
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelf-assembly of achiral building blocks into chiral cyclophanes using non-directional interactions†
Yuan
Zhang‡
a,
Benjamin
Ourri‡
 b,
Pierre-Thomas
Skowron
a,
Emeric
Jeamet
b,
Titouan
Chetot
b,
Christian
Duchamp
b,
Ana M.
Belenguer
c,
Nicolas
Vanthuyne
a,
Olivier
Cala
d,
Elise
Dumont
ef,
Pradeep K.
Mandal
g,
Ivan
Huc
g,
Florent
Perret
b,
Laurent
Vial
*b and
Julien
Leclaire
*b
b,
Pierre-Thomas
Skowron
a,
Emeric
Jeamet
b,
Titouan
Chetot
b,
Christian
Duchamp
b,
Ana M.
Belenguer
c,
Nicolas
Vanthuyne
a,
Olivier
Cala
d,
Elise
Dumont
ef,
Pradeep K.
Mandal
g,
Ivan
Huc
g,
Florent
Perret
b,
Laurent
Vial
*b and
Julien
Leclaire
*b
aAix Marseille Univ, CNRS, Centrale Marseille, iSm2, Marseille, France
bUniv Lyon, Univ Lyon 1, CNRS, INSA, CPE, ICBMS, F-69622 Lyon, France. E-mail: laurent.vial@univ-lyon1.fr; julien.leclaire@univ-lyon1.fr
cYusuf Hamied Department of Chemistry, University of Cambridge, Lensfield Road, Cambridge CB2 1EW, UK
dInstitut des Sciences Analytiques, UMR 5280 CNRS, Université Claude Bernard Lyon, Lyon, France
eENSL, CNRS, Laboratoire de Chimie UMR 5182, 46 allée d’Italie, 69364 Lyon, France
fUniversité Côte d’Azur, CNRS, Institut de Chimie de Nice, UMR 7272, 06108 Nice, France
gDepartment of Pharmacy and Center for Integrated Protein Science, Ludwig-Maximilians-Universität, Butenandtstr., 5–13, 81377 München, Germany
First published on 24th May 2023
Abstract
The diastereoselective assembly of achiral constituents through a single spontaneous process into complex covalent architectures bearing multiple stereogenic elements still remains a challenge for synthetic chemists. Here, we show that such an extreme level of control can be achieved by implementing stereo-electronic information on synthetic organic building blocks and templates and that non-directional interactions (i.e., electrostatic and steric interactions) can transfer this information to deliver, after self-assembly, high-molecular weight macrocyclic species carrying up to 16 stereogenic elements. Beyond the field of supramolecular chemistry, this proof of concept should stimulate the on-demand production of highly structured polyfunctional architectures.
Introduction
From bioactive compounds to materials, the stereochemical features of molecules are known to be critical to their properties. With the rise of organometallic and organic catalysis, the current synthetic toolbox allows chemists to set on demand the configuration of almost any stereogenic element of small drug-like molecules during their construction. Yet, molecular precursors and synthetic intermediates still have to be assembled step by step, each of which generally requires a tailored stereochemical input (which may involve a complex formulation of catalysts and additives) and a tedious purification procedure.1 At the other end of the spectrum of chemicals which meet a strong demand lie stereoregular organic polymers. If they can advantageously be produced by a one-pot approach, the trade-off is their inevitable stereo-dispersity and the narrow functional and configurational variety, the latter being limited to either iso-, syndio- or a-tacticity, far from the level of control that can be reached for small molecules.2In between these two worlds of small molecules and polymers lie (cyclo-)oligomeric architectures, which are the objects of the supramolecular world. Two macrocyclic species formed from achiral constituents currently stand as blockbuster host molecules: pillararenes and cucurbiturils.3,4 Assembling their respective flat or curved monomeric units enables steric repulsion to guide elongation and/or ring closure towards the least hindered stereo-regular species (Fig. 1i).5,6 If this selectivity is high, accessing alternative diastereoisomers remains a challenge, as the harsh assembling conditions limit the alternative interactions which may be used to gather the intermediates around a molecular partner and divert the stereochemical outcome of the synthetic process. This is where dynamic combinatorial chemistry (DCC), which proposes to reversibly assemble building blocks under mild conditions, should find all its use.7 Within the pool of reversible linkages used in DCC, the disulfide bond—a critical moiety in protein folding—is known to adopt two enantiomeric stable conformations due to lone pair repulsion between the sulfur atoms.8 Despite its central biological role, the mechanism by which it relays stereochemical information between asymmetrically folded peptide domains remains to be elucidated. At the molecular level, Feringa recently showed that the central chirality of a neighboring amino acid could induce, thanks to a hydrogen-bond relay, the asymmetric folding of the disulfide linkage within a 5-membered ring in an organic medium (Fig. 1ii).9 At the supramolecular level, some of us recently reported the template-free diastereoselective assembling of helical foldamers into homochiral disulfide-based cyclo-dimers.10 When we re-examined a posteriori the structure obtained in silico and by X-ray crystallography, we noticed that the disulfide bridges played a non-innocent role in the propagation of stereochemical information. In fact, the stereochemistry of this linkage (P/M) matches that of the neighbouring oligoamide building blocks (Fig. 1iii).
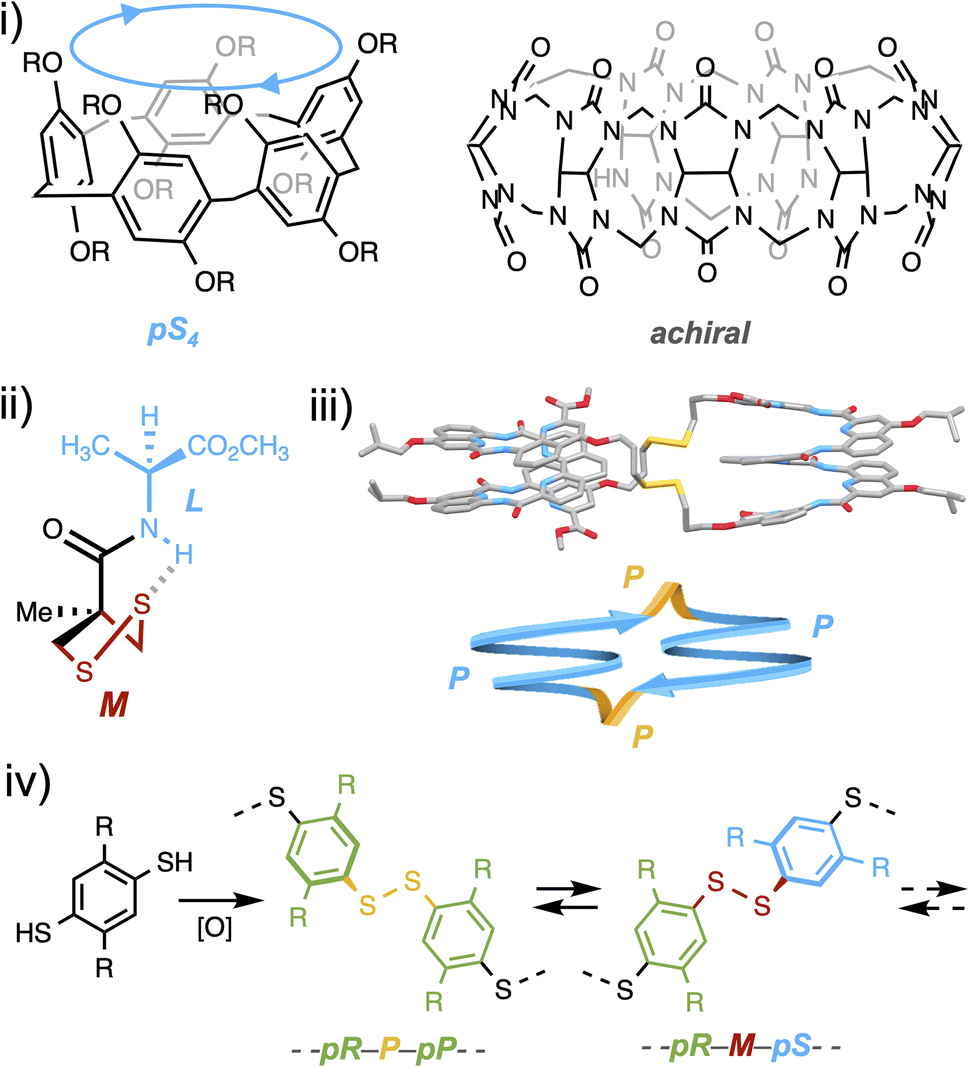 | ||
| Fig. 1 (i) Isotactic (pR)n/(pS)n and all-C facial configurations in pillararenes and cucurbiturils, respectively, (ii) hydrogen-bond mediated asymmetric folding of a disulfide linkage within a 5-membered ring, (iii) stereochemical match in disulfide-based cyclodimers, and (iv) creation of two classes of stereogenic elements during the self-assembling of dyn[n]arenes. | ||
Yet, the rationale for enabling to program on demand the configuration and conformation of an oligomeric species by exploiting the stereochemical features of components (building blocks, linkages and, if any, templates) has remained out of reach so far but has clearly been identified as one of the next major challenges in supramolecular chemistry.11 In particular, the role and relative configuration of the disulfide linkage between two stereogenic monomeric units have never been fully explored, although it spans well beyond supramolecular chemistry and may dramatically influence protein structures and functions.12 In the field of disulfide based macrocycles, which can be assembled by dynamic combinatorial strategies, examples of stereoselective assembling remain scarce and almost systematically involve chiral building blocks.13 During the past decade, some of us have designed and studied disulfide-based analogues of pilla[n]rarenes named dyn[n]arenes.14 Interestingly, thiol to disulfide cyclo-oligomerization of 1,4-bisthiophenols is accompanied by the creation of two classes of stereogenic elements: monomeric faces and the disulfide bridges linking them (Fig. 1iv).15 A central question is how to program their respective stereochemistry beforehand. In the present study, we demonstrate that non-directional attractive and repulsive interactions can be exploited as a reliable input to control on demand the configurational and conformational identities of dynamic covalent cyclophanes in water.
Results and discussion
Template-assisted stereochemical information transfer
Recently, we showed that the diversity of the libraries generated from building block A (Fig. 2), both in terms of macrocycle ring size and facial diastereoisomers, is largely underestimated by chromatographic analyses.16 Although this technique is universally adopted for disulfide-based dynamic combinatorial libraries, it only enables the detection of a trace amount of the homochiral tetramer A4 (i.e., the strict configurational analogue of pillar[4]arenes of pR4/pS4 configuration) within a mixture which is in reality, highly dominated by the homochiral trimer A3 and heterochiral tetramers A4 (each including at least one pR–M–pS sequence as in Fig. 1iv).17,18Fig. 2 gathers the possible combinations of pR and pS configurations arising from the positioning of prochiral monomer faces in- or outward with respect to the cavity (horizontal coordinates, A4-n (n = 1–4)) with P and M conformations of the disulfide bridges (vertical coordinates, conformers A4-x (x = a–c)) to afford a virtual combinatorial system of 16 tetramers A4. The first question that we addressed within the framework of the present study was to assess whether the stereogenic elements of these self-assembled cyclophanes (i.e., facial pR/pS configurations vs. M/P disulfide bridge conformations) were interrelated. This issue is truly central, as it probes the potential propagation of stereochemical information between monomer faces and linkages during the reversible oligomerization process. The structures of the cyclo-tetramers of pR4/pS4, pR3pS/pS3pR, and (pR)2(pS)2 configurations noted respectively in A4-a, A4-b and A4-c in Fig. 2 (no (pSpR)2 tetramer could be detected upon thiol-to-disulfide oxidation of A) were modelled by a combination of DFT and MD calculations (see ESI, section II.1.e†). These two classes of simulations respectively yielded the populations of the n most stable conformers (A4-xn (x = a–c and n = 1–4)) corresponding to each configuration, as well as an evaluation of their conformational flexibility (Fig. 2). The structures obtained and the repartition of the dihedral angles of the disulfide bridges of each species are gathered in Tables S4–S9.† The first main piece of information which could be extracted from the calculations was that heterochiral species A4-b and A4-c have a marked tendency to adopt a flattened conformation wherein the cavity is too narrow to be accessed by any guest whatever its size (Fig. S10 and S11†). This fully agrees with the external binding mode observed experimentally with polyamine partners.16 This also indicates that the formation of a pseudo-rotaxane by complex guest inclusion does not represent a sufficient energetic benefit to compensate for the unfavourable flattened-to-open induced fit. The second main observation we made is that all diastereoisomers A4-x (x = a–c), despite splitting each into several conformers A4-xn (n = 1–4) of various stabilities, share a common configurational-conformational inter-dependence: pR–P and pS–M facial–dihedral combinations seem to be systematically the most stable for steric reasons. When conflicts arise (a disulfide bridge surrounded by a pR and a pS face), it forces the –S–S– linkage to adopt a cis conformation, which eventually explains the flattened shape observed on heterochiral species A4-b and A4-c.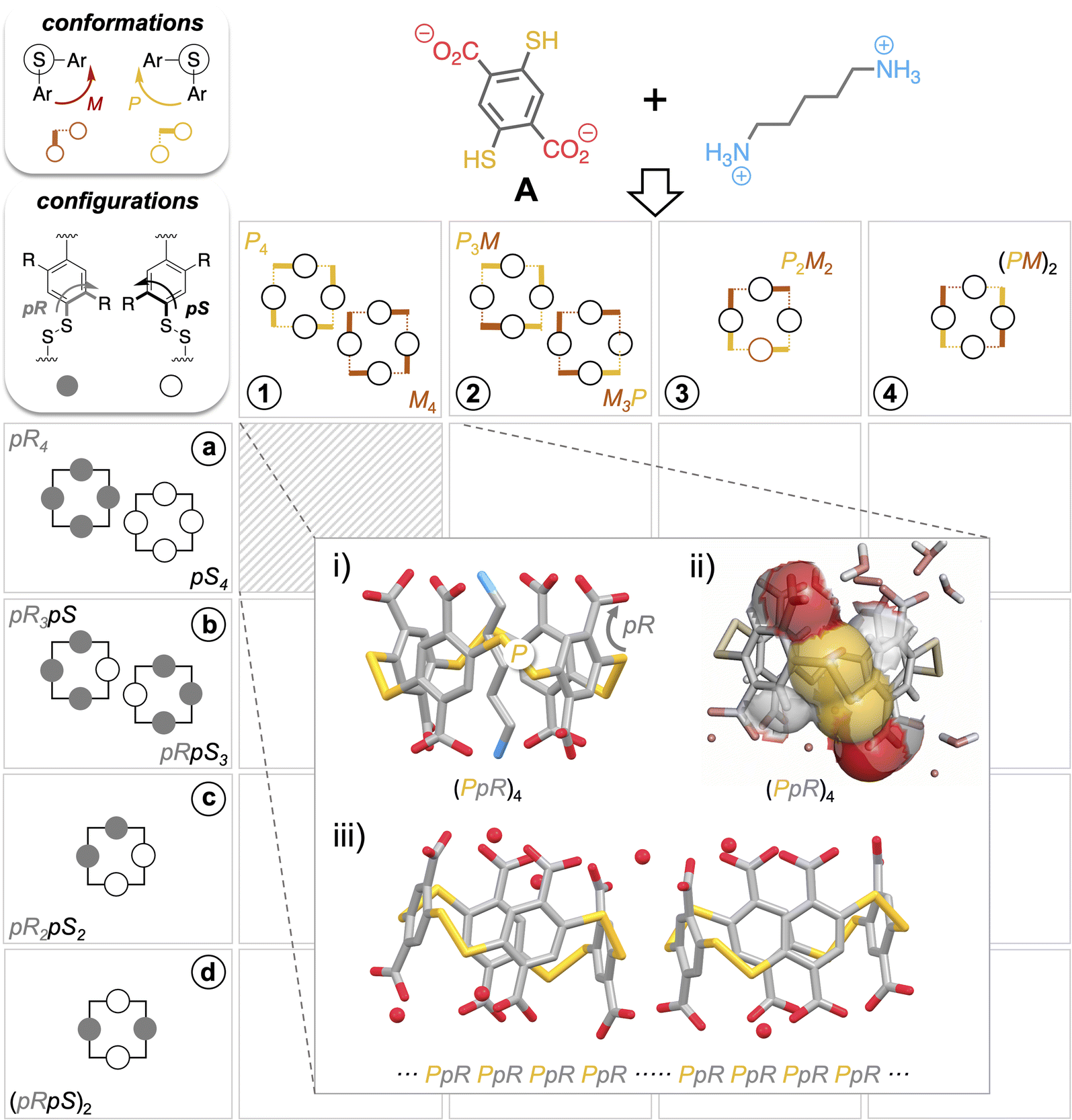 | ||
| Fig. 2 Combinations of pR and pS configurations arising from the positioning of prochiral monomer faces in- or outward with respect to the cavity (horizontal coordinates) with P and M conformations of the disulfide bridges (vertical coordinates) to afford a virtual combinatorial system of 16 tetramers A4. Templating by polyamines quantitatively yields a (pS–P)4 and (pR–M)4 racemic mixture: (i) crystallographic structure of its complex with cadaverine, (ii) crystallographic structure of the free macrocycle displaying van der Waals contacts between the substituents of the phenyl rings and the sulfur atoms, and (iii), conglomerate crystallization of the free macrocycle wherein homochiral packing occurs. | ||
While the virtual sub-library of cyclotetramers potentially encompasses 16 combinations of configurations and conformations, this stereochemical inter-dependence guides the real library toward the (xn) = (a1), (b2), (c3) diagonal on the gridded landscape in Fig. 2 (see ESI, section S.II.1.e†). The disulfide bond is not simply an a ± 90° bridging unit between phenylic monomers; it also actively relays the facial stereochemical information between neighbouring units (i.e., P and M bonds being respectively a relay between pR only and pS only faces).
The interconnection between the two levels of stereochemical information borne by dyn[4]arenes was confirmed experimentally on the homochiral A4 of pR4/pS4 configuration and P4/M4 conformation (x = a, n = 1). As previously reported, this cavitand can be quantitatively and diastereoselectively self-assembled, at the expense of other ring sizes and conformations, using biogenic polyamines as templates (Fig. S4 and S5†).19,20 Such an object combines the ideal cavity size and carboxylate group layout (at the vertices of its parallelepipedal structure) to optimize the binding of poly-ammonium through the formation of salt bridges (Fig. 2i).21 Our previously published optimized procedures enable on demand either the pure pseudo-rotaxane complex or the free host upon guest removal to be obtained.18 Both architectures were submitted to a crystallogenesis campaign, which eventually afforded samples eligible for structural elucidation by X-ray diffraction analyses. Guest-containing and free structures (Fig. S6–S8†) support and validate a posteriori the modelling data.
As expected, the template molecules imprint their main stereo-electronic feature (a linear backbone terminated by two electron poor moieties) into the assembled host (a parallelepipedal cavitand displaying a stereoregular layout of carboxylate groups, Fig. 2i). While the guest intermolecularly transfers its stereochemical information into the host by selecting its size and facial configuration, this host configuration then intramolecularly selects the optimal disulfide conformation. In fact, the steric hindrance of the carboxylate groups propagates along the vertical edges of the parallelepipedal cavity, positioning each S–S linkage right in the axis formed by the two neighbouring carboxylates (Fig. 2ii).22 Eventually, only the pR–P and pS–R pairs of stereoisomers are observed, in full agreement with the computational model.
Remarkably, not only simple guest molecules can determine up to 8 stereochemical elements on their self-assembled host along 24 bonds, but this information is recorded (i.e., both configurations and conformations remain implemented into the host upon guest departure) as confirmed using the X-ray structure of the free host (Fig. S6†). Fortuitously, the informational input initiated by the guest propagates beyond the individual host during subsequent crystallogenesis, as a conglomerate (i.e., crystals in which cyclophanes of (pR–P)4 and (pS–M)4 segregate) is obtained (Fig. 2iii). A third stereoselective self-assembling process takes place consecutively to the templating step: the self-sorting of nearest neighbour cavitands of identical stereochemistry.
Template-free stereochemical information transfer
From this first example of guest-to-host transfer of 8-bits of stereochemical information, we envisaged the possibility of extending the concept to any possible guest-free selection mode in DCC (i.e., folding and stacking). To do so, information should be encoded into the building blocks themselves through their stereochemical features exclusively. In the first generation of dynarenes based on A, the stereochemical information was able to propagate along the edges of the monomeric faces through steric interactions (Fig. 2ii). In a second-generation system, the same class of challenging non-directional interactions was conserved but implemented at different positions of the building blocks B and C (Fig. 3). The three-dimensional structure of these building blocks was inspired by Schraders' molecular tweezers which are equipped with benzene walls and norbornadiene edges.23 With this design, three groups differing in bulkiness are borne by the cis and trans staircase-shaped monomers B and C: the pair of axial C–Ha bonds involving the carbon at the junction of the norbornene and cyclobutene rings is the bulkiest class (cyan), the methylene CH2b bridge of the norbornene is slightly less hindered and ranks second (green), and finally the lateral C-Hc groups (grey), which are parallel to the C–S bond or the thiol group are the least hindered. Yet, these were hypothesized to be sufficiently bulky to conformationally freeze the disulfide bridge on the NMR timescale. When the bulkiest CHa and CHb units are borne on different faces (such as in monomer B), thiol to disulfide cyclo-oligomerization should in principle stereoselectively deliver convex dyn[n]arenes of (In)n configuration (Fig. 3i and ii). In this case, the bulkiest C-Ha group prevents concave ring closure. In addition, disulfide bridges should adopt M–P alternate conformations to avoid contact between protons Hb and Hc borne by nearest neighbours. In contrast, cyclo-oligomerization should be prohibited when both types of bulky units Ha and Hb are present on each face such as in monomer C.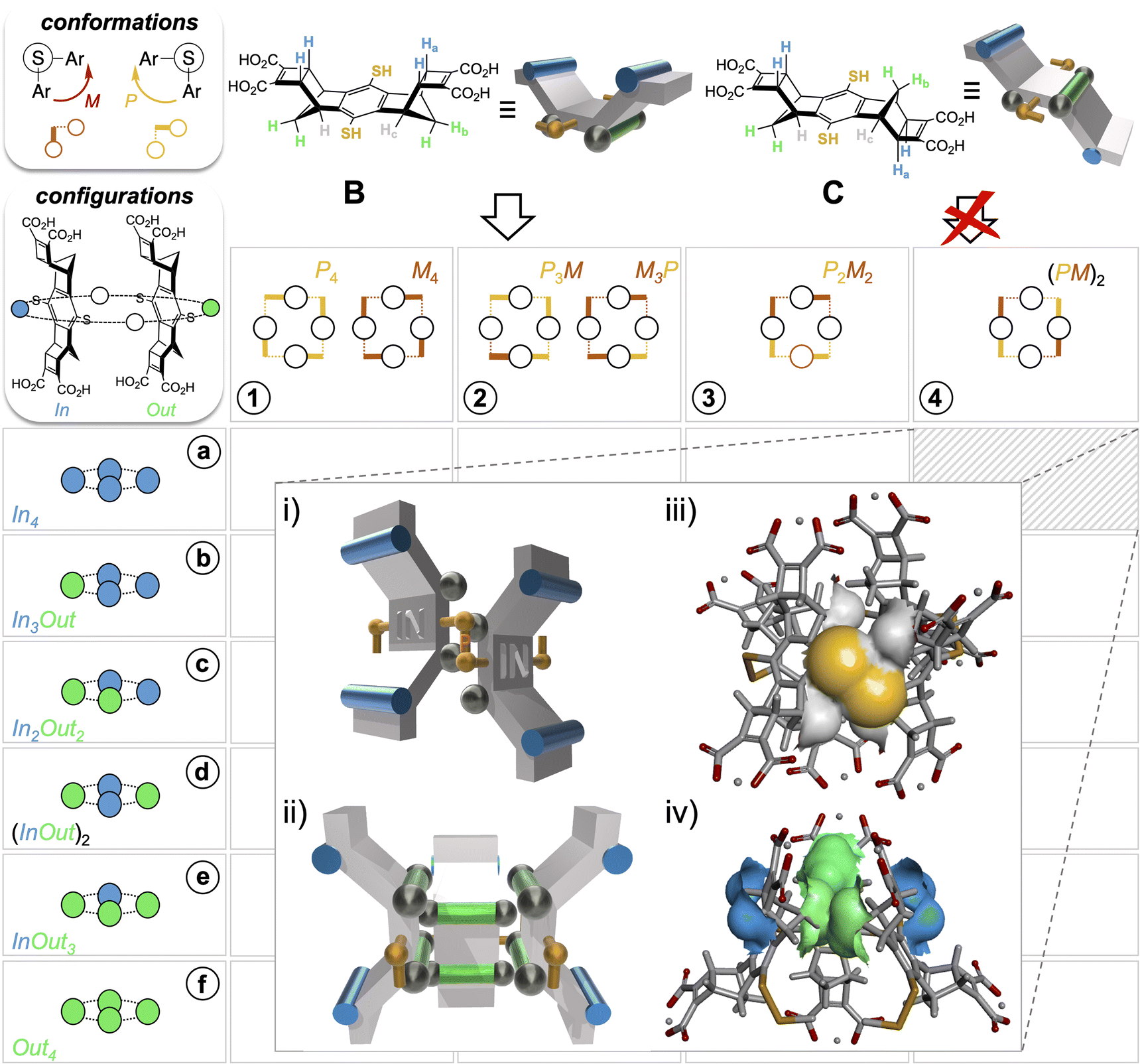 | ||
| Fig. 3 Principle of stereochemical information encoding by self-assembling of hindered bisthiophenol monomers B and C: (i and ii) schematic representation showing axial protons (cyan: methine C-Ha; green: methylene C-Hb) hindering the concave face of B and both faces of C (preventing the cyclo-oligomerization of the latter), and axial C-Hb (green) and equatorial C-Hc (grey) protons restricting disulfide dihedral rotation, and (iii and iv) DFT modelling at the M06 level with van der Waals contacts supporting the design. | ||
The synthesis of these new monomers B and C relied on a sequence of successive metal-free and metal-catalyzed cycloadditions, starting from simple hydroquinone (see Scheme S1 and ESI, section S.II.2.a†). Diastereoisomer separation of quinone-based intermediates was conducted by chiral chromatographic means at the middle of this synthetic sequence (see ESI, section S.I.2†). To enable the quantitative formation of discrete cyclic species rather than polymers,24 libraries were set in the millimolar range from building block B or C. Slightly acidic pH of 5.5 was chosen so that the monomeric units were mono(de)protonated at each dicarboxylic end (Fig. 4iv), thereby yielding soluble oligomers within which hydrogen bonds may potentially be shared between the nearest monomeric neighbours. This intermediate protonation state was verified a posteriori by the potentiometric titration of the isolated cyclo-oligomers obtained (Fig. S108 and S109†). Monitoring of the aerobic thiols-to-disulfide based cyclo-oligomerization by (−)-ESI-MS allowed us to qualitatively validate our design. With building block B, disulfide-based linear oligomers formed in increasing amounts during the first 4 hours. These oligomers then progressively decayed as they converted into their macrocyclic homologues (Fig. S85–S88†). A first phase of thiol-to-disulfide-based chain growth was similarly observed on C, but the scenario diverged during the second step. In fact, ring closure was replaced by overoxidation of the disulfide linkages (into thiolsulfinates) on all linear strands produced. The resulting species subsequently underwent cleavage into smaller linear fragments, which themselves were further overoxidized (from sulfenic to sulfonic acids, Fig. S89–S93†). Remarkably, no trace of such overoxidized species could be detected by ESI-MS in libraries formed from B. Beyond qualitatively validating our design, this preliminary analysis indicates that disulfide-based species may correspond to metastable redox states in some circumstances and therefore may require to be further stabilized to truly become thermodynamic products. Under the present conditions (absence of template and self-aggregation), stabilization must originate from intramolecular non-covalent interactions within the macrocyclic species Bn.
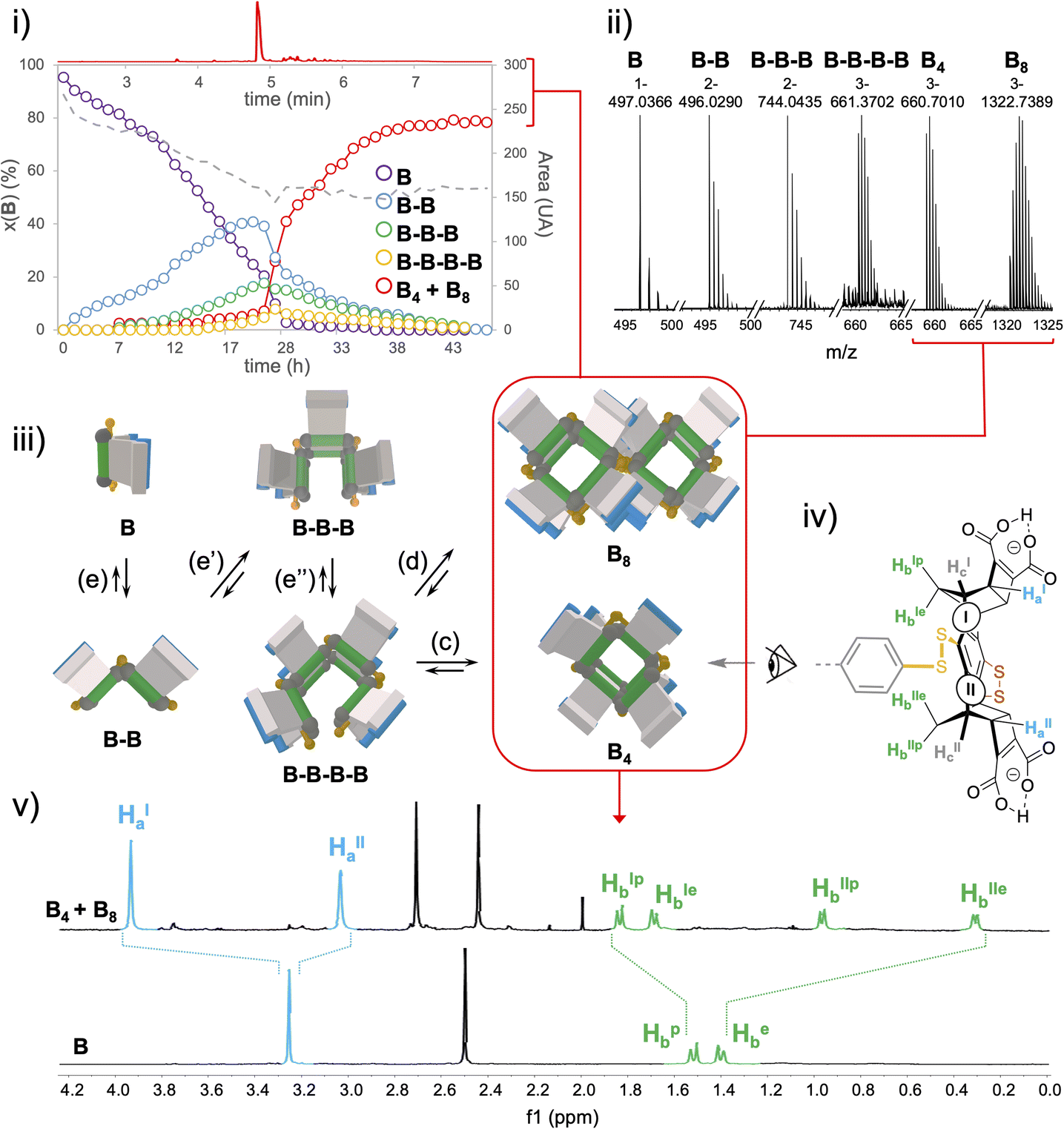 | ||
| Fig. 4 UPLC-MS (i) and ESI-MS (ii) analysis of the dynamic combinatorial library generated from monomer B (4 mM) in 50 mM AcONH4 at pH 5.5, (iii) schematic representation of the linear and cyclic species quantified and characterized by UPLC-UV and ESI-MS, (iv) monomeric unit displaying two sets of Ha–c protons (noted I and II), and (v) snapshot of the 1H NMR spectra of starting material B and final product B4 + B8. | ||
In-depth compositional and structural analyses of the architectures formed from monomer B were conducted by a combination of computational and experimental techniques. UPLC-UV/ESI-MS monitoring over time first provided a quantitative assessment of the populations of linear and cyclic objects produced throughout the oxidation process (Fig. 4i and ii and S94–S97†). It appeared that linear species of increasing chain length (from dimer B–B to tetramer B–B–B–B) are progressively generated from monomer B during the first 24 hours while the cyclic species, although detectable in ESI-MS by direct infusion (vide infra), remain limited to trace amounts. In fact, macrocyclization, which may occur upon ring closure of a single linear strand or dimerization of two single strands (eqn (c) and (d), Fig. 4iii) effectively takes over the elongation phase (eqn (e)–(e′′), Fig. 4iii) after 24 h, yielding quantitatively a single cyclo-tetrameric product noted B4. Intriguingly, the corresponding mass spectra indicated the presence of [B8-3H]3− and [B8-5H]5− ions (Fig. S104†). Careful examination of the isotopic patterns corresponding to [B4-2H]2− and [B4-3H]3− revealed that each of these respectively interfered with those of [B8-4H]4−and [B8-6H]6− (Fig. S103†). Given the octa-anionic nature of B4 at pH 5.5, formation of a non-covalent dimeric B4–B4 complex which may survive the ionization process seemed highly unlikely. Although a single and relatively sharp peak was obtained in UPLC after equilibration of the library (and remains stable over more than two weeks), it was ascribed to the co-elution of the cyclic tetramer B4 and octamer B8 (Fig. 4i–iii and S96†).
MS/MS fragmentation of the [B8-3H]3− ion systematically delivered the [B–H]− monomeric species, which is consistent with a covalent octamer (Fig. S105†). Despite a thorough screening of all currently available chromatographic phases, any attempt to separate these two species, which are presumably obtained from the same tetrameric linear parent B–B–B–B by ring closure (Fig. 4iii, eqn (c)) or cyclo-dimerization (Fig. 4iii, eqn (d)), remained fruitless. Similarly, our generic procedure of dynarene isolation by precipitation in an acidic medium proved ineffective in separating both species. The mixture obtained upon equilibration was thus analyzed as such by multi-nuclear and multi-dimensional NMR (ESI, section S.II.2.f†).
Quantitative 1H and 13C spectra recorded in the presence of an internal standard indicated the presence of a single well-resolved major species (approx. 80% molar, Fig. S120†), involving two sets of Ha–c magnetically different protons (noted I and II, see Fig. 4iv and v). COSY and NOESY experiments confirmed that these two sets are geometrically isolated, while heteronuclear 2D experiments (HSQC and HMBC) enabled the assignment of the signals within each set to one of the Ha–c classes (Fig. S112–S117†).
Finally, DOSY confirmed that these two sets belong to the same molecular entity, which displays a hydrodynamic radius of 9.08 ± 0.75 Å (as a comparison, A4-a's radius is 8.30 ± 0.80 Å and the radius of monomer B is measured by the same technique which is around 5.71 ± 0.07 Å under the same conditions, see Fig. S76 and S118, Tables S10 and S11†). These analyses enabled the scope of B4 cyclotetramer candidates (those displaying two geometrically distinct Ha-c units) to be narrowed to the meso-(In-M–In-P)2 and the meso (Out-M–Out-P)2 species and the racemic (In-P–Out-P)2 (In-M–Out-M)2 homochiral mixture. These three stereochemical combinations are located on the gridded landscape of all possible configurations and conformations summarized in Fig. 3 at the (a, 4), (f, 4) and (d, 1) coordinates respectively. Structurally speaking, they strongly differ regarding the degree to which the Ha and Hb protons are exposed to the outer solution.
Experimentally, it appears that macrocyclization of B induces the shielding and deshielding of Ha and Hb protons respectively, with a splitting within each class (Fig. 4v). This supports the hypothesis of a B4 diastereoisomer bearing Ha (and Hb protons) which all point outward (respectively inward) with respect to the macrocyclic cavity, wherein two distinct local environments can be found.
Assessment of the stability of each diastereoisomer of cyclotetramer B4, all of which are gathered on the map displayed in Fig. 3, was performed using DFT calculations (structure optimization with three alternative functionals, followed by single point (SP) energy calculation, see Section S.I.8 and Table S12†). Comparison of the energy levels, which is displayed in Fig. S127† unambiguously reveals that the (In-M–In-P)2 diastereoisomer is the most stable species and should experimentally dominate the sub-library of B4 stereoisomers obtained from B. In addition to complementing and refining the NMR analyses, this modelling campaign also provided some insight on the main contributions to the stability of the species. In agreement with our design, it appears that orienting the Ha-bearing concave faces toward the cavity (i.e., the Out configuration) destabilizes the corresponding architectures for steric reasons. Stabilizing London dispersion forces follow the same evolution (increasing in strength with the number of Out facial configurations) but their amplitude is insufficient and fails to compensate for the steric repulsion. To minimize steric clash between Hb protons of facing monomeric units (which are in van der Waals contact, see Fig. 3iv), the macrocyclic structure adopts a distorted octahedral shape. Finally, Hb and Hc protons play their ascribed roles as disulfide bridge instructors. Energy calculation validates a posteriori the encoding process, i.e., the effective translation of the stereochemical information contained in B by the assembling algorithm (the disulfide bond formation/exchange operating under the guidance of steric interactions). DFT modelling indeed confirms that the MPMP conformation is systematically favoured within each configurational series. In the case of the (In)4 configuration, which is effectively selected, this conformation minimizes the hindrance between Hb protons from neighbouring monomeric units. Finally, molecular dynamics confirmed that Hc provides sufficient hindrance in the vicinity of the S–S bridges to freeze and lock their conformation (see Fig. 3i and ii), at least on the millisecond NMR timescale.
At this stage, it should be mentioned that NMR spectra of equilibrated libraries either analysed directly (in situ) or after acid-induced precipitation and redissolution in a neutral medium (ex situ) both display sharp signals corresponding to meso-(In-M–In-P)2-B4 which account for 80% of the total material, while the remaining 20% imputed to the B8 adduct detected by ESI-MS are dispatched into broad signals buried in the baseline (Fig. S119 and 120†). We first investigated the impact of the amphoteric salt used for pH regulation on the outcome of the library, by changing either its anion or cation while maintaining the pH of the library at 5.5. UPLC-analysis did not reveal any deviation from the scenario of B4/B8 selective co-production (Fig. S96†). To rule out the possibility of an interconversion between B4 and B8, which would be driven by precipitation and therefore remain undetectable upon redissolution, we directly analysed the solid material collected by precipitation by MALDI-MS spectroscopy and solid-state NMR (Fig. S106 and S107†). Apart from a line broadening phenomenon, solid and liquid state spectra can be superimposed, thereby ruling out the aforementioned hypothesis. As B4 and B8 should result from the same B–B–B–B precursors through a monomolecular (ring closure) or bimolecular (dimerization) pathway, we reasoned that experimental parameters known to affect aggregating processes such as temperature, stirring rate or concentration may deviate the final proportions of B4 and B8 within the libraries from an 80/20 ratio.
Increasing incubation temperature (from 25 °C to 75 °C) or the stirring rate (from 0 to 1000 rpm) accelerated both elongation and cyclization phases, resulting in faster equilibration but the final mixtures remained identical in terms of composition (see Fig. S97† for UPLC-MS analyses). Increasing the concentration of the starting material from 4 to 32 mM induced a similar accelerating effect but also resulted in the formation of a solid material after several hours (above 15 mM, see Fig. S84†). This solid, which was filtered and dried, accounted for at best 10% of the total mass. It proved to be insoluble in all conventional solvents and no clear data could be extracted from ESI-MS or liquid-state NMR analyses upon partial dissolution. Solid state NMR delivered 13C CP-MAS spectra displaying broad peaks (Fig. S126†), indicating that this sample differs from the B4 + B8 fraction collected upon acidification but could not be identified.
As the analyses of the supernatant by ESI-MS and multinuclear NMR were similarly inconclusive (detection of the [B8-3H]3−, [B8-5H]5− and [B4-3H]3− ions for the former and faint broad signals merged with the baseline accompanying the sharp peaks corresponding to B4 for the latter, see Fig. S111 and S119†), we performed some semi-quantitative ESI-MS sampling during the assembling and precipitating phase. Quantification of the intensities and areas of the isotopic patterns of identical m/z values corresponding to [B4-nH]n− and [B8-2nH]2n− with time was performed after a preliminary validation by calibration (Fig. S121–S124†), i.e. injection of precise and increasing amounts of B4/B8 mixtures whose proportions had first been determined by NMR analyses (Fig. S120†). For libraries whose concentration in B was below the 15 mM threshold value, this kinetic monitoring revealed that the B8/B4 ratio increases with time during the cyclization regime, reaching a 0.25 asymptotic value which agrees well with NMR. In contrast, more concentrated samples led to higher proportions of B8 (up to 0.45), which abruptly dropped to the same asymptote amount (0.25) as soon as precipitation occurred (Fig. S125†).
These observations provided the framework for a crystallogenesis campaign based on supersaturated solutions prepared by re-dissolution of the solid B4 + B8 material collected after the full sequence of (1) thiol to disulfide assembling under dilute conditions and (2) acid precipitation. To our delight, single crystals eligible for structural elucidation by X-ray diffraction analysis could be collected after screening (Fig. S129†). It should be noted that any attempt to isolate them from the mother liquor led to their collapse and prevented us from recording MS or NMR spectra in the liquid state. The three-dimensional structure of this crystalline material obtained from supersaturated mixtures of B4 and B8 is shown in Fig. 5. In agreement with ESI-MS monitoring during the assembling step and with CP-MAS NMR of the solid precipitate, this analysis confirms that the insoluble material corresponds to a covalent octamer B8 (and rules out the unlikely hypothesis of a complex between two B4 species). The shared filiation of B4 and B8 with B–B–B–B is confirmed by the eight-shaped structure of B8 whose octahedral subunits (Fig. 5i, grey) almost perfectly superimpose with B4 (Fig. 5i, pink). This solid-state structure also reveals how the molecular information we encoded into the monomer is handled during the dimerization of B–B–B–B into B8, which competes with the ring closure into B4, i.e. when two linear tetramers are to be fused. As with B4, the stereochemical information encoded in the monomeric unit is stereoselectively translated into the cyclo-octamer both at the configurational and conformational levels. An all-In facial configuration, wherein the bulkiest Ha protons point toward the outer medium (Fig. 5ii, cyan) and the slightly less bulky Hb (Fig. 5b, green) are oriented inward is indeed exclusively obtained. Regarding disulfide bridge conformation, it is confirmed that the M/P alternance is the only combination which accommodates the hindrance borne by the building blocks, even on a larger ring size (Fig. 5iii and S130†). As in B4, the unusual eight-shaped folding finds its origin in the interplay between intramolecular attractive and repulsive interactions. Yet, careful examination of the packing between (M-In–P-In)4-B8 homologues reveals that intermolecular interactions in the solid lattice should act here as an additional force which drives the three-dimensional folding. In the equatorial plane of the macrocyclic ring, contacts between B8 molecules occur at the tip of their long sides, i.e. between the monomeric units which are not engaged into the junctions between B4 subunits, despite unexpected stacking of their Ha-bearing cyclobutene moieties (Fig. 5iv). At each pole of the B8 structure, 2 monomers are paired through this unusual interaction with a different B8 neighbour (Fig. S132†). Repetition of this pattern along the xy plane yields cyclo-trimeric and hexameric superstructures displaying respectively triangular and circular cavities (Fig. 5e and S131†). In the z-direction, stacking of the B8-layers occurs through H-bond pairing between the carboxylic moieties of homologous monomers (Fig. 5iv, red). The carboxylic paring concerns the four central building blocks of the 8-shaped B8 ring, i.e. the zone resulting from the cyclodimerization of two linear B–B–B–B tetramers. This analysis reveals that stacking in the solid phase, especially along the z axis, may be the driving force underpinning the unexpected formation and selection of a large ring size (which would be otherwise entropically unfavoured25). Eventually, our building block design, which relies on the rational introduction of bulky groups of varying hindrances, effectively enables 8 and 16-bits of (In-M–In-P)n stereochemical sequences to be encoded into dyn[4]- and dyn[8]-arenes, respectively in solution and in the solid state.
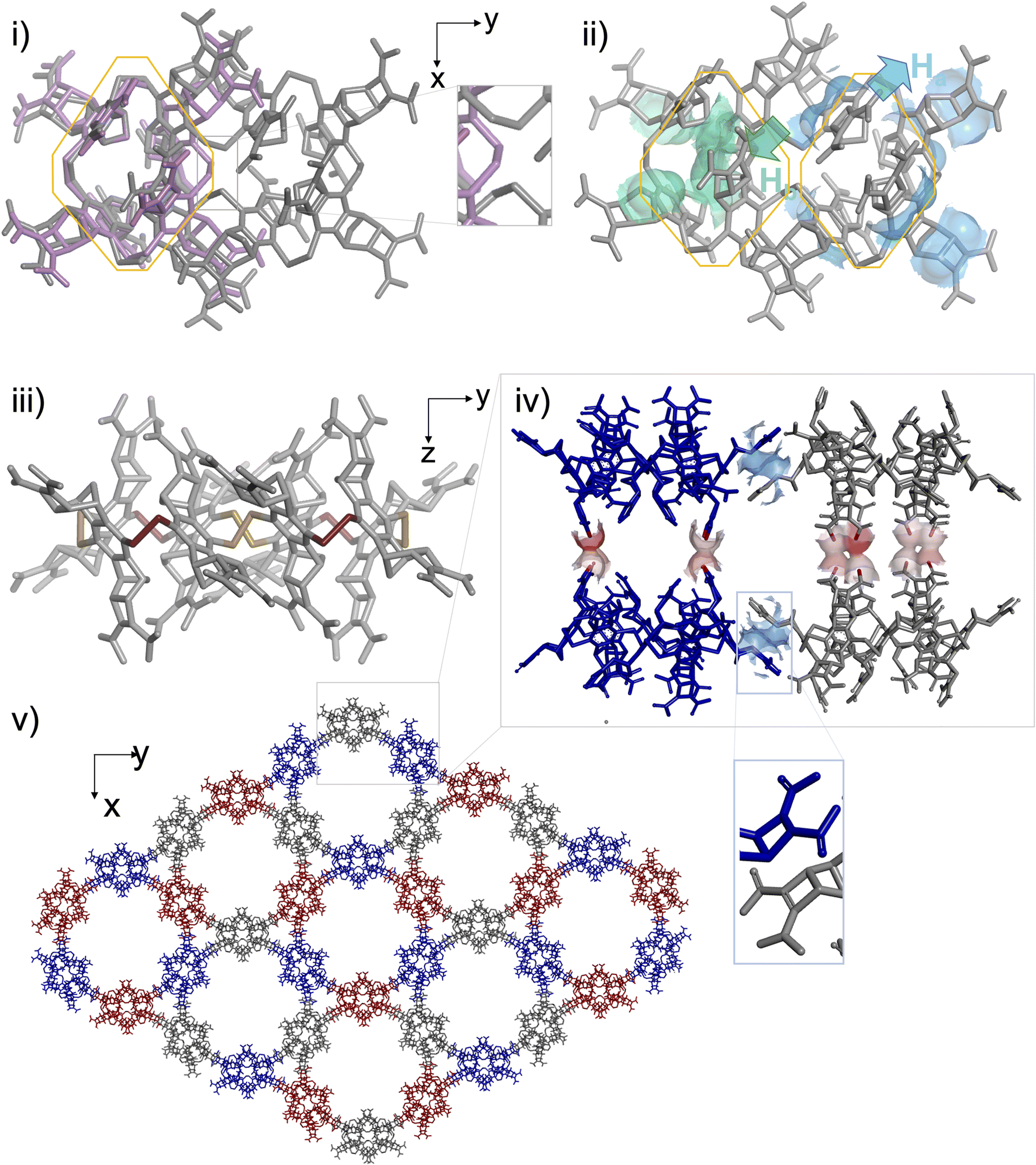 | ||
| Fig. 5 Structural elucidation of the B8 octamer by X-ray crystallography: (i) comparison of B4 and B8 structures obtained in silico and by X-ray diffraction respectively (pink: B4, grey: B8, and framed: zoom on the junction between tetrameric subunits), (ii) analysis of the facial configuration of the B8 structure (green: Ha and cyan: Hb), (iii) analysis of the disulfide conformation (yellow: P–SS and red: M–SS) of the B8 structure, and (iv) intermolecular packing between four B8 molecules (blue and red: van der Waals contacts between cyclobutene and carboxylic moieties) from two xy layers displayed in (v). | ||
Conclusions
We have provided here the proof of feasibility – through a rational design approach – of multi-bit stereochemical information transfer from achiral building blocks into disulfide-based cyclophanes using non-directional interactions. More specifically we showed that the steric hindrance borne by simple carboxylate or methylene moieties on 1,4-bisthiophenol building blocks can serve as an informational input. This input can be translated by a non-directional assembling algorithm (disulfide bridge formation and steric hindrance minimization), with or without the assistance of a template, into a specific stereochemical output (the configurational and conformational sequence of the assembled product). Unexpectedly, a third class of driving force, namely aggregation and crystal packing, can also influence the stereochemical selection by complementing (in the case of A4 with enantioselective discrimination upon conglomerate crystallization) or perturbing (in the case of B8) the information transfer. Overall, the scope of templating, folding and stacking, well known driving forces for compositional and size selection and amplification in DCC, was herein extended to stereochemistry.This conceptual toolbox should therefore be of great value to the broadest community. It may prompt molecular biologists to further decipher how stereochemical information propagates through disulfide bridges within protein architectures, especially those involved in misfolding and aggregating events.26,27 In the field of supramolecular chemistry, we believe that this study opens the way to the stereo-controlled assembly of macrocyclic, linear or interlocked architectures of unprecedented three-dimensional structures from very simple components.28 It may also prompt polymer chemists to bridge the gap between thermoplastics obtained with partial stereocontrol in a kinetic regime29 and reversible self-healing materials wherein the issue of tacticity has never been addressed. Finally, we are currently exploring the ability of various templates to induce a divergence in the stereochemical output from the A4 and B4 self-assembly. The results will be reported in due course.
Data availability
All data for this article are provided in the ESI.†Author contributions
J. L. designed the system. J. L., L. V., F. P. and I. H. designed the experiments. Y. Z., B. O., P.-T. S., and T.C. carried out the experimental work. A. M. B. designed the chromatographic methods and conducted the analyses. O. C. designed the NMR methods and conducted the analyses. N. V. designed the chiral chromatographic methods and conducted the analyses. P. K. M. conducted the X-ray diffraction analyses. C. D. designed and performed the MS experiments. E. D. designed the simulations. E. J. and B. O. performed the simulation. J. L. and L. V. wrote the paper. All authors contributed to revising the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the CCSM, CRMN and CCRMN for assistance with mass spectrometry, DOSY, and high field liquid and solid-state NMR analyses, respectively. We acknowledge the contribution of SFR Biosciences (UMS3444/CNRS, US8/Inserm, ENS de Lyon, UCBL) facilities: Protein Science Facility – F. Delolme for performing MALDI-TOF-MS analysis. We are grateful to S. Vidal for providing access to his UPLC facility (ANR-17-CE07-0054). We also would like to thank B. Kauffmann, S. Panneerselvam and I. Melnikov for assistance in crystallographic measurements at IECB, Bordeaux, P13 beamline operated by EMBL Hamburg at the PETRA III storage ring (DESY, Hamburg) and the ID30b beamline at ESRF, Grenoble. This work has benefited from the facilities and expertise of the Biophysical and Structural Chemistry platform (BPCS) at IECB, CNRS UMS3033, Inserm US001, and Bordeaux University. Financial support from the Chinese Scholarship Council (Y. Z scholarship) and UCB Biopharma (P. T. S. and B. O. scholarships) and the IR-RMN-THC Fr3050 CNRS is gratefully acknowledged. This work was supported by the LABEX iMUST (ANR-10-LABX-0064) of Université de Lyon, within the program “Investissements d'Avenir” operated by the French National Research Agency (ANR).Notes and references
- E. Massolo and M. Benaglia, in Organocatalysis: Stereoselective Reactions and Applications in Organic Synthesis, ed. M. Benaglia, De Gruyter, 2021, pp. 229–262 Search PubMed.
- J. C. Worch, H. Prydderch, S. Jimaja, P. Bexis, M. L. Becker and A. P. Dove, Nat. Rev. Chem., 2019, 3, 514–535 CrossRef CAS.
- T. Ogoshi, T. Yamagishi and Y. Nakamoto, Chem. Rev., 2016, 116, 7937–8002 CrossRef CAS PubMed.
- S. J. Barrow, S. Kasera, M. J. Rowland, J. del Barrio and O. A. Scherman, Chem. Rev., 2015, 115, 12320–12406 CrossRef CAS PubMed.
- For pillararenes, see: (a) P. D. Sala, R. D. Regno, L. D. Marino, C. Calabrese, C. Palo, C. Talotta, S. Geremia, N. Hickey, A. Capobianco, P. Neri and C. Gaeta, Chem. Sci., 2021, 12, 9952–9961 RSC; (b) S. Fa, T. Kakuta, T. Yamagishi, T. Ogoshi and T., Chem. Lett., 2019, 48, 1278–1287 CrossRef CAS; (c) M. Holler, N. Allenbach, J. Sonet and J.-F. Nierengarten, Chem. Commun., 2012, 48, 2576–2578 RSC.
- For cucurbiturils, see: (a) B. Vinciguerra, L. Cao, J. R. Cannon, P. Y. Zavalij, C. Fenselau and L. Isaacs, J. Am. Chem. Soc., 2012, 134, 13133–13140 CrossRef CAS PubMed; (b) M. Stancl, Z. Gargulakova and V. Sindelar, J. Org. Chem., 2012, 77, 10945–10948 CrossRef CAS PubMed; (c) L. Isaacs, S.-K. Park, S. Liu, Y. H. Ko, N. Selvapalam, Y. Kim, H. Kim, P. Y. Zavalij, G.-H. Kim, H.-S. Lee and K. Kim, J. Am. Chem. Soc., 2005, 127, 18000–18001 CrossRef CAS PubMed; (d) A. Day, A. P. Arnold, R. J. Blanch and B. Snushall, J. Org. Chem., 2001, 66, 8094–8100 CrossRef CAS PubMed.
- P. T. Corbett, J. Leclaire, L. Vial, K. R. West, J.-L. Wietor, J. K. M. Sanders and S. Otto, Chem. Rev., 2006, 106, 3652–3711 CrossRef CAS PubMed.
- B. Panijpan, J. Chem. Educ., 1977, 54, 670–672 CrossRef CAS.
- Q. Zhang, S. Crespi, R. Toyoda, R. Costil, W. R. Browne, D.-H. Qu, H. Tian and B. L. Feringa, J. Am. Chem. Soc., 2022, 144, 4376–4382 CrossRef CAS PubMed.
- C. Tsiamantas, X. de Hatten, C. Douat, B. Kauffmann, V. Maurizot, H. Ihara, M. Takafuji, N. Metzler-Nolte and I. Huc, Angew. Chem., Int. Ed., 2016, 55, 6848–6852 CrossRef CAS PubMed.
- Z. J. Kinney, V. C. Kirinda and C. S. Hartley, Chem. Sci., 2019, 10, 9057–9068 RSC.
- M. Carmack, J. Chem. Inf. Comput. Sci., 2004, 44, 286–288 CrossRef CAS PubMed.
- (a) T.-M. Gianga and G. D. Pantoș, Nat. Commun., 2020, 11, 3528 CrossRef CAS PubMed; (b) P. T. Corbett, L. H. Tong, J. K. M. Sanders and S. Otto, J. Am. Chem. Soc., 2005, 127, 8902–8903 CrossRef CAS PubMed; (c) A. T. ten Cate, P. Y. W. Dankers, R. P. Sijbesma and E. W. Meijer, J. Org. Chem., 2005, 70, 5799–5803 CrossRef CAS PubMed; (d) R. T. S. Lam, A. Belenguer, S. L. Roberts, C. Naumann, T. Jarrosson, S. Otto and J. K. M Sanders, Science, 2005, 308, 667–669 CrossRef CAS PubMed; (e) J. Li, P. Nowak and S. Otto, Angew. Chem., Int. Ed., 2015, 54, 833–837 CrossRef CAS PubMed.
- L. Vial, F. Perret and J. Leclaire, Eur. J. Org. Chem., 2022, e202101274 CAS.
- J.-F. Chen, J.-D. Ding and T.-B. Wei, Chem. Commun., 2021, 57, 9029–9039 RSC.
- M. Dumartin, J. Septavaux, M. Donnier-Maréchal, E. Jeamet, E. Dumont, F. Perret, L. Vial and J. Leclaire, Chem. Sci., 2020, 11, 8151–8156 RSC.
- Even if we acknowledge that dyn[n]arenes are inherently chiral, we decided to use the pS/pR descriptor for dyn[n]arenes by analogy with planar-chiral pillar[n]arenes, see: J.-F. Chen, J.-D. Ding and T.-B. Wei, Chem. Commun., 2021, 57, 9029–9039 RSC.
- For insights into issues associated with chirality and stereoisomerism, see: (a) Y. Wang, O. Allemann, T. Silviu Balaban, N. Vanthuyne, A. Linden, K. K. Baldridge and J. S. Siegel, Angew. Chem., Int. Ed., 2018, 57, 6470–6474 CrossRef CAS PubMed; (b) K. Mislow and J. Siegel, J. Am. Chem. Soc., 1984, 106, 3319–3328 CrossRef CAS.
- L. Vial, R. F. Ludlow, J. Leclaire, R. Pérez-Fernández and S. Otto, J. Am. Chem. Soc., 2006, 128, 10253–10257 CrossRef CAS PubMed.
- P.-T. Skowron, M. Dumartin, E. Jeamet, F. Perret, C. Gourlaouen, A. Baudouin, B. Fenet, J.-V. Naubron, F. Fotiadu, L. Vial and J. Leclaire, J. Org. Chem., 2016, 81, 654–661 CrossRef CAS PubMed.
- E. Jeamet, J. Septavaux, A. Héloin, M. Donnier-Maréchal, M. Dumartin, B. Ourri, P. Mandal, I. Huc, E. Bignon, E. Dumont, E. Bignon, E. Dumont, C. Morell, J.-P. Francoia, F. Perret, L. Vial and J. Leclaire, Chem. Sci., 2019, 10, 277–283 RSC.
- On related compounds from the same family, the kinetic barrier for the inversion of the disulfide bridge's dihedral angle was estimated to be around 75 kJ mol−1, see ref. 20. With carboxy substituents in positions 2 and 5, a higher value can be expected which should lead to signal splitting on the NMR timescale if multiple conformers coexist.
- M. Fokkens, T. Schrader and F.-G. Klärner, J. Am. Chem. Soc., 2005, 127, 14415–14421 CrossRef CAS PubMed.
- M. Holler, N. Allenbach, J. Soneta and J.-F. Nierengarten, Chem. Commun., 2012, 48, 2576–2578 RSC.
- P. T. Corbett, J. K. M. Sanders and S. Otto, Chem.–Eur. J., 2008, 14, 2153–2166 CrossRef CAS PubMed.
- Y. Li, J. Yan, X. Zhang and K. Huang, Proteins, 2013, 81, 1862–1873 CrossRef CAS PubMed.
- M. F. Mossuto, Int. J. Cell Biol., 2013, 318319 Search PubMed.
- H. V. Schröder, Y. Zhang and A. J. Link, Nat. Chem., 2021, 13, 850–857 CrossRef PubMed.
- A. J. Teator and F. A. Leibfarth, Science, 2019, 363, 1439–1443 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: All data supporting the findings of this study, including synthetic and analytical procedures. CCDC 1554744, 2003290, 2001333, and 2003509. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc01235b |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |