylium rings constituting CR1b and SQ1b calculated at GAIO UB3LYP/6-31G* are shown in red. | |
Correlation between band gaps and transition energy
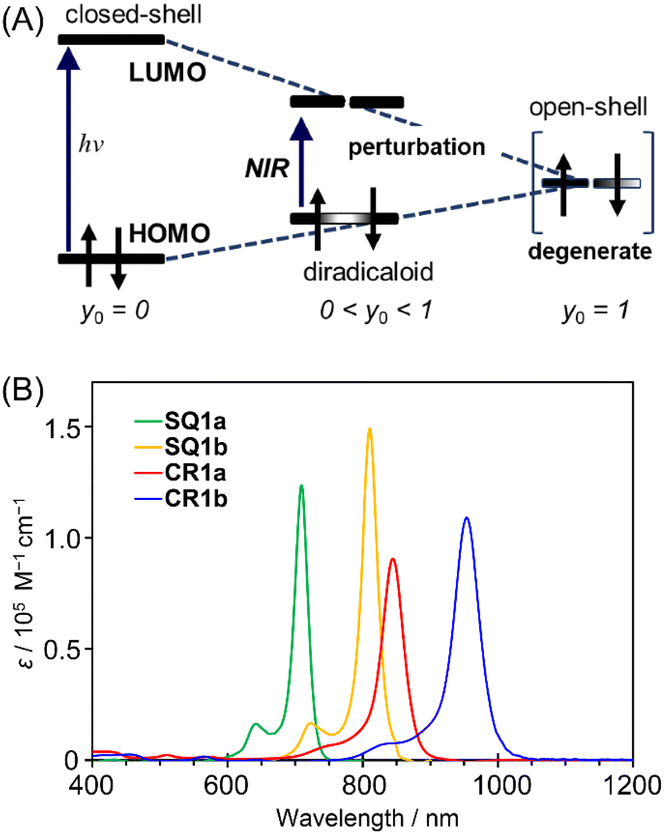
Diradical nature impacts not only the structure but also the energy level of a molecule. Based on a qualitative interpretation using simplistic perturbation principles, Fabian et al. stated that the energy gap between the HOMO and LUMO decreases; therefore the electronic transition energy decreases as the contribution of diradical nature increases (Fig. 8A).20 The intermediate electronic structures between the closed-shell and open-shell singlet (diradical) states, thus, help to realize the physical properties of functional dyes that have sufficiently reduced electronic transition energies corresponding to the NIR region.
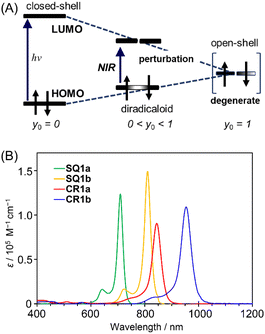 |
| | Fig. 8 (A) Relationship between transition energy and open-shell character. y0 indicates diradical character. Adapted from ref. 20 with some modifications. ©2003 John Wiley & Sons, Inc. (B) Electronic absorption spectra (CHCl3, c = 1 × 10−5 M) of CR1a (red), CR1b (blue), SQ1a (green), and SQ1b (orange). | |
HOMO–LUMO energy gaps were estimated from the oxidation and reduction potentials obtained by cyclic voltammetry of the dyes (Fig. S16†). As for open-shell singlet diradicaloids reported to date, CR1a–b and SQ1a–b have small bandgaps of <0.8 eV and <1.2 eV, respectively (Table S7†). The small HOMO–LUMO energy gaps are expected to lead the long wavelength electronic absorption spectra. Actually, CR1a and CR1b displayed intense NIR absorptions with maxima at 845 nm and 954 nm, respectively (Fig. 8B). SQ1a–b also exhibited sharp and intense absorptions on the higher energy side in comparison to croconaine dyes (710 nm for SQ1a and 810 nm for SQ1b).
A closer look at the absorption edges of CR1a and CR1b (Fig. S17†) shows broad shoulder peaks that exist at 900–1600 nm, which can be attributed to the low-lying double exciton states found in open-shell singlet diradicals.26 The absorption edges of SQ1a and SQ1b were somewhat broad and observed at around 800–1000 nm. The optical band gaps calculated from the onsets of absorption bands were roughly in agreement with the HOMO–LUMO gaps estimated by electrochemical measurements (Table S7†). The transition energy calculated at absorption maxima (λmax) decreases in the following order: SQ1a > SQ1b > CR1a > CR1b. This is in the same order as the decreasing ΔES–T values obtained from VT-ESR and SQUID measurements and is in the reverse order of their bond length differences at methine parts (ΔD) observed in the X-ray structure. Both ΔES–T and ΔD are correlated to the diradical character of the present dyes. Thus, according to the perturbation principle depicted in Fig. 8A, the contribution of diradical forms increased with a decrease in their transition energies.
Two-photon absorption
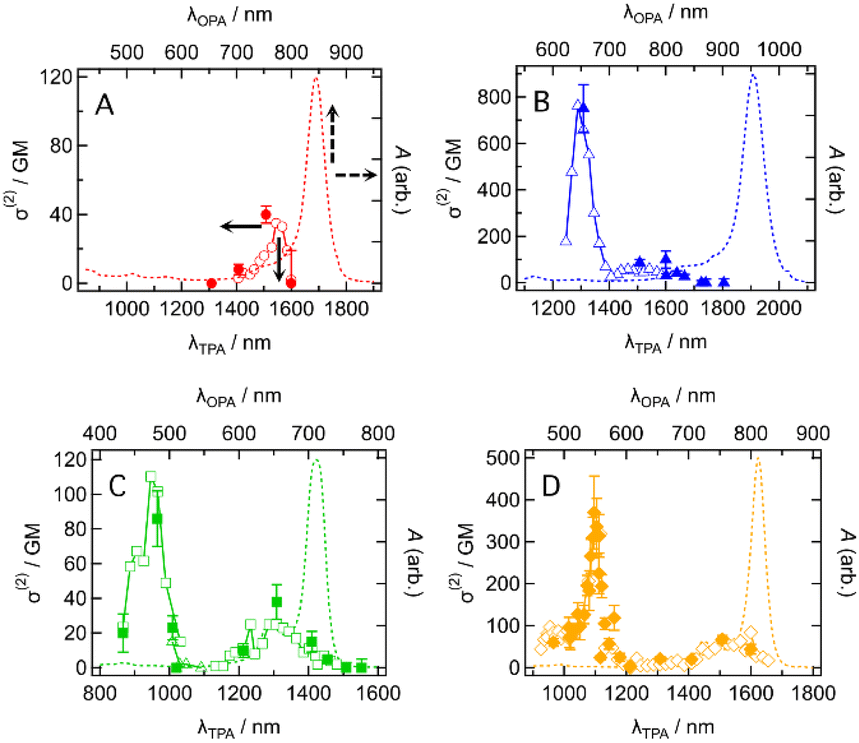
The intermediate open-shell character can also be understood as a diminishment of covalency at the π-bonds, allowing the electron distribution along the π-conjugated system to distort more easily. As a result, molecules with an intermediate open-shell character have an enhanced third-order nonlinear optical response and consequently two-photon absorption (TPA) activity.3,5 This motivated us to study the TPA properties of the present dyes. TPA spectra were measured by using the femtosecond Z-scan method (see the ESI†). Typical Z-scan traces are shown in Fig. S18–S21.† All dyes showed the TPA spectra in which a sharp and intense band was located at a shorter wavelength and a broad and weak band at longer wavelengths except for CR1a (Fig. 9). At wavelengths shorter than 1300 nm, CR1a showed clear saturable absorption (SA) of OPA (Fig. S22A†), which interfered with determining TPA cross-section σ(2). The other three dyes also showed SA (Fig. S22B–D†) but their wavelengths at which SA appeared were much shorter than that of CR1a. The sharp bands have relatively large σ(2) with values of 750 ± 100 GM (where 1 GM = 10−50 cm4 s molecule−1 photon−1) at 1310 nm for CR1b, 86 ± 15 GM at 965 nm for SQ1a, and 370 ± 90 GM at 1100 nm for SQ1b. The order is CR1b > SQ1b, > SQ1a. The croconaine dye has larger values than the squaraine dyes and the dye with a stronger electron-withdrawing group (X = S) has larger σ(2) than the dye with a weaker one (X = O) of the two squaraine dyes. The absolute values of these σ(2)s are not extraordinarily high; however, the peak σ(2) value of CR1b (750 GM at 1310 nm) was found to be of similar magnitude to that of the reported polymethine dye with a long π-conjugation chain and very strong acceptors (tricyanofuryl) at both ends (890 GM at 1590 nm, compound 2 in ref. 27). This fact suggests that the croconaine dye has unusually large σ(2) by taking account of the conjugation length and acceptor strength. The intermediate diradical character of the dyes may contribute to enhancing σ(2). In contrast to the sharp bands, the broad TPA bands were much weaker and in the order of tens of GMs (40 GM at around 1150 nm for CR1a, 60–100 GM at around 1400–1650 nm for CR1b, 30–40 GM at 1300 nm for SQ1a, and 70 GM at around 1500 nm for SQ1b). Interestingly, the transition energies of the broad band agreed with those of the 0–1 vibrational subpeaks of OPA. This correspondence suggests that the broad TPA bands can be assigned to partially allowed transitions because of vibronic coupling of the TPA-forbidden transition.28 The absence of such peaks in the simulated TPA spectra based on the quantum chemical calculations without considering the vibrational coupling (Fig. S23 and Section 14 in the ESI†) also supports this interpretation.
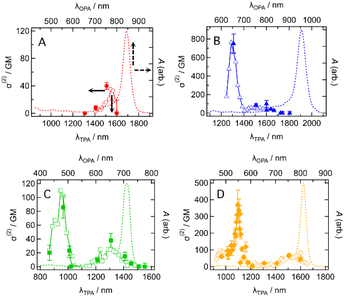 |
| | Fig. 9 Two-photon absorption (TPA) spectra in two-photon absorption cross section σ(2) (symbols, left and bottom axes) of (A) CR1a, (B) CR1b, (C) SQ1a, and (D) SQ1b in CHCl3 together with the corresponding one-photon absorption (OPA) spectra in absorbance A (dashed curves, right and top axes). λTPA and λOPA are the excitation wavelengths of TPA and OPA. They are plotted so that λTPA comes at the same horizontal position of 2 × λOPA, resulting in the same transition energies for TPA and OPA comes at the same horizontal position. Open symbols and the filled symbols with error bars mean that the σ(2) data were obtained by the different measurement procedures (wavelength scan and power scan, respectively; see the ESI† for details). | |
Estimation of diradical character y0
From the information on the peak position of TPA-allowed transition and ΔES–T by ESR measurements, diradical character y0 can be determined experimentally by using the relation,29| | 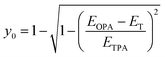 | (1) |
where EOPA is the energy level of the OPA-allowed excited state obtained from the OPA peak, ETPA is the energy level of the TPA-allowed excited state obtained from the sharp TPA peak, and ET is the energy level of the triplet state, i.e., the singlet–triplet energy-gap itself (=ΔES–T). The obtained y0 values are around 0.2 as listed in Table 2 and fall in the range of intermediate diradical character. The experimental y0 values obtained using eqn (1) are known to be approximately half the values of the theoretical calculations based on the occupation number of the natural orbitals.30 In fact, SQ1b falls on the correlation line perfectly and CR1b deviates more or less but is close to the correlation line in the plots of experimental y0 values with theoretical y0 values obtained from the literature (Fig. S24 and Table S9†).22a These facts also support that the croconaine and squaraine dyes have singlet diradical character. The number of reported data points of the experimental y0 is still limited, so these results are important to accumulate for a better understanding of the correlation.
Table 2 Excitation energy at one- and two-photon absorptions, energy of the triplet state and experimental y-values derived from eqn (1)
| Compd |
E
opa
(eV) |
E
T
(eV) |
E
TPA
(eV) |
y
0,exp
|
y
0,calc
|
|
Transition energy of the one-photon-allowed excited state from the one-photon absorption peak.
Energy level of the triplet state based on ESR measurements.
Energy level of the triplet state based on SQUID magnetometry measurements.
Transition energy of two-photon-allowed excited state. The value for CR1a is not available because the peak was not observed by saturable absorption.
Diradical character calculated using eqn (1).
Diradical character obtained by quantum chemical calculations in ref. 22a.
|
|
CR1a
|
1.46 |
0.129 |
NA |
NA |
0.63 |
|
CR1b
|
1.30 |
0.100 |
1.92 |
0.220 |
0.70 |
|
SQ1a
|
1.74 |
0.179c |
2.62 |
0.197 |
0.35 |
|
SQ1b
|
1.53 |
0.170 |
2.26 |
0.201 |
0.44 |
Conclusions
Four- and five-membered oxocarbon derivatives, croconaine dyes (CR1a–b) and squaraine dyes (SQ1a–b) bearing chalcogenopyrylium moieties with O and S chalcogen atoms were employed to demonstrate the intermediate open-shell character. The present dyes exhibit unique temperature-dependent magnetic properties in 1H-NMR, ESR spectra, and SQUID magnetometry. The singlet–triplet energy gaps (ΔES–T) estimated from the temperature dependence in the ESR study are small enough to allow thermal excitation to triplet states. Thus, the magnetic properties prove that they are intermediate open-shell singlet molecules with thermally accessible triplet states. The bond length analysis by using the difference in the averaged bond length along the oxyallyl part, ΔD, combined with theoretical calculations also indicates that the diradical form in which unpaired electrons were located on chalcogenopyrylium components contributes to the intermediate diradical nature. These dyes exhibited prominent NIR one-photon absorption. The transition energy is correlated to the ΔD and the ΔES–T values, indicating that the contribution of diradical forms for these dyes causes a decrease in their transition energy as predicted by the perturbation principle. The present dyes exhibit sharp and substantial two-photon absorption owing to a diminishment of covalency at the π bonds. We also successfully determined their diradical characters y0 by using the experimental values of the OPA and TPA state energies.
Croconaine and squaraine dyes have been recognized as closed-shell molecules so far, although they include oxyallyl diradical structures at oxocarbon units. This result not only clarifies that the present oxocarbon derivatives are a novel class of stable diradicaloids but also provides new insight by incorporating the concept of the intermediate open-shell character into the understanding of molecular properties in NIR absorbing functional dyes that have been recognized as covalent and closed-shell molecules. It might be necessary to revisit the theoretical framework regarding design, synthesis, structure, stability, optical properties, and magnetic properties of croconaine dyes and squaraine dyes from the viewpoint of singlet diradicaloids.
Data availability
All data associated with this article are available in the main text and ESI.†
Author contributions
T. M. and T. O. designed experiments, conducted synthesis of compounds, analysed the data, and wrote the manuscript. D. S. and H. F. conducted X-ray crystallographic analysis, collected ESR spectra, and analysed the data of ESR and SQUID measurements. N. S. and S. Y. conducted characterization of compounds. T. K. and K. K. collected and analysed OPA/TPA spectra. All authors reviewed the manuscript and agreed to its publication.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors sincerely thank Prof. Ryohei Kishi, Osaka University for helpful discussions about theoretical calculations. This work was supported by JSPS KAKENHI Grant Numbers JP16H06048, JP21K05215 (TM), and JP21H01887 (KK), the ENEOS Tonen General Research/Development Encouragement & Scholarship Foundation, and The Futaba Foundation (TM).
Notes and references
- For a review on diradicals, see M. Abe, Chem. Rev., 2013, 113, 7011–7088 CrossRef CAS PubMed.
- K. Yamaguchi, Chem. Phys. Lett., 1975, 33, 330–335 CrossRef CAS.
- N. Nakano, Chem. Rec., 2017, 17, 27–62 CrossRef PubMed.
- T. Kubo, A. Shimizu, M. Sakamoto, M. Uruichi, K. Yakushi, M. Nakano, D. Shiomi, K. Sato, T. Takui, Y. Morita and K. Nakasuji, Angew. Chem., Int. Ed., 2005, 44, 6564–6568 CrossRef CAS PubMed.
-
(a) K. Kamada, K. Ohta, T. Kubo, A. Shimizu, Y. Morita, K. Nakasuji, R. Kishi, S. Ohta, S.-i. Furukawa, H. Takahashi and M. Nakano, Angew. Chem., Int. Ed., 2007, 46, 3544–3546 CrossRef CAS PubMed;
(b) K. Kamada, S. Fuku-en, S. Minamide, K. Ohta, R. Kishi, M. Nakano, H. Matsuzaki, H. Okamoto, H. Higashikawa, K. Inoue, S. Kojima and Y. Yamamoto, J. Am. Chem. Soc., 2013, 135, 232–241 CrossRef CAS PubMed;
(c) T. Y. Gopalakrishna, W. Zeng, X. Lu and J. Wu, Chem. Commun., 2018, 54, 2186–2199 RSC.
- K. S. Mayer, D. J. Adams, N. Eedugurala, M. M. Lockart, P. Mahalingavelar, L. Huang, L. A. Galuska, E. R. King, X. Gu, M. K. Bowman and J. D. Azoulay, Cell Rep. Phys. Sci., 2021, 2, 100467 CrossRef CAS.
-
(a) T. Takahashi, K. Matsuoka, K. Takimiya, T. Otsubo and Y. Aso, J. Am. Chem. Soc., 2005, 127, 8928–8929 CrossRef CAS PubMed;
(b) A. Konishi, Y. Hirao, M. Nakano, A. Shimizu, E. Botek, B. Champagne, D. Shiomi, K. Sato, T. Takui, K. Matsumoto, H. Kurata and T. Kubo, J. Am. Chem. Soc., 2010, 132, 11021–11023 CrossRef CAS PubMed;
(c) A. Shimizu, R. Kishi, M. Nakano, D. Shiomi, K. Sato, T. Takui, I. Hisaki, M. Miyata and Y. Tobe, Angew. Chem., Int. Ed., 2013, 52, 6076–6079 CrossRef CAS PubMed;
(d) G. Li, Y. Han, Y. Zou, J. J. C. Lee, Y. Ni and J. Wu, Angew. Chem., Int. Ed., 2019, 58, 14319–14326 CrossRef CAS PubMed;
(e) H. Hayashi, J. E. Barker, A. C. Valdivia, R. Kishi, S. N. MacMillan, C. J. Gómez-García, H. Miyauchi, Y. Nakamura, M. Nakano, S.-I. Kato, M. M. Haley and J. Casado, J. Am. Chem. Soc., 2020, 142, 20444–20455 CrossRef CAS PubMed;
(f) J.-J. Shen, Y. Han, S. Dong, H. Phan, T. S. Herng, T. Xu, J. Ding and C. Chi, Angew. Chem., Int. Ed., 2021, 60, 4464–4469 CrossRef CAS PubMed.
-
(a) M. Bendikov, H. M. Duong, K. Starkey, K. N. Houk, E. A. Carter and F. Wudl, J. Am. Chem. Soc., 2004, 126, 7416–7417 CrossRef CAS PubMed;
(b) C.-N. Yeh and J.-D. Chai, Sci. Rep., 2016, 6, 30562 CrossRef CAS PubMed.
- L. Huang, N. Eedugurala, A. Benasco, S. Zhang, K. S. Mayer, D. J. Adams, B. Fowler, M. M. Lockart, M. Saghayezhian, H. Tahir, E. R. King, S. Morgan, M. K. Bowman, X. Gu and J. D. Azoulay, Adv. Funct. Mater., 2020, 30, 1909805 CrossRef CAS.
- Z. Chen, W. Li, M. A. Sabuj, Y. Li, W. Zhu, M. Zeng, C. S. Sarap, M. M. Huda, X. Qiao, X. Peng, D. Ma, Y. Ma, N. Rai and F. Huang, Nat. Commun., 2021, 12, 5889 CrossRef CAS PubMed.
- H. Hopf, Angew. Chem., Int. Ed., 2012, 124, 12111–12113 CrossRef.
-
(a)
R. West, in Oxocarbons, ed. R. West, Academic Press, Inc., New York, 1980, pp. 1–14 Search PubMed;
(b) R. West, H. Y. Niu, D. L. Powell and M. V. Evans, J. Am. Chem. Soc., 1960, 82, 6204–6205 CrossRef CAS;
(c) R. West and D. L. PoweIl, J. Am. Chem. Soc., 1963, 85, 2577–2579 CrossRef CAS;
(d) M. Ito and R. West, J. Am. Chem. Soc., 1963, 85, 2580–2584 CrossRef CAS.
- For a review on the quest for the synthesis of squaraine and croconaine dyes, see:
(a)
A. H. Schmidt, in Oxocarbons, ed. R. West, Academic Press, Inc., New York, 1980, pp. 185–231 Search PubMed;
(b) D. L. Lynch and D. G. Hamilton, Eur. J. Org. Chem., 2017, 3897–3911 CrossRef CAS;
(c) S. Sreejith, P. Carol, P. Chithra and A. Ajayaghosh, J. Mater. Chem., 2008, 18, 264–274 RSC;
(d) L. Beverina and P. Salice, Eur. J. Org. Chem., 2010, 1207–1225 CrossRef CAS.
- K.-Y. Law, Chem. Rev., 1993, 93, 449–486 CrossRef CAS.
-
(a) H.-J. Chang, M. V. Bondar, T. Liu, X. Liu, S. Singh, K. D. Belfield, A. Sheely, A. E. Masunov, D. J. Hagan and E. W. Van Stryland, ACS Omega, 2019, 4, 14669–14679 CrossRef CAS PubMed;
(b) S. Ohira, I. Rudra, K. Schmidt, S. Barlow, S. Chung, Q. Zhang, J. Matichak, S. R. Marder and J.-L. Brédas, Chem.–Eur. J., 2008, 14, 11082–11091 CrossRef CAS PubMed;
(c) S.-J. Chung, S. Zheng, T. Odani, L. Beverina, J. Fu, L. A. Padilha, A. Biesso, J. M. Hales, X. Zhan, K. Schmidt, A. Ye, E. Zojer, S. Barlow, D. J. Hagan, E. W. V. Stryland, Y. Yi, Z. Shuai, G. A. Pagani, J.-L. Brédas, J. W. Perry and S. R. Marder, J. Am. Chem. Soc., 2006, 128, 14444–14445 CrossRef CAS PubMed.
-
(a) G. Wei, S. Wang, K. Renshaw, M. E. Thompson and S. R. Forrest, ACS Nano, 2010, 4, 1924–1934 Search PubMed;
(b) F. Silvestri, M. D. Irwin, L. Beverina, A. Facchetti, G. A. Pagani and T. J. Marks, J. Am. Chem. Soc., 2008, 130, 17640–17641 CrossRef CAS PubMed;
(c) U. Mayerhöffer, K. Deing, K. Gruß, H. Braunschweig, K. Meerholz and F. Würthner, Angew. Chem., Int. Ed., 2009, 48, 8776–8779 CrossRef PubMed;
(d) G. Chen, H. Sasabe, T. Igarashi, Z. Hong and J. Kido, J. Mater. Chem. A, 2015, 3, 14517–14534 RSC.
- T. Maeda, A. Liess, A. Kudzus, A.-M. Krause, M. Stolte, H. Amitani, S. Yagi, H. Fujiwara and F. Würthner, Chem. Commun., 2020, 56, 9890–9893 RSC.
- G. M. Somashekharappa, C. Govind, V. Pulikodan, M. Paul, M. A. G. Namboothiry, S. Das and V. Karunakaran, J. Phys. Chem. C, 2020, 124, 21730–21739 CrossRef CAS.
- T. Maeda, S. Nitta, H. Nakao, S. Yagi and H. Nakazumi, J. Phys. Chem. C, 2014, 118, 16618–16625 CrossRef CAS.
- J. Fabian and R. Zahradník, Angew. Chem., 1989, 101, 693–710 (
Angew. Chem., Int. Ed. Engl.
, 1989
, 28
, 677–694
) CrossRef CAS.
-
(a) R. Gleiter and R. Hoffmann, Angew. Chem., Int. Ed. Engl., 1969, 8, 214–215 CrossRef CAS;
(b) T. Ichiro, S. M. Villano, A. J. Gianola, D. J. Goebbert, L. Velarde, A. Sanov, S. J. Blanksby, X. Zhou, D. A. Hrovat, W. Thatcher Borden and W. C. Lineberger, Angew. Chem., Int. Ed., 2009, 48, 8509–8511 CrossRef PubMed;
(c) B. M. Trost, Z. Huang and G. M. Murhade, Science, 2018, 362, 564–568 CrossRef CAS PubMed.
-
(a) D. López-Carballeira, D. Casanova and F. Ruipérez, ChemPhysChem, 2018, 19, 2224–2233 CrossRef PubMed;
(b) A. L. Puyad, G. K. Chaitanya, A. Thomas, M. Paramasivam and K. Bhanuprakash, J. Phys. Org. Chem., 2012, 26, 37–46 CrossRef;
(c) K. Srinivas, C. Prabhakar, C. L. Devi, K. Yesudas, K. Bhanuprakash and V. J. Rao, J. Phys. Chem. A, 2007, 111, 3378–3386 CrossRef CAS PubMed.
- The synthesis of the present dyes was reported in the following reference in which 13C-NMR data of dyes were not shown although compounds were characterized by MS, elemental analysis, and 1H-NMR. In the section on 1H-NMR data, it was mentioned that all proton signals in the aromatic region were broad. T. P. Simard, J. H. Yu, J. M. Zebrowski-Young, N. F. Haley and M. R. Detty, J. Org. Chem., 2000, 65, 2236–2238 CrossRef CAS PubMed.
- B. Bleaney and K. D. Bowers, Proc. R. Soc. London, Ser. A, 1952, 214, 451–465 CAS.
- P. von Ragué Schleyer, C. Maerker, A. Dransfeld, H. J. Jiao and N. J. R. von Eikema Hommes, J. Am. Chem. Soc., 1996, 118, 6317–6318 CrossRef PubMed.
- S. Di Motta, F. Negri, D. Fazzi, C. Castiglioni and E. V. Canesi, J. Phys. Chem. Lett., 2010, 1, 3334–3339 CrossRef CAS.
-
(a) Q. Bellier, N. S. Makarov, P.-A. Bouit, S. Rigaut, K. K. P. Feneyrou, G. Berginc, O. Maury, J. W. Perry and C. Andraud, Phys. Chem. Chem. Phys., 2012, 14, 15299–15307 RSC;
(b) M. Drobizhev, N. S. Makarov, S. E. Tillo, T. E. Hughes and A. Rebane, J. Phys. Chem. B, 2012, 116, 1736–1744 CrossRef CAS PubMed.
- M. Pawlicki, H. A. Collins, R. G. Denning and H. L. Anderson, Angew. Chem., Int. Ed., 2009, 48, 3244–3266 CrossRef CAS PubMed.
- K. Kamada, K. Ohta, A. Shimizu, T. Kubo, R. Kishi, H. Takahashi, E. Botek, B. Champagne and M. Nakano, J. Phys. Chem. Lett., 2010, 1, 937–940 CrossRef CAS.
-
K. Yamaguchi, in Self-Consistent Field: Theory and Applications, ed. R. Carbo and M. Klobukowski, Elsevier, Amsterdam, 1990, p. 727 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures and experimental details. CCDC 2194625 (SQ1a), 2194626 (SQ1b) and 2194627 (CR1b). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2sc06612b |
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Taishi
Oka
a,
Daisuke
Sakamaki
*a,
Taishi
Oka
a,
Daisuke
Sakamaki