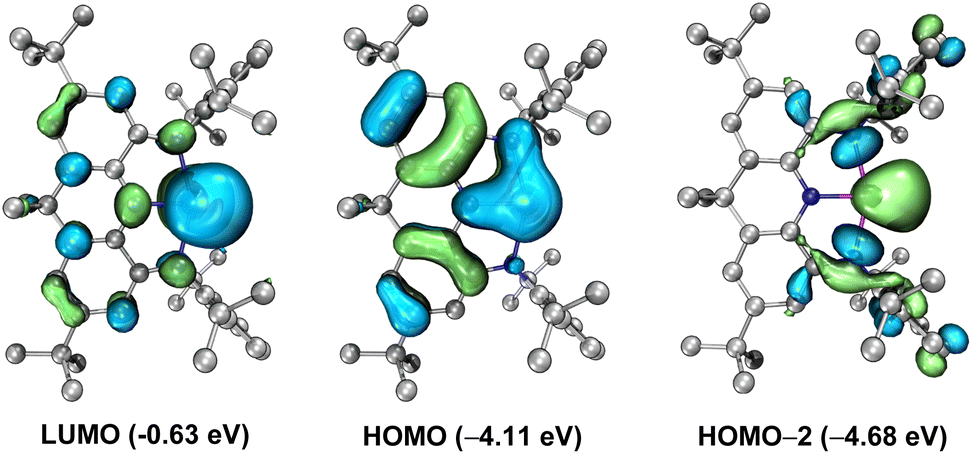DOI:
10.1039/D2SC07006E
(Edge Article)
Chem. Sci., 2023,
14, 5722-5727
A crystalline T-shaped planar group 14 anion†
Received
22nd December 2022
, Accepted 11th April 2023
First published on 3rd May 2023
Abstract
Isolable T-shaped planar pnictogen compounds R3Pn were reported more than three decades ago and have been attracting burgeoning interest in recent years; T-shaped planar group 14 anions, isoelectronic to R3Pn, however, are still unknown. Herein, we report the synthesis, full characterization, and reactivity of the first crystalline T-shaped planar group 14 anion 4 bearing a trinitrogen pincer ligand. DFT calculations indicate that the tricoordinate germanium center features both an unoccupied 4p orbital and two lone pairs of electrons. Its electron-rich nature allows for the nucleophilic attack on the methyl iodine giving methyl-substituted complex 5 and facile oxidation of the germanium center by elemental sulfur and selenium to furnish unpresented organic anions bearing terminal Ge![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Ch (Ch = S or Se) double bonds.
Ch (Ch = S or Se) double bonds.
Introduction
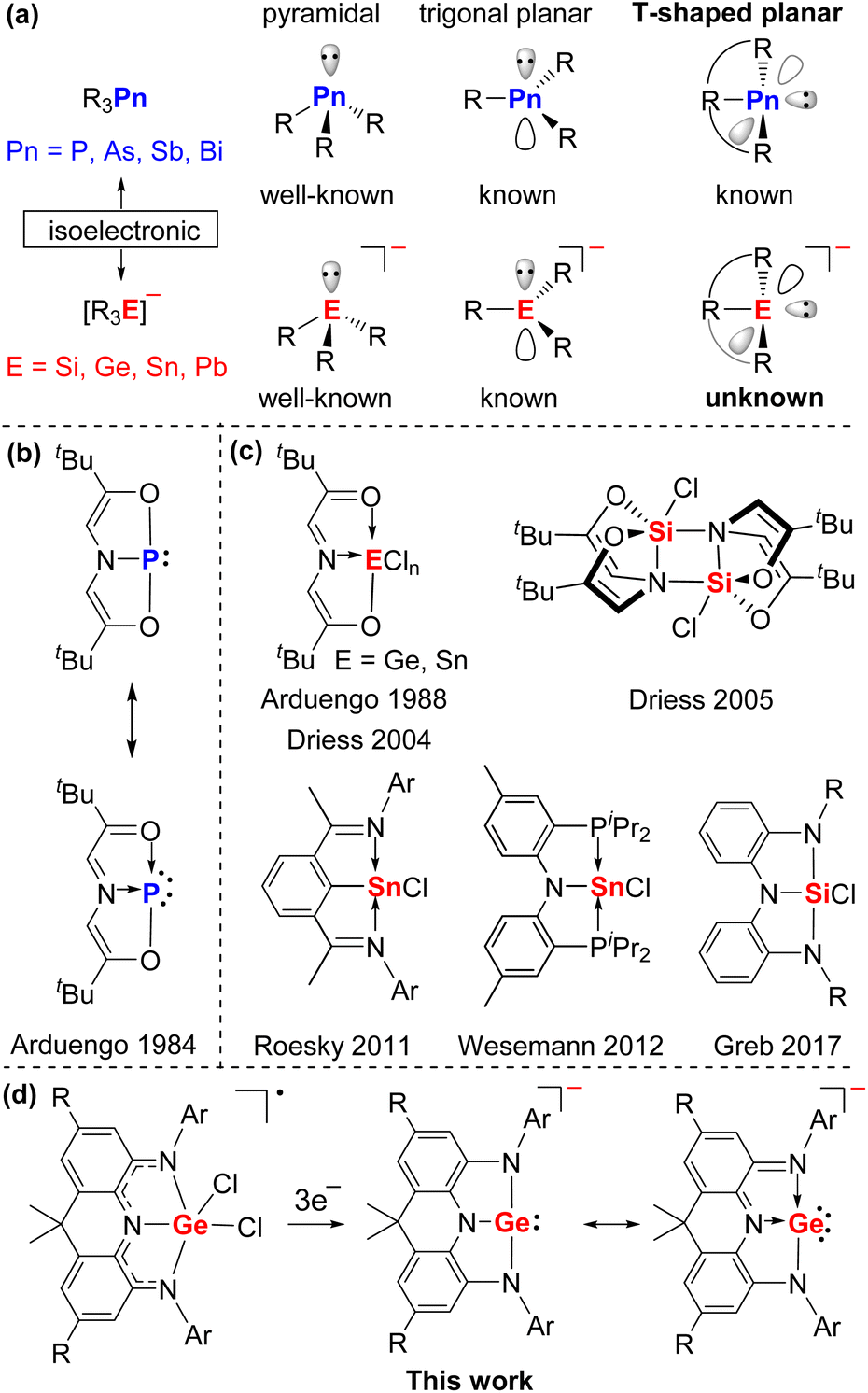
The heavier analogues of carboanions, [R3E]− (E = Si, Ge, Sn, Pb) are isoelectronic to the tricoordinate pnictogen compounds R3Pn (Pn = P, As, Sb, Bi). Both derivatives commonly feature a pyramidal or trigonal planar element center with a lone pair of electrons (Fig. 1a) and thus are widely used as nucleophiles or Lewis bases in organic synthesis and catalysis.1 Interestingly, in 1984 Arduengo and co-workers reported a novel T-shaped planar phosphorus compound bearing a geometrically constrained electron-rich ONO pincer ligand (Fig. 1b), which can transfer two electrons to the phosphorus center, leading to the unusual 10-electron 3-coordinated P atom (10-P-3) in the oxidation state of +1.2 Over decades, T-shaped planar group 15 compounds have attracted more and more attention owing to their peculiar electronic structures, fascinating reactivity, and potential applications in stoichiometric and even catalytic small molecule activation.3,4 Compared with the extensive studies on the geometrically constrained group 15 compounds, group 14 analogues are remarkably less investigated5,6 and T-shaped planar group 14 anions remain elusive despite initial calculations7 and great efforts on potential precursors by the groups of Arduengo, Driess, Roesky, Wessemann, and Greb (Fig. 1c).5 This mainly stems from the deficiency of suitable ligands and synthetic routes.
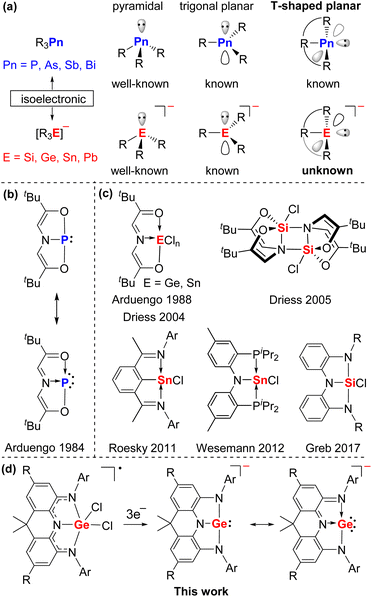 |
| | Fig. 1 (a) Molecular shapes of tricoordinate heavier group 14/15 compounds. (b) The first isolable T-shaped planar phosphorus compound. (c) Geometrically constrained group 14 precursors. (d) Present work. | |
Recently, we have reported a family of neutral and radical anionic T-shaped planar pnictogen species stabilized by a geometrically constrained and sterically bulky NNN pincer ligand 1 (Scheme 1).3i Herein, we present the successful implementation of this strategy into group 14 chemistry with the isolation, characterization, and reactivity of the first crystalline T-shaped planar group 14 anion salt (Fig. 1d).
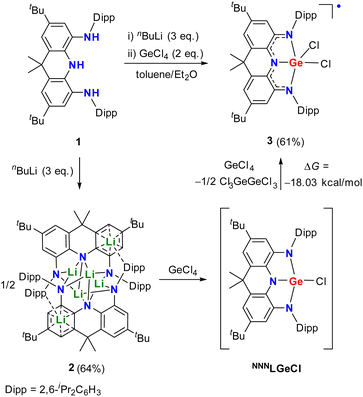 |
| | Scheme 1 Synthesis of the germanium precursor 3. | |
Results and discussion
Synthesis and characterization of the germanium precursor
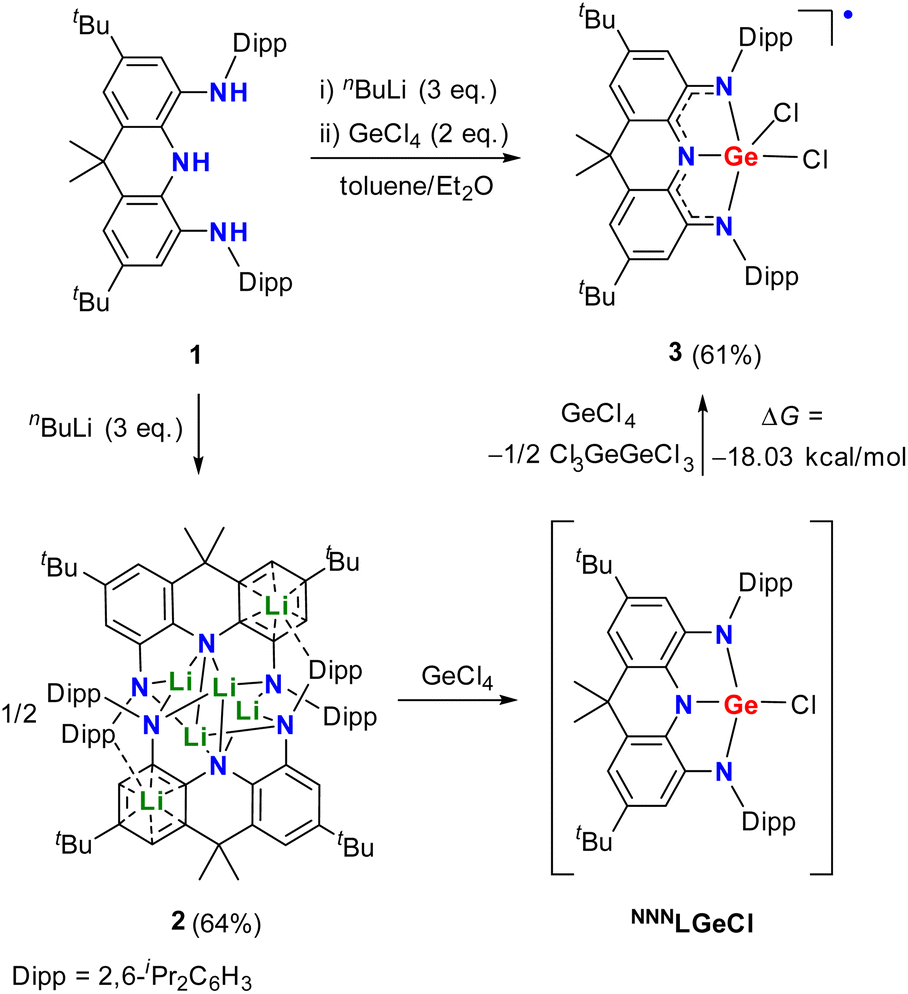
Reaction of the triamine 1 with three equivalents of nBuLi in toluene afforded the dimeric lithium salt 2 (Scheme 1 and Fig. 2a). Treatment of half equivalent of 2 with one equivalent of GeCl4 in diethyl ether resulted in a deep purple solution. After workup, dichloride complex 3 rather than the monochloride compound NNNLGeCl was unexpectedly isolated as a purple powder in 35% yield. It is proposed that the salt elimination between 2 and GeCl4 in 0.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio initially affords the intermediate NNNLGeCl, which is oxidized by another one equivalent of GeCl4 with formation of 3 and Cl3GeGeCl3 (Scheme 1). DFT calculations at the (U)B3LYP-D3BJ/6-311G(d) level demonstrated that the oxidation reaction occurs reasonably according to the negative Gibbs free energy change (ΔG) value of −12.39 kcal mol−1. Thus, direct addition of two equivalents of GeCl4 to the diethyl ether solution of 2 was carried out and 3 was isolated in a higher yield (61%). The EPR spectrum of 3 at room temperature displays a broad featureless signal centered at g = 1.9994 (Fig. S24†), supporting its radical character. Calculated spin density of 3 at the UB3LYP-D3BJ/6-311G(d) level mainly resides on the nitrogen atoms and the annulated phenyl rings with a small contribution from the germanium nucleus (Fig. S29†).
:1 ratio initially affords the intermediate NNNLGeCl, which is oxidized by another one equivalent of GeCl4 with formation of 3 and Cl3GeGeCl3 (Scheme 1). DFT calculations at the (U)B3LYP-D3BJ/6-311G(d) level demonstrated that the oxidation reaction occurs reasonably according to the negative Gibbs free energy change (ΔG) value of −12.39 kcal mol−1. Thus, direct addition of two equivalents of GeCl4 to the diethyl ether solution of 2 was carried out and 3 was isolated in a higher yield (61%). The EPR spectrum of 3 at room temperature displays a broad featureless signal centered at g = 1.9994 (Fig. S24†), supporting its radical character. Calculated spin density of 3 at the UB3LYP-D3BJ/6-311G(d) level mainly resides on the nitrogen atoms and the annulated phenyl rings with a small contribution from the germanium nucleus (Fig. S29†).
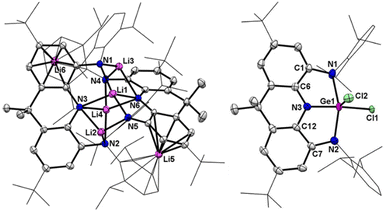 |
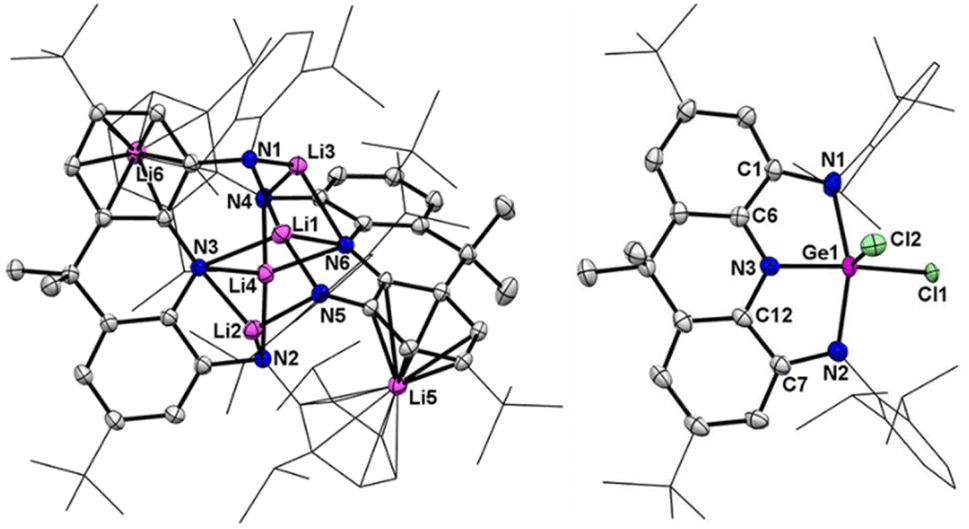
| | Fig. 2 Left: Solid-state structure of 2. Right: solid-state structure of 3. The tBu and Dipp groups are simplified as wireframes and hydrogen atoms are omitted for clarity. Thermal ellipsoids are set at the 30% probability level. Selected bond lengths (Å) and angles (°) for 2: Li1–N1 1.890(4), Li1–N3 2.254(4), Li1–N5 1.903(4), Li1–N6 2.236(4), Li2–N2 2.013(5), Li2–N3 2.133(4), Li2–N5 1.992(4), Li3–N1 1.978(4), Li3–N4 2.000(4), Li3–N6 2.157(4), Li4–N2 2.030(4), Li4–N3 2.092(4), Li4–N4 2.038(4), Li4–N6 2.073(4). Selected bond lengths (Å) and angles (°) for 3: Ge1–Cl1 2.091(2), Ge1–Cl2 2.163(2), Ge1–N1 2.011(2), Ge1–N2 2.039(2), Ge1–N3 1.872(2), C1–N1 1.387(3), C7–N2 1.379(3), C6–N3 1.349(3), C12–N3 1.362(3), C1–C6 1.404(3), C7–C12 1.402(3), N1–Ge1–N2 153.22(10), N1–Ge1–N3 80.34(8), N2–Ge1–N3 79.59(9), Cl1–Ge1–Cl2 104.09(10). | |
Crystals of 3 suitable for X-ray crystallographic studies were obtained from its saturated hexane solution. The molecular structure of 3 reveals a square pyramidal coordination of Ge1 by the basal N1, N2, N3, Cl2 atoms and the axial Cl2 atom (Fig. 2b). The Ge1–Cl1 bond (2.091(2) Å) is comparable to the Ge1–Cl2 bond (2.163(2) Å), while the Ge1–N1 (2.011(2) Å) and Ge1–N2 (2.039(3) Å) bond lengths are significantly longer than the Ge1–N3 bond distance (1.872(2) Å). The C–N (1.349(3)–1.387(3) Å) and C–C bond distances (1.404(3) and 1.402(3) Å) in two C2N2Ge rings are comparable to those of transition metal complexes featuring the NNN ligand in a diamido imino form.8 The UV-vis spectrum of 3 exhibits two absorption bands at 400 and 580 nm, the latter of which is also in the typical range for the dianionic NNN ligand (Fig. S25†).8
Synthesis, characterization, and DFT calculations of the T-shaped planar germanium anion salt
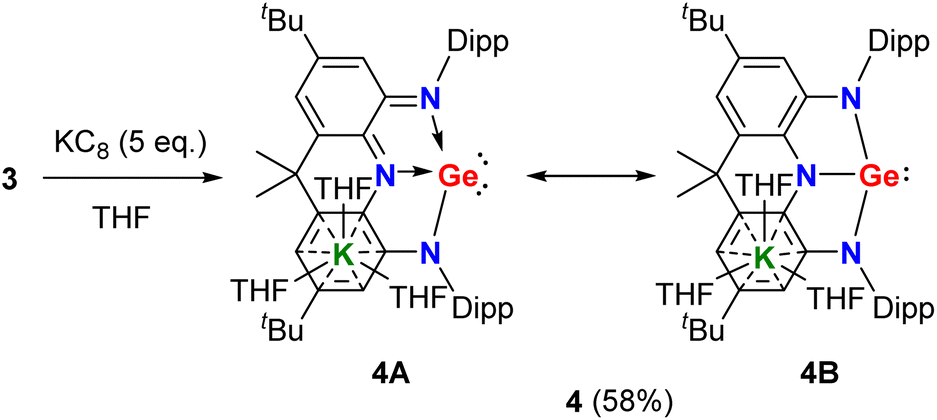
With the precursor 3 in hand, we investigated its reduction. Treatment of 3 with five equivalents of potassium graphite in THF under ambient conditions afforded, after work up, triaminogermanate salt 4 as a yellow powder in 58% yield (Scheme 2). The isolated compound 4 is extremely moisture- and oxygen-sensitive but is stable both in solution and in the solid state when stored under an inert atmosphere at room temperature. The 1H and 13C{1H} NMR spectra of 4 in THF-D8 show only one set of resonances for Dipp (Dipp = 2,6-iPr2C6H3) substituents and annulated arenes (Fig. S3 and S5†), suggesting the C2v symmetrical structure in solution.
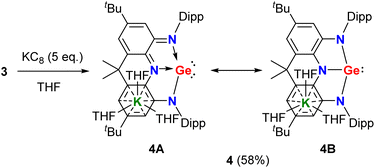 |
| | Scheme 2 Synthesis of the T-shaped planar germanium anion salt 4. | |
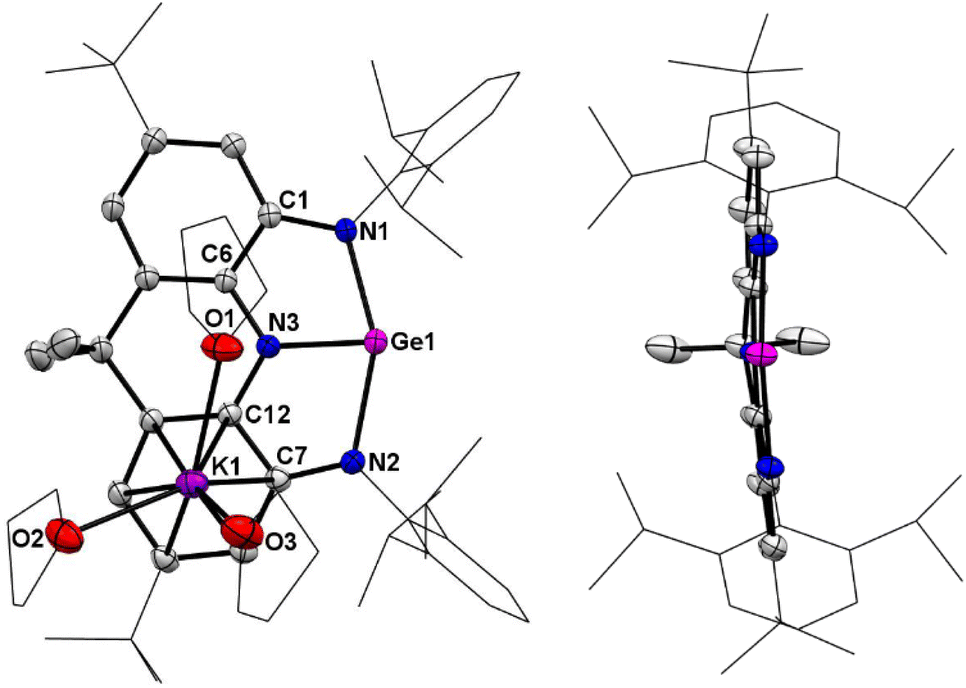
In the solid-state structure of 4, the solvated counter cation [K(THF)3]+ interacts with one annulated aryl ring in a η6 form and the distance between the germanium atom and potassium cation is 5.137 Å, which is larger than a sum of the van der Waals' radii of Ge and K (4.86 Å),9 suggesting no interaction between them (Fig. 3). The two fused C2N2Ge five-membered rings in 4 are almost coplanar with the sum of internal pentagon angles of 540° and 539.97°, respectively. Most strikingly, the germanium center in 4 is only coordinated by three nitrogen atoms to form a T-shaped planar geometry with a large N1–Ge1–N2 bond angle of 153.81(7)°. Upon reduction, the endocyclic N–Ge–N bond angles (78.23(7)° and 76.56(7)°) in 4 become more acute with respect to those (80.34(8)° and 79.59(9)°) of 3, thus reflecting the elongation of both the equatorial Ge1–N3 bond (1.9039(19) Å) and axial Ge1–N1/N2 bonds (2.0578(18) and 2.1633(18) Å) in 4. Additionally, the equatorial Ge1–N3 bond distance is shorter than those (1.925(7)–1.970(4) Å) in trinitrogen substituted germanides in pyramidal geometries,10 and the axial Ge1–N1/N2 bonds are longer, which are in good agreement with the calculated results reported by Arduengo and co-workers.7 Moreover, it is noteworthy that the average C–N (1.376(3) Å) and C–C (1.421(3) Å) bond distances in two C2N2Ge rings are between corresponding single and double bonds, implying that 4 is best viewed as a resonance of canonical forms 4A and 4B (Scheme 2), which represents an analogue of T-shaped bismuth(I/III) triamide3h and a rare example of valence tautomerism of p-block element compounds.11
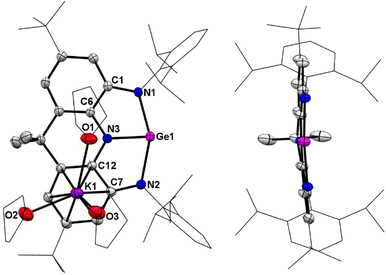 |
| | Fig. 3 Left: Solid-state structure of 4. Right: Side view of the anionic part of 4. The tBu and Dipp groups are simplified as wireframes and hydrogen atoms are omitted for clarity. Thermal ellipsoids are set at the 30% probability level. Selected bond lengths (Å) and angles (°): Ge1–N1 2.0578(18), Ge1–N2 2.1633(18), Ge1–N3 1.9039(19), C1–N1 1.372(3), C7–N2 1.359(3), C6–N3 1.388(3), C12–N3 1.384(3), C1–C6 1.414(3), C7–C12 1.428(3), N1–Ge1–N2 153.81(7), N1–Ge1–N3 78.23(7), N2–Ge1–N3 76.56(7). | |
To better understand the electronic structure of the anionic part of 4, DFT calculations at the B3LYP-D3BJ/6-311G(d) level in consideration of solvent(THF)-correction were carried out. Consistent with the experimental data, the N1–Ge1–N3 and N2–Ge1–N3 bond angles in 4 become narrower and the Ge–N bond lengths are elongated relative to 3. The natural bond orbital (NBO) analysis reveals that the average Wiberg bond index for the four C–N bonds in 4 is 1.16, demonstrating the partial double bond implied by the form 4A. Molecular orbital analysis of the anionic part of 4 shows that the lowest unoccupied molecular orbital (LUMO) is mainly the vacant 4p orbital of the Ge atom, while the highest occupied molecular orbital (HOMO) represents a diffuse π-type orbital mainly localized at the Ge center with partial delocalization over the annulated arene rings and nitrogen atoms and the HOMO–2 corresponds to the Ge-centered σ-type lone pair (Fig. 4). Therefore, the tricoordinate germanium center features both an unoccupied 4p orbital and two lone pairs of electrons. Note that the HOMO (−4.11 eV) and HOMO-2 (−4.68 eV) of 4 are comparable to that of the pyrimadal germanium anion [(NPh2)3Ge:]− (−4.32 eV, Fig. S32†) in energy and greatly higher than that of N-heterocyclic germylene [(DippNCH)2Ge:] (−5.24 eV, Fig. S33†), while the LUMO (−0.63 eV) of 4 is significantly higher than those of its neutral pnictogen analogues NNNLE (E = As: −2.37 eV; E = P: −2.16 eV, Fig. S34 and S35†) and [(DippNCH)2Ge:] (−1.24 eV), implying its relatively stronger electron-donating and weaker electron-withdrawing ability. The UV-vis spectrum of 4 in THF displays an absorption band at 396 nm (Fig. S26†), which is assigned to HOMO-1 → LUMO and HOMO → LUMO transitions according to the time-dependent DFT calculations (Table S2†).
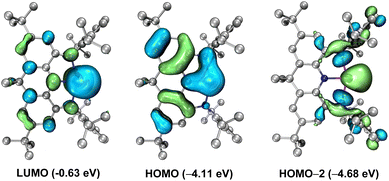 |
| | Fig. 4 Key frontier orbitals of the anionic part of 4. Left: LUMO. Middle: HOMO. Right: HOMO-2. | |
Reactivities of the T-shaped planar germanium anion salt
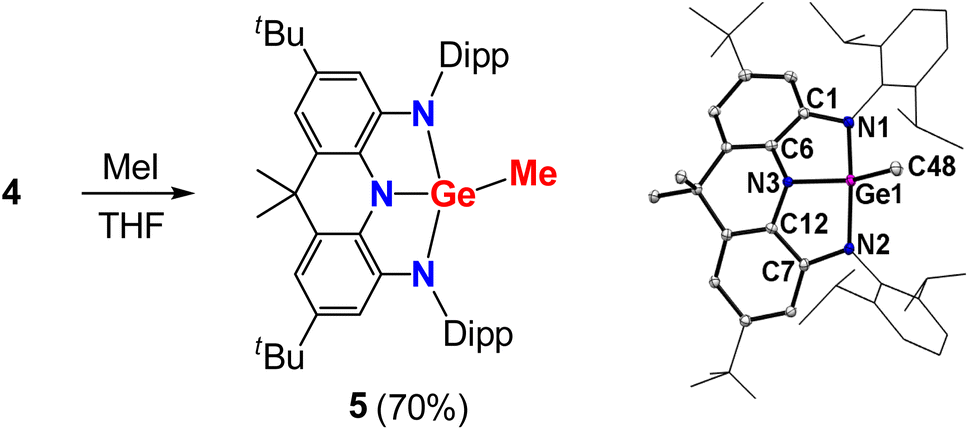
To get insight into the chemical properties of 4, its reactions were investigated. Treatments of 4 with typical nucleophilic reagents, such as 4-methylpyridine N-oxide, triethylphosphine oxide, KOtBu and LiMe, were performed at room temperature, and no reaction was observed, supporting its high LUMO in energy. However, reaction of 4 with one equivalent of methyl iodide in THF at room temperature completed within minutes to give compound 5 in 70% yield (Fig. 5). The 13C{1H} NMR spectrum of 5 displays a typical signal for the terminal methyl at −1.47 ppm (Fig. S10†). Single-crystal diffraction revealed a puckered, bicyclic C4N3Ge skeleton with a distorted tetrahedral Ge(IV) center.
 |
| | Fig. 5 Left: Reactivity of 4 with methyl iodide. Right: Solid-state structure of 5 (right). The tBu and Dipp groups are simplified as wireframes and hydrogen atoms are omitted for clarity. Thermal ellipsoids are set at the 30% probability level. Selected bond lengths (Å) and angles (°): Ge1–C48 1.9281(18), Ge1–N1 1.8908(15), Ge1–N2 1.8901(15), Ge1–N3 1.8425(14), C1–N1 1.409(2), C7–N2 1.412(2), C6–N3 1.401(2), C12–N3 1.399(2), C1–C6 1.392(2), C7–C12 1.396(2), N1–Ge1–C48 105.00(7), N2–Ge1–C48 104.84(7), N3–Ge1–C48 105.00(7), N1–Ge1–N2 141.84(6), N1–Ge1–N3 86.14(6), N2–Ge1–N3 86.59(6). | |
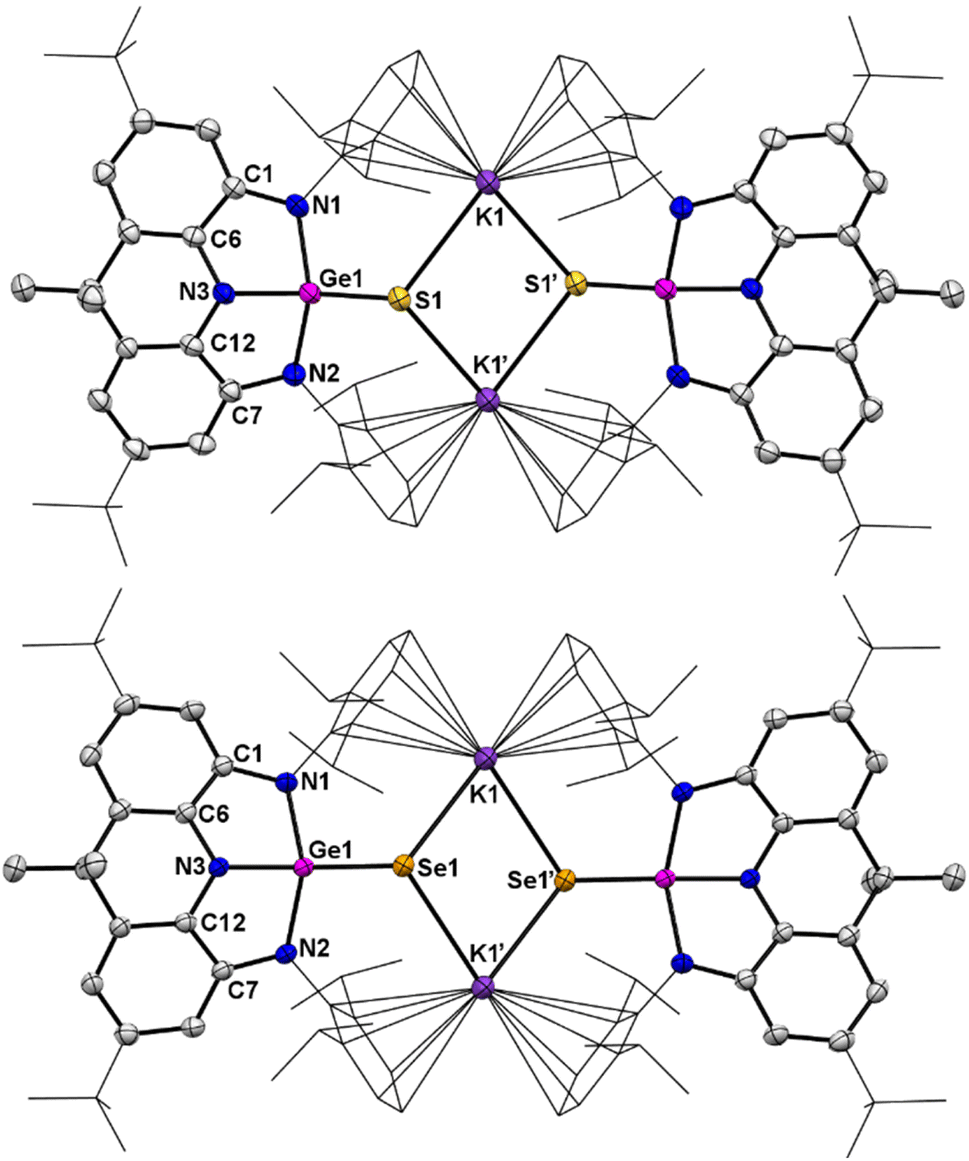
Moreover, treatment of a THF solution of 4 with stoichiometric amounts of sulfur at ambient temperature or selenium at 90 °C overnight produced, after removal of the solvent, 6 and 7, respectively, in moderate yields (Scheme 3). Spectroscopically, the 1H NMR spectra of 6 and 7 exhibit almost identical resonances for only one set of Dipp and annulated arenes (Fig. S13 and S18†), demonstrating their isostructural and symmetrical natures in solution. The 77Se NMR spectrum of 7 displays a singlet at −271.93 ppm (Fig. S23†), which is comparable to those of tetracoordinate germanium(IV) complexes with polar GeSe bonds.12 Structurally, 6 and 7 are also isostructural in the solid-state and exist as a dimer with the anionic units linked by the interactions between the potassium cation and the chalcogenide and aryl groups (Fig. 6). The germanium centers in 6 and 7 are coordinated in a distorted tetrahedral geometry by three nitrogen atoms and one chalcogenide. Upon oxidation, all of the Ge–N bond distances in 6 (avg. 1.897 Å) and 7 (avg. 1.911 Å) are contracted relative to those (avg. 1.974 Å) in 3 and are in the normal range for a germanium(IV) complex, while the C–N and C–C bond lengths in fused C2N2Ge five-membered rings are slightly elongated and shortened, respectively, and are consistent with the C–N single bonds and aromatic phenyl ring, indicating that the oxidation process occurs at the germanium center accompanied by somewhat intramolecular electron-transfer from the metal centers to the ligands. Notably, the terminal GeCh bonds (Ch = S, 2.1097(8) Å in 6; Ch = Se, 2.2595(12) Å in 7) are somewhat longer than those typical GeCh double bonds,13 but are significantly shorter than the Ge–Ch single bonds (∼2.26 Å for Ge–S and ∼2.39 Å for Ge–Se bonds)14 and comparable to the terminal GeCh double bonds (2.12 Å for [Ge4S10]4− and 2.247–2.287 Å for [Ge4Se10]4−) of inorganic adamantine-like anions [Ge4Ch10]4−,15 suggesting the existence of polarized (or formal) GeCh double bonds in 6 and 7. Notably, 6 and 7 represent the first examples of organic anions bearing terminal GeCh double bonds,15,16 which are isoelectronic to the well-known R3PnCh.17
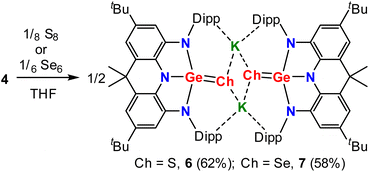 |
| | Scheme 3 Reactivity of 4 with elemental chalcogens. | |
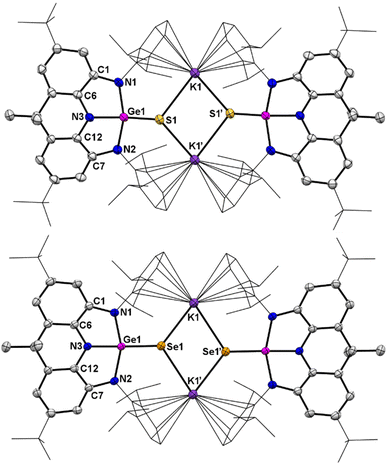 |
| | Fig. 6 Top: Solid-state structure of 6. Bottom: Solid-state structure of 7. The tBu and Dipp groups are simplified as wireframes and hydrogen atoms are omitted for clarity. Thermal ellipsoids are set at the 30% probability level. Selected bond lengths (Å) and angles (°) for 6: Ge1–S1 2.1097(8), Ge1–N1 1.917(3), Ge1–N2 1.916(3), Ge1–N3 1.857(3), K1–S1 3.1094(11), K1–S1′ 3.1491(10), C1–N1 1.394(4), C1–C6 1.394(4), C7–N2 1.396(4), C7–C12 1.395(4), C12–N3 1.403(4), N3–Ge1–S1 142.98(8), N1–Ge1–N2 139.16(11), N1–Ge1–N3 84.41(10), N2–Ge1–N3 84.41(10). Selected bond lengths (Å) and angles (°) for 7: Ge1–Se1 2.2595(12), Ge1–N1 1.928(3), Ge1–N2 1.934(3), Ge1–N3 1.872(3), K1–Se1 3.2541(17), K1–Se1′ 3.2477(17), C1–N1 1.410(4), C1–C6 1.399(4), C7–N2 1.406(4), C6–N3 1.395(3), C12–N3 1.400(3), C7–C12 1.401(4), N3–Ge1–Se1 143.51(8), N1–Ge1–N2 140.30(11), N1–Ge1–N3 84.52(10), N2–Ge1–N3 83.59(10). | |
Conclusions
More than three decades after the discovery of the T-shaped planar group 15 compound, this work demonstrates that its isoelectronic group 14 anion analog 4 is isolable as well. Compound 4 is prepared from the reduction of the germanium dichloride radical species 3 bearing a trinitrogen pincer ligand. X-ray diffraction studies and DFT calculations reveal that the germanium center in 4 possesses both a vacant 4p orbital and two lone pairs of electrons. The higher HOMO and LUMO of 4 in energy relative to its pnictogen analogues and the N-heterocyclic germylene suggest its relatively stronger nucleophilicity and weaker electrophilicity. Reaction of 4 with MeI gave the methyl-substituted complex 5 and oxidation of 4 with elemental chalcogens readily afforded unpresented organic anions 6 and 7 bearing terminal GeCh (Ch = S or Se) double bonds. Moreover, the trinitrogen pincer ligand exhibits three different oxidation states in the series of germanium complexes: radical anionic form in 3, diimine amido form in 4, and triamido form in 5–7, demonstrating its interesting redox-active properties, which is common for transition metal complexes but is rarely reported for main group elements. Based on these findings, this geometrically constrained noninnocent trinitrogen ligand could be viewed as a potentially suitable building block to stabilize other T-shaped planar group 14 species, which is in progress in our laboratory.
Data availability
Detailed experimental procedures and analytical data are available in the ESI.†
Author contributions
X. Liu performed the major synthesis work and properties study. Y. Dai performed DFT calculations. M. Bao and Y. Dai assisted with the NMR spectra and X-ray single crystallographic diffraction measurements. W. Wang recorded the UV-vis absorption spectra. Q. Li measured the mass spectra. C. Liu acquired some funding. Y. Su and X. Wang conceived the concept and prepared the manuscript. All the authors analysed and interpreted the results.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors greatly acknowledge financial support from the National Natural Science Foundation of China (Grants 22001184, Y. Su, 22231005, X. Wang, 11904425, C. Liu), the National Key R&D program of China (Grant 2018YFA0306004, X. Wang), the Natural Science Foundation of Jiangsu Province (Grant BK20200849), and the Entrepreneurship and Innovation Talent Program of Jiangsu Province (Y. Su, JSSCBS20210664, C. Liu). We thank the Shanxi Supercomputing Center of China and its Tianhe-2 system for performing calculations.
References
- Selected references are:
(a) H.-W. Lerner, Coord. Chem. Rev., 2005, 249, 781–798 CrossRef CAS;
(b) V. Y. Lee and A. Sekiguchi, Acc. Chem. Res., 2007, 40, 410–419 CrossRef CAS PubMed;
(c)
V. Y. Lee and A. Sekiguchi, Organometallic Compounds of Low-Coordinate Si, Ge, Sn and Pb: From Phantom Species to Stable Compounds, 2010, pp. 89–138 CrossRef;
(d) C. Prasang and D. Scheschkewitz, Struct. Bonding, 2013, 156, 1–47 CrossRef;
(e)
C. Marschner, Organosilicon Compounds: Theory and Experiment (Synthesis), 2017, vol. 1, pp. 295–360 Search PubMed.
-
(a) S. A. Culley and A. J. Arduengo, J. Am. Chem. Soc., 1984, 106, 1164–1165 CrossRef CAS;
(b) A. J. Arduengo, D. A. Dixon and D. C. Roe, J. Am. Chem. Soc., 1986, 108, 6821–6823 CrossRef CAS.
-
(a) M. Driess, N. Muresan, K. Merz and M. Päch, Angew. Chem., Int. Ed., 2005, 44, 6734–6737 CrossRef CAS PubMed;
(b) P. Šimon, F. de Proft, R. Jambor, A. Růžička and L. Dostál, Angew. Chem., Int. Ed., 2010, 49, 5468–5471 CrossRef PubMed;
(c) N. L. Dunn, M. Ha and A. T. Radosevich, J. Am. Chem. Soc., 2012, 134, 11330–11333 CrossRef CAS PubMed;
(d) S. M. McCarthy, Y.-C. Lin, D. Devarajan, J. W. Chang, H. P. Yennawar, R. M. Rioux, D. H. Ess and A. T. Radosevich, J. Am. Chem. Soc., 2014, 136, 4640–4650 CrossRef CAS PubMed;
(e) G. Zeng, S. Maeda, T. Taketsugu and S. Sakaki, Angew. Chem., Int. Ed., 2014, 53, 4633–4637 CrossRef CAS PubMed;
(f) A. Hentschel, A. Brand, P. Wegener and W. Uhl, Angew. Chem., Int. Ed., 2018, 57, 832–835 CrossRef CAS PubMed;
(g) F. Wang, O. Planas and J. Cornella, J. Am. Chem. Soc., 2019, 141, 4235–4240 CrossRef CAS PubMed;
(h) M. B. Kindervater, K. M. Marczenko, U. Werner-Zwanziger and S. S. Chitnis, Angew. Chem., Int. Ed., 2019, 58, 7850–7855 CrossRef CAS PubMed;
(i) M. K. Mondal, L. Zhang, Z. Feng, S. Tang, R. Feng, Y. Zhao, G. Tan, H. Ruan and X. Wang, Angew. Chem., Int. Ed., 2019, 58, 15829–15833 CrossRef CAS PubMed;
(j) M. Kořenková, M. Hejda, M. Erben, R. Jambor, A. Růžička, E. Rychagova, S. Ketkov and L. Dostál, Chem. – Eur. J., 2019, 25, 12884–12888 CrossRef PubMed.
-
(a) A. J. Arduengo and C. A. Stewart, Chem. Rev., 1994, 94, 1215–1237 CrossRef CAS;
(b) A. Brand and W. Uhl, Chem. – Eur. J., 2019, 25, 1391–1404 CrossRef CAS PubMed;
(c) J. Abbenseth and J. M. Goicoechea, Chem. Sci., 2020, 11, 9728–9740 RSC;
(d) S. Kundu, Chem. – Asian J., 2020, 15, 3209–3224 CrossRef CAS PubMed.
- For examples of geometrically constrained group 14 compounds, see:
(a) G. Bettermann and A. J. Arduengo, J. Am. Chem. Soc., 1988, 110, 877–879 CrossRef CAS;
(b) M. Driess, N. Dona and K. Merz, Chem. – Eur. J., 2004, 10, 5971–5976 CrossRef CAS PubMed;
(c) M. Driess, N. Muresan and K. Merz, Angew. Chem., Int. Ed., 2005, 44, 6738–6741 CrossRef CAS PubMed;
(d) S. Khan, R. Michel, J. R. Dietrich, R. A. Mata, H. W. Roesky, J.-P. Demers, A. Lange and D. Stalke, J. Am. Chem. Soc., 2011, 133, 17889–17894 CrossRef CAS PubMed;
(e) J. Henning, H. Schubert, K. Eichele, F. Winter, R. Pöttgen, H. A. Mayer and L. Wesemann, Inorg. Chem., 2012, 51, 5787–5794 CrossRef CAS PubMed;
(f) N. Kramer, C. Jöst, A. Mackenroth and L. Greb, Chem.–Eur. J., 2017, 23, 17764–17774 CrossRef CAS PubMed;
(g) F. Ebner and L. Greb, J. Am. Chem. Soc., 2018, 140, 17409–17412 CrossRef CAS PubMed;
(h) P. Ghana, J. Rump, G. Schnakenburg, M. I. Arz and A. C. Filippou, J. Am. Chem. Soc., 2021, 143, 420–432 CrossRef CAS PubMed;
(i) F. Ebner and L. Greb, Chem, 2021, 7, 2151–2159 CrossRef CAS PubMed;
(j) H. Ruppert, L. M. Sigmund and L. Greb, Chem. Commun., 2021, 57, 11751–11763 RSC;
(k) H. Ruppert and L. Greb, Angew. Chem., Int. Ed., 2022, 61, e202116615 CrossRef CAS PubMed.
- For examples of neutral T-shaped planar group 14 compounds, see:
(a) J. Flock, A. Sulianovic, A. Torvisco, W. Schoefberger, B. Gerke, R. Pöttgen, R. C. Fischer and M. Flock, Chem. – Eur. J., 2013, 19, 15504–15517 CrossRef CAS PubMed;
(b) T. Chu, L. Belding, A. Van Der Est, T. Dudding, I. Korobkov and G. I. Nikonov, Angew. Chem., Int. Ed., 2014, 53, 2711–2715 CrossRef CAS PubMed;
(c) M. T. Nguyen, D. Gusev, A. Dmitrienko, B. M. Gabidullin, D. Spasyuk, M. Pilkington and G. I. Nikonov, J. Am. Chem. Soc., 2020, 142, 5852–5861 CrossRef CAS PubMed.
- D. A. Dixon, A. J. Arduengo and M. F. Lappert, Heteroat. Chem., 1991, 2, 541–544 CrossRef CAS.
- A. I. Nguyen, K. J. Blackmore, S. M. Carter, R. A. Zarkesh and A. F. Heyduk, J. Am. Chem. Soc., 2009, 131, 3307–3316 CrossRef CAS PubMed.
- M. Mantina, A. C. Chamberlin, R. Valero, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. A, 2009, 113, 5806–5812 CrossRef CAS PubMed.
-
(a) A. Steiner and D. Stalke, J. Chem. Soc., Chem. Commun., 1993, 1702–1704 RSC;
(b) M. Veith, O. Schütt and V. Huch, Angew. Chem., Int. Ed., 2000, 39, 601–604 CrossRef CAS;
(c) L. Witteman, C. B. van Beek, O. N. van veenhuizen, M. Lutz and M.-E. Moret, Organometallics, 2019, 38, 231–239 CrossRef CAS PubMed.
- L. Greb, Eur. J. Inorg. Chem., 2022, e202100871 CAS.
-
(a) Y. Ding, Q. Ma, H. W. Roesky, I. Usón, M. Noltemeyer and H. Schmidt, Dalton Trans., 2003, 1094–1098 RSC;
(b) S. Nagendran and H. W. Roesky, Organometallics, 2008, 27, 457–492 CrossRef CAS;
(c) S. Sinhababu, R. K. Siwatch, G. Mukherjee, G. Rajaraman and S. Nagendran, Inorg. Chem., 2012, 51, 9240–9248 CrossRef CAS PubMed.
-
(a) N. Tokitoh, T. Matsumoto, K. Manmaru and R. Okazazi, J. Am. Chem. Soc., 1993, 115, 8855–8856 CrossRef CAS;
(b) T. Matsumoto, N. Tokitoh and R. Okazazi, Angew. Chem., Int. Ed., 1994, 33, 2316–2317 CrossRef;
(c) T. Matsumoto, N. Tokitoh and R. Okazazi, J. Am. Chem. Soc., 1999, 121, 8811–8824 CrossRef CAS.
-
(a) M. C. Kuchta and G. Parkin, J. Chem. Soc., Chem. Commun., 1994, 1351–1352 RSC;
(b) N. Tokitoh, T. Matsumoto and R. Okazazi, Bull. Chem. Soc. Jpn., 1999, 72, 1665–1684 CrossRef CAS;
(c) I. Saur, G. Rima, H. Gornitzka, K. Miqueu and J. Barrau, Organometallics, 2003, 22, 1106–1109 CrossRef CAS.
-
(a) J. Y. Pivan, O. Achak, M. Louër and D. Louër, Chem. Mater., 1994, 6, 827–830 CrossRef CAS;
(b) K.-Y. Wang, S. Zhang, H.-W. Liu, L. Cheng and C. Wang, Inorg. Chem., 2019, 58, 12832–12842 CrossRef CAS PubMed.
-
(a) Y. Xiong, S. Yao, S. Inoue, A. Berkefeld and M. Driess, Chem. Commun., 2012, 48, 12198–12200 RSC;
(b) D. Sarkar, C. Weetman, S. Dutta, E. Schubert, C. Jandl, D. Koley and S. Inoue, J. Am. Chem. Soc., 2020, 142, 15403–15411 CrossRef CAS PubMed.
-
(a)
G. Hua and J. D. Woollins, Selenium and Tellurium Chemistry: From Small Molecules to Biomolecules and Material, 2011, pp. 1–39 Search PubMed;
(b)
R. Davies and L. Patel, Handbook of Chalcogen Chemistry: New Perspectives in Sulfur, Selenium and Tellurium, 2nd edn, 2013, vol. 1, pp. 238–306 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *b and
Yuanting
Su
*b and
Yuanting
Su