
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDiscovery of reactive peptide inhibitors of human papillomavirus oncoprotein E6†
Xiyun
Ye‡
a,
Peiyuan
Zhang‡
 a,
Jason
Tao§
a,
John C. K.
Wang
b,
Amirhossein
Mafi
b,
Nathalie M.
Grob
a,
Anthony J.
Quartararo
a,
Hannah T.
Baddock
b,
Leanne J. G.
Chan
b,
Fiona E.
McAllister
b,
Ian
Foe
b,
Andrei
Loas
a,
Dan L.
Eaton
b,
Qi
Hao
b,
Aaron H.
Nile
*b and
Bradley L.
Pentelute
*acde
a,
Jason
Tao§
a,
John C. K.
Wang
b,
Amirhossein
Mafi
b,
Nathalie M.
Grob
a,
Anthony J.
Quartararo
a,
Hannah T.
Baddock
b,
Leanne J. G.
Chan
b,
Fiona E.
McAllister
b,
Ian
Foe
b,
Andrei
Loas
a,
Dan L.
Eaton
b,
Qi
Hao
b,
Aaron H.
Nile
*b and
Bradley L.
Pentelute
*acde
aDepartment of Chemistry, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, MA 02139, USA. E-mail: blp@mit.edu
bCalico Life Sciences LLC, 1170 Veterans Boulevard, South San Francisco, CA 94080, USA. E-mail: aaronnile@calicolabs.com
cThe Koch Institute for Integrative Cancer Research, Massachusetts Institute of Technology, 500 Main Street, Cambridge, MA 02142, USA
dCenter for Environmental Health Sciences, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, MA 02139, USA
eBroad Institute of MIT and Harvard, 415 Main Street, Cambridge, MA 02142, USA
First published on 25th October 2023
Abstract
Human papillomavirus (HPV) infections account for nearly all cervical cancer cases, which is the fourth most common cancer in women worldwide. High-risk variants, including HPV16, drive tumorigenesis in part by promoting the degradation of the tumor suppressor p53. This degradation is mediated by the HPV early protein 6 (E6), which recruits the E3 ubiquitin ligase E6AP and redirects its activity towards ubiquitinating p53. Targeting the protein interaction interface between HPV E6 and E6AP is a promising modality to mitigate HPV-mediated degradation of p53. In this study, we designed a covalent peptide inhibitor, termed reactide, that mimics the E6AP LXXLL binding motif by selectively targeting cysteine 58 in HPV16 E6 with quantitative conversion. This reactide provides a starting point in the development of covalent peptidomimetic inhibitors for intervention against HPV-driven cancers.
Introduction
High-risk forms of HPV are causative in multiple cancers, including cervical, vaginal, oropharyngeal, and potentially a subset of prostate cancers.1–4 Despite the introduction of HPV-directed vaccines over ten years ago, the lack of their widespread distribution, uptake by the general population and availability continues to make HPV positive (HPV+) cancers prevalent.4–7 Among ∼200 identified HPV strains, high-risk HPV16 and HPV18 are responsible for ∼75% of HPV-associated cervical cancers.8–11 Current treatment strategies of HPV cancers include radiation therapy,4 surgery,12 chemotherapy,13 monoclonal antibody (mAb)14 and checkpoint blockade.15–17 However, none of these approved options directly target HPV. The absence of this viral protein in uninfected tissues within the human proteome and its robust association with cancer makes it an attractive target for chemical intervention.HPV-encoded early protein 6 (E6) and early protein 7 (E7) are primary transforming viral proteins enhancing cancer cell proliferation and contributing to cancer progression.10,18 HPV E7 primarily binds and inactivates retinoblastoma protein (pRB) and related pocket proteins, p107 and p130, inducing their proteasome-dependent degradation and promoting cell cycle entry.19 Multiple host proteins interact with E6 via a leucine-rich LXXLL motif, including E6BP,20 IRF3,21 paxillin and tuberin;22 PDZ proteins such as MAGI-1;23 and other proteins such as p53, E6AP, MAML1, and p300/CBP.19,24 Nevertheless, the interaction of E6 with the E3 ubiquitin ligase, E6AP (encoded by the UBE3A gene), and p53 is thought to be a central transformative pathway of cell immortalization.25–28 E6 and E7 are active in different cell stages.18 E7 promotes cell entry into S phase,19 in turn, E6 prevents E7-induced apoptosis by degrading the apoptosis-inducing protein p53.29,30 HPV+ tumors typically harbor wild-type (WT) p53,31 as a result, silencing E6 can rescue p53 levels and initiate apoptosis in HPV+ cancer cell lines.32,33 Therefore, checkpoint networks are primed in HPV+ cells awaiting E6 disruption, providing support for its suitability as an oncology target.
HPV16 E6 (16E6) hijacks E6AP to form a complex with p53 that promotes ubiquitination of p53, leading to its subsequent proteasome-mediated degradation, while neither 16E6 nor E6AP interact with p53 alone.34–36 p53 mediates stress response, cell proliferation, and apoptosis, and its downregulation or mutation is a hallmark of carcinogenesis directly affecting efficacy of cancer therapy.37,38 It was reported that targeting the catalytic domain of the ubiquitin ligase E6AP (HECT domain) represses p53 ubiquitination in vitro.39 However, E6AP is a regulator of the proteostasis signaling network40 and has wide distribution patterns in humans.41 Mutations resulting in E6AP deficiency are linked to the Angelman syndrome, and there are potential associations with the Prader–Willi syndrome,42,43 diseases causing developmental defects. As such, targeting E6AP could result in on-target toxicity although acute inactivation has not been assessed. Given the potential toxicity associated with targeting E6AP and the lack of 16E6 in healthy human cells, targeting the viral 16E6 protein may provide a wide therapeutic index in HPV+ cancers to limit on-target side-effects.28
Efforts in the past decades have attempted to disrupt the 16E6/E6AP protein–protein interaction (PPI). Ribozymes and gene-silencing siRNA that reduce or remove the activity of the HPV E6 oncogene and its protein product have been shown to induce apoptosis in cancer cells.44–47 Polyhydroxy flavonoids48–50 display low micromolar E6 inhibition IC50 values and cytotoxicity in HPV+ cancer cells but have not been successful in clinical trials, possibly due to unclear structure–activity relationships (SAR), poor stability, off-target binding, and low specificity.27 Biomolecules, including E6-binding antibodies and mini-proteins with dissociation constants (KD) of 10–60 nM, have also been employed. However, their inhibitory effect is unsurprisingly hindered by poor cell penetration.51–53
The X-ray crystal structure of the ternary complex formed between HPV16 E6, an E6AP-derived LXXLL binding motif-containing peptide fused to MBP, and the p53 core domain was reported (PDB 4XR8, Fig. 1A).54 Investigation of the 16E6/E6AP interface revealed the main recognition domain as a peptide fragment of E6AP (residues 401–418).55 The peptide fragment contains a conserved LXXLL motif, critical for both peptide E6AP [401–418]56 and E6AP protein binding.55 The LXXLL motif identified on E6AP formed the basis of our starting point to design peptide inhibitors for 16E6.
 | ||
| Fig. 1 Rational design of E6AP-mimicking peptides. (A) Left: X-ray crystal structure of the ternary protein complex formed by 16E6 (white), MBP (yellow) fused to the E6AP LXXLL peptide (blue), and p53 (green). PDB 4XR8. Right: interaction interface of 16E6 and the LXXLL motif of E6AP. The E6AP LXXLL peptide binds the 16E6 hydrophobic groove and drives peptide recognition. (B) Rational design of a high-affinity E6 binding peptide. The N-terminal and C-terminal lysine side chain primary amines were modified with various small molecules listed in panel (C). (C) Chemical library of small molecules used for modifying E6AP-based peptides. Planar aromatic residues are in blue, hydrophobic residues in orange, other aromatic residues in black. (D) Table of N- and C-terminally modified E6AP peptide mimics. The apparent dissociation constant KD is measured by bio-layer interferometry (BLI) in competition mode, except for *KD measured by direct BLI. (E) BLI competition assay was used to measure the binding of 6′ and 6′-3L3A. Streptavidin tips were immobilized with 1-biotin and dipped into solutions of various concentrations of analyte and 16E6. (F) Direct BLI measurement of 6′-biotin, parameters are estimated to be: kon = 1.5 × 105 M−1 s−1, koff = 4.3 × 10−3 s−1, and KD = 3.0 ± 1.8 nM. | ||
Peptide mimics of E6AP are a promising strategy to overcome the challenges of non-specific binding of small molecules and poor penetration of large biomolecules.57 Peptides are of intermediate molecular weight between small molecules and proteins (2–5 kDa) and can be efficient at cell penetration while being endowed with high affinity and specificity to protein targets.58 A tri-cysteine peptide identified by phage display (KD = 118 nM) is prone to aggregation, poorly soluble and unable to compete E6AP-mimicking peptides.59–61 While an E6AP-mimicking peptide (KD = 4 μM)62 was reported to target 16E6, it exhibits insufficient affinity toward 16E6 to inhibit the PPI, likely due to a rapid koff rate.59–61
The addition of irreversible crosslinking groups is a suitable strategy for developing inhibitors of ‘undruggable’ proteins. Covalent inhibition, with koff values close to zero, is an effective strategy for overcoming resistance in the context of HIV-1 protease inhibitors,63 where high koff values are associated with drug resistance.64,65 Acrylamide-like warheads are popular in FDA-approved drugs owing to their mild and selective reactivity profile.66–68 Compared to other highly reactive electrophiles such as chloroacetamides, acrylamide warheads balance stability, reactivity, and substrate selectivity. Warhead-containing peptide inhibitors bind-and-react with targets to establish a covalent bond for irreversible inhibition. For example, the acrylamide modified BimBH3 peptide targets Cys55 on the Bcl-2 protein,69 and acrylamide-modified MCL-1 binding peptides crosslink to Cys55.69 These reactive peptides have multiple advantages compared to traditional peptide inhibitors, typically addressing potency, selectivity, toxicity, dosing, and target scope.66 Covalent small molecule inhibitors are also exemplified in this regard.
We developed high-affinity 16E6-binding peptides with single digit nanomolar affinity based on the LXXLL binding motif in E6AP, by introducing dehydroalanine (Dha) warheads to irreversibly disrupt the 16E6/E6AP interaction. The N- and C-termini of these peptides were appended to various chemical moieties in order to improve 16E6 binding affinity by nearly 1000-fold from ∼2.3 μM to 2.7 nM. The peptides were designed to crosslink with 16E6 at Cys58 located within the hydrophobic cavity of 16E6 that mediates engagement with the E6AP LXXLL peptide through a Dha warhead. We termed these warhead-containing peptide binders “reactides”. The engineered reactides form covalent peptide–16E6 conjugates, irreversibly binding the 16E6 pocket and preventing E6AP association. Based on our findings, we envision reactides as a possible strategy for further development of reactive variants to inhibit the HPV16 E6 protein.
Results
Rational design of high-affinity peptide binders to 16E6
As a starting point for our study, we examined the X-ray crystal structure of the ternary complex formed by 16E6, p53, and the LXXLL peptide of E6AP (PDB 4XR8). We selected a 17-mer peptide (1; sequence: IPESSELTLQELLGEER, Fig. 1 and S1A†) that covers residues 401–417 of E6AP and includes the LXXLL motif, as a template for affinity optimization.8 The binding affinity of peptide 1 was assessed by bio-layer interferometry (BLI) using recombinant MBP–16E64C4S protein (MBP–16E6), which contains a maltose-binding protein (MBP) tag and four cysteine to serine mutations to stabilize and solubilize HPV16 E6.70 The BLI binding assay was run in competition mode by immobilizing biotinylated 1 (1-biotin) onto streptavidin biosensor tips and immersing them across a serial dilution of unlabeled 1 until equilibrium.71 Peptide 1 competed MBP–16E6 binding to 1-biotin, revealing a competition KD of ∼2.3 μM (Fig. S1†). The competition KD values reported for the peptide variants discussed below were determined by BLI in a similar manner.While investigating our E6AP peptide designs, we serendipitously observed an increased binding affinity with N-terminal fluorescein isothiocyanate (FITC)-labeled peptide 1 (N-FITC) compared to the C-terminal FITC-labeled peptide 1 (C-FITC). The resulting N-FITC peptide showed a competition KD of 95 nM, while C-FITC peptide 1 had a competition KD of 640 nM. To identify critical residues, we performed alanine (Ala) scanning and synthesized 17 single Ala mutants of N-FITC, peptides A1 to A17, respectively (Table S1†). The alanine scanning identified hotspot residues Leu9, Leu12, and Leu13 to be critical for binding, as the A9, A12, and A13 peptides did not show measurable binding to MBP–16E6. This observation aligns with L12A and L13A single mutated peptides, which showed decreased E6 binding.72 Therefore, peptides simultaneously containing the L9A, L12A, and L13A mutations (or 3L3A triple mutants) were used as negative control peptides in this work.
We observe a 900-fold improvement in binding affinity to MBP–16E6 with N-terminal FITC modifications. To investigate the structure–activity relationships (SAR) contributing to the binding enhancement of terminal FITC modifications, we replaced FITC with a panel of small molecules at the C- or N-terminus of parent peptide 1 (Fig. 1B and C and Tables S2 and S3†). From 36 synthesized peptides, 8 terminally modified peptides displayed KD < 70 nM against MBP–16E6 (peptides N1, N2, N7, N9, N10, N19, C4, C9, and C10). At the N-terminus, planar and tricyclic arenes enhance binding by 50 to 250-fold relative to unmodified peptide 1, while bulky, hydrophobic, or aliphatic small molecules modestly improved affinity by less than 5-fold (Fig. 1C). The position of fluorene ring substitution has no effect on N- or C-terminal modifications. At the N-terminus, the largest ∼250-fold increase in binding affinity was observed with fluorene modification (N1). Removal of a phenyl ring from fluorene (N1, KD of 10 nM) to indane (N3, KD of 425 nM) decreases the binding by ∼42-fold. Compared to fluorene-modified N1, biphenyl-containing N21 (KD of 1412 nM) decreases the binding by ∼27-fold. These observations suggest that N-terminal planar tricyclic aromatic scaffolds are favorable motifs to enhance binding of parent peptide 1 to 16E6 (Fig. S2A†). The greatest improvements at the C-terminus, from 30- to 111-fold, were found with planar, tricyclic, and polyarenes, while bulky, non-conjugated aromatic or aliphatic small molecules did not significantly improve the binding compared to parent peptide 1 (Fig. 1C and Table S3†). The best performing C-terminal modification was anthracene, which showed a competition KD value of 22 nM, enhancing binding affinity to MBP–16E6 by ∼111-fold compared to the parent peptide 1 (Fig. S2B†).
We combined the N- and C-terminal modifications displaying the highest 16E6 binding affinity to generate double-modified peptide binders. These binders exhibited affinity improvements over single modifications, likely due to synergistic effects arising from binding features of the two terminal modifications (Fig. 1D). Peptide 6, containing N-terminal fluorene and C-terminal anthracene, showed the lowest competition KD of 3.7 nM to MBP–16E6 and was selected as our lead for further modifications. Additional optimization was achieved by substituting Ser5 to Ala, as previous Ala scan studies revealed a 2-fold increase in binding affinity (ESI Table S1†), resulting in peptide 6′ with a competition KD of 2.7 nM. To investigate the binding affinity and selectivity of 6′, biotinylated peptide 6′-biotin and its 3 L3A derivative 6′-3L3A were synthesized. 6′-3L3A displayed no observable binding towards MBP–16E6 (Fig. 1E). 6′-Biotin showed a direct binding KD of 3.0 ± 1.8 nM to MBP–16E6 (Fig. 1F). These results demonstrate that 6′ binding to MBP–16E6 is sequence-specific and requires the tri-leucine hotspot motif LXXLL. To assess target selectivity, 6′-biotin was immobilized and its affinity toward the unrelated proteins murine double minute 2 (MDM2) and an LXXLL motif-binding protein thyroid hormone receptor alpha (THRA)73 was measured by BLI. No appreciable signal during the association or dissociation steps was observed by BLI (Fig. S3B and C†), indicating that peptide 6′ selectively binds MBP–16E6.
Development of a dehydroalanine-modified 16E6 binding peptide that crosslinks to MBP–16E6
Irreversible covalent inhibition is an effective strategy to increase drug potency and selectivity toward inhibiting ‘undruggable’ targets.74 The binding interface of 16E6 contains the nucleophilic Cys58 residue in proximity to the E6AP LXXLL peptide binding pocket (Fig. 2A). To exploit this finding, we synthesized reactides E1–E11 based on E0, a truncated version of peptide 1, where a Cys-reactive acrylamide replaced Gly9, Glu10, or Glu10 of E0 (Fig. 2B and S4A†). We tested multiple electrophiles including phenylacrylamide (Phacr), acrylamide (Acr), propiolamide (Ppa), and Dha (Fig. S4B†).75 Liquid chromatography-mass spectrometry (LC-MS) monitored crosslinking between MBP–16E6 protein and the reactides, and the area under the total ion peak was used to estimate crosslinking yield. Gly9 was found to be the closest residue to Cys58 and its substitution with Dha gave the highest crosslinking yield of 74%, and it was thus chosen as our lead reactide scaffold E3 (Fig. 2C).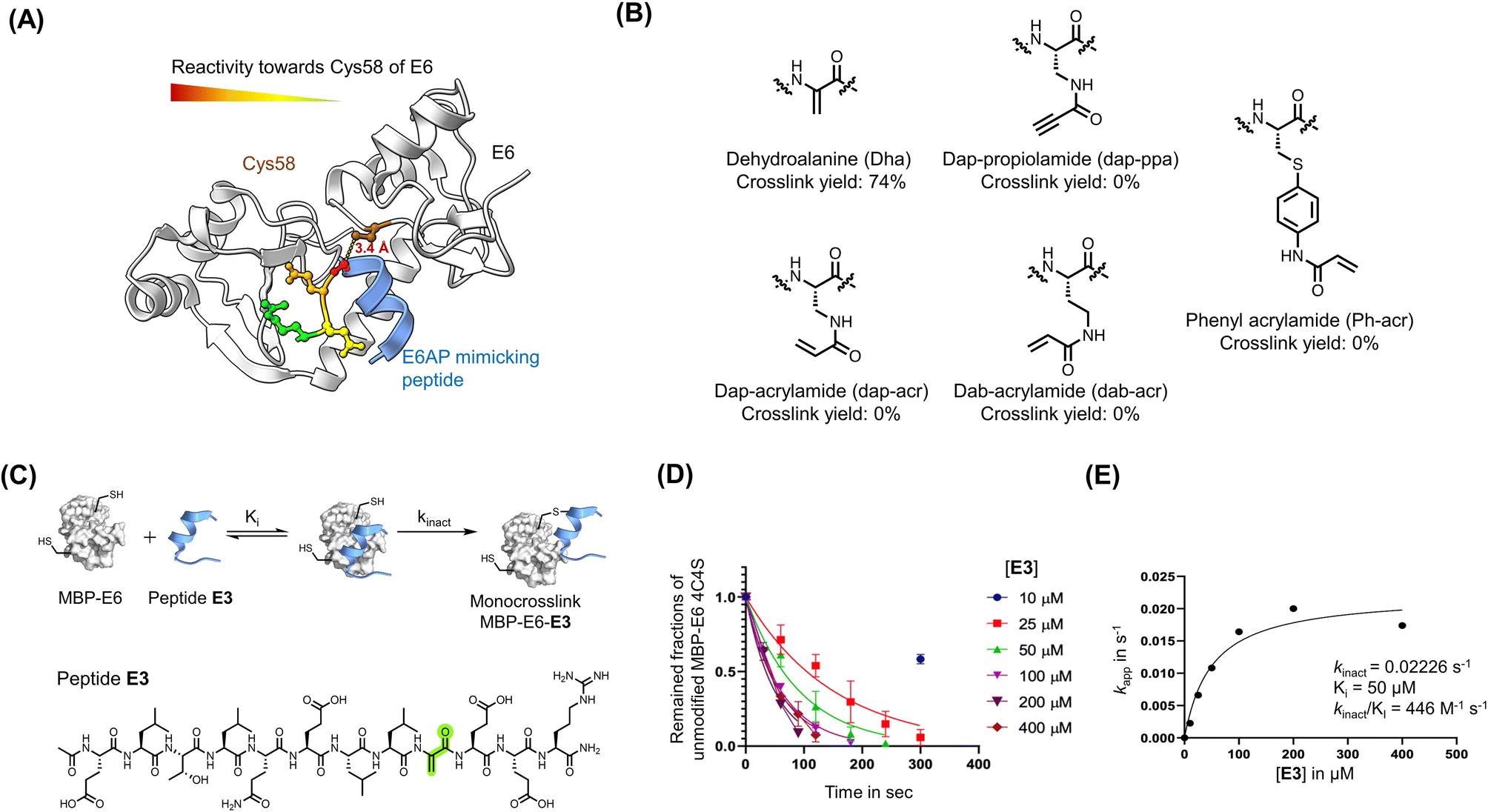 | ||
| Fig. 2 Dha was installed to generate reactive peptides. (A) Analysis of the main binding interface of 16E6 and E6AP. Cys58 of 16E6 is highlighted in brown. Residues of E6AP were colored based on their measured reactivity towards Cys58 of 16E6: Gly9 in red, Glu10 in orange, Glu11 in yellow, and Arg12 in Green. (B) Structures of electrophiles. (C) A scheme of the bind-and-react strategy. Structure of peptide E3 containing a Dha electrophile highlighted in green. (D) Apparent kinetic constant Kapp was calculated from a kinetic crosslinking study, where 50 nM of MBP–16E6 was incubated with different concentrations of E3. (E) A series of Kapp values were plotted against the corresponding E3 concentration to estimate kinact and Ki. | ||
Kinetic crosslinking studies were performed with E3 to estimate the binding constant Ki and first-order rate constant (kinact) involved in the two-stage bind-and-react strategy. Crosslinking yield was monitored over time by LC-MS (Fig. 2D and E). Kinetic parameters were estimated assuming a steady-state approximation, and a kinact value of 0.022 s−1 and Ki value of 50 μM were obtained. The kinact/Ki ratio was calculated to be 446 M−1 s−1, indicating that improvements in binding affinity will produce a more efficient covalent inhibitor.66 For comparison, the FDA-approved small molecular drug cysteine-targeted nirmatrelvir showed a kinact/Ki value of 55 mM−1 s−1.76
First, we installed Dha onto peptide 6′ to obtain reactide 7, which has a net charge of −3 that may be detrimental to cell permeability.77,78 To increase positive charge, improve solubility, retain binding affinity to 16E6, and further reduce molecular weight, we performed residue substitutions and truncations on 7. The LXXLL proximal RRNKK [417–421] segment from the E6AP protein was appended to the C-terminus of reactide 7 to increase the total charge from −3 to −1 (reactide 8). Next, Glu3Gln, Glu15Gln, and Glu16Ala mutations were applied to increase the total charge to +2, resulting in peptides 9 and 10. To reduce molecular weight, Ala13, Gln14, and Asn19 residues were omitted from peptide 10 to generate 11, 12, and 13. Reactides were evaluated by the BLI competition assay against 1-biotin to estimate their inhibitory constant, which we report here as an apparent Ki value. Peptides 8–13 showed comparable binding affinity to the parent reactide 7, with an apparent Ki ranging from 11 nM to 39 nM (Fig. S5†). Reactide 13 was selected for further characterization since it has the smallest molecular weight, highest net charge, and relatively low apparent Ki = 17 ± 3.9 nM (Fig. 3A–C). The negative control derivative of 13, (13-3L3A), showed no observable binding to MBP–16E6. Therefore, 13-3L3A was used as a negative control for subsequent peptides. A kinetic crosslinking study was performed with 13 and revealed a kinact of 0.027 s−1, a Ki value of 120 nM and a kinact/Ki ratio of 270 mM−1 s−1 (Fig. 3D and E). Reactide 13 represents a 504-fold improvement over E3.
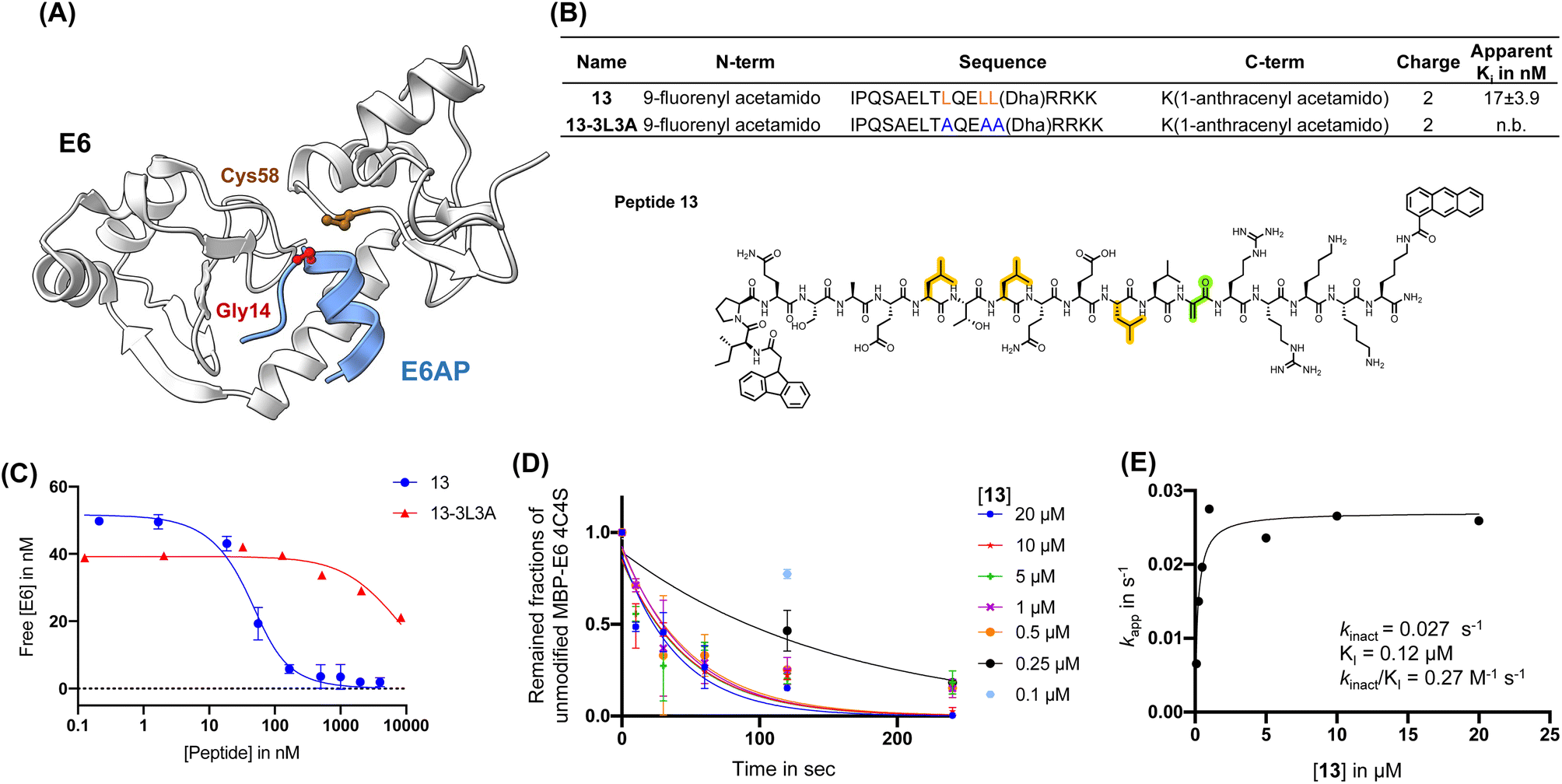 | ||
| Fig. 3 Affinity matured 16E6-binding peptide is endowed with improved reactivity. (A) Main binding interface of 16E6 and E6AP. Cys58 of 16E6 is highlighted in brown and targeted by an electrophile substituted at Gly14. (B) Sequence table of E6AP-mimicking 13 and 13-3L3A (control peptide). Apparent Ki is determined by BLI (n.b., non-binding). Peptides were in competition with immobilized 1-biotin following a 30 min incubation with 16E6. Dha: dehydroalanine. Structure of 13, Dha (green), tri-leucine (orange). (C) BLI competition assay measurement of 13 and 13-3L3A estimated apparent Ki in nM. (D) Apparent kinetic constant Kapp was calculated from a kinetic crosslinking study, where 10 nM of MBP–16E6 was incubated with different concentrations of 13. (E) Kapp was plotted against the corresponding concentrations of 13 to estimate kinact and Ki. | ||
Reactide 13 selectively crosslinks to MBP–16E6 in PBS
Reactide 13 uses a two-stage bind-and-react strategy to covalently crosslink to MBP–16E6 and block the E6AP binding site. When 13 (3 μM) and MBP–16E6 (1 μM) were incubated in PBS at 37 °C for 2 h, protein deconvolution mass spectra revealed >99% mono-crosslinking of 13 to MBP–16E6 (Fig. 4A). This was achieved despite the presence of 10 cysteine residues on MBP–16E6. To confirm site-selectivity, 13 was incubated with the MBP–16E6 mutant C58S for 12 h in PBS at 37 °C. No crosslinking was observed between 13 and MBP–16E6 C58S (Fig. 4B), which supports Cys58 as the sole site of MBP–16E6 modification. 13 showed no crosslink to THRA (Fig. S6†). Additionally, the negative control 13-3L3A showed no observable crosslinking to MBP–16E6 protein (Fig. 4C), supporting the selectivity of 13. To further investigate selectivity of 13, we synthesized 13-biotin which also demonstrated mono-crosslinking to MBP–16E6 (Fig. S7B†). The direct binding affinity of 13-biotin to MBP–16E6 was measured by BLI as 1.3 ± 0.6 nM (Fig. S7C†). 13-Biotin showed no observable association to MDM2 and THRA (Fig. S7D and E†). Collectively, these results indicate reactide 13 selectively binds and crosslinks to MBP–16E6 in PBS.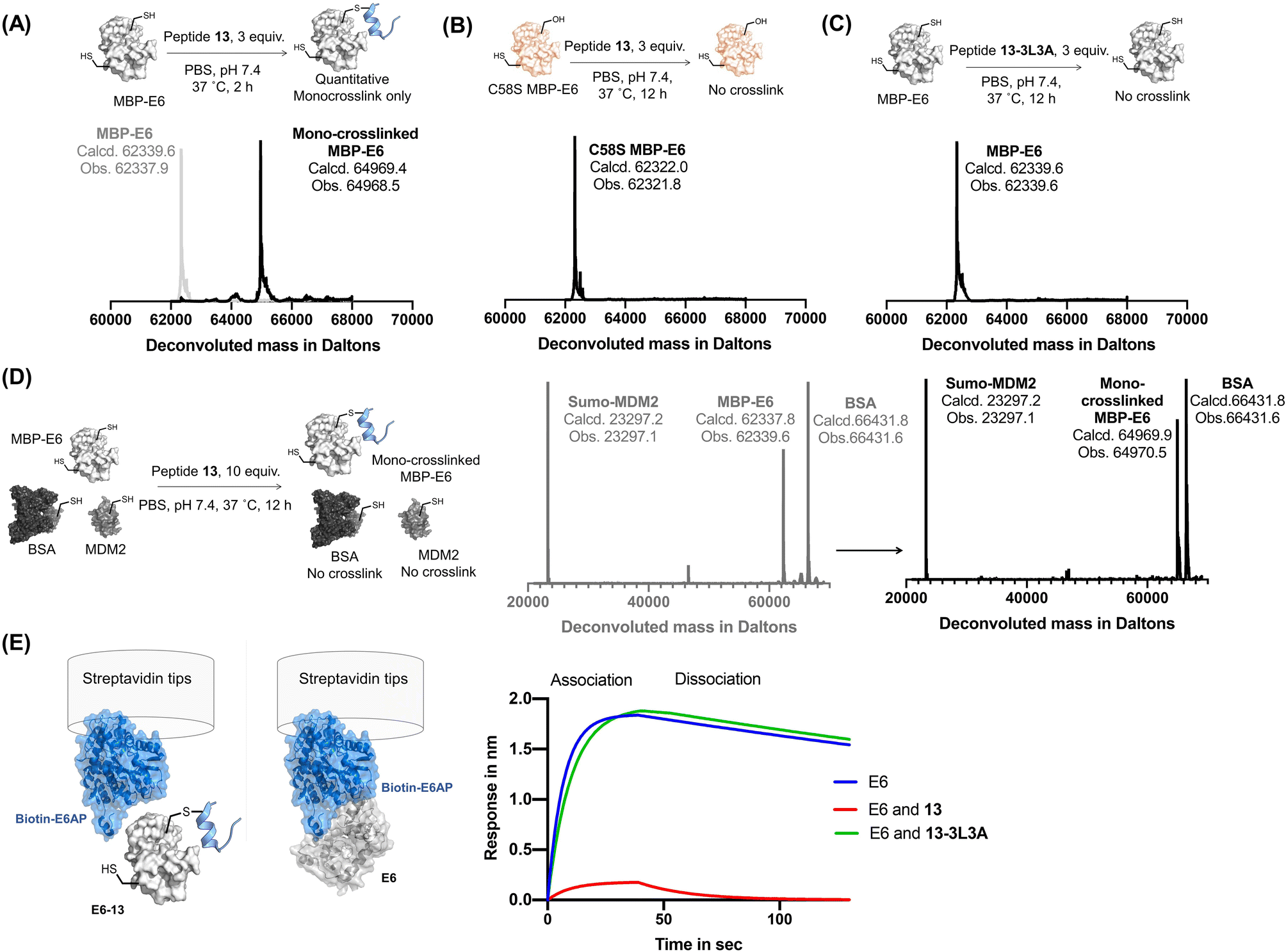 | ||
| Fig. 4 Reactive peptides selectively crosslink to MBP-16E6. (A) Crosslinking reaction of MBP–16E6 (1 μM) and reactide 13 (3 μM). Quantitative mono-crosslinked MBP–16E6–13 was observed at 2 h. (B) Crosslinking of MBP–16E6 C58S (1 μM) and peptide 13 (3 μM), no appreciable amount of reaction product was observed after 12 h. (C) Crosslinking of MBP–16E6 (1 μM) and peptide 13-3L3A (3 μM), no appreciable amount of reaction product was observed after 12 h. (D) Protein-selective intermolecular crosslinking of MBP–16E6 in a protein mixture. Only 16E6 was modified, as indicated by the LC-MS analysis. (E) BLI assay determined 16E6/E6AP complex formation. 1 μM of biotin–E6AP protein was immobilized on the streptavidin tip and dipped into 1 μM of MBP–16E6 only (blue), MBP–16E6 mixed with 1 μM of 13 (red) or MBP–16E6 mixed with 1 μM of 13-3L3A (green). | ||
Reactide 13 selectively crosslinks to MBP–16E6 in a protein mixture
To investigate whether 13 selectively crosslink to 16E6 in a protein mixture, we monitored modification of SUMO-MDM225-109 which contains one cysteine and bovine serum albumin (BSA) which has 35 cysteine residues. A mixture of the three proteins, 16E6 (1 μM), BSA (1 μM), and SUMO-MDM225-109 (1 μM) was incubated with 13 (10 μM) at 37 °C for 12 h and resolved by LC-MS (Fig. 4D). The results indicate 16E6 is mono-crosslinked with >95% conversion with no observable modifications to BSA or MDM2. These results further demonstrate crosslinking of 13 is specific to MBP–16E6 and depends on the hotspot motif LXXLL.To investigate selectivity of reactide 13 in the context of the proteome, we first incubated the HPV negative lysate from HT1080 cells supplemented with recombinant MBP–16E6 or MBP–16E6 C58S then spiked in 10 μM of 13-TAMRA at 4 °C overnight. After resolution of the lysate by SDS-PAGE, the gel was scanned in the TAMRA channel which displayed selective covalent binding for MBP–16E6 over MBP–16E6 C58S (Fig. S8†). Despite this high concentration used in the assay (>500-fold higher than 13's binding affinity to 16E6), only a modest degree of fluorescent signal was distributed across the proteome with a long exposure time suggesting that 13-TAMRA has a minor degree of non-specific covalent modifications (Fig. S8B†). As an orthogonal approach, 13-biotin (10 μM) or 13-3L3A-biotin (10 μM) were incubated with 50 μg of lysate and resolved by SDS-PAGE followed by western blot analysis probing with fluorescently labeled streptavidin which recognizes biotin (Fig. S9†). A signal at ∼60 kDa corresponding to MBP–16E6 was detected in the active 13-biotin reactide lane, but not in any of the other conditions, supporting 13-biotin's selectivity (Fig. S9A–C†). However, general background levels of signal were also observed in both 13-biotin and 13-3L3A-biotin at 10 μM, both of which contain the reactive Dha warhead, suggesting that 13-biotin and 13-3L3A-biotin at 10 μM have a modest degree of non-specific covalent modifications. Background signal was not observed in peptide-free conditions suggesting that the signal originates from the peptide (Fig. S9D†).
To gain insight into the identity of the reactome, we incubated either 13-biotin, 13-3L3A-biotin (5 μM) or DMSO in 2 mg of HPV16+ CaSki or HPV-HT1080 lysate overnight at 4 °C. Subsequently, we enriched the biotinylated peptides using streptavidin-conjugated magnetic beads followed by trypsin digestion and analysis by LC-MS/MS to identify peptide-associated proteins. Approximately 372 proteins were identified across all tested conditions in both the HT1080 and CaSki cells, with 13-biotin showing the highest magnitude of enrichment (Fig. S10A and ESI File S1†). Subsequently, we conducted pairwise analysis across the conditions for both HT1080 (Fig. S10B–D†) or HPV16+ CaSki cells (Fig. S10E–G†) which did not reveal clear enrichment patterns; however, a major signal was observed for Rpn1 which has been reported to interact with E6AP.79 Of note, we did not identify HPV16 E6 in CaSki cells which we attribute to the low abundance of this protein or inaccessibility of the peptide binding site in a cellular environment. Nevertheless, 13-biotin or 13-3L3A-biotin had only modest nonspecific enrichment of other proteins in both cell lines. Taken together, these data suggest that 13-biotin recognizes supplemented MBP–16E6 but not MBP–16E6 C58S with a minor to modest degree of non-specific conjugation to other proteins from lysate.
Disruption of 16E6/E6AP interaction by reactide 13
To assess the disruption of 16E6/E6AP interaction by reactide 13, we set up an 16E6/E6AP BLI binding assay by immobilizing biotinylated E6AP protein onto streptavidin tips. The 16E6/E6AP interaction was evaluated as the response (in nm) during the association step when dipped into recombinant MBP–16E6 solution (1 μM). MBP–16E6 crosslinked with reactide 13 had a significantly decreased BLI response signal to the immobilized E6AP protein, while the control reactide 13-3L3A had no impact (Fig. 4E). The decrease in BLI response signal indicates that the 16E6–13 conjugate does not bind E6AP, as its E6AP binding pocket was occupied by 13.Molecular modeling of the 16E6 and peptide-13 complex
Molecular modeling was conducted to understand the mechanism by which reactide 13 disrupts the 16E6/E6AP interaction using molecular docking and molecular dynamics (MD) simulations. The MBP tag may affect the conformation of the isolated E6AP-based peptide from the ternary complex structure with 16E6 and p53 (PDB 4XR8).8 Therefore, we removed the MBP tag and p53 protein from the structure and ran a 1.1 μs simulation of the 16E6-bound E6AP-LXXLL peptide. The peptide remained stable with an r.m.s.d. of 0.4, indicating α-helical features (Fig. S11†). This enabled us to build the model of 13 by incorporating the flexible N- and C-terminal sequences into the alpha helix of the E6AP LXXLL peptide and perform extensive conformational sampling calculations to optimize the reactide 13 model (Fig. S12B†). The covalent bond formed between 13 and 16E6 Cys58 was not considered when initiating the molecular docking to avoid any biases in the calculations. The docking result suggested that the primary driver of 13 binding to 16E6 are electrostatic and van der Waals interactions (Fig. 5A). By refining the top candidates obtained from the docking calculations using MD simulations (Fig. S12C†), we observed that the Dha (C atoms) resides within 3.7 Å from Cys58 (S atoms), confirming optimal placement for 13 to cross-link with 16E6. The thioether bond between 16E6 and Dha acts as a covalent lock, securing 13 at the 16E6/E6AP interface. Computational modeling confirms the importance of this bond as shown in Fig. S12D and S13.† Our trajectory r.m.s.d. analysis (Fig. 5B) for the 16E6 and 13 complex and 13 alone indicates that the covalent bond between Cys58 and Dha is instrumental in stabilizing 13 and effectively restricts the flexibility of the E6AP-LXXLL core sequence with an r.m.s.d. of 1.5 ± 0.3 Å. Taken together, our molecular simulations reveal that reactide 13 occupies the same position as the parent E6AP LXXLL peptide (r.m.s.d. = 1.9 Å, measured for Cα atoms), leading to the blocking of E6AP/16E6 binding (Fig. 5C and S14†). | ||
Fig. 5 Molecular modeling of the E6–peptide 13 conjugate. (A) Structural representation of 16E6 (white) bound to 13 (blue) featuring the Dha warhead (red), obtained through ∼120 ns MD simulation. The structure represents the averaged configurations of 13 within calculated clusters (see methods). (B) Trajectory analysis of root mean squared differences (r.m.s.d.) during MD simulations, showing stability and modest conformational changes of the complex. (C) Comparison of molecular modeling of the 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :16E6 complex with the X-ray structure of 16E6 (coral)–E6AP LXXLL peptide (yellow), p53 (plum) complex (PDB ID 4XR8). All r.m.s.d. were calculated for the Cα atoms. All structures were compared to the initial structure. (D) Intermolecular interactions between 16E6 (white) and 13 (blue), identified and analyzed from the MD simulation. Interactions shown in (D) are persistent during the ∼120 ns MD simulation. Dotted yellow lines represent hydrogen bonding. (E) Covered surfaces by the N-terminus within 5 Å: patch 1 in black [V38, Y39, C40, K41, R62, E63], patch 2 in yellow [C73, F76, Y77, I80, Y83, R84, H85, R136]. Left: E6 and 13 N-term detailed interactions. (F) Covered surfaces by the C-terminus within 5 Å: patch 1 [M8, F9, Q10, D11, P12, Q13, E14, R15, P16, R17, K18, L19, P20, Q21, D25]. Patch 2 in yellow [A53, D56, R55, L57, C58*]. Patch 3 in purple [Y99, K101, D105, L107, I108, R109, C110, C113, Q114, K115, P116, L117, R136, R138, W139, T140]. Left: E6 and C-terminal 13 detailed interactions. :16E6 complex with the X-ray structure of 16E6 (coral)–E6AP LXXLL peptide (yellow), p53 (plum) complex (PDB ID 4XR8). All r.m.s.d. were calculated for the Cα atoms. All structures were compared to the initial structure. (D) Intermolecular interactions between 16E6 (white) and 13 (blue), identified and analyzed from the MD simulation. Interactions shown in (D) are persistent during the ∼120 ns MD simulation. Dotted yellow lines represent hydrogen bonding. (E) Covered surfaces by the N-terminus within 5 Å: patch 1 in black [V38, Y39, C40, K41, R62, E63], patch 2 in yellow [C73, F76, Y77, I80, Y83, R84, H85, R136]. Left: E6 and 13 N-term detailed interactions. (F) Covered surfaces by the C-terminus within 5 Å: patch 1 [M8, F9, Q10, D11, P12, Q13, E14, R15, P16, R17, K18, L19, P20, Q21, D25]. Patch 2 in yellow [A53, D56, R55, L57, C58*]. Patch 3 in purple [Y99, K101, D105, L107, I108, R109, C110, C113, Q114, K115, P116, L117, R136, R138, W139, T140]. Left: E6 and C-terminal 13 detailed interactions. | ||
The modeling suggests important side chain interactions between 13 and 16E6 basic residues, including E6AP Glu6/16E6 Arg138 and E6AP Glu11/16E6 Arg11. 16E6 Arg138 is crucial as Arg138 mutation reduces E6AP recruitment and interactions substantially.80 In addition, 16E6 Arg17 forms a salt bridge with the C-terminus of the E6AP core peptide. The Glu6 carboxylate group is essential for 13 binding and establishes an additional network of hydrogen bonding with Ser81 and His85 from 16E6 (Fig. 5D). 16E6 Arg136 forms a hydrogen bond with Gln10 in the E6AP core. The keystone 16E6 Arg109 interacts with Leu13 through hydrophobic interactions, consistent with crystallographic observations.54 Notably, the alanine substitutions of both Arg13654 and Arg10980 led to impaired peptide binding, highlighting their crucial roles in stabilizing the 16E6/E6AP complex, though R136 exhibited conformational disorder54 and was oriented away from the peptide towards MBP. Collectively, peptide 13 targets all residues appearing in the binding pocket of E6 to disrupt the binding interface of 16E6 and E6AP.
The N- and C-terminal chemical moieties are located on flexible loops and dynamically bind to 16E6, making it challenging to identify static interactions similar to those found for the core LXXLL motif. However, despite their dynamic binding, the N- and C-terminal chemical moieties still interact with important surfaces of 16E6. Owing to their substantial interactions with the 16E6 surface, the N- and C-terminal chemical moieties likely enhance the peptide binding affinity (Fig. 5E and F).
Discussion
The affinity of a peptide binder to MBP–16E6 was improved from ∼2.3 μM to ∼2.7 nM, a nearly 900-fold improvement, by modifying its termini with chemical moieties. We and others have observed binding improvement by terminal fluorescein labeling. For example, Frank et al. reported an unexpected binding improvement with a fluorescein-labeled peptide inhibitor of replication protein A.81 Similarly, Torner et al. reported that tryptophan addition to the C-terminus of MDM2-binding peptide PMI has increased binding by 50-fold and is likely to engage a secondary binding pocket.82 Likewise, the terminal small molecule modifications on our E6AP-mimicking peptides have improved binding affinity, with retention of sequence specificity and selectivity for the E6AP/16E6 binding groove. Upon peptide anchoring to the binding groove through LXXLL residues, chemical moieties bearing arenes may adhere to adjacent surface patches to enhance binding. Although the detailed mechanism of interaction of the added aromatic moieties in the 16E6 binding groove remains to be determined, the observations reported in this work offer insights toward strategies of improving peptide binding affinity.The binding of 6′ and 13 to MBP–16E6 is sequence-specific, relying on the hotspot motif LXXLL. We conducted tests to confirm this specificity using the unrelated proteins MDM2 and the LXXLL motif-binding protein THRA. While the BLI signal in the latter studies was minimal, suggesting selective binding of peptides to MBP–16E6, there was a slight, yet detectable response with THRA at higher concentrations. It's worth acknowledging that the LXXLL motifs are prevalent in numerous nuclear receptor co-activators and repressors. This prevalence creates a complex landscape of potential interactions that should be explored further.
Despite increasing numbers of reports describing peptide-based covalent inhibitors, the strategy is under-exploited to target challenging protein–protein interactions.83 One major drawback for this strategy is off-target crosslinking66,68 which can be mitigated through a bind-and-react strategy. With this approach, the risks of non-specific crosslinking and non-selective inhibition are reduced through specific binding of the base peptide sequence which brings the reactive warhead and its target residue in close proximity. This strategy may allow for the use of less reactive groups to alleviate non-selective modification.
Dha is the simplest dehydroamino acid and is found in some microbial peptides. Due to its electrophilic nature and lack of geometric isomers resulting from the methylidene group, it has been utilized as a reactive probe targeting enzymatic mechanisms.84–88 To our knowledge, we exploit for the first-time Dha as an electrophilic warhead to generate a reactide targeting a cancer-relevant PPI. Reactivity of the reactide was fine-tuned by adjusting the electrophile and distance between the warhead and peptide backbone. Optimal crosslinking efficiency was achieved with Dha placed directly onto the peptide backbone to fit in the tight binding pocket at the target protein surface containing Cys58, as suggested by the co-crystal structure. The reactide is stable and not reactive to 0.4 mM free Cys and GST in cell media (Fig. S15†).
Although it is desirable to inactivate all pathogenic HPV subtypes with Cys58-targeted reactides, only a subset contain a cysteine within the LXXLL binding motif (Fig. S16†). Among high-risk types, only HPV35, HPV45 and HPV16 contain Cys58. Further research into effective HPV targeting sites should expand the window of opportunity for HPV+ diseases.
Conclusions
In this study, we investigated the inhibition of HPV16 E6 by utilizing peptides that mimic the native E6AP binding scaffold. Our findings from alanine scanning mutagenesis indicate that binding depends on the LXXLL sequence motif. Further affinity maturation with terminal small molecular chemical moieties endowed peptides with enhanced binding affinity towards 16E6 while maintaining stability and selectivity. Using these matured peptides, we designed high-affinity, selective, and potent peptide-based covalent inhibitors targeting the 16E6 oncoprotein using a cysteine-reactive acrylamide warhead. We demonstrate reactide 13 as an efficient and selective crosslinking tool and report the first covalent peptide for irreversible inhibition of HPV16 E6. As a result of the high potency of peptide 13, we anticipate that a bind-and-react strategy can be applied more broadly in the development of irreversible covalent peptide inhibitors. Continued advancement in covalent chemistry compatible with solid-phase peptide synthesis and biological display methods are expected to expand substrate scope and optimize crosslinking efficiency. This opens opportunities for the design of potent and selective inhibitors across a wide range of biological targets including challenging protein–protein interactions.Challenges remain for the development of reactides with covalent warheads, including peptide stability in a biological context, bioavailability, renal clearance, and biological barrier penetration properties. Possible solutions include the incorporation of unnatural amino acids, albumin-binding modifications, and cyclization methods.57 Further investigation is warranted into the cellular response, inhibitory mechanism, and cell penetration properties of this new class of 16E6 reactides. Nevertheless, our studies establish a foundation for the next generation of HPV-targeted therapeutics and put forward design principles to utilize in bind-and-react strategies for other high-risk HPV proteins and a broad variety of other oncogenic proteins.
Data availability
All data are available in the main text or the ESI.†Author contributions
B. L. P., A. H. N., Q. H. and X. Y. conceptualized the research; X. Y., P. Z., J. T., H. T. B., L. J. G. C., J. C. K. W. and A. M. performed experiments; N. M. G., A. J. Q., H. T. B., F. E. M., I. F., A. L., D. L. E., Q. H., A. H. N. and B. L. P. assisted with the design of experiments; X. Y. and P. Z. wrote the manuscript with input from all authors.Conflicts of interest
B. L. P. is a co-founder and/or member of the scientific advisory board of several companies focusing on the development of protein and peptide therapeutics. J. C. K. W., A. M., H. T. B., L. J. G. C., F. E. M., I. F., D. L. E., Q. H. and A. H. N. are employees of Calico Life Sciences LLC. A provisional patent disclosure was filed regarding the methodology and compounds described in this study.Acknowledgements
Calico Life Sciences LLC (to B. L. P.) provided financial support for this work.References
- I. Lombard, A. Vincent-Salomon, P. Validire, B. Zafrani, A. de la Rochefordière, K. Clough, M. Favre, P. Pouillart and X. Sastre-Garau, Human Papillomavirus Genotype as a Major Determinant of the Course of Cervical Cancer, J. Clin. Oncol., 1998, 16(8), 2613–2619, DOI:10.1200/JCO.1998.16.8.2613.
- A. Pal and R. Kundu, Human Papillomavirus E6 and E7: The Cervical Cancer Hallmarks and Targets for Therapy, Front. Microbiol., 2020, 10, 3116, DOI:10.3389/fmicb.2019.03116.
- S. V. Graham, Human Papillomavirus: Gene Expression, Regulation and Prospects for Novel Diagnostic Methods and Antiviral Therapies, Future Microbiol., 2010, 5(10), 1493–1506, DOI:10.2217/fmb.10.107.
- M. Schiffman, J. Doorbar, N. Wentzensen, S. De Sanjosé, C. Fakhry, B. J. Monk, M. A. Stanley and S. Franceschi, Carcinogenic Human Papillomavirus Infection, Nat. Rev. Dis. Primers, 2016, 2(1), 16086, DOI:10.1038/nrdp.2016.86.
- S. I. Pai and W. H. Westra, Molecular Pathology of Head and Neck Cancer: Implications for Diagnosis, Prognosis, and Treatment, Annu. Rev. Pathol., 2009, 4, 49–70, DOI:10.1146/annurev.pathol.4.110807.092158.
- R. Landy and A. K. Chaturvedi, HPV16 E6 Seropositivity and Oropharyngeal Cancer: Marker of Exposure, Risk, or Disease?, EBioMedicine, 2021, 103190, DOI:10.1016/j.ebiom.2020.103190.
- N. Brenner, A. J. Mentzer, M. Hill, R. Almond, N. Allen, M. Pawlita and T. Waterboer, Characterization of Human Papillomavirus (HPV) 16 E6 Seropositive Individuals without HPV-Associated Malignancies after 10 Years of Follow-up in the UK Biobank, EBioMedicine, 2020, 62, 103123, DOI:10.1016/j.ebiom.2020.103123.
- D. Martinez-Zapien, F. X. Ruiz, J. Poirson, A. Mitschler, J. Ramirez, A. Forster, A. Cousido-Siah, M. Masson, S. V. Pol, A. Podjarny and et al. , Structure of the E6/E6AP/P53 Complex Required for HPV-Mediated Degradation of P53, Nature, 2016, 529(7587), 541–545, DOI:10.1038/nature16481.
- F. X. Bosch, T. R. Broker, D. Forman, A.-B. Moscicki, M. L. Gillison, J. Doorbar, P. L. Stern, M. Stanley, M. Arbyn, M. Poljak and et al. , Comprehensive Control of Human Papillomavirus Infections and Related Diseases, Vaccine, 2013, 31, H1–H31, DOI:10.1016/j.vaccine.2013.10.003.
- A. A. McBride, Human Papillomaviruses: Diversity, Infection and Host Interactions, Nat. Rev. Microbiol., 2022, 20(2), 95–108, DOI:10.1038/s41579-021-00617-5.
- N. Muñoz, F. X. Bosch, S. de Sanjosé, R. Herrero, X. Castellsagué, K. V. Shah, P. J. F. Snijders and C. J. L. M. Meijer, Epidemiologic Classification of Human Papillomavirus Types Associated with Cervical Cancer, N. Engl. J. Med., 2003, 348(6), 518–527, DOI:10.1056/nejmoa021641.
- P. A. Cohen, A. Jhingran, A. Oaknin and L. Denny, Cervical Cancer, Lancet, 2019, 393(10167), 169–182, DOI:10.1016/S0140-6736(18)32470-X.
- R. G. Ghebre, S. Grover, M. J. Xu, L. T. Chuang and H. Simonds, Cervical Cancer Control in HIV-Infected Women: Past, Present and Future, Gynecol. Oncol. Rep., 2017, 21, 101–108, DOI:10.1016/j.gore.2017.07.009.
- Q. Wu, W. Qian, X. Sun and S. Jiang, Small-Molecule Inhibitors, Immune Checkpoint Inhibitors, and More: FDA-Approved Novel Therapeutic Drugs for Solid Tumors from 1991 to 2021, J. Hematol. Oncol., 2022, 15(1), 143, DOI:10.1186/s13045-022-01362-9.
- A. Hollebecque, T. Meyer, K. N. Moore, J.-P. H. Machiels, J. De Greve, J. M. López-Picazo, A. Oaknin, J. N. Kerger, V. Boni, T. R. J. Evans and et al. , An Open-Label, Multicohort, Phase I/II Study of Nivolumab in Patients with Virus-Associated Tumors (CheckMate 358): Efficacy and Safety in Recurrent or Metastatic (R/M) Cervical, Vaginal, and Vulvar Cancers, J. Clin. Oncol., 2017, 35(suppl. 15), 5504, DOI:10.1200/jco.2017.35.15_suppl.5504.
- Precision medicine meets cancer vaccines, Nat. Med., 2023, 29, 1287, DOI:10.1038/s41591-023-02432-2.
- H. C. Chung, J. H. M. Schellens, J.-P. Delord, R. Perets, A. Italiano, R. Shapira-Frommer, L. Manzuk, S. A. Piha-Paul, J. Wang, S. Zeigenfuss and et al. , Pembrolizumab Treatment of Advanced Cervical Cancer: Updated Results from the Phase 2 KEYNOTE-158 Study, J. Clin. Oncol., 2018, 36(suppl. 15), 5522, DOI:10.1200/jco.2018.36.15_suppl.5522.
- R. B. S. Roden and P. L. Stern, Opportunities and Challenges for Human Papillomavirus Vaccination in Cancer, Nat. Rev. Cancer, 2018, 18(4), 240–254, DOI:10.1038/nrc.2018.13.
- E. A. White, M. E. Sowa, M. J. A. Tan, S. Jeudy, S. D. Hayes, S. Santha, K. Münger, J. W. Harper and P. M. Howley, Systematic Identification of Interactions between Host Cell Proteins and E7 Oncoproteins from Diverse Human Papillomaviruses, Proc. Natl. Acad. Sci. U. S. A., 2012, 109(5), E260–E267, DOI:10.1073/pnas.1116776109.
- J. J. Chen, Y. Hong, E. Rustamzadeh, J. D. Baleja and E. J. Androphy, Identification of an α Helical Motif Sufficient for Association with Papillomavirus E6, J. Biol. Chem., 1998, 273(22), 13537–13544, DOI:10.1074/jbc.273.22.13537.
- L. V. Ronco, A. Y. Karpova, M. Vidal and P. M. Howley, Human Papillomavirus 16 E6 Oncoprotein Binds to Interferon Regulatory Factor-3 and Inhibits Its Transcriptional Activity, Genes Dev., 1998, 12(13), 2061–2072, DOI:10.1101/gad.12.13.2061.
- X. Tong and P. M. Howley, The Bovine Papillomavirus E6 Oncoprotein Interacts with Paxillin and Disrupts the Actin Cytoskeleton, Proc. Natl. Acad. Sci. U. S. A., 1997, 94(9), 4412–4417, DOI:10.1073/pnas.94.9.4412.
- L. E. Araujo-Arcos, S. Montaño, C. Bello-Rios, O. L. Garibay-Cerdenares, M. A. Leyva-Vázquez and B. Illades-Aguiar, Molecular Insights into the Interaction of HPV-16 E6 Variants against MAGI-1 PDZ1 Domain, Sci. Rep., 2022, 12(1), 1898, DOI:10.1038/s41598-022-05995-1.
- H. L. Howie, R. A. Katzenellenbogen and D. A. Galloway, Papillomavirus E6 Proteins, Virology, 2009, 384(2), 324–334, DOI:10.1016/j.virol.2008.11.017.
- C. A. Moody and L. A. Laimins, Human Papillomavirus Oncoproteins: Pathways to Transformation, Nat. Rev. Cancer, 2010, 10(8), 550–560, DOI:10.1038/nrc2886.
- K. Hoppe-Seyler, F. Bossler, J. A. Braun, A. L. Herrmann and F. Hoppe-Seyler, The HPV E6/E7 Oncogenes: Key Factors for Viral Carcinogenesis and Therapeutic Targets, Trends Microbiol., 2018, 158–168, DOI:10.1016/j.tim.2017.07.007.
- A. Kumar, E. Rathi, R. C. Hariharapura and S. G. Kini, Is Viral E6 Oncoprotein a Viable Target? A Critical Analysis in the Context of Cervical Cancer, Med. Res. Rev., 2020, 40(5), 2019–2048, DOI:10.1002/med.21697.
- O. Hassin and M. Oren, Drugging P53 in Cancer: One Protein, Many Targets, Nat. Rev. Drug Discovery, 2022, 1–18, DOI:10.1038/s41573-022-00571-8.
- D. L. Jones, D. A. Thompson and K. Münger, Destabilization of the RB Tumor Suppressor Protein and Stabilization of P53 Contribute to HPV Type 16 E7-Induced Apoptosis, Virology, 1997, 239(1), 97–107, DOI:10.1006/viro.1997.8851.
- K. Zerfass, A. Schulze, D. Spitkovsky, V. Friedman, B. Henglein and P. Jansen-Dürr, Sequential Activation of Cyclin E and Cyclin A Gene Expression by Human Papillomavirus Type 16 E7 through Sequences Necessary for Transformation, J. Virol., 1995, 69(10), 6389–6399, DOI:10.1128/jvi.69.10.6389-6399.1995.
- J. H. Lee, Y. S. Kang, J. W. Koh, S. Y. Park, B. G. Kim, E. D. Lee, K. H. Lee, K. B. Park and Y. L. Seo, P53 Gene Mutation Is Rare in Human Cervical Carcinomas with Positive HPV Sequences, Int. J. Gynecol. Cancer, 1994, 4(6), 371–378, DOI:10.1046/j.1525-1438.1994.04060371.x.
- X. Y. Niu, Z. L. Peng, W. Q. Duan, H. Wang and P. Wang, Inhibition of HPV 16 E6 Oncogene Expression by RNA Interference in Vitro and in Vivo, Int. J. Gynecol. Cancer, 2006, 16(2), 743–751, DOI:10.1111/j.1525-1438.2006.00384.x.
- J. T.-C. Chang, T. F. Kuo, Y. J. Chen, C. C. Chiu, Y. C. Lu, H. F. Li, C. R. Shen and A. J. Cheng, Highly Potent and Specific SiRNAs against E6 or E7 Genes of HPV16-or HPV18-Infected Cervical Cancers, Cancer Gene Ther., 2010, 17(12), 827–836, DOI:10.1038/cgt.2010.38.
- M. Scheffner, B. A. Werness, J. M. Huibregtse, A. J. Levine and P. M. Howley, The E6 Oncoprotein Encoded by Human Papillomavirus Types 16 and 18 Promotes the Degradation of P53, Cell, 1990, 63(6), 1129–1136, DOI:10.1016/0092-8674(90)90409-8.
- M. Scheffner, J. M. Huibregtse, R. D. Vierstra and P. M. Howley, The HPV-16 E6 and E6-AP Complex Functions as a Ubiquitin-Protein Ligase in the Ubiquitination of P53, Cell, 1993, 75(3), 495–505, DOI:10.1016/0092-8674(93)90384-3.
- T. Ansari, N. Brimer and S. B. Vande Pol, Peptide Interactions Stabilize and Restructure Human Papillomavirus Type 16 E6 To Interact with P53, J. Virol., 2012, 86(20), 11386–11391, DOI:10.1128/jvi.01236-12.
- A. Hafner, M. L. Bulyk, A. Jambhekar and G. Lahav, The Multiple Mechanisms That Regulate P53 Activity and Cell Fate, Nat. Rev. Mol. Cell Biol., 2019, 20(4), 199–210, DOI:10.1038/s41580-019-0110-x.
- C. C.-K. Chao, Mechanisms of P53 Degradation, Clin. Chim. Acta, 2015, 438, 139–147, DOI:10.1016/j.cca.2014.08.015.
- Y. Yamagishi, I. Shoji, S. Miyagawa, T. Kawakami, T. Katoh, Y. Goto and H. Suga, Natural Product-like Macrocyclic N-Methyl-Peptide Inhibitors against a Ubiquitin Ligase Uncovered from a Ribosome-Expressed de Novo Library, Chem. Biol., 2011, 18(12), 1562–1570, DOI:10.1016/j.chembiol.2011.09.013.
- H. Wang, Y. Wang, K. K. Kota, B. Kallakury, N. N. Mikhail, D. Sayed, A. Mokhtar, D. Maximous, E. H. Yassin, I. Gouda and et al. , Strong Association between Long and Heterogeneous Telomere Length in Blood Lymphocytes and Bladder Cancer Risk in Egyptian, Carcinogenesis, 2019, 40(6), 1284–1290, DOI:10.1093/CARCIN/BGZ072.
- R. Avagliano Trezza, M. Sonzogni, S. N. V. Bossuyt, F. I. Zampeta, A. M. Punt, M. van den Berg, D. C. Rotaru, L. M. C. Koene, S. T. Munshi, J. Stedehouder and et al. , Loss of Nuclear UBE3A Causes Electrophysiological and Behavioral Deficits in Mice and Is Associated with Angelman Syndrome, Nat. Neurosci., 2019, 22(8), 1235–1247, DOI:10.1038/s41593-019-0425-0.
- J. S. Sutcliffe, Y. H. Jiang, R. J. Galjaard, T. Matsuura, P. Fang, T. Kubota, S. L. Christian, J. Bressler, B. Cattanach, D. H. Ledbetter and et al. , The E6-AP Ubiquitin Protein Ligase (UBE3A) Gene Is Localized within a Narrowed Angelman Syndrome Critical Region, Genome Res., 1997, 7(4), 368–377, DOI:10.1101/gr.7.4.368.
- F. Offensperger, F. Müller, J. Jansen, D. Hammler, K. H. Götz, A. Marx, C. L. Sirois, S. J. Chamberlain, F. Stengel and M. Scheffner, Identification of Small-Molecule Activators of the Ubiquitin Ligase E6AP/UBE3A and Angelman Syndrome-Derived E6AP/UBE3A Variants, Cell Chem. Biol., 2020, 27(12), 1510–1520, DOI:10.1016/j.chembiol.2020.08.017.
- S. P. da Silva, J. M. O. Santos, V. F. Mestre, B. Medeiros-Fonseca, P. A. Oliveira, M. M. S. M. Bastos, R. M. Gil da Costa and R. Medeiros, Human Papillomavirus 16-Transgenic Mice as a Model to Study Cancer-Associated Cachexia, Int. J. Mol. Sci., 2020, 21(14), 1–17, DOI:10.3390/ijms21145020.
- K. Yamato, T. Yamada, M. Kizaki, K. Ui-Tei, Y. Natori, M. Fujino, T. Nishihara, Y. Ikeda, Y. Nasu, K. Saigo and et al., New Highly Potent and Specific E6 and E7 SiRNAs for Treatment of HPV16 Positive Cervical Cancer, Cancer Gene Ther., 2008, 15(3), 140–153, DOI:10.1038/sj.cgt.7701118.
- T. Sugo, M. Terada, T. Oikawa, K. Miyata, S. Nishimura, E. Kenjo, M. Ogasawara-Shimizu, Y. Makita, S. Imaichi, S. Murata and et al. , Development of Antibody-SiRNA Conjugate Targeted to Cardiac and Skeletal Muscles, J. Controlled Release, 2016, 237, 1–13, DOI:10.1016/j.jconrel.2016.06.036.
- L. M. Alvarez-Salas, A. E. Cullinan, A. Siwkowski, A. Hampel and J. A. Dipaolo, Inhibition of HPV-16 E6/E7 Immortalization of Normal Keratinocytes by Hairpin Ribozymes, Proc. Natl. Acad. Sci. U. S. A., 1998, 95(3), 1189–1194, DOI:10.1073/pnas.95.3.1189.
- J. J. Cherry, A. Rietz, A. Malinkevich, Y. Liu, M. Xie, M. Bartolowits, V. J. Davisson, J. D. Baleja and E. J. Androphy, Structure Based Identification and Characterization of Flavonoids That Disrupt Human Papillomavirus-16 E6 Function, PLoS One, 2013, 8(12), e84506, DOI:10.1371/journal.pone.0084506.
- C. H. Yuan, M. Filippova, J. L. Krstenansky and P. J. Duerksen-Hughes, Flavonol and Imidazole Derivatives Block HPV16 E6 Activities and Reactivate Apoptotic Pathways in HPV+ Cells, Cell Death Dis., 2016, 7(1), e2060, DOI:10.1038/cddis.2015.391.
- K. A. Malecka, D. Fera, D. C. Schultz, S. Hodawadekar, M. Reichman, P. S. Donover, M. E. Murphy and R. Marmorstein, Identification and Characterization of Small Molecule Human Papillomavirus E6 Inhibitors, ACS Chem. Biol., 2014, 9(7), 1603–1612, DOI:10.1021/cb500229d.
- J. Zhu, S. Kamara, Q. Wang, Y. Guo, Q. Li, L. Wang, J. Chen, Q. Du, W. Du, S. Chen and et al. , Novel Affibody Molecules Targeting the HPV16 E6 Oncoprotein Inhibited the Proliferation of Cervical Cancer Cells, Front. Cell Dev. Biol., 2021, 9, 1150, DOI:10.3389/fcell.2021.677867.
- W. Zhang, H. Shan, K. Jiang, W. Huang and S. Li, A Novel Intracellular Nanobody against HPV16 E6 Oncoprotein, Clin. Immunol., 2021, 225, 1521–6616, DOI:10.1016/j.clim.2021.108684.
- J. Ramirez, J. Poirson, C. Foltz, Y. Chebaro, M. Schrapp, A. Meyer, A. Bonetta, A. Forster, Y. Jacob, M. Masson and et al., Targeting the Two Oncogenic Functional Sites of the HPV E6 Oncoprotein with a High-Affinity Bivalent Ligand, Angew. Chem., 2015, 127(27), 8069–8073, DOI:10.1002/anie.201502646.
- K. Zanier, S. Charbonnier, A. O. M. h. O. Sidi, A. G. McEwen, M. G. Ferrario, P. Poussin-Courmontagne, V. Cura, N. Brimer, K. O. Babah, T. Ansari and et al. , Structural Basis for Hijacking of Cellular LxxLL Motifs by Papillomavirus E6 Oncoproteins, Science, 2013, 339(6120), 694–698, DOI:10.1126/science.1229934.
- J. M. Huibregtse, M. Scheffner and P. M. Howley, Localization of the E6-AP Regions That Direct Human Papillomavirus E6 Binding, Association with P53, and Ubiquitination of Associated Proteins, Mol. Cell. Biol., 1993, 13(8), 4918–4927, DOI:10.1128/mcb.13.8.4918-4927.1993.
- R. C. Elston, S. Napthine and J. Doorbar, The Identification of a Conserved Binding Motif within Human Papillomavirus Type 16 E6 Binding Peptides, E6AP and E6BP, J. Gen. Virol., 1998, 79(2), 371–374, DOI:10.1099/0022-1317-79-2-371.
- M. Muttenthaler, G. F. King, D. J. Adams and P. F. Alewood, Trends in Peptide Drug Discovery, Nat. Rev. Drug Discovery, 2021, 20(4), 309–325, DOI:10.1038/s41573-020-00135-8.
- P. E. Saw, X. Xu, S. Kim and S. Jon, Biomedical Applications of a Novel Class of High-Affinity Peptides, Acc. Chem. Res., 2021, 54(18), 3576–3592, DOI:10.1021/acs.accounts.1c00239.
- K. Zanier, C. Stutz, S. Kintscher, E. Reinz, P. Sehr, J. Bulkescher, K. Hoppe-Seyler, G. Travé and F. Hoppe-Seyler, The E6AP Binding Pocket of the HPV16 E6 Oncoprotein Provides a Docking Site for a Small Inhibitory Peptide Unrelated to E6AP, Indicating Druggability of E6, PLoS One, 2014, 9(11), e112514, DOI:10.1371/journal.pone.0112514.
- S. Dymalla, M. Scheffner, E. Weber, P. Sehr, C. Lohrey, F. Hoppe-Seyler and K. Hoppe-Seyler, A Novel Peptide Motif Binding to and Blocking the Intracellular Activity of the Human Papillomavirus E6 Oncoprotein, J. Mol. Med., 2009, 87(3), 321–331, DOI:10.1007/s00109-008-0432-1.
- C. Stutz, E. Reinz, A. Honegger, J. Bulkescher, J. Schweizer, K. Zanier, G. Travé, C. Lohrey, K. Hoppe-Seyler and F. Hoppe-Seyler, Intracellular Analysis of the Interaction between the Human Papillomavirus Type 16 E6 Oncoprotein and Inhibitory Peptides, PLoS One, 2015, 10(7), e0132339, DOI:10.1371/journal.pone.0132339.
- Y. Liu, Z. Liu, E. Androphy, J. Chen and J. D. Baleja, Design and Characterization of Helical Peptides That Inhibit the E6 Protein of Papillomavirus, Biochemistry, 2004, 43(23), 7421–7431, DOI:10.1021/bi049552a.
- C. F. Shuman, P. O. Markgren, M. Hämäläinen and U. H. Danielson, Elucidation of HIV-1 Protease Resistance by Characterization of Interaction Kinetics between Inhibitors and Enzyme Variants, Antiviral Res., 2003, 58(3), 235–242, DOI:10.1016/S0166-3542(03)00002-0.
- R. Mah, J. R. Thomas and C. M. Shafer, Drug Discovery Considerations in the Development of Covalent Inhibitors, Bioorg. Med. Chem. Lett., 2014, 24(1), 33–39, DOI:10.1016/j.bmcl.2013.10.003.
- R. A. Bauer, Covalent Inhibitors in Drug Discovery: From Accidental Discoveries to Avoided Liabilities and Designed Therapies, Drug Discovery Today, 2015, 20(9), 1061–1073, DOI:10.1016/j.drudis.2015.05.005.
- A. Abdeldayem, Y. S. Raouf, S. N. Constantinescu, R. Moriggl and P. T. Gunning, Advances in Covalent Kinase Inhibitors, Chem. Soc. Rev., 2020, 49(9), 2617–2687, 10.1039/c9cs00720b.
- V. Y. Berdan, P. C. Klauser and L. Wang, Covalent Peptides and Proteins for Therapeutics, Bioorg. Med. Chem., 2021, 29, 115896, DOI:10.1016/j.bmc.2020.115896.
- L. Boike, N. J. Henning and D. K. Nomura, Advances in Covalent Drug Discovery, Nat. Rev. Drug Discovery, 2022, 1–18, DOI:10.1038/s41573-022-00542-z.
- A. D. De Araujo, J. Lim, K. C. Wu, Y. Xiang, A. C. Good, R. Skerlj and D. P. Fairlie, Bicyclic Helical Peptides as Dual Inhibitors Selective for Bcl2A1 and Mcl-1 Proteins, J. Med. Chem., 2018, 61(7), 2962–2972, DOI:10.1021/acs.jmedchem.8b00010.
- Y. Nominé, S. Charbonnier, T. Ristriani, G. Stier, M. Masson, N. Cavusoglu, A. Van Dorsselaer, É. Weiss, B. Kieffer and G. Travé, Domain Substructure of HPV E6 Oncoprotein: Biophysical Characterization of the E6 C-Terminal DNA-Binding Domain, Biochemistry, 2003, 42(17), 4909–4917, DOI:10.1021/bi026980c.
- X. Ye, Y. C. Lee, Z. P. Gates, Y. Ling, J. C. Mortensen, F. S. Yang, Y. S. Lin and B. L. Pentelute, Binary Combinatorial Scanning Reveals Potent Poly-Alanine-Substituted Inhibitors of Protein-Protein Interactions, Commun. Chem., 2022, 5(1), 128, DOI:10.1038/s42004-022-00737-w.
- X. Be, Y. Hong, J. Wei, E. J. Androphy, J. J. Chen and J. D. Baleja, Solution Structure Determination and Mutational Analysis of the Papillomavirus E6 Interacting Peptide of E6AP, Biochemistry, 2001, 40(5), 1293–1299, DOI:10.1021/bi0019592.
- L. Ko, G. R. Cardona and W. W. Chin, Thyroid Hormone Receptor-Binding Protein, an LXXLL Motif-Containing Protein, Functions as a General Coactivator, Proc. Natl. Acad. Sci. U. S. A., 2000, 97(11), 6212–6217, DOI:10.1073/pnas.97.11.6212.
- R. Lonsdale and R. A. Ward, Structure-Based Design of Targeted Covalent Inhibitors, Chem. Soc. Rev., 2018, 47(11), 3816–3830, 10.1039/c7cs00220c.
- J. M. Chalker, S. B. Gunnoo, O. Boutureira, S. C. Gerstberger, M. Fernández-González, G. J. L. Bernardes, L. Griffin, H. Hailu, C. J. Schofield and B. G. Davis, Methods for Converting Cysteine to Dehydroalanine on Peptides and Proteins, Chem. Sci., 2011, 2(9), 1666–1676, 10.1039/c1sc00185j.
- H. Eng, A. L. Dantonio, E. P. Kadar, R. Scott Obach, L. Di, J. Lin, N. C. Patel, B. Boras, G. S. Walker, J. J. Novak and et al., Disposition of Nirmatrelvir, an Orally Bioavailable Inhibitor of SARS-CoV-2 3C-Like Protease, across Animals and Humans, Drug Metab. Dispos., 2022, 50(5), 576–590, DOI:10.1124/dmd.121.000801.
- H. de Jong, K. M. Bonger and D. W. P. M. Löwik, Activatable Cell-Penetrating Peptides: 15 Years of Research, RSC Chem. Biol., 2020, 1, 192–203, 10.1039/D0CB00114G.
- J. M. Wolfe, C. M. Fadzen, Z. N. Choo, R. L. Holden, M. Yao, G. J. Hanson and B. L. Pentelute, Machine Learning to Predict Cell-Penetrating Peptides for Antisense Delivery, ACS Cent. Sci., 2018, 4(4), 512–520, DOI:10.1021/acscentsci.8b00098.
- G. R. Buel, X. Chen, R. Chari, M. J. O’Neill, D. L. Ebelle, C. Jenkins, V. Sridharan, S. G. Tarasov, N. I. Tarasova, T. Andresson and et al. , Structure of E3 Ligase E6AP with a Proteasome-Binding Site Provided by Substrate Receptor HRpn10, Nat. Commun., 2020, 11(1), 1291, DOI:10.1038/s41467-020-15073-7.
- M. C. Conrady, I. Suarez, G. Gogl, D. I. Frecot, A. Bonhoure, C. Kostmann, A. Cousido-Siah, A. Mitschler, J. Lim, M. Masson and et al. , Structure of High-Risk Papillomavirus 31 E6 Oncogenic Protein and Characterization of E6/E6AP/P53 Complex Formation, J. Virol., 2020, 95(2), e00730, DOI:10.1128/jvi.00730-20.
- A. O. Frank, B. Vangamudi, M. D. Feldkamp, E. M. Souza-Fagundes, J. W. Luzwick, D. Cortez, E. T. Olejniczak, A. G. Waterson, O. W. Rossanese, W. J. Chazin and et al., Discovery of a Potent Stapled Helix Peptide That Binds to the 70N Domain of Replication Protein A, J. Med. Chem., 2014, 57(6), 2455–2461, DOI:10.1021/jm401730y.
- J. M. Torner, Y. Yang, D. Rooklin, Y. Zhang and P. S. Arora, Identification of Secondary Binding Sites on Protein Surfaces for Rational Elaboration of Synthetic Protein Mimics, ACS Chem. Biol., 2021, 16(7), 1179–1183, DOI:10.1021/acschembio.1c00418.
- F. M. Paulussen and T. N. Grossmann, Peptide-Based Covalent Inhibitors of Protein–Protein Interactions, J. Pept. Sci., 2023, 29(1), 1–12, DOI:10.1002/psc.3457.
- L. H. Jones, Dehydroamino Acid Chemical Biology: An Example of Functional Group Interconversion on Proteins, RSC Chem. Biol., 2020, 298–304, 10.1039/d0cb00174k.
- J. Dadová, S. R. Galan and B. G. Davis, Synthesis of Modified Proteins via Functionalization of Dehydroalanine, Curr. Opin. Chem. Biol., 2018, 46, 71–81, DOI:10.1016/j.cbpa.2018.05.022.
- R. W. MacKintosh, K. N. Dalby, D. G. Campbell, P. P. T. W. Cohen, P. P. T. W. Cohen and C. MacKintosh, The Cyanobacterial Toxin Microcystin Binds Covalently to Cysteine-273 on Protein Phosphatase 1, FEBS Lett., 1995, 371(3), 236–240, DOI:10.1016/0014-5793(95)00888-G.
- M. Runnegar, N. Berndt, S. M. Kong, E. Y. C. Lee and L. F. Zhang, In Vivo and In Vitro Binding of Microcystin to Protein Phosphatase 1 and 2A, Biochem. Biophys. Res. Commun., 1995, 216(1), 162–169, DOI:10.1006/bbrc.1995.2605.
- D. M. Downs and D. C. Ernst, From Microbiology to Cancer Biology: The Rid Protein Family Prevents Cellular Damage Caused by Endogenously Generated Reactive Nitrogen Species, Mol. Microbiol., 2015, 96(2), 211–219, DOI:10.1111/mmi.12945.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc02782a |
| ‡ These authors contributed equally to this work. |
| § Author Jason Tao was unable to be contacted following the changes at submission of the revised manuscript, therefore they do not take any responsibility for the changes in the revised manuscript. |
| This journal is © The Royal Society of Chemistry 2023 |