
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotoredox radical/polar crossover enables C–H gem-difunctionalization of 1,3-benzodioxoles for the synthesis of monofluorocyclohexenes†
Jiabao
Tian
and
Lei
Zhou
 *
*
School of Chemistry, Sun Yat-Sen University, Panyu District, Guangzhou 510006, China. E-mail: zhoul39@mail.sysu.edu.cn
First published on 12th May 2023
Abstract
A photocatalytic C–H gem-difunctionalization of 1,3-benzodioxoles with two different alkenes for the synthesis of highly functionalized monofluorocyclohexenes is described. Using 4CzIPN as the photocatalyst, the direct single electron oxidation of 1,3-benzodioxoles allows their defluorinative coupling with α-trifluoromethyl alkenes to produce gem-difluoroalkenes in a redox-neutral radical polar crossover manifold. The C–H bond of the resultant γ,γ-difluoroallylated 1,3-benzodioxoles was further functionalized via radical addition to electron-deficient alkenes using a more oxidizing iridium photocatalyst. The capture of in situ generated carbanions by an electrophilic gem-difluoromethylene carbon and consecutive β-fluoride elimination afford monofluorocyclohexenes. The synergistic combination of multiple termination pathways of carbanions enables rapid incorporation of molecular complexity via stitching simple and readily accessible starting materials together.
Introduction
Owing to the easy generation of diverse radical intermediates under mild conditions, visible-light photoredox catalysis has emerged as a powerful tool for organic synthesis.1 One of notable features of photoredox catalysis is its inherent ability to crossover from a radical to a polar pathway during the overall catalytic process, which has resulted in a wide array of novel synthetic methodologies, especially in the field of alkene functionalization.2 The carbon-centered radicals generated upon Giese-type radical addition to electron-deficient alkenes are readily single electron reduced to the corresponding carbanions.3 Although direct protonation is the most common pathway to terminate these carbanions (Scheme 1a), other processes have been developed in the last few years, including: (1) elimination of a neighbouring leaving group;4 when carbanions have β-substituted leaving groups, they tend to form a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond via E1cB-type elimination. In this context, our group5 and Molander6 have reported photocatalytic defluorinative coupling reactions of radical precursors and α-trifluoromethyl alkenes for the synthesis of diverse gem-difluoroalkenes (Scheme 1b). (2) Ring closure reactions;7 when the leaving groups are present at the distal position of carbanions, the intramolecular SN2 reaction would produce cyclic products (Scheme 1c). Although the formation of cyclopropanes via 3-exo-tet cyclization is often effective,7a the construction of other-membered rings is still considered to be challenging.7d (3) Trapping by a carbon-based electrophile;8 the capture of carbanions by electrophiles rather than a proton, such as CO2 and aldehydes, is a synthetically useful pathway to introduce a functional group into the molecules (Scheme 1d). Undoubtedly, the synergistic combination of multiple termination pathways of carbanions can rapidly increase molecular complexity through stitching structural fragments together. For such a purpose, it is crucial to find a radical precursor, which allows the generation of radicals at least twice and the subsequent radical addition to different alkenes in a controllable manner.9
C double bond via E1cB-type elimination. In this context, our group5 and Molander6 have reported photocatalytic defluorinative coupling reactions of radical precursors and α-trifluoromethyl alkenes for the synthesis of diverse gem-difluoroalkenes (Scheme 1b). (2) Ring closure reactions;7 when the leaving groups are present at the distal position of carbanions, the intramolecular SN2 reaction would produce cyclic products (Scheme 1c). Although the formation of cyclopropanes via 3-exo-tet cyclization is often effective,7a the construction of other-membered rings is still considered to be challenging.7d (3) Trapping by a carbon-based electrophile;8 the capture of carbanions by electrophiles rather than a proton, such as CO2 and aldehydes, is a synthetically useful pathway to introduce a functional group into the molecules (Scheme 1d). Undoubtedly, the synergistic combination of multiple termination pathways of carbanions can rapidly increase molecular complexity through stitching structural fragments together. For such a purpose, it is crucial to find a radical precursor, which allows the generation of radicals at least twice and the subsequent radical addition to different alkenes in a controllable manner.9
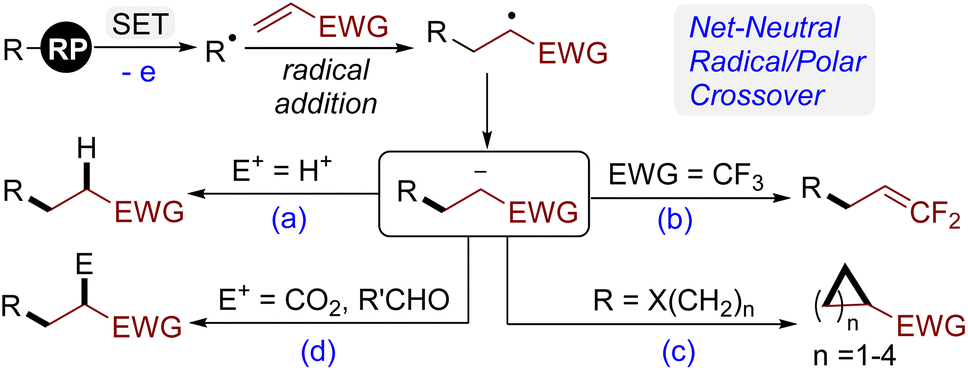 | ||
| Scheme 1 Termination pathways of carbanions. | ||
The dual C–F bond cleavage of α-trifluoromethyl alkenes has been demonstrated as a valuable alternative to the existing C–F bond formation protocols for the construction of mono-fluorinated carbo- or heterocyclic frameworks.10 Typically, it is accomplished by means of the classical base-mediated sequential SN2′/SNV reactions of binucleophiles with trifluoromethyl alkenes (Scheme 2a).11 To distinguish the reactivity of two nucleophilic sites, a fine control on the reaction conditions is always necessary. Taking advantage of readily formed α-amino alkyl radicals by photoredox catalysis, we have previously reported the visible light-mediated [3 + 3] annulation of tertiary amines with α-trifluoromethyl alkenes. When the alkyl groups of tertiary amines are different, an additional carboxylic group was installed to control the regioselectivity (Scheme 2b).12 Through the combination of photoredox catalysis and the intramolecular base-mediated SNV reaction, the defluorinative annulations of trifluoromethyl alkenes with radical precursors bearing a nucleophilic site have been developed, allowing the dual C–F bond cleavage of a CF3 group in a more controllable manner (Scheme 2c).13 To extend such a strategy to substrates without a nucleophilic site,14 our group recently reported a photocatalytic sulfonyl radical-triggered [3 + 2] annulation of α-CF3 alkenes with dihydrofuran (Scheme 2d).15 Due to the electron-deficient nature of the sulfonyl group, gem-difluoroalkenes generated by this three-component reaction can further undergo the SNV reaction to give fused pentenes in the presence of LiHMDS. However, the strong basic conditions of the SNV reaction not only limit the functional group tolerance but also suffer from some chemoselectivity issues because bases are potential nucleophiles to undergo the SNV reaction with gem-difluoroalkenes.16 Therefore, milder alternatives are highly sought after.
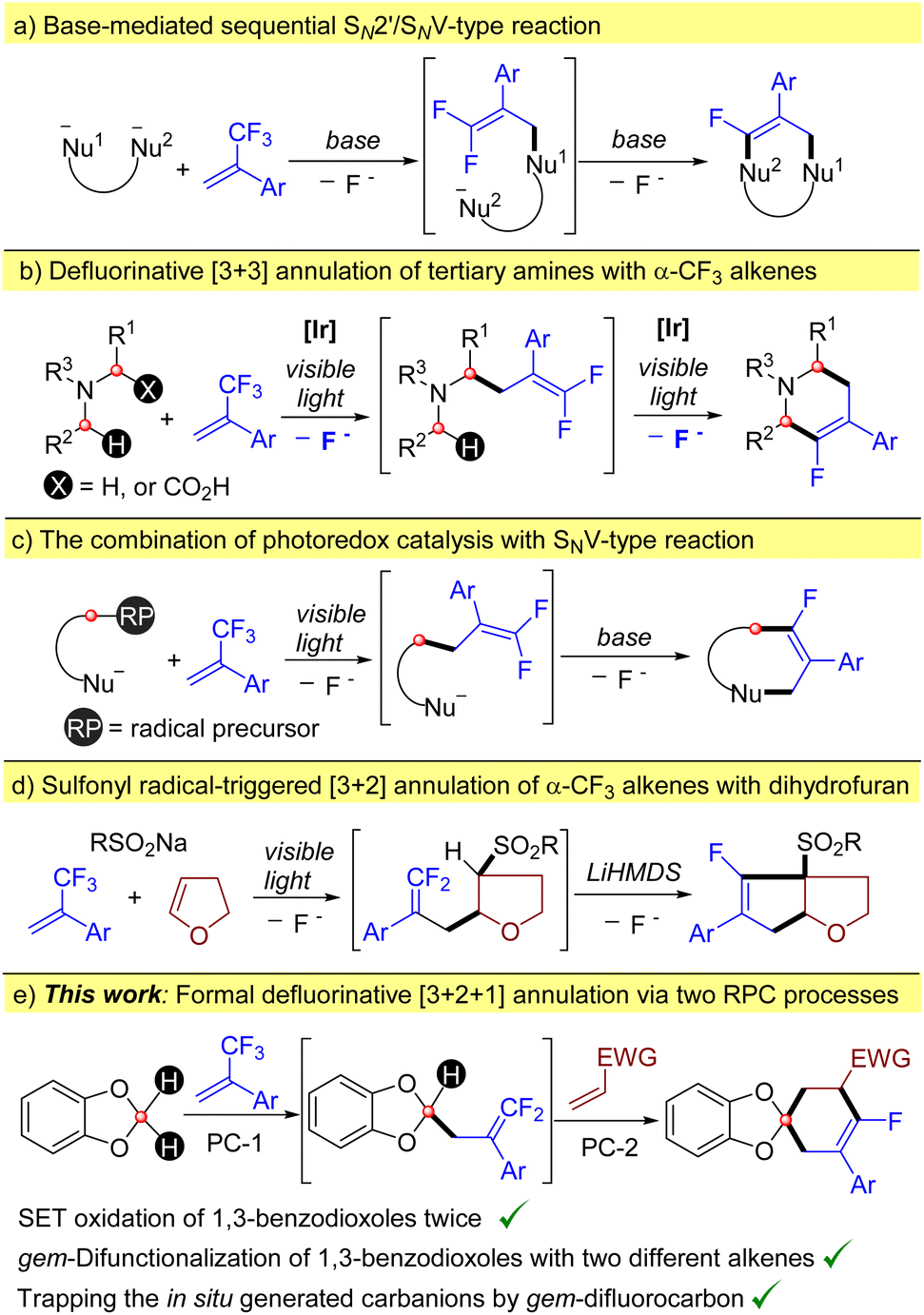 | ||
| Scheme 2 Cyclization via dual C–F bond cleavage of α-CF3 alkenes. | ||
Herein, we report the synergistic combination of multiple termination pathways of carbanions for the synthesis of spiro monofluorinated cyclohexenes via a formal defluorinative [3 + 2 + 1] annulation of 1,3-benzodioxoles, α-trifluoromethyl alkenes and electron-deficient alkenes (Scheme 2e). 1,3-Benzodioxole served as a biradical precursor, which allows the step-by step radical addition to α-CF3 alkenes and electron-deficient alkenes in a net RPC manner. The carbanions in situ generated in the second addition process can be trapped by the gem-difluoroalkenes intramolecularly to close the ring before they are protonated. Notably, although the radical addition of 1,3-benzodioxoles to alkenes has been extensively studied, the 1,3-benzodioxol-2-yl radicals involved in all these reactions are generated via the hydrogen-atom transfer (HAT) pathway in the presence of various HAT catalysts,17 such as (nBu4N)4[W10O32] (TBADT),17a–c (n-Bu4N)2S2O8,17d and quinuclidine,17e thus usually providing Giese-type alkylation products. To the best of our knowledge, the direct photochemical SET oxidation of 1,3-benzodioxoles and their 2-substituted analogues is hitherto not reported.
Results and discussion
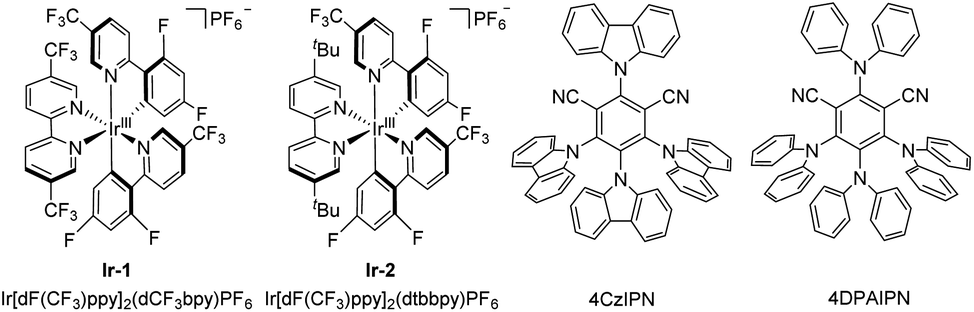
Initially, the reaction conditions for the defluorinative mono-γ,γ-difluoroallylation of 1,3-benzodioxole 1a with α-trifluoromethyl-(p-phenyl)styrene 2a were optimized. Using a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of 1a and 2a with Ir[dFCF3ppy]2(dCF3bpy)PF6 (Ir-1) as the photocatalyst and 2,4,6-collidine as the base, the reaction gave a mixture of mono and dual γ,γ-difluoroallylation products 3a and 3a′ in the yields of 51% and 14%, respectively (Table 1, entry 1). The yields of 3a and 3a′ varied when the reaction was performed in other solvents, such as CH2Cl2, DMF, THF and MeOH; but the total yields were lower than that in MeCN (Table 1, entries 2–5). On switching the ratio of 1a and 2a from 1:1 to 1:2, the reaction afforded dual functionalization product 3a′ in 90% yield and 3a was consumed completely (Table 1, entry 6). Other photocatalysts such as Ir[dF(CF3)ppy]2(dtbbpy)PF6 (Ir-2) and 4DPAIPN were found to be essentially ineffective (Table 1, entries 7 and 8). To our delight, 3a could be obtained as the major product with 4CzIPN as the photocatalyst, even using excess amounts of 2a (Table 1, entries 9 and 10). The reaction afforded 29% yield of 3a without 3a′ when the ratio of 1a and 2a was 2:1 (Table 1, entry 11). However, the defluorinative coupling of 3a with the unconsumed 1a would form mono-fluorinated alkene 3a′′ as the side product (see Scheme S1 in the ESI†). We found that the reaction provided the best yield of 3a after 13 h of irradiation as monitored by TLC (Table 1, entry 12). Further inspection of the conditions revealed that this reaction cannot proceed when Cs2CO3, DABCO and DBU were employed as the bases (Table 1, entries 13–15). LiOH is an effective base for the reaction; but it is no better than 2,4,6-collidine (Table 1, entry 16). The control experiments indicated that no product was observed in the absence of either light or the catalyst (Table 1, entries 17 and 18).
:1 ratio of 1a and 2a with Ir[dFCF3ppy]2(dCF3bpy)PF6 (Ir-1) as the photocatalyst and 2,4,6-collidine as the base, the reaction gave a mixture of mono and dual γ,γ-difluoroallylation products 3a and 3a′ in the yields of 51% and 14%, respectively (Table 1, entry 1). The yields of 3a and 3a′ varied when the reaction was performed in other solvents, such as CH2Cl2, DMF, THF and MeOH; but the total yields were lower than that in MeCN (Table 1, entries 2–5). On switching the ratio of 1a and 2a from 1:1 to 1:2, the reaction afforded dual functionalization product 3a′ in 90% yield and 3a was consumed completely (Table 1, entry 6). Other photocatalysts such as Ir[dF(CF3)ppy]2(dtbbpy)PF6 (Ir-2) and 4DPAIPN were found to be essentially ineffective (Table 1, entries 7 and 8). To our delight, 3a could be obtained as the major product with 4CzIPN as the photocatalyst, even using excess amounts of 2a (Table 1, entries 9 and 10). The reaction afforded 29% yield of 3a without 3a′ when the ratio of 1a and 2a was 2:1 (Table 1, entry 11). However, the defluorinative coupling of 3a with the unconsumed 1a would form mono-fluorinated alkene 3a′′ as the side product (see Scheme S1 in the ESI†). We found that the reaction provided the best yield of 3a after 13 h of irradiation as monitored by TLC (Table 1, entry 12). Further inspection of the conditions revealed that this reaction cannot proceed when Cs2CO3, DABCO and DBU were employed as the bases (Table 1, entries 13–15). LiOH is an effective base for the reaction; but it is no better than 2,4,6-collidine (Table 1, entry 16). The control experiments indicated that no product was observed in the absence of either light or the catalyst (Table 1, entries 17 and 18).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | PC |
1a:2a |
Solvent | Base | 3a (%) | 3a′ (%) |
| a All the reactions were carried out at the 0.2 mmol scale using 1a, 2a, photocatalyst (2.5 mol%) and base (3 equiv) under the irradiation of a 5 W blue LED at room temperature for 24 h. b Yield was determined by 19F NMR using 4-bromotrifluorobenzene as an internal standard. c Mono-fluorinated alkene 3a′′ by the reaction of 3a and 1a was isolated in 43% yield. d Reaction time: 13 h. e In the dark. | ||||||
| 1 | Ir-1 | 1:1 |
MeCN | Collidine | 51 | 14 |
| 2 | Ir-1 | 1:1 |
CH2Cl2 | Collidine | 38 | 16 |
| 3 | Ir-1 | 1:1 |
DMF | Collidine | 45 | 10 |
| 4 | Ir-1 | 1:1 |
THF | Collidine | 42 | 12 |
| 5 | Ir-1 | 1:1 |
MeOH | Collidine | 14 | 6 |
| 6 | Ir-1 | 1:2 |
MeCN | Collidine | 0 | 90 |
| 7 | Ir-2 | 1:1 |
MeCN | Collidine | 0 | 0 |
| 8 | 4DPAIPN | 1:1 |
MeCN | Collidine | 0 | 0 |
| 9 | 4CzIPN | 1:1 |
MeCN | Collidine | 56 | 2 |
| 10 | 4CzIPN | 1:2 |
MeCN | Collidine | 71 | 5 |
| 11 | 4CzIPN | 2:1 |
MeCN | Collidine | 29 | 0 |
| 12 | 4CzIPN | 2:1 |
MeCN | Collidine | 81 | 0 |
| 13 | 4CzIPN | 2:1 |
MeCN | Cs2CO3 | 0 | 0 |
| 14 | 4CzIPN | 2:1 |
MeCN | DABCO | 0 | 0 |
| 15 | 4CzIPN | 2:1 |
MeCN | DBU | 5 | 0 |
| 16 | 4CzIPN | 2:1 |
MeCN | LiOH | 27 | 0 |
| 17 | 4CzIPN | 2:1 |
MeCN | Collidine | 0 | 0 |
| 18 | None | 2:1 |
MeCN | Collidine | 0 | 0 |
|
||||||
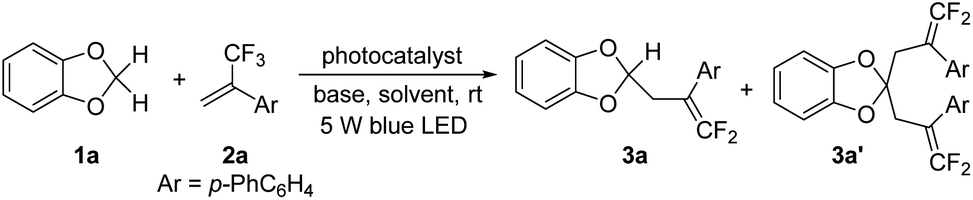
Having established the optimal reaction conditions (Table 1, entry 12), we next explored the scope of the mono-gem-difluoroallyation of 1,3-benzodioxoles with a variety of α-CF3 alkenes (Scheme 3). The reactions of 1a with α-CF3 styrenes bearing an electron-donating group on the phenyl ring, such as p-Me (3b), p-tBu (3c), m-Me (3g), and m-OMe (3h) occurred smoothly. The installation of an electron-withdrawing chloro or ester group at the para-position of α-CF3 styrene slightly disfavoured the reactions, affording 3d and 3e in moderate yields. α-CF3 (meta-bromo)styrene (3i) has similar efficiency as that without a substituent (3f). α-CF3 alkenes substituted by 3,4-ethylenedioxyphenyl (3j) and 2-naphthyl (3k) were also suitable substrates. Next, 2a was used to examine the scope of 1,3-benzodioxoles. We were delighted to find that 1,3-benzodioxoles bearing an aldehyde (3l) or a free alcohol (3m) motif underwent reactions with 2a smoothly, while these sensitive functional groups remained intact. In the case of 3n, only one of two symmetric 1,3-benzodioxole units was mono-functionalized. The reaction of 2a with 2-cyclohexyl 1,3-benzodioxole produced 3o in 66% yield, indicating that the interference of an alkyl group at the 2-position was weak.
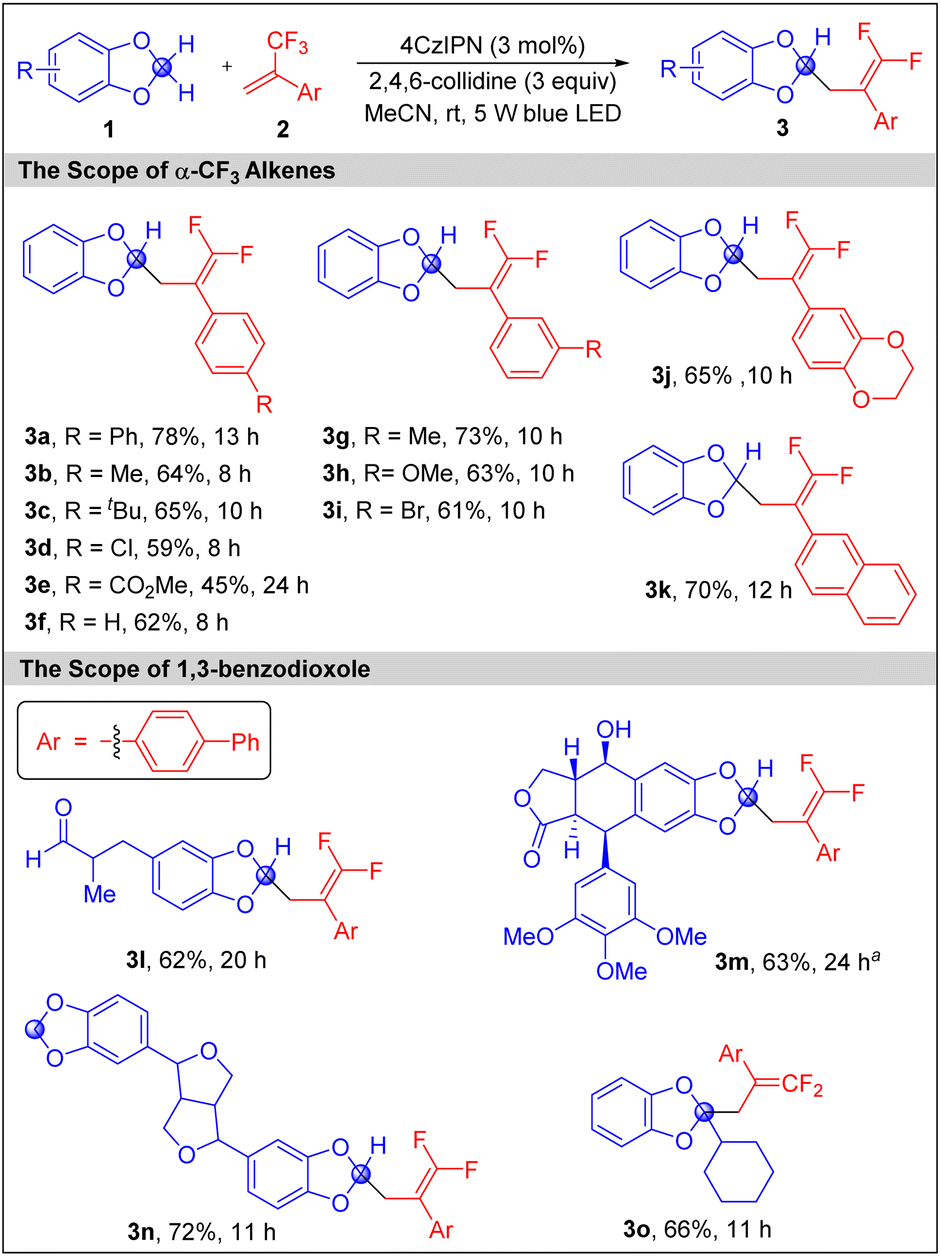 | ||
| Scheme 3 Mono-γ,γ-difluoroallyation of 1,3-benzodioxoles. Reaction conditions: 1,3-benzodioxole 1 (0.4 mmol), α-CF3 alkene 2 (0.2 mmol), 4CzIPN (2.5 mol%) and 2,4,6-collidine (3 equiv) under the irradiation of a 5 W blue LED at r.t. Isolated yields. aIr-1 was used as the photocatalyst. | ||
With various mono-γ,γ-difluoroallylated 1,3-benzodioxoles in hand, we then examined their reactions with alkenes for the construction of monofluorocyclohexenes via another C–F bond cleavage process. Because Ir[dFCF3ppy]2(dCF3bpy)PF6 (Ir-1) showed better catalytic ability than 4CzIPN for the dual functionalization of 1,3-benzodioxole 1a, this photocatalyst was used for the reactions of mono-γ,γ-difluoroallylated 1,3-benzodioxoles 3 with alkenes 4 (Scheme 4). We were delighted to find that the reaction of 3a and ethyl acrylate (4a) provided the anticipated monofluorinated cyclohexene 5a in 86% yield. Alkenes 4 with benzyl (5b) and (2-perfluorohexanyl)ethyl (5c) also provided cyclized products in good yields. N,N-dimethyl acrylamide was also a suitable alkene for the reaction (5d). Using acrolein as the radical acceptor, the desired monofluorinated cyclohexene 5e was obtained only in 38% yield, while direct protonation product 5e′ was isolated in the yield of 40%. Alkenes monosubstituted by other electron-withdrawing groups (EWGs), such as sulfonyl (5f), phosphoryl (5g), and 4-pyridinyl (5h) were found to be viable. It is noteworthy that the substrates were not limited to electron-deficient alkenes, as exemplified by the reaction of 3a with styrene to form 5i. To further demonstrate the generality of this protocol, 1,1- and 1,2-disubstituted alkenes were tested. α-Methyl acrylates underwent reactions with 3a to give products in good yields (5j, 5k). An epoxide in the ester motif of 5k can survive under the present weak basic conditions. Due to the chiral carbon in the epoxide, 5k was obtained as a 1.4:1 mixture of two diastereomers. Spiro product 5l was constructed in 63% yield by the reaction of 3a and α-methylene-γ-butyrolactone. When diethyl maleate (cis-1,2-substituted alkene) and diethyl fumarate (trans-1,2-substituted alkene) were employed as the alkenes, 5m was obtained with the trans-configuration in both cases, as determined by the H–H cosy spectra. In contrast, the reaction of indene and 3a delivered the cis-isomer of 5n in 61% yield. Remarkably, a fused bicycle bearing a bridgehead quaternary carbon (5o) was constructed from tri-substituted alkene methyl 1-cyclohexene-1-carboxylate in 63% yield with excellent stereoselectivity. The cyclization was found to not be significantly affected by the substituent on the phenyl ring attached to the CCF2 unit, providing monofluorinated cyclohexenes 5p–t in 71–81% yields.
 | ||
| Scheme 4 Synthesis of monofluorinated cyclohexenes via photocatalytic defluorinative reactions of 3 with various alkenes 4. Reaction conditions: mono-gem-difluoroallylated 1,3-benzodioxole 3 (0.2 mmol), alkene 4 (0.4 mmol), Ir[dFCF3ppy]2(dCF3bpy)PF6 (2.5 mol%) and 2,4,6-collidine (3 equiv) under the irradiation of a 5 W blue LED at r.t. for 48 h. Isolated yields. aAlkylation but direct protonation product 5e′ was isolated in 40% yield. bReaction time: 72 h. | ||
To demonstrate the practicality of this method, 3a and 5a were prepared in the gram-scale under standard conditions and their synthetic applications were demonstrated. As shown in Scheme 5a, treatment of mono-γ,γ-difluoroallylated 1,3-benzodioxole 3a with pyrrolidine (10 equiv) in MeCN at room temperature afforded amide 6 in 85% yield without any other additives.18 The oxygen of the carbonyl group might have come from the trace amount of water in the solvent. The catechol protecting group of 5a can be readily removed in 0.5 mL of 90% v/v aqueous trifluoroacetic acid (TFA) solution at room temperature.19 However, the deprotected cyclohexenone tends to undergo aromatization followed by aromatic nucleophilic substitution (SNAr) with the aid of an electron-withdrawing ester group, affording 1,4-diphenol 7 in 51% yield eventually (Scheme 5b).20 The hydrolysis of the cyclic acetal structure of 5a in aqueous AcOH/HCl solution at 120 °C led to the mono-acetyl substituted 1,4-diphenol 8 with the removal of the ester group.21 In this case, an acetate ion rather than H2O acted as the nucleophile to react with the Csp2–F bond, while another hydroxyl group generated by the deprotection and tautomerization of carbonyl remained intact (Scheme 5c). The reduction of 5a with Et3SiH can cleave one C–O bond of a cyclic acetal motif, providing ether 9 in 81% yield with >20:1 dr selectivity (Scheme 5d).22 The deprotection of 5j using the same conditions as described in Scheme 5b released the carbonyl group successfully, which afforded mono-fluorinated 3-cyclohex-1-one 10 in 41% yield (Scheme 5e). We also examined the synthesis of 5avia two steps in one-pot without the isolation of 3a. To our delight, 5a was isolated in 44% yield under slightly modified conditions (Scheme 5f). One-step reaction by mixing 1a, 2a and 4a together led to a complex mixture, despite the formation of 5a which could be detected by 19F NMR in 17% yield.
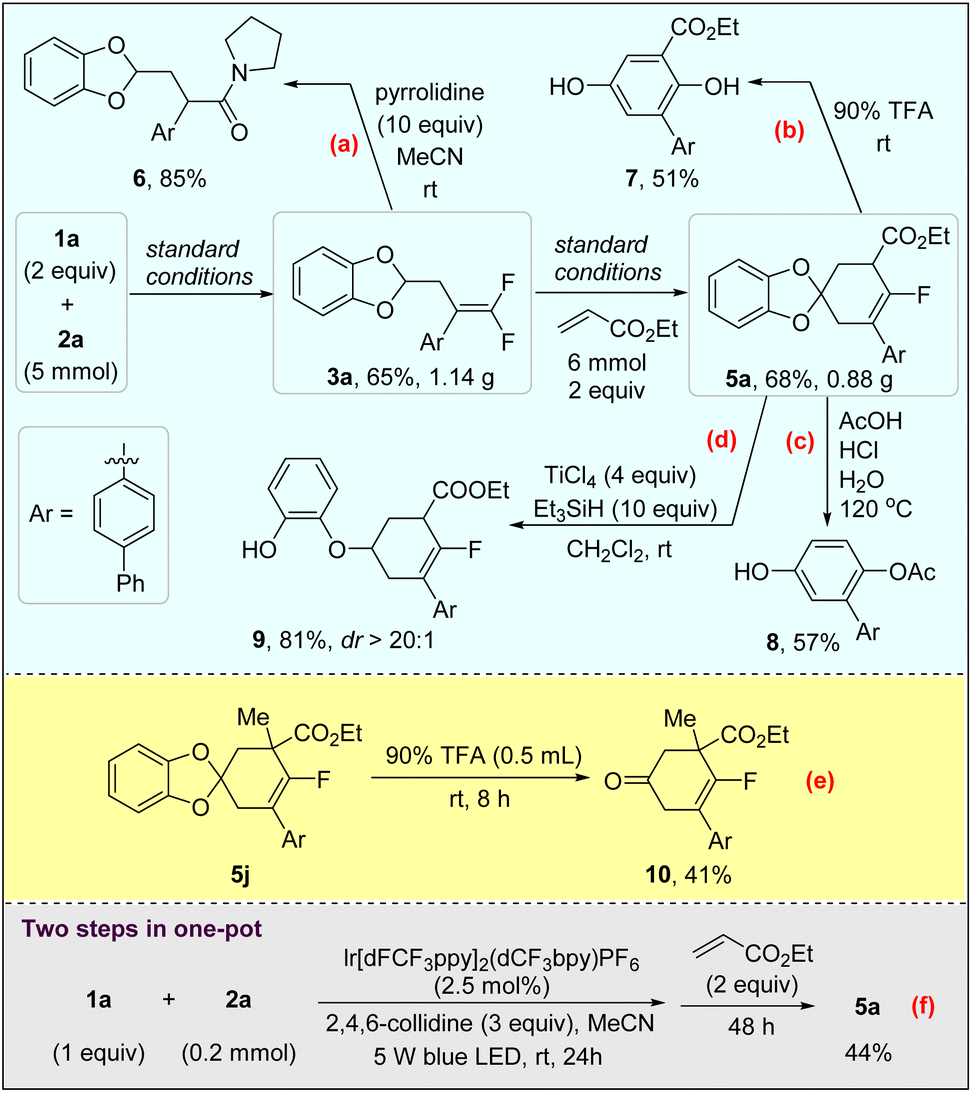 | ||
| Scheme 5 Gram scale reaction, synthetic applications of the products, and one-pot reaction. | ||
Preliminary mechanistic studies were carried out to probe the possible reaction pathway. When 2,2,6,6-tetramethylpiperidinooxyl (TEMPO) was added to the reaction of 1a and 2a under standard conditions (Table 1, entry 12), the formation of 3a was completely suppressed and an 1,3-benzodioxol-2-yl-TEMPO adduct 11 was detected by HRMS (Scheme 6a), suggesting the generation of a 1,3-benzodioxol-2-yl radical. The luminescence quenching experiments demonstrated that both 1a and 3a displayed luminescence quenching of the excited Ir-1 photocatalyst. By comparing the rates of the slope (Scheme 6b), we found that the quenching ability of 3a was lower than that of 1a, which provides the opportunity of dual functionalization of the CH2 group with two different alkenes. The cyclic voltammetry experiments (Fig. S3 in the ESI†) indicated that 3a has a higher oxidative potential (E1/2 = +1.53 V vs. SCE) than 1a (E1/2 = +1.16 V vs. SCE).23 Therefore, Ir-1 with stronger oxidative ability (E1/2(PC*/PC−) = +1.68 V vs. SCE)24 produced dual gem-difluoroallylation 3a′ efficiently, while 4CzIPN (E1/2(PC*/PC−) = +1.35 V vs. SCE)25 can restrain the further functionalization of 3a. A decrease in Ir-1* luminescence was not observed by adding 2,4,6-collidine, indicating that this reagent acted as a base only. In the gram scale reaction for the synthesis of 5j, 7% yield of protonated product 5j′ was isolated. When 5j′ was treated with 2,4,6-collidine, no cyclized product 5j can be detected (Scheme 6c). With pKaH 15.00,26 2,4,6-collidine is not strong enough to deprotonate the α-hydrogen of an electron-withdrawing group, for example, the C–H bond alpha to an ester group with a pKa of around 25.27 Therefore, a carbanion was generated in situ during the photoredox catalytic processes rather than deprotonation by a base.
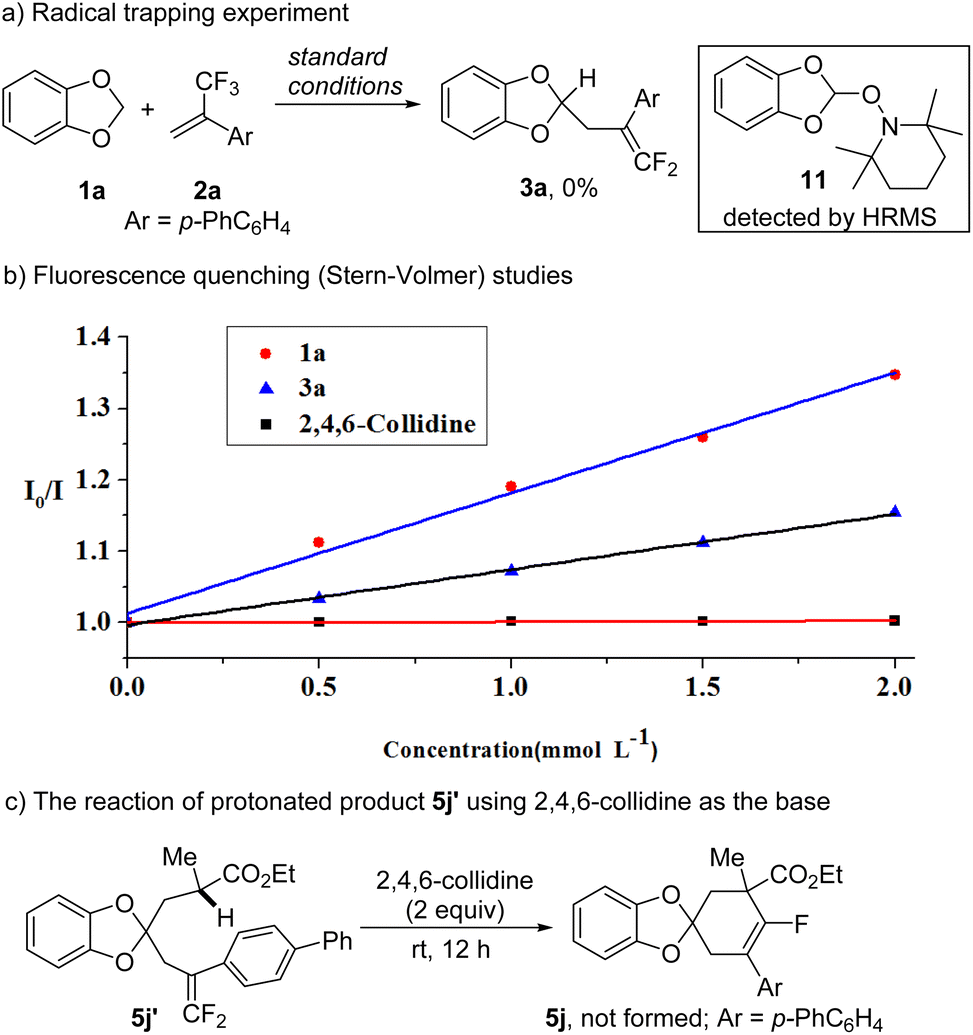 | ||
| Scheme 6 Mechanistic studies. | ||
On the basis of the above results, a plausible mechanism for this two-step formal [3 + 2 + 1] defluorinative annulation is proposed in Scheme 7. The single electron oxidation of 1,3-benzodioxole by the excited photocatalyst 4CzIPN* produces radical cation A. In the presence of a base, the deprotonation of A generates 1,3-benzodioxol-2-yl radical B, which undergoes addition to the α-CF3 alkene to give radical C. The reduction of C by 4CzIPN˙− forms α-CF3 carbanion D and regenerates the photocatalyst in its ground state. β-Fluoride elimination of D leads to mono-gem-difluoroallylated 1,3-benzodioxole 3. In the second photoredox catalytic cycle, carbanion G was generated with an Ir complex as the photocatalyst via similar processes for the formation of D. Due to the high electron deficiency on gem-difluoromethylene carbon, the in situ generated carbanion can be trapped by gem-difluoroalkene intramolecularly to give H (path a). Alternatively, intramolecular radical addition to gem-difluoroalkene followed by SET reduction is also a possible route to form H (path b). However, the presence of a single EWG imparts electrophilic characteristics to alkyl radicals, which disfavours their addition to the high electron deficient gem-difluoromethylene carbon.28 In contrast, alkyl radicals adjacent to an EWG are readily converted to carbanions through SET reduction.3a The observation of the direct protonation products, such as 5e′ and 5j′ indicated the formation of carbanion G. Therefore, path b is less likely for ring closure. Finally, monofluorocyclohexene 5 was produced via the β-fluoride elimination of intermediate H.
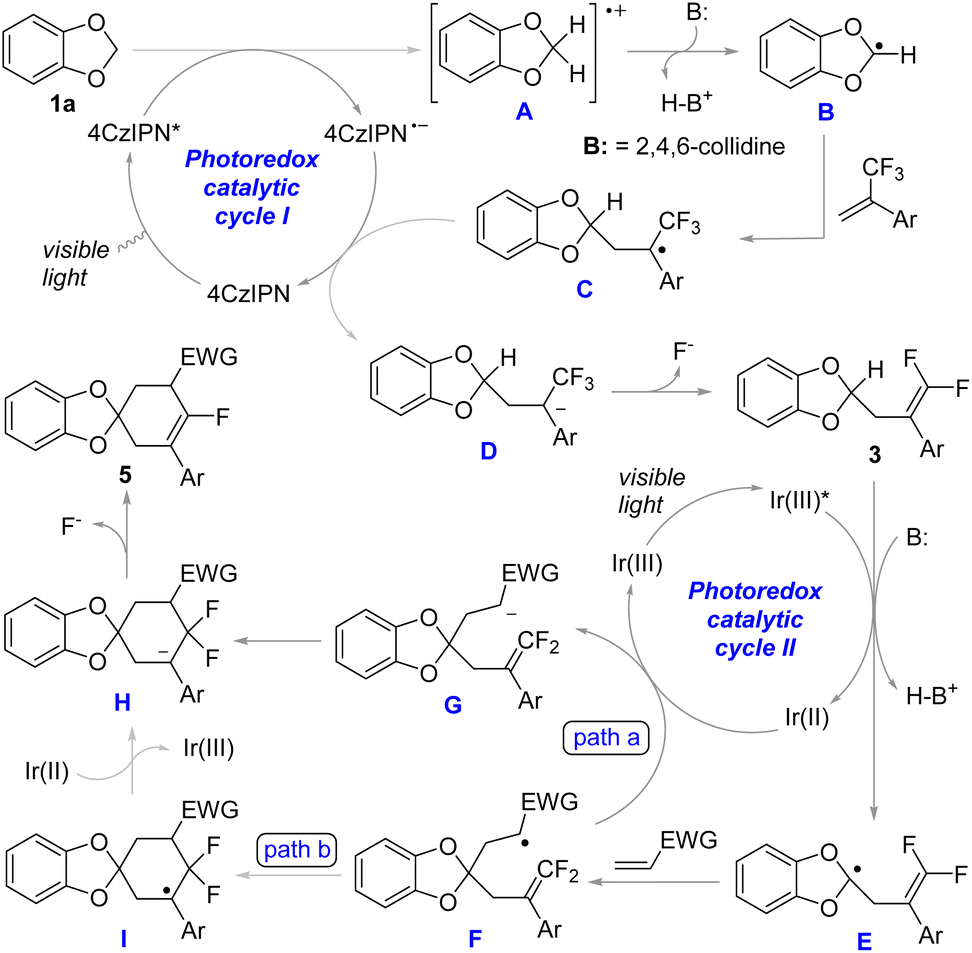 | ||
| Scheme 7 A plausible mechanism. | ||
Conclusions
In conclusion, we report that a photoredox radical/polar crossover enables gem-difunctionalization of 1,3-benzodioxoles with two different alkenes. The key 1,3-benzodioxol-2-yl radicals were produced by direct single electron oxidation rather than hydrogen atom abstraction. The photocatalytic defluorinative coupling of 1,3-benzodioxoles with α-trifluoromethyl alkenes cleaves the first C–F bond, while the second C–F bond cleavage was achieved via the annulation of the resultant γ,γ-difluoroallylated 1,3-benzodioxoles and electron-deficient alkenes. A functionalized monofluorocyclohexene framework was constructed in a formal [3 + 2 + 1] annulation process. Since α-trifluoromethyl alkenes were readily prepared from bulk industrial chemical 2-bromo-3,3,3-trifluoropropene (BTP), we wish that this dual C–F bond cleavage protocol can be a valuable alternative to the existing C–F bond formation reactions for the synthesis of interesting fluorinated compounds.29Data availability
The data underlying this study are available in the ESI.†Author contributions
J. T. discovered the reaction, performed the experiments and analysed the data. L. Z. conceived and directed the project. L. Z. prepared the manuscript while J. T prepared the ESI.† All authors discussed the experimental results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21871300 and 22271318).Notes and references
- C. Stephenson, T. Yoon and D. W. C. MacMillan, Visible Light Photocatalysis in Organic Chemistry, Wiley-VCH, German, 2018 Search PubMed.
- (a) T. Courant and G. Masson, J. Org. Chem., 2016, 81, 6945–6952 CrossRef CAS PubMed; (b) T. Koike and M. Akita, Chem, 2018, 4, 409–437 CrossRef CAS; (c) M. Patel, B. Desai, A. Sheth, B. Z. Dholakiya and T. Naveen, Asian J. Org. Chem., 2021, 10, 3201–3232 CrossRef CAS; (d) A. L. Gant Kanegusuku and J. L. Roizen, Angew. Chem., Int. Ed., 2021, 60, 21116–21149 CrossRef CAS PubMed.
- For reviews on radical/polar crossover reactions, see: (a) S. Sharma, J. Singh and A. Sharma, Adv. Synth. Catal., 2021, 363, 3146–3169 CrossRef CAS; (b) R. J. Wiles and G. A. Molander, Isr. J. Chem., 2020, 60, 281–293 CrossRef CAS PubMed; (c) L. Pitzer, J. L. Schwarz and F. Glorius, Chem. Sci., 2019, 10, 8285–8291 RSC.
- (a) L. Zhou and D. Anand, Visible light-mediated C−F bond activation, in Late-Stage Fluorination of Bioactive Molecules and Biologically-Relevant Substrates, ed. A. Postigo, Elsevier, Amsterdam, Netherlands, 2019, vol. 109, pp. 159–181 Search PubMed; (b) F. Tian, G. Yan and J. Yu, Chem. Commun., 2019, 55, 13486–13505 RSC; (c) L. Zhou, Molecules, 2021, 26, 7051 CrossRef CAS PubMed; (d) G. Yan, K. Qiu and M. Guo, Org. Chem. Front., 2021, 8, 3915–3942 RSC; (e) F. Zhao, W. Zhou and Z. Zuo, Adv. Synth. Catal., 2022, 364, 234–267 CrossRef CAS.
- T. Xiao, L. Li and L. Zhou, J. Org. Chem., 2016, 81, 7908–7916 CrossRef CAS PubMed.
- S. B. Lang, R. J. Wiles, C. B. Kelly and G. A. Molander, Angew. Chem., Int. Ed., 2017, 56, 15073–15077 CrossRef CAS PubMed.
- (a) J. P. Phelan, S. B. Lang, J. S. Compton, C. B. Kelly, R. Dykstra, O. Gutierrez and G. A. Molander, J. Am. Chem. Soc., 2018, 140, 8037–8047 CrossRef CAS PubMed; (b) C. Shu, R. S. Mega, B. J. Andreassen, A. Noble and V. K. Aggarwal, Angew. Chem., Int. Ed., 2018, 57, 15430–15434 CrossRef CAS PubMed; (c) T. Guo, L. Zhang, X. Liu, Y. Fang, X. Jin, Y. Yang, Y. Li, B. Chen and M. Ouyang, Adv. Synth. Catal., 2018, 360, 4459–4463 CrossRef CAS; (d) W. Luo, Y. Yang, Y. Fang, X. Zhang, X. Jin, G. Zhao, L. Zhang, Y. Li, W. Zhou, T. Xia and B. Chen, Adv. Synth. Catal., 2019, 361, 4215–4221 CrossRef CAS; (e) C. Shu, A. Noble and V. K. Aggarwal, Angew. Chem., Int. Ed., 2019, 58, 3870–3874 CrossRef CAS PubMed.
- (a) V. R. Yatham, Y. Shen and R. Martin, Angew. Chem., Int. Ed., 2017, 56, 10915–10919 CrossRef CAS PubMed; (b) J. Hou, A. Ee, H. Cao, H.-W. Ong, J.-H. Xu and J. Wu, Angew. Chem., Int. Ed., 2018, 57, 17220–17224 CrossRef CAS PubMed; (c) A. Hu, Y. Chen, J.-J. Guo, N. Yu, Q. An and Z. Zuo, J. Am. Chem. Soc., 2018, 140, 13580–13585 CrossRef CAS PubMed; (d) T. Ju, Y.-Q. Zhou, K.-G. Cao, Q. Fu, J.-H. Ye, G.-Q. Sun, X.-F. Liu, L. Chen, L.-L. Liao and D.-G. Yu, Nat. Catal., 2021, 4, 304–311 CrossRef CAS; (e) J.-H. Ye, T. Ju, H. Huang, L.-L. Liao and D.-G. Yu, Acc. Chem. Res., 2021, 54, 2518–2531 CrossRef CAS PubMed.
- (a) N. Katta, Q.-Q. Zhao, T. Mandal and O. Reiser, ACS Catal., 2022, 12, 14398–14407 CrossRef CAS PubMed; (b) L. Leng, Y. Fu, P. Liu and J. M. Ready, J. Am. Chem. Soc., 2020, 142, 11972–11977 CrossRef CAS PubMed.
- (a) D. Ge and X.-Q. Chu, Org. Chem. Front., 2022, 9, 2013–2055 RSC; (b) F. Tian, G. Yan and J. Yu, Chem. Commun., 2019, 55, 13486–13505 RSC.
- (a) K. Fuchibe, M. Takahashi and J. Ichikawa, Angew. Chem., Int. Ed., 2012, 51, 12059–12062 CrossRef CAS PubMed; (b) J. Yang, A. Mao, Z. Yue, W. Zhu, X. Luo, C. Zhu, Y. Xiao and J. Zhang, Chem. Commun., 2015, 51, 8326–8329 RSC; (c) J. Yang, X. Zhou, Y. Zeng, C. Huang, Y. Xiao and J. Zhang, Chem. Commun., 2016, 52, 4922–4925 RSC; (d) T. Fujita, M. Takazawa, K. Sugiyama, N. Suzuki and J. Ichikawa, Org. Lett., 2017, 19, 588–591 CrossRef CAS PubMed; (e) H. Zeng, H. Li, H. Jiang and C. Zhu, Sci. China: Chem., 2022, 65, 554–562 CrossRef CAS.
- (a) L. Li, T. Xiao, H. Chen and L. Zhou, Chem.–Eur. J., 2017, 23, 2249–2254 CrossRef CAS PubMed; (b) H. Chen, T. Xiao, L. Li, D. Anand, Y. He and L. Zhou, Adv. Synth. Catal., 2017, 359, 3642–3647 CrossRef CAS.
- (a) H. Chen, Y. He and L. Zhou, Org. Chem. Front., 2018, 5, 3240–3244 RSC; (b) Y. He, D. Anand, Z. Sun and L. Zhou, Org. Lett., 2019, 21, 3769–3773 CrossRef CAS PubMed; (c) Z. Sun and L. Zhou, J. Org. Chem., 2022, 87, 4801–4812 CrossRef CAS PubMed.
- (a) T. Ichitsuka, T. Fujita, T. Arita and J. Ichikawa, Angew. Chem., Int. Ed., 2014, 53, 7564–7568 CrossRef CAS PubMed; (b) T. Fujita, T. Arita, T. Ichitsuka and J. Ichikawa, Dalton Trans., 2015, 44, 19460–19463 RSC.
- W. Li, X. Chen and L. Zhou, Org. Lett., 2022, 24, 5946–5950 CrossRef CAS PubMed.
- (a) X. Zhang and S. Cao, Tetrahedron Lett., 2017, 58, 375–392 CrossRef CAS; (b) T. Fujita, K. Fuchibe and J. Ichikawa, Angew. Chem., Int. Ed., 2019, 58, 390–402 CrossRef CAS PubMed; (c) S. Koley and R. A. Altman, Isr. J. Chem., 2020, 60, 313–339 CrossRef CAS PubMed; (d) M. O. Zubkov, M. D. Kosobokov and A. D. Dilman, Russ. J. Org. Chem., 2021, 57, 1017–1035 CrossRef CAS.
- (a) D. Ravelli, A. Albini and M. Fagnoni, Chem.–Eur. J., 2011, 17, 572–579 CrossRef CAS PubMed; (b) J. J. Murphy, D. Bastida, S. Paria, M. Fagnoni and P. Melchiorre, Nature, 2016, 532, 218–222 CrossRef CAS PubMed; (c) A. Prieto and M. Taillefer, Org. Lett., 2021, 23, 1484–1488 CrossRef CAS PubMed; (d) W. C. Chan, J. K. Vinod and K. Koide, J. Org. Chem., 2021, 86, 3674–3682 CrossRef CAS PubMed; (e) Y.-Q. Guo, Y. Wu, R. Wang, H. Song, Y. Liu and Q. Wang, Org. Lett., 2021, 23, 2353–2358 CrossRef CAS PubMed.
- S. Li, P. W. Davies and W. Shu, Chem. Sci., 2022, 13, 6636–6641 RSC.
- A. T. Tran, N. P. West, W. J. Britton and R. J. Payne, Elucidation of mycobacterium tuberculosis type II dehydroquinase Inhibitors using a fragment elaboration strategy, ChemMedChem, 2012, 7, 1031–1043 CrossRef CAS PubMed.
- Q.-K. Kang, Y. Lin, Y. Li, L. Xu, K. Li and H. Shi, Catalytic SNAr hydroxylation and alkoxylation of aryl fluorides, Angew. Chem., Int. Ed., 2021, 60, 20391–20399 CrossRef CAS PubMed.
- J. J. Pak, J. L. Mayo and E. Shurdha, Tetrahedron Lett., 2006, 47, 233–237 CrossRef CAS.
- X. Ariza, O. Pineda, J. Vilarrasa, G. W. Shipps, Y. Ma and X. Dai, Org. Lett., 2001, 3, 1399–1401 CrossRef CAS PubMed.
- (a) J. Vacek, E. Vrublová, M. K. M. Janovská, M. Fojta, E. Šimková, J. Stýskala, J. Skopalová, J. Hrbáč and J. Ulrichová, Electroanalysis, 2011, 23, 1671–1680 CrossRef CAS; (b) S. Thiruvottriyur Shanmugam, R. Van Echelpoel, G. Boeye, J. Eliaerts, M. Samanipour, H. Y. V. Ching, A. Florea, S. Van Doorslaer, F. Van Durme, N. Samyn, M. Parrilla and K. De Wael, ChemElectroChem, 2021, 8, 4826–4834 CrossRef CAS.
- G. J. Choi, Q. Zhu, D. C. Miller, C. J. Gu and R. R. Knowles, Nature, 2016, 539, 268–271 CrossRef CAS PubMed.
- J. Luo and J. Zhang, ACS Catal., 2016, 6, 873–877 CrossRef CAS.
- (a) F. An, B. Maji, E. Min, A. R. Ofial and H. Mayr, J. Am. Chem. Soc., 2020, 142, 1526–1547 CrossRef CAS PubMed; (b) S. Tshepelevitsh, A. Kütt, M. Lõkov, I. Kaljurand, J. Saame, A. Heering, P. G. Plieger, R. Vianello and I. Leito, Eur. J. Org. Chem., 2019, 6735–6748 CrossRef CAS.
- Internet Bond-energy Databank (pKa and BDE)−iBonD Home Page, https://ibond.nankai.edu.cn Search PubMed.
- F. Parsaee, M. C. Senarathna, P. B. Kannangara, S. N. Alexander, P. D. E. Arche and E. R. Welin, Nat. Rev. Chem., 2021, 5, 486–499 CrossRef CAS PubMed.
- During the review process of this work, a similar photocatalyzed cascade cyclization of gem-difluoroalkenes and α,β-unsaturated carbonyl compounds for the synthesis of monofluorocyclohexenes was reported: Z. Li, Y. Zhang, Y. Zhang, X. He and X. Shen, Angew. Chem., Int. Ed., 2023, e202303218 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc00912b |
| This journal is © The Royal Society of Chemistry 2023 |