
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent advances in permeable polymersomes: fabrication, responsiveness, and applications
Yanyan
Zhu†
a,
Shoupeng
Cao†
 b,
Meng
Huo
*c,
Jan C. M.
van Hest
*d and
Hailong
Che
*a
b,
Meng
Huo
*c,
Jan C. M.
van Hest
*d and
Hailong
Che
*a
aDepartment of Chemical Engineering, School of Environmental and Chemical Engineerin, Shanghai University, Shanghai 200444, China. E-mail: hche@shu.edu.cn
bMax Planck Institute for Polymer Research, Mainz 55128, Germany
cDepartment of Chemistry, Key Laboratory of Surface & Interface Science of Polymer Materials of Zhejiang, Zhejiang Sci-Tech University, Hangzhou 310018, China. E-mail: huomeng@zstu.edu.cn
dDepartment of Chemical Engineering and Chemistry, Department of Biomedical Engineering, Institute for Complex Molecular Systems, Eindhoven University of Technology, Eindhoven 5600 MB, The Netherlands. E-mail: J.C.M.v.Hest@tue.nl
First published on 21st June 2023
Abstract
Polymersomes are vesicular nanostructures enclosed by a bilayer-membrane self-assembled from amphiphilic block copolymers, which exhibit higher stability compared with their biological analogues (e.g. liposomes). Due to their versatility, polymersomes have found various applications in different research fields such as drug delivery, nanomedicine, biological nanoreactors, and artificial cells. However, polymersomes prepared with high molecular weight components typically display low permeability to molecules and ions. It hence remains a major challenge to balance the opposing features of robustness and permeability of polymersomes. In this review, we focus on the design and strategies for fabricating permeable polymersomes, including polymersomes with intrinsic permeability, the formation of nanopores in the membrane bilayers by protein insertion, and the construction of stimuli-responsive polymersomes. Then, we highlight the applications of permeable polymersomes in the fields of biomimetic nanoreactors, artificial cells and organelles, and nanomedicine, to underline the challenges in the development of polymersomes as soft matter with biomedical utilities.
Introduction
Molecular self-assembly to afford complex structures is ubiquitously found in nature, such as the formation of a phospholipid bilayer, DNA folding into the double helix structure, and polypeptides assembling into active proteins.1,2 This has inspired scientific communities to create synthetic analogues by mimicking biological complexity and function.3 For instance, assemblies based on amphiphilic block copolymers constructed from water-soluble and water-insoluble segments have drawn considerable attention from communities active in nanomedicine and biomimicry.4,5 Advances in synthetic chemistry have enriched the diversity of polymer building blocks to the extent that there is almost no limit to the variety of building blocks (i.e., monomers) in a polymer structure.6,7 Thus, a wide range of tailor-made and functional polymeric self-assemblies can be created with regard to physical features (i.e., size and morphology), flexibility, surface chemistry, and stability (including both disassembly kinetics and stimuli-responsive pathways).8 For instance, amphiphilic block copolymers can assemble into various well-defined architectures such as spherical vesicles,9–15 non-spherical vesicles,16–25 spherical micelles,26 and worm-like micelles.27,28 This can be facilely realized via altering the compositions of the building blocks, self-assembly approaches, as well as by exploiting environmental changes such as the temperature and ionic concentrations.Polymersomes (also known as polymeric vesicles) are hollow structures surrounded by bilayers self-assembled from amphiphilic block copolymers. By altering the composition and length of the block copolymers, both the chemical and physical properties of polymersome membranes can be tailored to meet specific application requirements.29 The versatility of polymer chemistry also allows adjustment of the molecular modules in a facile manner and tailors responsiveness to specific environmental conditions.30 Polymersome bilayers contain a hydrophilic cavity which is capable to encapsulate and protect both hydrophilic components (such as proteins, RNAs, and enzymes). In addition, the hydrophobic membrane bilayers can load hydrophobic cargoes (such as emissive agents and chemotherapeutic drugs) by hydrophilic–hydrophilic interactions. This enables polymersomes to be used in a range of applications in the biomedical field such as nanocarriers used in drug delivery systems.31 The presence of a membrane structure separates the polymersome internal environment, or lumen, from the external medium and modulates internal chemical processes via regulated molecular transport. Polymersomes are also utilized as biomimetic nanoreactors by encapsulating catalytic species such as active enzymes. This is an exciting way to mimic the structural complexity and function of biological compartments found in living systems and makes it feasible to engineer polymersomes as active synthetic organelles.32–34
Compared with liposomes, polymersomes prepared from higher molecular weight components typically display higher structural stability and durability, but they are also less leaky, and usually display limited permeability to molecules and ions, due to the hydrophobic interactions and entanglement of the polymer chain in polymersome bilayers.34,35 For example, polymersomes consisting of poly(2-methyl-2-oxazoline)-block-poly(dimethylsiloxane)-block-(2-methyl-2-oxazoline) (PMOXA-b-PDMS-b-PMOXA) or polyethyleneoxide–polyethylethylene (PEO–PEE) showed poor permeability to common ions.36,37 Small organic molecules and even water have difficulty in penetrating the bilayer membrane of polymersomes.38 Membrane permeability is essential for polymersomes to be applied as controlled release vehicles and biomimetic nanoreactors, as the permeability control plays a crucial role in regulating the effective membrane transportation of information signals, substrates, and products to and from the catalytic center.39–42 In general, the porousness, hydrodynamic size, and the hydrophilic and hydrophobic properties of the membranes can regulate the transportation of cargos through the polymersomes. In addition, adjusting the interactions between the polymersome membranes and loaded molecules can also realize the controlled release of cargos. However, in most cases, molecular transportation and information exchange across the polymeric membranes usually relies on passive diffusion or involves mechanisms that result in the reversible loss of polymersome structural integrity. This is not conducive to mimicking intracellular information exchange and selective transport of different substances, nor is it conducive to making polymersomes available for a wider range of applications. To approach this challenge, facile strategies are needed to fabricate polymersomes with tunable membrane permeability. In recent years, the following approaches have been proposed to be effective strategies for obtaining polymersomes with controlled membrane permeability: (1) integrating hydrophobic blocks containing crystalline or semi-crystalline polymers such as poly(lactic acid) (PLA), poly(caprolactone) (PCL), and poly(3-(isocyano-L-alanylaminoethyl)thiophene) (PIAT) in the formation of permeable or semipermeable polymersomes.42 (2) Adding pore-forming proteins to induce the formation of channels in the polymeric membranes which enhances the polymersome permeability and allows for selective molecule exchange.43 (3) Utilizing stimuli-responsive constituents to endow the polymersomes with triggered-induced (such as pH, CO2, light, temperature, enzyme, etc.44–48) permeability control. (4) Formation of polyion complex vesicles (PICsomes) from polyelectrolyte building blocks. Additionally, the permeability of the polymersomes can also be controlled and regulated by specific molecular design, while maintaining their structural integrity.35
In this review, we will summarize the state of the art concerning permeable polymersomes as functional synthetic compartments (Fig. 1). Polymersomes with inherent permeability or permeability enhanced with biomolecule insertion will be first described. The examples that utilize environmental changes or external stimuli to tailor the membrane permeability will be discussed next. Subsequently, polymersomes with bio-related applications such as drug release, biomimetic nanoreactors, and artificial organelles in vitro and in vivo will be highlighted. Finally, the future opportunities and challenges of permeable polymersomes will be discussed. The chemical structures, names, abbreviations, and polymerization methods of common polymers used for permeable polymersomes in this review are summarized in Table 1.
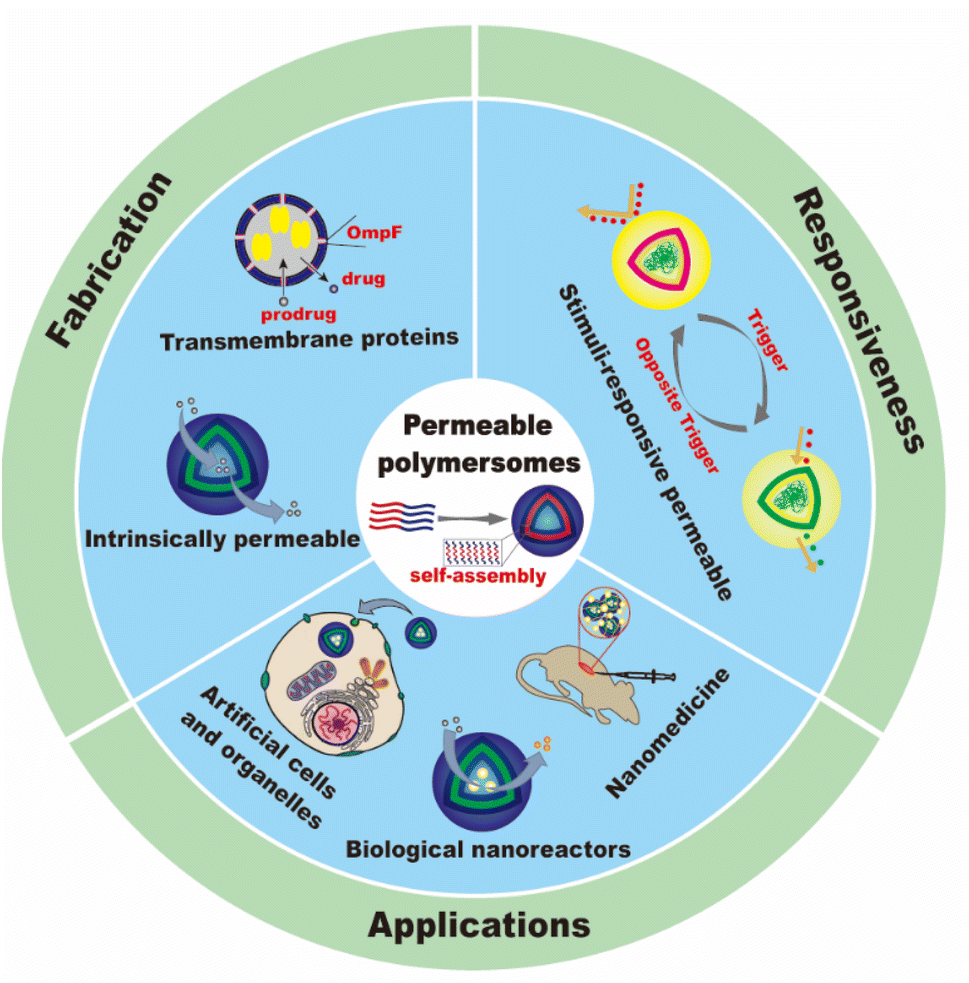 | ||
| Fig. 1 Schematic illustration of permeable polymersomes for diverse biomimetic applications. | ||
| Polymer structures | Names | Abbreviations | Polymerization method |
|---|---|---|---|
|
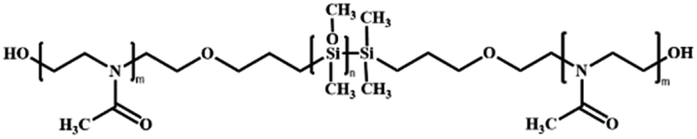
Poly(2-methyl-2-oxazoline)-block-poly(dimethylsiloxane)-block-(2-methyl-2-oxazoline) | PMOXA-b-PDMS-b-PMOXA | Cationic ring opening living polymerization (CROP) |
|
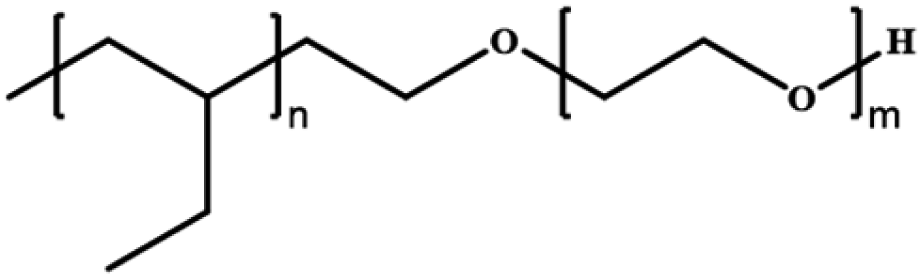
Polyethyleneoxide-block-polyethylethylene | PEO–PEE | Anionic polymerization (AP) |

|
Poly(ethylene glycol)-block-poly(lactic acid) | PEG-b-PLA | Ring opening polymerization (ROP) |
|
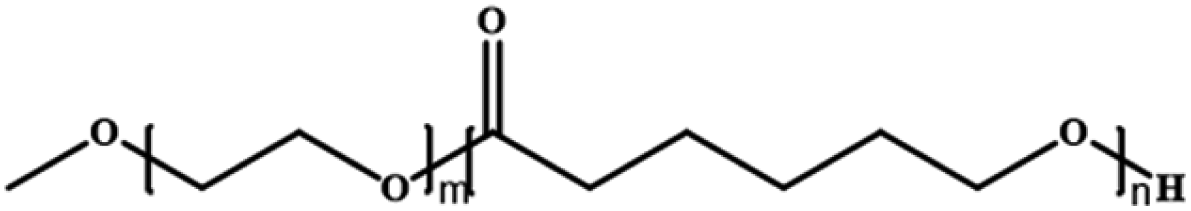
Poly(ethylene glycol)-block-poly(ε-caprolactone) | PEG-b-PCL | ROP |
|
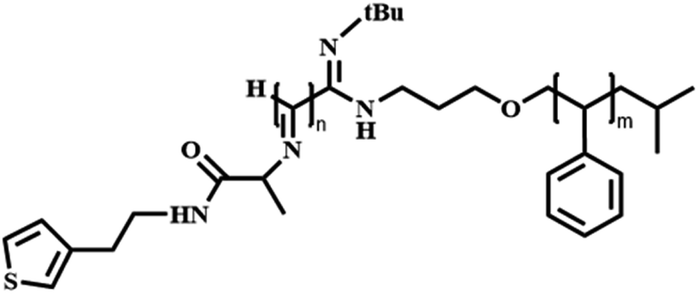
Polystyrene-block-poly(L-isocyanoalanine (2-thiophen-3-ylethyl)amide) | PS-b-PIAT | Atom transfer radical polymerization (ATRP) |
|
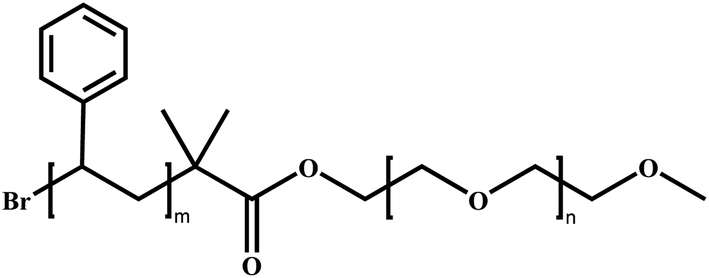
Poly(ethylene glycol)-block-poly(styrene) | PEG-b-PS | ATRP |
|
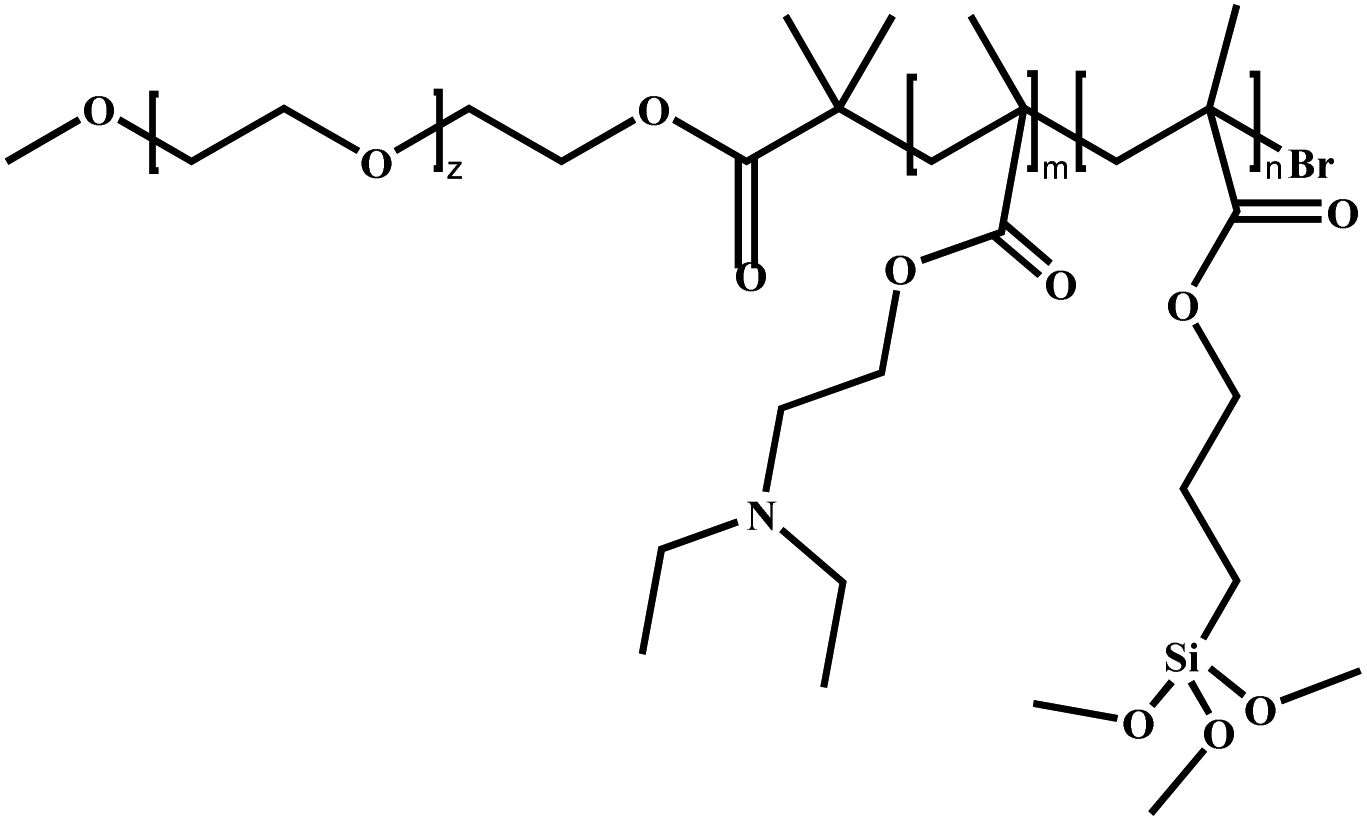
Poly(ethylene oxide)-block-poly[2-(diethylamino)ethyl methacrylate-stat-3-(trimethoxysilyl)propyl methacrylate] | PEO-b-P(DEA-stat-TMSPMA) | ATRP |

|
Poly(ethylene glycol)-block-poly[(2-(piperidin-1-yl)ethyl methacrylate)-co-(7-(3-methacryloyloxyethoxy)-4-methylcoumarin)] | PEG-b-P(PEMA-co-CMA) | Reversible addition–fragmentation chain transfer (RAFT) |

|
Poly(ethylene oxide)-block-PSPA, (SPA is spiropyran-based monomer containing a unique carbamate linkage) | PEO-b-PSPA | RAFT |
|
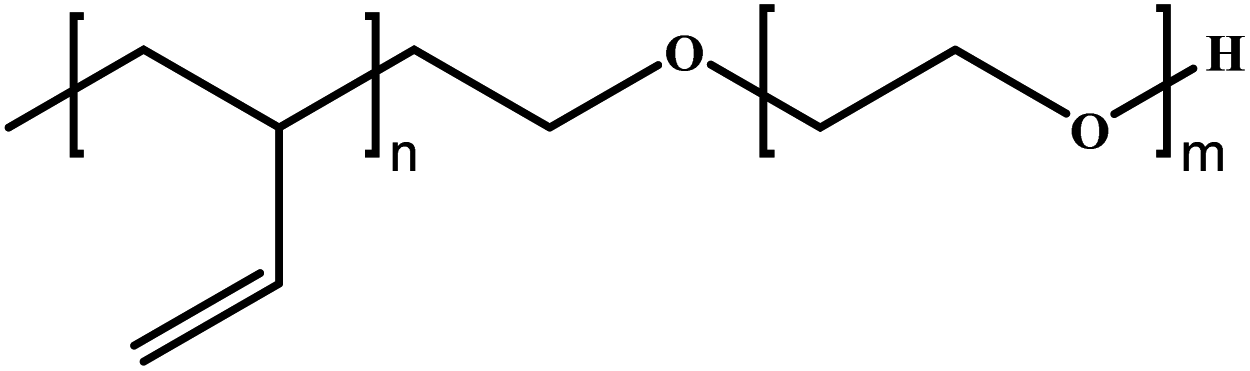
Poly(ethylene oxide)-block-poly(butadiene) | PEO-b-PB | AP |

|
Poly(ethylene oxide)-block-poly(o-azidomethyl benzoyl glycerol methacrylate) | PEO-b-PAGMA | RAFT |
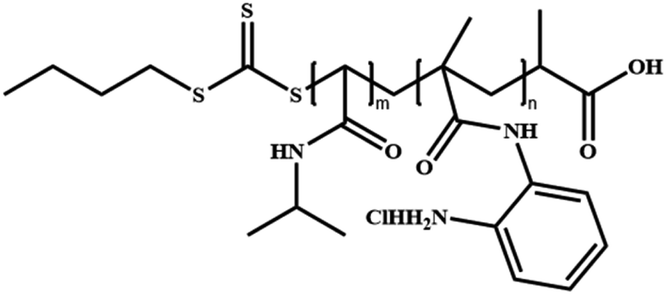
|
Poly[N-isopropyl acrylamide]-co-poly[N-(2-aminophenyl)methacrylamide] | P(NIPAM-co-NAPMA) | RAFT |

|
Poly[N-isopropyl acrylamide]-co-2-[3-(2-aminophenyl)ureido)ethyl methacrylate] | P(NIPAM-co-APUEMA) | RAFT |
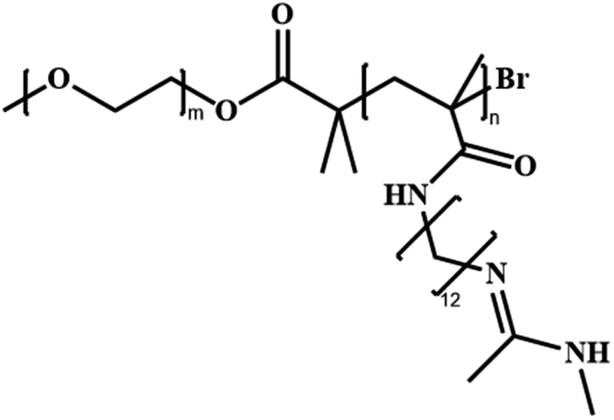
|
Poly(ethylene glycol)-block-poly(N-amidino)dodecyl acrylamide | PEG-b-PAD | RAFT |

|
Methoxy poly(ethylene glycol)-poly(L-cysteine) | PEG-PLCC | ROP |
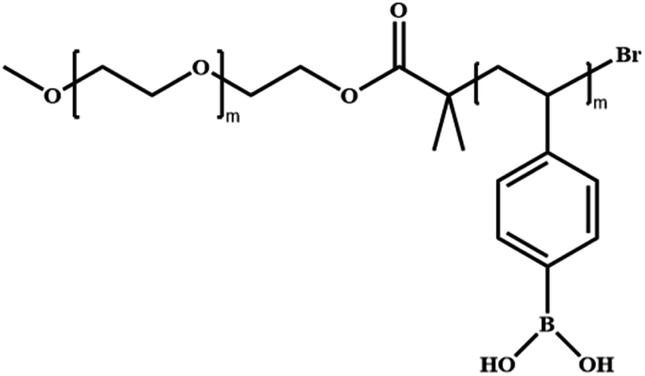
|
Poly(ethylene glycol)-block-poly(styrene boronic acid) | PEG-b-PSBA | ATRP |
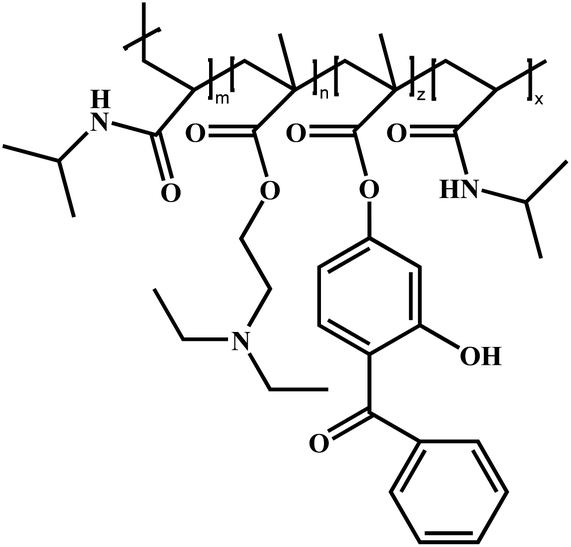
|
Poly(N-isopropylacrylamide)-block-poly[2-(diethylamino)ethyl methacrylate-co-2-hydroxy-4-(methacryloyloxy)benzophenone]-block-poly(N-isopropylacrylamide) | PNIPAM-b-PDEAEMA-co-PBMA-b-PNIPAM (PN-DB-N) | ATRP |
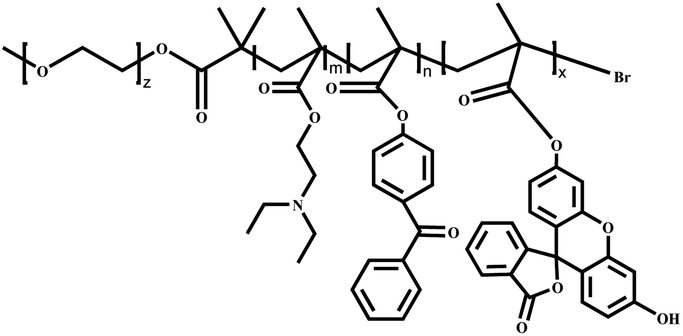
|
Poly(ethylene glycol)-block-poly[2-(diethylamino)ethyl methacrylate-co-2-hydroxy-4-(methacryloyloxy)benzophenone-co-fluorescein O-methacrylate] | PEG-b-P(DEAEMA-co-BMA-co-FMA) | ATRP |

|
Poly(ethylene glycol)-b-poly(caprolactone-gradient-trimethylene carbonate) | PEG-PCLgTMC | ROP |
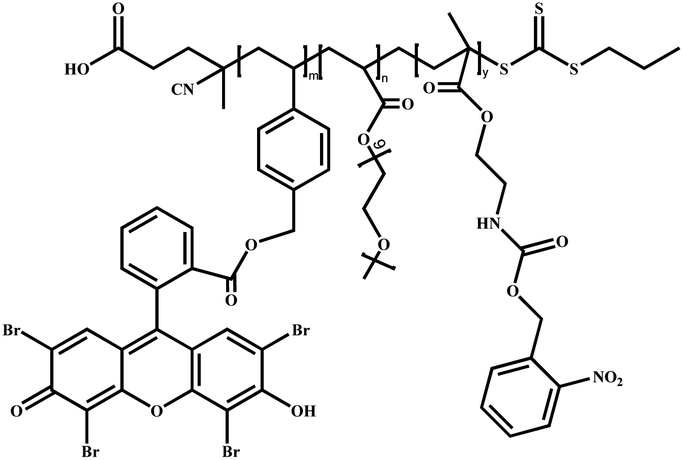
|
Poly(oligo(ethylene glycol)monomethyl ether methacrylate-co-eosin Y)-block-poly(2-nitrobenzy loxycarbonylaminoethylmethacrylat) | P(OEGMA-co-EOS)-b-PNBOC | ATRP |
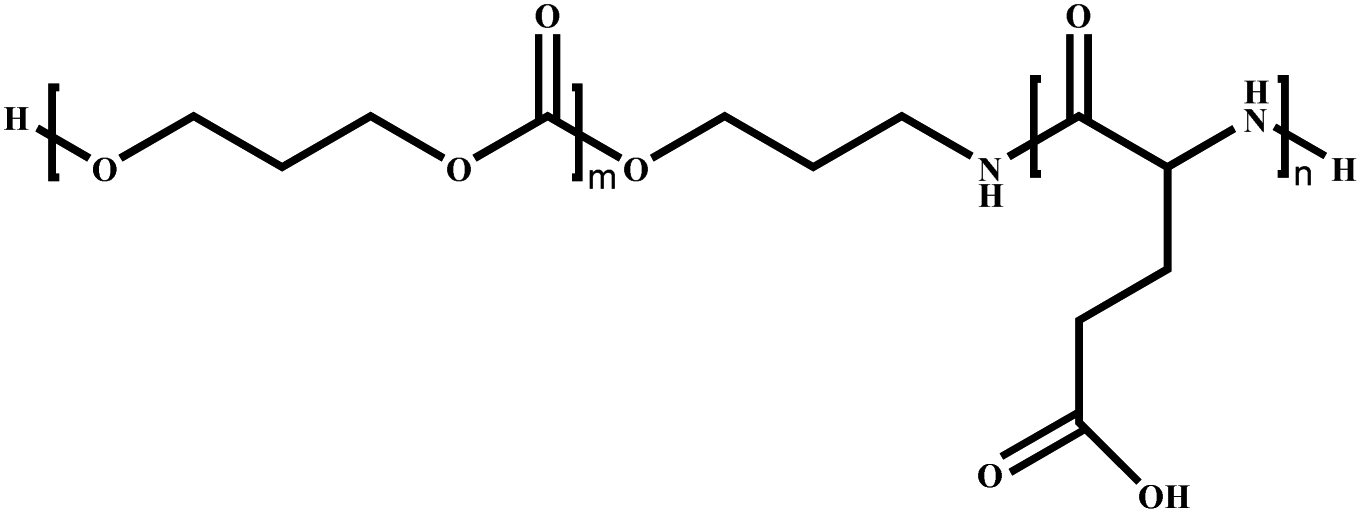
|
Poly(trimethylene carbonate)-block-poly(L-glutamic acid) | PTMC-b-PGA | ROP |
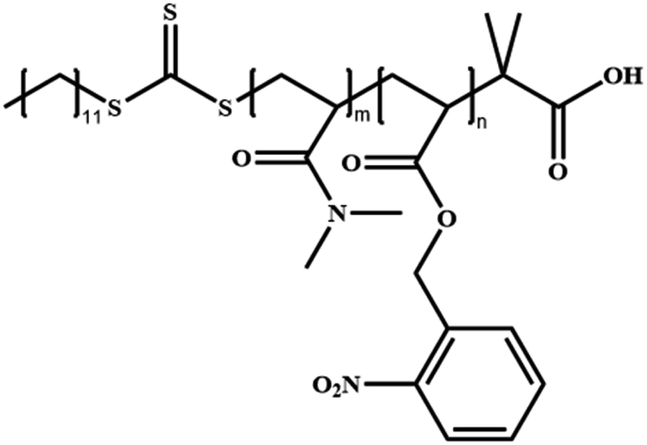
|
Poly(N,N′-dimethylacrylamide)-block-poly(o-nitrobenzyl acrylate) | PDMA-b-PNBA | RAFT |
Strategies for the fabrication of permeable polymersomes
Intrinsically permeable polymersomes
Since polymersomes were reported, only a few examples of polymersomes were found to be permeable. Poly(ethylene glycol) (PEG) is a water-soluble polymer with hydroxyl functional groups on its molecular chain, exhibiting excellent biocompatibility and low toxicity. It can be copolymerized with other hydrophobic polymers to form permeable polymersomes through self-assembly. For example, polymersomes made from the biodegradable copolymers poly(ethylene glycol)-block-poly(lactic acid) (PEG-b-PLA) and poly(ethylene glycol)-block-poly(caprolactone) (PEG-b-PCL) displayed semi-permeable features.49 Polymersomes constructed from PMOXA-b-PDMS-b-PMOXA are permeable to oxygen.33 In addition, polymersomes with a shell composed of a polyelectrolyte or fabricated from block copolymer polystyrene40-b-poly(L-isocyanoalanine(2-thiophen-3-ylethyl)amide)50 (PS40-b-PIAT50) displayed intrinsic permeability.42 For instance, Nolte et al. constructed polymersomes from PS40-b-PIAT50, which is an amphiphilic structure of which the hydrophilic PIAT block containing thiophene adopts a rigid rod-shaped helical conformation (coil) and hydrophobic PS block adopts a flexible rod-shaped conformation (rod). Rod-coil diblock copolymers have slightly different polarities and amphiphilic properties in both organic and aqueous solvents, enabling them to self-assemble into polymersomes. In addition, the presence of highly organized stacks of thiophene in the polymersome membranes may endow them with permeability. PS40-b-PIAT50 polymersomes were loaded with Candida antarctica Lipase B (CALB), and served as biomimetic nanoreactors (Fig. 2a).50 The substrate 6,8-difluoro-4-methylumbelliferyl octanoate (DiFMU octanoate) could penetrate through the bilayer membrane of these polymersomes, and trigger catalytic activity inside. The efficiency of the polymersome nanoreactors was evaluated by the fluorescence intensity of individual polymersomes, which was stronger than that produced by the autohydrolysis of the DiFMU ester. This suggested that the trapped enzyme remained active and polymersomes displayed permeability. Notably, the catalytic product did not leak out from the polymersomes since the product was more polar than the substrate, which demonstrated the selective permeability of the PS-b-PIAT polymersomes. Van Hest et al. reported the construction of two polymersomes with different permeability composed of PS-b-PEG and PS-b-PIAT, respectively, and investigated their utility for enzymatic cascade reactions inside the polymersome nanoreactors (Fig. 2b).42 The permeability of these two polymersomes was evaluated by testing the activity of the loaded enzymes copper-zinc superoxide dismutase (SOD1) and catalase (CAT). The corresponding activity profiles of SOD1 in the free and encapsulated states for both polymersomes were similar, which indicates that the loading process did not affect enzymatic activity and the superoxide radical substrate could pass both membranes equally fast. Upon the addition of H2O2, oxygen content increased immediately in the CAT-loaded polymersomes. In addition, they demonstrated that PS-b-PIAT polymersomes displayed higher permeability and hence higher activity to H2O2 than PS-b-PEG polymersomes. Though these polymersomes exhibit certain permeability to some substances, they still are far from meeting the requirements for expanding potential applications. The polymersomes should be more intelligent and adjustable in terms of their behaviors and applications.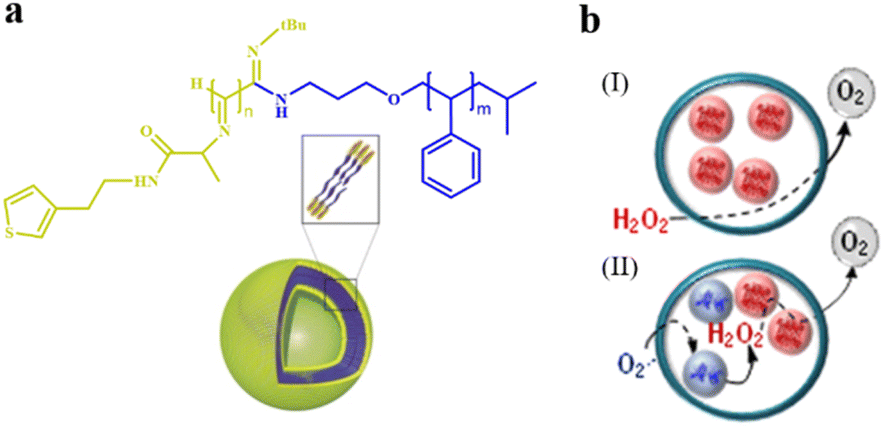 | ||
| Fig. 2 (a) Schematic representation of a polystyrene-block-poly(L-isocyanoalanine (2-thiophen-3-ylethyl)amide) (PS-b-PIAT) polymersome. Yellow is PIAT and blue is PS. Reproduced with permission from ref. 50. Copyright 2003, Wiley-VCH. (b) The enzymatic activities of PS-b-PEG and PS-b-PIAT polymersome nanoreactors were assessed by the addition of H2O2. (I): PEG-b-PS, (II): PS-b-PIAT. Reproduced with permission from ref. 42. Copyright 2013, American Chemical Society. | ||
Polymersomes with transmembrane proteins
In addition to the intrinsically permeable polymersomes discussed above, permeable polymersomes can also be formed by inserting protein nanopores into the membrane of the polymersome.51–53 For example, permeable polymersomes can be constructed by combining multifunctional biological components such as transmembrane proteins, pore proteins, and enzymes with polymersomes.52 This method involves first forming a complex between the target polymer material and the protein, followed by adjusting the concentration of the protein in the complex to control the permeability of the polymersomes. This approach utilizing proteins to construct permeable polymersomes has great potential for a wide range of applications, particularly in drug delivery. For instance, Van Gelder et al. introduced channel proteins to the polymer membranes to selectively control the transportation of guest molecules.54 The polymersomes were prepared by the self-assembly of amphiphilic ABA type triblock copolymers PMOXA-b-PDMS-b-PMOXA in an aqueous solution. Two different bacterial outer membrane protein porins (ompF and Tsx), were incorporated into the polymersome membranes to form protein channels (Fig. 3a). They found that the permeability of PMOXA-b-PDMS-b-PMOXA polymersomes was significantly improved by introducing the channel proteins. Small molecular substrates crossed the PMOXA-b-PDMS-b-PMOXA polymersomes through the channel protein, which were then activated by the encapsulated prodrug-activating enzyme (e.g. purine-specific nucleoside hydrolase of Trypanosoma vivax (TvNH)); the formed products were released to the exterior environment through the protein channel. | ||
| Fig. 3 (a) Schematic representation of poly(2-methyl-2-oxazoline)-block-poly(dimethylsiloxane)-block-(2-methyl-2-oxazoline) (PMOXA-b-PDMS-b-PMOXA) polymersomes as a functionalized nanoreactor. Reprinted from ref. 54. Copyright 2005, American Chemical Society. (b) Schematic representation of the nanoreactor system. (c) Channel- and pH-dependent nanoreactor activity in confocal microscopy (Magnification 400×; Zeiss ConfoCor 2, HAL 100, HBO 100, Software LSM 510 Zeiss): active nanoreactors with ompF pores show strong fluorescent activity at pH = 5 (I), but show no significant fluorescent activity without the ompF pores (II); active nanoreactors with ompF (III) and without ompF (IV) pores show no fluorescent activity at pH = 7.5. The substrate concentration in this experiment was 75 μM. Reprinted from ref. 55. Copyright 2006, American Chemical Society. | ||
In follow-up research, Hunziker et al. demonstrated that PMOXA-b-PDMS-b-PMOXA polymersomes integrated with ompF gave rise to the diffusion of substrates in a size-selective manner, leading to the activation of enzymatic reactions. (Fig. 3b).55 In addition, the enzymatic reaction inside the polymersome nanoreactors showed catalytic activity controlled by external conditions. For instance, the enzyme was active between pH 4–7 but displayed lower activity at higher or lower pH values (Fig. 3c). The functionality of the polymersome nanoreactors was accomplished via the enzymatic hydrolysis process of nonfluorescent water-soluble phosphatase substrates, which were converted to a water-insoluble product with enhanced fluorescent emissions.
The use of protein-based channels is an effective strategy to enhance the polymersome permeability, which can be well controlled by adjusting the structure and function of proteins. However, the size of protein-based channels often enabled the undesired exchange of small molecules. In addition, protein-based channels are fragile and can be altered in a complex cellular environment, which may limit their biomedical applications.
Stimuli–responsive polymersomes
Polymersomes with stimuli–responsiveness are of great importance because their chemical and physical features such as size, morphology adaption, surface characteristics, and assembly–disassembly transition can be well-regulated and controlled in response to environmental changes or external stimuli.56,57 This allows them to be used effectively in a variety of applications such as controlled drug release, imaging, diagnostics, smart actuators, self-healing materials, and recyclable catalysis.58,59 A specific research approach to the construction of polymersomes with permeability regulation is the introduction of stimuli-responsive building blocks to polymersomes, which has become the most intensively investigated route toward permeable polymersomes.60 In general, three categories of stimuli are utilized, including physical triggers (e.g. light,61,62 temperature,63–65 and electric field66), chemical stimulations (e.g. pH,67–70 redox potential,46,71,72 glucose,48,66 and gas73,74), and biological factors (e.g. enzymes,75,76 adenosine triphosphate (ATP),77 and DNA78). In this section, we discuss recent progress on stimuli-responsive polymersomes with controlled membrane permeability.pH–responsive polymersomes
pH–responsive polymersomes are the most studied stimuli–responsive system. The mechanism involves the pH-induced modulation of the hydrophilicity of polymer chains, which results in permeability regulation of the polymersome membrane.79,80 For example, Armes et al. reported pH-responsive polymersomes consisting of poly(ethylene oxide)-block-poly[2-(diethylamino) ethyl methacrylate-stat-3-(trimethoxysilyl) propyl methacrylate].81 The polymersomes were crosslinked to ensure the membrane integrity upon pH change (Fig. 4a). When the pH was increased with the addition of triethylamine, deprotonation of DEA occurred; this resulted in the formation of polymersomes with increased size because of some degree of vesicle reorganization. Smaller polymersomes were obtained by employing higher EDA content under the same conditions (Fig. 4b). The pH-switched permeability of the polymersomes was proven by a fluorescence experiment via rapidly mixing hollow vesicles and rhodamine B dye in aqueous solutions under different pH conditions. The results showed that the fluorescence intensity in the solution at pH = 2 was much lower than that at pH = 12, due to the self-quenching effect of the rhodamine B dye inside the polymersomes.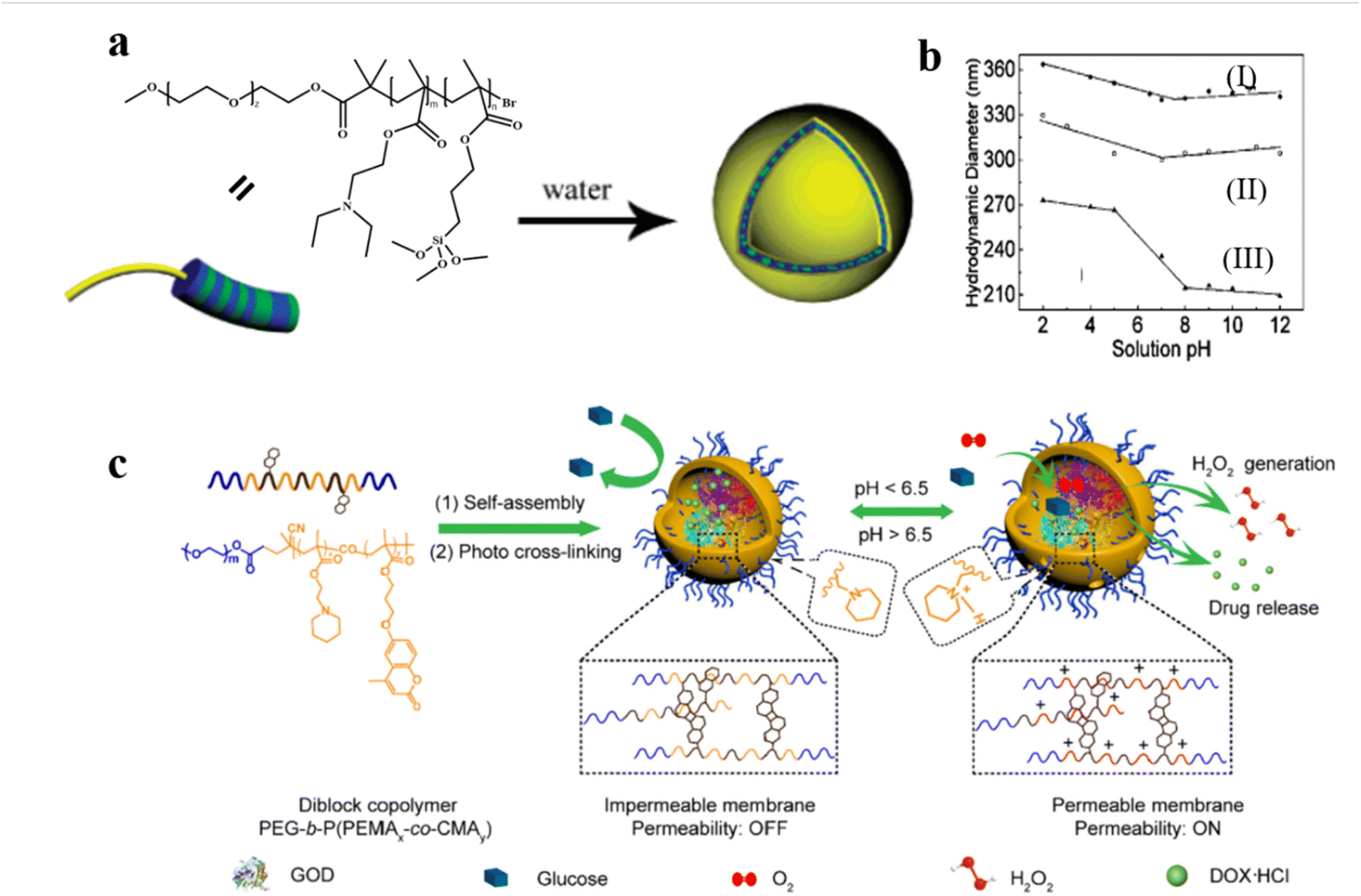 | ||
| Fig. 4 (a) Schematic representation of the formation of pH-responsive poly(ethylene oxide)-block-poly[2-(diethylamino) ethyl methacrylate-stat-3-(trimethoxysilyl) propyl methacrylate] [PEO-b-P(DEA-stat-TMSPMA)] polymersomes. (b) Variation of hydrodynamic vesicle diameter with solution pH: polymersomes prepared (I) with PEO43-b-P(DEA40-stat-TMSPMA40) and (II) without triethylamine; and (III) PEO43-b-P (DEA60-stat-TMSPMA)10, prepared with triethylamine. Reprinted from ref. 81. Copyright 2005, American Chemical Society. (c) Schematic representation of DOX/GOD@MCL-polymersomes as intelligent nanoreactors and drug delivery vehicles. Reprinted from ref. 61. Copyright 2020, Elsevier. | ||
pH–responsive polymersomes can also be constructed by incorporating acid-cleavable bonds into the block copolymer to improve permeability.61 For instance, Ge et al. synthesized the diblock copolymer poly(ethylene glycol)-block-poly[(2-(piperidin-1-yl)ethyl methacrylate)x-co-(7-(3-methacryloyloxyethoxy)-4-methylcoumarin)y] PEG-b-P(PEMAx-co-CMAy) which are able to form polymersomes of which the membrane is crosslinked after light irradiation. Small molecular doxorubicin hydrochloride (DOX) and large-sized glucose oxidase (GOD) were loaded inside the polymersomes.61 At low pH, the protonation of the PPEMA moieties induced a bilayer hydrophobic-to-hydrophilic transition which increased the permeability of the polymersomes, allowing for selective diffusion of small molecules. In addition, the pH-responsive segments endowed the cross-linked polymersomes with enhanced membrane permeability upon a pH change from neutral 7.4 to acidic 6.5, which is similar to the tumor microenvironment pH. The permeability of the polymersomes was tested by a colorimetric cascade reaction of the GOD-catalyzed oxidation of glucose in the presence of O2 (Fig. 4c). The glucose oxidation produced H2O2, which induced oxidation of 3,3′,5,5′-tetramethylbenzidine (TMB) into UV-vis detectable oxidized TMB (oxTMB) in the presence of Horse Radish Peroxidase (HRP). The results showed that polymersomes became permeable at pH ∼6.5. Interestingly, they found that variations in the cross-linking degree also significantly altered the permeability of the polymersomes, which provides another strategy to realize the fine-tuning of polymersome permeability.
Photo-responsive polymersomes
Photo-sensitive systems have gained considerable research interest because of attractive advantages such as non-invasiveness, easy controllability, and spatial control.82 Introducing photo-responsive groups to amphiphilic copolymers can endow the self-assembled polymersomes with photo-responsive features. Thus, the structure and function of photo-responsive polymersomes can be precisely regulated and controlled upon exposure to light.83 For example, organic photochromes such as azobenzene84,85 and spiropyran86 are usually incorporated into light-responsive polymersomes; light irradiation can adjust the polarity and conformational shape of the polymersomes, as well as enable the regulation of permeability.Recently, Bruns et al. synthesized block copolymers containing poly(pentafluorophenyl methacrylate) as the hydrophobic block via reversible addition–fragmentation chain-transfer (RAFT) radical polymerization and conjugated donor–acceptor stenhouse adducts (DASA) via the activated esters.62 These block copolymers self-assembled into polymersomes and the permeability of the resultant polymersomes was modulated by visible light irradiation. They found that two types of enzyme-loaded polymersomes containing different DASA units can selectively respond to lower wavelength (green) or longer wavelength (red) light irradiation (Fig. 5a). Light irradiation led to the formation of hydrophilic DASA blocks, which endowed the polymersomes with enhanced permeability. In another example, Liu et al. developed a photochromic polymersome from amphiphilic PEO-b-PSPA based diblock copolymers, where SPA is a spiropyran (SP)-derived monomer.87 The photochromic group SP underwent reversible isomerization between hydrophobic spiropyran (SP, λ2 > 450 nm irradiation) and zwitterionic merocyanine (MC, λ1 < 420 nm irradiation) states by light irradiation under different wavelengths. This converted impermeable SP polymersomes to permeable MC polymersomes, due to enhanced noncovalent interactions including π–π stacking interactions and hydrogen bonding interactions (Fig. 5b). Therefore, the polymersome permeability could be regulated in a controlled manner via altering the irradiation of λ1/λ2 light.
 | ||
| Fig. 5 (a) DASA-functionalized visible light-responsive nanoreactors. Reprinted from ref. 62. Copyright 2018, American Chemical Society. (b) Amphiphilic poly(ethylene oxide)-block-poly(spiropyran) (PEO-b-PSPA) diblock copolymers self-assemble into polymersomes with hydrophobic bilayers containing carbamate-based hydrogen bonding motifs. Reprinted from ref. 87. Copyright 2015, American Chemical Society. | ||
Additionally, Bruns et al. developed light-responsive nanoreactors from polymersomes composed of three kinds of amphiphilic block copolymers: α,ω-hydroxy-end-capped PMOXA-b-PDMS-b-PMOXA, α,ω-acrylate-end-capped (PMOXA-b-PDMS-b-PMOXA), and poly(ethylene oxide)-block-poly(butadiene) (PEO-b-PB).88 The polymersomes were incorporated with 2-hydroxy-4′-2-(hydroxyethoxy)-2-methylpropiophenone (PP-OH) to enable UV-light responsiveness. UV-light irradiation induced the formation of primary radicals (ketyl and alcohol), which reacted with polymeric constituents in the membranes. In all three cases, this led to chemical modification of the polymer chain with the hydrophilic PP-OH, which increased the permeability of membranes without changing the size and shape of the polymersomes (Fig. 6a). They encapsulated HRP into the polymersomes and determined the permeability of polymersomes by the enzymatic reaction with 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) (ABTS) as a model substrate. The results showed that the absorption intensity of product was significantly increased in the polymersomes treated with the PP-OH and UV-light irradiation, compared to the polymersomes without PP-OH treatment or UV light irradiation (Fig. 6b).
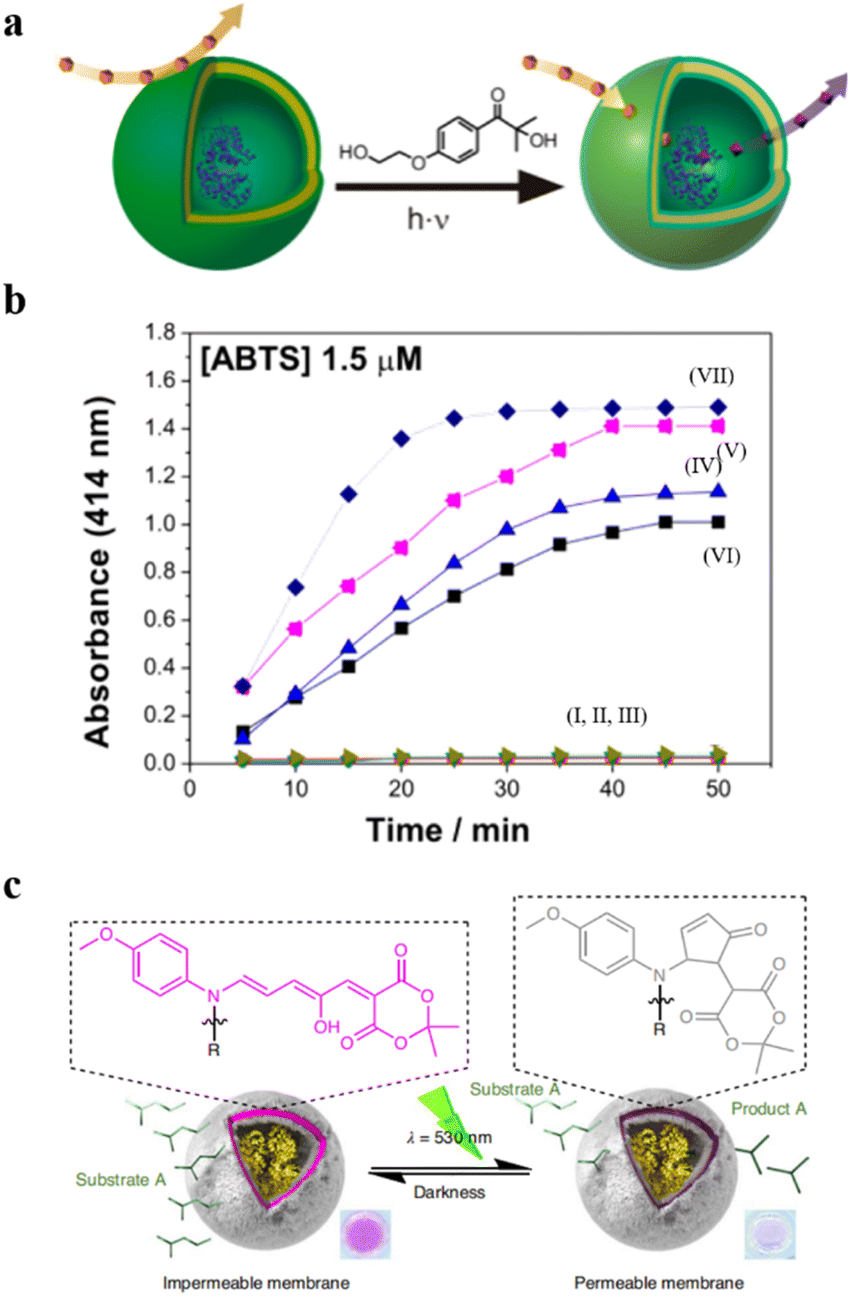 | ||
| Fig. 6 (a) Schematic representation of UV-responsive nanoreactors. (b) HRP activity test. (I–III) HRP-filled polymersomes before photoreaction with 2-hydroxy-4′-2-(hydroxyethoxy)-2-methylpropiophenone (PP-OH) and (IV–VII) after photoreaction with PP-OH. (IV) A-PMOXA-b-PDMS-b-PMOXA-A-HRP-PP-OH. (V) PMOXA-b-PDMS-b-PMOXA-HRP-PP-OH. (VI) PEO-b-PB-HRP-PP-OH. (VII) Free HRP. Reprinted from ref. 88. Copyright 2013, American Chemical Society. (c) The catalytic activity of the DASA esterase nanoreactor is activated by green light irradiation and deactivated by fading light irradiation. Reprinted from ref. 89. Copyright 2023, Springer Nature. | ||
The permeability of the DASA polymersome bilayers created by Stevens et al. can also be enhanced under green light irradiation, providing the possibility for information exchange and control.89 They introduced a non-equilibrium nanosystem consisting of two types of polymersomes, in which the enzyme-containing polymersomes can undergo photo-gated chemical communication. Specifically, esterase was encapsulated into photo-responsive DASA polymersomes. The permeability of polymersome bilayers was enhanced under green light irradiation, allowing the substrate ethyl acetate to enter the polymersomes and react with esterase to produce acetic acid, leading to a decrease in pH. As the pH decreased, the pH-sensitive pigment methyl red (MR) was converted into a green light-absorbing substance, absorbing green light and deactivating the photo-switching ability of the DASA nanoreactor (Fig. 6c). Utilizing light-mediated pH fluctuation, non-equilibrium communication between nanoreactors was achieved, which could be applied in artificial organelles, protocells and soft robotics.
Gas–responsive polymersomes
Physiologically relevant gases such as CO2, NO, and H2S have been exploited to construct gas–responsive polymer assemblies.90 The stimulus intensity of gas-sensitive systems can be precisely modulated by gas flow, which is useful for investigating the controlled permeability change of polymersomes. In this regard, gas–responsive polymersomes are of great interest to the scientific community because of the various potential biomedical applications such as intracellular imaging, diagnostics, and drug delivery.91For instance, Yan et al. developed polymersomes consisting of poly(ethylene oxide)-block-poly(o-azidomethyl benzoyl glycerol methacrylate) (PEO-b-PAGMA) and a class of particular block copolymers containing o-azidomethylbenzoate (AzMB) to enable H2S-responsiveness.92 The AzMB pendants in the polymer chain were cleaved in the presence of H2S, leading to the disassembly of the polymersomes. H2S-responsive polymersomes could open up new approaches to fulfill the need for various biomedical applications such as intracellular targeted release and therapy. In another example, Boyer et al. developed NO and temperature dual-responsive polymersomes consisting of poly[N-isopropyl acrylamide]-co-poly[N-(2-aminophenyl) methacrylamide] (P(NIPAM-co-NAPMA)) and poly[N-isopropyl acrylamide]-co-2-[3-(2-aminophenyl)ureido)ethyl methacrylate] (P(NIPAM-co-APUEMA)).93 The NAPMA and APUEMA moieties reacted with NO under mild conditions; this manipulated the polymer phase transition and controlled their self-assembly behaviors. They found that the lower critical solution temperature (LCST) values of P(NIPAM-co-NAPMA) copolymers gradually increased upon increasing NO concentrations, while the LCST values of P(NIPAM-co-APUEMA) copolymers decreased with increasing NO concentrations.
It is worth noting that CO2 is one of the most widely used “on-and-off” switches and CO2-responsive systems have a wide range of potential biomedical applications. CO2 possesses good biocompatibility and biomembrane permeability. In addition, CO2 is one of the important metabolites in cells, and it can react selectively with various functional groups (such as tertiary amine, amidine, and guanidine groups). This feature is also utilized in the membrane permeability control of polymersomes.91 For instance, Yuan et al. synthesized amphiphilic block copolymers consisting of PEG as the hydrophilic block and CO2-responsive poly(N-amidino)dodecyl acrylamide (PAD) as the hydrophobic block.45 PEG-b-PAD copolymers self-assembled into spherical polymersomes and the permeability of the polymersome membrane was regulated via CO2 stimulation. This also gave rise to a structural change in the polymersome PAD chain (Fig. 7a). To investigate the polymersome permeability adjusted by CO2, they encapsulated two kinds of dye-decorated hyperbranched poly(ethylene imine) nanoparticles (PEI-5 and PEI-25) in polymersomes. The selective polymersome permeability was evaluated by tracking the release of PEI-5 and PEI-25. The release behavior of polymersomes encapsulating PEI-5 and PEI-25 demonstrated that a higher concentration of CO2 resulted in a more effective release. With increased CO2 concentration, polymersomes swelled and the size of the polymersome membrane channels increased, while polymersomes were almost impermeable when no stimulus was applied (Fig. 7b). These results showed that the permeability of polymersome membranes is highly dependent on the environmental CO2 concentration.
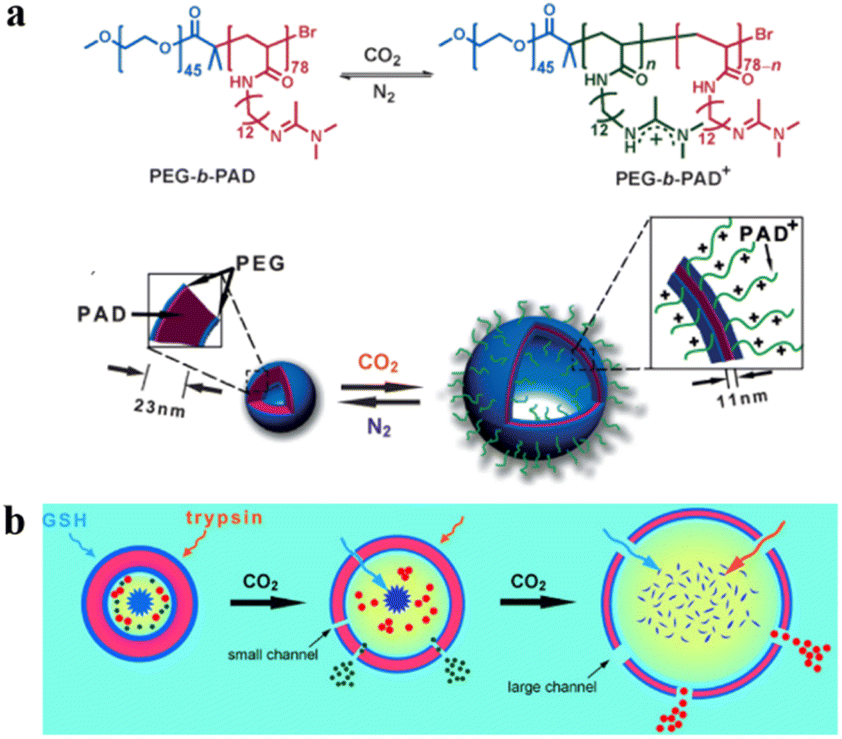 | ||
| Fig. 7 (a) Switchable chemical structural changes of poly(ethylene glycol)-block-poly(N-amidino) dodecyl acrylamide (PEG-b-PAD) copolymers. (b) Gas-controlled selective release of cargos from the polymersomes. Reprinted from ref. 45. Copyright 2013, Wiley-VCH. | ||
Oxidation–responsive polymersomes
The permeability of polymersomes can also be adjusted via the presence of reactive oxygen species (ROS). This is normally realized via the chemical structure transition of polymer compositions stimulated by ROS. The abnormal redox states in tumor and inflammatory tissues can thus be considered as targets for site-specific delivery of therapeutics using ROS–responsive systems. For instance, disulfide bonds are usually introduced into polymersomes as cross-linkers, which can be reduced to two thiols under reductive conditions. The disulfide bond cleavage can further lead to the dissociation of the polymersomes.66Recently, Ding et al. prepared ROS–responsive polypeptide vesicles from cholesterol-decorated methoxy poly(ethylene glycol)-poly(L-cysteine) copolymers (mPEG-PLCC). The permeability of the polymersome membrane was well-tuned by the ordered transition of the secondary conformations.94 The structural organization of mPEG-PLCC transformed from a β-sheet to α-helix upon ROS stimulation, which resulted in a decrease in membrane wall thickness along with changes in the permeability of the polymersomes (Fig. 8a). They found that during oxidation the appearance of characteristic minima at 207 and 222 nm in the CD spectra and a gradual shift of the amide I region from 1630 cm−1 to 1649 cm−1 in the FTIR spectra when the polymersomes were treated with H2O2. This confirmed the transformation of copolymers from β-sheet to α-helix and demonstrates that oxidation alters the secondary structure of MPEG-PLCC. To investigate the influence of membrane changes on the permeability of polymersomes, hydrophilic rhodamine 6G (R6G) was encapsulated in the polymersomes, which were then treated with H2O2. The self-quenching effect of R6G remains unchanged over time in the absence of an oxidizing environment, however, the dye was released rapidly into the aqueous media, and the fluorescence intensity recovered by more than 82% within 12 h in an oxidative environment (Fig. 8b and c).
 | ||
| Fig. 8 (a) Schematic representation of conformation-regulated permeability of methoxy poly(ethylene glycol)-poly(L-cysteine) (mPEG-PLCC) polymersomes. (b) and hydrophilic drug R6G (c) from MPEG-PLCC assemblies in media with or without 10% H2O2. Reprinted from ref. 94. Copyright 2021, Wiley-VCH. | ||
Temperature–responsive polymersomes
Temperature–responsive polymersomes generally consist of thermosensitive polymers, which can undergo volume phase transition upon temperature changes. They have a lower critical solution temperature (LCST) or upper critical solution temperature (USCT). However, most of the present research on temperature–responsive polymersomes is mainly focused on the temperature-induced irreversible disassembly of polymersomes, which further leads to the release of encapsulated cargoes.39 In this case, polymersomes lose their structural integrity upon a temperature change, which limits their application as nanoreactors.Kharlampieva et al. synthesized an amphiphilic triblock copolymer poly(N-vinylcaprolactam)n-poly(dimethylsiloxane)65-poly(N-vinylcaprolactam)n (PVCLn-PDMS65-PVCLn) by RAFT polymerization.47 These copolymers self-assembled into temperature-responsive polymersomes at room temperature. PVCL chains collapsed when the temperature increased, owing to the destruction of hydrogen bonds between VCL and water, and enhanced hydrophobic interactions between PVCL segments, which increased the polymersome permeability (Fig. 9a). The length of PVCL was utilized to control the release of the loaded anticancer drug doxorubicin (DOX) (Fig. 9b). Importantly, the temperature-induced structural change of polymersomes was reversible, and the permeability of polymersomes was regulated by modulating the temperature between 37 °C and 42 °C, without disrupting the structural integrity of the polymersomes.
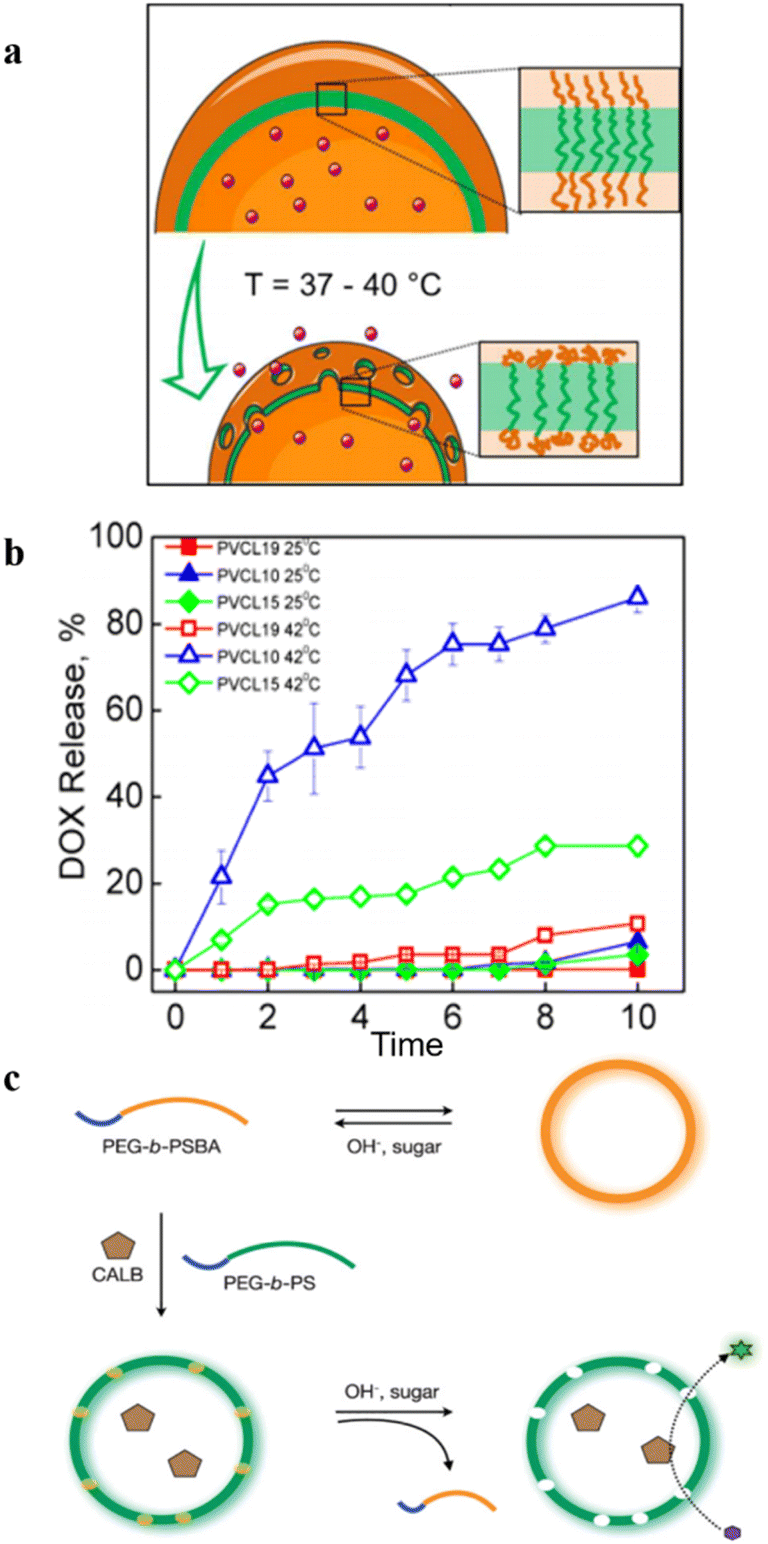 | ||
| Fig. 9 (a) Schematic representation of the emulsion-centrifugation process producing polymersomes. (b) The plot of vitro DOX release (weight%). Reprinted from ref. 47. Copyright 2013, American Chemical Society. (c) Schematic representation of the formation of permeable polymersomes utilizing pH–responsive and glucose–responsive block copolymers. Reprinted from ref. 48. Copyright 2009, Wiley-VCH. | ||
In addition to the above-mentioned single stimuli-responsive models, there are also some reports on dual or triply-responsive polymersomes to regulate their permeabilities. For instance, Kim et al. constructed pH and glucose dual-responsive polymersomes, consisting of amphiphilic block copolymers poly(ethylene glycol)-b-poly(styrene boronic acid) (PEG-b-PSBA) and PEG-b-PS (Fig. 9c).48 They found that the change in pH and glucose concentrations in the aqueous media had a significant effect on the solubility of the PSBA block when polymersomes were prepared from a mixture of PEG-b-PS and PEG-b-PSBA. The responsive PSBA block in PEG-b-PSBA and the nonpolar PS block in PEG-b-PS are prone to phase separation during the formation of the bilayer, and the phase-separated PSBA domains were dispersed in the PS matrix. These PSBA domains were easily extracted from the PS matrix by changing the pH and the sugar concentrations, enabling the permeability of the polymersomes to be tuned via these two stimuli.
Voit et al. reported an example of temperature and pH dual-responsive hollow capsules.95 They synthesized ABA-type triblock copolymer brushes poly(N-isopropylacrylamide)-block-poly[2-(diethylamino)ethylmethacrylate-co-2-hydroxy-4(methacryloyloxy) benzophenone]-block-poly(N-isopropylacrylamide) PNIPAM-b-PDEAEMA-co-PBMA-b-PNIPAM(PN-DB-N) rapidly on a silica template via surface-initiated single electron transfer living radical polymerization (SI-SET-LRP). Hollow capsules with photo-cross-linked membranes were obtained after removing the template. The PNIPAM layer in the central membrane layer collapsed when the temperature was above the LCST, leading to an impermeable membrane bilayer. Moreover, the photo-cross-linked PDEAEMA chain in the central membrane layer changed from a deprotonated, hydrophobic entangled state at a high pH value to a protonated, hydrated, repulsive state at a low pH value, which led to a change in polymersome permeability. With this dual responsiveness hollow capsules were impermeable at 40 °C, semi-permeable at 25 °C at pH = 8, and permeable at 25 °C at pH = 6 (Fig. 10a and b). Therefore, the permeability of hollow capsules could be adjusted and modulated precisely via control of temperature and pH.
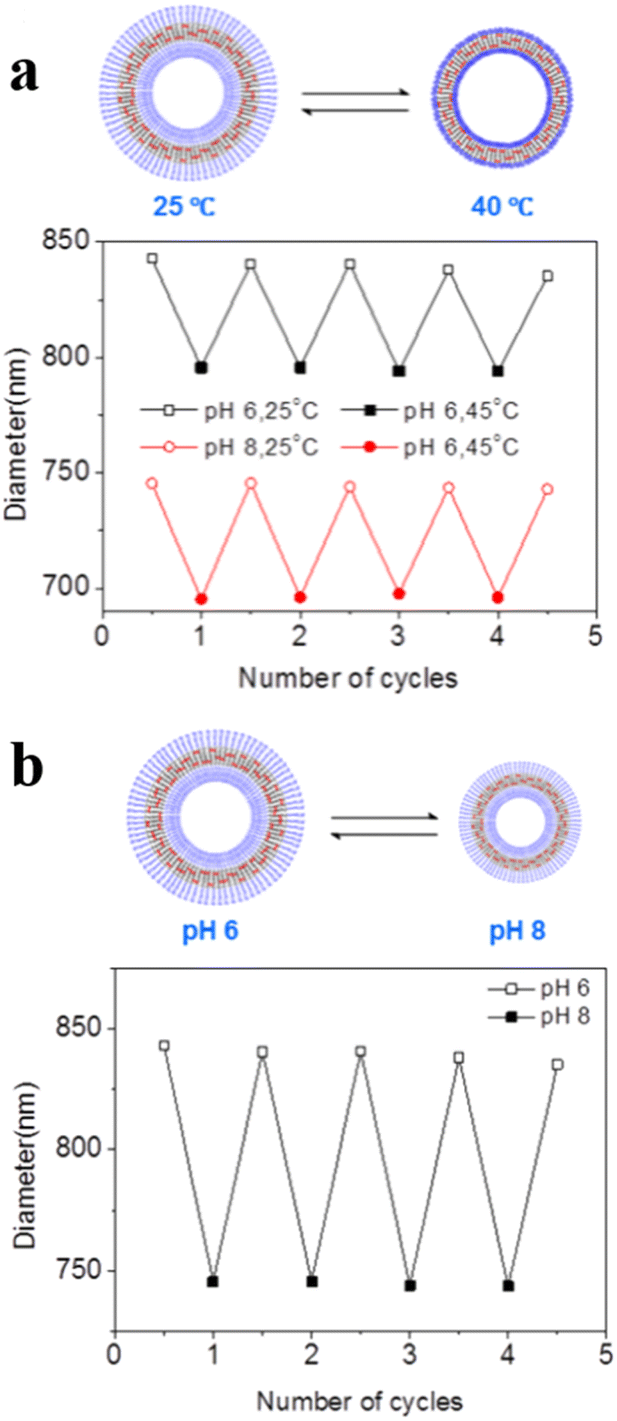 | ||
| Fig. 10 (a) Reversible swelling–shrinking of hollow capsules upon switching between 25 and 40 °C at different pH. (b) Reversible swelling–shrinking of hollow capsules upon switching between pH = 6 and 8 at 25 °C. The diameter was measured by Dynamic Light Scattering (DLS). Reprinted from ref. 95. Copyright 2018, American Chemical Society. | ||
In addition to the use of stimuli-responsive polymers, there are several other strategies for the formation of permeable polymersomes. For example, the use of polyelectrolytes to construct polyion complex vesicles (PICsomes),96,97 and post-modification of polymersomes,98 have also been shown as effective approaches for modulating the polymersome permeability. The recent advances in construction of stimuli-responsive polymersomes demonstrate that the permeability of polymersomes can be well regulated via changes of the environmental signals. The incorporation of stimuli-responsiveness in the polymersomes enabled phase transitions or structural changes under specific conditions, thereby controlling the permeability of polymersomes. This strategy provides a promising way for precise control and release of cargos, which are promising candidates for the construction of nanoreactors and artificial organelles, as well as nanocarriers for nanomedicine.
Applications of permeable polymersomes
Permeable polymersomes that are biocompatible and degradable can be employed in a range of different fields including nanoreactors, artificial cells and organelles.60,99 In addition, they are potential carriers for drug loading and delivery due to their high loading capacity, selective release, and stability. Scientists are committed to designing and developing such polymersomes to be applied in many fields.Polymersomes as biologically active nanoreactors
Permeable polymersomes can be transformed into nanoreactors via encapsulating catalytic species such as enzymes inside the polymersome lumen or the hydrophobic membrane.100,101 They have shown great potential in confined catalysis by separating catalytic species from each other and protecting catalysts from undesired influences from the environment.99,102,103Recently, amphiphilic block copolymers with two kinds of different DASAs in their hydrophobic blocks were synthesized by using Meldrum's acid-based furan adduct (MELD) and five-membered ring pyrazolone-based furan adduct (PYRA), which self-assembled into MELD and PYRA polymersomes, respectively (Fig. 11a and b).62 HRP was encapsulated into photo-responsive MELD polymersomes and the biocatalytic activity was switched “on” under light conditions and “off” under dark conditions due to a change in permeability (Fig. 11c). The catalytic reactions of PYRA polymersomes encapsulating Glucose Oxidase (GOx) was similar to that of MELD polymersomes (Fig. 11d). Therefore, DASA polymersome nanoreactors are useful vehicles for on-demand catalytic reactions, which allow the diffusion control of the reactants across the polymer membrane via light irradiation. Interestingly, the GOx-HRP cascade reaction could be facilely achieved by mixing the two types of polymersome nanoreactors, and the on/off switch of the nanoreactors was controlled via alternate red-green light irradiation (Fig. 11e).
 | ||
| Fig. 11 (a) Preparation of the block copolymers. (b) Schematic representation of Meldrum's acid-based furan adduct (MELD) polymersomes and pyrazolone-based furan adduct (PYRA) polymersomes. (c) UV-vis absorbance indicates product formation upon light irradiation of HRP loaded into MELD polymersomes. (d) UV-vis absorbance indicating product formation upon light irradiation of GOx loaded into PYRA polymersomes. Continuous irradiation with white light (blue and pink), alternating light and dark cycle (orange), dark (black), and non-catalytic blank reaction (red). (e) Cascade pyrroliol reaction catalyzed by a mixture of MELD-HRP and PYRA-GOx nanoreactors, alternately deactivated in the dark and activated with red light (λ = 630 nm)(red), dark and green light (λ = 525 nm) (green), dark(black). Reprinted from ref. 62. Copyright 2018, American Chemical Society. | ||
We recently developed self-regulated “breathing” polymersome nanoreactors composed of poly(ethylene glycol)-block-poly[2-(diethylamino) ethyl methacrylate-co-2-hydroxy-4-(methacryloyloxy) benzophenone-co-fluorescein O-methacrylate] PEG-b-P(DEAEMA-co-BMA-co-FMA) block copolymers, which showed time-controlled and fuel-driven biocatalysis (Fig. 12a).104 Polymersome nanoreactors shrank owing to deprotonation of the pH-sensitive PDEAEMA block when they were dispersed in a base buffer, creating an “off” state due to the impermeable polymersome membrane. The pH value of the solution decreased after the addition of chemical fuel (HCl and urea), resulting in an increased size of the polymersome nanoreactors and enhanced polymersome permeability. Subsequently, urea was converted to ammonia, which led to a gradual pH increase. As a result, polymersome nanoreactors turned back to the “off” state. HRP and urease labelled with Rhodamine-B (RhB) were coencapsulated into polymersome nanoreactors. Then samples were incubated in a pH 9.0 buffer, and no absorbance was observed at 416 nm. However, samples with the addition of HCl and urea resulted in a gradual increase in the absorbance at 416 nm over time which was then stabilized to a constant value. The higher the urea concentration, the faster the catalysis reached its endpoint. More importantly, with the continuous addition of urea three consecutive nanoreactors “OFF–ON–OFF” cycles were achieved (Fig. 12b).
 | ||
| Fig. 12 (a) Schematic representation of feedback-induced temporal control of polymersome nanoreactors. (b) Reversible nanoreactor “ON–OFF” regulation in time following repeated additions of urea. Reprinted from ref. 104. Copyright 2018, American Chemical Society. | ||
Permeable polymersomes are able to encapsulate catalysts or reactant molecules, and control reaction rate and selectivity by adjusting their permeability. These polymersomes have great potential in the field of nanoreactors, providing new solutions for biocatalytic reactions.
Polymersomes as artificial cells and organelles
Polymersome nanoreactors have also been applied as artificial organelles in multi-compartmentalized systems. The multiple subcompartments were incorporated in giant polymersomes to perform a wide variety of biochemical reactions similar to a biological cell.105 These systems offer greater flexibility and more complex cell mimetic behavior compared to single-compartment systems. For example, Lecommandoux et al. investigated the effects of multi-compartmentalization and functional control on a multistep reaction pathway in polymersomes-in-polymersomes systems.106 Phenylacetone monooxygenase (PAMO) was used to oxidize a ketone into an ester, and the ester was subsequently hydrolyzed by a lipase, candida antarctica lipase B (CalB). Finally, the resulting hydroxy-functional profluorescent compound was oxidized by alcohol dehydrogenase (ADH), which led to the formation of a fluorescent product. The artificial organelles were composed of intrinsically permeable PS-PIAT polymersomes. CalB and ADH were encapsulated in these nanoreactors and these were loaded together with PAMO and the reagents into micrometer-sized PB-b-PEO polymersomes (Fig. 13a). The validity of the three-enzyme cascade was first tested without the encapsulation in the PB-b-PEO polymersomes, with either all enzymes in solution or with CalB and ADH enzymes encapsulated in PS-b-PIAT nanoreactors (Fig. 13b). When CalB was replaced by alcalase, a protease, the activity of the free three-enzyme cascade was considerably decreased due to the proteolytic effect of alcalase on the free enzymes. When alcalase was encapsulated in PS-b-PIAT nanoreactors, the physical separation ensured a more efficient conversion (Fig. 13c).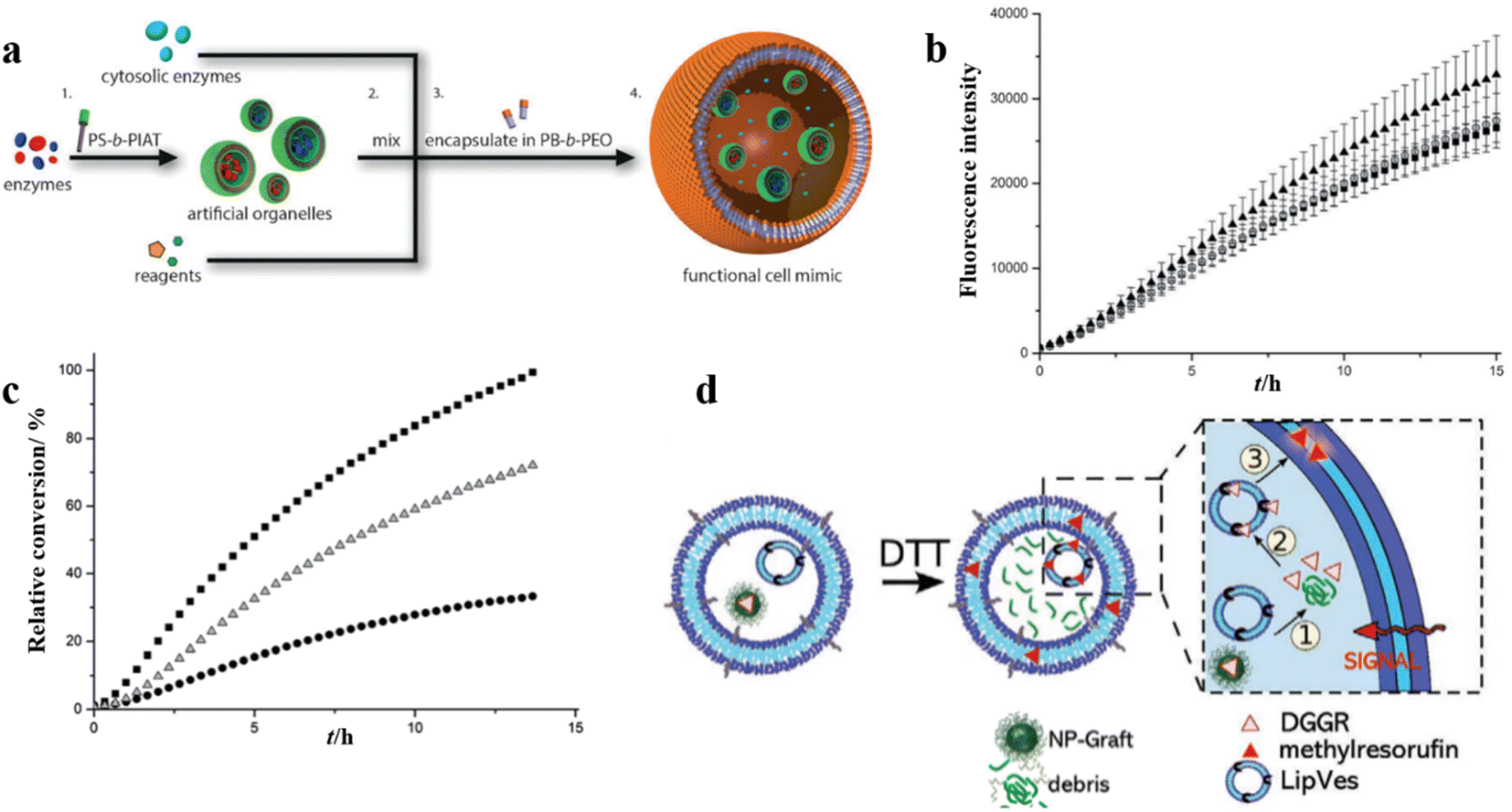 | ||
| Fig. 13 (a) Schematic representation of polymersomes-in-polymersome microreactor. (b) Ensemble fluorescence measurements of the cascade reaction with all of the enzymes free in solution (▲), CalB in polystyrene-block-poly(L-isocyanoalanine (2-thiophen-3-ylethyl)amide) (PS-b-PIAT) nanoreactors (■), or CalB and ADH in PS-b-PIAT nanoreactors (O). (c) Ensemble fluorescence measurements of the cascade reaction with CalB in PS-b-PIAT nanoreactors (■), alcalase in PS-b-PIAT nanoreactors(Δ), free alcalase (●). Reprinted from ref. 106. Copyright 2014, Wiley-VCH. (d) The schematic representation of the enzymatic reaction of lipase substrate (DGGR) loaded on nano-particles and LipVes loaded with lipase enzyme, and these polymersomes are encapsulated as subcompartments into a polymer GUV. Reprinted from ref. 107. Copyright 2020, Wiley-VCH. | ||
In another example, Meier et al. created a self-assembled vesicular multicompartment system in which complex signal transduction was achieved via a cascade involving two different types of artificial organelles.107 These systems contained structurally different compartments, which were composed of a polymeric membrane (giant unilamellar vesicle (GUV)) encapsulating artificial organelles consisting of a mixture of PMOXA5-b-PDMS58-b-PMOXA5 and PDMS65-b-heparin copolymers. The lipase substrate 1,2-Di-O-lauryl-rac-glycero-3-(glutaric acid 6-methylresorufin ester) (DGGR) was incorporated in the stimuli-responsive artificial organelle (NPGraft) based on the graft copolymer (poly(2-methyl-2-oxazoline)88-graft(SS)-poly(ε-caprolactone)238 (PMOXA88-g(SS)-PCL238), and the lipase enzyme was entrapped in the secondary artificial organelles (LipVes), then these two kinds of artificial organelles were loaded into GUVs, which formed a stimuli-responsive multicompartment system. Fluorescence was not observed in a nonreductive environment, but DGGR was released from the subcompartment NP-graft under reductive conditions and interacted freely with enzymes in LipVes forming the fluorescent product after adding the signalling molecule DTT to the GUVs (Fig. 13d). The lipase reaction was successfully triggered via the signal transduction between two types of artificial organelles within a giant vesicle with multiple internal subcompartments.
Enzyme-based polymersome bio-nanoreactors are potentially useful for the treatment of specific diseases associated with organelle defects.108–111 Recently, van Hest and coworkers reported an assembly of a rudimentary multi-compartmentalized structure by the spontaneous encapsulation of poly(ethylene glycol)-b-poly(caprolactone-gradient-trimethylene carbonate) (PEG-PCLgTMC) polymersomes inside cell-sized protocells.112 The hierarchical self-assembly of the protocells was achieved by membranization of coacervate microdroplets with a tailor-made poly(glutamic acid) (PGA) containing terpolymer PEG-PCLgTMC-PGA (Fig. 14a). Three types of polymersome sub-organelles (via loading different fluorescent proteins) were encapsulated and distributed evenly inside the protocell. This demonstrated that the protocell system allowed for the spatial organization of macromolecular cargo (Fig. 14b). Moreover, the protocell membrane protected the sequestered FITC-BSA from degradation by the external proteolytic enzyme (e.g. proteinase K) (Fig. 14c). Such ability of spatial organization and protein protection represents an important step forward towards the development of complex artificial organelles and cells.
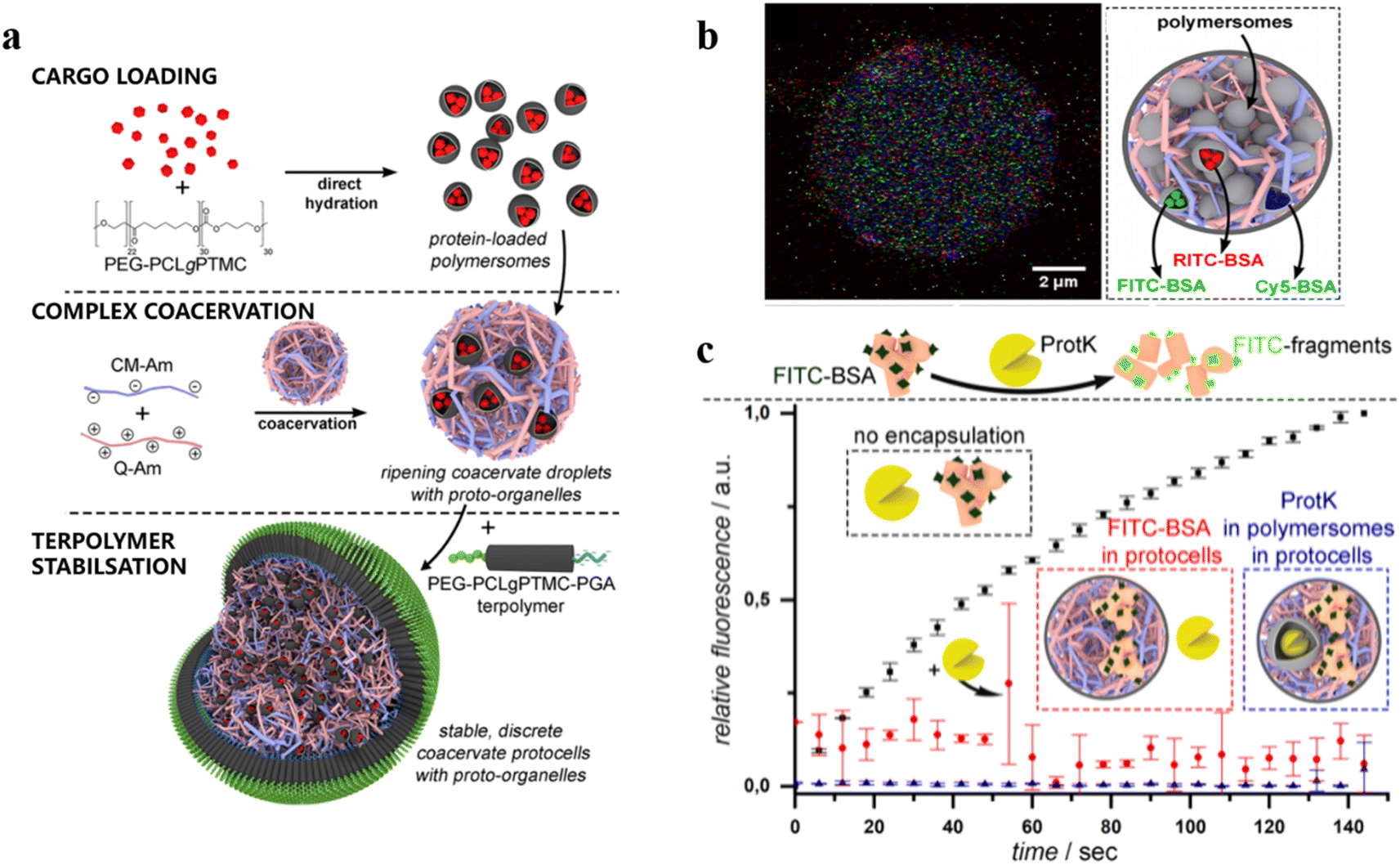 | ||
| Fig. 14 (a) Schematic representation of a hierarchical protocell. (b) Hierarchical protocell sequestering three kinds of proto-organelles loaded with different fluorescently labelled proteins. (c) Bulk fluorescence emission spectroscopy of self-assembled systems. BSA is labelled with fluorescent probes, which are quenched when BSA remains intact. Upon protease K (protK) activity fluorescence is observed. Encapsulation thus protects BSA from hydrolysis. Reprinted from ref. 112. Copyright 2019, American Chemical Society. | ||
Stimuli-responsive artificial organelles (AOs) were also applied in in vivo research.113 Palivan et al. created PMOXA6-b-PDMS44-b-PMOXA6 AOs with endogenous stimuli-triggered enzymatic activity and used them as cellular implants in zebrafish embryos (ZFE).114 Polymeric membranes with inserted protein gates (OmpF porin) were responsive to changes in glutathione (GSH) concentration in intracellular environments and triggered the in situ HRP enzymatic reaction, while maintaining the structural integrity of the nanocompartment (Fig. 15a). To investigate the cellular internalization and intracellular localization, HRP labelled with Atto488 (HRPAtto488) and Atto647 (HRP-Atto647) was loaded into AOs with and without OmpF, respectively (Fig. 15b). AOs did not co-locate with lysosomes, indicating intracellular endosomal escape. Notably, the permeability of AOs with OmpF was enhanced, which allowed substrate to pass through the polymersome membranes to be converted by HRP into a resorufin-like product (RLP). They found that AOs with OmpF displayed a significant increase in the intracellular fluorescence, compared to AOs without OmpF. Moreover, they injected AOs into ZFE and did not observe acute toxicity in vivo after 24 h. Cellular internalization of AOs was successful in vivo into the early immune system of ZFE, and AOs were able to remain intact in ZFE macrophages due to the stable and robust polymeric membrane (Fig. 15c).
 | ||
| Fig. 15 (a) Schematic representation of modified OmpF acting as a gate in catalytic nanocompartments. (b) Molecular representation of the OmpF-M cysteine mutant. (c) ZFE biodistribution in vivo and activity of AOs (lateral view of the ZFE crosssection). Blue: ZFE melanocytes. Green: HRP-Atto488. Red: RZLP. Reprinted from ref. 114. Copyright 2018, Nature Publishing Group. | ||
In another work, Palivan and co-workers constructed microscale bioinspired molecular factories (MFs) which were composed of giant plasma membrane vesicles (GPMVs) with the same membrane and cytosol environment as the cells and loaded with functional AOs, to mimic the architecture and functionality of eukaryotic cells.115 Firstly, AOs based on PMOXA6-b-PDMS44-b-PMOXA6 were prepared by the thin film rehydration method, then they were preloaded into donor cells. A vesiculation buffer was added to the cells after internalization of AOs to induce the formation of GPMV. Induced by the vesiculation buffer, the cytosol with the artificial cargos was transferred to the GPMV to form the newly equipped-GPMVs (E-GPMVs). The membrane and inner cavity of E-GPMVs were modified with CellTracker Deep Red Dye (CDTR) and membrane protein LcK tyrosine kinase GFP (Lck-GFP), which allowed specific localization of the artificial cargos. PMOXA6-b-PDMS44-b-PMOXA6 polymersomes loaded with sulforhodamine B (SRB) solution were taken up by HepG2 cells, and then transferred to the inner cavity of E-GPMVs inserted with Lck-GFP to create a robust multicompartment cell-like architecture (Fig. 16a). Amplex UltraRed as a nonfluorescent substrate was encapsulated into GPMVs and HRP was encapsulated into polymersomes with or without OmpF. The results showed that only GPMVs with OmpF showed an increase in fluorescence (Fig. 16b and c). The experiments with the ZFE model showed the MFs were nontoxic and were able to function in vivo for a long time with structural and functional integrity (Fig. 16d). This work presents the feasibility of using MFs with complex composition as artificial cell mimics in vivo, and open new perspectives for in vitro diagnostic and in vivo therapeutic solutions.
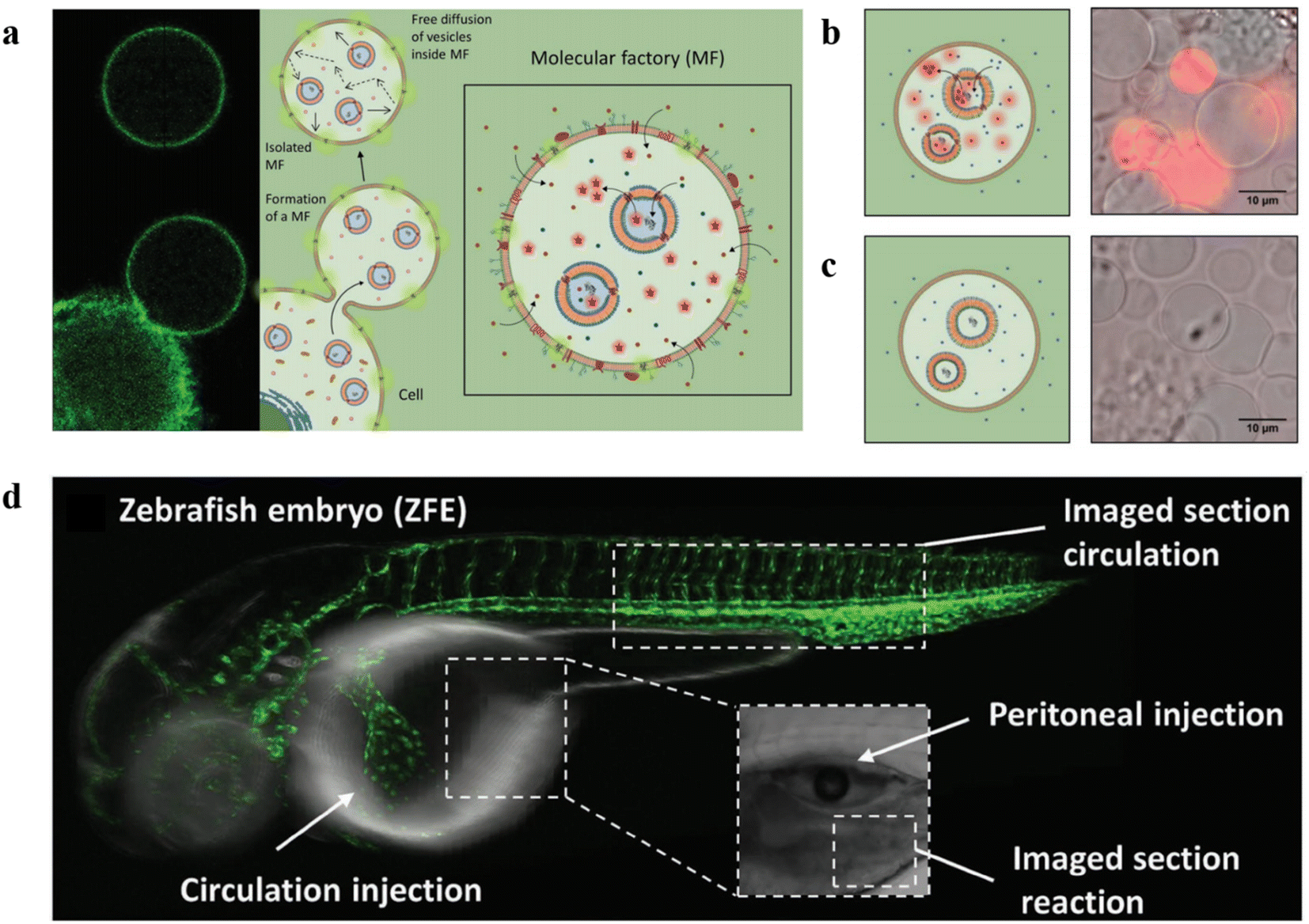 | ||
| Fig. 16 (a) Schematic representation of the formation of molecular factories (MFs). (b) Left: Schematic representation of MFs containing AOs with OmpF. Right: Relative fluorescence intensity recordings of MFs containing AOs with OmpF. (c) Left: Schematic representation of MFs containing AOs without OmpF. Right: Relative fluorescence intensity recordings of MFs containing AOs without OmpF. (d) Schematic representation of ZFE injection and imaging. Reprinted from ref. 115. Copyright 2020, Wiley-VCH. | ||
Compartments based on photoactivated giant polymersomes (GPs) are an attractive option for designing robust cell-like systems with controlled molecular membrane transport.116 Recently, Cao et al. prepared GPs with photoactivated membrane permeability, which served as active compartments for engineering cell-like systems.117 A photo-transducer composed of a photo-responsive spiropyran (SP) was integrated into PB22-b-PEO12 polymersome membranes to tune the polymersome permeability. The polymersome permeability was increased by the photoisomerization of SP, which allowed small modules to pass through the polymeric membrane while macromolecular cargo was retained in the compartments (Fig. 17a). β-Galactosidase (β-gal) was encapsulated into SP-GPs by the double emulsion method to construct light-activated microreactors. The microreactors were placed in a solution containing the non-fluorescent substrate β-gal-FITC. They found that fluorescence intensity at 520 nm gradually increased after 20 s (Fig. 17b and c). Additionally, artificial cells constructed using SP-GPs were used to mimic cell-like co-localization of biomolecules in biomolecular condensates, which served as membrane-less compartments to concentrate biomolecules (Fig. 17d). These interesting findings in this study demonstrated that polymeric compartments can construct dynamic systems which exhibit excellent biomimetic utility.
 | ||
| Fig. 17 (a) Schematic representation of design and construction of the giant polymersomes (GPs). (b) UV-induced uptake of β-gal-FITC. (c) Fluorescence intensity of SP-GPs microreactors at different conditions. (d) Schematic representation of the coacervation process inside SP-GPs which could concentrate biomolecules. Reprinted from ref. 117. Copyright 2022, Wiley-VCH. | ||
Permeable polymersomes are used as artificial compartments to simulate the functions of natural organelles due to their ability to encapsulate and selectively transport molecules. This is well illustrated in the examples given in this review. However, more efforts are needed to study how to assemble multiple types and functions of permeable polymersomes into more complex artificial organelle systems and integrate them with natural biological entities. These issues need to be further addressed to promote the development of permeable polymersomes in the field of artificial organelles.
Polymersomes in nanomedicine
Polymersomes can be utilized as nanovectors to deliver drugs to the target site safely and accurately.118,119 Unfortunately, in most cases, the burst release due to the structural disruption of polymersomes may result in a diminished treatment effect.34 Therefore, it would be beneficial to develop permeable polymersomes to achieve “on-demand” drug release by regulating the permeability of polymersomes while maintaining their structural stability.120Using a photo-triggered cross-linking reaction, Ju and coworkers fabricated a biomimetic polymersome, which achieved sustained drug release for a certain period under NIR light. The amphiphilic block copolymer P(OEGMA-co-EOS)-b-PNBOC (POPN), was synthesized from the hydrophobic monomer 2-nitrobenzyloxycarbonylaminoethylmethacrylate (NBOC) and the hydrophilic monomers oligo(ethylene glycol) monomethyl ether methacrylate (OEGMA) and the methacrylate derivative of eosin Y, and was assembled into spherical polymersomes (Fig. 18a).121 Upon NIR irradiation, the primary amine was uncaged in PNBOC, which cross-linked to neighboring ester groups through an amidation reaction. This resulted in a hydrophobic-to-hydrophilic transition of the building blocks and increased polymersome permeability (Fig. 18b). They encapsulated the hypoxia-activated prodrug AQ4N and upconversion nanoparticles (PEG-UCNP) into the polymersome and tracked the change of drug release before and after NIR irradiation. They found that the polymersomes were impermeable in the absence of NIR irradiation. However, the polymersome permeability was highly improved and the encapsulated AQ4N diffused across the polymersome membrane under NIR light irradiation (Fig. 18c). Moreover, the permeability of the polymersome could be adjusted by the NIR irradiation time, resulting in a controlled release rate of AQ4N.
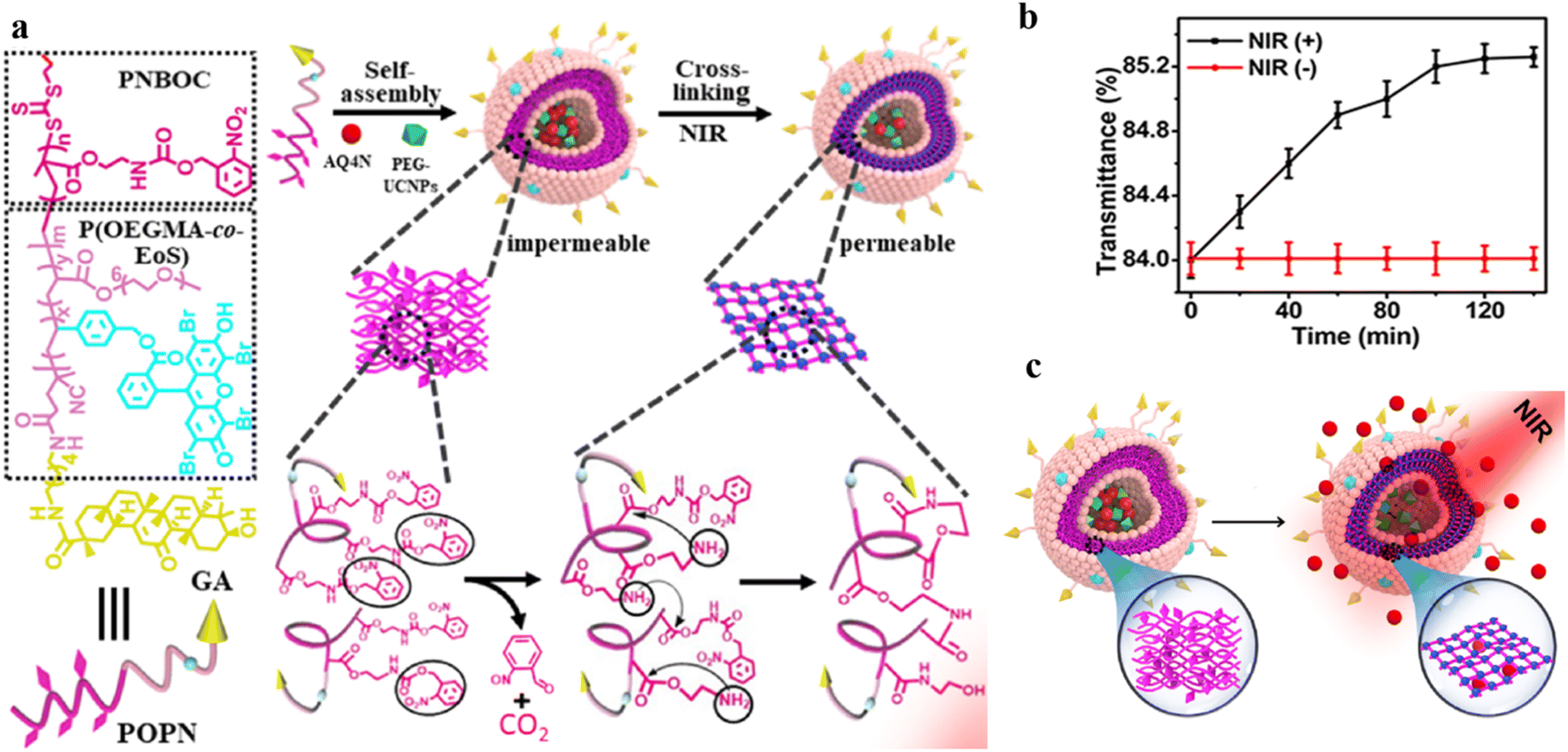 | ||
| Fig. 18 (a) Structure and composition of the amphiphilic diblock copolymer poly(oligo(ethylene glycol) monomethyl ether methacrylate-co-eosin Y)-block-poly(2-nitrobenzyloxycarbonylaminoethylmethacrylate) (P(OEGMA-co-EOS)-b-PNBOC) (POPN) and the hypoxia-activated prodrug/PEG ligand (AQ4N/PEG-UCNPs) polymersome assembly. (b) Optical transmission of the PEG-UCNPs polymersomes, depending on the time of NIR irradiation (in the presence of NIR (+)) and in the absence of (NIR (−)) irradiation. (c) Schematic representation of continuous release of the hydrophilic agent AQ4N in the polymersomes after NIR irradiation. Reprinted from ref. 121. Copyright 2021, American Chemical Society. | ||
Lecommandoux et al. prepared polymersomes-in-polymersomes structures (PiPs, also referred to as vesosomes), which were obtained by the self-assembly of amphiphilic block copolymers in aqueous solution via free-energy minimization.122 The inner polymersomes were formed from poly(trimethylene carbonate)-b-poly(L-glutamic acid) (PTMC-b-PGA) by nanoprecipitation. Then a suspension of these polymersomes was loaded into larger PB-b-PEO polymersomes by emulsion-centrifugation, resulting in giant polymeric vesosomes containing multiple compartments (Fig. 19a). The specific process was divided into three steps: a small part of polymersome suspension containing PTMC-b-PGA was poured onto the interface between toluene and 380 mOsm glucose aqueous solution, and PB-b-PEO copolymers were dissolved in toluene to stabilize the emulsion droplets at the interface. Finally, these droplets passed through the interface by centrifugal forced and were packaged by PB-b-PEO polymersomes, causing forming polymer vesosomes. The release curve of DOX as a model drug proved that PiPs significantly reduced the release rate of the anticancer drug DOX when encapsulated in the inner polymersomes. The release rate of DOX loaded in PTMC-b-PGA polymersomes (nano-DOX) was about twice that of polymer vesosomes loaded with nano-DOX as inner polymersomes (veso-DOX) (Fig. 19b). The polymersome-in-polymersome structures with slower membrane diffusion open up a new avenue for controlled drug release.
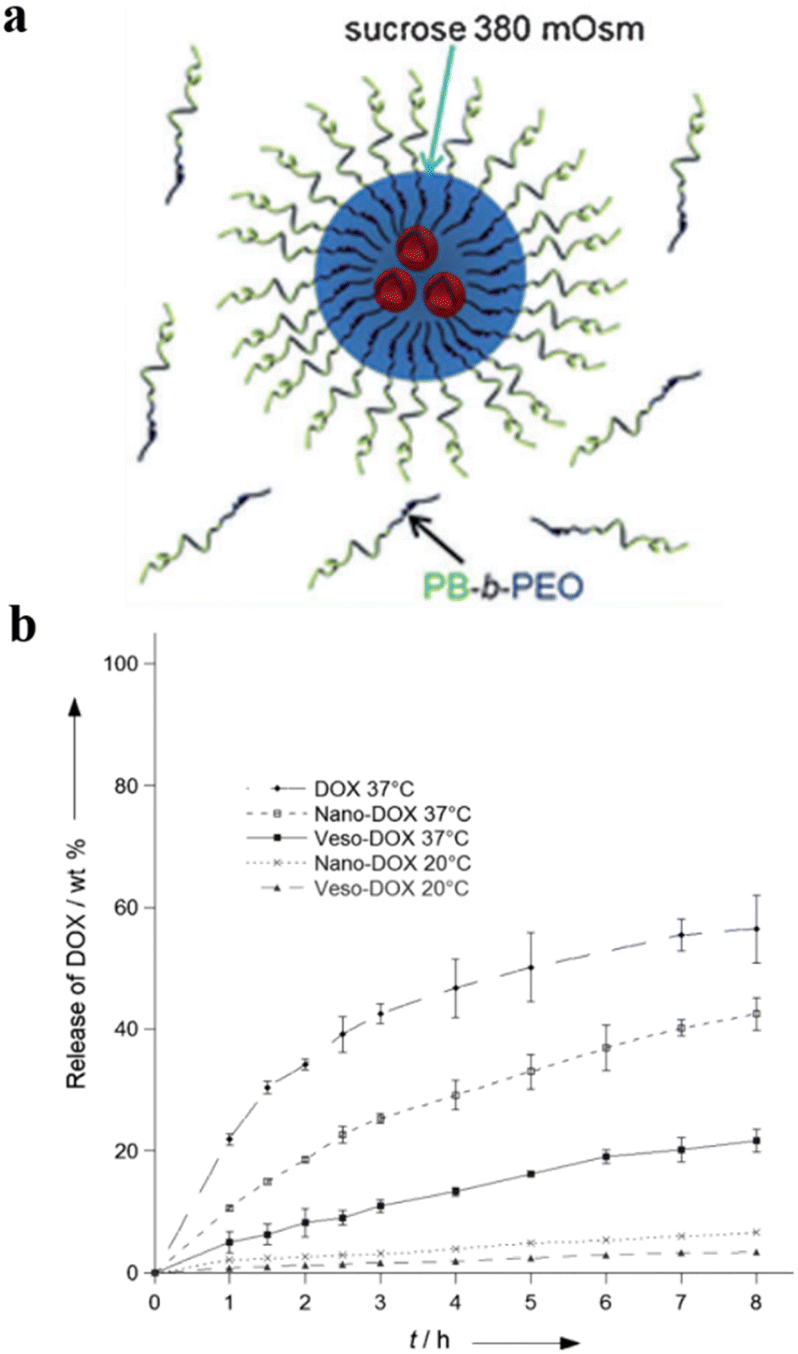 | ||
| Fig. 19 (a) Schematic representation of the emulsion-centrifugation process producing giant polymer vesosomes. The droplets containing poly(trimethylene carbonate)-block-poly(L-glutamic acid) (PTMC-b-PGA) which were poured onto the interface between toluene and 380 mOsm glucose aqueous solution passed through the interface by centrifugal forced and were packaged by PB-b-PEO polymersomes in toluene. (b) The plot of in vitro DOX release (weight%), (I) free DOX at 37 °C, (II) nano-DOX at 37 °C, (III) veso-DOX at 37 °C, (IV) nano-DOX at 20 °C and (V) veso-DOX at 20 °C. Reprinted from ref. 122. Copyright 2012, Wiley-VCH. | ||
Gu et al. demonstrated that the photocleavage reaction of o-nitrobenzyl groups in photo-responsive polymersomes consisting of poly(N,N′-dimethylacrylamide)30-b-poly(o-nitrobenzyl acrylate)97 (PDMA30-b-PNBA97) induced the dissociation of polymersomes and gave rise to the co-release of both hydrophilic and hydrophobic drugs encapsulated inside the polymersomes (Fig. 20a).123 They co-loaded hydrophilic drug DOX and hydrophobic Nile red (NR) into polymersomes and tracked the release of both cargoes. They found that the fluorescence intensity of NR did not change in the absence of UV irradiation but decreased sharply upon UV irradiation (Fig. 20b and c). As shown in Fig. 20d, only a small amount of DOX was released from polymersomes in the absence of UV irradiation, while the release rate of DOX was significantly increased over time in the presence of UV irradiation. This suggests that the dissociation of polymersomes led to the photo-controlled release of both hydrophilic and hydrophobic cargoes.
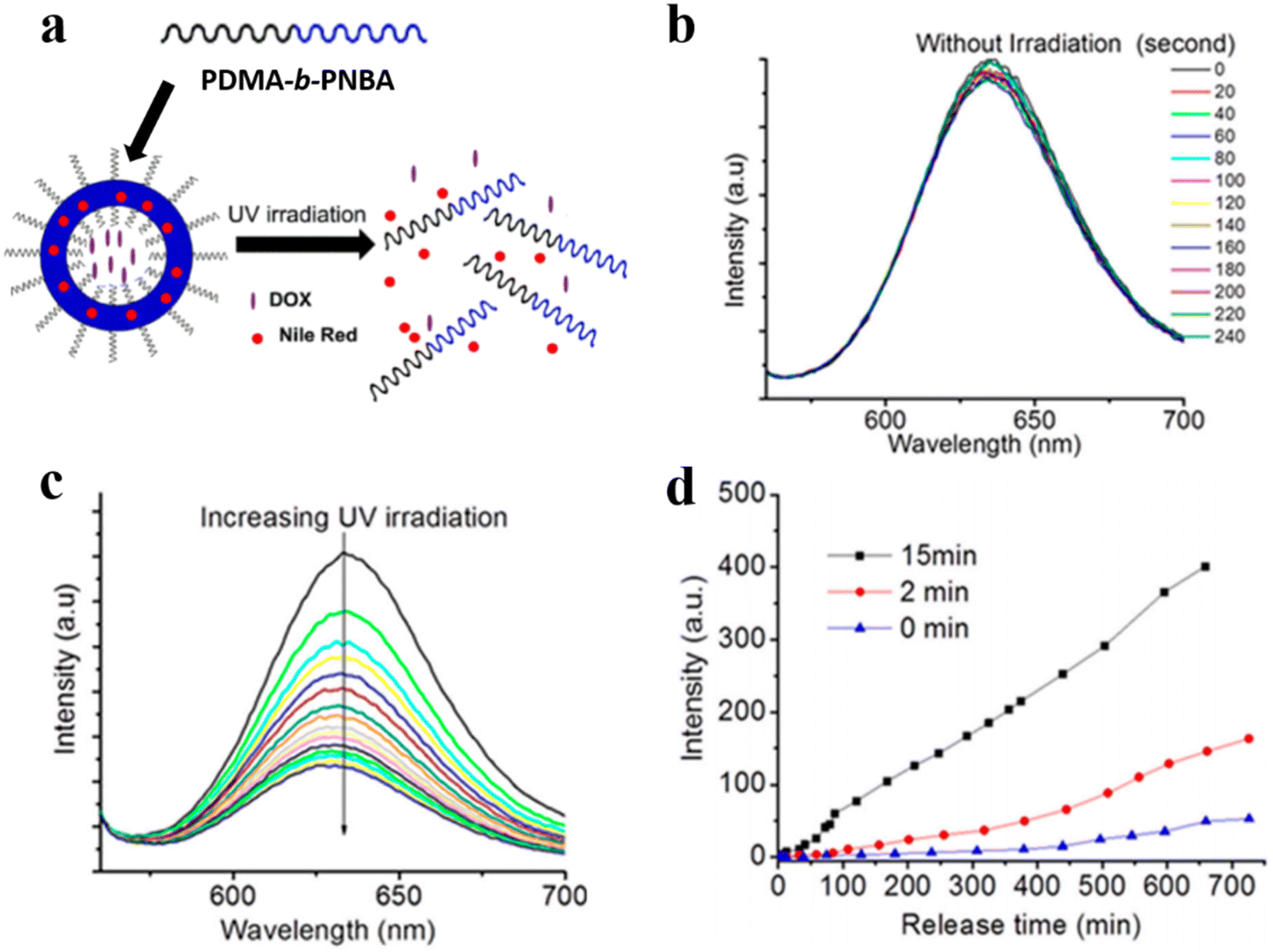 | ||
| Fig. 20 (a) Schematic representation of poly(N,N′-dimethylacrylamide)-block-poly(o-nitrobenzyl acrylate) (PDMA-b-PNBA) polymersomes for the release of drugs. (b) NR release spectra without UV irradiation. (c) NR emission spectra under UV irradiation. (d) DOX release upon UV irradiation at different times. Reprinted from ref. 123. Copyright 2020, Multidisciplinary Digital Publishing Institute. | ||
Polymersomes with SP are capable of converting into MC polymersomes upon UV irradiation, and this transition can reversibly regulate the polymeric membranes from impermeable to permeable (Fig. 21a).87 A hydrophilic anticancer drug, 2′-deoxy-5-fluorouridine (5-dFu) was encapsulated into the polymersome aqueous lumen, and the release behavior of 5-dFu from polymersomes was monitored by UV-vis spectroscopy. Over 90% of 5-dFu was released from MC polymersomes after 8 h UV irradiation, while the released 5-dFu from SP polymersomes was less than 10%. Under dark conditions, MC polymersomes reverted to SP polymersomes and the release of drugs could be actuated by short UV irradiation (Fig. 21b). Effective release of 5-dFu was observed when SP polymersomes were converted to MC polymersomes upon UV light irradiation, while the release of 5-dFu was almost prohibited upon visible light irradiation (Fig. 21c). Thus, on-demand and switchable release of drugs from polymersomes was obtained via alternate UV/vis light irradiation.
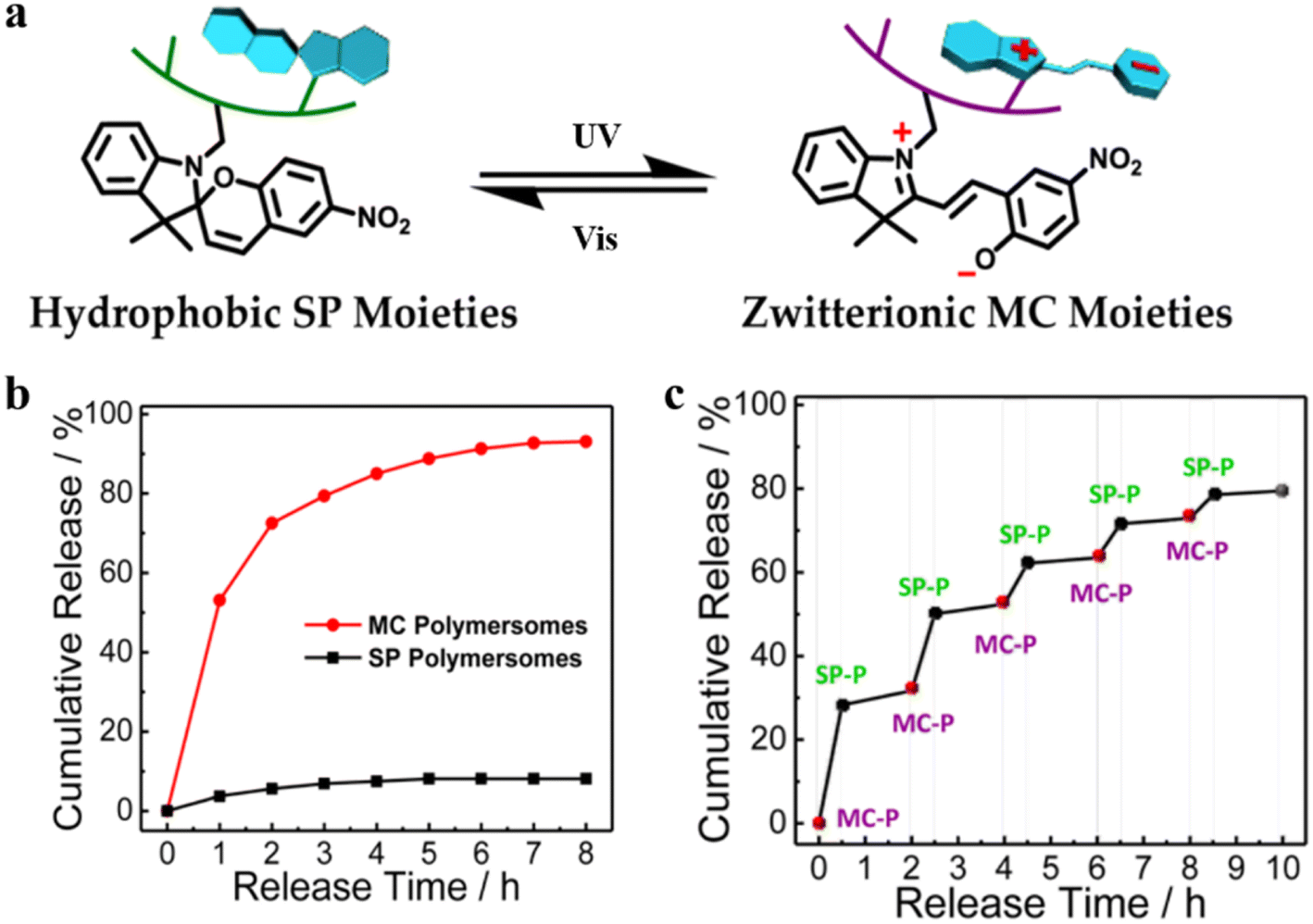 | ||
| Fig. 21 (a) Schematic representation of SP-to-MC photochromic transition. (SP-to-MC: the transition from impermeable SP polymersomes to permeable MC polymersomes. SP: Spiropyran. MC: Zwitterionic merocyanine.) (b) The release of 5-dFu from SP and MC polymersomes upon UV irradiation. (c) Ladder-type controlled release profiles of 5-dFu from polymersomes under alternated UV-vis light irradiation. Reprinted from ref. 87. Copyright 2015, American Chemical Society. | ||
Recent studies have demonstrated that polymersome nanoreactors are emerging as a powerful vehicle to be utilized in the biomedical field.124,125 For example, enzyme-loaded polymersome nanoreactors are able to convert model prodrugs into active drugs in situ for tumor therapy. For instance, semi-permeable, biodegradable and biocompatible nanoreactors with effective cellular integration have been coined artificial organelles with therapeutic potential in vitro. Van Hest et al. developed semi-permeable polymersomes containing PEG-PCLgTMC block copolymers. The polymersomes were prepared via a direct hydration methodology which facilitated molecular encapsulation and avoided the use of an organic solvent (Fig. 22a).126 The surface of the polymersomes was decorated with cell-penetrating peptides (CPPs), which can effectively promote cellular uptake of the nano-reactor. Their studies showed that TAT, a CPP derived from human immunodeficiency virus (HIV), and the cyclic variant of TAT (cTAT) influenced the cell uptake of polymersome nanoreactors. Flow cytometry revealed that peptide-modified polymersomes displayed a higher cell uptake efficiency compared with unmodified polymersomes. In addition, cellular uptake of cTAT-modified polymersomes was higher than TAT-modified polymersomes when the % cell-penetrating peptide was up to 5% (Fig. 22b). Next, they assessed the function of polymersome nanoreactors in vitro by catalytic decomposition of reactive oxygen species. Catalase was encapsulated inside the TAT-polymersomes, to obtain an antioxidant nanoreactor (AON). To examine the ROS-shielding ability of those AONs, cells were pulse-treated with the specified concentration of AONs followed by washing and subjecting to an external H2O2. The crystal violet analysis showed that AONs containing catalase could protect the cell against the deleterious consequence of external H2O2 (Fig. 22c). In addition, AONs also displayed a shielding effect in human-complex-I-deficient fibroblasts against the detrimental effects of H2O2, highlighting their therapeutic potential in bio-relevant applications.
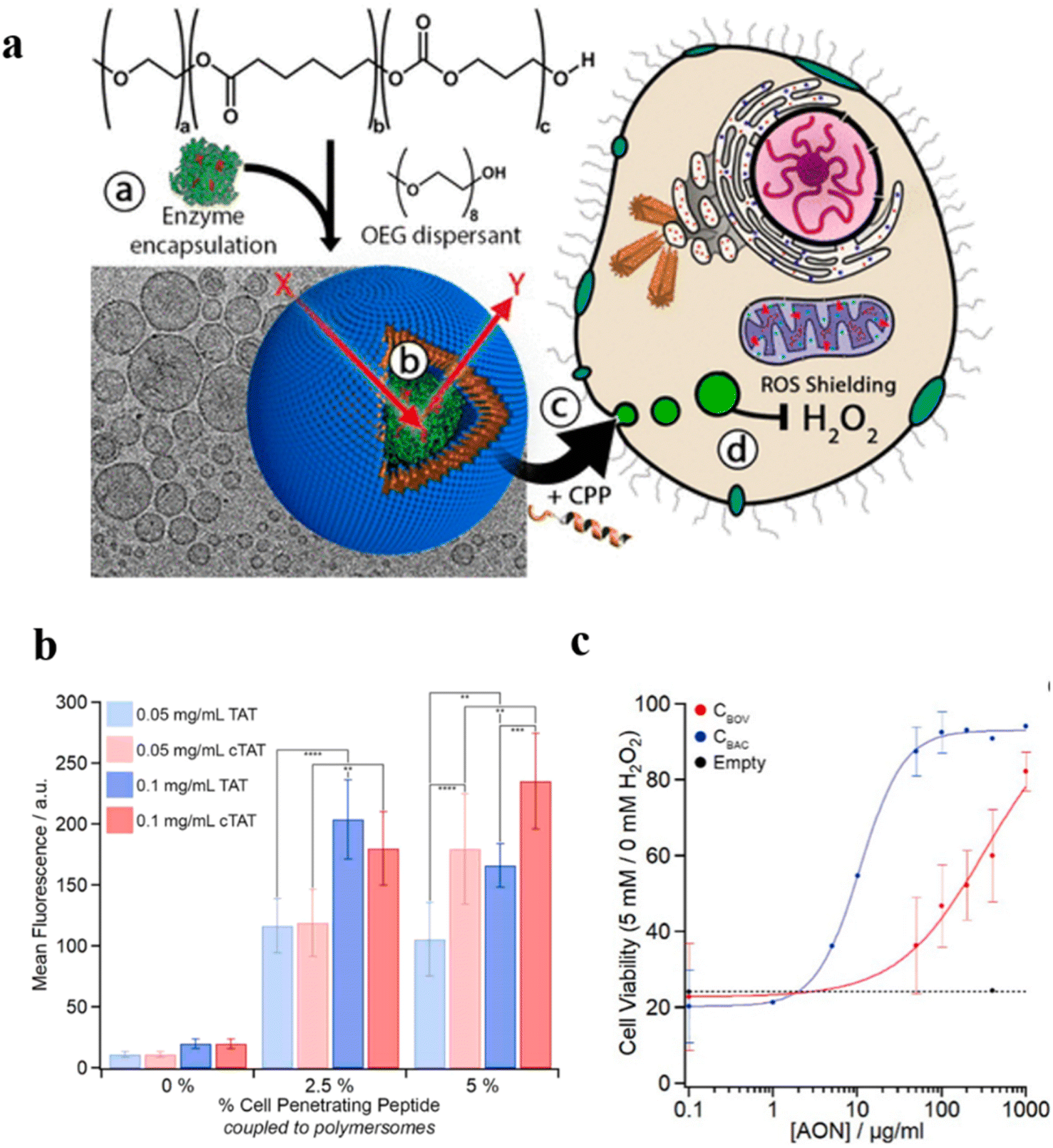 | ||
| Fig. 22 (a) Schematic representation of TAT-polymersomes protecting the cell against H2O2. Scale bar = 100 nm. (b) Uptake of NBD-labelled polymersomes after 24 h at different polymersome concentrations (0.05–0.1 mg mL−1) and with different percentages of cell penetrating peptide (CPP) (0–5%). (c) The shielding effect of AONs on exogenous H2O2 in cells. CBOC and CBAC are both catalase. Reprinted from ref. 126. Copyright 2018, American Chemical Society. | ||
Kataoka et al. prepared semipermeable and enzyme-loaded polyion complex vesicles (PICsomes) by a facile vortex and self-assembly method.127 PEG-based aniomers (PEG-b-PAsp), catiomers (homo-P(Asp-AP)), and β-gal were simply mixed together to obtain β-gal-loaded PICsomes (β-gal@PICsomes) with a diameter of 100 nm (Fig. 23a). β-gal@Cy5-PICsomes and β-gal were intravenously injected into the tail vein of mice bearing C26 tumors and a model prodrug HMDER-β-gal was given after 96 h. The results showed that only tumor tissue of mice treated with β-gal@Cy5-PICsomes showed significant fluorescence corresponding to HMDER due to the delivery of β-gal by the PICsomes and the in situ catalytic conversion of HMDER-β-gal into HMDER. Free β-gal was cleared quickly in vivo compared to β-gal@Cy5-PICsomes. Almost no HMDER fluorescence was observed when HMDER-β-Gal was injected into the mice in the absence of enzyme (Fig. 23b). Ex vivo fluorescence imaging demonstrated that the HMDER fluorescence in major organs (liver, lung, spleen, kidney) was negligible, which indicated that most of the HMDER fluorescence was observed from tumor tissue (Fig. 23c).
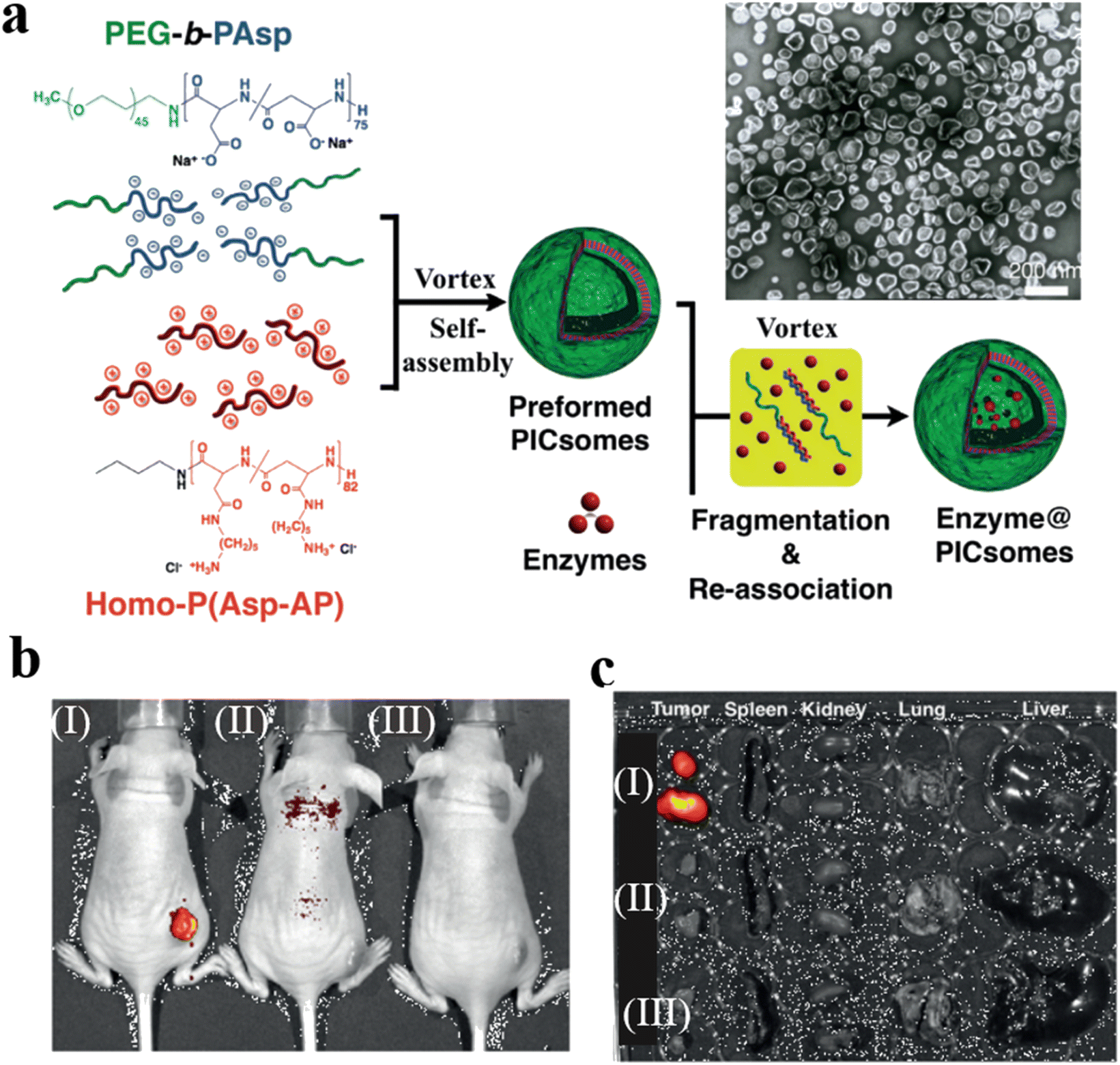 | ||
| Fig. 23 (a) Schematic representation of PICsomes and enzyme@PICsomes via preformed PICsomes. Scale bar = 200 nm. (b) In vivo imaging of C26 tumor-bearing mice. The tumor site of the mouse was treated with (I) β-Gal@Cy5-PICsomes, (II) free β-Gal, and (III) HMDER-β-Gal. (c) The fluorescence imaging of organs ex vivo. Reprinted from ref. 127. Copyright 2016, Wiley-VCH. | ||
Ding et al. reported the construction of polypeptide vesicles self-assembled from cholesterol-decorated methoxy poly(ethylene glycol)-poly(L-cysteine) (mPEG-PLCC) copolymers, and their membrane permeability was well controlled upon a ROS-induced secondary conformational change.94 The mPEG-PLCC polymersome nanoreactors were prepared by encapsulating glucose oxidase (GOD), and the presence of glucose triggered the insulin secretion in vivo. (Fig. 24a). The enzyme and FITC-labelled insulin (FITC-insulin) were loaded in the polymersomes (E + I@MPEG-PLCC group), which were subsequently injected into streptozotocin (STZ)-induced type 1 diabetic and healthy mice. The results showed that free FITC-insulin was cleared quickly and became undetectable after 24 h, while insulin-loaded polymersomes were slower to be cleared and showed stronger fluorescence spreading in the mice after 24 h. Furthermore, the insulin release from polymersomes when exposed to a higher level of glucose in diabetic mice resulted in faster clearance of glucose by the E + I@MPEG-PLCC group from diabetic mice than that from healthy mice. Subsequently, an equivalent amount of free insulin, and E + I@MPEG-PLCC were injected into diabetic mice, which decreased blood glucose levels quickly within 1 h. There was however no control of blood glucose level in the free insulin group after 1 h, while the blood glucose levels of the E + I@MPEG-PLCC group were kept in a normoglycemic range for 4 h (Fig. 24b). An enzyme-linked immunosorbent assay (ELISA) indicated that mice injected with E + I@MPEG-PLCC showed a consistently higher plasma insulin level than the mice injected with free insulin. This demonstrated polymersome-based vehicles displayed a longer circulation, which is beneficial for long-term control of blood glucose levels (Fig. 24c).
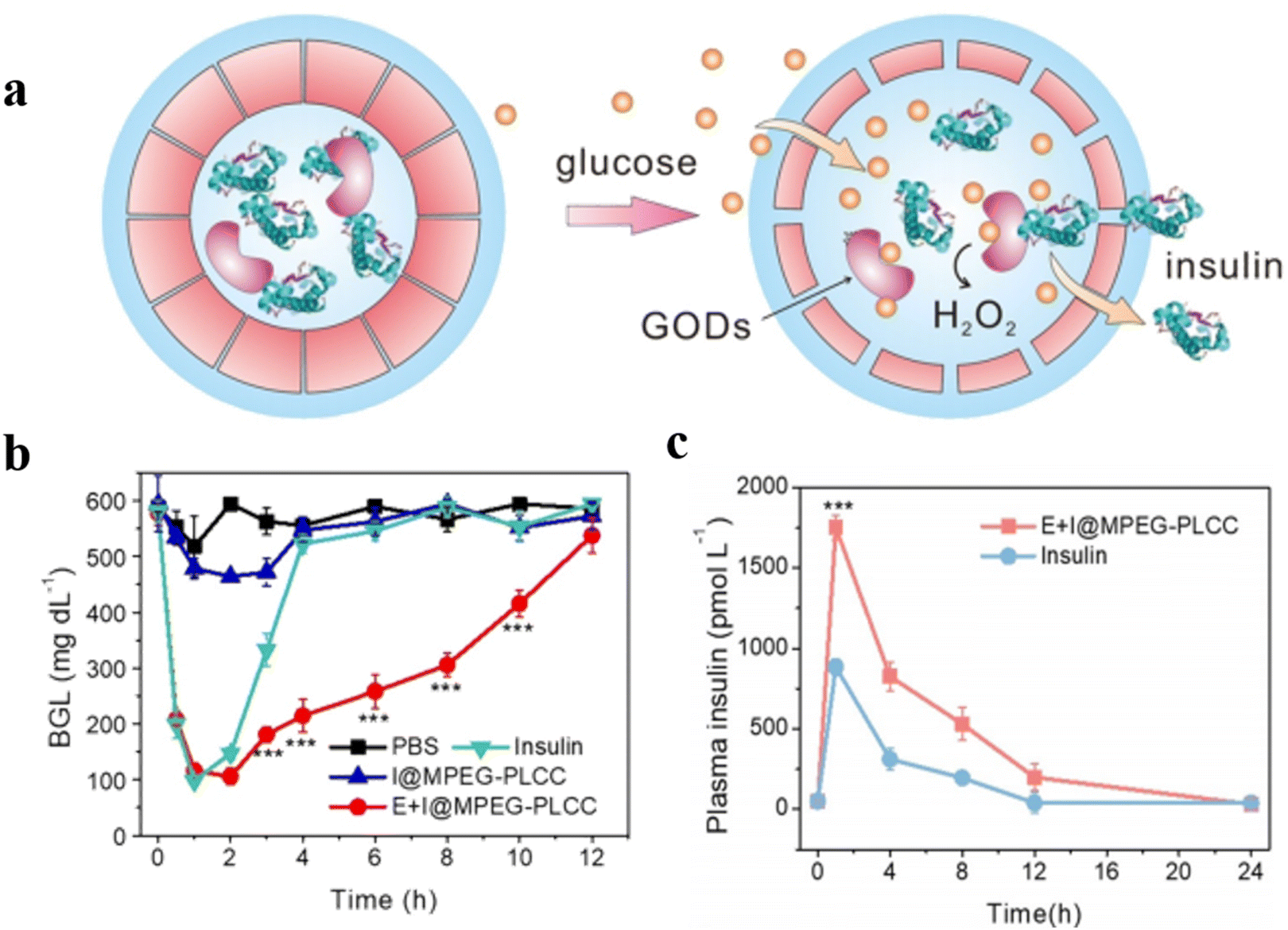 | ||
| Fig. 24 (a) Schematic representation of glucose-responsive insulin delivery based on GOD-loaded methoxy poly(ethylene glycol)-poly(L-cysteine) (mPEG-PLCC) polymersomes. (b) Blood glucose levels in STZ-induced diabetic mice after PBS, E + I@MPEG-PLCC, I@MPEG-PLCC, and insulin injection at different times. (c) Plasma human insulin levels in STZ-induced diabetic mice after E + I@MPEG-PLCC and insulin injection for different times. Reprinted from ref. 94. Copyright 2021, Wiley-VCH. | ||
Permeable polymersomes can protect bio-active species and drugs from degradation, achieve precise release, and increase their bioavailability, thereby improving therapeutic efficacy and reducing side effects. However, some challenges need to be addressed. For example, the biocompatibility of permeable polymersomes needs to be optimized for clinical applications. In addition, how to improve the cellular uptake of polymersomes to enhance their therapeutic effect is still an urgent issue to be addressed.
Conclusion and outlook
Compartmentalization inside living systems allows a high degree of control over a range of complex biological processes via various chemical/enzymatic networks. This has inspired scientists to create synthetic analogues that mimic biological complexity to yield desirable functions. The past decades have witnessed great progress in utilizing polymersomes as a facile platform for the engineering of biomimetic nano-compartments. The development of functional polymersomes requires a multi-interdisciplinary approach, which integrates synthesis, self-assembly, catalysis, nanoscience, and medicine to provide novel functionalities.In this review, we have summarized various strategies for the formation of permeable polymersomes, such as utilizing specific amphiphilic copolymers (PEG-b-PLA, PMOXA-b-PDMSPMI, PS-b-PIAT), inserting transmembrane proteins in the polymersome membranes, and developing stimuli-responsive polymers. These approaches to engineering permeable polymersomes enable membrane permeability control while also allowing maintaining their structural integrity. We also discussed the application of permeable polymersomes as nanoreactors, biomimetic artificial organelles, and drug-delivery vehicles. The recent promising applications of permeable polymersomes in in vivo biomedical applications are highlighted.
Although exciting progress has been made in the field of permeable polymersomes and numerous applications, specifically in the fields of nanomedicine and artificial organelle research, have been shown, challenges still exist in the construction of functional permeable polymersomes (Fig. 25). Firstly, increasing attentions should be paid to the biocompatibility of the degradation products of these building blocks, which would be a key aspect governing the success to apply polymersomes as drug delivery systems. To meet the fundamental criteria for in vivo application, the degradation products of polymersomes must not have cytotoxicity or cause immune responses, inflammations and carcinogenesis. In principle, the use of biodegradable components for the construction of permeable polymersomes should be ubiquitously considered due to the need for biocompatibility. For instance, PLA and PLGA are commonly used to fabricate non-ionic polymersomes because of their good biocompatibility and degradability. PEGylation is often incorporated into charged polymersomes, where PEG can cover the positively charged surface thus improving the biocompatibility and overcoming toxicity issues. More in vitro and in vivo experiments are needed to evaluate the safety and efficacy of permeable polymersomes. Secondly, the incompatibility of reaction cascades with selective mass transport across the polymersome membranes is still a substantial challenge. The reaction cascade requires multiple transfers between the inside and outside of polymersomes, and the uncontrolled interactions and interferences in these processes may affect the structure and performance of polymersomes. In this regard, the selective mass transfer on the polymersome membranes needs to be carried out in different chemical environments. Furthermore, the low encapsulation efficiency of bioactive species hinders their utility as more effective nanoreactors. Most importantly, the mechanism of permeable polymersomes as polymer nanomedicine and artificial organelles when implanted in complex in vivo microenvironments has not been fully understood and investigated, which can also be considered an important task for future researchers.
 | ||
| Fig. 25 A concluding scheme for the challenges of permeable polymersomes. | ||
Author contributions
All authors contributed to the writing and revision of the Manuscript.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (52203175 and 21905171), Shanghai Pujiang Program of China (22PJ1403600), and the ERC Advanced Grant Artisym (694120). S.C. thanks the Alexander von Humboldt Foundation for a fellowship and financial grant (3.5-CHN-1222717-HFST-P).References
- C. Fong, T. Le and C. J. Drummond, Chem. Soc. Rev., 2012, 41, 1297–1322 RSC.
- H. Li, J. D. Carter and T. H. LaBean, Mater. Today, 2009, 12, 24–32 CrossRef CAS.
- H. A. Klok and S. Lecommandoux, Adv. Mater., 2001, 13, 1217–1229 CrossRef CAS.
- M. Elsabahy, G. S. Heo, S. M. Lim, G. R. Sun and K. L. Wooley, Chem. Rev., 2015, 115, 10967–11011 CrossRef CAS PubMed.
- Y. Tu, F. Peng, A. Adawy, Y. Men, L. K. E. A. Abdelmohsen and D. A. Wilson, Chem. Rev., 2016, 116, 2023–2078 CrossRef CAS PubMed.
- M. Elsabahy and K. L. Wooley, Chem. Soc. Rev., 2012, 41, 2545–2561 RSC.
- Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969–5985 RSC.
- Y. Zhu, B. Yang, S. Chen and J. Du, Prog. Polym. Sci., 2017, 64, 1–22 CrossRef CAS.
- Y. Men, F. Peng, Y. Tu, J. C. M. van Hest and D. A. Wilson, Polym. Chem., 2016, 7, 3977–3982 RSC.
- R. Salva, J. F. Le Meins, O. Sandre, A. Brulet, M. Schmutz, P. Guenoun and S. Lecommandoux, ACS Nano, 2013, 7, 9298–9311 CrossRef CAS PubMed.
- Z. Deng, Y. Qian, Y. Yu, G. Liu, J. Hu, G. Zhang and S. Liu, J. Am. Chem. Soc., 2016, 138, 10452–10466 CrossRef CAS PubMed.
- J. Du, Y. Tang, A. L. Lewis and S. P. Armes, J. Am. Chem. Soc., 2005, 127, 17982–17983 CrossRef CAS PubMed.
- V. Ladmiral, M. Semsarilar, I. Canton and S. P. Armes, J. Am. Chem. Soc., 2013, 135, 13574–13581 CrossRef CAS PubMed.
- J. Zhu, S. Zhang, K. Zhang, X. Wang, J. Mays, K. Wooley and D. Pochan, Nat. Commun., 2013, 4, 2297 CrossRef PubMed.
- M. J. Derry, L. A. Fielding and S. P. Armes, Prog. Polym. Sci., 2016, 52, 1–18 CrossRef CAS.
- S. A. Meeuwissen, K. T. Kim, Y. Chen, D. J. Pochan and J. C. M. van Hest, Angew. Chem., Int. Ed., 2011, 50, 7070–7073 CrossRef CAS PubMed.
- L. Abdelmohsen, D. S. Williams, J. Pille, S. G. Ozel, R. S. M. Rikken, D. A. Wilson and J. C. M. van Hest, J. Am. Chem. Soc., 2016, 138, 9353–9356 CrossRef CAS PubMed.
- A. Blanazs, J. Madsen, G. Battaglia, A. J. Ryan and S. P. Armes, J. Am. Chem. Soc., 2011, 133, 16581–16587 CrossRef CAS PubMed.
- K. T. Kim, J. H. Zhu, S. A. Meeuwissen, J. Cornelissen, D. J. Pochan, R. J. M. Nolte and J. C. M. van Hest, J. Am. Chem. Soc., 2010, 132, 12522–12524 CrossRef CAS PubMed.
- Y. Men, W. Li, G. J. Janssen, R. S. M. Rikken and D. A. Wilson, Nano Lett., 2018, 18, 2081–2085 CrossRef CAS PubMed.
- C. K. Wong, A. F. Mason, M. H. Stenzel and P. Thordarson, Nat. Commun., 2017, 8, 1240 CrossRef.
- C. K. Wong, A. D. Martin, M. Floetenmeyer, R. G. Parton, M. H. Stenzel and P. Thordarson, Chem. Sci., 2019, 10, 2725–2731 RSC.
- C. K. Wong, M. H. Stenzel and P. Thordarson, Chem. Soc. Rev., 2019, 48, 4019–4035 RSC.
- Y. Men, W. Li, Y. Tu, F. Peng, G. J. A. Janssen, R. J. M. Nolte and D. A. Wilson, ACS Nano, 2019, 13, 12767–12773 CrossRef CAS PubMed.
- S. Varlas, S. B. Lawrenson, L. A. Arkinstall, R. K. O'Reilly and J. C. Foster, Prog. Polym. Sci., 2020, 107, 101278 CrossRef CAS.
- Y. Lu, J. Lin, L. Wang, L. Zhang and C. Cai, Chem. Rev., 2020, 120, 4111–4140 CrossRef CAS PubMed.
- T. Shimada, N. Sakamoto, R. Motokawa, S. Koizumi and M. Tirrell, J. Phys. Chem. B, 2012, 116, 240–243 CrossRef CAS PubMed.
- L. M. Walker, Curr. Opin. Colloid Interface Sci., 2001, 6, 451–456 CrossRef CAS.
- C. Martin, N. Aibani, J. F. Callan and B. Callan, Ther. Delivery, 2016, 7, 15–31 CrossRef CAS PubMed.
- E. Rideau, R. Dimova, P. Schwille, F. R. Wurm and K. Landfester, Chem. Soc. Rev., 2018, 47, 8572–8610 RSC.
- J. S. Lee and J. Feijen, J. Controlled Release, 2012, 161, 473–483 CrossRef CAS PubMed.
- H. Che and J. C. M. van Hest, ChemNanoMat, 2019, 5, 1092–1109 CrossRef CAS.
- R. Peters, I. Louzao and J. C. M. van Hest, Chem. Sci., 2012, 3, 335–342 RSC.
- H. Deng, L. Lin, S. Wang, G. Yu, Z. Zhou, Y. Liu, G. Niu, J. Song and X. Chen, Adv. Mater., 2019, 31, 1903443 CrossRef PubMed.
- X. Wang, G. Liu, J. Hu, G. Zhang and S. Liu, Angew. Chem., Int. Ed., 2014, 53, 3138–3142 CrossRef CAS PubMed.
- C. Martino, S. H. Kim, L. Horsfall, A. Abbaspourrad, S. J. Rosser, J. Cooper and D. A. Weitz, Angew. Chem., Int. Ed., 2012, 51, 6416–6420 CrossRef CAS PubMed.
- L. Quan, H. Ding, C. Pan, Y. Wei and Z. Xie, Colloids Surf., B, 2018, 161, 156–161 CrossRef CAS PubMed.
- D. E. Discher and A. Eisenberg, Science, 2002, 297, 967–973 CrossRef CAS PubMed.
- F. Meng, Z. Zhong and J. Feijen, Biomacromolecules, 2009, 10, 197–209 CrossRef CAS PubMed.
- N. Ben-Haim, P. Broz, S. Marsch, W. Meier and P. Hunziker, Nano Lett., 2008, 8, 1368–1373 CrossRef CAS PubMed.
- D. M. Vriezema, P. M. L. Garcia, N. S. Oltra, N. S. Hatzakis, S. M. Kuiper, R. J. M. Nolte, A. E. Rowan and J. C. M. van Hest, Angew. Chem., Int. Ed., 2007, 46, 7378–7382 CrossRef CAS.
- I. Louzao and J. C. M. van Hest, Biomacromolecules, 2013, 14, 2364–2372 CrossRef CAS PubMed.
- M. G. Gutierrez, F. Jalali-Yazdi, J. Peruzzi, C. T. Riche, R. W. Roberts and N. Malmstadt, Small, 2016, 12, 5256–5260 CrossRef CAS.
- M. S. Kim and D. S. Lee, Chem. Commun., 2010, 46, 4481–4483 RSC.
- Q. Yan, J. Wang, Y. Yin and J. Yuan, Angew. Chem., Int. Ed., 2013, 52, 5070–5073 CrossRef CAS PubMed.
- Z. Sun, G. Liu, J. Hu and S. Liu, Biomacromolecules, 2018, 19, 2071–2081 CrossRef CAS PubMed.
- F. Liu, V. Kozlovskaya, S. Medipelli, B. Xue, F. Ahmad, M. Saeed, D. Cropek and E. Kharlampieva, Chem. Mater., 2015, 27, 7945–7956 CrossRef CAS.
- K. T. Kim, J. Cornelissen, R. J. M. Nolte and J. C. M. van Hest, Adv. Mater., 2009, 21, 2787–2791 CrossRef CAS.
- M. Kumar, M. Grzelakowski, J. Zilles, M. Clark and W. Meier, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 20719–20724 CrossRef CAS PubMed.
- D. M. Vriezema, J. Hoogboom, K. Velonia, K. Takazawa, P. C. M. Christianen, J. C. Maan, A. E. Rowan and R. J. M. Nolte, Angew. Chem., Int. Ed., 2003, 42, 772–776 CrossRef CAS PubMed.
- S. May, M. Andreasson-Ochsner, Z. K. Fu, Y. X. Low, D. Tan, H. P. M. de Hoog, S. Ritz, M. Nallani and E. K. Sinner, Angew. Chem., Int. Ed., 2013, 52, 749–753 CrossRef CAS PubMed.
- E. Amado, R. Schops, W. Brandt and J. Kressler, ACS Macro Lett., 2012, 1, 1016–1019 CrossRef CAS PubMed.
- T. Nishimura and K. Akiyoshi, Adv. Sci., 2018, 5, 1800801 CrossRef.
- A. Ranquin, W. Versees, W. Meier, J. Steyaert and P. Van Gelder, Nano Lett., 2005, 5, 2220–2224 CrossRef CAS PubMed.
- P. Broz, S. Driamov, J. Ziegler, N. Ben-Haim, S. Marsch, W. Meier and P. Hunziker, Nano Lett., 2006, 6, 2349–2353 CrossRef CAS PubMed.
- G. Qin, C. Hu, Y. Jiang, S. Dong, L. Liu and H. Zhao, J. Polym. Sci., 2021, 59, 1958–1971 CrossRef CAS.
- J. Zhuang, M. R. Gordon, J. Ventura, L. Li and S. Thayumanavan, Chem. Soc. Rev., 2013, 42, 7421–7435 RSC.
- Z. Ge, D. Xie, D. Chen, X. Jiang, Y. Zhang, H. Liu and S. Liu, Macromolecules, 2007, 40, 3538–3546 CrossRef CAS.
- S. Guragain, B. P. Bastakoti, V. Malgras, K. Nakashima and Y. Yamauchi, Chem.–Eur. J., 2015, 21, 13164–13174 CrossRef CAS PubMed.
- H. Che and J. C. M. van Hest, J. Mater. Chem. B, 2016, 4, 4632–4647 RSC.
- A. A.-W. M. M. Japir, N. Lu, Y. Wang, J. F. Mukerabigwi, Q. Zhou and Z. Ge, Eur. Polym. J., 2020, 138, 109982 CrossRef.
- O. Rifaie-Graham, S. Ulrich, N. F. B. Galensowske, S. Balog, M. Chami, D. Rentsch, J. R. Hemmer, J. R. de Alaniz, L. F. Boesel and N. Bruns, J. Am. Chem. Soc., 2018, 140, 8027–8036 CrossRef CAS.
- V. Kozlovskaya, F. Liu, Y. M. Yang, K. Ingle, S. Qian, G. V. Halade, V. S. Urban and E. Kharlampieva, Biomacromolecules, 2019, 20, 3989–4000 CrossRef CAS.
- W. Zhang, C. Hong and C. Pane, ACS Appl. Mater. Interfaces, 2017, 9, 15086–15095 CrossRef CAS PubMed.
- Z. Al-Ahmady and K. Kostarelos, Chem. Rev., 2016, 116, 3883–3918 CrossRef CAS.
- X. Hu, Y. Zhang, Z. Xie, X. Jing, A. Bellotti and Z. Gu, Biomacromolecules, 2017, 18, 649–673 CrossRef CAS PubMed.
- B. Iyisan, J. Kluge, P. Formanek, B. Voit and D. Appelhans, Chem. Mater., 2016, 28, 1513–1525 CrossRef CAS.
- E. Gallon, T. Matini, L. Sasso, G. Mantovani, A. A. de Benito, J. Sanchis, P. Caliceti, C. Alexander, M. J. Vicent and S. Salmaso, Biomacromolecules, 2015, 16, 1924–1937 CrossRef CAS PubMed.
- S. Bazban-Shotorbani, M. M. Hasani-Sadrabadi, A. Karkhaneh, V. Serpooshan, K. I. Jacob, A. Moshaverinia and M. Mahmoudi, J. Controlled Release, 2017, 253, 46–63 CrossRef CAS PubMed.
- F. Wang, J. Gao, J. Xiao and J. Du, Nano Lett., 2018, 18, 5562–5568 CrossRef CAS PubMed.
- M. Karimi, S. M. M. Basri, M. Vossoughi, P. S. Pakchin, H. Mirshekari and M. R. Hamblin, Curr. Org. Chem., 2016, 20, 2949–2959 CrossRef CAS.
- H. Sun, F. Meng, R. Cheng, C. Deng and Z. Zhong, Antioxid. Redox Signaling, 2014, 21, 755–767 CrossRef CAS.
- B. Yan, D. Han, O. Boissiere, P. Ayotte and Y. Zhao, Soft Matter, 2013, 9, 2011–2016 RSC.
- K. Jie, Y. Zhou, Y. Yao, B. Shi and F. Huang, J. Am. Chem. Soc., 2015, 137, 10472–10475 CrossRef CAS PubMed.
- D. Guo, K. Wang, Y. Wang and Y. Liu, J. Am. Chem. Soc., 2012, 134, 10244–10250 CrossRef CAS PubMed.
- G. J. Habraken, M. Peeters, P. D. Thornton, C. E. Koning and A. Heise, Biomacromolecules, 2011, 12, 3761–3769 CrossRef CAS PubMed.
- C. Zheng, Y. Zhao and Y. Liu, Chin. J. Polym. Sci., 2018, 36, 322–346 CrossRef CAS.
- J. Liu, V. Postupalenko, S. Lorcher, D. Wu, M. Chami, W. Meier and C. G. Palivan, Nano Lett., 2016, 16, 7128–7136 CrossRef CAS.
- J. Gaitzsch, D. Appelhans, L. Wang, G. Battaglia and B. Voit, Angew. Chem., Int. Ed., 2012, 51, 4448–4451 CrossRef CAS PubMed.
- X. Liu, D. Appelhans, Q. Wei and B. Voit, Adv. Sci., 2017, 4, 1600308 CrossRef PubMed.
- J. Du and S. P. Armes, J. Am. Chem. Soc., 2005, 127, 12800–12801 CrossRef CAS PubMed.
- C. Yao, X. Wang, G. Liu, J. Hu and S. Liu, Macromolecules, 2016, 49, 8282–8295 CrossRef CAS.
- W. Sun, M. Parowatkin, W. Steffen, H. J. Butt, V. Mailander and S. Wu, Adv. Healthcare Mater., 2016, 5, 467–473 CrossRef CAS PubMed.
- C. Yao, P. Wang, X. Li, X. Hu, J. Hou, L. Wang and F. Zhang, Adv. Mater., 2016, 28, 9341–9348 CrossRef CAS PubMed.
- H. Chen, L. Xiao, Y. Anraku, P. Mi, X. Liu, H. Cabral, A. Inoue, T. Nomoto, A. Kishimura, N. Nishiyama and K. Kataoka, J. Am. Chem. Soc., 2014, 136, 157–163 CrossRef CAS PubMed.
- K. M. Herbert, S. Schrettl, S. J. Rowan and C. Weder, Macromolecules, 2017, 50, 8845–8870 CrossRef CAS.
- X. Wang, J. Hu, G. Liu, J. Tian, H. Wang, M. Gong and S. Liu, J. Am. Chem. Soc., 2015, 137, 15262–15275 CrossRef CAS PubMed.
- M. Spulber, A. Najer, K. Winkelbach, O. Glaied, M. Waser, U. Pieles, W. Meier and N. Bruns, J. Am. Chem. Soc., 2013, 135, 9204–9212 CrossRef CAS PubMed.
- O. Rifaie-Graham, J. Yeow, A. Najer, R. Wang, R. Sun, K. Zhou, T. N. Dell, C. Adrianus, C. Thanapongpibul, M. Chami, S. Mann, J. R. de Alaniz and M. M. Stevens, Nat. Chem., 2022, 15, 110–118 CrossRef.
- Q. Zhang, L. Lei and S. Zhu, ACS Macro Lett., 2017, 6, 515–522 CrossRef CAS PubMed.
- Q. Yan and Y. Zhao, Chem. Commun., 2014, 50, 11631–11641 RSC.
- Q. Yan and W. Sang, Chem. Sci., 2016, 7, 2100–2105 RSC.
- J. Hu, M. R. Whittaker, H. Duong, Y. Li, C. Boyer and T. P. Davis, Angew. Chem., Int. Ed., 2014, 53, 7913–7918 CrossRef.
- Y. Zheng, Z. Wang, Z. Li, H. Liu, J. Wei, C. Peng, Y. Zhou, J. Li, Q. Fu, H. Tan and M. Ding, Angew. Chem., Int. Ed., 2021, 60, 22529–22536 CrossRef CAS PubMed.
- X. Liu, D. Appelhans and B. Voit, J. Am. Chem. Soc., 2018, 140, 16106–16114 CrossRef CAS PubMed.
- X. Wang, C. Yao, G. Zhang and S. Liu, Nat. Commun., 2020, 11, 1524 CrossRef CAS PubMed.
- A. Kishimura, S. Liamsuwan, H. Matsuda, W. F. Dong, K. Osada, Y. Yamasaki and K. Kataoka, Soft Matter, 2009, 5, 529–532 RSC.
- W. Chiang, Y. Lan, Y. Huang, Y. Chen, Y. Huang, S. Lin, C. Chern and H. Chiu, Polymer, 2012, 53, 2233–2244 CrossRef CAS.
- L. Klermund and K. Castiglione, Bioprocess Biosyst. Eng., 2018, 41, 1233–1246 CrossRef CAS PubMed.
- Y. Altay, A. Llopis-Lorente, L. K. E. A. Abdelmohsen and J. C. M. van Hest, Macromol. Chem. Phys., 2022, 224, 2200269 CrossRef.
- S. Moreno, B. Voit and J. Gaitzsch, Colloid Polym. Sci., 2020, 299, 309–324 CrossRef.
- T. Einfalt, R. Goers, I. A. Dinu, A. Najer, M. Spulber, O. Onaca-Fischer and C. G. Palivan, Nano Lett., 2015, 15, 7596–7603 CrossRef CAS PubMed.
- J. Gaitzsch, X. Huang and B. Voit, Chem. Rev., 2016, 116, 1053–1093 CrossRef CAS PubMed.
- H. Che, S. Cao and J. C. M. van Hest, J. Am. Chem. Soc., 2018, 140, 5356–5359 CrossRef CAS PubMed.
- D. Gaur, N. C. Dubey and B. P. Tripathi, Adv. Colloid Interface Sci., 2022, 299, 102566 CrossRef CAS PubMed.
- R. J. Peters, M. Marguet, S. Marais, M. W. Fraaije, J. C. van Hest and S. Lecommandoux, Angew. Chem., Int. Ed., 2014, 53, 146–150 CrossRef CAS PubMed.
- A. Belluati, S. Thamboo, A. Najer, V. Maffeis, C. Planta, I. Craciun, C. G. Palivan and W. Meier, Adv. Funct. Mater., 2020, 30, 2002949 CrossRef CAS.
- A. Larrañaga, M. Lomora, J. R. Sarasua, C. G. Palivan and A. Pandit, Prog. Mater. Sci., 2017, 90, 325–357 CrossRef.
- P. Tanner, P. Baumann, R. Enea, O. Onaca, C. Palivan and W. Meier, Acc. Chem. Res., 2011, 44, 1039–1049 CrossRef CAS PubMed.
- S. Thamboo, A. Najer, A. Belluati, C. von Planta, D. L. Wu, I. Craciun, W. Meier and C. C. Palivan, Adv. Funct. Mater., 2019, 29, 1904267 CrossRef.
- H. Gumz, S. Boye, B. Iyisan, V. Kronert, P. Formanek, B. Voit, A. Lederer and D. Appelhans, Adv. Sci., 2019, 6, 1801299 CrossRef PubMed.
- A. F. Mason, N. A. Yewdall, P. L. W. Welzen, J. X. Shao, M. van Stevendaal, J. C. M. van Hest, D. S. Williams and L. Abdelmohsen, ACS Cent. Sci., 2019, 5, 1360–1365 CrossRef CAS PubMed.
- M. Godoy-Gallardo, M. J. York-Duran and L. Hosta-Rigau, Adv. Healthcare Mater., 2018, 7, 1700917 CrossRef.
- T. Einfalt, D. Witzigmann, C. Edlinger, S. Sieber, R. Goers, A. Najer, M. Spulber, O. Onaca-Fischer, J. Huwyler and C. G. Palivan, Nat. Commun., 2018, 9, 1127 CrossRef CAS PubMed.
- T. Einfalt, M. Garni, D. Witzigmann, S. Sieber, N. Baltisberger, J. Huwyler, W. Meier and C. G. Palivan, Adv. Sci., 2020, 7, 1901923 CrossRef CAS.
- I. Yildiz, S. Impellizzeri, E. Deniz, B. McCaughan, J. F. Callan and F. M. Raymo, J. Am. Chem. Soc., 2011, 133, 871–879 CrossRef CAS PubMed.
- S. Cao, L. C. da Silva and K. Landfester, Angew. Chem., Int. Ed., 2022, 61, e202205266 CAS.
- M. Machtakova, H. Therien-Aubin and K. Landfester, Chem. Soc. Rev., 2022, 51, 128–152 RSC.
- R. H. Staff, M. Gallei, M. Mazurowski, M. Rehahn, R. Berger, K. Landfester and D. Crespy, ACS Nano, 2012, 6, 9042–9049 CrossRef CAS.
- C. G. Palivan, R. Goers, A. Najer, X. Zhang, A. Car and W. Meier, Chem. Soc. Rev., 2016, 45, 377–411 RSC.
- Y. He, S. Guo, Y. Zhang, Y. Liu and H. Ju, ACS Appl. Mater. Interfaces, 2021, 13, 14951–14963 CrossRef CAS PubMed.
- M. Marguet, L. Edembe and S. Lecommandoux, Angew. Chem., Int. Ed., 2012, 51, 1173–1176 CrossRef CAS PubMed.
- W. Hou, R. Liu, S. Bi, Q. He, H. Wang and J. Gu, Molecules, 2020, 25, 5147 CrossRef PubMed.
- C. G. Palivan, O. Fischer-Onaca, M. Delcea, F. Itel and W. Meier, Chem. Soc. Rev., 2012, 41, 2800–2823 RSC.
- D. Zhou, Z. Fei, L. Jin, P. Zhou, C. Li, X. Liu and C. Zhao, J. Mater. Chem. B, 2021, 9, 801–808 RSC.
- L. van Oppen, L. Abdelmohsen, S. E. van Emst-de Vries, P. L. W. Welzen, D. A. Wilson, J. A. M. Smeitink, W. J. H. Koopman, R. Brock, P. Willems, D. S. Williams and J. C. M. van Hest, ACS Cent. Sci., 2018, 4, 917–928 CrossRef CAS.
- Y. Anraku, A. Kishimura, M. Kamiya, S. Tanaka, T. Nomoto, K. Toh, Y. Matsumoto, S. Fukushima, D. Sueyoshi, M. R. Kano, Y. Urano, N. Nishiyama and K. Kataoka, Angew. Chem., Int. Ed., 2016, 55, 560–565 CrossRef CAS PubMed.
Footnote |
| † Yanyan Zhu and Shoupeng Cao contribute this work equally. |
| This journal is © The Royal Society of Chemistry 2023 |