
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFine tuning of electrosynthesis pathways by modulation of the electrolyte solvation structure
Florian
Dorchies
 ab and
Alexis
Grimaud
*abc
ab and
Alexis
Grimaud
*abc
aChimie du Solide et de l’Energie, UMR 8260, Collège de France, 75231 Paris Cedex 05, France. E-mail: alexis.grimaud@bc.edu
bRéseau sur le Stockage Electrochimique de l’Energie (RS2E), CNRS FR3459, 80039 Amiens Cedex, France
cDepartment of Chemistry, Merkert Chemistry Center, Boston College, 2609 Beacon Street, Chestnut Hill, MA 02467, USA
First published on 23rd May 2023
Abstract
Electrosynthesis is a method of choice for designing new synthetic routes owing to its ability to selectively conduct reactions at controlled potentials, high functional group tolerance, mild conditions and sustainability when powered by renewables. When designing an electrosynthetic route, the selection of the electrolyte, which is composed of a solvent, or a mixture of solvents, and a supporting salt, is a prerequisite. The electrolyte components, generally assumed to be passive, are chosen because of their adequate electrochemical stability windows and to ensure the solubilization of the substrates. However, very recent studies point towards an active role of the electrolyte in the outcome of electrosynthetic reactions, challenging its inert character. Particular structuring of the electrolyte at nano- and micro-scales can occur and impact the yield and selectivity of the reaction, which is often overlooked. In the present Perspective, we highlight how mastering the electrolyte structure, both in bulk and at electrochemical interfaces, introduces an additional level of control for the design of new electrosynthetic methods. For this purpose, we focus our attention on oxygen-atom transfer reactions using water as the sole oxygen source in hybrid organic solvent/water mixtures, these reactions being emblematic of this new paradigm.
Introduction
When powered by renewables, electrochemical oxygenation reactions are regarded as a promising alternative to conventional organic synthesis for more environmentally friendly functionalization of C–H and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds.1 Among the numerous electrosynthetic routes that have been devised,1–3 an emerging approach for increasing their sustainability consists in using water as the sole oxygen source. Along with its environmental benefit, this strategy provides a means to avoid in situ generation of hazardous oxidants such as Cl2 or H2O2 but also dispenses with the use of molecular catalysts or redox mediators, simplifying the reaction medium and facilitating the isolation of the final product. The synthetically relevant reactions developed in this context include the lactonization of ketones,4 epoxidation of alkenes,5–8 oxidation of sulfides9,10 and oxidation of C–H and CC bonds to carbonyls.11,12
C bonds.1 Among the numerous electrosynthetic routes that have been devised,1–3 an emerging approach for increasing their sustainability consists in using water as the sole oxygen source. Along with its environmental benefit, this strategy provides a means to avoid in situ generation of hazardous oxidants such as Cl2 or H2O2 but also dispenses with the use of molecular catalysts or redox mediators, simplifying the reaction medium and facilitating the isolation of the final product. The synthetically relevant reactions developed in this context include the lactonization of ketones,4 epoxidation of alkenes,5–8 oxidation of sulfides9,10 and oxidation of C–H and CC bonds to carbonyls.11,12
Developing electrochemical oxygen-atom functionalization reactions with water serving as the oxygen source requires solubilization/dispersion of both the organic substrate and water, which are generally immiscible. This can be achieved by the use of multiphasic or monophasic electrolytes. While multiphase approaches can effectively disperse organic molecules and water via the use of ultrasound or via chemical strategies such as microemulsions,13 recent studies pioneering the use of water as the reactant in electrosynthesis rather made use of monophasic mixtures of an organic solvent and water, denoted as “hybrid electrolytes”. The chosen organic solvent is miscible with water and ideally allows for the solubilization of a wide range of organic substrates. Acetonitrile (ACN)/water mixtures are the most commonly used electrolytes for such synthetic applications due to the large electrochemical window of ACN and its low toxicity. One can note that a hybrid electrolyte made of a mixture of ethanol and water has recently been used for the epoxidation of alkenes with water as the sole oxygen source8 but once highly apolar alkene substrates are used, the resulting mixture is a multiphasic system.
The oxygen-atom transfer reactions using water as the sole oxygen source in hybrid organic solvent/water mixtures are still in their infancy. Developing their full potential is intimately linked to recent advances made in the field of electrocatalysis. Indeed, further developing these synthetic routes requires the control of surface intermediates to promote the oxygen-atom transfer to the organic substrate, calling upon electrocatalyst engineering. Most importantly, following recent realization that supporting salt ions play an important role in the selectivity and kinetics of electrocatalytic reactions such as the CO2 reduction reaction (CO2RR),14,15 hydrogen evolution reaction (HER)16,17 and OER,18,19 we recently showed a similar effect on the epoxidation of cyclooctene.7 Finally, ACN/water mixtures, the most commonly used electrolytes for the oxygenation reactions at stake, are currently employed to tune both the cathodic and anodic reactivity of water via confinement effects.16,17,20,21 Indeed, while being macroscopically homogeneous, these mixtures exhibit a nanoscale structuring, which can be tuned by the nature of the supporting salt.17 Realizing that ACN/water mixtures are not the only synthetically relevant mixtures of solvents exhibiting nanoscale structuring, further development of electrolyte mixtures offers a new means for modulating electrosynthetic reactions.
In the present Perspective, we aim at highlighting potential research focuses for designing new electrosynthetic reactions, with a special emphasis on the role of the bulk electrolyte dynamics and structure and its impact on the specific interactions at the electrochemical interface. The new class of electrochemical oxygen-atom transfer reactions in hybrid electrolytes serves as a basis for the discussion, which is enriched with results from other (electro)chemical reactions and fundamental studies on electrolyte structuring. To provide the reader with the current developments in the electrochemical reactions at the core of this Perspective, the state of the art is first presented. Means to tune the electrolyte structure, both in bulk and at the electrochemical interface, are then discussed. Doing so, we highlight how it constitutes an additional degree of control for the design of new electrosynthetic methods, which is often overlooked.
State of the art
In organic and hybrid electrolytes, the direct electro-oxidation of C–H and CC bonds occurs at very high potentials (>2 V vs. the standard hydrogen electrode, SHE) and produces carbon-centered radicals and/or cations. Therefore, two strategies have been explored to develop electrochemical oxygenation reactions in hybrid organic/water electrolytes. The first one consists in generating highly reactive radical or cationic intermediates via the direct oxidation of the organic substrate. These intermediates subsequently react chemically with water to form carbon–oxygen bonds. This strategy, successfully applied for the oxidation of sulfides9,10 and the activation of benzylic C–H bonds,11 requires the use of very high potentials which can hinder chemoselectivity when using complex organic substrates. Furthermore, it is non-catalytic in nature as the substrate activation occurs via outer-sphere electron transfer. The second strategy consists in harnessing the partial oxidation/activation of water into *OH or *O intermediate species (* denotes a surface site) at lower potentials than those required for the direct electro-oxidation of C–H and CC bonds. For that, appropriate electrocatalysts are needed to generate these surface species that subsequently react with the organic substrate to form the desired chemical functions. This strategy was applied for the electrosynthesis of lactones4 and epoxides5–8 (Scheme 1). However, *O intermediates can be further oxidized and lead to the undesired oxygen evolution reaction (OER). In short, the OER, which shares similar *OH and *O intermediates with the oxygenation reaction, is competing, asking for a fine tuning of both the catalyst and its interaction with liquid electrolytes in order to make this approach viable.
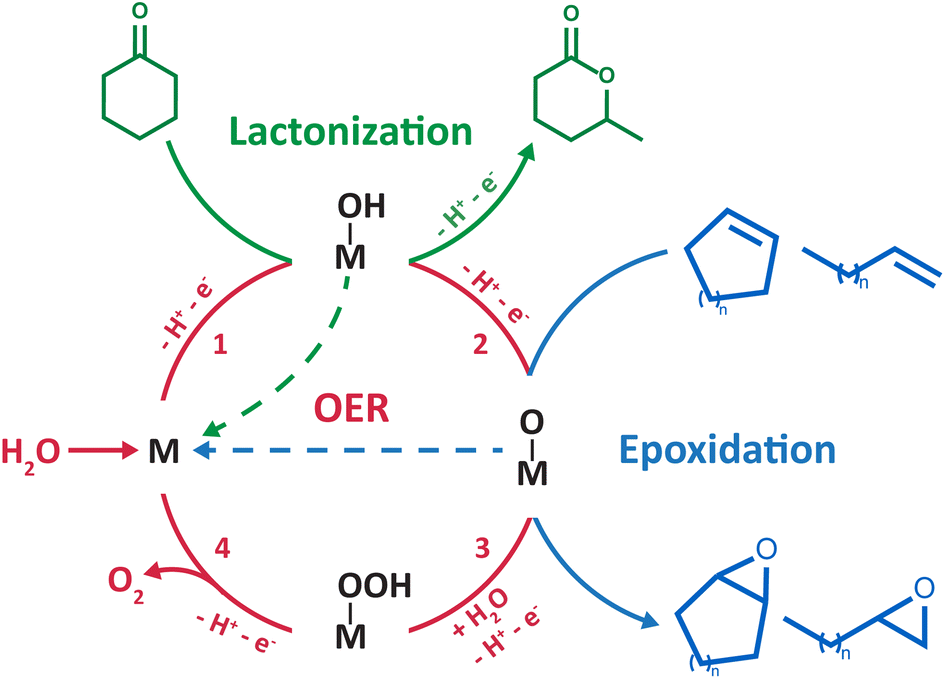 | ||
| Scheme 1 Catalytic cycle showing in red the four commonly accepted steps for the OER at the surface of noble metals/transition metal oxides (simplified as an “M” site).28 The “M–OH” and “M–O” intermediates were proposed in the literature to be shared with electrochemical lactonization4 (green) and epoxidation5–8 (blue) reactions developed in hybrid electrolytes, respectively. | ||
Manthiram and coworkers recently devised an electrochemical route for the lactonization of cyclic ketones in ACN/water mixtures at platinum electrodes, with water serving as the sole oxygen source.4 Based on the dependence of the reactants on the yield and the distribution of the observed products, the authors proposed that the reaction proceeds first by the activation of water at the surface of platinum, forming Pt–OH intermediates at very high potentials (>2.6 V vs. SHE). The ketone molecule subsequently reacts with Pt–OH surface species, forming a tetrahedral intermediate that leads to lactone products after two additional steps. The faradaic efficiency (FE) of the reaction is limited to ∼20%, suffering from the competition with the OER and the direct oxidation of the ketone substrate owing to the very high potentials required to activate water at platinum electrodes.
Most importantly, using similar ACN/water mixtures, the same group pioneered the inner sphere activation of water at heterogeneous catalysts for electrochemical epoxidation of liquid alkenes.5 Drawing a parallel between this epoxidation strategy and the OER, the authors selected manganese oxide nanoparticles as an electrocatalyst among the many candidates that could be explored for this new reaction. Indeed, reactive high-valent MnIVO intermediate species at the surface of nanoparticles were shown to be key intermediates of the OER, leading the authors to hypothesize that these intermediates can act as oxygen atom donors to alkene substrates to form the corresponding epoxides (Scheme 1). The epoxidation operates at milder potentials (>1.8 V vs. SHE) than the lactonization reaction previously described. Cyclooctene was initially chosen as a model substrate for the design of the epoxidation route, which was successfully extended to a wide range of cyclic and linear alkenes, demonstrating the broad application of the method. However, the applicability of this strategy is yet to be demonstrated for gaseous alkene substrates such as ethylene and propylene, which is critical for industrial relevance as ethylene oxide and propylene oxide are among the most abundantly produced chemicals globally. Several studies report the feasibility of electrochemically oxidizing gaseous alkanes and alkenes in aqueous electrolytes using *O intermediates generated during the OER,22–26 which calls for further research examining the benefits of using hybrid electrolytes with gaseous substrates. Furthermore, limited FEs of ∼30% were initially obtained due to competition with the OER and epoxide overoxidation to ketones. Focusing on improving the electrocatalyst, a FE of ∼ 50% was later obtained by fine-tuning the manganese oxide nanoparticles with single Ir atom dopants.6 As a matter of comparison, a significantly higher FE of ∼71% is maintained for hours at industrially relevant current densities of 300 mA cm−2 for the chloride-mediated electrosynthesis of ethylene oxide.27 Therefore, to make this strategy industrially viable, further research is required on flow-cell type electrolyzers to investigate the competition with the OER at industrially relevant current densities, with the aim to maintain sufficiently high epoxide FE. Similarly, taking inspiration from the optimization of OER electrocatalysts, Li and coworkers recently studied RuO2 as an electrocatalyst for the epoxidation of cyclooctene and proposed, using density functional theory calculations, the involvement of metal oxo intermediates in the oxygen-atom transfer to the organic substrate.8 Building on these developments, we recently demonstrated that the mechanism of the epoxidation of cyclooctene is dependent on the electrocatalyst surface.7 For that, we aimed for a single surface that can possess, or not, oxygen ligands as a function of cycling conditions. Gold was selected as, in ACN/water mixtures, water reactivity at metallic gold surfaces is decoupled from that at gold oxide surfaces formed at high potential. Using this singular property, it was shown that at metallic gold surfaces, the epoxidation of cyclooctene proceeds via the activation of the substrate by in situ formation of a homogeneous cationic metal catalyst following anodic dissolution. Instead, at gold oxide surfaces, the epoxidation mechanism shares similar reaction intermediates with the OER, as previously suggested for other metal oxides5,6,8 (Scheme 1).
As observed by different groups, not only the nature of the electrocatalyst but also the electrolyte composition affects the FE of electrochemical oxygenation reactions. Manthiram and coworkers optimized the ACN to water ratio to obtain the best FE while maintaining the solubility of cyclooctene in these mixtures. An optimal FE was found for the following electrolyte composition: ACN containing 5 M water, 0.1 M supporting salt and 200 mM cyclooctene.5 Several studies reported that the nature of the supporting salt, and especially of the cation, dramatically influences electrocatalytic reactions such as the OER and the HER. While strong Lewis acids such as Li+ activate water reduction by weakening the O–H bonds in water, hydrophobic organic cations such as tetrabutylammonium (TBA+) were found to hinder the accessibility of water to the electrochemical interface at the cathode.16,17 More surprisingly, TBA+ was also found to slow down water oxidation at the anode,18 while strong Lewis acid cations are detrimental to the OER, due to strong cation–OH2 interactions.18,19 Bearing in mind the intimate link between the OER and oxygen-atom transfer reactions enabled by the inner-sphere activation of water, we recently explored the effect of supporting salt cations on the epoxidation of cyclooctene. The epoxidation mechanism at gold oxide was shown to occur at very high potentials (>1.9 V vs. SHE) and to be cation-independent. This observation is in line with previous studies showing that cations only influence the kinetics of the best performing catalysts for the OER and HER, i.e. those requiring lower driving force.29,30 In contrast, the epoxidation mechanism at metallic gold, which is homogeneous in nature and occurs at lower potentials (∼1.5 V vs. SHE), is drastically impacted by the nature of the supporting salt cations. Using TBA+ instead of inorganic cations such as Na+ and Li+ resulted in an improved selectivity towards cyclooctene epoxidation. These results were rationalized by molecular dynamics (MD) simulations both in bulk and at electrified interfaces which showed that changing the nature of the supporting salt cation results in the modification of the hydrophilicity of the electrode/electrolyte interface. The interface is found more hydrophobic in the presence of the organic cation TBA+, which hinders the formation of a passivating oxide layer at the surface of the electrode and in turn favors anodic gold dissolution, a phenomenon at the origin of the in situ formation of a homogeneous cationic metal catalyst.7
Overall, the early developments made in electrochemical oxygen-atom transfer reactions harnessing the inner-sphere activation of water highlight the intimacy between the mechanistic investigations of complex electrosynthetic reactions and the electrocatalysis field.31 A major focus of future research lies in the optimization of the electrocatalysts to improve the epoxidation FE. For that, descriptors previously developed for the OER under alkaline or acidic conditions that encompass the metal–oxygen bond covalency and/or the involvement of the oxygen ligand in the O–O bond formation may serve to design better oxygen atom transfer catalysts.32,33 Nevertheless, it is yet to be demonstrated if physical descriptors developed for the OER will translate to epoxidation reactions (and other electrochemical oxygenation reactions). Furthermore, as revealed by studying gold surfaces, drastic surface decomposition and reformation can be expected in hybrid electrolytes, complicating the study of the true active sites and catalytic pathway. This raises an important question regarding the stability of metal oxides in organic solvents, for which the redeposition process is expected to be drastically different from water-based systems, and asking for the study of Pourbaix diagrams in hybrid electrolytes. Furthermore, the epoxidation mechanism as currently proposed relies on the reaction of ligand oxygen with alkenes. However, if the rate for ligand exchange is not perfectly matched with that of oxygen vacancies refilled by water, drastic dissolution can be expected. Once again, further research is needed to understand if such rates differ in hybrid electrolytes when compared to water-based electrolytes. Hence, not only the electrocatalysts must be optimized, but mastering the interaction of the electrolyte with the electrocatalyst surface has emerged as a critical factor to improve electrocatalytic and electrosynthetic reactions. In light of the above considerations and the knowledge gathered in the past few years in the electrocatalysis field, future research efforts focusing on the improvement of electrochemical oxygenation reactions in hybrid electrolytes, and more generally electrosynthetic reactions, are summarized in Fig. 1. In the following, we focus in detail on the effects related to the electrolyte dynamics and structure in bulk and at electrochemical interfaces.
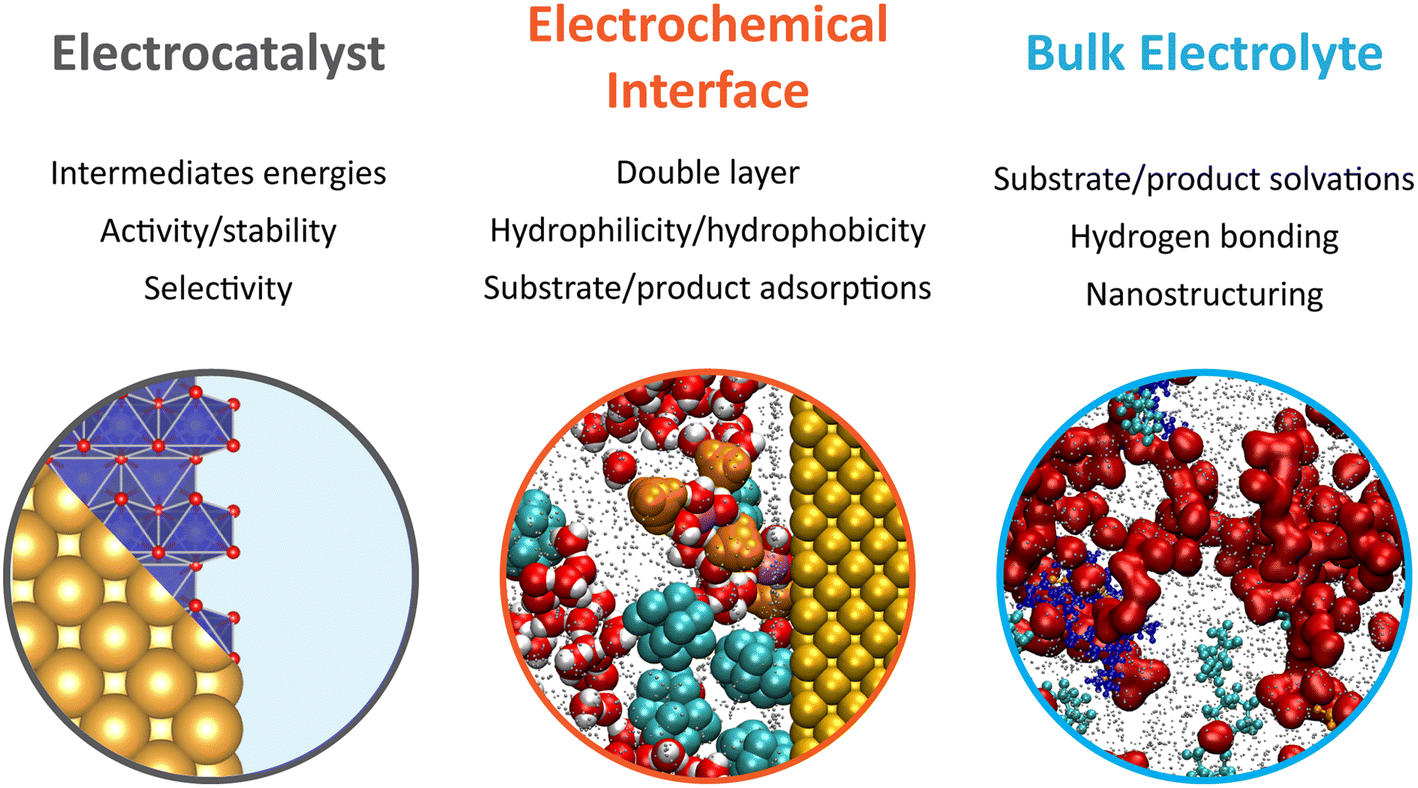 | ||
| Fig. 1 Schematic representation of future research efforts focusing on the design of new electrosynthetic routes, taking inspiration from oxygen-atom transfer reactions enabled by the inner-sphere activation of water at heterogeneous catalysts in hybrid electrolytes. | ||
Bulk and interfacial electrolyte structuring for modulation in electrosynthesis
The electrolyte, which is composed of a single solvent or a mixture of solvents and a supporting salt, can affect the outcome of organic electrosynthesis. When selecting a solvent/supporting salt pair, important factors to consider comprise their respective electrochemical stability windows and the solvent's ability to dissolve and dissociate the supporting salt and to solubilize the substrates. Moreover, several studies reported that the chemical nature of the solvent, itself, can impact electro–organic reactions.34 For example, the proticity of the solvent used for the decarboxylative dimerisation of carboxylic acids (known as the Kolbe coupling) controls the nature of the electrochemically generated intermediate (radical or cationic), with protic solvents favoring the formation of the radical intermediate and therefore promoting the desired Kolbe dimerization.34 The solvent deprotonation free energy is also known to control the selectivity in organic halide electrocarboxylation, where solvents with high deprotonation energies limit the competing hydrogenolysis reaction.35Also revealing are the anodic phenol/arene C–C cross-coupling reactions using 1,1,1,3,3,3-hexafluoropropan-2-ol (HFIP) as solvent (structure given in Fig. 3a), for which the addition of water or methanol was shown to dramatically improve the yield and selectivity of the targeted reaction.36,37 HFIP is used not only because of its large electrochemical window but also because of its ability to strongly solvate the aryl radicals generated at the anode. However, in pure HFIP, the undesired product from the homocoupling of the arene is generally obtained. This is because phenols, and especially electron-rich phenols, are strongly shielded in pure HFIP due to their ability to engage in hydrogen bonding. This shielding prevents nucleophilic attack on the generated radical cations of the arene. Polar additives such as methanol disrupt the phenol-HFIP solvate and facilitate the deprotonation of phenols, which favors in most cases cross-coupling.36,37
This example hints that beyond the nature of the solvent and supporting salt, the structuring of the electrolyte can be pivotal for the outcome of an electro-organic reaction, introducing an additional level of control for the design of new electrosynthetic methods. Inspired by fundamental studies on the nanoscale structuring of ACN/water mixtures, this consideration is at the forefront of the study of the emerging class of electrochemical oxygenation reactions described in the previous section. Using these reactions as a starting point, we discuss how controlling the structure of the electrolyte in bulk and, most importantly, at the electrochemical interface, i.e. where the reaction takes place, can help optimize the yield and the selectivity of electrochemical reactions.
Bulk electrolyte
While being homogeneous at the macroscale, the ACN/water mixtures used for the electrochemical oxygenation reactions enabled by the inner sphere activation of water exhibit heterogeneity at the nanoscale. Upon addition of water into ACN, clustering of water molecules occurs and, for sufficiently high water content (∼10 wt%; ∼5 M), the formation of aqueous-rich and organic nanodomains is observed, both in small angle X-ray scattering (SAXS) and MD simulations.7,17 Upon addition of cyclooctene into these mixtures, the analysis of bulk MD simulations showed that, to no surprise, cyclooctene is located in the organic nanodomains (Fig. 2a) and does not disrupt the number and size of the aqueous nanodomains.7 Alike the reactant (cyclooctene), the product of the epoxidation reaction, cyclooctene oxide, is located in the organic nanodomains (Fig. 2b and c).38 However, combined angular/radial distribution functions (CDFs) analysis shows that cyclooctene oxide, while being in the organic nanodomains, is engaged in hydrogen bonding with water through its epoxide moiety (Fig. 2d and e).38 Cyclooctene oxide is thus primarily located at the interface between the organic and aqueous nanodomains, thereby showing a preferential orientation inside the organic domains, in contrast to cyclooctene. Following this observation, opportunities arise to potentially modify the reactivity of organic substrates by controlling their interaction with aqueous domains inside the nanoheterogeneities, via the introduction of hydrophilic functional groups.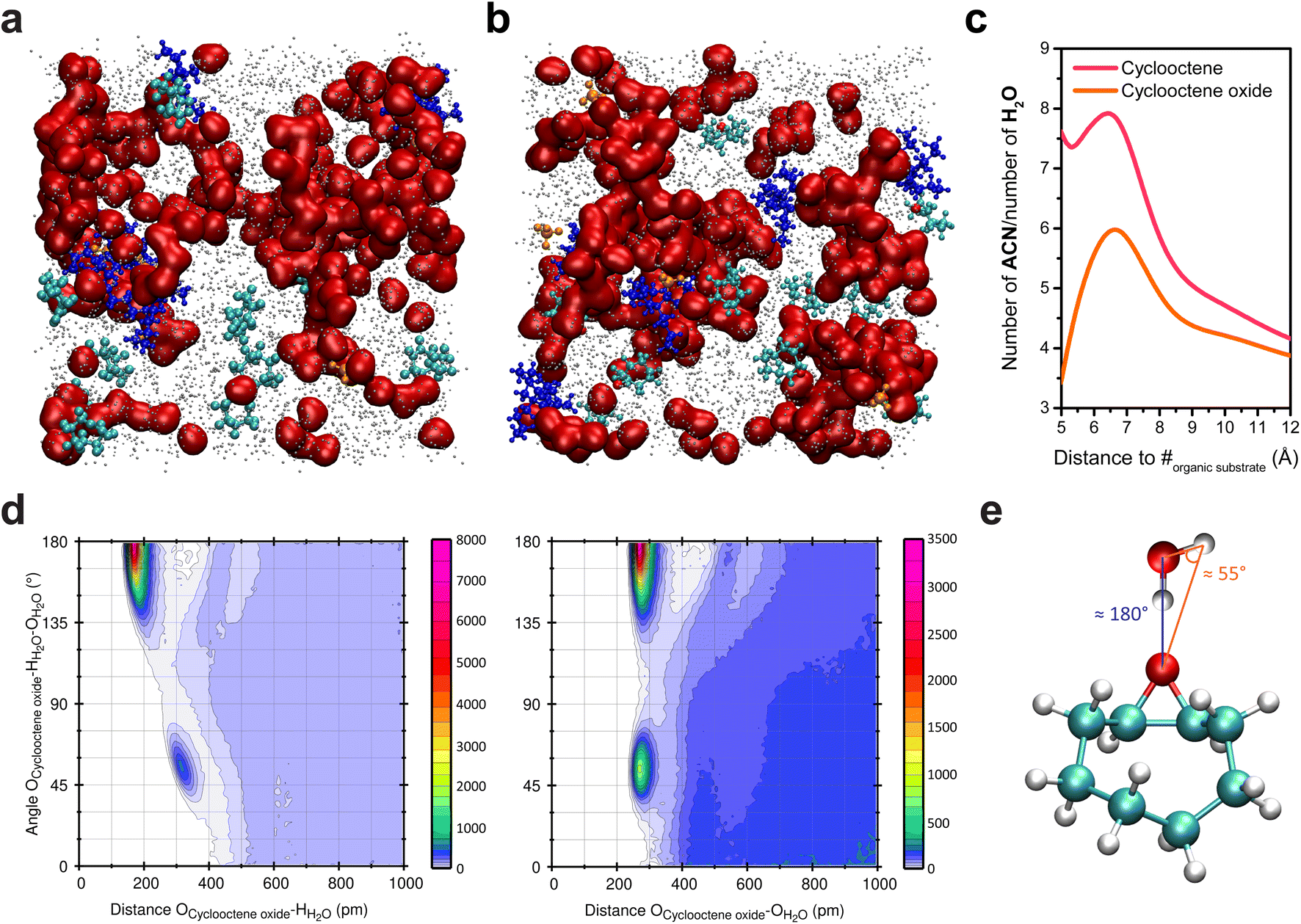 | ||
| Fig. 2 Snapshots of bulk MD simulations in a hybrid ACN/5 M water (0.1 M TBAClO4) electrolyte with (a) cyclooctene and (b) cyclooctene oxide. Water molecules are represented in red, ACN in grey (the size of which was decreased for clarity), ClO4− in orange, TBA+ in deep blue, and cyclooctene and cyclooctene oxide in cyan (the oxygen of the latter is shown in red). (c) Ratio of the number of ACN and H2O molecules surrounding cyclooctene and cyclooctene oxide as a function of the distance to their mass center (#), showing that both are located in the organic nanodomains (i.e. ratio much greater than unity). The ratio at distances lower than 5 Å is irrelevant as the first solvent molecules surrounding the substrates are found at >5 Å from their mass center. At long distances, the ratio tends towards 3.34, which is the ratio of ACN and H2O molecules in the MD simulation box. (d) Combined angular/radial distribution functions (CDFs) for the angle Ocycloocteneoxide–HH2O–OH2O and the distances Ocycloocteneoxide–HH2O (left) and Ocycloocteneoxide–OH2O (right). The maxima in probability distributions are found for two values of the angle Ocycloocteneoxide–HH2O–OH2O (55° and 180°) associated with two Ocycloocteneoxide–HH2O distances (175.0 and 318.5 pm) and one Ocycloocteneoxide–OH2O distance (275.0 pm). In the SPC/E model, which was used in the simulations, the HH2O–OH2O distance is 1.0 Å and the angle HH2O–OH2O–HH2O is 109.47°. The angles and distances values obtained with the CDFs thus show the establishment of hydrogen bonding39 between cyclooctene oxide and water molecules, as depicted in (e). Panel (a) is reproduced from ref. 7 with permission. Copyright 2022 American Chemical Society. | ||
A parallel can be established between ACN/water mixtures and mixtures of HFIP with polar protic molecules such as water, H2O2 or methanol, which also display heterogeneities at the nanoscale while being macroscopically homogeneous.40,41 As discussed above, these mixtures are widely studied for electrochemical phenol/arene C–C cross-couplings and also for chemical epoxidation of alkenes and lactonization of ketones with H2O2.42–44 The heterogeneity at the nanoscale results from the segregation of the polar –OH groups of HFIP and the added polar protic molecule (water, H2O2 or methanol) from the (CF3)2CH– moieties of HFIP. The groups of Waldvogel and Kirchner rationalized the impact of such heterogeneity at the nanoscale using MD simulations performed on a reaction of interest: the chemical epoxidation of cyclooctene using H2O2 as the oxidant.40 While H2O2 incorporates into the polar microphase, the authors found that cyclooctene molecules alter the hydrogen bond network in solution and forms non-polar clusters. These clusters are surrounded by the fluorinated moieties of HFIP, themselves surrounded by the polar microphase, giving rise to a triphasic layered micelle structure (Fig. 3b). Their finding therefore shows that the oxidant has to penetrate the fluorinated barrier to react with the substrate and that the reaction, previously assumed to be a homogeneous catalytic process, is rather figuratively a phase transfer or an interfacial reaction. Adapting this chemical epoxidation route to electrochemical epoxidation reactions with water as the sole oxygen source might be of significant interest. The shielding effect of the fluorinated barrier around the cluster of apolar alkene substrates could prevent the direct substrate oxidation at the anode, which is a competing reaction at high potentials. Because the triphasic shielding structure is obtained with apolar aprotic substrates (cyclooctene), structuring at the nanoscale might be drastically different for substrates bearing additional functional groups such as polar groups or, most importantly in this particular case, fluorinated groups. As suggested for ACN/water mixtures, this consideration could offer a means to control bulk structuring of organic substrates inside nanoheterogeneities, potentially impacting electrochemical reactions after the migration of the substrate to the electrochemical interface.
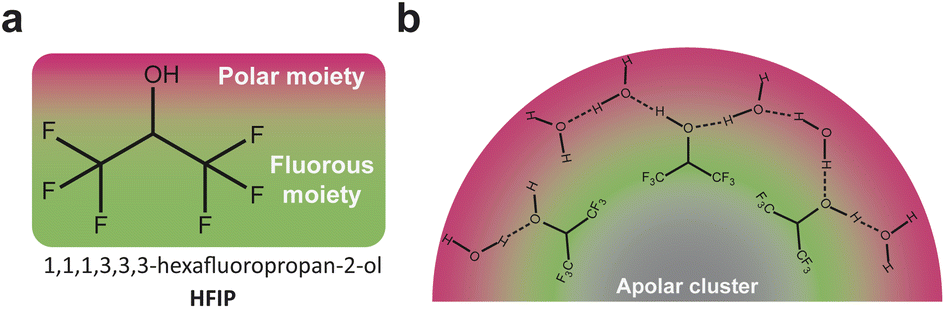 | ||
| Fig. 3 (a) Chemical structure of 1,1,1,3,3,3-hexafluoropropan-2-ol (HFIP). (b) Nonpolar clusters formed from cyclooctene molecules, surrounded by the fluorous moieties of HFIP themselves surrounded by the polar microphase, giving rise to a triphasic layered micelle structure. Adapted from ref. 40. | ||
Moreover, translating this chemical epoxidation route into its electrochemical counterpart would require the use of a supporting salt, which might significantly impact the nanoscale structuring. Indeed, it was shown for ACN/water mixtures that the number and size of the aqueous domains can be tuned by the nature of the supporting salt and its concentration. In ACN containing 10 wt% water, increasing the concentration of LiClO4 as the supporting salt was found to enhance the size of the aqueous nanodomains formed in the organic electrolyte.17 In addition to the strong Li+–OH2 interaction which promotes the cleavage of the water O–H bond, larger aqueous nanodomains were found to correlate with an increased reactivity of water towards the HER at platinum and gold electrodes, as the larger the domains, the easier the hydroxide ions generated during the HER will diffuse away from the interface. This result highlights that the electrolyte structure, both at short and long ranges, must be considered to capture HER kinetics. Several studies also report that, for the OER, strong Lewis acids such as Li+ are detrimental in aqueous electrolytes due to strong cation–OH2 interactions18,19 and that decreasing the amount of water in ACN/water mixtures increases water activity towards the OER due to the disruption of the hydrogen bonding network.21,45 A more comprehensive study on the effect of short range cation/anion–water interactions and the size of aqueous nanodomains in hybrid electrolytes on the anodic reactivity of water is however currently needed. Such a study would provide a means for tuning the activity of water for more efficient oxygen-atom transfer to organic substrates. In hybrid electrolytes, the short- and long-range structuring of water dramatically depends on the nature and the concentration of the supporting salt, which impacts both its cathodic and anodic reactivity.
A similar question arises for organic substrates. In ACN/5 M water mixtures, cyclooctene’ solvation shell is unaffected by the nature of the supporting salt cation (Li+, Na+ or TBA+), although a slight preferential interaction between cyclooctene and the organic cation TBA+ exists.7 Similarly, the solvation structure of cyclooctene oxide is not affected by the nature of the supporting salt cation, which suggests that, in contrast to water, the nature of the supporting salts is unlikely to modify the solvation structure of organic substrates in hybrid ACN/water electrolytes. For a given supporting salt, we can however expect that changing its concentration will affect the solvation structure of organic substrates as it affects the size and number of the nanodomains in the electrolyte. For HFIP/water mixtures, the effect of the concentration and nature of the supporting salt is certainly massive. In particular, the use of a salt containing organic ions and/or fluorinated species is expected to disrupt the biphasic layered structure (or triphasic in the presence of an apolar aprotic substrate such as cyclooctene) which can significantly modulate electrosynthetic reactions performed in these mixtures.
Electrochemical interface
Electrochemical reactions taking place at the electrochemical interface, its structuring and dynamics are pivotal for the outcome of electrosynthetic reactions. With this in mind, pulsed electrosynthesis appears as a method of choice to control the dynamics of the electrochemical interface. Indeed, pulsed electrosynthesis, a method relying on the application of pulses of voltage/current to drive a given reaction separated by resting periods at a lower (in absolute value) voltage/current,46 allows for the cyclic renewal of the diffusion layer.47 The dynamic renewal of the diffusion layer alters the chemical environment in the vicinity of the working electrode, which can improve the chemo- and product selectivities compared to traditional constant current/potential electrosynthesis.46,48–51 For example, pulsed electrolysis has recently been used to overcome the poor functional group tolerance of the strongly oxidative Kolbe coupling.50 Under constant current electrolysis, a locally acidic pH is generated around the anode (coming from the oxidation of solvent, water, or the substrate itself) which prevents the formation of carboxylate anions. Because the direct oxidation of carboxylic acids is not favored, the oxidation of other functional groups such as alkenes or arenes outcompetes the decarboxylative reaction, leading to poor Kolbe dimer selectivity when other functional groups are present on the substrate. In contrast, during pulsed electrosynthesis, the anodic period does not last long enough to create a persistent acidic environment around the electrode. As a result, the oxidation of the carboxylate occurs in preference to other functional groups. Attempts to purposely functionalize the electrode surface to tune the properties of the electrochemical interface were also reported. For instance, non-covalent functionalization of electrode surfaces with ionic liquid thin films was shown to alter the interfacial hydrogen bonding network, impacting the kinetics of the nitrogen and oxygen reduction reactions.52–54 In contrast, the use of cationic surfactants was shown to allow for the control of interfacial water content.55 Aside from these engineering approaches, structuring of the electrochemical interface offers an exciting approach for fine tuning of electro-synthetic reactions.Knowing that bulk nanoheterogeneities present in ACN/water and HFIP/polar solvent mixtures were shown to be conserved at the electrochemical interface,17,41 preferential orientation inside nanoheterogeneities can potentially be used to modulate the geometry of the reactants approaching the electrode surface, thereby inducing particular chemo- and/or stereo-selectivities. Moreover, studying the cross-coupling reaction between 2-methoxy-4-methylphenol A and 1,2,4-trimethoxybenzene B at boron-doped diamond (BDD) electrodes in HFIP/methanol mixtures (Fig. 4a), the presence of nanodomains was shown to result in the modification of the spin density on the phenoxyl radical A* generated at the BDD electrode.41 Through particular hydrogen bonding with both solvents in these domains, radical formation on the C6 carbon of A* is favored, which is critical for the formation of the targeted AB compound. Combined with an enrichment of both substrates at the electrode surface aided by HFIP, the presence of interfacial nanodomains (Fig. 4b) was thus shown to promote selectivity towards the targeted cross-coupling product, which shows that interfacial structuring can also impact electrogenerated intermediates and therefore influence the synthetic pathway. In addition to the enrichment of substrates at the electrochemical interface, substrates can also chemically interact with the surface. Substrates A and B were found to adsorb at the electrode surface, due to attractive lipophilic–lipophilic interactions between the substrates and the BDD electrode. When studying the epoxidation of cyclooctene in ACN/5 M water electrolytes, MD simulations at electrified interfaces showed that the substrate, cyclooctene, adsorbs at metallic gold anodes (Fig. 4c and d), which contributes to increasing the hydrophobicity of the electrochemical interface. This observation pinpoints that specific interactions with the electrode surface drastically impact the chemical environment in the vicinity of the electrode surface and must be considered.
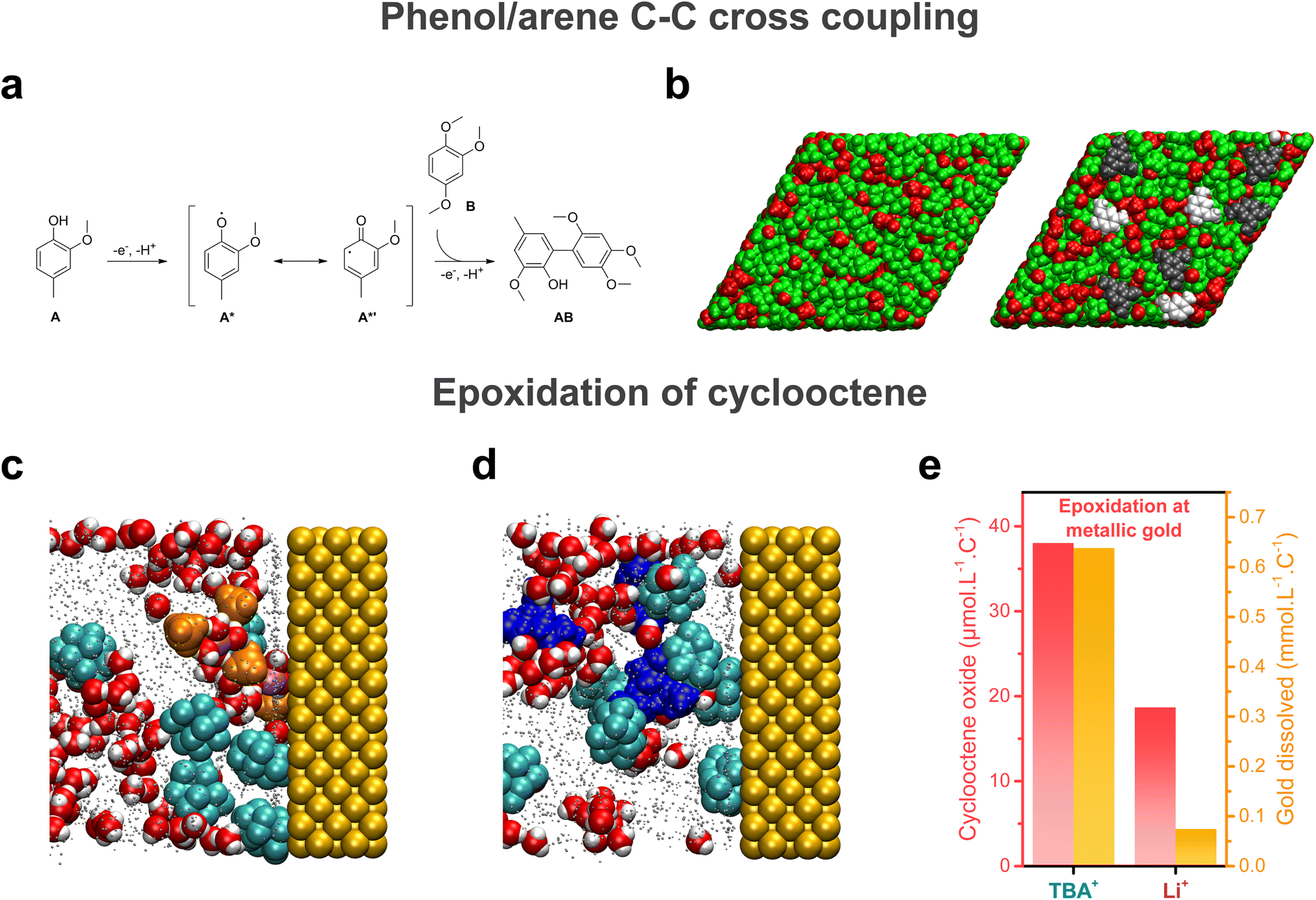 | ||
| Fig. 4 (a) Cross-coupling reaction between 2-methoxy-4-methylphenol A and 1,2,4-trimethoxybenzene B at boron-doped diamond (BDD) electrodes in HFIP/methanol mixtures, as studied in ref. 41. (b) MD snapshot of the liquid structure at the BDD electrode interface in a HFIP/methanol mixture, showing the interfacial domains. The MD simulation box contained either only the HFIP/methanol mixture (left) or the HFIP/methanol with A and B. Methanol molecules and the hydroxyl groups of HFIP are represented in red, the CF3–CH–CF3 moieties of HFIP in green, A in light grey and B in dark grey. In both cases, the electrode is not represented and lies behind the molecules in both panels. Reproduced from ref. 41 with permission. Copyright 2019 American Chemical Society. MD snapshots of the liquid structure in the vicinity of a gold anode in an ACN/water mixture containing 5 M water, 200 mM cyclooctene and (c) 0.1 M LiClO4 or (d) 0.1 M TBAClO4 as the supporting salt. Reproduced from ref. 7 with permission. Copyright 2022 American Chemical Society. (e) Quantity of cyclooctene oxide produced and gold dissolved during electrolysis at metallic gold for the TBA+- and Li+-containing electrolytes. Adapted from ref. 7. | ||
Beyond the presence of interfacial domains, a universal phenomenon in electrosynthetic reactions is the migration of supporting salt ions to the electrode surface in the presence of an electric field. The nature and the concentration of the supporting salt ions will therefore impact the structure of the electrochemical interface, offering further modulation pathways for electrosynthetic reactions. Independently of the nature of supporting salt ions, tuning the electrochemical double layer screening efficiency by varying the supporting salt concentration can significantly influence the kinetics and FE of electrosynthetic reactions. It was recently shown that higher CO2 reduction rates and enhancement of CO2 to CO FE were obtained when using intermediate concentrations of ionic liquids in ACN, resulting from a more efficient screening of the electrochemical double layer.56 In dilute and concentrated electrolytes, a small number of ions and excessive ionic correlations, respectively, are responsible for the formation of thick double layers. At intermediate concentrations, however, the screening length approaches ionic sizes, forming a thinner double layer and creating a large potential gradient. The magnitude of the local potential gradient is key in determining electrochemical reaction rates by creating polarized environments that help stabilize intermediates. Accordingly, strongly screening cases obtained at intermediate salt concentrations were found to enhance CO2RR activity. This effect of salt concentration is not ion- or solvent-specific and can therefore be explored for other electrochemical reactions.
Further modulation of electrosynthetic reactions can be obtained by tuning the nature of supporting salt ions. For instance, the use of large organic hydrophobic cations such as TBA+ results in the formation of a hydrophobic electrochemical double layer at the cathode in ACN/water mixtures, hindering the reduction of water.16,17 More surprisingly, tetramethylammonium (TMA+) and TBA+ cations were also shown to alter the OER in alkaline electrolytes.18 In ACN/5 M water mixtures, the solvation structure of organic substrates inside the nanodomains was found to be independent on the nature of supporting salt cations. However, for the epoxidation of cyclooctene at metallic gold, which proceeds by the in situ formation of an homogeneous cationic metal catalyst following anodic dissolution, a stark effect of the nature of the supporting salt on the outcome of the reaction was found, with a higher yield and selectivity obtained when using TBA+ instead of inorganic cations (Na+ and Li+) (Fig. 4e).7 Combining rotating disk electrode measurements with inductively coupled plasma mass spectrometry (ICP-MS) and electrochemical quartz crystal microbalance (EQCM) experiments, the presence of TBA+ in the electrolyte was correlated with a greater anodic dissolution of gold, which in turn favored the formation of cyclooctene oxide. This result was rationalized by performing MD simulations at electrified interfaces which revealed that, in the presence of TBA+, the electrochemical interface is more hydrophobic compared to Li+ or Na+, thus favoring gold dissolution over the passivation of the electrode surface with the formation of a gold oxide layer (Fig. 4c and d). A similar cationic effect was observed on the selectivity of cathodic reactions, such as the electrohydrodimerization of acrylonitrile in aqueous electrolytes.57 Large cations such as Cs+ limit the availability of water at the electrochemical interface. This effect hinders the competing HER and limits the reaction of water with the electrogenerated acrylonitrile radical anions and therefore increases the selectivity towards adiponitrile at low current densities. However, at high current densities, when mass transport limitations dominate, smaller cations such as Na+ improve the stability of the electrogenerated acrylonitrile radical anions, which increases their lifetime and promotes the reaction pathway to adiponitrile formation. Thus, there is a competing effect on reaction selectivity, leading to a maximum adiponitrile selectivity with cations having an intermediate size, i.e. K+, at higher current densities. The latter example pinpoints that for a given electrolyte, the distribution of charged species at the interface is dependent on the applied potential and on current density, which entails that selectivity induced by the structuring of the electrochemical interface will also depend on the operating conditions. This consideration is of prime importance, especially for implementation in industrial processes which require high current densities and might necessitate adaptability to variable operating current densities to adjust to the electricity surplus generated by renewable energy sources.49,58
Interestingly, supporting salt ion effects were also recently shown for homogeneous electrosynthetic routes such as the catalytic reduction of benzyl chloride with metal tetraphenylporphyrin.59 The hydrodynamic radius of the supporting salt cation (Li+, TMA+ or TBA+) modulates the stability of the metal–alkyl intermediate involved. For large cations such as TBA+, the charged intermediate is efficiently stabilized by the soft solvent–ion shell surrounding it. In contrast, small cations such as Li+ having a more compact solvation shell likely destabilize the metal–alkyl intermediate, in turn promoting catalysis via the cleavage of the metal–alkyl bond. The metal–alkyl intermediate arises from a chemical reaction between the alkyl chloride substrate and the electro-generated active oxidation state of the catalyst. Consequently, a critical question to answer in future research is whether the (de)stabilizing effect of supporting salt cations on the intermediate occurs in bulk or at the electrochemical interface. Such research would allow for establishing a general framework linking electrolyte effects observed for homogeneous and heterogeneous electrosynthetic reactions and help the rational design of electrolytes for targeted applications.
Conclusion and future outlook
In conclusion, electrolyte structuring at nano- and micro-scales can drastically impact the yield and the selectivity of electrosynthetic reactions, as revealed by recent reports. Such studies are still scarce. However, they hint that mastering this structuring, both in bulk and at the electrochemical interface, can provide additional chemical space for the rational design of synthetic routes. The knowledge gathered in the past few years in the electrocatalysis field on the effect of the electrolyte structuring on the kinetics and selectivity of reactions such as the CO2RR, HER or OER can help select the best electrolyte for a given electrosynthesis. This consideration can be further extended to reactions performed in ionic liquids, as they are widely used for synthetic applications and show very particular bulk and interfacial structuring.60–63The use of mixtures of miscible solvents, such as ACN/water, HFIP/methanol or HFIP/water, in electrosynthesis is particularly promising, as they allow for fine-tuning the interactions of each reactant. The interactions of organic substrates with the different domains formed in those solvent systems greatly depend on their functional groups. A preferential orientation inside these nanoheterogeneities can arise and be harnessed to induce chemo- and/or stereo-selectivity. The establishment of such nanoheterogeneities has implications beyond electrosynthesis. Knowing that some supporting salts are known to induce demixing of ACN and water64 while others maintain the nanoheterogeneity without inducing phase separation, fundamental work is currently devoted to understanding salt-induced liquid–liquid phase separation phenomena.65,66 Such phenomena are ubiquitous, with implications in various fields such as biology, chemical extraction or batteries. However, not all synthetically relevant mixtures of solvents exhibit structuring, like mixtures of N,N-dimethylformamide (DMF) and water. Although in these mixtures DMF and water molecules exhibit some tendency for self-association, molecular dynamics simulations show that no microheterogeneity occurs.67 This pinpoints that fundamental studies dedicated to the understanding of the structuring of electrolytes, even those not targeting synthetic applications, can enable the rational selection of electrolytes for a given reaction.
Moreover, the vision of supporting salt ions as solely spectators is outdated. Indeed, salt ions can be selected to tune the chemical environment in the vicinity of the electrode/electrocatalyst surface. Upon the application of an electric field, the supporting salt ions migrate to the electrode surface, forming an electrochemical double layer. The size of the supporting salt cations and anions and their chemical properties influence the chemical composition of the double layer; they must be chosen in accordance with the synthetic objectives. Going beyond the organic and inorganic salts traditionally used in electrochemistry, a particular focus of future research lies in the exploration and the synthesis of supporting salt ions with complex and tailored structures. For instance, using chiral cations and anions,68,69 either as a supporting salt or additives, could constitute a promising yet unexplored strategy for inducing stereoselectivity in electrosynthesis.70,71
Along with traditional electrochemical measurements such as cyclic voltammetry, advanced techniques such as EQCM,7,72operando spectroscopy techniques such as surface enhanced Raman and FTIR73,74 or laser-jump measurements75 are necessary to study the electrode–electrolyte interface. Information on the nano- to micro-scale structuring of bulk electrolytes can also be accessed via SAXS measurements.17,40,76 In addition to these experimental techniques, thorough understanding of the electrolyte structure heavily relies on correlating the experimental results with theoretical/computational studies such as density functional theory (DFT) calculations77,78 and MD simulations,7,17 with the latter being a computational technique of choice to account for the different interactions at the nano- and micro-scales. The advent of DFT calculations with explicit solvent models79 and ab initio MD simulations80 will help get a full picture of how the solvation structure affects electron transfer at electrochemical interfaces. Collaborations between experts in electrochemistry, physical chemistry and organic synthesis are therefore required to master the electrolyte structuring towards synthetic applications. We believe that such synergy between the different fields could lead to the discovery of new synthetically relevant chemical routes, as recently suggested in the literature.81
Author contributions
F. D. conducted data curation and wrote the manuscript under the supervision of A. G.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors thank Dr Alessandra Serva and Prof. Mathieu Salanne for help with MD simulations. F. D. thanks the École normale supérieure Paris-Saclay for his PhD scholarship.Notes and references
- G. M. Tomboc, Y. Park, K. Lee and K. Jin, Chem. Sci., 2021, 12, 8967–8995 RSC.
- M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230–13319 CrossRef CAS PubMed.
- E. J. Horn, B. R. Rosen and P. S. Baran, ACS Cent. Sci., 2016, 2, 302–308 CrossRef CAS PubMed.
- J. H. Maalouf, K. Jin, D. Yang, A. M. Limaye and K. Manthiram, ACS Catal., 2020, 10, 5750–5756 CrossRef CAS.
- K. Jin, J. H. Maalouf, N. Lazouski, N. Corbin, D. Yang and K. Manthiram, J. Am. Chem. Soc., 2019, 141, 6413–6418 CrossRef CAS PubMed.
- M. Chung, K. Jin, J. S. Zeng, T. N. Ton and K. Manthiram, J. Am. Chem. Soc., 2022, 144, 17416–17422 CrossRef CAS PubMed.
- F. Dorchies, A. Serva, D. Crevel, J. De Freitas, N. Kostopoulos, M. Robert, O. Sel, M. Salanne and A. Grimaud, J. Am. Chem. Soc., 2022, 144, 22734–22746 CrossRef CAS PubMed.
- X. Lin, Z. Zhou, Q. Li, D. Xu, S.-Y. Xia, B.-L. Leng, G.-Y. Zhai, S.-N. Zhang, L.-H. Sun, G. Zhao, J.-S. Chen and X.-H. Li, Angew. Chem., Int. Ed., 2022, 61, e202207108 CAS.
- L. Ma, H. Zhou, M. Xu, P. Hao, X. Kong and H. Duan, Chem. Sci., 2021, 12, 938–945 RSC.
- J. K. Park and S. Lee, J. Org. Chem., 2021, 86, 13790–13799 CrossRef CAS PubMed.
- Y. Sun, X. Li, M. Yang, W. Xu, J. Xie and M. Ding, Green Chem., 2020, 22, 7543–7551 RSC.
- C. Li, Q. Cheng, C. Wu, Q. Wang, W. Hu, L. Zou, K. Wen and H. Yang, Chem. Commun., 2022, 58, 10496–10499 RSC.
- F. Marken and J. D. Wadhawan, Acc. Chem. Res., 2019, 52, 3325–3338 CrossRef CAS PubMed.
- S. Ringe, E. L. Clark, J. Resasco, A. Walton, B. Seger, A. T. Bell and K. Chan, Energy Environ. Sci., 2019, 12, 3001–3014 RSC.
- B. Pan, Y. Wang and Y. Li, Chem. Catal., 2022, 2, 1267–1276 CrossRef CAS.
- N. Dubouis, A. Serva, E. Salager, M. Deschamps, M. Salanne and A. Grimaud, J. Phys. Chem. Lett., 2018, 9, 6683–6688 CrossRef CAS PubMed.
- N. Dubouis, A. Serva, R. Berthin, G. Jeanmairet, B. Porcheron, E. Salager, M. Salanne and A. Grimaud, Nat. Catal., 2020, 3, 656–663 CrossRef CAS.
- C. Yang, O. Fontaine, J. M. Tarascon and A. Grimaud, Angew. Chem., Int. Ed., 2017, 56, 8652–8656 CrossRef CAS PubMed.
- M. Görlin, J. Halldin Stenlid, S. Koroidov, H.-Y. Wang, M. Börner, M. Shipilin, A. Kalinko, V. Murzin, O. V. Safonova, M. Nachtegaal, A. Uheida, J. Dutta, M. Bauer, A. Nilsson and O. Diaz-Morales, Nat. Commun., 2020, 11, 6181 CrossRef PubMed.
- A. Serva, N. Dubouis, A. Grimaud and M. Salanne, Acc. Chem. Res., 2021, 54, 1034–1042 CrossRef CAS PubMed.
- J. C. Hidalgo-Acosta, M. D. Scanlon, M. A. Méndez, P. Peljo, M. Opallo and H. H. Girault, ChemElectroChem, 2016, 3, 2003–2007 CrossRef CAS.
- H. Zhang, C. Li, Q. Lu, M. J. Cheng and W. A. Goddard, J. Am. Chem. Soc., 2021, 143, 3967–3974 CrossRef CAS PubMed.
- C. Hsieh, Y. Tang, Y. Ho, W. Shao and M. Cheng, J. Phys. Chem. C, 2023, 127, 308–318 CrossRef CAS.
- K. Otsuka, T. Ushiyama, I. Yamanaka and K. Ebitani, J. Catal., 1995, 157, 450–460 CrossRef CAS.
- S. Iguchi, M. Kataoka, R. Hoshino and I. Yamanaka, Catal. Sci. Technol., 2022, 12, 469–473 RSC.
- X. C. Liu, T. Wang, Z. M. Zhang, C. H. Yang, L. Y. Li, S. Wu, S. Xie, G. Fu, Z. Y. Zhou and S. G. Sun, J. Am. Chem. Soc., 2022, 144, 20895–20902 CrossRef CAS PubMed.
- W. R. Leow, Y. Lum, A. Ozden, Y. Wang, D. H. Nam, B. Chen, J. Wicks, T. T. Zhuang, F. Li, D. Sinton and E. H. Sargent, Science, 2020, 368, 1228–1233 CrossRef CAS PubMed.
- W. T. Hong, M. Risch, K. A. Stoerzinger, A. Grimaud, J. Suntivich and Y. Shao-Horn, Energy Environ. Sci., 2015, 8, 1404–1427 RSC.
- R. Zhang, P. E. Pearce, Y. Duan, N. Dubouis, T. Marchandier and A. Grimaud, Chem. Mater., 2019, 31, 8248–8259 CrossRef CAS.
- Y. Duan, N. Dubouis, J. Huang, D. A. Dalla Corte, V. Pimenta, Z. J. Xu and A. Grimaud, ACS Catal., 2020, 10, 4160–4170 CrossRef CAS.
- L. F. T. Novaes, J. Liu, Y. Shen, L. Lu, J. M. Meinhardt and S. Lin, Chem. Soc. Rev., 2021, 50, 7941–8002 RSC.
- A. Grimaud, W. T. Hong, Y. Shao-Horn and J. M. Tarascon, Nat. Mater., 2016, 15, 121–126 CrossRef CAS PubMed.
- A. Grimaud, O. Diaz-Morales, B. Han, W. T. Hong, Y. L. Lee, L. Giordano, K. A. Stoerzinger, M. T. M. Koper and Y. Shao-Horn, Nat. Chem., 2017, 9, 457–465 CrossRef CAS PubMed.
- L. Schulz and S. Waldvogel, Synlett, 2019, 30, 275–286 CrossRef CAS.
- N. Corbin, G. P. Junor, T. N. Ton, R. J. Baker and K. Manthiram, J. Am. Chem. Soc., 2023, 145, 1740–1748 CrossRef CAS PubMed.
- A. Kirste, B. Elsler, G. Schnakenburg and S. R. Waldvogel, J. Am. Chem. Soc., 2012, 134, 3571–3576 CrossRef CAS PubMed.
- B. Elsler, A. Wiebe, D. Schollmeyer, K. M. Dyballa, R. Franke and S. R. Waldvogel, Chem.–Eur. J., 2015, 21, 12321–12325 CrossRef CAS PubMed.
- F. Dorchies and A. Grimaud, Molecular dynamics (MD) simulations methods and results on cyclooctene oxide in ACN/water hybrid electrolytes, v1.0, Zenodo, 2023, DOI:10.5281/zenodo.7799839.
- S. J. Grabowski, in Understanding Hydrogen Bonds, The Royal Society of Chemistry, 1st edn, 2020, pp. 1–40 Search PubMed.
- O. Hollóczki, A. Berkessel, J. Mars, M. Mezger, A. Wiebe, S. R. Waldvogel and B. Kirchner, ACS Catal., 2017, 7, 1846–1852 CrossRef.
- O. Hollóczki, R. Macchieraldo, B. Gleede, S. R. Waldvogel and B. Kirchner, J. Phys. Chem. Lett., 2019, 10, 1192–1197 CrossRef PubMed.
- A. Berkessel and J. A. Adrio, J. Am. Chem. Soc., 2006, 128, 13412–13420 CrossRef CAS PubMed.
- A. Berkessel, J. A. Adrio, D. Hüttenhain and J. M. Neudörfl, J. Am. Chem. Soc., 2006, 128, 8421–8426 CrossRef CAS PubMed.
- A. Berkessel, M. R. M. Andreae, H. Schmickler and J. Lex, Angew. Chem., Int. Ed., 2002, 41, 4481–4484 CrossRef CAS PubMed.
- J. C. Hidalgo-Acosta, M. A. Méndez, M. D. Scanlon, H. Vrubel, V. Amstutz, W. Adamiak, M. Opallo and H. H. Girault, Chem. Sci., 2015, 6, 1761–1769 RSC.
- S. Rodrigo, D. Gunasekera, J. P. Mahajan and L. Luo, Curr. Opin. Electrochem., 2021, 28, 100712 CrossRef CAS.
- A. J. Bard and L. R. Faulkner, Electrochemical Methods: Fundamentals and Applications, Wiley, 2nd edn, 2001 Search PubMed.
- Y. Kawamata, K. Hayashi, E. Carlson, S. Shaji, D. Waldmann, B. J. Simmons, J. T. Edwards, C. W. Zapf, M. Saito and P. S. Baran, J. Am. Chem. Soc., 2021, 143, 16580–16588 CrossRef CAS PubMed.
- D. E. Blanco, B. Lee and M. A. Modestino, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 17683–17689 CrossRef CAS PubMed.
- Y. Hioki, M. Costantini, J. Griffin, K. C. Harper, M. P. Merini, B. Nissl, Y. Kawamata and P. S. Baran, Science, 2023, 380, 81–87 CrossRef CAS PubMed.
- R. Casebolt, K. Levine, J. Suntivich and T. Hanrath, Joule, 2021, 5, 1987–2026 CrossRef CAS.
- Y. Li, A. Malkani, R. Gawas, S. Intikhab, B. Xu, M. Tang and J. Snyder, ACS Catal., 2023, 13, 382–391 CrossRef CAS.
- M. A. Ortuño, O. Hollóczki, B. Kirchner and N. López, J. Phys. Chem. Lett., 2019, 10, 513–517 CrossRef PubMed.
- T. Wang, Y. Zhang, B. Huang, B. Cai, R. R. Rao, L. Giordano, S. G. Sun and Y. Shao-Horn, Nat. Catal., 2021, 4, 753–762 CrossRef CAS.
- A. K. Pennathur, C. Tseng, N. Salazar and J. M. Dawlaty, J. Am. Chem. Soc., 2023, 145, 2421–2429 CrossRef CAS PubMed.
- B. Liu, W. Guo and M. A. Gebbie, ACS Catal., 2022, 12, 9706–9716 CrossRef CAS.
- D. E. Blanco, R. Atwi, S. Sethuraman, A. Lasri, J. Morales, N. N. Rajput and M. A. Modestino, J. Electrochem. Soc., 2020, 167, 155526 CrossRef CAS.
- A. Wiebe, B. Riehl, S. Lips, R. Franke and S. R. Waldvogel, Sci. Adv., 2017, 3, 1–8 Search PubMed.
- D. Boucher, Z. A. Nguyen and S. Minteer, Faraday Discuss., 2023 10.1039/D3FD00054K , accepted manuscript..
- J. P. Hallett and T. Welton, Chem. Rev., 2011, 111, 3508–3576 CrossRef CAS PubMed.
- R. Elfgen, O. Hollóczki and B. Kirchner, Acc. Chem. Res., 2017, 50, 2949–2957 CrossRef CAS PubMed.
- R. Hayes, G. G. Warr and R. Atkin, Phys. Chem. Chem. Phys., 2010, 12, 1709–1723 RSC.
- F. Endres, O. Höfft, N. Borisenko, L. H. Gasparotto, A. Prowald, R. Al-Salman, T. Carstens, R. Atkin, A. Bund and S. Zein El Abedin, Phys. Chem. Chem. Phys., 2010, 12, 1724–1732 RSC.
- M. Li, B. Zhuang, Y. Lu, L. An and Z. G. Wang, J. Am. Chem. Soc., 2021, 143, 773–784 CrossRef CAS PubMed.
- D. Degoulange, R. Pandya, M. Deschamps, D. A. Skiba, B. M. Gallant, S. Gigan, H. B. de Aguiar and A. Grimaud, Proc. Natl. Acad. Sci., 2023, 120, e2220662120 CrossRef CAS PubMed.
- N. Dubouis, A. France-Lanord, A. Brige, M. Salanne and A. Grimaud, J. Phys. Chem. B, 2021, 125, 5365–5372 CrossRef CAS PubMed.
- V. Koverga, Á. Juhász, D. Dudariev, M. Lebedev, A. Idrissi and P. Jedlovszky, J. Phys. Chem. B, 2022, 126, 6964–6978 CrossRef CAS PubMed.
- R. J. Phipps, G. L. Hamilton and F. D. Toste, Nat. Chem., 2012, 4, 603–614 CrossRef CAS PubMed.
- M. P. Walsh, J. M. Phelps, M. E. Lennon, D. S. Yufit and M. O. Kitching, Nature, 2021, 597, 70–76 CrossRef CAS PubMed.
- M. Ghosh, V. S. Shinde and M. Rueping, Beilstein J. Org. Chem., 2019, 15, 2710–2746 CrossRef CAS PubMed.
- D. Il Park, S. Jung, H. J. Yoon and K. Jin, Electrochim. Acta, 2021, 397, 139271 CrossRef.
- E. Bendadesse, A. V. Morozov, A. M. Abakumov, H. Perrot, J. M. Tarascon and O. Sel, ACS Nano, 2022, 16, 14907–14917 CrossRef CAS PubMed.
- I. Ledezma-Yanez and M. T. M. Koper, J. Electroanal. Chem., 2017, 793, 18–24 CrossRef CAS.
- P. Cao, Y. Sun and R. Gu, J. Raman Spectrosc., 2005, 36, 725–735 CrossRef CAS.
- I. Ledezma-Yanez, W. D. Z. Wallace, P. Sebastián-Pascual, V. Climent, J. M. Feliu and M. T. M. Koper, Nat. Energy, 2017, 2, 1–7 Search PubMed.
- M. Bier, J. Mars, H. Li and M. Mezger, Phys. Rev. E, 2017, 96, 1–10 CrossRef PubMed.
- M. C. O. Monteiro, F. Dattila, B. Hagedoorn, R. García-Muelas, N. López and M. T. M. Koper, Nat. Catal., 2021, 4, 654–662 CrossRef CAS.
- M. C. O. Monteiro, F. Dattila, N. López and M. T. M. Koper, J. Am. Chem. Soc., 2022, 144, 1589–1602 CrossRef CAS PubMed.
- A. Kopač Lautar, J. Bitenc, T. Rejec, R. Dominko, J.-S. Filhol and M.-L. Doublet, J. Am. Chem. Soc., 2020, 142, 5146–5153 CrossRef PubMed.
- K. G. Reeves, A. Serva, G. Jeanmairet and M. Salanne, Phys. Chem. Chem. Phys., 2020, 22, 10561–10568 RSC.
- Y. Kawamata and P. S. Baran, Joule, 2020, 4, 701–704 CrossRef.
| This journal is © The Royal Society of Chemistry 2023 |