
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrochemical intercalation of rubidium into graphite, hard carbon, and soft carbon†
Daisuke
Igarashi
 ,
Ryoichi
Tatara
,
Ryusei
Fujimoto
,
Tomooki
Hosaka
and
Shinichi
Komaba
*
,
Ryoichi
Tatara
,
Ryusei
Fujimoto
,
Tomooki
Hosaka
and
Shinichi
Komaba
*
Department of Applied Chemistry, Tokyo University of Science, Shinjuku, Tokyo 162-8601, Japan. E-mail: komaba@rs.tus.ac.jp
First published on 4th October 2023
Abstract
The electrochemical insertion of Rb into carbonaceous materials, including graphite, was achieved herein. Rubidium ions were reversibly inserted into and extracted from graphite via electrochemical processes using different non-aqueous electrolytes containing rubidium bis(trifluoromethanesulfonyl)amide (RbTFSA) salts in carbonate esters, glymes, and ionic liquids, similar to the process used for other lighter alkali metal ions such as Li+ and K+. The chemical compositions of the rubidiated graphite were determined to be RbC8, RbC24, and RbC36 at each step of the electrochemical reduction process. Graphite underwent a phase transition to RbC8 exhibiting a stage-1 structure, with stage-3 RbC36 and stage-2 RbC24 as intermediates, as confirmed by ex situ and in situ X-ray diffraction and ex situ Raman spectroscopy, similar to the electrochemical phase evolution of staged potassium graphite intercalation compounds (K-GICs). Furthermore, Rb was reversibly inserted into and extracted from graphitizable and non-graphitizable carbons such as pitch-derived soft carbon and commercial hard carbon, along with other alkali metals such as Li, Na, and K.
Introduction
Graphite is known to form alkali metal-graphite intercalation compounds (A-GICs) in which alkali metals are incorporated into the interlayer spaces of graphite via chemical or electrochemical processes.1,2 A-GICs have characteristic layered structures called “stage structures,” where layers of intercalated chemical species called “intercalants” are regularly located at intervals along a fixed number of graphene layers. In many studies on GICs, the layered structures are denoted as ‘stage n,’ where n is the number of graphene layers sandwiched between the intercalant layers. The stage structures can be controlled by varying parameters such as the temperature, pressure, chemical potential of the alkali metals, and electrochemical potential of the host graphite electrode. Due to their controllable structures and various physical properties, which depend on the intercalants and stage numbers, GICs have been studied for application as chemical reagents, catalysts, conducting materials, hydrogen storage materials, and battery materials.1The first GICs of heavy alkali metals such as K, Rb, and Cs were discovered in 1926,3 followed by the discovery of Li-GICs in 1975.4 In the early days of such research, GIC samples were often prepared through direct chemical reaction of the host graphite with elemental alkali metals. The typical method for synthesizing GICs involved a chemical process called the ‘two-bulb method,’ in which the host graphite was in contact with the vapor of an alkali metal in a vacuum tube.1 Other methods such as mechanical mixing,5 pressurization,4 and catalytic processes6 have also been used to synthesize alkali metal GICs.
Several electrochemical processes have been developed for the synthesis of Li-GICs using poly(ethylene oxide)-based electrolytes7 and ethylene carbonate-containing electrolytes.8 Furthermore, the electrochemistry of Li-GICs has been extensively investigated, geared towards applications in rechargeable Li-ion batteries with negative electrode reactions.9 The effects of differences in the host graphite,10,11 electrolyte composition,12,13 reaction temperature,14,15 and other factors16 on the formation of Li-GICs via electrochemical processes and the resultant structures have been studied in detail, although Li-GICs were discovered much later than other alkali-metal GICs.
Recently, three research groups, including ours, reported electrochemical K intercalation into graphite at room temperature.17–19 In these reports, the stepwise electrochemical formation of stage-1, stage-2, and stage-3 K-GIC phases was successfully confirmed using ex situ X-ray diffraction (XRD) and Raman spectroscopy techniques. Since then, K-GIC has been widely researched as a promising candidate negative electrode material for K-ion batteries (an alternative to Li-ion batteries) because of the much higher earth-abundance of potassium compared to lithium.20
However, the electrochemistry of Rb- and Cs-GICs has not been as actively studied as those of Li- and K-GICs, plausibly because of the lack of application of the former as battery materials. In addition to our earlier work on Rb-GICs,21 Yadav et al. recently evaluated electrochemical Rb storage in graphite. However, the reversible capacity for Rb storage obtained by Yadav et al. was approximately 200 mA h g−1, which is lower than the theoretical limit of 279 mA h g−1, corresponding to stage-1 RbC8, and is insufficient to definitively confirm Rb intercalation.22 Herein, for the first time, we demonstrate the electrochemical intercalation of Rb into graphite with a high reversible capacity that approaches the theoretical capacity of 279 mA h g−1, assuming the chemical composition of RbC8, based on comparative electrochemical and structural analysis of K-GICs.20 Moreover, the electrochemical insertion of alkali metals (Li, Na, K, and Rb) into amorphous carbon materials such as soft and hard carbon is systematically compared and studied.
Experimental
Synthesis of RbTFSA salt
RbTFSA, as a Rb+ source in the electrolytes, was synthesized via a simple neutralization reaction, as described in previous studies.23,24 A 25 g portion of hydrogen bis(trifluoromethanesulfonyl)amide (HTFSA; Tokyo Chemical Industry) was dissolved in 250 mL of ethanol. Rb2CO3 powder (10 mol% excess stoichiometric ratio of RbTFSA, Sigma-Aldrich) was added to the solution and stirred overnight. After confirming complete neutralization by checking the pH, the excess Rb2CO3 powder remaining in the solution was filtered, and ethanol was removed under vacuum using a rotary evaporator. The resulting mixture of RbTFSA and a small amount of residual Rb2CO3 was added to diethyl carbonate (DEC). Because of the difference in solubility, RbTFSA dissolved in DEC, whereas Rb2CO3 remained undissolved. The RbTFSA/DEC solution and residual Rb2CO3 were separated by vacuum filtration, and the RbTFSA powder was obtained as the target product by evaporating the DEC solvent from the filtrate using a rotary evaporator.Electrode preparation and electrochemical measurements
To prepare the graphite electrode, natural graphite powder (SNO3, SEC Carbon) and a carboxymethylcellulose binder (Daicel Miraizu) at a weight ratio of 95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 were mixed in deionized water (as a dispersion medium) to form a slurry, which was applied onto Cu or Al foil. Lab-made soft carbon and commercial hard carbon (Carbotron P(J), Kureha) electrodes consisting of 90% active material, 5% acetylene black (Li-400, Denka), and 5% sodium polyacrylate binder (Kishida Chemical) were prepared in a similar manner. Mass loading of the carbon electrodes was approximately 1.5 mg cm−2. Soft carbon powder was prepared by heating needle coke at 1200 °C for 2 h under steady Ar flow in a tubular furnace.
:5 were mixed in deionized water (as a dispersion medium) to form a slurry, which was applied onto Cu or Al foil. Lab-made soft carbon and commercial hard carbon (Carbotron P(J), Kureha) electrodes consisting of 90% active material, 5% acetylene black (Li-400, Denka), and 5% sodium polyacrylate binder (Kishida Chemical) were prepared in a similar manner. Mass loading of the carbon electrodes was approximately 1.5 mg cm−2. Soft carbon powder was prepared by heating needle coke at 1200 °C for 2 h under steady Ar flow in a tubular furnace.
The electrolytes were prepared by mixing appropriate amounts of the alkali metal salts with the solvents. All the salts and solvents were stored in an Ar-filled glove-box. Lithium bis(fluorosulfonyl)amide (LiTFSA), potassium bis(fluorosulfonyl)amide (KTFSA), N-methyl-N-propylpyrrolidinium bis(fluorosulfonyl)amide ([C3C1pyrr][FSA]), and N-methyl-N-propylpyrrolidinium bis(trifluoromethanesulfonyl)amide ([C3C1pyrr][TFSA]) were purchased from Kanto Chemical. Sodium bis(trifluoromethanesulfonyl)amide (NaTFSA), ethylene carbonate (EC), propylene carbonate (PC), diethyl carbonate (DEC), triglyme (G3), and tetraglyme (G4) were purchased from Kishida Chemical. Cesium bis(trifluoromethanesulfonyl)amide (CsTFSA) was purchased from Tokyo Chemical Industry. All alkali metal salts were vacuum-dried at 110 °C before use, and all the solvents were used as received.
Electrochemical measurements of the graphite and amorphous carbon electrodes were performed using a 2032-coin type cell (Hohsen) or a three-electrode cell (Toyo System). A glass-fiber filter (GB-100R, Advantec) was used as the separator. Disc-shaped and ring-shaped samples of the elemental alkali metals were used as the counter and reference electrodes, respectively, in the three-electrode cells. Reagent-grade Li (>99.8%), Na (>99%), K, (>97%), and Rb (>99.6%) metal was purchased from Honjo Chemical, Kanto Chemical, Nacalai Tesque, and Aldrich, respectively. As-purchased Li metal foil was punched into discs of appropriate size for use. Other metals were purchased in lumps and were pressed into foil using a seam roller.25 Metal discs for counter electrodes were made by punching the metal foil, and metal rings for reference electrodes of three-electrode system were prepared by cutting the foil into thin strips and rounding them.
To measure the electrode potential of each alkali metal, a Ag+/Ag reference electrode was used in a two-compartment three-electrode cell (SB1A, EC Frontier) without separators. The Ag+/Ag reference was composed of a Ag wire soaked in Ag0.02[C3C1pyrr]0.98[TFSA]0.2[FSA]0.8 electrolyte, a mixture of AgTFSA (Tokyo Chemical Industry), [C3C1pyrr][TFSA], and [C3C1pyrr][FSA] in a molar ratio of 2:18:80. Activated carbon (YP50F, Kuraray Co., Ltd.) mixed with Ketjen black (EC600JD, Lion Specialty Chemicals Co., Ltd.) and a polytetrafluoroethylene binder (PTFE, F-104, Daikin Industries, Ltd.) in a weight ratio of 80:10:10 was pressed onto a Ti mesh for use as the counter electrode for the SB1A cell. The illustration of the two-compartment cell is shown in Fig. S1.† For the operando X-ray diffraction (XRD) measurements, a custom-made three-electrode cell (EC Frontier) with a Be X-ray transmission window was used. Open-circuit voltage (OCV) measurements of carbon electrodes were performed using coin-type cells in the following procedure: a constant current of 12.4 mA g−1 for the Li cells and 9.3 mA g−1 for the other cells was applied for 30 min, followed by a relaxation time of 3 h, after which the OCV was recorded. This process was repeated until the voltage reached 0 V for the reduction process and 2 V for the oxidation process. To eliminate the effect of electrolyte decomposition during the first few cycles, OCV measurements were performed in the sixth cycle after pre-cycling for the first five cycles. All electrochemical measurements were performed at room temperature (20–30 °C).
Material characterization
The structures of the active material powder and composite electrodes were characterized using X-ray diffractometers (SmartLab for powder and ex situ measurements and MultiFlex for operando measurements, Rigaku Corporation). A homemade airtight sample holder was used to prevent exposure of the samples to air during ex situ XRD analysis of the graphite electrodes post electrochemical testing. The morphologies of the active materials were observed by scanning electron microscopy (SEM, JCM-6000, JEOL Limited). Raman spectra of the graphite electrodes were recorded using a laser Raman spectrometer (Raman 11i, Nanophoton). A 532 nm green laser was used as the excitation source for the spectroscopic analysis. During the ex situ Raman spectroscopy measurements, an electrode sample was placed between a slide glass and a thin cover glass, and vacuum grease was applied between the slide and cover glasses to prevent exposure of the sample to air.Results and discussions
Electrochemistry of elemental alkali metals
Before electrochemical analysis of the graphite electrode in the Rb cells, we investigated the electrochemistry of Rb metal as a reference and counter electrode. In the case of Na and K systems, it is known that Na and K metal electrodes exhibit remarkably unstable potential, making them unreliable as reference electrodes,25–27 necessitating optimization of the electrolyte composition for each alkali metal. In this study, we selected an ionic liquid containing the FSA anion as the standard electrolyte because it has been reported that the surface of alkali metals is more stable28 and the intercalation reactions of Li or K into graphite are more reversible in ionic liquid electrolytes of such compositions than in conventional carbonate-ester-based organic electrolytes.29 Although we previously used a mixture of KFSA and [C3C1pyrr][FSA] as the electrolyte for electrochemical K intercalation into graphite,29 the RbFSA salt is not commercially available, to the best of our knowledge. Therefore, we used the RbTFSA salt, which is more chemically stable30 and easier to synthesize than the RbFSA salt,23,31 as the source of Rb+ ions in the electrolyte. The ionic liquid electrolyte was prepared by mixing 20 mol% of the alkali metal TFSA salt with 80 mol% of [C3C1pyrr][FSA]. However, in the case of the Cs system, the CsTFSA salt did not dissolve completely in [C3C1pyrr][FSA]. Thus, saturated CsTFSA/[C3C1pyrr][FSA] was used as the electrolyte in the Cs system for comparison with the Li, Na, K, and Rb systems in later experiments.Fig. 1a shows the potentials of the Cu substrates on which the alkali metals were electrochemically plated in the A0.2[C3C1pyrr]0.8[TFSA]0.2[FSA]0.8 electrolytes. In this experiment, the open-circuit voltage of the Cu working electrode was monitored for 24 h after electrochemical plating of the alkali metal by applying a current density of −25 μA cm−2 for 5 h. The observed potentials of the Li+/Li, Na+/Na, K+/K, Rb+/Rb, and Cs+/Cs redox couples were 0, +0.13, −0.23, −0.27, and −0.39 V vs. Li+/Li, respectively. A trend in which heavier alkali metals have lower electrode potentials was confirmed, except in the case of Li+/Li and Na+/Na. In a previous study by Yamamoto et al. on the electrode potentials of alkali metals in ionic liquid electrolytes containing a pyrrolidinium cation and a (fluorosulfonyl)(trifluoromethanesulfonyl)amide anion,32 similar values were obtained with corresponding trends. The electrode potential of each alkali metal had a stable, constant value for at least 24 h. Thus, Rb metal can be used as a reliable reference electrode with the present ionic liquid-based electrolytes.
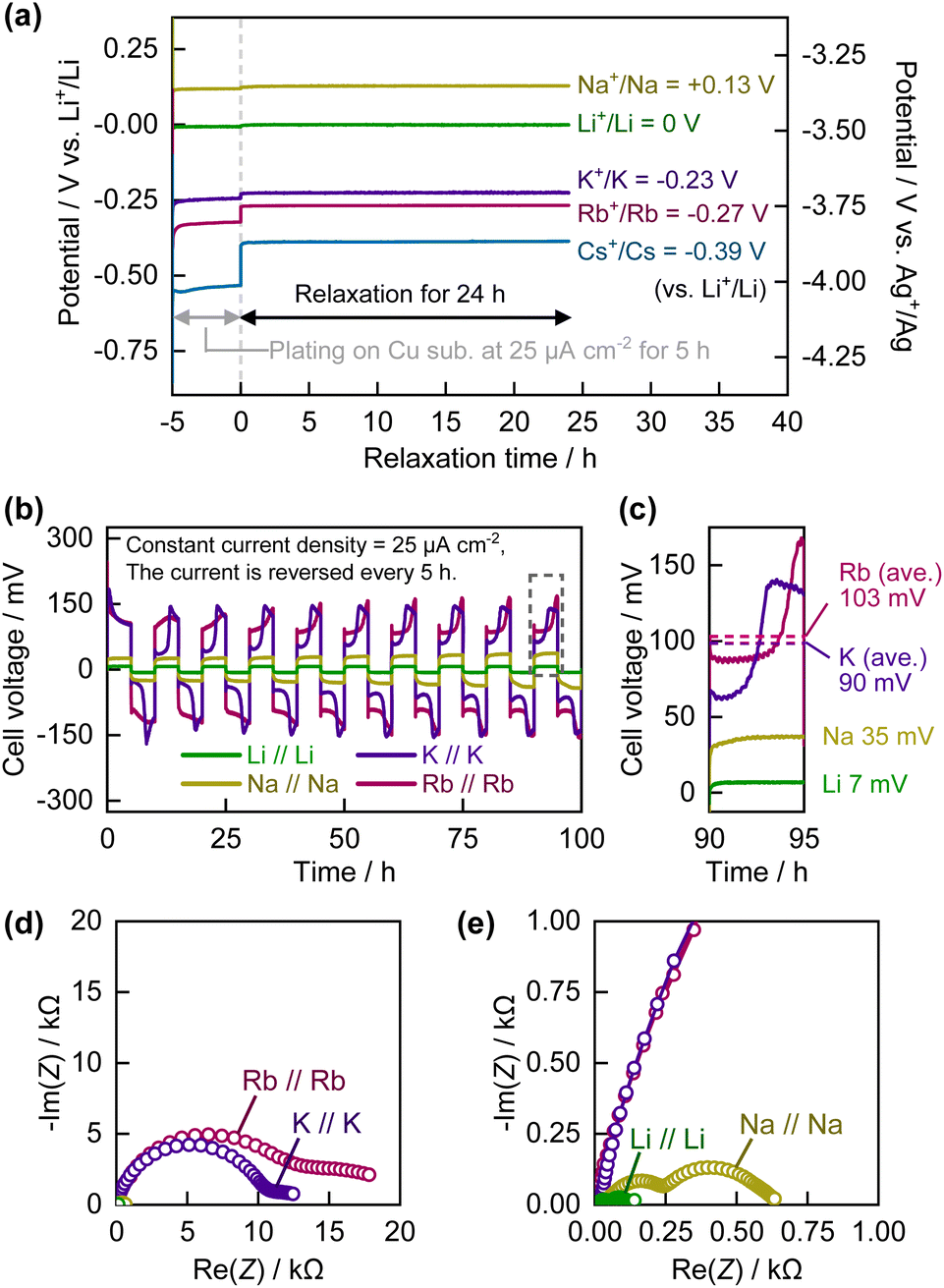 | ||
| Fig. 1 (a) Plot of potential versus time during/after electrochemical plating of alkali metals on Cu foil at 25 μA cm−2. (b) Cell voltage of alkali-metal symmetric cells under reverse constant current of ±25 μA cm−2. (c) Enlarged view of a part of (b) enclosed by dotted line. (d) Nyquist plots for alkali-metal symmetric cells. (e) Enlarged view of (d). | ||
In subsequent experiments, we used commercially available elemental alkali metals, including metallic Rb, to prepare alkali metal electrodes. Rb metal was carefully and safely handled from glass ampoules in an Ar-filled glove-box (see Fig. S2†) owing to its extremely high reactivity, low melting point (39 °C), and hazardous nature. Fig. 1b and c show the cell voltage curves of the A//A symmetric cell, to which a current density of 25 μA cm−2 was applied. Although there was a trend toward greater polarization in the case of the heavier alkali metals, the average cell voltage of Rb//Rb (∼103 mV) was not significantly higher than that of K//K (∼99 mV), which is consistent with the Nyquist plots from the electrochemical impedance spectroscopy (EIS) data for the A//A symmetric cells (see Fig. 1d and e). Semicircles were observed in the plots for each cell and their combinations. The resistance includes the resistance of the alkali metal–electrolyte interface. The Li//Li and Na//Na cells showed relatively small resistances of approximately 0.14 and 0.63 kΩ, respectively. In comparison, the interface resistance of the K//K and Rb//Rb cells was more than one order of magnitude higher, with very large values of 12 and 18 kΩ, respectively. The overall trend is consistent with the previously reported EIS data for Li//Li, Na//Na, and K//K symmetric cells with carbonate ester-based electrolytes.27,33 However, the difference in resistance between the K//K and Rb//Rb cells was only a few tens of percent. Thus, the effect of this difference in resistance on the overpotential of the metal plating/stripping reaction was not significant when Rb was used as the counter/reference electrode in the two-electrode system. From the above results, we conclude that Rb metal can be used not only as a reference electrode, but also as a counter electrode in two- or three-electrode systems, analogous to the use of K-metal cells in studies on electrode materials for K-ion batteries.20
Electrochemical Rb intercalation into graphite
Galvanostatic reduction/oxidation testing of the graphite composite electrode was performed using a three-electrode cell with an Rb reference electrode and Rb counter electrode, at a current density of 27.9 mA g−1 (corresponding to C/10), in the potential range of 0.005–2 V vs. Rb+/Rb. The graphite powder used in this study comprised natural graphite flakes with a small particle size of approximately 3 μm (Fig. S3†). The potential versus capacity profile (the same as the charge/discharge profile for battery testing) is shown in Fig. 2a. A reversible oxidation capacity of 276 mA h g−1 was obtained in the 1st cycle, which corresponds to the chemical composition of Rb0.99C8 (=RbC8.09), i.e., the formation of stage-1 Rb-GIC and RbC8 in the fully reduced state via an electrochemical process. The Coulombic efficiency in the 1st cycle was 86.5%. The remaining irreversible capacity of 13.5% is possibly due to cathodic electrolyte decomposition and formation of a surface layer on the electrode, which is called solid electrolyte interphase (SEI) in the chemistry of battery materials.34,35 The SEI layer on the graphite electrode should conduct Rb+ ions and be electron insulating to passivate the electrode.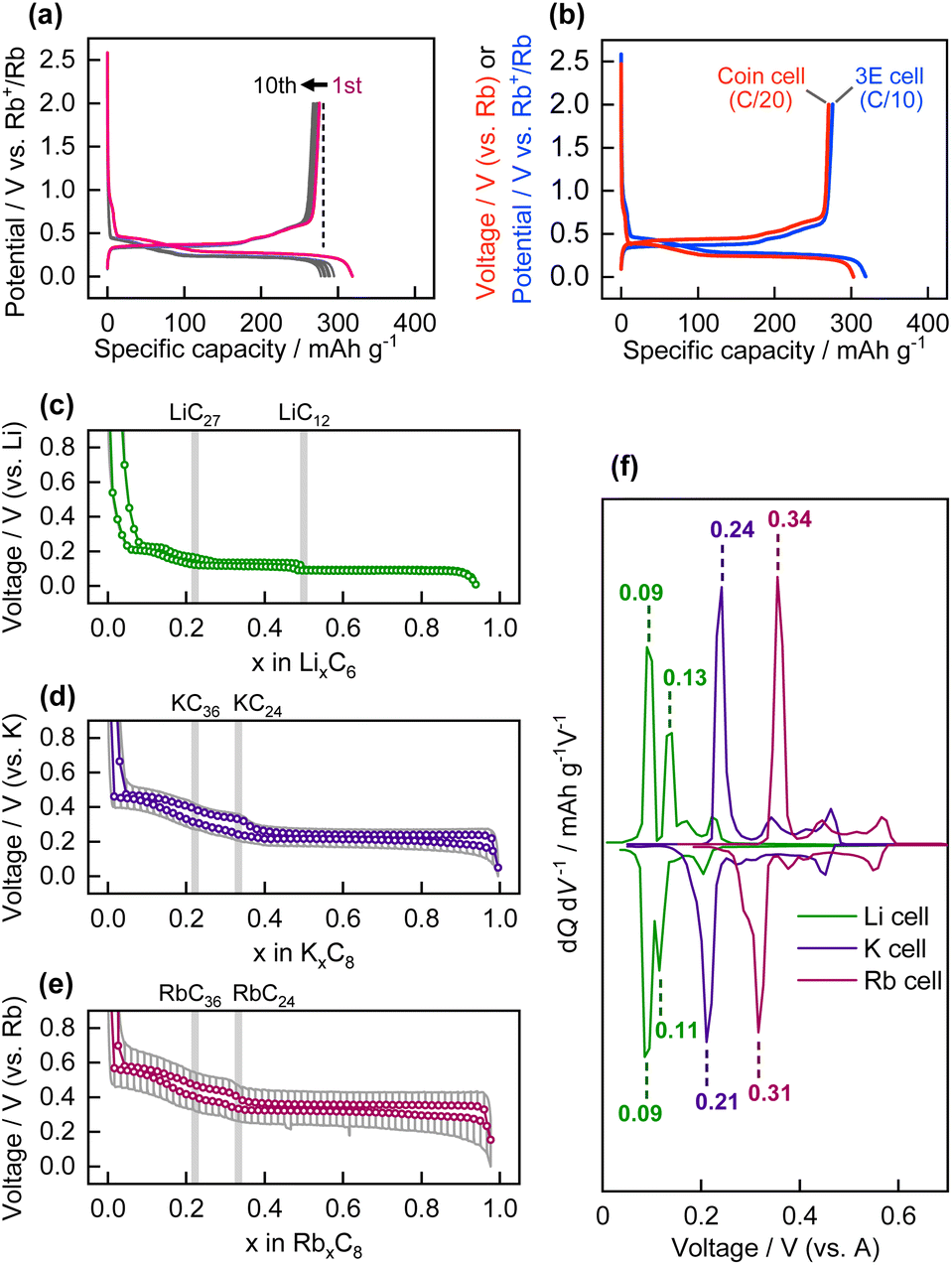 | ||
| Fig. 2 (a) Galvanostatic reduction/oxidation curves of graphite electrode in three-electrode cell. (b) Comparison of reduction/oxidation curves of graphite electrodes in three-electrode cell and coin-type cell. OCV profiles of graphite electrodes in (c) Li, (d) K, and (e) Rb cells. The OCV values are plotted as colored circles and the cell voltage profiles during polarization are drawn as solid gray lines. (f) Differential curves of OCV profiles in (c), (d), and (e). | ||
Galvanostatic tests were also performed on the graphite electrode in a coin-type cell equipped with a Rb counter electrode, at a lower current density of C/20. The reduction/oxidation profile is shown in Fig. 2b and compared with that of the three-electrode cell. Despite the greater polarization of the Rb counter electrode (vide supra), the graphite working electrode exhibited almost the same reversible capacity as the three-electrode cell. The results show that the coin-type cell with the two-electrode system can also be used for electrochemical evaluation of “Rb cells” at a sufficiently small current density. In addition, the galvanostatic reduction–oxidation curves of graphite electrodes with different current collectors, Cu or Al, are shown in Fig. S4.† It is generally known that Al current collector cannot be used for Li-GICs since Li forms alloys with Al via an electrochemical process. In contrast, Al current collector is available for K-GICs due to the absence of K–Al alloys,19 and the same is true for Rb-GICs.
The galvanostatic reduction/oxidation profiles of graphite in Li, Na, K, and Rb cells, measured using coin-type cells, are shown in Fig. S5.† In our previous report, graphite in Li and K cells delivered reversible capacities corresponding to the formation of stage-1 Li-GIC (LiC6) and stage-1 K-GIC (KC8), whereas the formation of Na-GICs by graphite in Na cells was limited, and thus provided a negligible reversible capacity.36Fig. 2c–f display the open-circuit voltage (OCV) as a function of the molar fraction of alkali metals in the A-GICs and the differential OCV vs. voltage plot for graphite in the Li, K, and Rb cells. Although the steps were ill-defined in the OCV profiles of the K- and Rb-GICs but a stepwise profile was observed for the Li-GICs, the plateau-like potential profiles indicate that two-phase reactions occurred during the alkali ion (de)intercalation process. These reactions involve two adjacent stage structures as two different phases; that is, a pair comprising stage-3 and stage-2, or a pair comprising stage-2 and stage-1.37 Notably, the reversible capacity and overall shape of the OCV profiles of the K and Rb cells were almost identical, except for the plateau potential values. The well-matched OCV profiles suggest that the phase transitions of K- and Rb-GICs proceed via the same transition mechanism. In Fig. 2c–e, the chemical compositions corresponding to the boundaries of the adjacent plateaus are LiC27, LiC12, and LiC6 in the Li cell; KC36, KC24, and KC8 in the K cells; and RbC36, RbC24, and RbC8 in the Rb cells. It has been reported that heavy alkali metal GICs form stage-1 and stage-n stage structures with respective chemical compositions of AC8 AC12n, where n > 1.1
Characterization of Rb-inserted graphite
To confirm the stage structure of “rubidiated” graphite, the graphite electrode was subjected to ex situ analysis at 0.4, 0.35, and 0.001 V in the reduction process and at 2 V after reoxidation. The potentials of 0.4 and 0.35 V correspond to the plateau boundary of RbC36 and RbC24, respectively (Fig. 2e), whereas the potentials of 0.001 and 2 V correspond to the fully reduced and re-oxidized states, respectively. The optical images of the graphite electrodes at each potential are shown in Fig. 3a. The graphite composite electrodes maintained a black coloration during the rubidiation process until the potential reached 0.4 V, but turned dark blue at 0.35 V. Eventually, the fully rubidiated electrode became a bright orange color. These color changes are the same as those observed for chemically prepared Rb-GICs and other heavy alkali-metal GICs in previous literature.1,5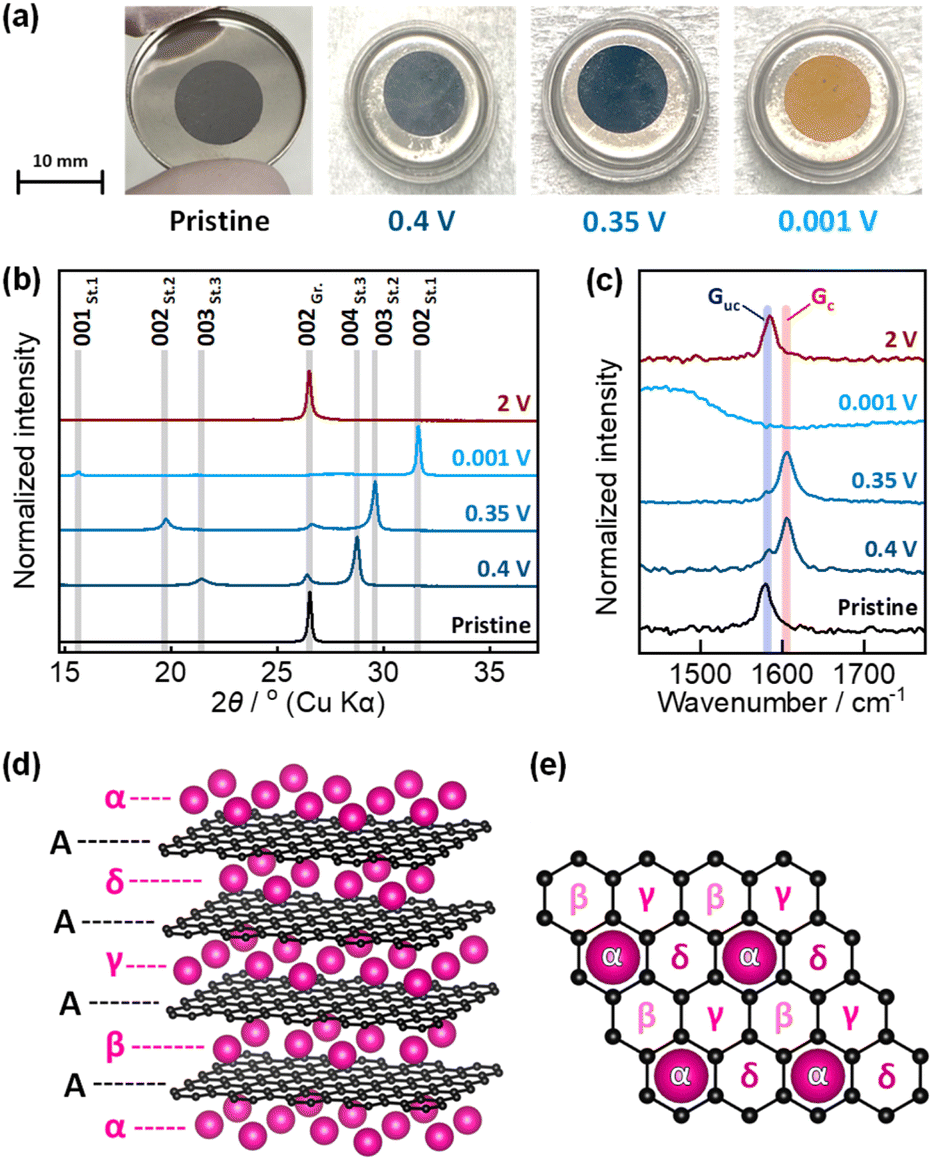 | ||
| Fig. 3 (a) Photographs of graphite electrodes electrochemically reduced in Rb cells. (b) Ex situ XRD patterns and (c) ex situ Raman spectra of graphite electrodes. (d) Crystal structure model of stage-1 Rb-GIC, RbC8, based on consideration of the in-plane Rb arrangement. (e) Plausible in-plane Rb arrangement in RbC8. The Greek letters from α to δ represent equivalent Rb positions. | ||
The ex situ XRD patterns and Raman spectra of the graphite electrodes that were reduced or reoxidized to a given potential are shown in Fig. 3b and c. The pattern of the electrochemically reduced graphite electrode showed two sharp diffraction peaks at lower and higher angles than the 002 diffraction peak of pristine graphite. These peaks are assigned to the 00n and 00(n + 1) diffraction peaks of stage-n Rb-GICs, where n = 3, 2, or 1 based on previous studies of the chemical preparation of Rb-GICs. Note that the diffraction peaks of Rb-GICs were indexed without accounting for the in-plane ordering of the Rb atoms, despite the fact that the three-dimensional arrangement of stage-1 Rb-GIC (RbC8) was estimated to be AαAβAγAδ in a previous report,1 where A represents the graphene layers, and α, β, γ, and δ represent the in-plane ordered Rb atoms, as illustrated in Fig. 4d and e. In this stage-1 crystal structure, considering the in-plane ordering of Rb, the diffraction peaks of stage-1 Rb-GIC should be indexed as 004 or 008 in crystallography. The 002 diffraction peak of graphite was restored when the electrode was re-oxidized to 2 V, reflecting the reversibility of electrochemical Rb intercalation, as observed in the electrochemical test described above.
 | ||
| Fig. 4 (a) Operando XRD patterns of graphite electrodes during initial reduction processes in Li, K, and Rb cells. Operando XRD patterns as heat maps and corresponding reduction/oxidation curves of graphite electrodes during 2nd cycle in (b) Li, (c) K, and (d) Rb cells. The black line in the heat map of (d) is the area where diffraction caused by irregular orientation of a cell component was observed (see Fig. S9†). | ||
The ex situ Raman spectra of the pristine graphite electrode (Fig. 2c) showed a single peak at ca. 1580 cm−1, corresponding to the E2g mode of sp2 graphitic carbon,24–27 often called the “G band.” The intensity of the G band decreased upon electrochemical reduction to 0.4 and 0.35 V, while a new peak appeared at ca. 1610 cm−1 and gradually gained intensity. The new peak originates from the charged graphene layers adjacent to the inserted Rb layer38 (see illustrations in Fig. S6†), and is thus often called the “Gc band”, whilst the original G band of uncharged graphene is called the “Guc band” in this context.39 The peak intensity ratio of the Gc and Guc bands (corresponding to the ratio of the number of uncharged and charged graphene layers) was ca. 2 at 0.4 V and <2 at 0.35 V. The data suggest the formation of a stage-3 structure at 0.4 V and stage-2 structure at 0.35 V, which is consistent with ex situ XRD measurements. The spectrum of the fully reduced electrode showed a broadened peak over a wide wavenumber range of ∼1200–1600 cm−1, whereas the Guc and Gc bands disappeared. This result is unexpected based on the model shown in Fig. S6,† but is a commonly observed feature of stage-1 A-GICs.18,40 The Guc band was recovered by reoxidation at 2 V, which corresponds to the ex situ XRD data. The electrochemical measurements and ex situ characterization results confirmed the formation of stage-3 RbC36, stage-2 RbC24, and stage-1 RbC8 during the reduction process. Since these observations provide sufficient evidence demonstrating the electrochemically reversible intercalation of Rb into graphite, the phase evolution was studied in detail using operando XRD.
Phase transition of A-GICs via electrochemical process
Operando XRD was used to study the dynamic electrochemical phase evolution of the A-GICs. The potential–capacity plots of the graphite electrodes are shown in Fig. S7;†in situ XRD data were acquired for the electrodes employed in our custom-made three-electrode cell. The graphite electrodes in each cell delivered a sufficient reversible capacity compared to those in the coin cells, despite the difference in the cell configuration. The diffraction patterns of the graphite electrodes obtained during the early alkaliation process (in the 1st cycle) are shown in Fig. 4a. For the Li cell, the 002 diffraction peak of graphite continuously shifted to the peak position corresponding to stage-8, followed by a biphasic phase change to a lower-stage structure. This observation is in agreement with previous studies.37,41 The continuous peak shift, which appears to be associated with a solid-solution reaction, indicates the formation of a structure called ‘dilute stage-1’ before reaching the higher stage, around stage 8. In contrast, the intensity of the 002 diffraction peaks in the patterns of the K and Rb cells continuously decreased, whereas the diffraction angle was maintained at approximately 26.5°. The broad peak attributed to higher-stage structures, such as stage 8 (judging from the diffraction angle), gradually became more intense. In our previous study,37 we regarded the initial K+ intercalation process in K-GICs as the formation of higher-stage K-GICs with many stacking disorders, in which K+ layers are randomly distributed in the graphite host. We termed such structures as a ‘disorderly stacked high stage’. Based on the operando XRD results, the initial process of Rb intercalation observed in this study was identical to the K intercalation process in K-GICs; thus, Rb-GICs were found to form a disorderly stacked high stage in the initial rubidiation process.The diffraction patterns of the graphite electrodes in each cell after the 1st dealkaliation up to 2 V (vs. A+/A) are shown in Fig. S8,† along with those of the electrodes in the pristine state. The 002 diffraction peaks in the XRD patterns of dealkaliated graphite in the cells with the heavier alkali metals (K and Rb) tended to have lower intensity and were broader, indicating that the phase transition of the heavier alkali metal GICs is reversible, but is accompanied by a decrease in crystallinity. The operando XRD patterns of the graphite electrodes in the Li, K, and Rb cells, collected at the 2nd cycle of the alkaliation and dealkaliation process, are displayed in Fig. 4b–d as heat maps, and in Fig. S9† as waterfall plots. A clear biphasic phase transition was observed in the Li cell during the major stages of both lithiation and delithiation. As a structure unique to Li-GIC, the so-called ‘stage 2L’ phase, which appeared only in the delithiation process due to some kinetic effect,14,42–45 was observed between stage 2 and stage 3 in the Li cell. The observed overall phase transition is in agreement with previous reports.37,42,43 Some recent reports suggest that there should be a more complicated phase evolution in electrochemical alkali ion (de)intercalation processes, where the presence of the stage-2 KC16 phase with a KC8-type in-plane K arrangement was proposed by Liu et al.46 and our group.37 This is an interesting hysteresis of the structure and chemical composition, unique to K-GICs, which was not observed in the Li system. Given the similarities in the electrochemistry and structural changes of the K- and Rb-GICs, as described above, it is expected that the same hysteresis will appear for Rb-GIC due to the stage-2 structure, as observed in previous studies on K-GICs.37,46 In this study, however, it was difficult to accurately determine and discuss the chemical composition of RbCx and the hysteresis accompanying the structural change because the in situ XRD system used herein was not effectively hermetically sealed compared to common battery-test cells, such as coin-type cells, leading to atmospheric contamination; thus, the extremely reactive Rb metal and/or Rb-GIC electrode suffered from irreversible side reactions upon galvanostatic cycling in the in situ cell.
The effect of the type of alkali metal on the structural changes of the GICs is reflected in the width of the diffraction lines; that is, the crystallinity and coherent length of the GICs. Compared with the Li cell, the diffraction peaks observed in the XRD patterns of the K and Rb cells were relatively broad, and the broadening was even greater in the case of the Rb cell. This observation implies that the coherence along the stacking axes is reduced in heavier-alkali-metal GICs. The intercalation of alkali ions with large ionic radii causes drastic structural changes in the host graphite material. The tails of the diffraction peaks derived from adjacent stages in the K and Rb cells appeared to be combined because of a progressive phase transition, as in the case of K-GIC.37 Thus, the phase changes were ill-defined and did not appear to involve clear two-phase reactions, especially in the transition between higher stages, such as from stage 4 to 3 or from stage 3 to 2. These observations are consistent with the Daumas–Hérold model, which is the most widely accepted structural phase transition model for alkali-metal GICs.2 In the Daumas–Hérold model, partial deformation of the graphene sheets owing to alkali metal intercalation is assumed, and the GICs are assumed to be composed of several stage-structured domains. The staging reaction proceeds via the movement of these deformations of the graphene sheets and stage domains along the in-plane direction. Therefore, the intercalation of larger alkali metal ions induces pronounced deformation and strain in the graphene sheets, as assumed in the Daumas–Hérold model (see Fig. S10†).
Effect of electrolyte
Various non-aqueous solutions were also evaluated as electrolytes for the electrochemical Rb intercalation reactions based on the knowledge of electrolytes for Li-, Na-, and K-ion batteries.20 The galvanostatic reduction/oxidation profiles of the graphite electrode in ionic liquid electrolytes, namely, Rb0.2[C3C1pyrr]0.8[TFSA]0.2[FSA]0.8 (referred to as “FSA-based IL” here) and Rb0.2[C3C1pyrr]0.8[TFSA]1.0 (referred to as “TFSA-based IL”), are shown in Fig. 5a. Almost no reversible capacity was obtained for the TFSA-based IL, whereas the formation of RbC8 was confirmed for the FSA-based IL, as discussed above. A similar trend to that observed for Li-GICs in a previous study47,48 was observed, which can be explained further by the ability of the FSA anion to form a more suitable SEI layer on the surface of the graphite and Rb metal electrodes through reductive decomposition, compared to the TFSA anion.28,49,50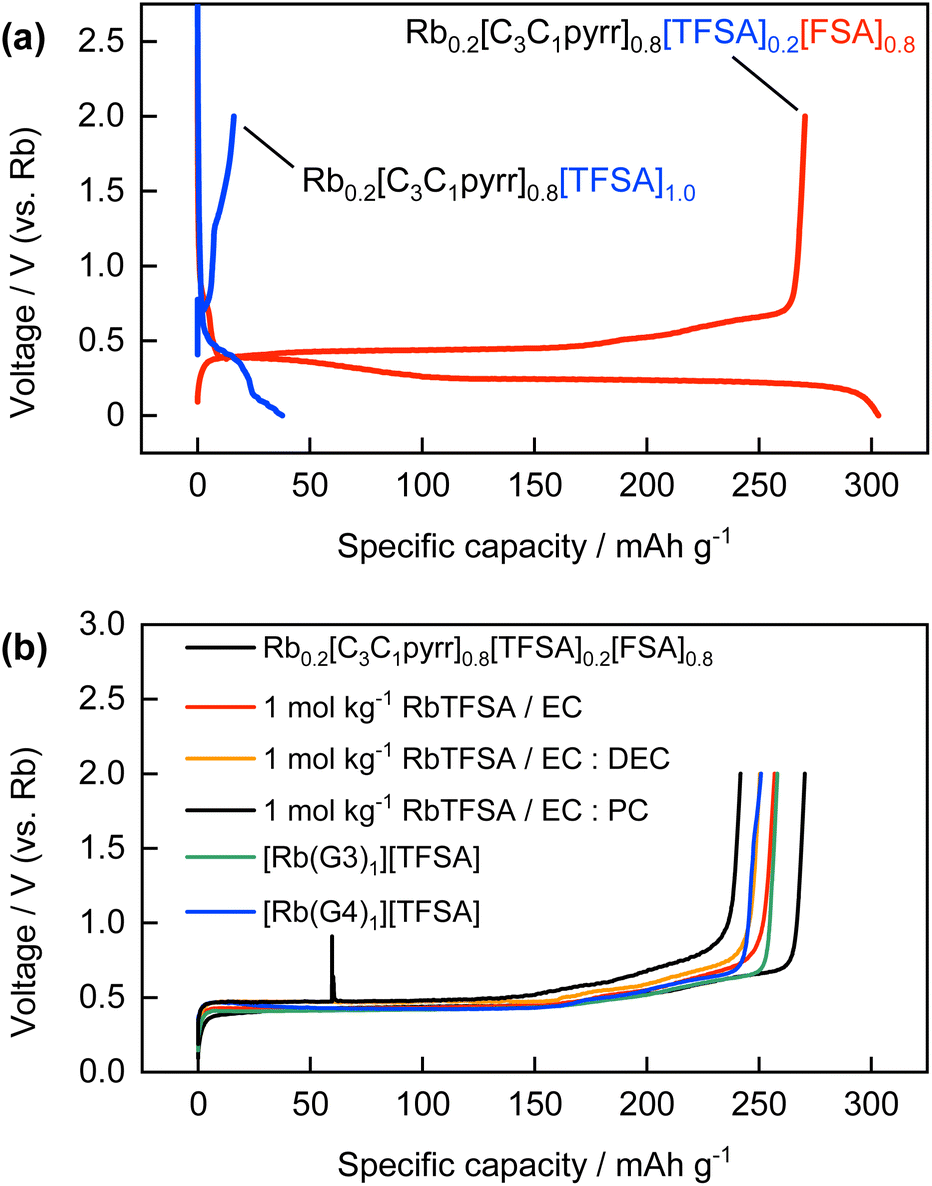 | ||
| Fig. 5 (a) Galvanostatic reduction/oxidation curves of graphite electrodes in Rb0.2[C3C1pyrr]0.8[TFSA]0.2[FSA]0.8 and Rb0.2[C3C1pyrr]0.8[TFSA]1.0 electrolyte. (b) Galvanostatic oxidation curves of graphite electrodes in non-aqueous organic electrolytes. | ||
Conventional organic solvent-based electrolytes such as carbonate esters and glymes were further evaluated because ethylene carbonate (EC)-based electrolytes are typically used and glyme-based electrolytes that contain highly concentrated alkali metal salts have recently received much attention in the study of Li-GICs12,51 and K-GICs.52,53 Interestingly, in these experiments, we found that equimolar mixtures of RbTFSA and G3 or G4 were in the liquid state, whereas those of NaTFSA or KTFSA and G3 or G4 were in the solid state at room temperature.54 The relationship between the species of alkali metals, including Rb and Cs, and the chain length of the glyme solvents is currently under study and has been reported elsewhere. Fig. 5b presents a comparison of the derubidiation curves of the graphite electrode in certain organic solvent-based electrolytes with those of the FSA-based IL. The reduction/oxidation curves for each electrolyte are shown in Fig. S10 and S11.† Similar reversible capacities and potential changes corresponding to the formation of stage 1 Rb-GICs were obtained for both conventional EC-based and concentrated glyme-based electrolytes. The reversible Rb intercalation obtained in EC-based electrolytes, despite the absence of FSA anions, plausibly originates from SEI formation through EC decomposition. A large irreversible capacity was observed, which was possibly due to the continuous electrolyte decomposition and exfoliation of graphite particles in the first reduction process in the case of 1 mol kg−1 RbTFSA/PC, as shown in Fig. S9b.† This tendency is well-known in the Li system13 and has also been observed in the K system.37 Reversible Rb intercalation was also observed in the glyme-based concentrated RbTFSA electrolyte in the absence of either the FSA anion or EC. This can be explained by the inversion of the co-intercalation potential of [Rb(glyme)]+ and the intercalation potential of desolvated Rb+ in highly concentrated electrolytes, referred to as ‘solvate ionic liquids’,55 as proposed by Moon et al. for the Li-GIC system,51 or may be due to specific SEI formation in concentrated electrolytes even in the absence of the FSA anion. In the case of moderately concentrated (1 mol kg−1) electrolytes (Fig. S12†), the formation of RbC8 was not confirmed from the rubidiation/derubidiation curves. Instead, some reversible reactions proceeded at a far higher potential of approximately 1 V vs. Rb+/Rb, which corresponds to the co-intercalation of alkali metal–glyme complexes of [A(glyme)]+, as commonly observed for the systems of Li, Na, and K and ether solvents including glymes56–58 and other organic solvents such as dimethyl sulfoxide.59 Although the mechanism of the co-intercalation reaction and the exact and detailed structures of the corresponding ternary GICs are not known and are under debate, even for Li, Na, and K systems, the electrode reaction or structure of ternary [Rb(glyme)]+-GICs was first demonstrated in this study. Detailed characterization is also in progress and will be reported elsewhere.
Electrochemical Rb insertion into amorphous carbons
We further studied electrochemical Rb insertion into soft carbon (SC) and hard carbon (HC), also known as ‘graphitizable carbon’ and ‘non-graphitizable carbon,’ respectively, because these carbon materials show high redox activity as Li, Na, or K insertion hosts.17,60,61 The XRD patterns and SEM images of the SC and HC are shown in Fig. 6a–c. The diffraction patterns of SC and HC used in this study were typical for coke-derived carbon heated at approximately 1200 °C, which can be converted into graphite by further heating,62 and non-graphitizable carbon composed of pseudo-graphitic nanodomains and nanovoids, respectively, based on the broadened diffraction lines, intensity ratio of the 002graphite and 100/101graphite peaks, and the intensity of scattering in the low-angle region. Both materials comprised unoriented, ruggedly shaped particles with diameters of a few to several tens of micrometers.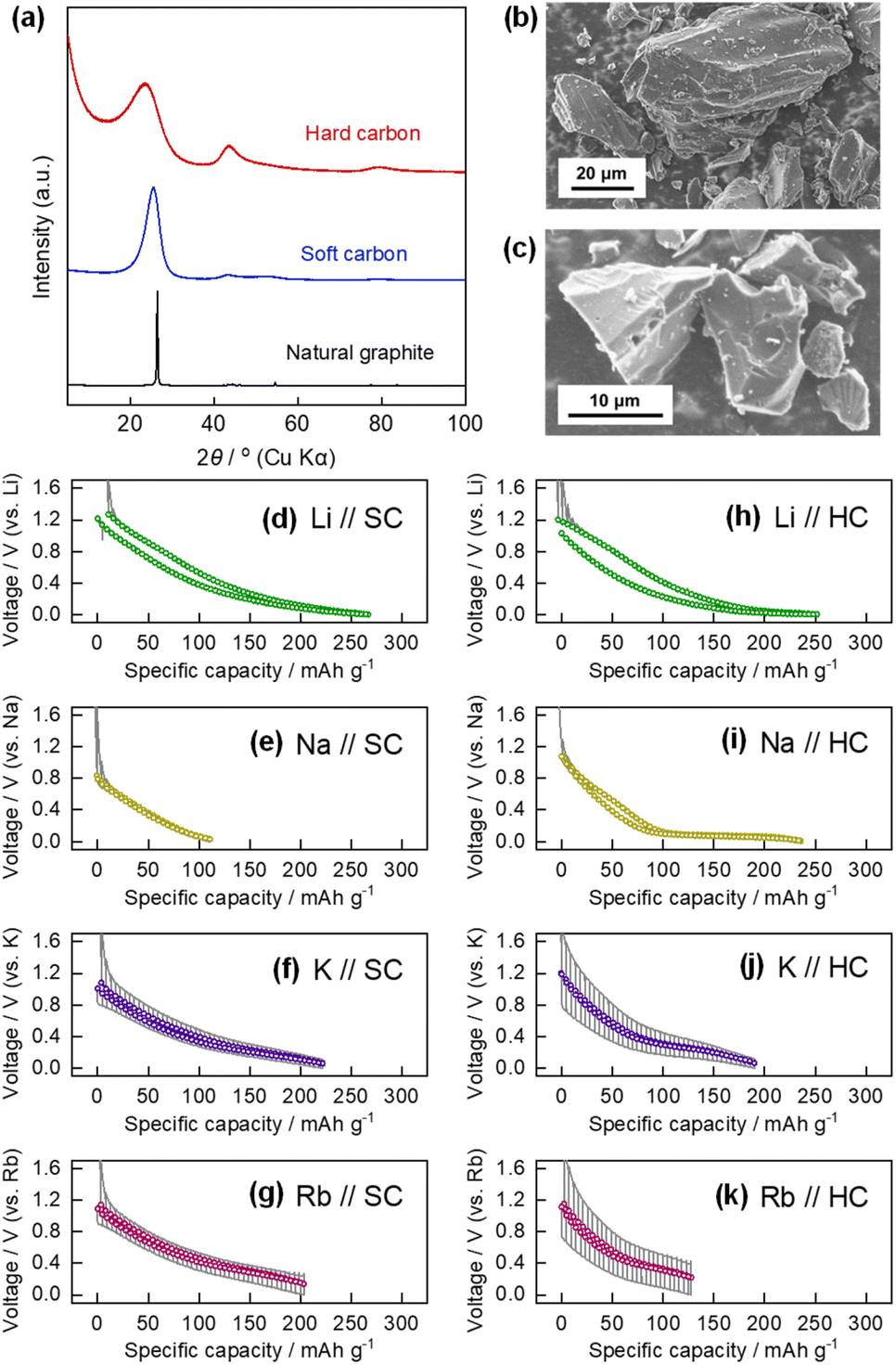 | ||
| Fig. 6 (a) Powder XRD patterns of natural graphite, SC, and HC. SEM images of (b) SC and (c) HC powder. OCV profiles of (d) Li//SC, (e) Na//SC, (f) K//SC, (g) Rb//SC, (h) Li//HC, (i) Na//HC, (j) K//HC, and (k) Rb//HC cells. The OCV values are plotted as colored circles and the cell voltage profiles during polarization are drawn as solid gray lines. | ||
The constant-current reduction/oxidation curves of the SC in the Li, Na, K, and Rb cells are shown in Fig. S13a–d,† and the corresponding OCV profiles are shown in Fig. 6d–g, respectively. The reversible capacities of the SC in the Li, Na, K, and Rb cells in the 1st cycle were 269, 121, 231, and 241 mA h g−1, respectively. Based on the reversible capacity values, the chemical compositions of the alkali-metal-inserted SCs were estimated to be LiC8.3, NaC18.4, KC9.7, and RbC9.3. The potential–capacity profiles of all the cells showed slopes with no potential plateaus, although the capacities varied with the alkali metal species. Because typical SC materials have graphite-like layered structures containing turbostratic graphene layers and various types of defect sites, it has been widely reported that Li+, Na+, and K+ are electrochemically stored both by adsorption on the defect sites and intercalation into the interlayer space; thus, the potential profiles had gentle slopes due to the complicated alkali insertion and adsorption process.60,63,64 The present experiments show that the sodiation capacity is much smaller than that of the other alkali metals. The mechanism of alkali metal insertion into the SC is more similar to that of graphite than that of alkali metal insertion into HC (see below). Thus, alkali metals that can be inserted into the interlayer of graphite should also be similarly inserted into the SC. The potassiation and rubidiation capacities of SC were almost equivalent, which may be associated with the similar chemical compositions and (de)intercalation mechanisms of K- and Rb-GICs.
The reduction/oxidation curves of HC in the Li, Na, K, and Rb cells are shown in Fig. S13e–h,† and the corresponding OCV profiles are shown in Fig. 6h–k. The reversible capacities of HC in the Li, Na, K, and Rb cells during the 1st cycle were 263, 249, 188, and 160, respectively. Based on the reversible capacities, the estimated compositions of the alkali-metal-containing HCs are LiC8.5, NaC9.0, KC11.9, and RbC13.9. The potential profiles of the Li and Na cells consist of a slope in the higher potential region between 1.0 and 0.1 V and a plateau at lower potentials close to the standard electrode potential of the alkali metal. Although the mechanism of alkali metal insertion into HC is still under debate, leading studies on HCs as negative electrode materials for Li- or Na-ion batteries suggest that adsorption on defect sites, intercalation into the interlayer of graphite-like domains, and formation of quasi-metallic Li or Na clusters in nanopores occur in the stated order from high to low potential, close to A+/A.63,65–75 The formation of quasi-metallic clusters at lower potentials explains why some HC materials exhibit significantly large Na storage capacities that exceed the stoichiometric composition of NaC6.73 The mechanism by which K is inserted into HC has been less investigated and is less understood than that of the Li and Na systems, and no direct evidence of the formation of quasi-metallic K clusters, as in the case of Li and Na, has been reported, to the best of our knowledge. Although the OCV profiles of the K//HC and Rb//HC cells were similar, including the absence of a low-potential plateau, the remarkable polarization upon (de)alkaliation was more pronounced in the Rb cell than in the K//HC cell. The difference in the polarization between the K//HC and Rb//HC cells was more noticeable than that between the K//graphite and Rb//graphite cells, as shown in Fig. 2d and e, or K//SC and Rb//SC cells in Fig. 6f and g, which accounts for not only the difference in the plating/stripping overvoltage between the K and Rb metal counter electrodes, but also for some intrinsic variation in the insertion mechanism of K and Rb into the HC materials.
In this study, we systematically investigated the electrochemical insertion of rubidium into graphite and disordered carbon electrodes in several electrolyte media at room temperature by comparing the insertion of Li, Na, and K, which we have previously studied. In the future, the interface and interphase chemistry of Rb-inserted carbon electrodes should be studied to understand the charge-transfer process, including the desolvation of solvated Rb+ ions at the electrolyte/electrode interface. We emphasize the importance of not only battery technology, but also fundamental studies on the intercalation chemistry of group 1 alkali metals in various host materials.
Conclusions
The reversible electrochemical insertion of Rb into carbonaceous materials was demonstrated and systematically compared with the corresponding insertion of Li, Na, and K. Analysis of the galvanostatic reduction/oxidation of the graphite electrode showed that a reversible capacity of nearly 279 mA h g−1 was obtained for the insertion of Rb+ ions into graphite. The chemical composition of the electrode in the fully reduced state, as calculated from the reversible capacity, corresponded to RbC8. Electrochemically synthesized Rb-GICs showed similar behavior to other alkali metal GICs, but different characteristics that were clearly affected by the ionic radius of the inserted cation. The stepwise potential profile of the graphite electrode and ex situ characterization of the electrode employed in the Rb cell indicated that the Rb insertion reaction consisted of several biphasic reactions, in which two different stage structures coexisted. The electrochemical structural changes in Li-, K-, and Rb-GICs were investigated and compared using operando XRD techniques, demonstrating that the formation of GICs proceeds with more structural defects along the stacking direction for alkali metal GICs with larger atomic sizes. This observation supports the conventional Daumas–Hérold model, which considers partial deformation of graphene sheets owing to the insertion of intercalants. Additionally, it was confirmed that solvated Rb+ can be directly and electrochemically inserted into graphite in an ether-based electrolyte and that Rb can be inserted into graphite and amorphous carbon materials such as SC and HC. While this study of Rb-GICs is not directly useful for the development of energy devices similar to those of Li- and K-GICs, we believe that our work contributes to the progress of systematic research on GICs.Data availability
The datasets in the current study are available from the corresponding author on reasonable request.Author contributions
D. I., R. T., and R. F. contributed to the experimental design. D. I. performed the electrochemical measurements and experiments for characterizing the graphite and other carbon electrodes with the advice of R. T. and T. H., R. F. and R. T. synthesized and characterized RbTFSA. D. I. wrote the first draft of the manuscript with the advice of R. T., S. K. supervised the study. All authors reviewed and revised the manuscript in several steps and approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was partially funded by the MEXT Program: Data Creation and Utilization Type Materials Research and Development Project (JPMXP1121467561), JST-CREST (grant no. JPMJCR21O6), and JSPS KAKENHI (grant no. JP20H02849, JP21K14724, JP21K20561, JP22K14772, and JP23K13829). DI thanks JST for establishing university fellowships for the Creation of Science and Technology Innovation (grant no. JPMJFS2144). RT thanks the TEPCO Memorial Foundation research grant (basic research), ECSJ Kanto branch research grant, and Takahashi Industrial and Economic Research Foundation. Schematic illustrations of the crystal structures used in this study were drawn using VESTA.76References
- M. S. Dresselhaus and G. Dresselhaus, Adv. Phys., 1981, 30, 139–326 CrossRef CAS.
- Y. Li, Y. Lu, P. Adelhelm, M.-M. Titirici and Y.-S. Hu, Chem. Soc. Rev., 2019, 48, 4655–4687 RSC.
- K. Fredenhagen and G. Cadenbach, Z. Anorg. Allg. Chem., 1926, 158, 249–263 CrossRef CAS.
- D. Guerard and A. Herold, Carbon, 1975, 13, 337–345 CrossRef CAS.
- D. M. Ottmers and H. F. Rase, Carbon, 1966, 4, 125–127 CrossRef CAS.
- H.-F. Klein, J. Gross and J. O. Besenhard, Angew Chem. Int. Ed. Engl., 1980, 19, 491–492 CrossRef.
- R. Yazami and P. Touzain, J. Power Sources, 1983, 9, 365–371 CrossRef CAS.
- R. Fong, U. von Sacken and J. R. Dahn, J. Electrochem. Soc., 1990, 137, 2009 CrossRef CAS.
- J. Asenbauer, T. Eisenmann, M. Kuenzel, A. Kazzazi, Z. Chen and D. Bresser, Sustainable Energy Fuels, 2020, 4, 5387–5416 RSC.
- A. Satoh, N. Takami and T. Ohsaki, Solid State Ionics, 1995, 80, 291–298 CrossRef CAS.
- J. R. Dahn, T. Zheng, Y. Liu and J. S. Xue, Science, 1995, 270, 590–593 CrossRef CAS.
- Y. Yamada and A. Yamada, J. Electrochem. Soc., 2015, 162, A2406 CrossRef CAS.
- S. J. An, J. Li, C. Daniel, D. Mohanty, S. Nagpure and D. L. Wood, Carbon, 2016, 105, 52–76 CrossRef CAS.
- C. Schmitt, A. Kube, N. Wagner and K. A. Friedrich, ChemElectroChem, 2022, 9, e202101342 CrossRef CAS.
- H. Oka, Y. Makimura, T. Uyama, T. Nonaka, Y. Kondo and C. Okuda, J. Power Sources, 2021, 482, 228926 CrossRef CAS.
- H. L. Andersen, L. Djuandhi, U. Mittal and N. Sharma, Adv. Energy Mater., 2021, 11, 2102693 CrossRef CAS.
- Z. Jian, W. Luo and X. Ji, J. Am. Chem. Soc., 2015, 137, 11566–11569 CrossRef CAS PubMed.
- W. Luo, J. Wan, B. Ozdemir, W. Bao, Y. Chen, J. Dai, H. Lin, Y. Xu, F. Gu, V. Barone and L. Hu, Nano Lett., 2015, 15, 7671–7677 CrossRef CAS PubMed.
- S. Komaba, T. Hasegawa, M. Dahbi and K. Kubota, Electrochem. Commun., 2015, 60, 172–175 CrossRef CAS.
- T. Hosaka, K. Kubota, A. S. Hameed and S. Komaba, Chem. Rev., 2020, 120, 6358–6466 CrossRef CAS PubMed.
- D. Igarashi, R. Fujimoto, R. Tatara, T. Hosaka and S. Komaba, 48th Annu. Meet. Carbon Soc. Jpn., 2021, 1G38 Search PubMed.
- A. Yadav, H. Kobayashi, T. Yamamoto and T. Nohira, Electrochemistry, 2023, 91, 017002 CrossRef CAS.
- R. Hagiwara, K. Tamaki, K. Kubota, T. Goto and T. Nohira, J. Chem. Eng. Data, 2008, 53, 355–358 CrossRef CAS.
- S. Terada, T. Mandai, R. Nozawa, K. Yoshida, K. Ueno, S. Tsuzuki, K. Dokko and M. Watanabe, Phys. Chem. Chem. Phys., 2014, 16, 11737–11746 RSC.
- M. Hamada, R. Tatara, K. Kubota, S. Kumakura and S. Komaba, ACS Energy Lett., 2022, 7, 2244–2246 CrossRef CAS.
- S. Komaba, T. Ishikawa, N. Yabuuchi, W. Murata, A. Ito and Y. Ohsawa, ACS Appl. Mater. Interfaces, 2011, 3, 4165–4168 CrossRef CAS.
- T. Hosaka, S. Muratsubaki, K. Kubota, H. Onuma and S. Komaba, J. Phys. Chem. Lett., 2019, 10, 3296–3300 CrossRef CAS PubMed.
- T. Hosokawa, K. Matsumoto, T. Nohira, R. Hagiwara, A. Fukunaga, S. Sakai and K. Nitta, J. Phys. Chem. C, 2016, 120, 9628–9636 CrossRef CAS.
- H. Onuma, K. Kubota, S. Muratsubaki, T. Hosaka, R. Tatara, T. Yamamoto, K. Matsumoto, T. Nohira, R. Hagiwara, H. Oji, S. Yasuno and S. Komaba, ACS Energy Lett., 2020, 5, 2849–2857 CrossRef CAS.
- N. Xiao, G. Gourdin and Y. Wu, Angew. Chem., Int. Ed., 2018, 57, 10864–10867 CrossRef CAS PubMed.
- K. Kubota, T. Nohira and R. Hagiwara, J. Chem. Eng. Data, 2010, 55, 3142–3146 CrossRef CAS.
- T. Yamamoto, S. Nishijima and T. Nohira, J. Phys. Chem. B, 2020, 124, 8380–8387 CrossRef CAS.
- D. I. Iermakova, R. Dugas, M. R. Palacín and A. Ponrouch, J. Electrochem. Soc., 2015, 162, A7060 CrossRef CAS.
- E. Peled, J. Electrochem. Soc., 1979, 126, 2047 CrossRef CAS.
- E. Peled and S. Menkin, J. Electrochem. Soc., 2017, 164, A1703 CrossRef CAS.
- K. Kubota, M. Dahbi, T. Hosaka, S. Kumakura and S. Komaba, Chem. Rec., 2018, 18, 459–479 CrossRef CAS PubMed.
- H. Onuma, K. Kubota, S. Muratsubaki, W. Ota, M. Shishkin, H. Sato, K. Yamashita, S. Yasuno and S. Komaba, J. Mater. Chem. A, 2021, 9, 11187–11200 RSC.
- S. A. Solin and N. Caswell, J. Raman Spectrosc., 1981, 10, 129–135 CrossRef CAS.
- M. Shimizu, T. Koya, A. Nakahigashi, N. Urakami, T. Yamakami and S. Arai, J. Phys. Chem. C, 2020, 124, 13008–13016 CrossRef CAS.
- P. C. Eklund, G. Dresselhaus, M. S. Dresselhaus and J. E. Fischer, Phys. Rev. B: Solid State, 1977, 16, 3330–3333 CrossRef CAS.
- H. Fujimoto, M. Murakami, T. Yamanaka, K. Shimoda, H. Kiuchi, Z. Ogumi and T. Abe, J. Electrochem. Soc., 2021, 168, 080508 CrossRef CAS.
- J. R. Dahn, Phys. Rev. B: Condens. Matter Mater. Phys., 1991, 44, 9170–9177 CrossRef CAS PubMed.
- T. Ohzuku, Y. Iwakoshi and K. Sawai, J. Electrochem. Soc., 1993, 140, 2490 CrossRef CAS.
- C. Didier, W. K. Pang, Z. Guo, S. Schmid and V. K. Peterson, Chem. Mater., 2020, 32, 2518–2531 CrossRef CAS.
- H. Fujimoto, S. Takagi, K. Shimoda, H. Kiuchi, K. Okazaki, T. Murata, Z. Ogumi and T. Abe, J. Electrochem. Soc., 2021, 168, 090515 CrossRef CAS.
- J. Liu, T. Yin, B. Tian, B. Zhang, C. Qian, Z. Wang, L. Zhang, P. Liang, Z. Chen, J. Yan, X. Fan, J. Lin, X. Chen, Y. Huang, K. P. Loh and Z. X. Shen, Adv. Energy Mater., 2019, 9, 1900579 CrossRef.
- M. Ishikawa, T. Sugimoto, M. Kikuta, E. Ishiko and M. Kono, J. Power Sources, 2006, 162, 658–662 CrossRef CAS.
- T. Sugimoto, Y. Atsumi, M. Kikuta, E. Ishiko, M. Kono and M. Ishikawa, J. Power Sources, 2009, 189, 802–805 CrossRef CAS.
- I. A. Shkrob, T. W. Marin, Y. Zhu and D. P. Abraham, J. Phys. Chem. C, 2014, 118, 19661–19671 CrossRef CAS.
- S. Terada, H. Susa, S. Tsuzuki, T. Mandai, K. Ueno, K. Dokko and M. Watanabe, J. Phys. Chem. C, 2018, 122, 16589–16599 CrossRef CAS.
- H. Moon, R. Tatara, T. Mandai, K. Ueno, K. Yoshida, N. Tachikawa, T. Yasuda, K. Dokko and M. Watanabe, J. Phys. Chem. C, 2014, 118, 20246–20256 CrossRef CAS.
- T. Hosaka, K. Kubota, H. Kojima and S. Komaba, Chem. Commun., 2018, 54, 8387–8390 RSC.
- T. Hosaka, T. Matsuyama, K. Kubota, R. Tatara and S. Komaba, J. Mater. Chem. A, 2020, 8, 23766–23771 RSC.
- T. Mandai, K. Yoshida, S. Tsuzuki, R. Nozawa, H. Masu, K. Ueno, K. Dokko and M. Watanabe, J. Phys. Chem. B, 2015, 119, 1523–1534 CrossRef CAS.
- C. Zhang, K. Ueno, A. Yamazaki, K. Yoshida, H. Moon, T. Mandai, Y. Umebayashi, K. Dokko and M. Watanabe, J. Phys. Chem. B, 2014, 118, 5144–5153 CrossRef CAS.
- B. Jache and P. Adelhelm, Angew. Chem., Int. Ed., 2014, 53, 10169–10173 CrossRef CAS PubMed.
- H. Kim, J. Hong, G. Yoon, H. Kim, K.-Y. Park, M.-S. Park, W.-S. Yoon and K. Kang, Energy Environ. Sci., 2015, 8, 2963–2969 RSC.
- H. Kim, G. Yoon, K. Lim and K. Kang, Chem. Commun., 2016, 52, 12618–12621 RSC.
- J. O. Besenhard, H. Möhwald and J. J. Nickl, Carbon, 1980, 18, 399–405 CrossRef CAS.
- Z. Jian, C. Bommier, L. Luo, Z. Li, W. Wang, C. Wang, P. A. Greaney and X. Ji, Chem. Mater., 2017, 29, 2314–2320 CrossRef CAS.
- K. Kubota, S. Shimadzu, N. Yabuuchi, S. Tominaka, S. Shiraishi, M. Abreu-Sepulveda, A. Manivannan, K. Gotoh, M. Fukunishi, M. Dahbi and S. Komaba, Chem. Mater., 2020, 32, 2961–2977 CrossRef CAS.
- M. Ou, Y. Zhang, Y. Zhu, C. Fan, S. Sun, J. Feng, X. Sun, P. Wei, J. Xu, J. Peng, X. Wu, G. Jiang, Q. Li, C. Fang and J. Han, ACS Appl. Mater. Interfaces, 2021, 13, 28261–28269 CrossRef CAS PubMed.
- D. A. Stevens and J. R. Dahn, J. Electrochem. Soc., 2001, 148, A803 CrossRef CAS.
- H. Tan, R. Zhou and B. Zhang, J. Power Sources, 2021, 506, 230179 CrossRef CAS.
- K. Tatsumi, J. Conard, M. Nakahara, S. Menu, P. Lauginie, Y. Sawada and Z. Ogumi, J. Power Sources, 1999, 81–82, 397–400 CrossRef CAS.
- S. E. Hayes, R. A. Guidotti, W. R. Even, P. J. Hughes and H. Eckert, J. Phys. Chem. A, 2003, 107, 3866–3876 CrossRef CAS.
- H. Fujimoto, A. Mabuchi, K. Tokumitsu, N. Chinnasamy and T. Kasuh, J. Power Sources, 2011, 196, 1365–1370 CrossRef CAS.
- Y. Fang, K. Peuvot, A. Gratrex, E. V. Morozov, J. Hagberg, G. Lindbergh and I. Furó, J. Mater. Chem. A, 2022, 10, 10069–10082 RSC.
- R. Morita, K. Gotoh, M. Fukunishi, K. Kubota, S. Komaba, N. Nishimura, T. Yumura, K. Deguchi, S. Ohki, T. Shimizu and H. Ishida, J. Mater. Chem. A, 2016, 4, 13183–13193 RSC.
- X. Dou, I. Hasa, D. Saurel, C. Vaalma, L. Wu, D. Buchholz, D. Bresser, S. Komaba and S. Passerini, Mater. Today, 2019, 23, 87–104 CrossRef CAS.
- K. Gotoh, T. Yamakami, I. Nishimura, H. Kometani, H. Ando, K. Hashi, T. Shimizu and H. Ishida, J. Mater. Chem. A, 2020, 8, 14472–14481 RSC.
- Y. Morikawa, S. Nishimura, R. Hashimoto, M. Ohnuma and A. Yamada, Adv. Energy Mater., 2020, 10, 1903176 CrossRef CAS.
- Y. Youn, B. Gao, A. Kamiyama, K. Kubota, S. Komaba and Y. Tateyama, npj Comput. Mater., 2021, 7, 1–8 CrossRef.
- J. M. Stratford, P. K. Allan, O. Pecher, P. A. Chater and C. P. Grey, Chem. Commun., 2016, 52, 12430–12433 RSC.
- J. M. Stratford, A. K. Kleppe, D. S. Keeble, P. A. Chater, S. S. Meysami, C. J. Wright, J. Barker, M.-M. Titirici, P. K. Allan and C. P. Grey, J. Am. Chem. Soc., 2021, 143, 14274–14286 CrossRef CAS PubMed.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc03281g |
| This journal is © The Royal Society of Chemistry 2023 |