DOI:
10.1039/D3SC03514J
(Review Article)
Chem. Sci., 2023,
14, 9996-10024
Liquid electrolyte chemistries for solid electrolyte interphase construction on silicon and lithium-metal anodes†
Received
8th July 2023
, Accepted 11th August 2023
First published on 11th August 2023
Abstract
Next-generation battery development necessitates the coevolution of liquid electrolyte and electrode chemistries, as their erroneous combinations lead to battery failure. In this regard, priority should be given to the alleviation of the volumetric stress experienced by silicon and lithium-metal anodes during cycling and the mitigation of other problems hindering their commercialization. This review summarizes the advances in sacrificial compound-based volumetric stress-adaptable interfacial engineering, which has primarily driven the development of liquid electrolytes for high-performance lithium batteries. Besides, we discuss how the regulation of lithium-ion solvation structures helps expand the range of electrolyte formulations and thus enhance the quality of solid electrolyte interphases (SEIs), improve lithium-ion desolvation kinetics, and realize longer-lasting SEIs on high-capacity anodes. The presented insights are expected to inspire the design and synthesis of next-generation electrolyte materials and accelerate the development of advanced electrode materials for industrial battery applications.
 Sewon Park | Sewon Park received his PhD degree from the School of Energy and Chemical Engineering at the Ulsan National Institute of Science and Technology (UNIST), South Korea, under the supervision of Prof. Nam-Soon Choi. He is currently a post-doctoral researcher in Nam-Soon Choi's group at the Korea Advanced Institute of Science and Technology (KAIST), South Korea. His research focuses on the development of electrolytes for lithium-ion batteries. |
 Saehun Kim | Saehun Kim is currently a PhD candidate in the Department of Chemical & Biomolecular Engineering at the Korea Advanced Institute of Science and Technology (KAIST), South Korea, under the supervision of Prof. Nam-Soon Choi. His research focuses on the development of functional electrolyte additives for lithium-metal batteries. |
 Jeong-A Lee | Jeong-A Lee is currently a PhD candidate in the Department of Chemical & Biomolecular Engineering at the Korea Advanced Institute of Science and Technology (KAIST), South Korea, under the supervision of Prof. Nam-Soon Choi. Her research focuses on the development of functional electrolyte additives for lithium-metal batteries. |
 Makoto Ue | Makoto Ue is currently a Guest Professor at the Research Organization for Nano & Life Innovation, Waseda University. He obtained his M.S. (1981) and PhD (1995) from the University of Tokyo. He studied at the University of Pittsburgh and Lawrence Berkeley National Laboratory (1988–1990). He was a Special Researcher at the National Institute for Materials Science (2018–2020), a Vice President at Samsung SDI (2012–2017), and a Fellow at Mitsubishi Chemical Corporation (2007–2012). He has authored approximately 200 papers and 300 patents, as well as received eight academic awards including the IBA Research Award (1997), ECS Battery Technology Award (2004), and ECSJ Achievement Award (2022). |
 Nam-Soon Choi | Nam-Soon Choi is currently an Associate Professor in the Department of Chemical & Biomolecular Engineering at the Korea Advanced Institute of Science and Technology (KAIST), South Korea. She received her PhD (2004) from the Department of Chemical and Biomolecular Engineering at KAIST. She was a Professor at the Ulsan National Institute of Science and Technology (UNIST) (2010–2021). She is also an Associate Editor for ACS Applied Materials & Interfaces (since 2022). Her current research interests include the mechanistic analysis of electrode–electrolyte interfaces and the development of tailored electrolyte systems for rechargeable batteries. |
1. Introduction
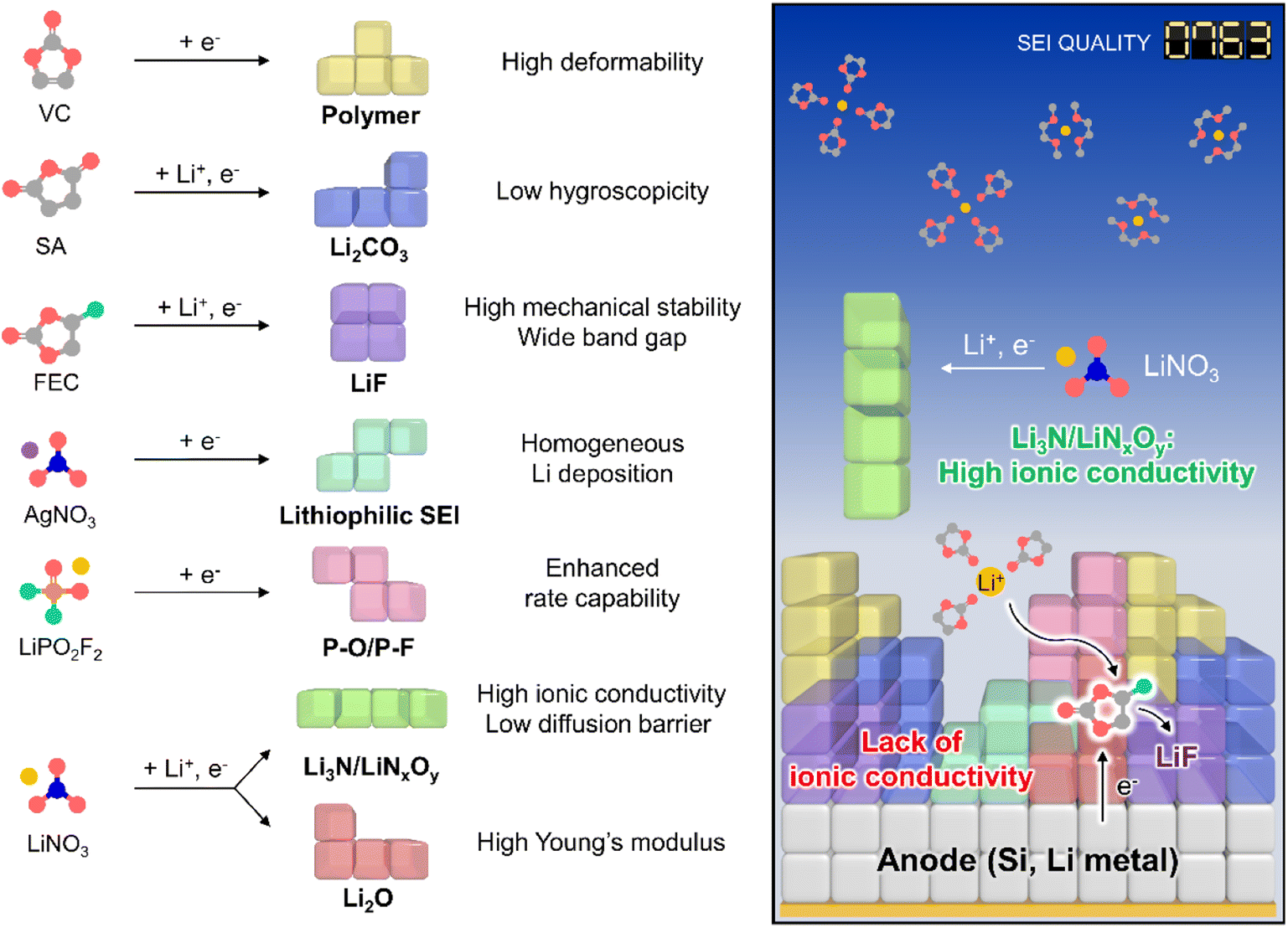
The creation of solid electrolyte interphases (SEIs) on the anodes of lithium chemistry-exploiting rechargeable batteries is important for their commercialization and performance enhancement, allowing for the high-energy-densities required by emerging applications such as electric vehicles. In this regard, high-capacity anode materials including silicon and lithium-metal dominate next-generation battery systems but suffer from large cycling-induced volume changes,1,2 which result in the mechanical fracture of protective SEI layers and thus cause persistent side reactions with electrolyte components.3 In turn, the undesired electrolyte decomposition at the anode during cycling parasitically consumes lithium-ions and solvents and gives rise to progressive SEI growth to increase the internal cell resistance.4 Although ethylene carbonate (EC) forms an SEI on graphite anodes,5 further SEI strengthening is essential to ensure the long lifespan of high-energy batteries containing silicon and lithium-metal anodes over wide temperature and current density ranges.4,6 The importance of interfacial chemistry has been amplified through the use of vinylene carbonate (VC)7 and fluoroethylene carbonate (FEC)8 for graphite and silicon anodes, respectively.9,10 Controlled SEI formation on anodes aided by ionic11 and nonionic reductive additives can prevent electrolyte decomposition and enhance electrochemical kinetics even under extreme conditions, which is something not easily achieved for SEI-free anodes (Fig. 1, Tables S1 and S2†).12,13
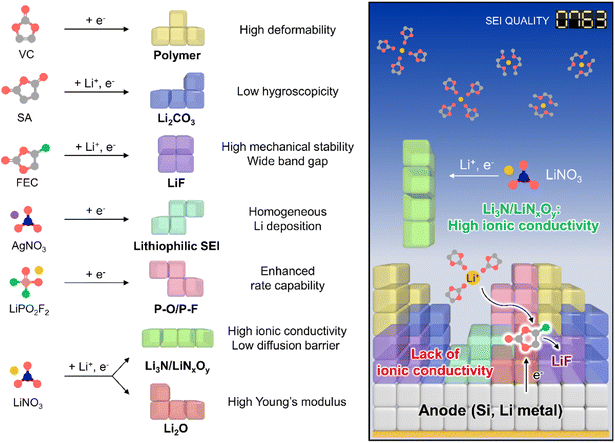 |
| | Fig. 1 Schematic illustration of SEI design considering individual component properties. | |
The inclusion of high-capacity silicon anodes into lithium-ion batteries (LIBs) is limited by the dynamic volume change-induced mechanical fracture of silicon during electrochemical (de)lithiation. This fracture results in the spatial rearrangement of silicon anode particles and thus causes the loss of electrical connection and the exposure of active silicon to induce irrevocable electrolyte decomposition. Consequently, strategies involving volumetric stress-adaptable interfacial engineering with regard to electrolyte formulation are required to enhance the electrochemical reversibility of silicon anodes. Unlike silicon anodes, which are chemically stable against electrolytes prior to electrochemical cycling, lithium-metal anodes are highly reactive toward electrolyte components and suffer from lithium dendrite formation and low coulombic efficiency (CE) due to the irreversible electrolyte decomposition-induced consumption of active lithium-metal and SEI thickening. Uncontrolled lithium dendrite formation poses safety (e.g., short-circuiting and explosion) risks and thus hinders the realization of practical lithium-metal batteries (LMBs). Lithium-metal anodes for LMBs engage in spontaneous chemical reactions with electrolyte components to form uncontrolled interlayers that do not effectively mitigate the electrochemical decomposition of electrolytes. This drawback highlights the need to modify the content and type of SEI formers, which regulate anode-electrolyte interfaces, and liquid electrolyte formulations including salts and solvents, which alter the (de)solvation chemistry of lithium-ions. Based on an understanding of existing SEI chemistries, this review highlights important findings that have enabled considerable improvements in the interfacial stability of silicon and lithium-metal anodes and establishes SEI structure–property relationships to suggest new research directions for liquid electrolyte chemistry. Therefore, the present summary is expected to generate significant ripple effects in the field of next-generation high-energy battery electrolytes.
2. Understanding the SEI structure
2.1 Microstructure
The SEI structure and nature are the key parameters determining the cyclability of silicon and lithium-metal anodes. The multiple functions of SEIs include the inhibition of undesired reactions between the anode and the electrolyte after initial cycling and the regulation of the lithium-ion flux to realize uniform lithiation. SEI characterization can be performed on scales ranging from molecular arrangements to millimeter-range macroscopic objects. A comprehensive understanding of structural and morphological SEI changes at the microscale can be achieved by optical and scanning electron microscopy (SEM) imaging (Fig. 2a and d). The coverage ability and (electro)chemical/mechanical integrity of SEIs directly affect the interfacial stability of anodes. SEIs with a coverage suitable for limiting the irrevocable damage to the anode surface directly in contact with the electrolyte conserve the uniform lithium-ion supply path, limit SEI thickening, and enable the reversible electrochemical reaction of the anode without a large overpotential.14 In addition, strain-adaptable SEIs sustaining repeated anode expansion and contraction ensure the conservation of the long-range anode microstructure with the use of minimal amounts of SEI-forming compounds.20,21
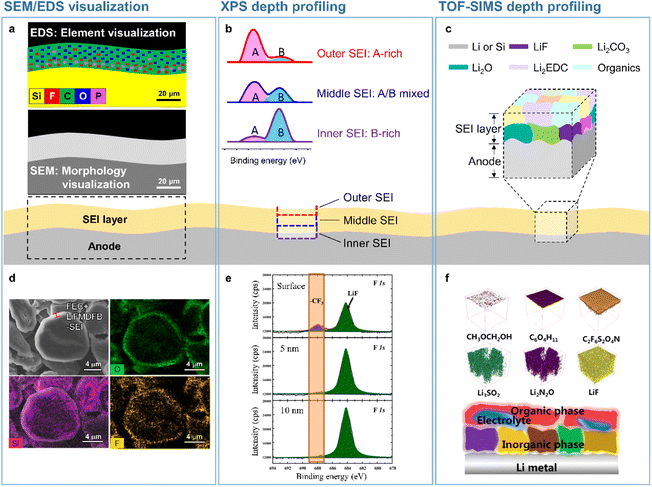 |
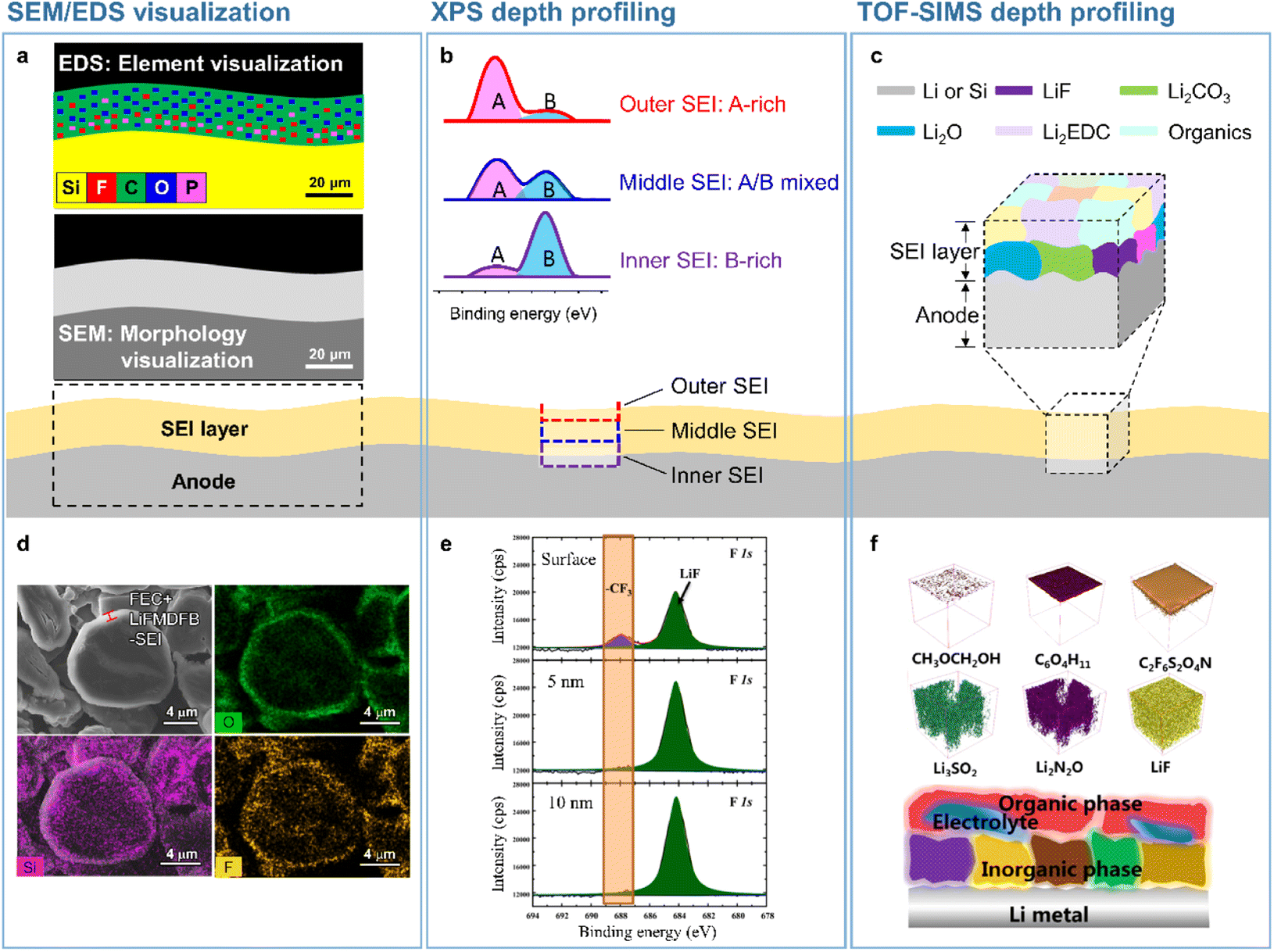
| | Fig. 2 Analytical techniques used to probe the SEI microstructure. (a) SEM/EDS visualization, (b) XPS depth profiling, and (c) TOF-SIMS depth profiling. (d) SEM/EDS analysis of SEIs formed on a silicon-embedded graphite anode. Reproduced from ref. 14 with permission of the Royal Society of Chemistry, copyright 2018. (e) XPS depth profiles of SEIs formed on a lithium-metal anode. Reproduced from ref. 15 with permission of Elsevier, copyright 2021. (f) TOF-SIMS 3D mapping of SEI fragments on the lithium-metal anode. Reproduced from ref. 16 with permission of American Chemical Society, copyright 2022. | |
Microscale visual observations of SEIs provide information on the effects of their chemical composition, thickness, and morphology on anode performance in batteries. Further information such as the crystallinity of SEI constituents and their spatial arrangement can be provided by nanoscale analysis. The spectroscopic methods used to identify the chemical structure of SEIs include infrared spectroscopy,22,23 Raman spectroscopy,24 X-ray photoelectron spectroscopy (XPS),23,25 time-of-flight secondary-ion mass spectrometry (TOF-SIMS), and energy-dispersive X-ray spectroscopy (EDS).26 Depth profiling techniques involving sputtering using X-rays or ion beams have been used to achieve a comprehensive understanding of the 3D chemical structure of SEIs. Among the above methods, XPS and TOF-SIMS profiling are the ones most effective for the 3D rendering of the SEI chemical structure (Fig. 2b, c, e, and f).15,27,28 The XPS depth profiling of silicon anodes exposed to an electrolyte containing FEC revealed that the corresponding SEI mainly consisted of LiF and polyene compounds.27 Additionally, TOF-SIMS depth profiling showed that LiF and organic species were abundantly present in both the outer and inner SEI layers, while Li2O was predominantly present in the inner SEI layer.28 The 3D chemical structure of SEIs on lithium-metal anodes has also been actively explored. SEIs produced on lithium-metal anodes benefit from mechanically robust inorganic species in the inner layer and therefore efficiently suppress lithium dendrite formation and lithium isolation, improving the electrochemical reversibility of lithium-metal.15 Mechanically fragile and organic-rich SEIs with low moduli decrease the CE and cycling lifetime of LMBs, whereas inorganic-rich SEIs with LiF, Li3N, LixNOy, and Li2O in the inner layer exert opposite effects.15
2.2 Nanostructure
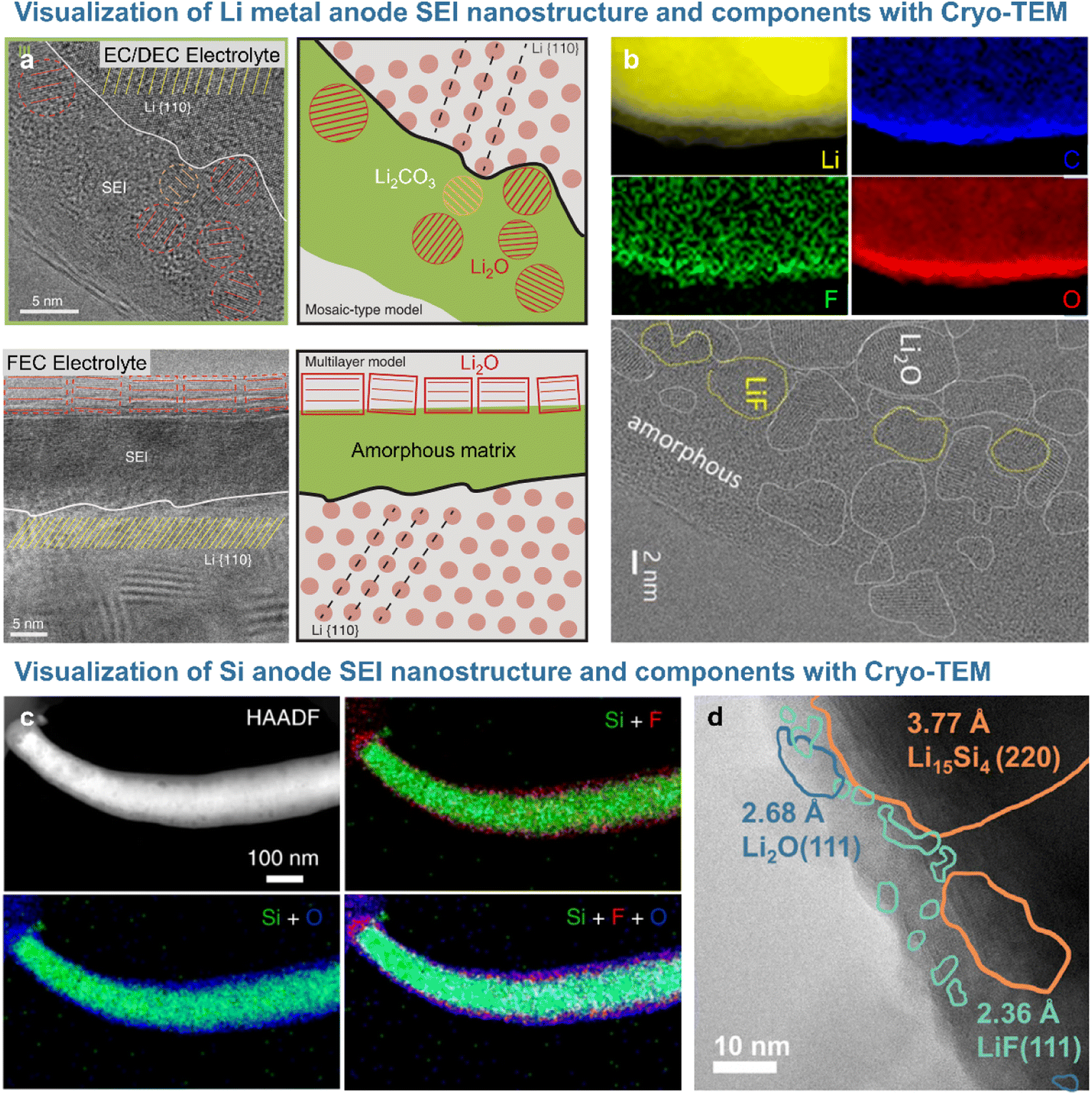
Short-range features and local structures can be characterized at the atomic level by transmission electron microscopy (TEM). Given that active electrode materials and SEIs with chemically sensitive species are likely to be damaged by the TEM beam, cryogenic sample preparation techniques (e.g., focused ion beam) and analysis methods such as cryo-TEM are used for non-destructive SEI analysis. The Cui17 group analyzed the morphology and crystal structure of lithium dendrites and SEI nanostructure using cryo-TEM to avoid beam-induced damage. In particular, the beam used for standard high-resolution TEM imaging damaged lithium dendrites, whereas cryo-TEM allowed the damage-free high-resolution analysis of lithium and SEI morphology and the crystal structure.17 The SEI formed on lithium dendrites in electrolytes containing carbonate solvents and lithium hexafluorophosphate (LiPF6) consisted of 3 nm Li2O and Li2CO3 particles randomly dispersed in an amorphous matrix (Fig. 3a). The SEI produced after the addition of 10 vol% FEC exhibited an ordered multi-layer structure featuring randomly distributed inorganic particles and organic compounds, with Li2O particles of ∼15 nm forming the outer layer and an amorphous polymer matrix forming the inner layer. However, LiF, which strongly impacts the performance of lithium-metal anodes, was not observed on the lithium dendrite surface.17,29 The Cui group revealed that lithium electrodeposited on a copper current collector in 1 M LiPF6 in [EC/DEC + 10% FEC] as an electrolyte was covered with an organic inner layer and an inorganic outer layer containing Li2O and LiF. Cryo-TEM and electron energy loss spectroscopy (EELS) analyses demonstrated that the 15 nm-thick compact SEI on lithium-metal was rich in Li2O and that LiF precipitated as particles larger than 100 nm on the surface of the highly conductive copper current collector.29 The Lucht and Gu groups showed that nanostructured LiF particles precipitated on the lithium-metal surface and realized a uniform diffusion field at the microscale to suppress the formation of lithium dendrites in FEC-supplemented electrolytes (Fig. 3b).18,30 The nanostructure of SEIs formed on silicon-based anodes can be visualized through cryogenic analysis (Fig. 3c and d). The Wang group demonstrated that the SEI formed on a silicon anode in the presence of FEC consisted of inert LiF nanoparticles in a thin (∼26 nm) amorphous layer (Fig. 3d).19 This result was largely consistent with the findings of Jaumann et al., who showed that 5 nm nanocrystalline LiF particles are produced at silicon nanoparticle anodes upon the addition of FEC.31 In contrast, the thick (∼41 nm) SEI formed on a silicon anode in an additive-free electrolyte was composed of electrochemically active Li2O, which forms resistive LixSiOy species (Li2O + Si + e− → LixSiOy + Li+) during delithiation. LiF nanocrystals not only enhanced the mechanical integrity of the dense SEI on the silicon anode, but also induced interstitial defects in this layer and facilitated the movement of lithium-ions through the same.31
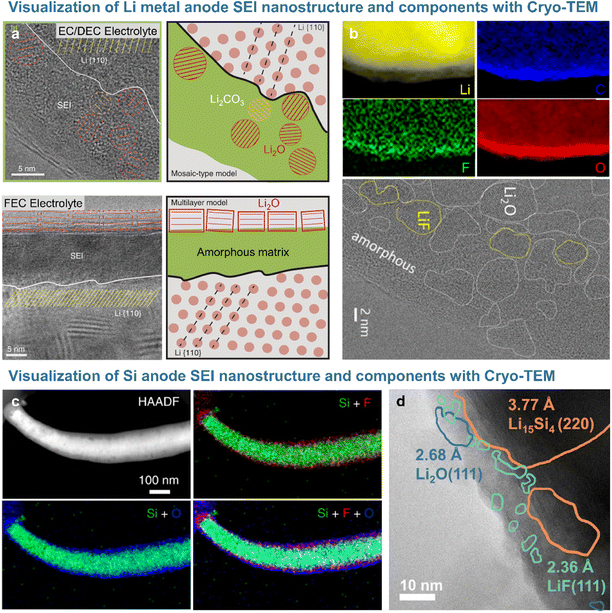 |
| | Fig. 3 (a) Cryo-TEM image of the SEI formed on lithium-metal with EC/DEC electrolyte and FEC electrolyte. Reproduced from ref. 17 with permission of AAAS, copyright 2017. (b) EELS mapping and Cryo-TEM images of the SEI on lithium-metal. Reproduced from ref. 18 with permission of Cell Press, copyright 2021. (c) Cryo-STEM image and EDS mappings of the silicon anode and SEI cycled in FEC-containing electrolyte. Reproduced from ref. 4 with permission of Springer Nature, copyright 2021. (d) Cryo-TEM image of the silicon anode after a cycle. Reproduced from ref. 19 with permission of Cell Press, copyright 2021. | |
2.3 Li-ion transport mechanisms
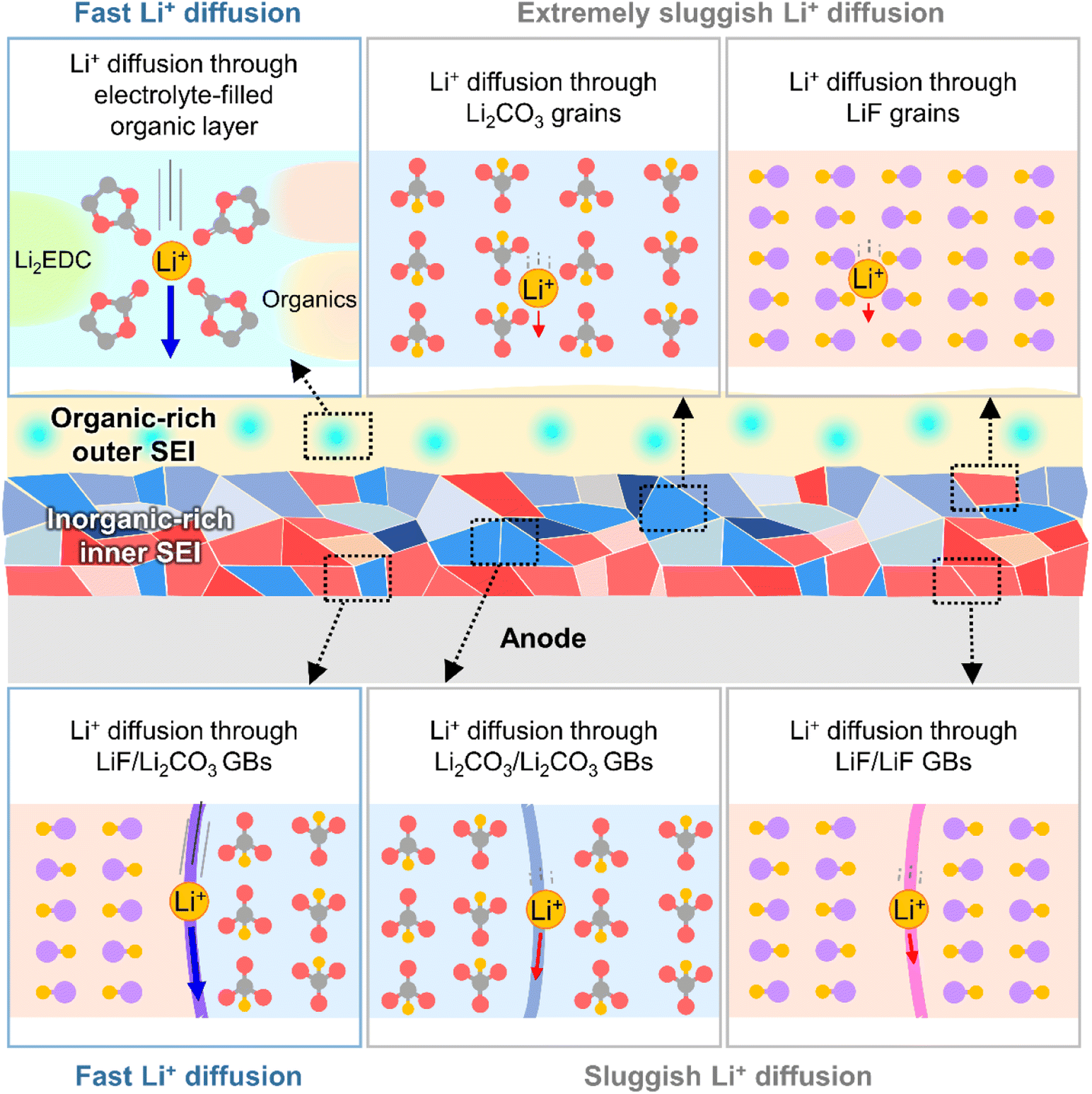
The kinetics of lithium-ion transport in the SEI influences the charge/discharge rate capability of batteries and lithiation/delithiation reversibility without undesired metallic lithium plating or vertical lithium growth in certain anode regions. The SEI exists in a swollen state, wetted by the electrolyte,32 and lithium-ions rapidly diffuse through the electrolyte-filled porous organic layer (Fig. 4).33 Diffusion through the dense inorganic SEI composed of polycrystalline materials is the rate-determining step.34 The diffusion of lithium-ions through the grains of inorganic species (LiF, Li2CO3, Li2O, and LiOH) is based on direct hopping, knock-off, vacancy diffusion and concerted exchange.35–37 First-principle calculations on each diffusion mechanism revealed that the migration barriers for lithium-ions in Li2CO3, Li2O, and LiOH crystals are one order of magnitude lower than those in LiF crystals, which indicates that lithium-ion diffusion through bulk Li2CO3, Li2O, and LiOH crystals is much faster than that through LiF crystals.35–37 However, lithium-ion diffusion through grain boundaries is orders of magnitude faster than that through bulk grains.37,38 In contrast to homogeneous grain boundaries (LiF/LiF, Li2O/Li2O, and Li2CO3/Li2CO3), heterogeneous ones composed of LiF and other inorganic species (LiF/Li2O and LiF/Li2CO3) facilitate lithium-ion transport by providing low energy barriers and short diffusion distances (Fig. 4).34,39,40 Nanocrystalline LiF generated from fluorine-donating additives such as FEC on silicon and lithium-metal anodes enhances the lithium-ion conductivity of the corresponding SEIs and enables the fabrication of batteries with high rate capabilities.31
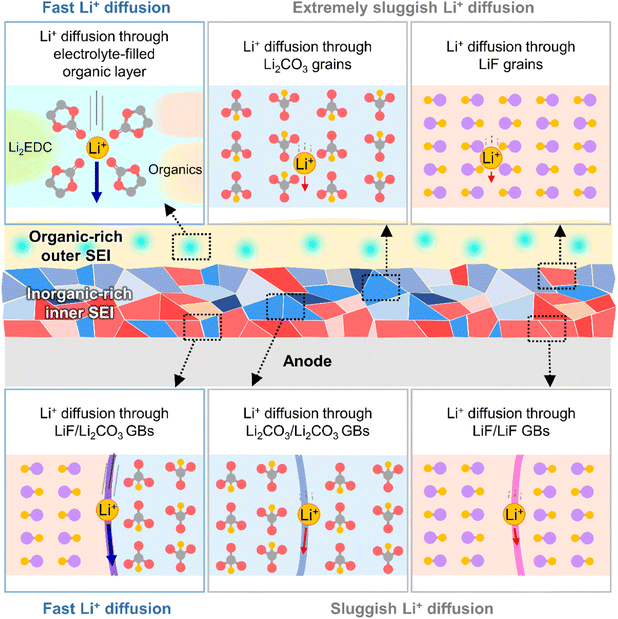 |
| | Fig. 4 Schematic illustration of lithium-ion transport at SEIs. GBs = grain boundaries. | |
2.4 Reactive species-driven instability of SEIs
2.4.1 Corrosive species.
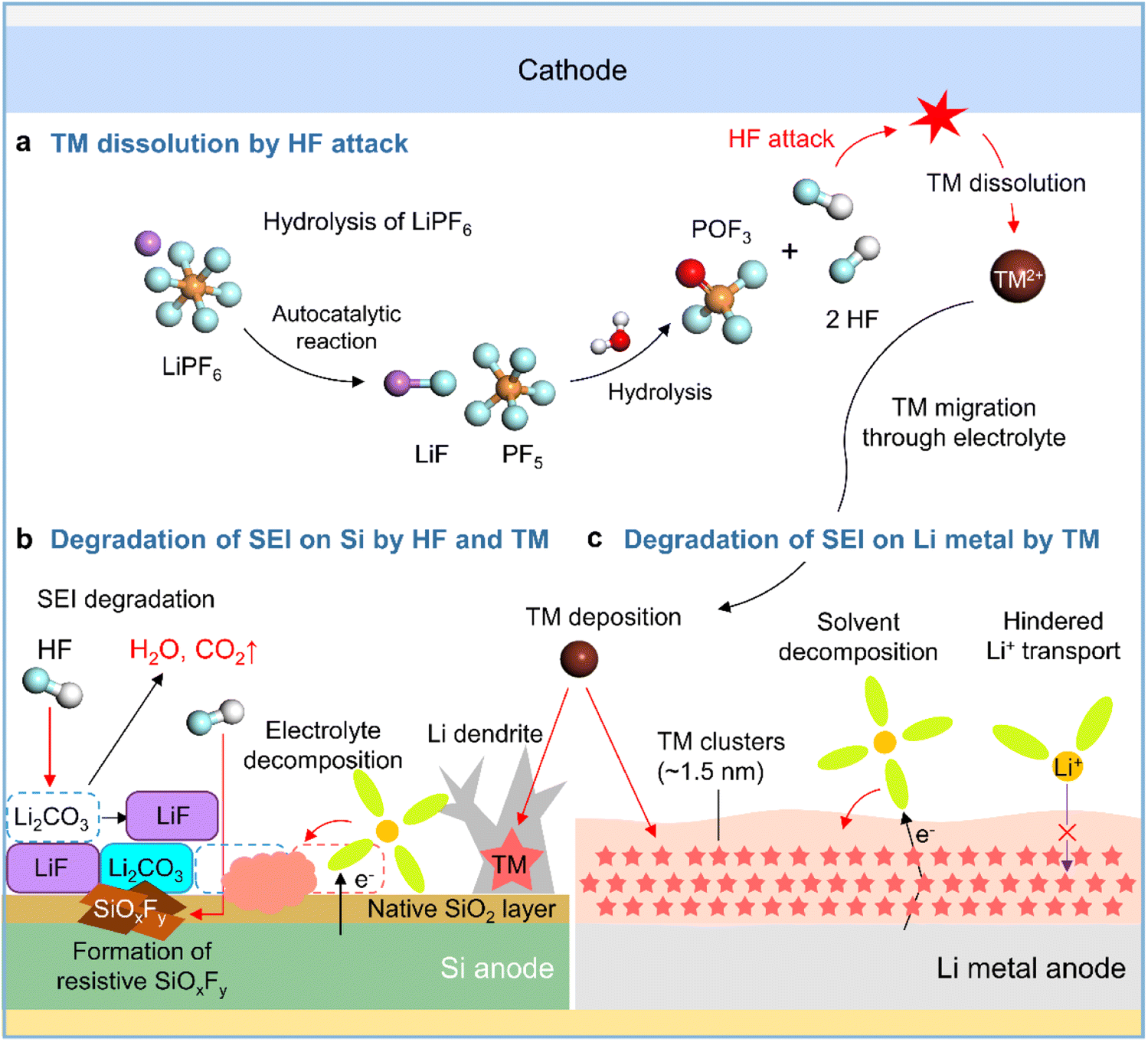
Although lithium bis(trifluoromethanesulfonyl)imide (LiTFSI) is hygroscopic, it is not hydrolyzed easily owing to the presence of strong C–F bonds. In contrast, LiPF6 in commercial electrolytes is highly susceptible to irreversible hydrolysis by traces of moisture inside the battery (Fig. 5a).41,42 Thus, the generated corrosive acidic compounds such as HF, HPO2F2, H2PO3F, and H3PO4 cause the leaching of transition metals from the cathode, carbonate solvent decomposition producing gases such as CO2, and the chemical/physical degradation of the anode SEI.43 SEI constituents such as Li2CO3, Li2O, LiOCO2R, and LiOR are decomposed by HF attack (Li2CO3 + 2HF → 2LiF + H2O + CO2, Li2O + 2HF → 2LiF + H2O, LiOR + HF → LiF + ROH, and LiOCO2R + HF → LiF + ROH + CO2) to form CO2 gas, which provokes cell swelling, and H2O, which causes parasitic reactions in electrolytes (Fig. 5b).58,59 HF reacts with the native oxide layer (SiO2) on the surface of silicon anodes to form SiOxFy (Fig. 5b).43 SEIs formed on anodes exposed to LiPF6-based electrolytes are apt to be thick and porous because of the repeated destruction/reconstruction caused by HF attack.60,61 In addition, HF induces the decomposition of binders such as sodium carboxymethyl cellulose (R–COONa + HF → R–COOH + NaF) in silicon-based anodes, resulting in weakened bonding between silicon particles and electrically conductive agents.62
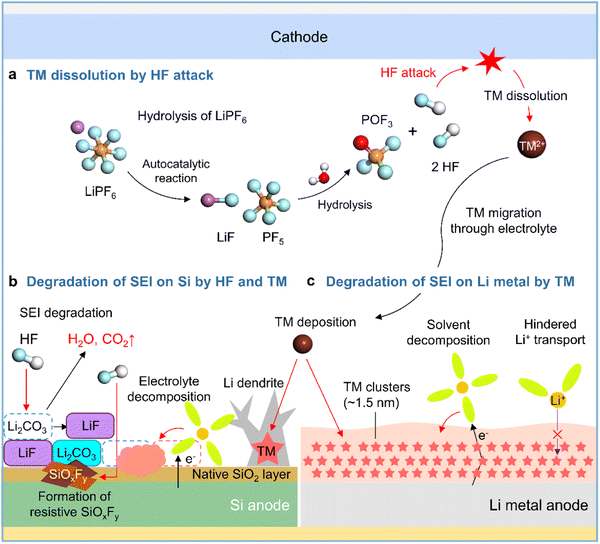 |
| | Fig. 5 Schematic illustration of (a) LiPF6 hydrolysis and HF-triggered transition metal (TM) dissolution from the cathode, (b) degradation of the SEI on the silicon anode caused by HF attack and TM deposition, and (c) degradation of the SEI on the lithium-metal anode caused by TM deposition. | |
2.4.2 Transition metal deposition on anodes.
Transition metal leaching from nickel-rich layered oxides (LiNixCoyMnzO2, x ≥ 0.5, x + y + z = 1) and high-voltage spinel cathodes (LiMn2O4 and LiNi0.5Mn1.5O4) is a major cause of performance degradation in LIBs.63,64 High-valence Ni4+ species formed in the deeply charged states of nickel-rich layered cathodes are reduced by electron-rich electrolyte components to Ni3+ and Ni2+, causing problems such as lattice oxygen loss, cation mixing, and nickel-ion leaching.63,65 Crystal structure distortion due to the Jahn–Teller effect and the disproportionation of Mn3+ to form the electrolyte-soluble Mn2+ aggravates Mn2+ leaching from the cathode and provokes Mn deposition on the anode, which hinders anode lithiation and increases anode impedance.64,66,67 Additionally, transition metal leaching from nickel-rich layered cathodes and high-voltage spinel cathodes is promoted by the corrosive HF generated via the hydrolysis of LiPF6 in commercial electrolytes.43 Several studies have reported the catalytic decomposition of electrolyte components caused by transition metals deposited on graphite anodes and dendritic lithium growth (Fig. 5b).64,68,69 However, the mechanism of the interfacial degradation of silicon and lithium-metal anodes caused by transition metal deposition was first examined only in 2019.70,71 Research on various cathode–anode combinations and electrolyte compositions aids the advancement of silicon and lithium-metal anodes. Zhang et al. demonstrated the effects of transition metals leached from LiNi0.90Mn0.05Co0.05O2 cathodes on SiOx/graphite anodes,70 showing that higher contents of dissolved transition metals in the electrolyte resulted in the thickening of the SEI on the SiOx/graphite anode and thus adversely affected its performance. Furthermore, TOF-SIMS analysis of the SiOx/graphite anode surface after 100 cycles revealed that the presence of 20 mM Ni2+, Co2+, or Mn2+ in the electrolyte significantly increased the amount of organic C2HO− species produced by the decomposition of carbonate solvents involving an increase in inorganic PO2− and LiF2− ions produced by the decomposition of LiPF6. Furthermore, transition metals deposited on silicon anodes can catalyze electrolyte decomposition. Cui et al. used cryo-TEM to show that electrolytes with dissolved Ni2+ promote the formation of nanoscale nickel clusters with a median diameter of 1.5 nm inhomogeneously distributed in the SEI on lithium-metal anodes, revealing that the SEI with these clusters was more than four times thicker than that without nickel clusters (Fig. 5c).72 Moreover, the nickel-rich SEI led to the continuous reduction of the electrolyte by providing a pathway for electron transport and hindered lithium-ion migration, thus resulting in large ohmic polarization that induced unwanted metallic lithium plating on the anode below 0 V vs. Li/Li+ (Fig. 5c).
3. Regulation of SEI chemistry on silicon and lithium-metal anodes
3.1 Balanced heterostructured SEIs
3.1.1 Organic-rich SEIs.
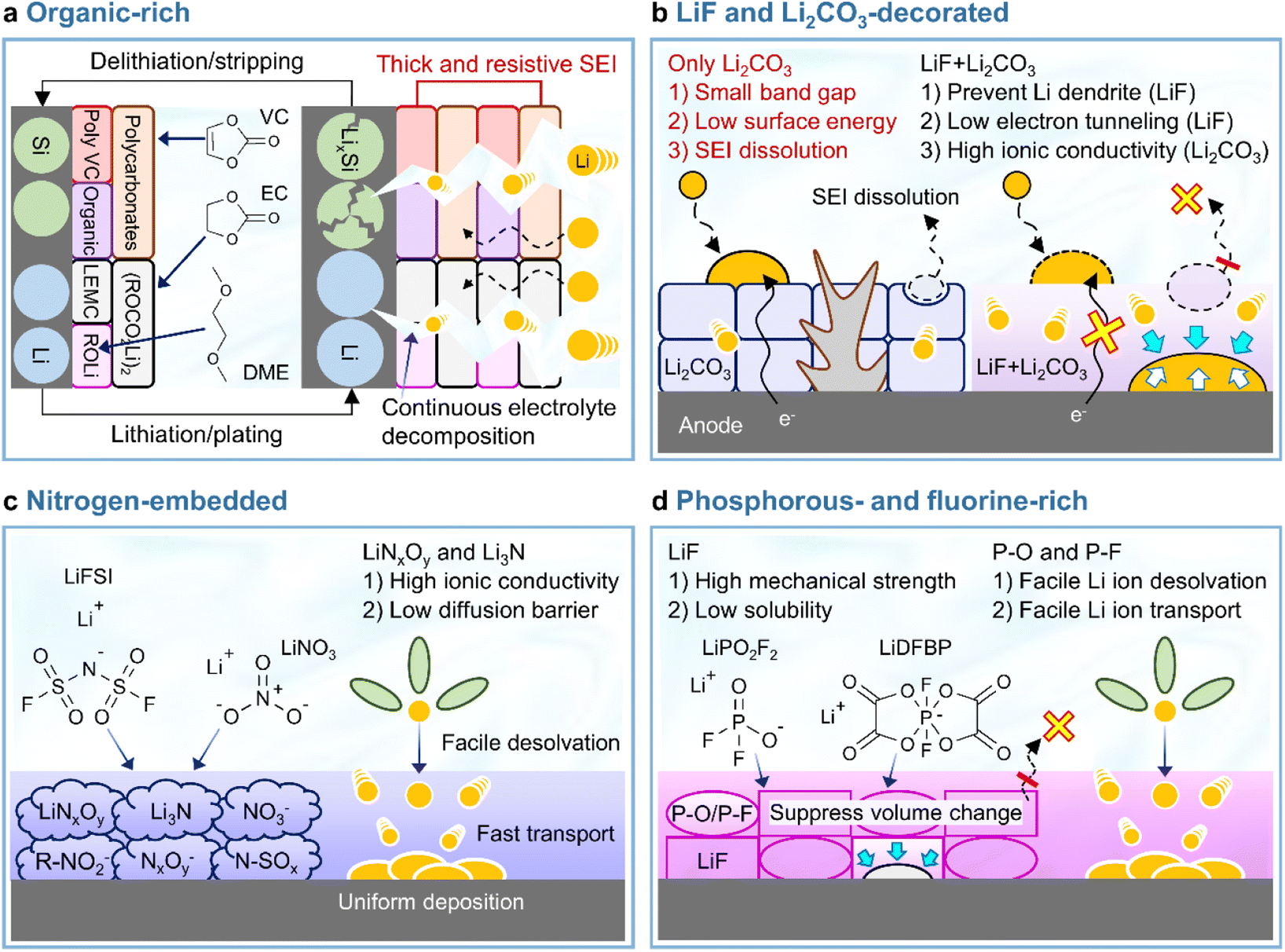
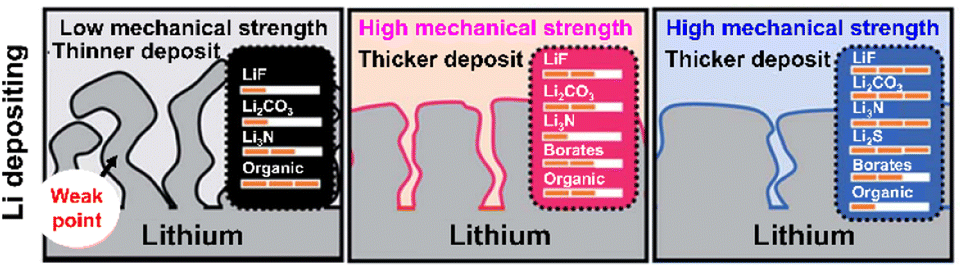
In view of its positive impact on graphite anodes, VC has also been assertively employed as an electrolyte additive for silicon anodes.73–75 In the late 2000s and early 2010s, VC was demonstrated to form carbon- and oxygen-rich SEIs with low amounts of phosphorus and fluorine generated by the decomposition of LiPF6 on thin film silicon anodes without carbon sources such as binders and conducting agents.73,75 The VC-driven formation of organic-rich SEIs containing poly(VC), polycarbonates, and Li2CO3 mitigates electrolyte decomposition (which results in a inhomogeneous SEI structure) and enables uniform lithiation across the entire surface of silicon with less mechanical fracturing (Fig. 6a). However, as the thickness of the silicon thin film increased from 100 to 500 nm, the VC-derived SEI with high impedance showed a capacity decrease.74 Moreover, when silicon nanoparticles with a large surface area and carbon conducting agents were used to assure the high electronic conductivity of anodes, the contents of carbon, oxygen, phosphorus, and fluorine components in the VC-derived SEI were similar to those in the VC-free electrolyte after cycling.76 This similarity was ascribed to the low strain adaptability of VC-derived SEIs for silicon lithiation and delithiation accompanied by large volume changes and the occurrence of uncontrolled electrolyte decomposition during repeated cycling (Fig. 6a). Moreover, spatially dense poly(VC)-rich SEIs act as resistive layers impeding lithium-ion transport and thus result in poor rate capability.31,77 Similar behavior was observed when succinic anhydride (SA) was used as an additive. On silicon thin film anodes, SA formed a hydrocarbon- and Li2CO3-based SEI that inhibited salt decomposition and improved anode cyclability.78 However, when high-surface-area silicon/mesoporous carbon anodes were used, SA formed a thick and resistive organic-rich SEI and resulted in performance similar to that observed in the absence of electrolyte additives.79 In this regard, one can consider the complementary usage of mechanically robust and electrically insulating inorganic compounds such as LiF and adhesive deformable organic species in the SEI to realize high-structural-stability silicon anodes without well-defined spaces (such as those observed for graphite anodes) and facile lithium-ion transport at silicon anodes while mitigating electrolyte decomposition. Early studies on SEIs produced on lithium-metal anodes mainly focused on the formation and identification of organic-rich SEIs.80–82 The generation of organic-rich SEIs is ascribed to two reasons. First, as the Fermi energy level of metallic lithium exceeds the reduction potential of organic solvents, the lithium-metal anode engages in spontaneous chemical reactions with the organic solvent-containing electrolyte, which causes aggressive SEI formation.83 Second, in the case of conventional electrolytes with an inorganic salt concentration of 1 M, lithium-ions are coordinated by the nucleophilic oxygen atoms of solvent molecules, while anions are present in bare form, i.e., are relatively uninvolved in the formation of solvation sheaths.83 Under such conditions, anion depletion occurs in the vicinity of the lithium-metal anode, as most mobile anions migrate to the cathode during charging. Consequently, the SEI is mostly composed of byproducts generated by the reductive decomposition of organic solvents. The composition of organic-rich SEIs depends on the type of solvent used. For instance, ROCO2Li, (ROCO2Li)2, lithium ethylene monocarbonate (LEMC), dilithium ethylene dicarbonate (Li2EDC), lithium methyl carbonate (LMC), and RCO3Li are produced in carbonate-based solvents, while ethereal solvents generate lithium alkoxides (ROLi) (Fig. 6a).44,81,83 The formation of organic SEIs is easier in carbonate-based solvents than in ethereal solvents because of the higher reactivity of the former toward lithium-metal. However, recent research on the interfaces of lithium-metal anodes has not dealt solely with the formation of organic-rich SEIs because of their limitations.84 Organic-rich SEIs strongly adhere to the lithium-metal anode and are therefore stably anchored, although their low mechanical strength and interfacial energy result in poor resistance to volume changes during repeated lithium plating and stripping resulting in the formation of damaged and less dense SEIs (Fig. 6a and Table 1). In addition to this mechanical disintegrity, lithium-ion diffusion to the SEI–lithium interface is uneven, leading to vertical lithium penetration and ultimately resulting in lithium dendrite growth. These processes cause irreversible lithium consumption, low CE, and a rapid increase in interfacial resistance. In severe cases, the current collector may become disconnected from the lithium-metal anode and large amounts of inactive lithium residuals can be formed. Finally, the practical application of organic SEIs alone is hindered by the risks posed by the formation of lithium dendrites and the flammability of conventional organic solvents. For these reasons, researchers strive to improve the cyclability of LMBs by mixing or layering inorganic SEI components with organic ones rather than by solely forming organic SEIs on lithium-metal anodes. The Mullins group demonstrated that the relative proportions of organic and inorganic species in the SEI influence its mechanical strength (Fig. 7),85 revealing that high-mechanical-strength multicomponent inorganic-rich SEIs can be realized on lithium-metal anodes.
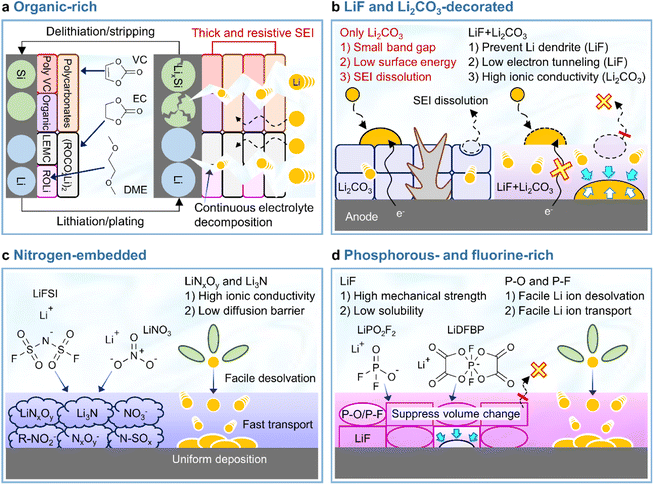 |
| | Fig. 6 Schematic illustration of balanced heterostructured SEIs. (a) Organic-rich SEI, (b) LiF and Li2CO3-decorated SEI, (c) nitrogen-embedded SEI, and (d) phosphorus- and fluorine-rich SEI. | |
Table 1 Characteristics of SEI components
|
|
Organics |
LiF |
Li2O |
Li2CO3 |
Li3N |
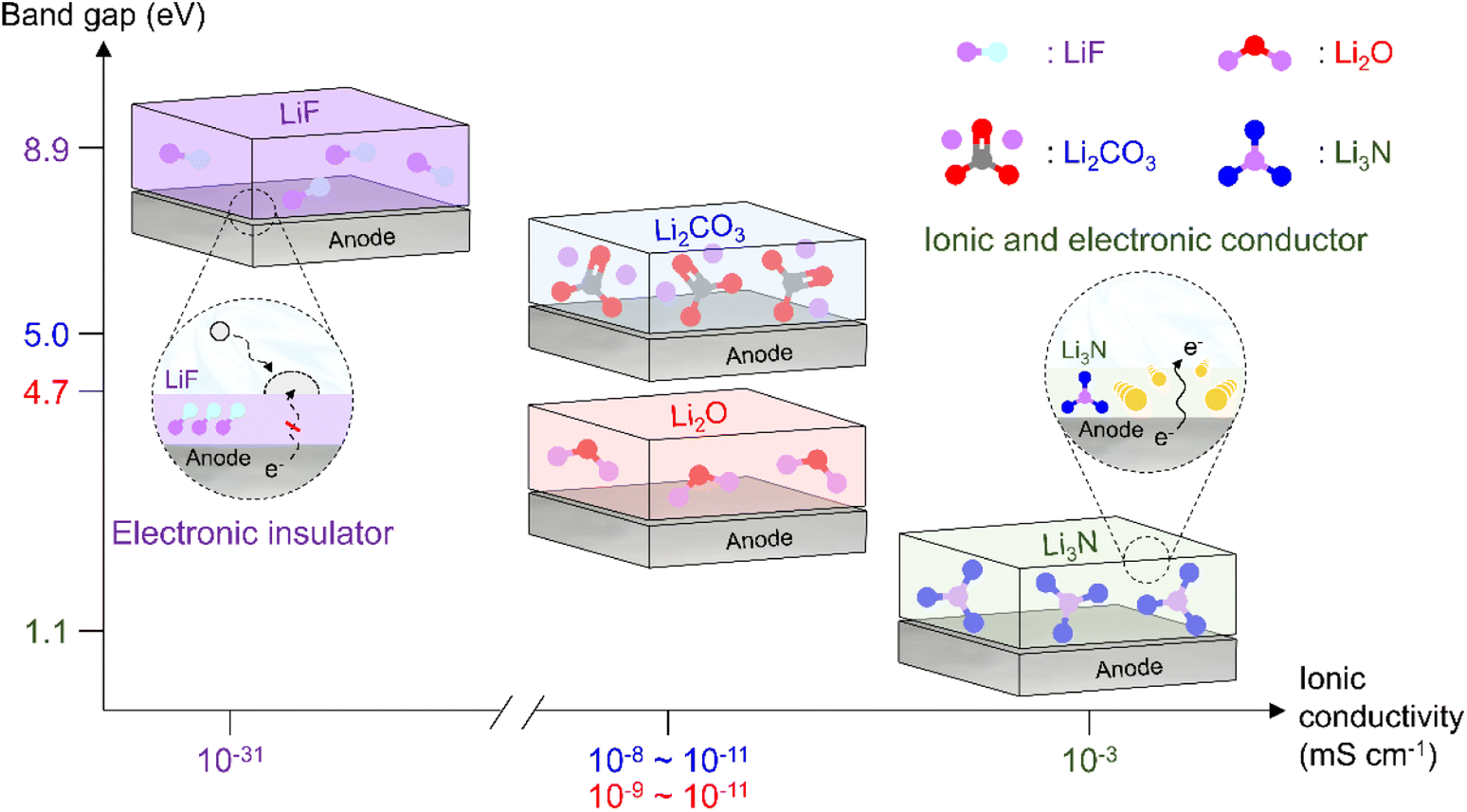
| Band gap (eV) |
|
8.9 (ref. 44) |
4.7 (ref. 44) |
5.0 (ref. 44) |
1.1 (ref. 44) |
| Shear modulus (GPa) |
<1.0 (ROCO2Li)45 |
55.6 (ref. 45) |
45.6 (ref. 45) |
28.9 (ref. 45) |
|
| Young's modulus (GPa) |
5.8 (LiEC),46 8.9 (LiMC),46 18.9 (Li2EDC)46 < 1.0 (ROCO2Li)47 |
65.0 (ref. 47) |
169.0 (ref. 47) |
75.0 (ref. 47) |
48.0 (ref. 48) |
| Diffusion barrier (eV) |
0.6 (Li2EDC)49 0.8 (Li alkyl carbonates)47 |
0.6–0.7 (ref. 49 and 50) |
0.2 (ref. 50) |
0.2–0.5 (ref. 50) |
0.007–0.038 (ref. 50) |
| Electrochemical stability window (V vs. Li/Li+) |
|
0–6.4 (ref. 51) |
∼3.0 (ref. 51) |
|
0.4 (ref. 51) |
| Ionic conductivity (S cm−1) |
10−9 (Li2EDC, LMC)50,52 6.4 × 10−6 (LEMC)47 |
10−31 (ref. 47) |
10−9–10−11 (ref. 47) |
10−8–10−11 (ref. 47) |
10−3 (ref. 53) |
| Chemical stability |
|
Low solubility54 |
|
Low hygroscopicity44 |
Poor chemical stability under ambient moist conditions56 |
| High solubility55 |
| Interfacial energy (meV Å−2) |
|
73.3 (ref. 57) |
38.7 (ref. 57) |
59.2 (ref. 57) |
|
| Lithium dendrite suppression ability (eV Å−2 MPa, Section S1) |
|
5129.0 (ref. 57) |
3018.0 (ref. 57) |
3731.0 (ref. 57) |
|
 |
| | Fig. 7 Schematic effects of organic and inorganic species on the mechanical strength of SEIs on lithium-metal anodes. Reproduced from ref. 85 with permission of the Royal Society of Chemistry, copyright 2019. | |
3.1.2 LiF and Li2CO3-decorated SEIs.
Although various structures have been proposed as interface modifiers for silicon and lithium-metal anodes, LiF remains the dominant material for realizing highly stable SEIs. The formation of LiF-based SEIs on lithium-metal anodes has been extensively studied to inhibit the formation of lithium dendrites owing to the high chemical stability of these SEIs, their low solubility in electrolytes, high shear modulus, high mechanical strength, and wide electrochemical stability window (Table 1).51,54,86 Unlike Li2CO3, which has a narrow band gap and low surface energy, LiF has a wide band gap and therefore does not allow electron tunneling to the top layer of the SEI.87 The lower solubility of LiF in carbonate solvents compared to that of Li2CO3 helps inhibit SEI dissolution and enables long-term anode stability.88 However, the poor ionic conductivity of LiF (∼10−31 S cm−1) leads to sluggish lithium-ion diffusion kinetics and generates significant overpotentials upon lithium deposition and stripping.89 Li2CO3 features a higher ionic conductivity (10−8–10−11 S cm−1) and lower density (2.11 g cm−3) than LiF (2.64 g cm−3) and may therefore facilitate the diffusion of lithium-ions through the grain boundaries of SEI components.90,91 Nonetheless, the low mechanical strength and surface energy of Li2CO3 cannot prevent the generation of lithium dendrites, making it an unsuitable SEI component for LMBs (Fig. 6b).57,92 Another important factor to consider for the formation of high-quality anode SEIs is their solubility in the employed electrolytes during cycling or at elevated temperatures, as SEI dissolution accelerates the side reactions of electrolytes at anodes. In this regard, LiF and Li2CO3-decorated SEIs have complementary features that can compensate for the corresponding individual shortcomings (Fig. 6b). SEIs containing both LiF and Li2CO3 can exhibit high ionic conductivity while featuring an electrically insulating nature, high interfacial energy, and high mechanical strength, thus helping in suppressing dendritic lithium creation and improve lithium-ion diffusion to lithium-metal anodes.40,93 The synergistic effect of LiF and Li2CO3 was demonstrated by Wu et al.40 According to their calculations, a heterostructured SEI decorated with LiF and Li2CO3 featured a higher lithium-ion adsorption energy (0.66 eV) than Li2CO3-rich (0.47 eV) and LiF-rich (0.49 eV) SEIs, thus exhibiting a higher affinity for lithium-ions and leading to a more uniform lithium-ion flux and lithium deposition. Additionally, the difficulty of electron transport through the LiF and Li2CO3-decorated SEI allowed lithium-ions to be reduced at the SEI–lithium interface and not on the SEI surface, which inhibited the formation of lithium dendrites. Stable SEI formation on lithium-metal anodes can be achieved through the appropriate combination of several SEI components with different characteristics rather than through the use of a single SEI component.94 Multilayer SEI structuring is further discussed in Section 3.3.4. Importantly, the synergistic effects of LiF and Li2CO3 as SEI components have also been reported for silicon-based anodes.91,95,96 An engineered LiF and Li2CO3-containing SEI on a silicon thin film anode was shown to be less resistive during lithiation and delithiation while preventing electrolyte reduction because of its superior electronically insulating nature.91 Icosafluoro-15-crown-5-ether, which forms resistive LiF impeding the electrochemical reaction of anodes, was combined with ion-conductive Li2CO3 to expand the lifespan of a SiO-graphite anode.95 The defects present at the LiF/Li2CO3 interface can not only cause electron depletion near Li2CO3 and thus enhance its insulating properties, but also provide a pathway for lithium-ion transport and thus increase lithium-ion conductivity.
3.1.3 Nitrogen-embedded SEIs.
Lewis-basic nitrogen-containing species such as LiNxOy, Li3N, N–SOx, NO3−, NxOy−, and R–NO2− can support facile lithium-ion transport through the SEI while minimizing the localization of the lithium-ion flux (Fig. 6c).61,97 In particular, LiNO3, which is a representative substance for constructing nitrogen-embedded SEIs, can be reduced to LiNxOy and Li3N (LiNO3 + 8Li+ + 8e− → Li3N + 3Li2O). Additionally, Li3N can be generated by the decomposition of LiNxOy.61,98–100 Both LiNxOy and Li3N, which are the main products of LiNO3 reduction, exhibit high ion conductivities of ∼10−3 S cm−1 at room temperature and help preserve a uniform lithium-ion flux at the lithium-metal anode even at high current densities (Table 1).101,102 To take advantage of nitrogen-containing SEI formation on the lithium-metal anode, one should pair the use of LiNO3 as an electrolyte additive with the use of high-donor number (high-DN) ethereal solvents such as 1,2-dimethoxyethane and tetraethylene glycol dimethyl ether.103–105 However, as the DN (21.1 kcal mol−1) of the NO3− anion exceeds those of carbonate solvents (16.4 kcal mol−1 for EC, 17 kcal mol−1 for dimethyl carbonate, 16.0 kcal mol−1 for diethyl carbonate (DEC), and 9.1 kcal mol−1 for FEC), LiNO3 is insufficiently dissociated in conventional carbonate-based electrolytes.61,105 This limited dissociation can be enhanced through the use of solubilizers such as tris(pentafluorophenyl)borane, indium trifluoromethanesulfonate, and lithium tetrafluoroborate, additives with different cations, and organic nitrates such as isosorbide dinitrate.61,97,106–112 A Lewis acid–base adduct forming a dative bond with LiNO3via its 1-(trimethylsilyl)imidazole moiety was developed to increase the solubility of LiNO3 in carbonate-based electrolytes.113 Early studies on LiNO3 as an electrolyte additive primarily focused on its role in the formation of nitrogen-decorated SEIs during the operation of LMBs and the influence of this salt on SEI properties.114 Besides, LiNO3 tends to weaken the ion–dipole interaction between lithium-ions and solvents and enhances lithium-ion desolvation kinetics, particularly at extremely low temperatures.115 The construction of nitrogen-containing SEIs in LMBs can be achieved by directly introducing LiNO3, nitrates, or solvents capable of generating Li3N-based SEIs. For instance, 2,2,2-trifluoro-N,N-dimethylacetamide (FDMA; FDMA + 6Li+ + 6e− → LiC2OF3 + 2LiCH3 + Li3N and FDMA + 7Li+ + 7e− → LiCF3 + LiCO + 2LiCH3 + Li3N) can result in the formation of Li3N-containing SEIs.116 In the same manner, nitrogen-containing SEIs with high ionic conductivity species such as Li3N enable fast lithium-ion transport at silicon anodes.117–119 Silicon nitrides (Si3N4 and SiNx) feature high mechanical stability (Young's modulus > 1.1 GPa) and can act as structural buffers for the volume change of silicon during repeated lithiation and delithiation, enabling the formation of Li3N for ion-transmittable SEI generation in carbonate-based electrolytes.117–119 Thus, the coating of silicon anode particles with silicon nitride can be a good option for the formation of nitrogen-containing SEIs without using LiNO3, which is poorly soluble in carbonate-based electrolytes. Interfacial engineering using nitrogen-donating compounds offers versatile routes to the construction of such SEIs.
3.1.4 Phosphorus- and fluorine-rich SEIs.
Phosphate-based additives forming polar P–O motifs as SEI constituents tune the channels for facile lithium-ion movement through LiF-rich SEIs providing grain boundaries as dominant lithium-ion pathways (Fig. 6d).120 In particular, lithium difluorophosphate (LiDFP) forms ionically conductive SEIs composed of LiF and polar P–O species on graphite anodes and is therefore commonly chosen as an electrolyte additive for EV-adaptable high-power batteries over wide temperature ranges.121 Moreover, this additive also effectively works for silicon-based anodes.122 When employed in conjunction with FEC, LiDFP forms P–O species along with the LiF and polymeric species (polyene and poly(VC)) formed by FEC and reduces the SEI impedance of silicon-based anodes.122 Lithium difluoro(bisoxalato) phosphate (LiDFBOP) forms spatially flexible SEIs that comprise LiF and phosphorus-/fluorine-containing organics and can tolerate the volume changes due to silicon lithiation and delithiation.122,123 The building blocks of phosphorus- and fluorine-rich SEIs can be formed by the (electro)chemical decomposition of phosphorus- and fluorine-donating compounds. LiDFP,120 LiDFBOP, and ethoxy(pentafluoro)cyclotriphosphazene (EFPN) were reported to dictate the distribution of phosphorus in the SEI.124 Polar P–F and P–O species facilitate the permeation of lithium-ions through the SEI by promoting their desolvation and uniform supply while mitigating the localized current distribution across the anode surface (Fig. 6d). The prevalence of LiF in the SEI enables a homogeneous lithium-ion flux, suppresses dendrite formation, and restrains the dissolution of the SEI into the electrolyte;86 moreover, high mechanical strength125 and low solubility can be achieved using fluorine-donating compounds such as fluorinated solvents and salts with fluorine-releasing anions.54
3.2 Fluorine chemistry for SEIs on silicon anodes
3.2.1 Fluorinated structures.
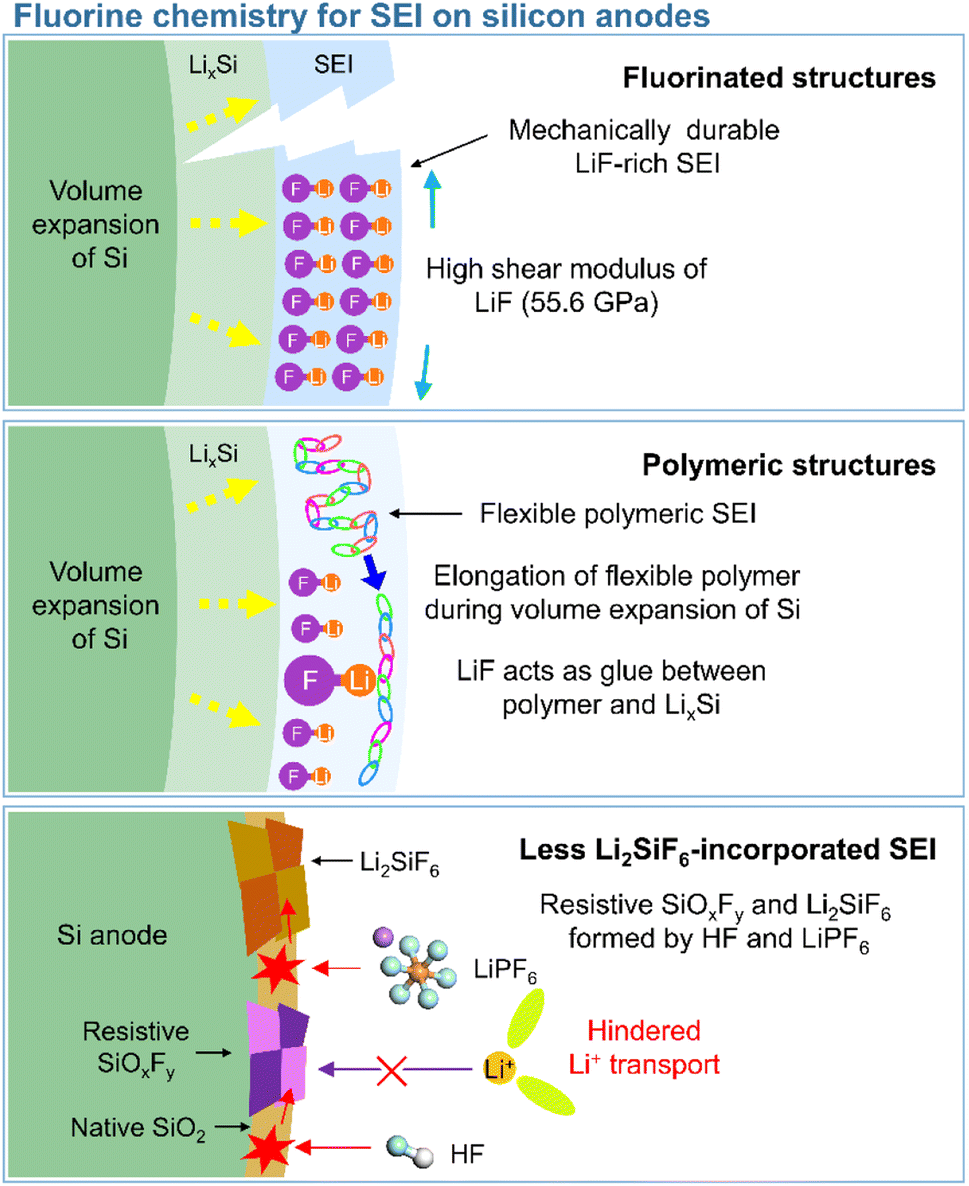
In view of their relatively low lowest unoccupied molecular orbital (LUMO) energy due to the high electron affinity of fluorine, fluorine-containing electrolyte additives form fluorine-rich SEIs including LiF at the surface of silicon anodes.126 Given the need to assure SEI stability, primarily to withstand silicon volume changes during lithiation and delithiation, LiF, which features an extremely low solubility in electrolyte solvents (<0.4 mM in DEC) and high electrochemical robustness, is considered an effective SEI component for preventing solvent molecules from coming into contact with the electrode surface and reacting.54 In addition, the high mechanical stability (shear modulus = 55.6 GPa) of LiF helps increase the structural stability of SEIs on silicon anodes and their resistance to large volume changes during cycling (Fig. 8).127 As fluorine has the highest electronegativity among all elements, LiF formed by the decomposition of fluorine-containing additives at electron-rich anodes during charging engages in strong bonding (via its fluorine atoms) with the lithium of the lithium–silicon alloy (Li⋯F bond length = 1.78 Å), thus securing SEI anchoring on the anode.128 In addition, the fluorine in LiF is thought to strongly bind to the lithium-ions of the Li2EDC aggregate, which is an organic SEI component generated by EC reduction. Thus, LiF acts as glue between the organic SEI layer and the lithium–silicon alloy (Fig. 8). To investigate the effect of LiF on silicon anodes, Haruta et al. deposited a 4 nm-thick LiF layer on a 100 nm-thick silicon thin film using radiofrequency sputtering.129 The LiF-coated silicon-film electrode without additives showed improved CE during initial cycling. LiF was found to effectively bind to lithiated silicon and lithium-containing organic SEI components, stably maintaining the anchoring state of the SEI and thus ensuring the reversible lithiation and delithiation of silicon anodes. In addition, despite the low ionic conductivity of LiF, facile lithium-ion transport is expected in LiF-enriched SEIs, as the contact between LiF and other SEI components such as Li2CO3 results in space charge accumulation along their interfaces.91 Compared to dense VC-derived SEIs comprising poly(VC), FEC-derived SEIs composed of LiF nanocrystals provide defects and interfaces between LiF and other SEI components to facilitate lithium-ion transport.31
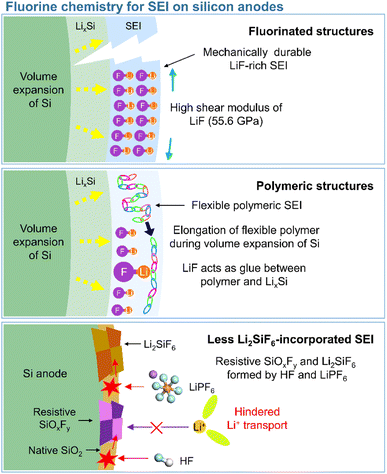 |
| | Fig. 8 Schematic illustration of fluorinated structures, polymeric structures, and less Li2SiF6-containing SEIs on silicon anodes. | |
3.2.2 Polymeric structures.
This section deals with the beneficial impact of strain-adaptive polymeric SEI species on silicon anodes. The build-up of spatially deformable polymeric SEIs has been intensively studied to alleviate SEI collapse due to severe volume changes during repeated silicon lithiation and delithiation.130–133 Organic SEI components such as Li2EDC, LiOCO2CH3, and LiOCO2C2H5 formed by the decomposition of carbonate solvents are more soluble in conventional carbonate-based electrolytes than inorganic SEI components, which complicates the protection of anodes against parasitic reactions with electrolytes.134 Unlike that of short-chain organic species, the solubility of polymeric species in electrolytes is limited. Thus, polymeric SEIs remain stably anchored on silicon anodes while effectively inhibiting electrolyte decomposition by blocking electron supply. In addition, electrolyte additives releasing fluorine to form LiF, which can act as glue for maintaining the connectivity of SEI components and simultaneously form flexible polymeric SEIs, have been developed to enhance the adhesion between polymer species and silicon anodes. 5-Methyl-4-((trifluoromethoxy)methyl)-1,3-dioxol-2-one (DMVC-OCF3) was shown to contribute to the formation of a spatially deformable polymeric SEI along with VC and 5-methyl-4-((trimethylsilyloxy)methyl)-1,3-dioxol-2-one.132 In addition, the OCF3− anion formed by the reduction of DMVC-OCF3 led to an improvement in adhesion between the polymeric SEI and silicon-containing anode by forming LiF. 4-(Allyloxy)phenyl fluorosulfate produced a polymeric SEI comprising LiF to accommodate the large volume changes of silicon-based anodes and secure morphological stability (Fig. 8).133 Linear compounds with –CF3 end groups change their adsorption orientation on the amorphous silicon surface at open-circuit voltage and enable better protection of silicon anodes by fluorinated polymeric or organic species-based SEIs.135
3.2.3 Less Li2SiF6-incorporated SEIs.
Unlike LiF, Li2SiF6, which is formed by the chemical reaction between silicon and LiPF6 (Si + 2LiPF6 → Li2SiF6 + 2PF3) or HF (Si + 6HF + 2Li+ + 2e− → Li2SiF6 + 3H2), imposes a high interfacial resistance on the silicon anode and provokes the loss of active silicon as a lithium-storage site (Fig. 8).139–142 Yu et al. demonstrated the generation of PF3 in a reaction between a LiPF6-based electrolyte and a silicon anode inside a reactor at 90 °C and confirmed the formation of crystalline Li2SiF6 on the silicon anode by mass spectrometry and X-ray diffraction analysis.143 The electrically insulating Li2SiF6 (conductivity <10−10 S cm−1) formed on the surface of silicon anodes impedes both lithium-ion and electron transport (Fig. 8).144 The silicon anodes can be coated with TiN, TiO2, SiO2, and SiC to suppress the generation of Li2SiF6via an undesired reaction with LiPF6.143,145–147 However, the carbon coating layer on silicon may catalyze its reaction with LiPF6 and aggravate the formation of Li2SiF6.139,147 To inhibit Li2SiF6 formation, coatings such as titanium nitride and silicon carbide have been proposed.143,147 Another method of mitigating Li2SiF6 generation and thus securing a long cycle life is the replacement of LiPF6 with LiFSI or LiTFSI.
3.3 Layered SEIs on lithium-metal anodes
3.3.1 Lithiophilic materials.
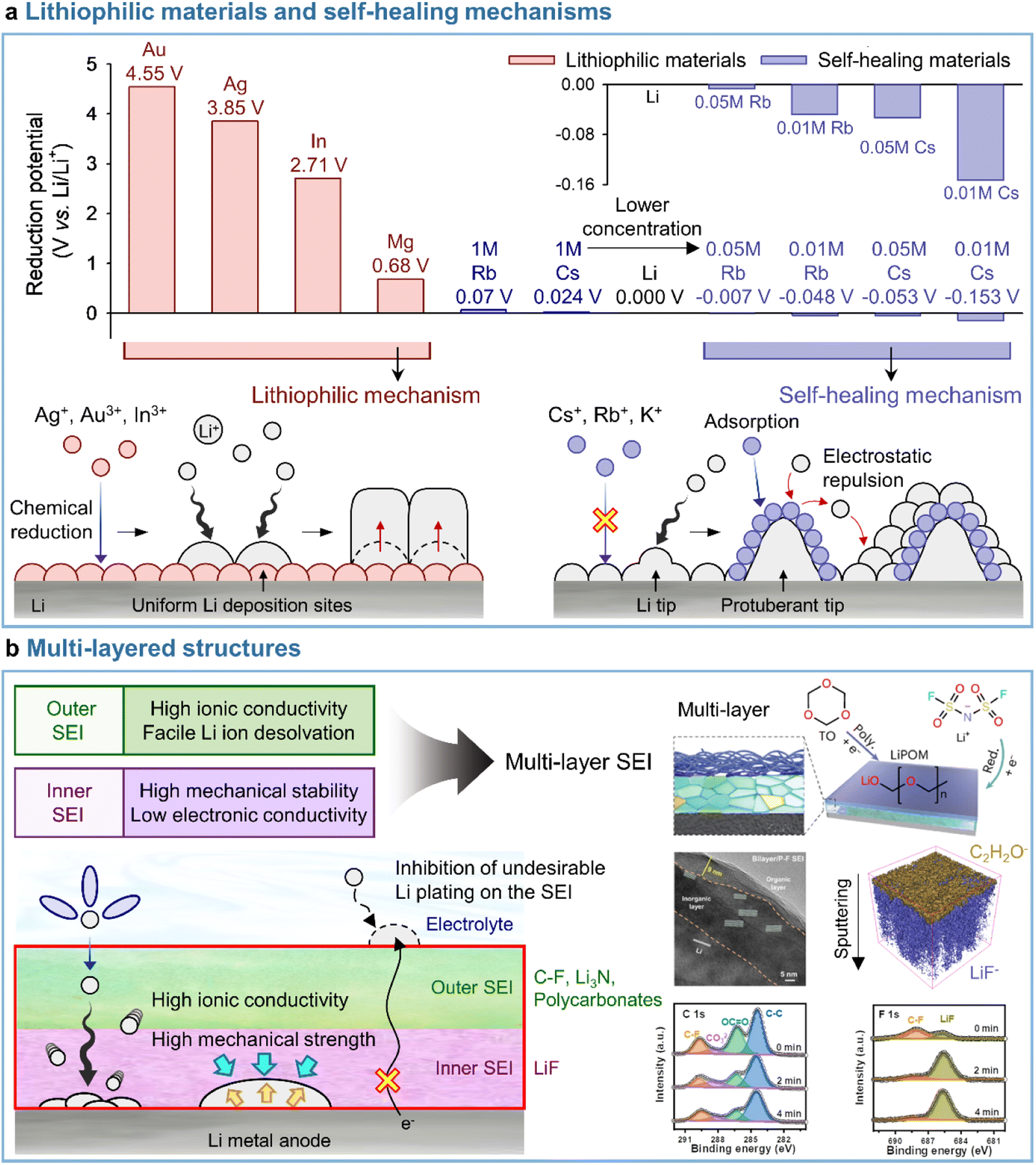
A uniform distribution of lithiophilic sites on the anode is required to achieve a dendrite-free morphology with dense lithium. Metal cations such as Ag+ (Ag+ + e− → Ag; 0.8 V vs. Li/Li+), In3+ (In3+ + 3e− → In; −0.34 V vs. Li/Li+), and Mg2+ (Mg2+ + 2e− → Mg; −2.37 V vs. Li/Li+), which undergo reduction at higher potentials than Li+ (Li+ + e− → Li; −3.04 V vs. Li/Li+), are preferentially deposited on the anode side prior to lithium plating and serve as lithiophilic sites, leading to the reduction of overpotential and the alleviation of vertical lithium plating within a specific zone (Fig. 9a). Lithiophilic materials with low energy barriers for lithium-ion diffusion and high lithium binding energies provide a uniform lithium-ion flux across the entire electrode surface, establish evenly distributed active sites, and ensure high SEI quality to enhance the reversibility of lithium-metal anodes in batteries. Lithiophilic SEI structures can be fabricated using (i) ex situ techniques and solutions containing lithiophilic (e.g., Ag,158,159 In,160 Au,158 and Sb161) species or (ii) in situ strategies by introducing metal cation-based lithiophilic compounds into the electrolyte for stable SEI formation without prior surface treatment.109,162 Instead of ex situ SEI formation, InF3-containing carbonate electrolytes afforded an SEI containing a lithium–indium alloy (LixIny) with a high bulk diffusion coefficient (DLi ≈ 10−8 to 10−6 cm2 s−1) and a lithium halide and favored a nondendritic and compact lithium-plating morphology.160 The generation of the two phases (LixIny and LiF) in the SEI can be described by eqn (1) and (2).| | | 3Li + InF3 → In + 3LiF | (1) |
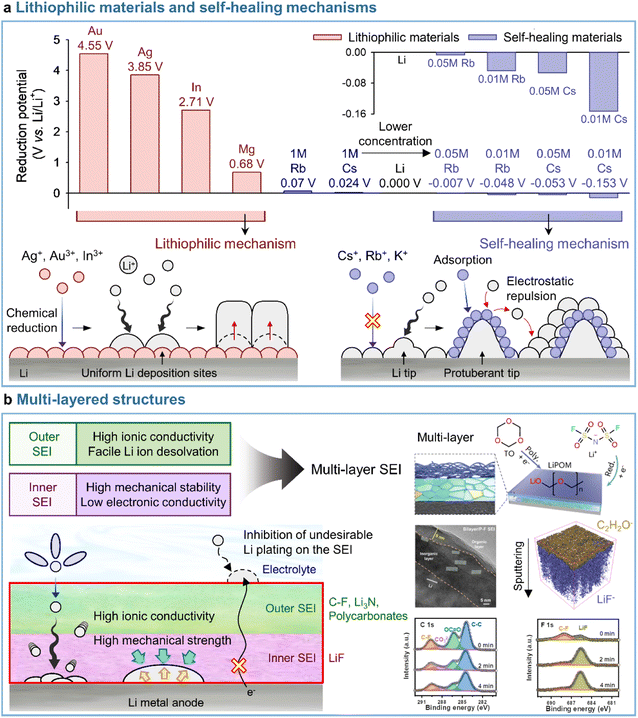 |
| | Fig. 9 Representative models for (a) lithiophilic materials and self-healing mechanisms and (b) multi-layered SEI structures. Reproduced from ref. 136, 137, and 138 with permission of American Chemical Society, copyright 2013, John Wiley & Sons, Inc., copyright 2021, and Springer Nature, copyright 2023, respectively. | |
In addition to those on electrolyte additives containing metal cations, some studies deal with the loading of lithiophilic seeds into the SEI through the introduction of small amounts of nonmetallic additives such as tetrachloro-1,4-benzoquinone (TCBQ) and 2-fluoropyridine (2-FP).163,164 The incorporation of TCBQ affords lithiophilic species such as the quinone lithium salt (Li2TCBQ), which possesses a high lithium-ion affinity and promotes uniform lithium plating owing to the favorable attraction of lithium-ions to the carbonyl oxygen in the Li2TCBQ structure. Similarly, lithiophilic 2-FP leads to the generation of a Lewis-basic pyridinic nitrogen-containing SEI that exhibits a high ionic conductivity and provides lithium nucleation sites. The above evidence indicates that lithiophilic SEIs, which can be constructed using both metallic and nonmetallic additives, are critical for the stable operation of LMBs. However, the excessive introduction of lithiophilic materials into electrolytes causes undesired volume expansion during repetitive cycling due to lithium-metal cation alloying, which can undermine the stable operation of LMBs.176 Consequently, more stable SEIs for the reliable operation of LMBs can be realized by introducing optimal amounts of lithiophilic materials and leveraging their synergistic effects with other interfacial modifiers. This topic is discussed in more detail in Section 4.4.
3.3.2 Self-healing mechanisms.
Most studies focus on the formation of stable SEIs by introducing electrolyte additives (i.e., through mechanical barrier formation) rather than by addressing the fundamental part of lithium dendrite formation. Interestingly, the self-healing electrostatic shield (SHES) mechanism suggests a different direction for the inhibition of lithium dendrite formation.137 In the case of conventional electrolytes, a protuberant tip is created on the lithium-metal anode during the initial lithium deposition. With continuing electrodeposition, the current density at the tip increases, which mainly results in lithium electrodeposition toward the tip to form lithium dendrites.177 The introduction of electrolyte additives containing nonlithium-cations following the SHES mechanism shows a different aspect (Fig. 9a). More specifically, during the initial electrodeposition, the lithium-ions and nonlithium additive cations are absorbed onto the lithium-metal anode at a potential slightly lower than that of lithium-ion reduction and higher than that of additive reduction. At this point, as the electrolyte additive is not reductively decomposed to form an SEI, the cations of this additive are not electrodeposited on the tip but accumulate in its vicinity to form an electrostatic shield. This shield prevents the electrodeposition of lithium in the protruding area, shifting it to nearby areas and thus enabling deposition uniformity. The SHES mechanism is applicable when electrolyte additive cations have a lower standard reduction potential than lithium-ions. According to the Nernst equation, metal cations such as Cs+ and Rb+ at low concentrations (∼0.05 M) show lower standard reduction potentials than 1 M Li+ and are therefore not electrodeposited on the lithium-metal anode, i.e., lithium alloying does not occur.
Conversely, cations such as Mg2+, Al3+, Zn2+, Ga3+, In3+, and Sn2+ cannot be applied, as their standard reduction potentials exceed that of Li+, i.e., these cations can be reduced prior to Li+. Zhang's group first established the SHES mechanism and realized the stable operation of LMBs by introducing electrolyte additives such as CsPF6 and RbPF6.137 Furthermore, the fact that not only Cs+ and Rb+ but also K+ can follow the SHES mechanism inspires further research.178
3.3.3 Multi-layered structures.
A deep understanding of electrode–electrolyte interfaces is crucial for improving LMB performance, as they are one of the biggest obstacles hindering practical LMBs applications.6 Since Peled defined the electrochemically insulating and ionically conducting passivation layer, named SEI, in 1979,165 numerous studies have contributed to its fundamental understanding and exploration by diverse analytical methods. One effective strategy is to control the SEI composition and structure, as they strongly impact the cyclability of LMBs.179 However, this control is challenging to realize, as SEIs contain complex products of reductive electrolyte decomposition. To improve SEI stability in accordance with the above, researchers have examined electrolyte additives capable of forming LiF-based SEIs, which feature high mechanical stability, low lithium-ion diffusion barriers, and large band gaps. However, single-layer LiF-based SEIs composed of small LiF particles may be destroyed under extreme conditions by the extrusion of lithium dendrites formed on lithium-metal anodes after numerous cycles.138 To maximize the functionality of LiF-based SEIs and prolong the cycle life of LMBs, one should ensure a homogeneous lithium-ion supply in the long term and delay the unwanted exposure of lithium-metal to the electrolyte through an ionically conductive outer SEI. Even when SEIs with high ionic conductivity, such as those containing Li3N, are introduced as outer SEIs, they cannot ensure the long-term stability of LMBs because of the low band gap and narrow electrochemical stability window of this nitride. To overcome these limitations, researchers have introduced the concept of multi-layer SEIs, which combine the advantages of each SEI composed of both organic and inorganic materials.169 More organized multi-layer structures are preferred, as they can promote homogeneous lithium plating and stripping and thus reduce the risk of dendrite formation (Fig. 9b).180 Interestingly, in the case of multi-layer SEI structures, the outer layer is composed of species facilitating the desolvation of lithium-ions or enhancing ionic conductivity, e.g., C–F bonds,136 Li3N,21 Li polyoxymethylene,138 and polycarbonates.181 C–F bonds are electron-rich and highly polar, while polycarbonates are lithiophilic and therefore enhance ion conduction through the SEI. The inner layer features components (such as LiF) that can accommodate the severe volume fluctuation of the lithium-metal anode during cycling. Our recent results reveal that the introduction of fluorine- and nitrogen-donating electrolyte additives forming LiF and Li3N, respectively, significantly improves the cyclability of LMBs.21 The presence of multilayer SEI structures on lithium-metal anodes was confirmed by XPS depth profiling according to sputtering time or by XPS analysis at a variable charging voltage. Cyclic voltammetry analysis demonstrated the sequential occurrence of decomposition peaks for each electrolyte component, thereby confirming the build-up of a multilayer SEI.
4. Design rules for desirable SEIs on silicon and lithium-metal anodes
4.1 Key factors for SEI build-up and conservation
SEI formation in lithium batteries is a complex process due to (electro)chemical reactions between the anode (silicon or lithium-metal) and electrolyte. Therefore, direct contact between the electrolyte and lithium-metal results in an instantaneous and uncontrolled reaction affording a primitive SEI that contains insoluble multiphase organic and inorganic species.
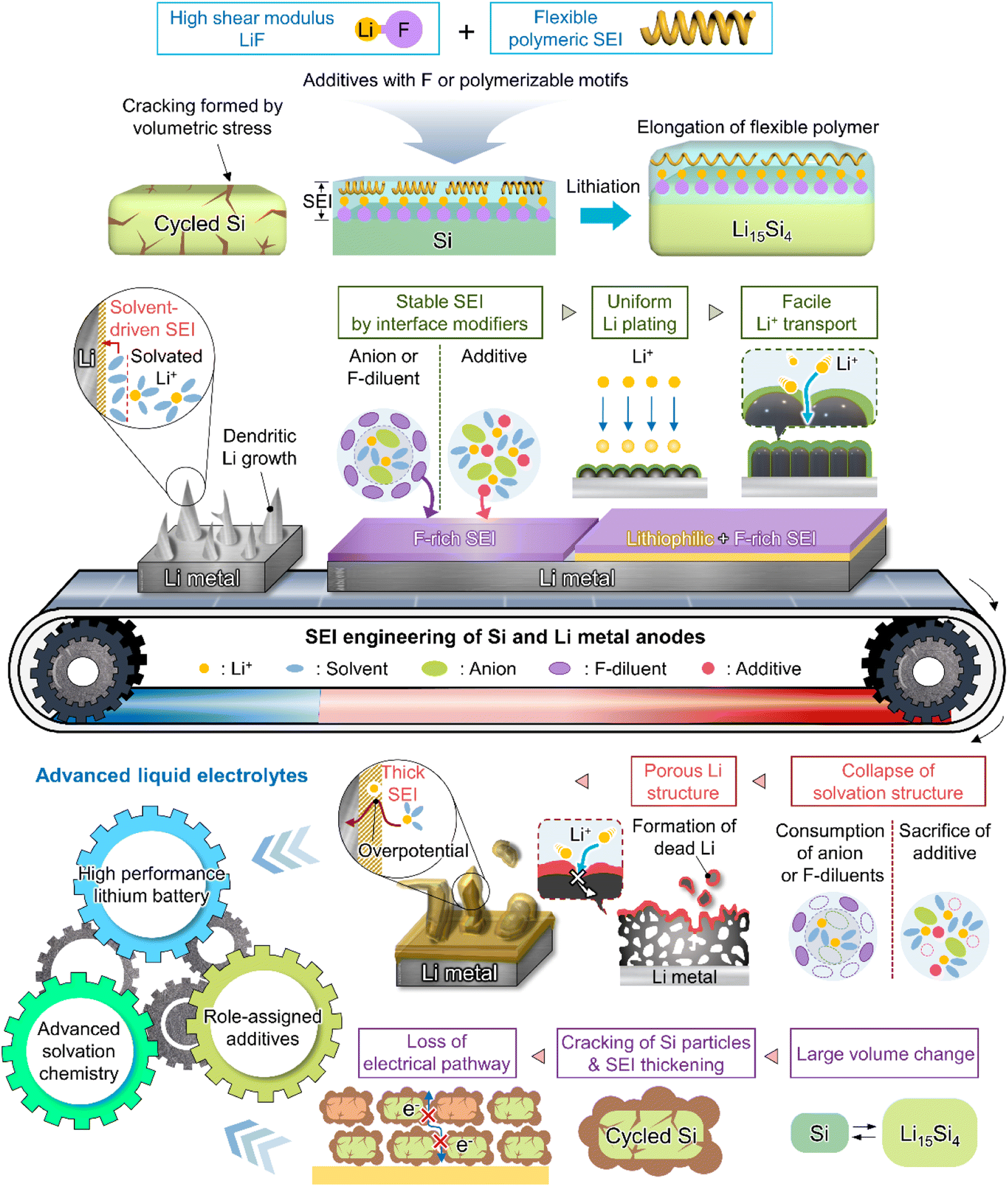
As the battery starts to operate under an applied electric field, the electrolyte and primitive SEI undergo further decomposition to generate an SEI containing organic components such as lithium alkyl carbonates and lithium alkoxides as well as inorganic components such as LiF, Li2O, and Li2CO3.182 The thus formed SEI acts as a physical barrier preventing irreversible electrolyte decomposition and, hence, battery performance degradation. The SEI composition and thickness depend on numerous factors such as electrolyte type, operation temperature, and charge/discharge conditions (Fig. 10 and 11). Since the SEI concept was first proposed by Peled in 1979, various SEI aspects, including composition and properties, have been unveiled.165 As a starting point, the Peled group proposed a mosaic structure model in 1997, while the Aurbach group relied on the results of postmortem (e.g., infrared spectroscopy and XPS) analyses and suggested that the SEI is composed of various organic and inorganic components forming a multi-layer structure.81,166 In 2017, the Cui group used cryo-TEM to show that the SEI features both mosaic and multi-layer structures.17 Research on the overall SEI structure, which has advanced along with analysis methods, amplifies interest in SEI components.183 In this regard, a deep understanding of SEI constituents enables strategic structuring that suits the desired SEI, helps find materials guaranteeing high-performance and long-term stability of lithium batteries, and is essential for electrolyte structure design. In particular, the continuous decomposition and restoration cycle of the SEI caused by the repetitive volume expansion and contraction of the lithium-metal anode requires further consideration. This repetitive decomposition and restoration cycle leads to the continuous consumption of the electrolyte and lithium-metal anode, resulting in the recurring formation of a new type of SEI. Therefore, effective SEI formation is crucial for long-term durability. Therefore, a deep understanding of the characteristics of representative organic and inorganic species such as LiF, Li2O, Li2CO3, and Li3N is necessary for the future enhancement of SEI quality (Fig. 12 and Table 1).
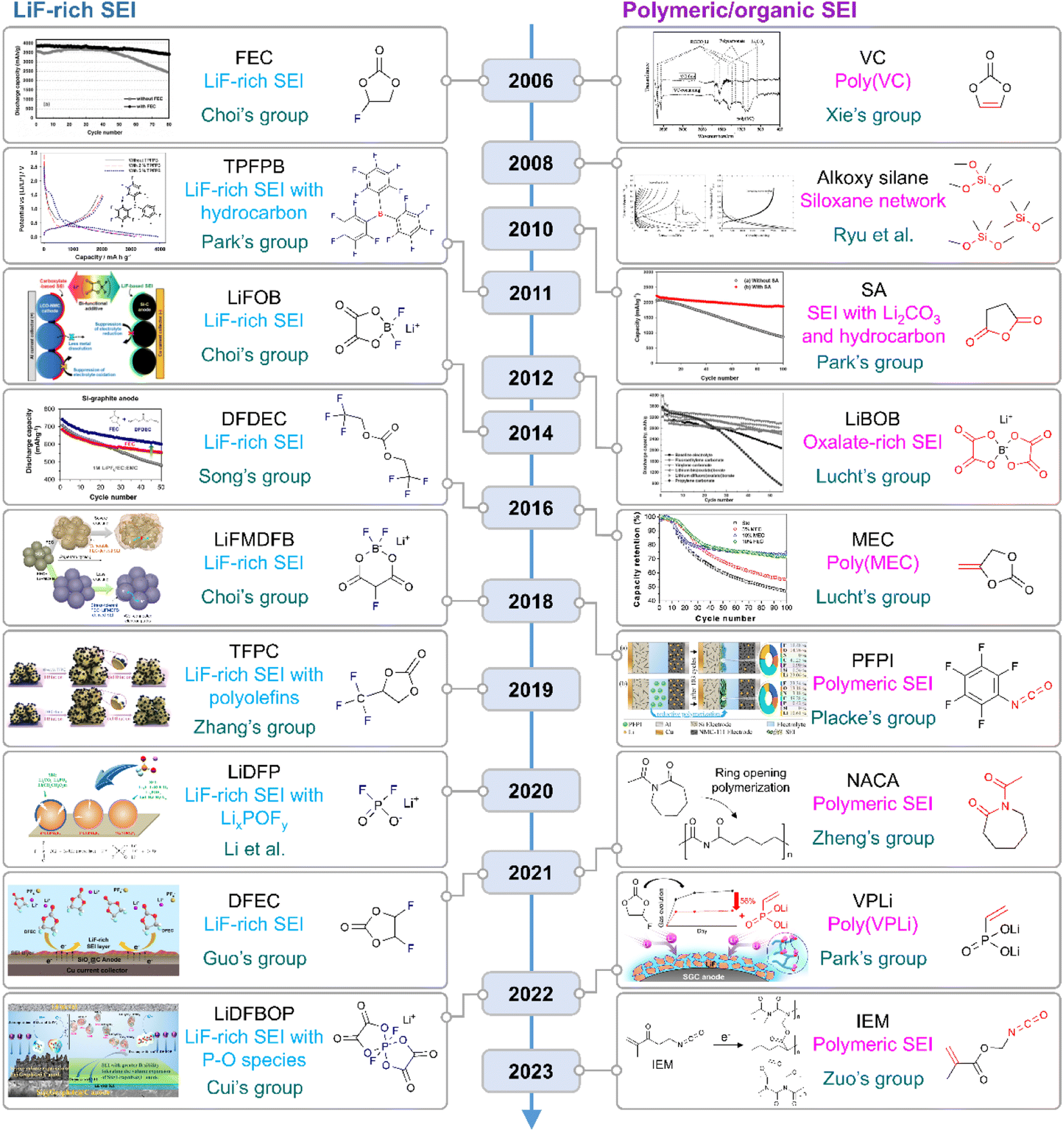
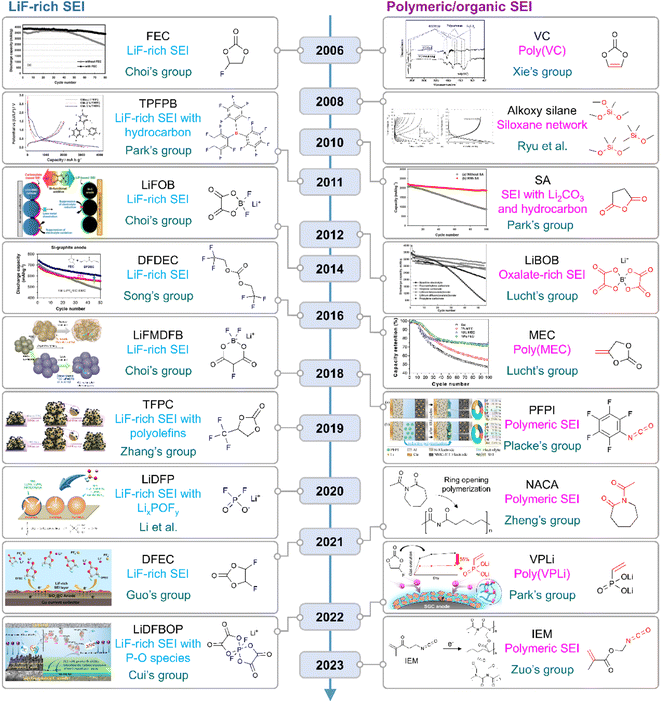 |
| | Fig. 10 Flow of research for SEIs on silicon anodes. TPFPB: tris(pentafluorophenyl)borane, LiFOB: lithium difluoro(oxalato)borate, LiFMDFB: lithium fluoromalonato(difluoro)borate, TFPC: trifluoropropylene carbonate, LiBOB: lithium bis(oxalato)borate, MEC: methylene ethylene carbonate, PFPI: pentafluorophenyl isocyanate, NACA: N-acetylcaprolactam, VPLi: dilithium vinylphosphonate, and IEM: 2-isocyanatoethyl methacrylate. Reproduced from ref. 10, 14, 75, 78, 123, 130, 131 and 148–157 with permission of Elsevier, copyright 2006, Royal Society of Chemistry, copyright 2018, IOP Publishing, copyright 2012, Elsevier, copyright 2010, American Chemical Society, copyright 2022, 2018, Elsevier, copyright 2016, American Chemical Society, copyright 2016, IOP Publishing, copyright 2008, Elsevier, copyright 2011, 2014, John Wiley & Sons, Inc., copyright 2019, 2020, American Chemical Society, copyright 2021, 2021, 2022, and Elsevier, copyright 2023, respectively. | |
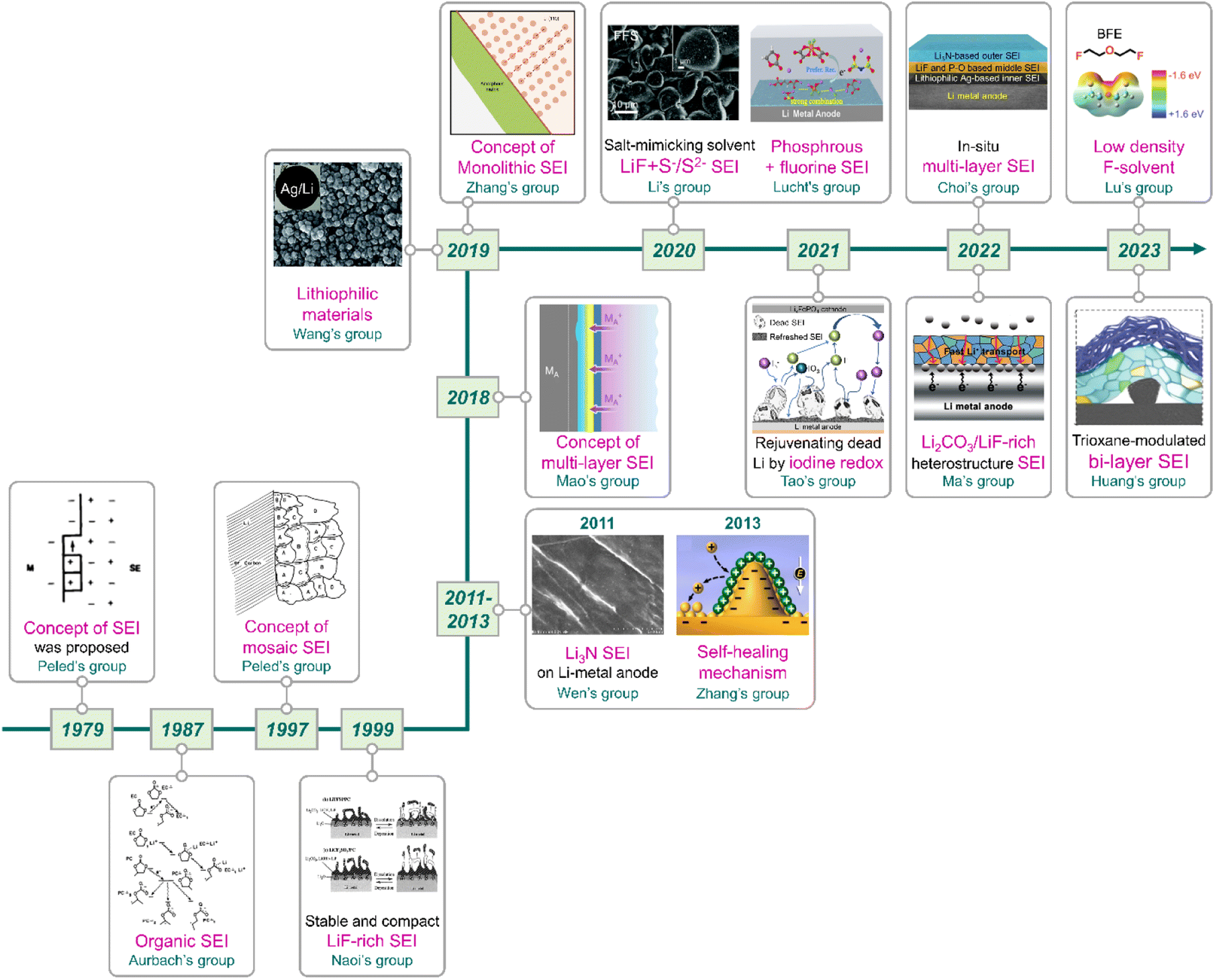
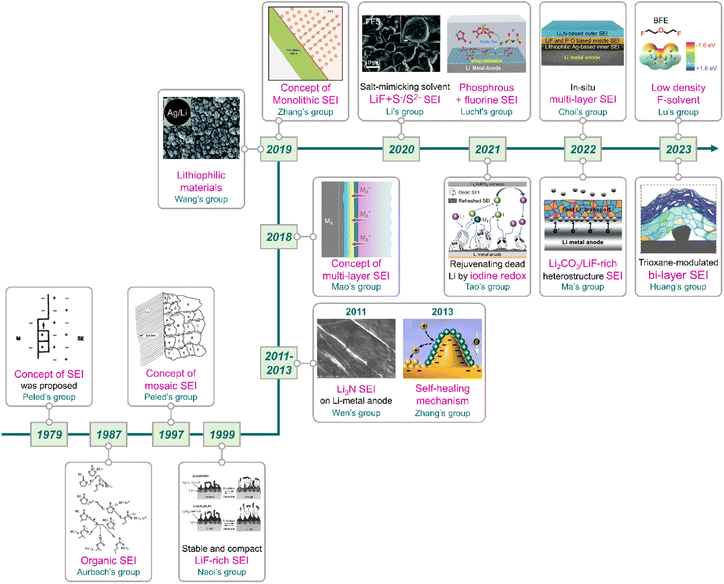 |
| | Fig. 11 Flow of research for SEIs on lithium-metal anodes. Reproduced from ref. 40, 81, 137, 138, 159 and 165–175 with permission from John Wiley & Sons, Inc., copyright 2022, IOP Publishing, copyright 1987, Royal Society of Chemistry, copyright 2019, American Chemical Society, copyright 2013, IOP Publishing, copyright 1979, Springer Nature, copyright 2018, 2023, IOP Publishing, copyright 1997, Springer Nature, copyright 2019, 2023, Royal Society of Chemistry, copyright 2020, American Chemical Society, copyright 2021, Springer Nature, copyright 2021, IOP Publishing, copyright 1999, Elsevier, copyright 2011, and American Chemical Society, copyright 2020, respectively. | |
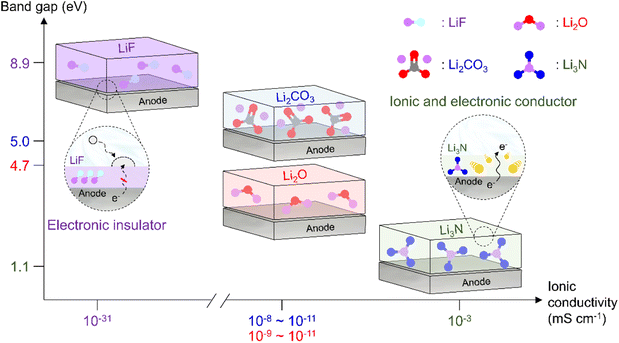 |
| | Fig. 12 Comparison of band gaps and ionic conductivities of inorganic SEI species (LiF, Li2CO3, Li2O, and Li3N). | |
4.2 Salt-mimicking solvents and additives
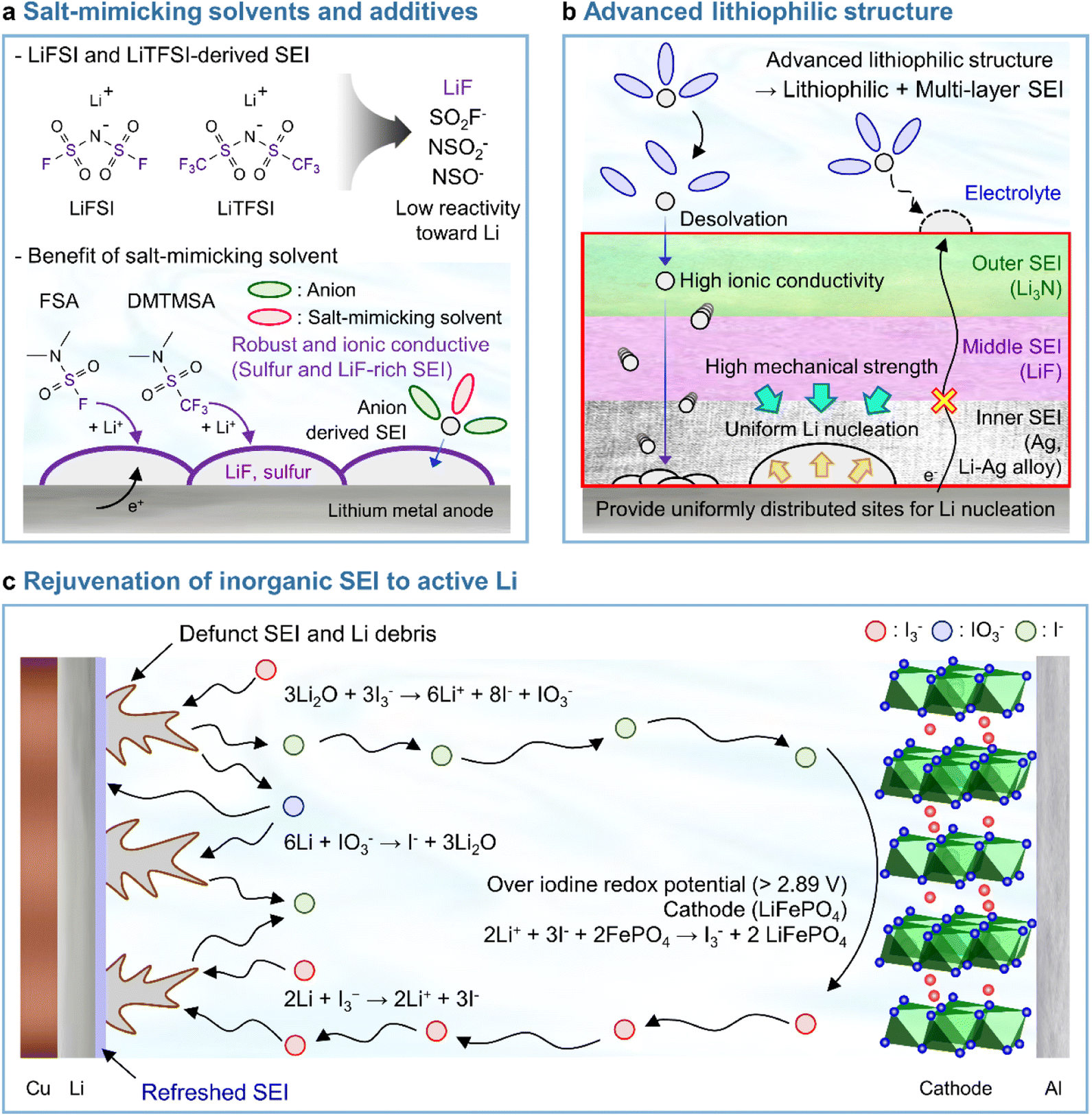
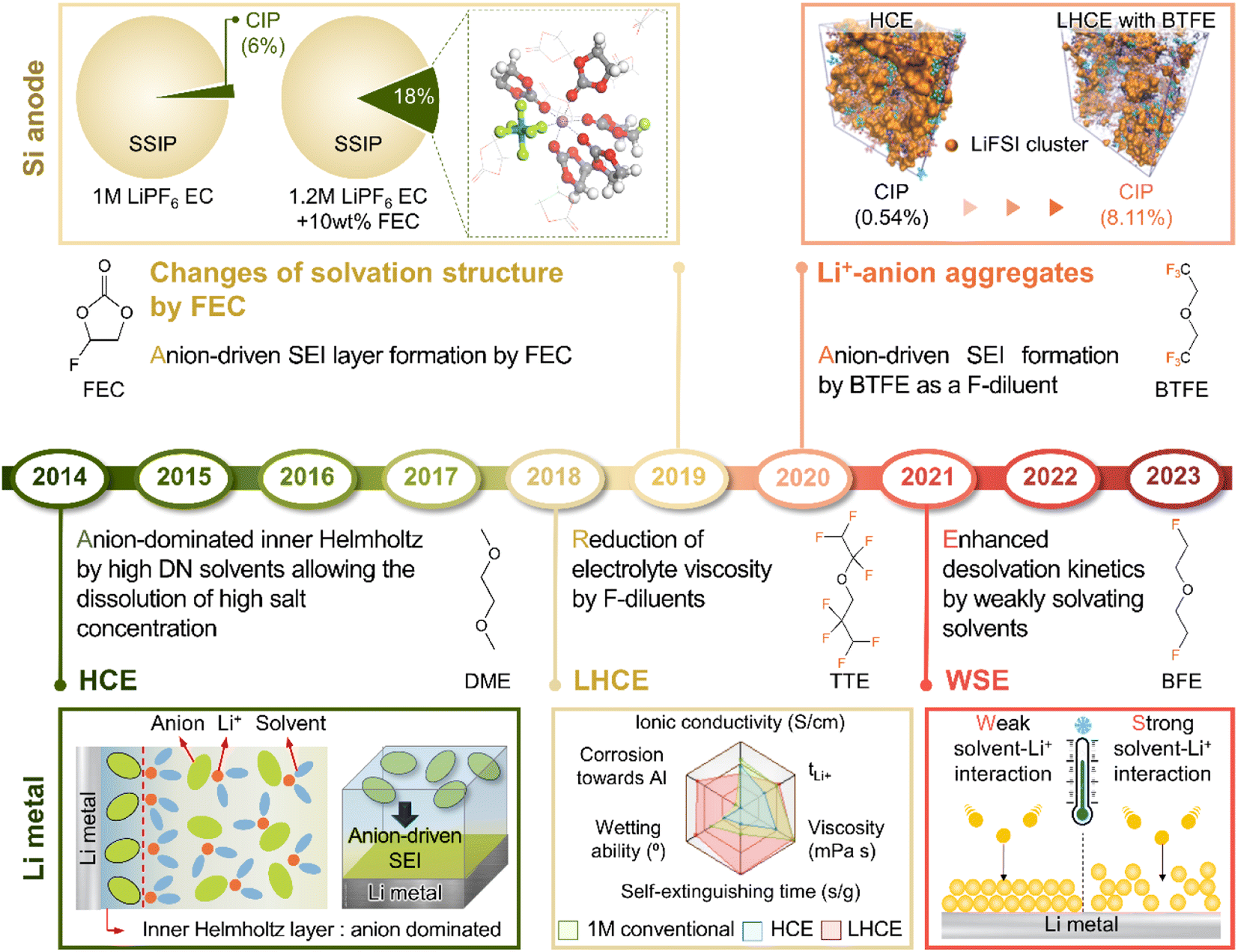
LiPF6 has been widely used as an electrolyte salt in LIBs because of its high solubility and good ionic conductivity.185 However, when used in LMBs, LiPF6 can decompose at the lithium-metal anode to form a thick and unstable SEI, which causes poor performance and safety issues due to lithium dendrites.186 To resolve the above problems, researchers have examined lithium salts such as LiFSI and LiTFSI as potential replacements for LiPF6 in LMBs.187 LiFSI and LiTFSI form anion-derived SEIs that consist of species (LiF, SO2F−, NSO2−, and NSO−) with high ionic conductivities and low reactivity toward lithium-metal anodes.188 Furthermore, the importance of anion-derived SEI formation is emphasized by the development of electrolyte solvation structures, as exemplified by highly concentrated electrolytes (HCEs), localized highly concentrated electrolytes (LHCEs), and weakly solvating electrolytes (WSEs).189–192 Previous research strategies relying on the introduction of excess lithium salts into the electrolyte to form anion-derived SEIs improve the cyclability and CE of LMBs but cause problems such as the increased viscosity and decreased ionic conductivity of electrolytes. Therefore, materials that can realize similar effects with anion-derived SEIs are highly desired for lithium-metal anodes. In line with this necessity, researchers have designed solvents with salt-mimicking properties, such as fluorosulfonamide (FSA) and N,N-dimethyltrifluoromethane-sulfonamide (DMTMSA), which inherit the beneficial properties of FSI− and TFSI−, respectively (Fig. 13a).171,193 FSA, an FSI-inspired solvent with one fluorosulfonamide group and two methyl substituents, has a better LiF formation ability than FEC and generates stable lithium–electrolyte interfaces. In addition, low-valence sulfur species (S− and S2−) formed by FSA decomposition can enhance cation mobility because of the high atomic polarizability of sulfide ions, thus helping in forming lithium-ion conductive SEIs and reduce the overall interfacial resistance of anodes. Furthermore, DMTMSA, which possesses a TFSI− moiety, enables the operation of Li|NCM811 full cells at high-voltages owing to its wide electrochemical stability window and ability to form stable SEIs. In addition to solvent-level studies, Jiang et al. presented insights into various FSI− moiety-based additives with different structural designs, revealing that nitrogen-containing sulfamoyl fluorides outperform their non-nitrogenated analogs.194 In conclusion, the discovery of solvents and additives containing FSI− or TFSI− moieties suggests the possibility of expanding from conventional ethereal- and carbonate-based systems to sulfonamide-based ones while maintaining stable SEI formation, high ionic conductivity, and the wide electrochemical stability window of electrolytes. An electrolyte additive with a FSI−-inspired fluorinated sulfonyl group generated a stable SEI on a silicon-based anode.133 4-(Allyloxy)phenyl fluorosulfate in combination with VC formed a multi-layered SEI with a sulfur-rich inner layer, deformable polymeric middle layer, and LiF-rich outer layer. The fluorosulfate additive-derived layer facilitated lithium-ion transport in the SEI on the silicon-based anode and assured the structural stability of the SEI withstanding the large volume changes of silicon upon lithiation and delithiation.
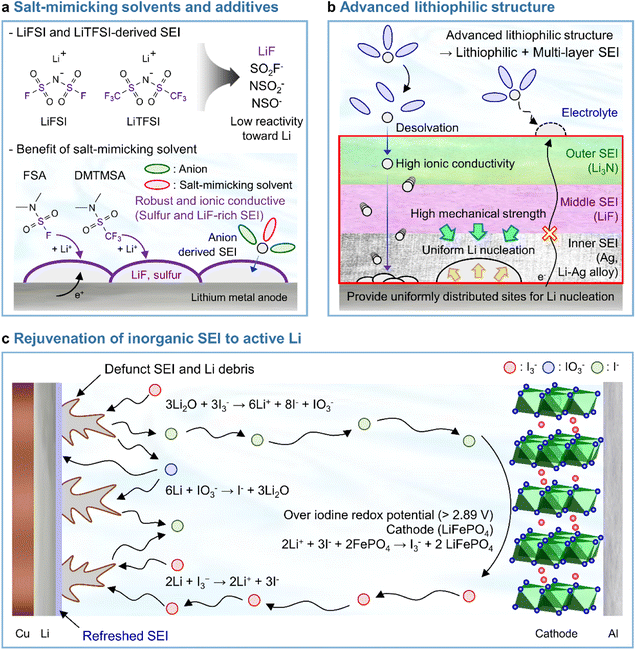 |
| | Fig. 13 Recent research on SEIs deposited on lithium-metal anodes. (a) LiF + lower-valence sulfur species (S−/S2−)-containing an SEI constructed using salt-mimicking solvents and additives. Reproduced from ref. 171 with permission of the Royal Society of Chemistry, copyright 2020. (b) Advanced lithiophilic structure. (c) Rejuvenation of inorganic SEIs to active lithium. Reproduced from ref. 173 with permission of Springer Nature, copyright 2021. | |
4.3 Fluorinated solvents and salts
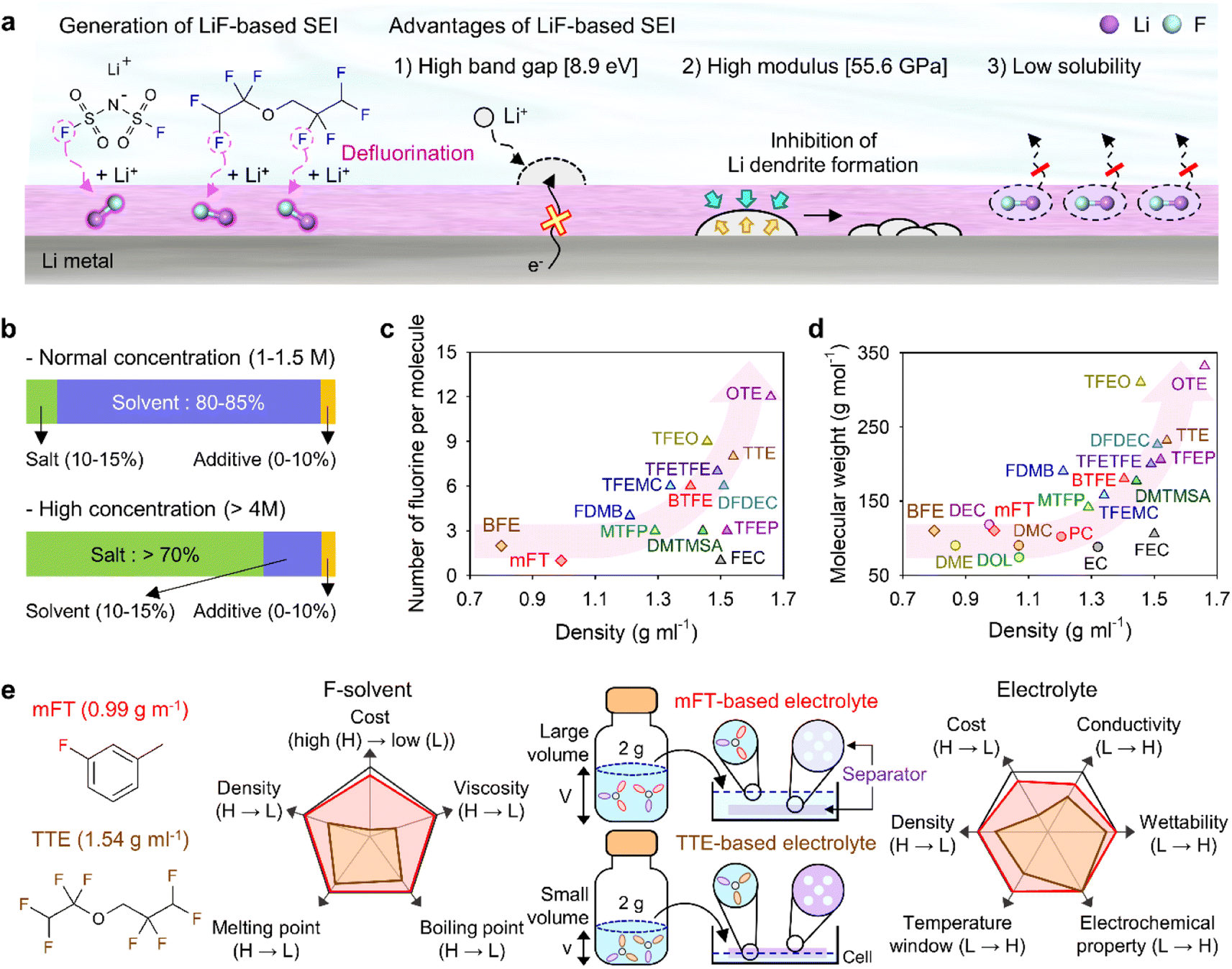
Theoretically, fluorinated solvents or additives have a lower LUMO energy than conventional solvents because of the strongly electron-withdrawing nature of fluorinated functional groups.195 This energy level reduction facilitates the formation of LiF-based SEIs (Fig. 14a), which offer the advantage of an electronically insulating nature due to the high band gap and high mechanical strength of LiF. Owing to the fluorine-donating character of fluorinated solvents helping to form fluorine-rich SEIs, the fluorination of carbonates has been suggested to mechanically and electrochemically strengthen the SEI on silicon anodes. Fluorinated EC, known as FEC, is the most successful SEI-forming additive for silicon-based anodes.10,196,197 The use of FEC, which is more prone to reductive decomposition than EC, leads to the formation of a LiF-rich SEI accommodating the large volume changes of silicon anodes with less spatial deformation.14,198 Indeed, the synergy between (i) the lower reactivity of FEC toward lithiated silicon compared to that of EC and (ii) the FEC-derived SEI alleviating electrolyte decomposition at anodes drastically enhances the cycle performance of silicon nanoparticle and silicon nanowire anodes.77,199–203 Furthermore, in view of its low-lying highest occupied molecular orbital, FEC contributes to the improved anodic stability of electrolytes and enables the use of high-voltage cathodes (≥4.4 V vs. Li/Li+).14,198,204–206 Thus, the replacement of EC with FEC as an electrolyte solvent is an effective strategy for enhancing the performance of high-energy-density batteries with silicon-based anodes and high-voltage cathodes. However, FEC is thermally unstable in LiPF6-based electrolytes, undergoing defluorination promoted by Lewis acids such as PF5 at high temperatures to generate corrosive HF and other acidic compounds.43,207 The introduction of LiPF6 stabilizers or HF scavengers should be considered to mitigate this problem. Fluorinated linear carbonates have been explored as additives to achieve the interfacial stability of silicon anodes.151,208–210 When di(2,2,2-trifluoroethyl) carbonate (DFDEC) was employed as a coadditive along with FEC, the cycling performance of silicon-graphite composite anodes exceeded that observed for the electrolyte supplemented with FEC only. The SEI created by the combination of DFDEC and FEC contained a higher amount of LiF than that formed using FEC alone. The increasing attention drawn to LHCEs for LMBs has inspired the active development of electrolytes containing F-diluents for LIBs with silicon anodes. Compounds such as bis(2,2,2-trifluoroethyl) ether (BTFE), 1,1,2,2-tetrafluoroethyl 2,2,3,3-tetrafluoropropyl ether (TTE), methyl 2,2,2-trifluoroethyl carbonate (TFEMC), and 1H,1H,5H-octafluoropentyl 1,1,2,2-tetrafluoroethyl ether (OTE) have been used as F-diluents, enhancing the electrochemical stability of LIBs with silicon anodes.211–214 F-Diluents, which do not generally enter the primary solvation sheath, not only act as fluorine donors, aiding the formation and maintenance of LiF-rich SEIs and the repair of damaged SEIs on the silicon anode surface, but also induce the formation of partial highly concentrated regions in electrolytes, involving anions in the solvation structure and thereby forming anion-derived inorganic-rich SEIs. Changes in the solvation structure and the formation of anion-derived SEIs, which lead to interfacially stable anodes, are further discussed in Section 4.6. Similar to the use of fluorine chemistry for silicon anodes, LiF-enriched SEIs have drawn much attention as a promising alternative to conventional ones.215,216 LiF-rich SEIs feature the benefits of outstanding chemical stability and high surface energy and can therefore prevent the growth of lithium dendrites, which is a significant safety concern in LMBs.217 LiF-based SEIs on lithium-metal anodes can be fabricated following three main pathways. The first approach relies on the artificial (ex situ) formation of LiF-based SEIs to protect lithium-metal anodes against side reactions with electrolytes. Among the related methods, the direct coating of artificial LiF layers on lithium-metal anodes is cheaper than the use of fluorinated solvents and additives.218 The benefits of ex situ SEI creation were exploited to substantially increase the interfacial stability of lithium-metal anodes even in carbonate-based electrolytes. Artificial LiF-based SEIs can be constructed through the treatment of lithium-metal anodes with solutions containing polyvinylidene fluoride + dimethylformamide or ammonium hydrogen difluoride + dimethyl sulfoxide,218,219 nitrogen trifluoride gas (NF3; 2NF3 + 6Li → 6LiF + N2) or fluorinated precursors (gaseous Freon R134a (1,1,1,2-tetrafluoroethane)),220,221 LiPF6 hydrolysis (LiPF6 + H2O → LiF + 2HF + POF3),222 atomic layer deposition (lithium tert-butoxide, titanium fluoride, and HF/pyridine),223,224 and physical vapor deposition.225 However, these SEIs may undergo mechanical fracture during cycling, which causes continued electrolyte decomposition at the exposed active lithium-metal and thus results in thick SEI formation and electrolyte depletion.220 Therefore, when considering ex situ SEI formation, structural optimization ensuring the mechanical integrity of the SEI is vital for securing the long-term stability of LiF-based SEIs along with (electro)chemical stability. The second approach relies on the formation of anion-driven SEIs through the control of the electrolyte solvation structure.190,226 Lithium salts such as LiFSI and LiTFSI can donate fluorine atoms to build LiF-based SEIs, particularly HCEs with small portions of free solvent molecules, as anions are mainly involved in the formation of a solvation sheath around lithium-ions. Such anion-derived SEIs show good compatibility with high-voltage cathode materials, and the above method has emerged as an effective SEI generation strategy for the stable operation of LMBs.227 However, under practical conditions, the introduction of excess lithium salts to increase the electrolyte concentration also increases electrolyte viscosity and cost. To address this issue, researchers have examined LHCEs that contain F-diluents capable of securing the formation of anion-based SEIs in HCEs while not interfering with the dissolution of lithium salts and reducing the overall electrolyte viscosity. The fluorinated solvent can form a LiF-based SEI, which can improve CE of LMBs.228,229 Moreover, the fluorinated solvent can repair the damaged LiF-based SEI during repetitive cycles.230 Furthermore, researchers consider the formation of anion-driven SEIs through the introduction of solvents that have low solubilizing power and involve more anions in the formation of a solvation sheath around lithium-ions.231 The third strategy used to construct LiF-based SEIs on lithium-metal anodes involves the use of fluorine-containing electrolyte solvents or additives. FEC is commonly used to form fluorinated SEIs to ensure the stable operation of LMBs. Alternatively, LiF-rich SEIs170 can be generated using electrolytes exclusively composed of fluorinated solvents232 and supplements such as hexafluoroisopropyl trifluoromethanesulfonate (HFPTf), trimethylsilyl(fluorosulfonyl)(n-nonafluorobutanesulfonyl)imide (TMS-FNFSI), 2,2,2-trifluoroethyl trifluoroacetate (TFE), 2-fluoropyridine (2-FP), difluoroethylene carbonate (DFEC), 2,2,2-trifluoroacetate (TFA), and 1,1,1,3,3,3-hexafluoroisopropyl methyl ether (HFPM).233–237 Despite the advantages of fluorinated solvents and salts, their widespread use for cell operation under practical conditions is hindered by several problems238 such as cost-effectiveness, scalability for large-scale production, stability during long-duration storage, environmental concerns, and the inherently high density of fluorinated solvents.239,240 Fluorinated salts and solvents are expensive and currently unsuitable for mass production. In the Li|LiNixCoyMnzO2 (x + y + z = 1) full cell design for high-energy-density (high-capacity cathode: 4 mA h cm−2, thin lithium foil: 8 mA h cm−2, and lean amount of electrolyte: 2.4 g A h−1), the electrolyte accounts for 23.6% of the system weight and thus has the second-highest relative content after the cathode (49.2%).241 Additionally, the electrolyte cost accounts for ∼15% of the total battery cost.2 In general, electrolytes with a normal concentration of 1–1.5 M are composed of 12.6 wt% lithium salt, 0–10 wt% electrolyte additive, and 80–85 wt% solvent (Fig. 14b).242 This implies that the cost of lithium salt and electrolyte solvent are the main factors determining electrolyte cost. In HCEs (>4 M), the expensive lithium salt has a content of >70 wt%, significantly increasing electrolyte cost and making electrolyte use not economically feasible (Fig. 14b).243 Furthermore, the various reported ethereal fluorinated solvents such as TTE, BTFE, and tris(2,2,2-trifluoroethyl)orthoformate (TFEO) are relatively expensive and have a high density of ∼1.5 g cm−3, which may cause a small volume of the electrolyte to be injected in practical full cell fabrication and thus lead to problems such as poor separator wetting and performance degradation due to the lack of electrolyte to be evenly spread across the electrode surfaces and separator micropores (Fig. 14c and d and Table S3†).244 Unlike high-density fluorinated solvents such as TTE and BTFE, monofluoride bis(2-fluoroethyl) ether (BFE) and m-fluorotoluene (mFT) can minimize the issue of electrolyte wetting owing to their relatively low-densities of 0.98 and 0.99 g cm−3, respectively, while retaining the advantages of fluorine-enriched SEI formation on the anode (Fig. 14e).175,184 Moreover, currently reported fluorinated ethers are expensive and not commercially available, and their large-scale production is hindered by the lack of correspondingly optimized synthetic methods.245 As the price of conventional carbonate-based solvents has decreased after decades of synthesis optimization for mass production, the price of various recently developed fluorinated solvents could also decrease after the optimization of the production process for electrolyte design.2 However, at present, cost-competitiveness is a key concern that cannot be negated in electrolyte design. Environmental issues also need to be considered.246,247 In the case of fluorinated solvents, the solvent itself is rich in fluorine atoms, which substitute hydrogen atoms, and the thus generated stable C–F bonds are difficult to degrade naturally. Thus, C–F bond-containing per and polyfluoroalkyl substances (PFASs) increase the burden on the environment and pose problems such as global warming.248,249 These solvents have also been reported to exhibit toxic effects on human health and the environment, which has raised concerns about their safety. The stability of fluorinated solvents during long-term storage and battery operation is yet another concern, as they can undergo decomposition and release toxic gases over time.239 Finally, fluorinated solvents used as diluents weaken the polarity of the solvating group interacting with lithium-ions because of the electron-withdrawing nature of the –F group, allowing it to function as a diluent that does not dissociate the salt.2,250 Therefore, if fluorinated solvents are used alone, the dissociation ability of lithium salts decreases, which can limit the practical level of electrolyte design by decreasing ionic conductivity and thus causing high ohmic polarization. Hence, even though the formation of a stable LiF-rich SEI may improve the long-term interfacial stability of lithium-metal anodes and LMB performance, research on nonfluorinated salts and solvents should also be carried out to facilitate the practical utilization of the developed electrolytes.
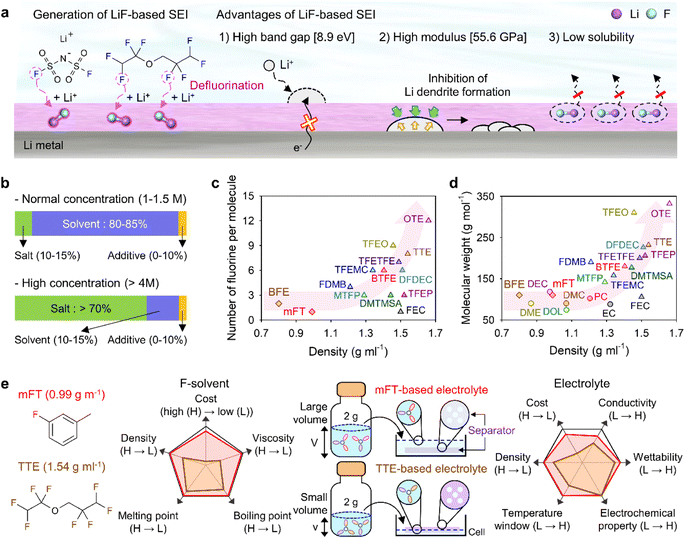 |
| | Fig. 14 (a) Schematic illustration of LiF-based SEI construction on lithium-metal anodes and advantages of such SEIs. (b) Contents of lithium salt, solvent, and additives in electrolytes with normal (1–1.5 M) and high (>4 M) concentrations. Comparison of electrolyte solvent (mainly fluorinated solvent) properties. (c) Per-molecule number of fluorine atoms and density of conventional fluorinated solvents. (d) Molecular weight and density of conventional nonfluorinated and fluorinated solvents. EC: ethylene carbonate, PC: propylene carbonate, DMC: dimethyl carbonate, DEC: diethyl carbonate, DME: 1,2-dimethoxyethane, DOL: 1,3-dioxolane, FEC: fluoroethylene carbonate, BTFE: bis(2,2,2-trifluoroethyl) ether, TTE: 1,1,2,2-tetrafluoroethyl 2,2,3,3-tetrafluoropropyl ether, OTE: 1H,1H,5H-octafluoropentyl 1,1,2,2-tetrafluoroethyl ether, TFEO: tris(2,2,2-trifluoroethyl)orthoformate, MTFP: methyl 3,3,3-trifluoropropionate, DMTMSA: N,N-dimethyltrifluoromethane-sulfonamide, FDMB: fluorinated 1,4-dimethoxylbutane, TFEMC: methyl 2,2,2-trifluoroethyl carbonate, DFDEC: di(2,2,2-trifluoroethyl) carbonate, TFEP: 2-(2,2,2-trifluoroethoxy)-1,3,2-dioxaphospholane-2-oxide, and TFETFE: 1,1,2,2-tetrafluoroethyl 2,2,2-trifluoroethyl ether. (e) Importance of fluorinated solvent (F-solvent) density considering low amounts of electrolytes for high-energy-density LMBs. H and L indicate high and low in a graph, respectively. Reproduced from ref. 184 with permission of American Chemical Society, copyright 2023. | |
4.4 Advanced lithiophilic structure design
In Section 3.3.1, we discussed the application of lithiophilic materials in forming in situ lithiophilic SEIs and the associated benefits of uniform lithium deposition, reduced plating overpotential, and decreased interfacial resistance. In view of the above, initial studies on the creation of lithiophilic SEI species were carried out by introducing electrolyte additives. Recent years have witnessed progress in the fabrication of lithiophilic structures based on the combination of lithiophilic SEI formers with other SEI-forming additives. Silver cations possess a higher reduction potential than lithium-cations and are therefore readily reduced at the lithium-metal anode to generate silver nanoparticles. A Ag + Li3N-based SEI formed using AgPF6 and LiNO3 as additives251 and a Ag–Li3N–LiF interlayer created by combining silver trifluoromethanesulfonate and LiNO3 were reported to enable stable LMB operation.252 In particular, our group reported that AgNO3 as the sole lithiophilic additive cannot ensure the long lifespan of LMBs and demonstrated that high-electrochemical-reversibility lithium-metal anodes in LMBs can be realized through hierarchical SEI creation based on differences in the electron-receiving tendencies and adsorption energies on lithium-metal (Fig. 13b).174
4.5 Rejuvenation of inorganic SEIs to active lithium
The main cause of LMB capacity degradation and insufficient lifespan is the formation of inactive (dead, i.e., electrically isolated) lithium.84 The thermal runaway caused by dead lithium and lithium dendrites hinders the practical application of LMBs.253
Recent studies have presented compelling evidence that SEIs formed on lithium-metal anodes are primarily composed of Li2O rather than LiF.17 However, the mechanical integrity and passivation function of a Li2O-dominant SEI are compromised because of the inherent volume fluctuation associated with lithium plating and stripping. As a result, dead lithium provokes the degradation of battery performance. Advanced artificial SEI structures and SEI-regulating electrolyte additives have been proposed to improve the performance of LMBs.174,254 However, SEIs are susceptible to breaking because of the severe volume fluctuation during lithium plating and stripping, which causes the exposure of fresh lithium to the electrolyte and the generation of new SEI species. This recurring breakage and restoration of the SEI limits its durability. Furthermore, the relationship between a defunct SEI and isolated lithium-metal debris remains uncertain, making it challenging to demonstrate the inhibition of dead lithium formation to prevent battery failure. Therefore, research on recovering dead lithium is crucial for improving the long-term cyclability and safety of LMBs. A triiodide/iodide (I3−/I−) redox couple can be utilized to reactivate inactive lithium (Fig. 13c).173 The reduction of I3− leads to the conversion of inactive lithium into soluble LiI, while the oxidation of LiI by the delithiated cathode restores lithium-ions in the cathode from the inactive lithium. The I3− regenerated through oxidation further drives redox reactions. The reactions involved in the above processes are presented below.
Reactions on the lithium-metal anode:173
| | | 3Li2O + 3I3− → 6Li+ + 8I− + IO3− | (3) |
| | | 6Li + IO3− → I− + 3Li2O | (4) |
| | | 2Li + I3− → 2Li+ + 3I− | (5) |
The I3− additive removes inactive lithium sources from the SEI, which results in the formation of soluble LiI and IO3− species (eqn (3)). LiIO3 can be reduced by lithium to form Li2O and LiI (eqn (4)). The deposited Li2O becomes part of the healthy SEI on the lithium-metal anode. The oxygen from the Li2O in the defunct SEI is transported to reconstruct the SEI on the lithium-metal via a resolvable LiIO3-based oxygen carriage medium. Additionally, I3− reacts with the lithium debris to produce LiI (eqn (5)). All lithium released from the defunct SEI or dead lithium exists as soluble LiI and is transported to the cathode through a LiI-based lithium carriage medium, finally returning to the lithium during the following charging.
Reactions on the cathode:
| | | LiFePO4: 2Li+ + 3I− + 2FePO4 → I3− + 2LiFePO4 | (6) |
| | | LiNi0.5Co0.2Mn0.3O2: 2xLi+ + 3xI− + 2Li1−xNi0.5Co0.2Mn0.3O2 → xI3− + 2LiNi0.5Co0.2Mn0.3O2 | (7) |
The LiI transferred to the cathode can react with the delithiated cathode and regenerate I3− at >2.89 V (oxidation potential of I− to I3−, eqn (6) and (7)).255 This regenerated I3− diffuses back to the lithium and continues the rejuvenation of the defunct SEI or dead lithium. The above rejuvenation mechanism has been extensively exploited to enhance the long-term reversibility of lithium-metal anodes.173,256,257 In addition, SEI stability can be improved by forming a Li3N-rich SEI through the rejuvenation of Li2O, which is unavoidably generated by the decomposition of LiNO3 (LiNO3 + 8Li+ + 8e− → Li3N + 3Li2O).103,258 This allows the formation of SEIs with superb ionic conductivity due to the increased content of highly ion-conductive Li3N (∼10−3 S cm−1) and enables epitaxy-like planar lithium deposition on lithium-metal anodes, thereby enhancing SEI stability.
4.6 Solvation chemistry
The tuning of solvation structures in liquid electrolytes modifies the composition and properties of SEIs on anodes by changing the LUMO energy (and hence, the electron-accepting ability) of solvents, salts, and additives, which are present in the solvation sheath (Fig. 15).14 Dilute electrolytes contain solvent-separated ion pairs (SSIPs) in which lithium-ions are coordinated by polar solvent molecules. Organic SEI species generated by the reductive decomposition of solvent molecules as promoters forming a primary solvation sheath in dilute electrolytes may facilitate the spatial relaxation of SEIs on anodes. However, at high-areal-capacities leading to large volume changes of silicon and lithium-metal anodes, mechanically fragile organic SEI species fail to prevent the direct contact of electrolytes with anodes, which results in the further consumption of electrolytes and the deactivation of anode materials covered with electrically resistive SEI species. The proportion of SSIPs decreases with increasing salt concentrations, and cation–anion aggregates (AGGs) emerge in the solvation sheath in HCEs (>3 M). FEC, which is employed as a reducible additive, induces contact ion pair (CIP) generation and the reductive decomposition of electrolytes by altering the solvation structure through the formation of dative bonds between lithium-ions and PF6− anions or FEC. The increased number of CIPs in the solvation structure lowers the LUMO energy of solvates and largely modifies the mechanism of SEI construction.259 Compared to SSIPs, CIPs comprising anions and lithium-cations facilitate the formation of inorganic species such as LiF, which effectively buffers the large strain induced by lithiated silicon. OTE and BTFE employed as F-diluents with DNs (<10) lower than those of main electrolyte solvents (Fig. 15) lead to the emergence of CIPs and weaken the strong dative bonds between lithium-ions and EC solvent molecules. Hence, these species hinder the decomposition of EC by minimizing the probability of its direct contact with the anode during charging and enabling the generation of inorganic-rich SEIs.213,260 Additionally, the replacement of EC with a mixture of tetrahydrofuran (THF) and 2-methyltetrahydrofuran (MTHF) (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) resulted in the formation of a LiF-rich SEI on the silicon anode in an electrolyte containing 2 M LiPF6.261 The latter salt formed CIPs and AGGs via Li+–PF6− aggregation and thus experienced a reduction potential increase. Importantly, as THF and MTHF have lower reduction potentials than Li+–PF6− aggregates, they are stable in electrolytes during cycling. The formation of LiF by the decomposition of PF6− anions effectively extended the lifespan of silicon anodes at an areal-capacity of 2.5 mA h cm−2. Similarly, the formation of LiF by FSI− and TFSI− decomposition in CIPs and AGGs enhances the interfacial properties of lithium-metal anodes. As the participation of anions in the solvation structure at elevated salt concentrations in electrolytes creates anion-derived SEIs, the structural optimization of anions can maximize the quality of SEIs comprising electrochemically stable inorganic species. HCEs commonly contain polar solvents with high dielectric constants and DNs sufficient to achieve high salt concentrations (>3 M) without solid precipitation. HCEs may be referred to as solvate ionic liquids (SILs) – a new class of ionic liquids consisting of a coordinating solvent and salt and possessing very similar properties to those of ionic liquids.195 Glyme–lithium salt SILs show good thermal stability, unique lithium-ion transport through ligand exchange between chelating complexes, poorly dissolving SEI components, improved oxidation durability, and aluminum corrosion inhibition. These facilitate the future application of SILs as next-generation lithium battery electrolytes. At such high salt concentrations, anions start to enter the primary solvation sheath of lithium-ions and the formation of CIPs and AGGs becomes more feasible. CIPs and AGGs decrease the proportion of free solvent molecules and facilitate the generation of an inner Helmholtz layer on the lithium-metal surface (Fig. 15). Instead of a solvent-derived SEI, an anion-derived inorganic SEI capable of enduring the drastic volumetric stress due to the lithiation and delithiation of silicon and lithium-metal anodes is constructed. However, the practical usage of HCEs is hindered by their high viscosity, poor wettability, and high cost. The addition of fluorinated solvents to HCEs affords less viscous electrolytes denoted as LHCEs. These fluorinated solvents are miscible with strongly solvating solvents but do not enter the primary solvation sheath. In particular, since the trifluoromethyl group (CF3) is a functional group with strong electron substituents, it can weaken the interaction between lithium-ions and fluorinated solvent molecules.240 In this manner, fluorinated solvents reduce electrolyte viscosity by enhancing the mutual interaction between solvent–lithium-ion complexes while preserving the overall solvation structure of the HCE, including CIPs and AGGs. Even though small amounts of salts (1–1.2 M) are introduced into the electrolyte, anion-derived SEIs can be formed by CIPs and AGGs. Thus, when LHCEs enabling the creation of anion-derived SEIs are used, side reactions between the electrolyte and lithium-metal can be effectively controlled during lithium stripping/plating. Furthermore, fluorinated solvent molecules allow LMBs to be operated even in extreme environments, e.g., at elevated or extremely low temperatures. Ren et al. developed sulfone-based LHCEs with a fluorinated ethereal solvent (TTE) allowing the stable operation of LMBs at −10 °C.262 Since the discovery of the important effects of fluorinated solvents on changes in the solvation structure and the manipulation of SEI properties, the development of these solvents has drawn much attention (Fig. 15). However, fluorinated solvents remain costly, and the initial solvation structures may not be preserved during cycling because of the consumption of fluorinated solvents for SEI construction upon first lithium plating. Furthermore, for the primary solvation sheath of lithium-ions in LHCEs, the strong interaction between these ions and polar solvent molecules favors the reductive decomposition of solvent molecules in the vicinity of the anode and causes the sluggish desolvation of lithium-ions at the lithium-metal anode. The interaction between lithium-ions and polar solvents can be weakened by decreasing the solvating power of the latter through oxygen atom removal or increasing the steric hindrance in solvent molecules based on the use of longer alkyl chains.263–265 This change in the molecular structure of polar solvents not only affords weakly solvating solvents, but also allows for CIP and AGG formation even at low concentrations (1 M).
:1, v/v) resulted in the formation of a LiF-rich SEI on the silicon anode in an electrolyte containing 2 M LiPF6.261 The latter salt formed CIPs and AGGs via Li+–PF6− aggregation and thus experienced a reduction potential increase. Importantly, as THF and MTHF have lower reduction potentials than Li+–PF6− aggregates, they are stable in electrolytes during cycling. The formation of LiF by the decomposition of PF6− anions effectively extended the lifespan of silicon anodes at an areal-capacity of 2.5 mA h cm−2. Similarly, the formation of LiF by FSI− and TFSI− decomposition in CIPs and AGGs enhances the interfacial properties of lithium-metal anodes. As the participation of anions in the solvation structure at elevated salt concentrations in electrolytes creates anion-derived SEIs, the structural optimization of anions can maximize the quality of SEIs comprising electrochemically stable inorganic species. HCEs commonly contain polar solvents with high dielectric constants and DNs sufficient to achieve high salt concentrations (>3 M) without solid precipitation. HCEs may be referred to as solvate ionic liquids (SILs) – a new class of ionic liquids consisting of a coordinating solvent and salt and possessing very similar properties to those of ionic liquids.195 Glyme–lithium salt SILs show good thermal stability, unique lithium-ion transport through ligand exchange between chelating complexes, poorly dissolving SEI components, improved oxidation durability, and aluminum corrosion inhibition. These facilitate the future application of SILs as next-generation lithium battery electrolytes. At such high salt concentrations, anions start to enter the primary solvation sheath of lithium-ions and the formation of CIPs and AGGs becomes more feasible. CIPs and AGGs decrease the proportion of free solvent molecules and facilitate the generation of an inner Helmholtz layer on the lithium-metal surface (Fig. 15). Instead of a solvent-derived SEI, an anion-derived inorganic SEI capable of enduring the drastic volumetric stress due to the lithiation and delithiation of silicon and lithium-metal anodes is constructed. However, the practical usage of HCEs is hindered by their high viscosity, poor wettability, and high cost. The addition of fluorinated solvents to HCEs affords less viscous electrolytes denoted as LHCEs. These fluorinated solvents are miscible with strongly solvating solvents but do not enter the primary solvation sheath. In particular, since the trifluoromethyl group (CF3) is a functional group with strong electron substituents, it can weaken the interaction between lithium-ions and fluorinated solvent molecules.240 In this manner, fluorinated solvents reduce electrolyte viscosity by enhancing the mutual interaction between solvent–lithium-ion complexes while preserving the overall solvation structure of the HCE, including CIPs and AGGs. Even though small amounts of salts (1–1.2 M) are introduced into the electrolyte, anion-derived SEIs can be formed by CIPs and AGGs. Thus, when LHCEs enabling the creation of anion-derived SEIs are used, side reactions between the electrolyte and lithium-metal can be effectively controlled during lithium stripping/plating. Furthermore, fluorinated solvent molecules allow LMBs to be operated even in extreme environments, e.g., at elevated or extremely low temperatures. Ren et al. developed sulfone-based LHCEs with a fluorinated ethereal solvent (TTE) allowing the stable operation of LMBs at −10 °C.262 Since the discovery of the important effects of fluorinated solvents on changes in the solvation structure and the manipulation of SEI properties, the development of these solvents has drawn much attention (Fig. 15). However, fluorinated solvents remain costly, and the initial solvation structures may not be preserved during cycling because of the consumption of fluorinated solvents for SEI construction upon first lithium plating. Furthermore, for the primary solvation sheath of lithium-ions in LHCEs, the strong interaction between these ions and polar solvent molecules favors the reductive decomposition of solvent molecules in the vicinity of the anode and causes the sluggish desolvation of lithium-ions at the lithium-metal anode. The interaction between lithium-ions and polar solvents can be weakened by decreasing the solvating power of the latter through oxygen atom removal or increasing the steric hindrance in solvent molecules based on the use of longer alkyl chains.263–265 This change in the molecular structure of polar solvents not only affords weakly solvating solvents, but also allows for CIP and AGG formation even at low concentrations (1 M).
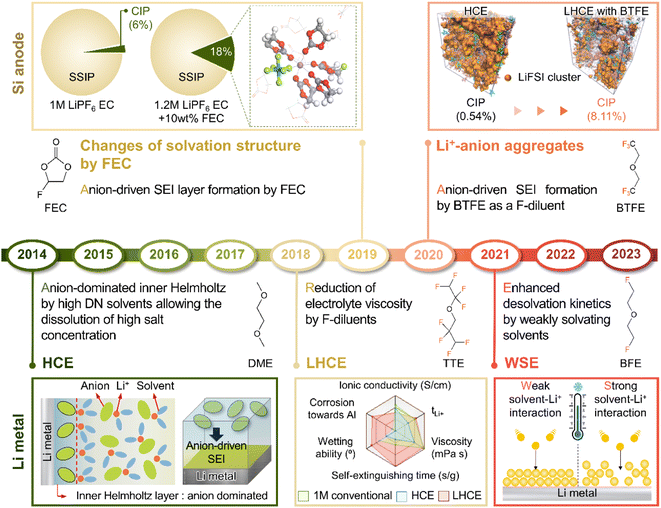 |
| | Fig. 15 Research trends in electrolyte solvation structure for silicon and lithium-metal anodes. Reproduced with ref. 222, 251, and 252 with permission of American Chemical Society, copyright 2020, and Elsevier, copyright 2019, 2022, respectively. | |
Fluorination is another effective method of regulating the steric hindrance of solvent molecules. Zhang et al. reported the effects of the fluorination degree on solvent properties.175 Compared to its trifluoro (–CF3) and difluoro (–CHF2) counterparts, the monofluoro (–CH2F) group allowed for high ionic conductivity and good compatibility with lithium-metal anodes and nickel-rich cathodes. The regulation of the interaction between lithium-ions and solvent molecules enabled desolvation kinetics enhancement and helped suppress the parasitic reactions of electrolytes at the lithium-metal anode, which resulted in a dense and less dendritic morphology of the deposited lithium.
5. Summary and outlook
The widespread use of silicon and lithium-metal anodes highlights the need to make their high lithium-storage performance well-reversible, which is hard to achieve because of the extreme volume fluctuations causing structural and compositional SEI changes during cycling. To make strain-adaptable and long-lasting SEIs feasible, we envision a platform that seamlessly integrates the design of cheap and ubiquitous electrolytes, electrolyte formulation, and accurate in-depth analysis for the realization of high-performance silicon and lithium-metal anodes. The regulation of the chemical environment of SEIs at anode-electrolyte interfaces is believed to enable the realization of stable-cycling-performance batteries with silicon and lithium-metal anodes. The combined use of two or three species such as high-modulus LiF, lithiophilic materials, and spatially deformable organic species for homogeneous distribution in single-layer and hierarchical (bi- or multilayer) structures can meet the versatile specifications for extending the health of batteries over wide temperature ranges, which is not readily attained using monocomponent SEIs. Moreover, the desirable target SEI species should maintain their interconnections for satisfactory mechanical integrity to endure the volume fluctuation of silicon and lithium-metal anodes upon lithiation and delithiation. Although anion-derived or solvent-derived SEIs can enhance anode performance, the consumption of salt anions and solvents precludes the preservation of initial lithium-ion solvation structures driven by the competitive coordination of solvents and anions to lithium-cations during cycling (Fig. 16). In this regard, the incorporation of sacrificial interface modifiers, which mitigate the unnecessary consumption of electrolyte solvents and salts ensuring ion conduction in cells, can be a practical option. Although the developed additives enable the creation of stable SEIs on anodes, side effects such as solvation structure change and solvent/salt decomposition should be thoroughly examined prior to application to batteries. Another important aspect of additive development is the establishment of optimal charging protocols (C-rates, temperatures, and pulse voltage rise) for the formation of the desired SEIs, as the charging protocol greatly influences the decomposition pathways of additives with different reduction kinetics and target SEI components may not be created. More importantly, the deep understanding of the functions of existing materials through in situ and ex situ analyses should pave the way for the discovery of new research directions in the field of liquid electrolytes for silicon and lithium-metal anodes. The optimized design of interface modifiers along with solvents and salts providing elements or motifs for the creation of stable SEIs will move silicon and lithium-metal anodes closer to industrial applications, especially considering minimal environmental issues, cost-effectiveness (especially <$78 kg−1 for additives), and practical conditions such as the high mass loading (≥3–4 mA h cm−2) of silicon and the use of thinner lithium metal (anode-to-cathode capacity ratio ≤2) to achieve high-energy-density batteries (Fig. 16). Furthermore, to be applicable to commercial lithium rechargeable batteries, the developed electrolyte materials should have a reasonably low density of <1.4 g cm−3 (to ensure that electrolyte injection during battery manufacturing does not cause electrolyte wetting-related problems) and long-term storage stability (low melting point of <−20 °C and a high boiling point of >100 °C) with less gas evolution over a wide temperature range of −40–80 °C. Despite the multitude of available electrolyte systems, common reference electrolytes for lithium-metal anodes meeting all industrial requirements remain unknown. If LiFSI is employed as the main salt, moderate salt concentrations should be considered to mitigate aluminum corrosion upon cell operation, especially at elevated temperatures or high voltages. In the case of silicon-containing anodes, FEC and FEC + VC combinations are still considered interface modifiers, and their content is manipulated by the amount of silicon in the LIB anode, as the few additives electrochemically outperforming FEC and VC are overly costly for commercial applications. A deep understanding of the developed SEIs will offer new insights into the establishment of liquid electrolyte chemistry to realize long-lasting batteries with silicon or lithium-metal anodes without any significant increase in cost.
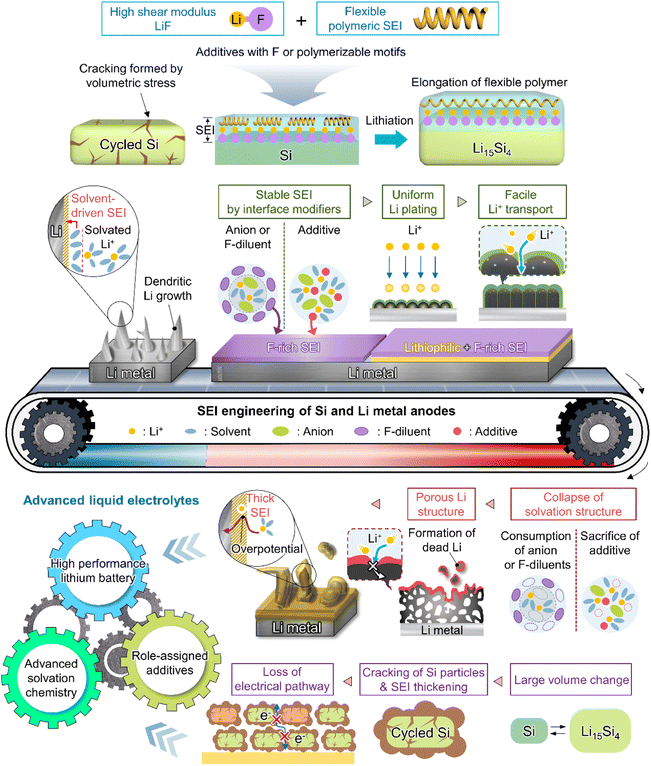 |
| | Fig. 16 Necessity of advanced liquid electrolyte chemistries for high-energy lithium batteries with silicon and lithium-metal anodes. | |
Author contributions
S. Park and S. Kim contributed equally to this study. Conceptualization: S. Park, S. Kim and N.-S. Choi; writing the original draft: S. Park, S. Kim and J.-A. Lee; validation: M. Ue and N.-S. Choi; review and editing: M. Ue and N.-S. Choi.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This study was supported by LG Energy Solution-KAIST Frontier Research Laboratory, the Technology Development Program (2022M3J1A1085404, Development of Key Technologies for Lithium–Air Batteries with High Energy Density Based on Tectonic Electrode Designs) funded by the Ministry of Science, ICT & Future Planning, and the Technology Innovation Program (20012216, Development of Ultrathin Anode Technology for High Energy Density Batteries) funded by the Ministry of Trade, Industry & Energy.
References
- S. Chae, M. Ko, K. Kim, K. Ahn and J. Cho, Joule, 2017, 1, 47–60 CrossRef CAS.
- H. Wang, Z. Yu, X. Kong, S. C. Kim, D. T. Boyle, J. Qin, Z. Bao and Y. Cui, Joule, 2022, 6, 588–616 CrossRef CAS.
- H. Wu, H. Jia, C. Wang, J. G. Zhang and W. Xu, Adv. Energy Mater., 2021, 11, 2003092 CrossRef CAS.
- Y. He, L. Jiang, T. Chen, Y. Xu, H. Jia, R. Yi, D. Xue, M. Song, A. Genc and C. Bouchet-Marquis, Nat. Nanotechnol., 2021, 16, 1113–1120 CrossRef CAS PubMed.
- K. Xu, Nat. Energy, 2021, 6, 763 CrossRef CAS.
- A. Wang, S. Kadam, H. Li, S. Shi and Y. Qi, npj Comput. Mater., 2018, 4, 15 CrossRef.
-
B. Simon and J.-P. Boeuve, Societe des Accumulateurs Fixes et de Traction SA, FR Pat., FR2719161B1, Priority, 1994.
-
R. S. McMillan, D. J. Worsfold, J. J. Murray, I. Davidson and Z. X. Shu, National Research Council of Canada, US Pat., US6506524B1, Priority, 1996.
- D. Aurbach, K. Gamolsky, B. Markovsky, Y. Gofer, M. Schmidt and U. Heider, Electrochim. Acta, 2002, 47, 1423–1439 CrossRef CAS.
- N.-S. Choi, K. H. Yew, K. Y. Lee, M. Sung, H. Kim and S.-S. Kim, J. Power Sources, 2006, 161, 1254–1259 CrossRef CAS.
-
R. Oshita, M. Fujimoto, T. Noma and K. Nishio, Sanyo Electric Co. Ltd, JP Pat., JP3439085B2, Priority, 1997.
- L. Li, W. Lv, J. Chen, C. Zhu, S. Dmytro, Q. Zhang and S. Zhong, ACS Appl. Energy Mater., 2022, 5, 11900–11914 CrossRef CAS.
- B. Zhang, M. Metzger, S. Solchenbach, M. Payne, S. Meini, H. A. Gasteiger, A. Garsuch and B. L. Lucht, J. Phys. Chem. C, 2015, 119, 11337–11348 CrossRef CAS.
- J.-G. Han, J. B. Lee, A. Cha, T. K. Lee, W. Cho, S. Chae, S. J. Kang, S. K. Kwak, J. Cho, S. Y. Hong and N.-S. Choi, Energy Environ. Sci., 2018, 11, 1552–1562 RSC.
- S. Yuan, S. Weng, F. Wang, X. Dong, Y. Wang, Z. Wang, C. Shen, J. L. Bao, X. Wang and Y. Xia, Nano Energy, 2021, 83, 105847 CrossRef CAS.
- C. Ma, F. Xu and T. Song, ACS Appl. Mater. Interfaces, 2022, 14, 20197–20207 CrossRef CAS PubMed.
- Y. Li, Y. Li, A. Pei, K. Yan, Y. Sun, C.-L. Wu, L.-M. Joubert, R. Chin, A. L. Koh, Y. Yu, J. Perrino, B. Butz, S. Chu and Y. Cui, Science, 2017, 358, 506–510 CrossRef CAS PubMed.
- B. Han, X. Li, S. Bai, Y. Zou, B. Lu, M. Zhang, X. Ma, Z. Chang, Y. S. Meng and M. Gu, Matter, 2021, 4, 3741–3752 CrossRef CAS.
- X. Zhang, S. Weng, G. Yang, Y. Li, H. Li, D. Su, L. Gu, Z. Wang, X. Wang and L. Chen, Cell Rep. Phys. Sci., 2021, 2, 100668 CrossRef CAS.
- X. Q. Zhang, X. Chen, X. B. Cheng, B. Q. Li, X. Shen, C. Yan, J. Q. Huang and Q. Zhang, Angew. Chem., Int. Ed., 2018, 57, 5301–5305 CrossRef CAS PubMed.
- S. Kim, S. O. Park, M.-Y. Lee, J.-A. Lee, I. Kristanto, T. K. Lee, D. Hwang, J. Kim, T.-U. Wi and H.-W. Lee, Energy Storage Mater., 2022, 45, 1–13 CrossRef.
- K.-C. Möller, H. Santner, W. Kern, S. Yamaguchi, J. Besenhard and M. Winter, J. Power Sources, 2003, 119, 561–566 CrossRef.
- Q. Wang, J. Yang, X. Huang, Z. Zhai, J. Tang, J. You, C. Shi, W. Li, P. Dai and W. Zheng, Adv. Energy Mater., 2022, 12, 2103972 CrossRef CAS.
- M. Piernas-Muñoz, A. Tornheim, S. Trask, Z. Zhang and I. Bloom, Chem. Commun., 2021, 57, 2253–2256 RSC.
- M. Nie, D. P. Abraham, Y. Chen, A. Bose and B. L. Lucht, J. Phys. Chem. C, 2013, 117, 13403–13412 CrossRef CAS.
- S. Pan, J. Han, Y. Wang, Z. Li, F. Chen, Y. Guo, Z. Han, K. Xiao, Z. Yu and M. Yu, Adv. Mater., 2022, 34, 2203617 CrossRef CAS PubMed.
- H. Nakai, T. Kubota, A. Kita and A. Kawashima, J. Electrochem. Soc., 2011, 158, A798 CrossRef CAS.
- K. Schroder, J. Alvarado, T. A. Yersak, J. Li, N. Dudney, L. J. Webb, Y. S. Meng and K. J. Stevenson, Chem. Mater., 2015, 27, 5531–5542 CrossRef CAS.
- W. Huang, H. Wang, D. T. Boyle, Y. Li and Y. Cui, ACS Energy Lett., 2020, 5, 1128–1135 CrossRef CAS.
- Z. L. Brown, S. Jurng, C. C. Nguyen and B. L. Lucht, ACS Appl. Energy Mater., 2018, 1, 3057–3062 CrossRef CAS.
- T. Jaumann, J. Balach, U. Langklotz, V. Sauchuk, M. Fritsch, A. Michaelis, V. Teltevskij, D. Mikhailova, S. Oswald and M. Klose, Energy Storage Mater., 2017, 6, 26–35 CrossRef.
- Z. Zhang, Y. Li, R. Xu, W. Zhou, Y. Li, S. T. Oyakhire, Y. Wu, J. Xu, H. Wang and Z. Yu, Science, 2022, 375, 66–70 CrossRef CAS PubMed.
- S. Shi, P. Lu, Z. Liu, Y. Qi, L. G. Hector Jr, H. Li and S. J. Harris, J. Am. Chem. Soc., 2012, 134, 15476–15487 CrossRef CAS PubMed.
- A. Ramasubramanian, V. Yurkiv, T. Foroozan, M. Ragone, R. Shahbazian-Yassar and F. Mashayek, J. Phys. Chem. C, 2019, 123, 10237–10245 CrossRef CAS.
- A. M. F. A. Soto, F. El-Mellouhi and P. B. Balbuena, Chem. Mater., 2018, 30, 3315–3322 CrossRef.
- S. Lorger, R. Usiskin and J. Maier, J. Electrochem. Soc., 2019, 166, A2215–A2220 CrossRef CAS.
- A. S. Kulathuvayal and Y. Su, ACS Appl. Energy Mater., 2023, 6, 5628–5645 CrossRef CAS.
- C. Wang, S. Wang, Y.-B. He, L. Tang, C. Han, C. Yang, M. Wagemaker, B. Li, Q.-H. Yang, J.-K. Kim and F. Kang, Chem. Mater., 2015, 27, 5647–5656 CrossRef CAS.
- X.-X. Ma, X. Shen, X. Chen, Z.-H. Fu, N. Yao, R. Zhang and Q. Zhang, Small Struct., 2022, 3, 2200071 CrossRef CAS.
- D. Wu, J. He, J. Liu, M. Wu, S. Qi, H. Wang, J. Huang, F. Li, D. Tang and J. Ma, Adv. Energy Mater., 2022, 12, 2200337 CrossRef CAS.
- D. Aurbach, A. Zaban, Y. Ein-Eli, I. Weissman, O. Chusid, B. Markovsky, M. Levi, E. Levi, A. Schechter and E. Granot, J. Power Sources, 1997, 65, 91–98 CrossRef.
- L. Terborg, S. Nowak, S. Passerini, M. Winter, U. Karst, P. R. Haddad and P. N. Nesterenko, Anal. Chim. Acta, 2012, 714, 121–126 CrossRef CAS PubMed.
- J. G. Han, K. Kim, Y. Lee and N.-S. Choi, Adv. Mater., 2019, 31, e1804822 CrossRef PubMed.
- X. Shan, Y. Zhong, L. Zhang, Y. Zhang, X. Xia, X. Wang and J. Tu, J. Phys. Chem. C, 2021, 125, 19060–19080 CrossRef CAS.
- J. Qian, W. Xu, P. Bhattacharya, M. Engelhard, W. A. Henderson, Y. Zhang and J.-G. Zhang, Nano Energy, 2015, 15, 135–144 CrossRef CAS.
- H. Shin, J. Park, S. Han, A. M. Sastry and W. Lu, J. Power Sources, 2015, 277, 169–179 CrossRef CAS.
- Y. Wang and F. Hao, J. Electrochem. Energy Convers. Storage, 2022, 19, 040801 CrossRef CAS.
- K. Chen, R. Pathak, A. Gurung, E. A. Adhamash, B. Bahrami, Q. He, H. Qiao, A. L. Smirnova, J. J. Wu, Q. Qiao and Y. Zhou, Energy Storage Mater., 2019, 18, 389–396 CrossRef.
- P. Zhai, L. Liu, X. Gu, T. Wang and Y. Gong, Adv. Energy Mater., 2020, 10, 2001257 CrossRef CAS.
- Z. Li, Y. Chen, X. Yun, P. Gao, C. Zheng and P. Xiao, Adv. Funct. Mater., 2023, 2300502 CrossRef CAS.
- Y. Zhu, X. He and Y. Mo, ACS Appl. Mater. Interfaces, 2015, 7, 23685–23693 CrossRef CAS PubMed.
- L. Schafzahl, H. Ehmann, M. Kriechbaum, J. Sattelkow, T. Ganner, H. Plank, M. Wilkening and S. A. Freunberger, Chem. Mater., 2018, 30, 3338–3345 CrossRef CAS.
- K. Park and J. B. Goodenough, Adv. Energy Mater., 2017, 7, 1700732 CrossRef.
- J. Jones, M. Anouti, M. Caillon-Caravanier, P. Willmann and D. Lemordant, Fluid Phase Equilib., 2009, 285, 62–68 CrossRef CAS.
- B. Han, Z. Zhang, Y. Zou, K. Xu, G. Xu, H. Wang, H. Meng, Y. Deng, J. Li and M. Gu, Adv. Mater., 2021, 33, e2100404 CrossRef PubMed.
- Y. Sun, Y. Li, J. Sun, Y. Li, A. Pei and Y. Cui, Energy Storage Mater., 2017, 6, 119–124 CrossRef.
- X. Fan, X. Ji, F. Han, J. Yue, J. Chen, L. Chen, T. Deng, J. Jiang and C. Wang, Sci. Adv., 2018, 4, eaau9245 CrossRef CAS PubMed.
- Y. Liu, L. Hong, R. Jiang, Y. Wang, S. V. Patel, X. Feng and H. Xiang, ACS Appl. Mater. Interfaces, 2021, 13, 57430–57441 CrossRef CAS PubMed.
- H. Kang, M. Song, M. Yang and J.-w. Lee, J. Power Sources, 2021, 485, 229286 CrossRef CAS.
- Z. Piao, P. Xiao, R. Luo, J. Ma, R. Gao, C. Li, J. Tan, K. Yu, G. Zhou and H. M. Cheng, Adv. Mater., 2022, 34, e2108400 CrossRef PubMed.
- S. Liu, X. Ji, N. Piao, J. Chen, N. Eidson, J. Xu, P. Wang, L. Chen, J. Zhang, T. Deng, S. Hou, T. Jin, H. Wan, J. Li, J. Tu and C. Wang, Angew. Chem., Int. Ed., 2021, 60, 3661–3671 CrossRef CAS PubMed.
- J. Shin, T.-H. Kim, Y. Lee and E. Cho, Energy Storage Mater., 2020, 25, 764–781 CrossRef.
- S. S. Zhang, Energy Storage Mater., 2020, 24, 247–254 CrossRef.
- C. Zhan, T. Wu, J. Lu and K. Amine, Energy Environ. Sci., 2018, 11, 243 RSC.
- S. Y. Park, S. Park, H. Y. Lim, M. Yoon, J. H. Choi, S. K. Kwak, S. Y. Hong and N.-S. Choi, Adv. Sci., 2023, 10, e2205918 CrossRef PubMed.
- Y. Kim, J. Lim and S. Kang, Int. J. Quantum Chem., 2013, 113, 148–154 CrossRef CAS.
- T. Liu, A. Dai, J. Lu, Y. Yuan, Y. Xiao, L. Yu, M. Li, J. Gim, L. Ma, J. Liu, C. Zhan, L. Li, J. Zheng, Y. Ren, T. Wu, R. Shahbazian-Yassar, J. Wen, F. Pan and K. Amine, Nat. Commun., 2019, 10, 4721 CrossRef CAS PubMed.
- C. Jayawardana, N. Rodrigo, B. Parimalam and B. L. Lucht, ACS Energy Lett., 2016, 6, 3788–3792 CrossRef.
- W. Li, J. Electrochem. Soc., 2020, 167, 090514 CrossRef CAS.
- X. Zhang, Z. Cui and A. Manthiram, Adv. Energy Mater., 2022, 12, 2103611 CrossRef CAS.
- J. Betz, J.-P. Brinkmann, R. Nölle, C. Lürenbaum, M. Kolek, M. C. Stan, M. Winter and T. Placke, Adv. Energy Mater., 2019, 9, 1900574 CrossRef.
- R. A. Vilá, W. Huang and Y. Cui, Cell Rep. Phys. Sci., 2020, 1, 100188 CrossRef.
- L. Chen, K. Wang, X. Xie and J. Xie, J. Power Sources, 2007, 174, 538–543 CrossRef CAS.
- M. Ulldemolins, F. L. Cras, B. Pecquenard, V. P. Phan, L. Martin and H. Martinez, J. Power Sources, 2012, 206, 245–252 CrossRef CAS.
- S. Dalavi, P. Guduru and B. L. Lucht, J. Electrochem. Soc., 2012, 159, A642–A646 CrossRef CAS.
- C. C. Nguyen and B. L. Lucht, J. Electrochem. Soc., 2014, 161, A1933–A1938 CrossRef.
- Y. Jin, N. H. Kneusels, L. E. Marbella, E. Castillo-Martinez, P. Magusin, R. S. Weatherup, E. Jonsson, T. Liu, S. Paul and C. P. Grey, J. Am. Chem. Soc., 2018, 140, 9854–9867 CrossRef CAS.
- G.-B. Han, M.-H. Ryou, K. Cho, Y. Lee and J.-K. Park, J. Power Sources, 2010, 195, 3709–3714 CrossRef CAS.
- A. Rezqita, M. Sauer, A. Foelske, H. Kronberger and A. Trifonova, Electrochim. Acta, 2017, 247, 600–609 CrossRef CAS.
- G. Zhuang, K. Xu, H. Yang, T. R. Jow and P. R. Jr, J. Phys. Chem. B, 2005, 109, 17567–17573 CrossRef CAS PubMed.
- D. Aurbach, M. L. Daroux, P. W. Faguy and E. Yeager, J. Electrochem. Soc., 1987, 134, 1611–1620 CrossRef CAS.
- H. Ota, Y. Sakata, Y. Otake, K. Shima, M. Ue and J.-i. Yamaki, J. Electrochem. Soc., 2004, 151, A1778–A1788 CrossRef CAS.
- X. Zheng, L. Huang, X. Ye, J. Zhang, F. Min, W. Luo and Y. Huang, Chem, 2021, 7, 2312–2346 CAS.
- C. Fang, J. Li, M. Zhang, Y. Zhang, F. Yang, J. Z. Lee, M. H. Lee, J. Alvarado, M. A. Schroeder, Y. Yang, B. Lu, N. Williams, M. Ceja, L. Yang, M. Cai, J. Gu, K. Xu, X. Wang and Y. S. Meng, Nature, 2019, 572, 511–515 CrossRef CAS PubMed.
- H.-H. Sun, A. Dolocan, J. A. Weeks, R. Rodriguez, A. Heller and C. B. Mullins, J. Mater. Chem. A, 2019, 7, 17782–17789 RSC.
- C. Monroe and J. Newman, J. Electrochem. Soc., 2004, 151, A880–A886 CrossRef CAS.
- J. Tan, J. Matz, P. Dong, J. Shen and M. Ye, Adv. Energy Mater., 2021, 11, 2100046 CrossRef CAS.
- L. A. Ma, A. J. Naylor, L. Nyholm and R. Younesi, Angew Chem. Int. Ed. Engl., 2021, 60, 4855–4863 CrossRef CAS PubMed.
- J. Pan and Y.-T. Cheng, Phys. Rev. B: Condens.
Matter Mater. Phys., 2015, 91, 134116 CrossRef.
- Z. Liu, Y. Qi, Y. X. Lin, L. Chen, P. Lu and L. Q. Chen, J. Electrochem. Soc., 2016, 163, A592–A598 CrossRef CAS.
- Q. Zhang, J. Pan, P. Lu, Z. Liu, M. W. Verbrugge, B. W. Sheldon, Y. T. Cheng, Y. Qi and X. Xiao, Nano Lett., 2016, 16, 2011–2016 CrossRef CAS PubMed.
- S. Liu, X. Ji, J. Yue, S. Hou, P. Wang, C. Cui, J. Chen, B. Shao, J. Li, F. Han, J. Tu and C. Wang, J. Am. Chem. Soc., 2020, 142, 2438–2447 CrossRef CAS PubMed.
- S. Jiang, X. Xu, J. Yin, H. Wu, X. Zhu and Y. Gao, ACS Appl. Mater. Interfaces, 2022, 14, 38758–38768 CrossRef CAS PubMed.
- S. Jiao, X. Ren, R. Cao, M. H. Engelhard, Y. Liu, D. Hu, D. Mei, J. Zheng, W. Zhao, Q. Li, N. Liu, B. D. Adams, C. Ma, J. Liu, J.-G. Zhang and W. Xu, Nat. Energy, 2018, 3, 739–746 CrossRef CAS.
- M. G. Oh, S. Kwak, K. An, Y. H. T. Tran, D. G. Kang, S. J. Park, G. Lim, K. Kim, Y. S. Lee and S. W. Song, Adv. Funct. Mater., 2023, 33, 2212890 CrossRef CAS.
- Z. Ahmad, V. Venturi, H. Hafiz and V. Viswanathan, J. Phys. Chem. C, 2021, 125, 11301–11309 CrossRef CAS.
- X. Wang, S. Li, W. Zhang, D. Wang, Z. Shen, J. Zheng, H. L. Zhuang, Y. He and Y. Lu, Nano Energy, 2021, 89, 106353 CrossRef CAS.
- Y. Ren, J. S. Ko, R. M. Kasse, X. Song, M. F. Toney and J. N. Weker, J. Electrochem. Energy Convers. Storage, 2020, 17, 041009 CrossRef.
- Q. Shi, Y. Zhong, M. Wu, H. Wang and H. Wang, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 5676–5680 CrossRef CAS PubMed.
- H. Liu, X. Yue, X. Xing, Q. Yan, J. Huang, V. Petrova, H. Zhou and P. Liu, Energy Storage Mater., 2019, 16, 505–511 CrossRef.
- U. v. ALPEN, J. Solid State Chem., 1978, 29, 379–392 CrossRef.
- H. D. Nguyen, T. D. Pham, A. B. Faheem, H. M. Oh and K. K. Lee, Batteries Supercaps, 2023, 6, e202200453 CrossRef.
- G. G. Eshetu, X. Judez, C. Li, O. Bondarchuk, L. M. Rodriguez-Martinez, H. Zhang and M. Armand, Angew Chem. Int. Ed. Engl., 2017, 56, 15368–15372 CrossRef CAS PubMed.
- T. Chen, Z. Jin, Y. Liu, X. Zhang, H. Wu, M. Li, W. Feng, Q. Zhang and C. Wang, Angew Chem. Int. Ed. Engl., 2022, 61, e202207645 CrossRef CAS PubMed.
- C. Liao, L. Han, W. Wang, W. Li, X. Mu, Y. Kan, J. Zhu, Z. Gui, X. He, L. Song and Y. Hu, Adv. Funct. Mater., 2023, 33, 2212605 CrossRef CAS.
- S. Li, W. Zhang, Q. Wu, L. Fan, X. Wang, X. Wang, Z. Shen, Y. He and Y. Lu, Angew Chem. Int. Ed. Engl., 2020, 59, 14935–14941 CrossRef CAS PubMed.
- W. Zhang, Z. Shen, S. Li, L. Fan, X. Wang, F. Chen, X. Zang, T. Wu, F. Ma and Y. Lu, Adv. Funct. Mater., 2020, 30, 2003800 CrossRef CAS.
- Z. Guo, X. Song, Q. Zhang, N. Zhan, Z. Hou, Q. Gao, Z. Liu, Z. Shen and Y. Zhao, ACS Energy Lett., 2022, 7, 569–576 CrossRef CAS.
- S. H. Lee, J. Y. Hwang, J. Ming, Z. Cao, H. A. Nguyen, H. G. Jung, J. Kim and Y. K. Sun, Adv. Energy Mater., 2020, 10, 2000567 CrossRef CAS.
- R. Zhao, X. Li, Y. Si, W. Guo and Y. Fu, ACS Appl. Mater. Interfaces, 2021, 13, 40582–40589 CrossRef CAS PubMed.
- Z. Wang, L. P. Hou, Z. Li, J. L. Liang, M. Y. Zhou, C. Z. Zhao, X. Zeng, B. Q. Li, A. Chen and X. Q. Zhang, Carbon Energy, 2023, 5, e283 CrossRef CAS.
- Q.-K. Zhang, S.-Y. Sun, M.-Y. Zhou, L.-P. Hou, J.-L. Liang, S.-J. Yang, B.-Q. Li, X.-Q. Zhang and J.-Q. Huang, Angew. Chem., Int. Ed., 2023, e202306889 Search PubMed.
- V. A. K. Adiraju, O. B. Chae, J. R. Robinson and B. L. Lucht, ACS Energy Lett., 2023, 8, 2440–2446 CrossRef CAS.
- D. Aurbach, E. Pollak, R. Elazari, G. Salitra, C. S. Kelley and J. Affinito, J. Electrochem. Soc., 2009, 156, A694–A702 CrossRef CAS.
- X.-Q. Zhang, X. Chen, L.-P. Hou, B.-Q. Li, X.-B. Cheng, J.-Q. Huang and Q. Zhang, ACS Energy Lett., 2019, 4, 411–416 CrossRef CAS.
- Q. Wang, Z. Yao, C. Zhao, T. Verhallen, D. P. Tabor, M. Liu, F. Ooms, F. Kang, A. Aspuru-Guzik, Y. S. Hu, M. Wagemaker and B. Li, Nat. Commun., 2020, 11, 4188 CrossRef PubMed.
- R. C. de Guzman, J. Yang, M. Ming-Cheng Cheng, S. O. Salley and K. Y. S. Ng, J. Mater. Chem. A, 2014, 2, 14577–14584 RSC.
- Z. Xiao, C. Lei, C. Yu, X. Chen, Z. Zhu, H. Jiang and F. Wei, Energy Storage Mater., 2020, 24, 565–573 CrossRef.
- S. Mei, S. Guo, B. Xiang, J. Deng, J. Fu, X. Zhang, Y. Zheng, B. Gao, P. K. Chu and K. Huo, J. Energy Chem., 2022, 69, 616–625 CrossRef CAS.
- H. Zheng, H. Xiang, F. Jiang, Y. Liu, Y. Sun, X. Liang, Y. Feng and Y. Yu, Adv. Energy Mater., 2020, 10, 2001440 CrossRef CAS.
- K.-E. Kim, J. Y. Jang, I. Park, M.-H. Woo, M.-H. Jeong, W. C. Shin, M. Ue and N.-S. Choi, Electrochem. Commun., 2015, 61, 121–124 CrossRef CAS.
- X. Zheng, G. Fang, Y. Pan, Q. Li and M. Wu, J. Power Sources, 2019, 439, 227081 CrossRef CAS.
- J. Wang, S. Li, J. Zhang, L. Song, H. Dong, N. Zhang, P. Wang, D. Zhao, L. Zhang and X. Cui, ACS Sustainable Chem. Eng., 2022, 10, 15199–15210 CrossRef CAS.
- Q. Liu, Z. Chen, Y. Liu, Y. Hong, W. Wang, J. Wang, B. Zhao, Y. Xu, J. Wang, X. Fan, L. Li and H. B. Wu, Energy Storage Mater., 2021, 37, 521–529 CrossRef.
- Y. Ozhabes, D. Gunceler and T. A. Arias, Mater. Sci., 2015, 1504, 05799 Search PubMed.
- N. Xu, J. Shi, G. Liu, X. Yang, J. Zheng, Z. Zhang and Y. Yang, J. Power Sources Adv., 2021, 7, 100043 CrossRef CAS.
- J. Zhao, L. Liao, F. Shi, T. Lei, G. Chen, A. Pei, J. Sun, K. Yan, G. Zhou, J. Xie, C. Liu, Y. Li, Z. Liang, Z. Bao and Y. Cui, J. Am. Chem. Soc., 2017, 139, 11550–11558 CrossRef CAS PubMed.
- Y. Okuno, K. Ushirogata, K. Sodeyama and Y. Tateyama, Phys. Chem. Chem. Phys., 2016, 18, 8643–8653 RSC.
- M. Haruta, Y. Kijima, R. Hioki, T. Doi and M. Inaba, Nanoscale, 2018, 10, 17257–17264 RSC.
- R. Nolle, A. J. Achazi, P. Kaghazchi, M. Winter and T. Placke, ACS Appl. Mater. Interfaces, 2018, 10, 28187–28198 CrossRef PubMed.
- C. C. Nguyen and B. L. Lucht, Electrochem. Commun., 2016, 66, 71–74 CrossRef CAS.
- S. Park, S. Y. Jeong, T. K. Lee, M. W. Park, H. Y. Lim, J. Sung, J. Cho, S. K. Kwak, S. Y. Hong and N.-S. Choi, Nat. Commun., 2021, 12, 838 CrossRef CAS PubMed.
- H. Moon, H. Nam, M. P. Kim, S. M. Lee, H. Kim, M. H. Jeon, Y. S. Lee, K. Kim, J. H. Chun, S. K. Kwak, S. Y. Hong and N.-S. Choi, Adv. Funct. Mater., 2023, 33, 2303029 CrossRef CAS.
- F. Aupperle, N. von Aspern, D. Berghus, F. Weber, G. G. Eshetu, M. Winter and E. Figgemeier, ACS Appl. Energy Mater., 2019, 2, 6513–6527 CrossRef CAS.
- Y. Horowitz, H. L. Han, W. T. Ralston, J. R. de Araujo, E. Kreidler, C. Brooks and G. A. Somorjai, Adv. Energy Mater., 2017, 7, 1602060 CrossRef.
- F. Li, J. He, J. Liu, M. Wu, Y. Hou, H. Wang, S. Qi, Q. Liu, J. Hu and J. Ma, Angew Chem. Int. Ed. Engl., 2021, 60, 6600–6608 CrossRef CAS PubMed.
- F. Ding, W. Xu, G. L. Graff, J. Zhang, M. L. Sushko, X. Chen, Y. Shao, M. H. Engelhard, Z. Nie, J. Xiao, X. Liu, P. V. Sushko, J. Liu and J. G. Zhang, J. Am. Chem. Soc., 2013, 135, 4450–4456 CrossRef CAS PubMed.
- Q.-K. Zhang, X.-Q. Zhang, J. Wan, N. Yao, T.-L. Song, J. Xie, L.-P. Hou, M.-Y. Zhou, X. Chen, B.-Q. Li, R. Wen, H.-J. Peng, Q. Zhang and J.-Q. Huang, Nat. Energy, 2023, 8, 725–735 CrossRef CAS.
- Z. Xiao, C. Yu, X. Lin, X. Chen, C. Zhang, H. Jiang and F. Wei, Catal. Today, 2021, 364, 61–66 CrossRef CAS.
- A. Poulon-Quintin, E. Ogden, A. Large, M. Vaudescal, C. Labrugere, M. Bartala and C. Bertrand, Dent. Mater., 2021, 37, 832–839 CrossRef CAS PubMed.
- W. Lu, W. Pu, K. Xie and C. Zheng, ACS Appl. Energy Mater., 2019, 3, 733–742 CrossRef.
- W. Lu, S. Xiong, W. Pu, K. Xie and C. Zheng, ChemElectroChem, 2017, 4, 2012–2018 CrossRef CAS.
- C. Yu, X. Lin, X. Chen, L. Qin, Z. Xiao, C. Zhang, R. Zhang and F. Wei, Nano Lett., 2020, 20, 5176–5184 CrossRef CAS PubMed.
- J. Xie, L. Tong, L. Su, Y. Xu, L. Wang and Y. Wang, J. Power Sources, 2017, 342, 529–536 CrossRef CAS.
- Z. Xiao, C. Yu, X. Lin, X. Chen, C. Zhang, H. Jiang, R. Zhang and F. Wei, Nano Energy, 2020, 77, 105082 CrossRef CAS.
- J. Xie, H. Zhang, J. Chu, W. Shen, R. Chen and J. Yu, J. Alloys Compd., 2018, 769, 1072–1079 CrossRef CAS.
- C. Yu, X. Chen, Z. Xiao, C. Lei, C. Zhang, X. Lin, B. Shen, R. Zhang and F. Wei, Nano Lett., 2019, 19, 5124–5132 CrossRef CAS PubMed.
- Y.-G. Ryu, S. Lee, S. Mah, D. J. Lee, K. Kwon, S. Hwang and S. Doo, J. Electrochem. Soc., 2008, 155, A583 CrossRef CAS.
- G.-B. Han, J.-N. Lee, J. W. Choi and J.-K. Park, Electrochim. Acta, 2011, 56, 8997–9003 CrossRef CAS.
- S. J. Lee, J.-G. Han, Y. Lee, M.-H. Jeong, W. C. Shin, M. Ue and N.-S. Choi, Electrochim. Acta, 2014, 137, 1–8 CrossRef CAS.
- H. Jo, J. Kim, D.-T. Nguyen, K. K. Kang, D.-M. Jeon, A. R. Yang and S.-W. Song, J. Phys. Chem. C, 2016, 120, 22466–22475 CrossRef CAS.
- Z. Hu, L. Zhao, T. Jiang, J. Liu, A. Rashid, P. Sun, G. Wang, C. Yan and L. Zhang, Adv. Funct. Mater., 2019, 29, 1906548 CrossRef CAS.
- C. Li, W. Zhu, B. Lao, X. Huang, H. Yin, Z. Yang, H. Wang, D. Chen and Y. Xu, ChemElectroChem, 2020, 7, 3743–3751 CrossRef CAS.
- G. Liu, T. Jiao, Y. Cheng, K. Zhou, Y. Zou, M. Wang, Y. Yang and J. Zheng, ACS Appl. Energy Mater., 2021, 4, 10323–10332 CrossRef CAS.
- L.-B. Huang, G. Li, Z.-Y. Lu, J.-Y. Li, L. Zhao, Y. Zhang, X.-D. Zhang, K.-C. Jiang, Q. Xu and Y.-G. Guo, ACS Appl. Mater. Interfaces, 2021, 13, 24916–24924 CrossRef CAS PubMed.
- H. Kang, H. Kim, C. Yeom and M. J. Park, ACS Appl. Energy Mater., 2022, 5, 4673–4683 CrossRef CAS.
- R. Li, C. Fu, B. Cui, C. Cui, X. Mu, Y. Gao, G. Yin and P. Zuo, J. Power Sources, 2023, 554, 232337 CrossRef CAS.
- F. Guo, C. Wu, H. Chen, F. Zhong, X. Ai, H. Yang and J. Qian, Energy Storage Mater., 2020, 24, 635–643 CrossRef.
- T. Liu, Q. Hu, X. Li, L. Tan, G. Yan, Z. Wang, H. Guo, Y. Liu, Y. Wu and J. Wang, J. Mater. Chem. A, 2019, 7, 20911–20918 RSC.
- S. Choudhury, Z. Tu, S. Stalin, D. Vu, K. Fawole, D. Gunceler, R. Sundararaman and L. A. Archer, Angew Chem. Int. Ed. Engl., 2017, 56, 13070–13077 CrossRef CAS PubMed.
- A. Hu, W. Chen, X. Du, Y. Hu, T. Lei, H. Wang, L. Xue, Y. Li, H. Sun, Y. Yan, J. Long, C. Shu, J. Zhu, B. Li, X. Wang and J. Xiong, Energy Environ. Sci., 2021, 14, 4115–4124 RSC.
- F. Chu, J. Hu, J. Tian, X. Zhou, Z. Li and C. Li, ACS Appl. Mater. Interfaces, 2018, 10, 12678–12689 CrossRef CAS PubMed.
- X. Shen, H. Ji, J. Liu, J. Zhou, C. Yan and T. Qian, Energy Storage Mater., 2020, 24, 426–431 CrossRef.
- Z. Xie, Z. Wu, X. An, X. Yue, A. Yoshida, X. Du, X. Hao, A. Abudula and G. Guan, Chem. Eng. J., 2020, 393, 124789 CrossRef CAS.
- E. Peled, J. Electrochem. Soc., 1979, 126, 2047 CrossRef CAS.
- E. Peled, D. Golodnitsky and G. Ardel, J. Electrochem. Soc., 1997, 144, L208 CrossRef CAS.
- K. Naoi, M. Mori, Y. Naruoka, W. M. Lamanna and R. Atanasoski, J. Electrochem. Soc., 1999, 146, 462 CrossRef CAS.
- M. Wu, Z. Wen, Y. Liu, X. Wang and L. Huang, J. Power Sources, 2011, 196, 8091–8097 CrossRef CAS.
- Y. Gu, W. W. Wang, Y. J. Li, Q. H. Wu, S. Tang, J. W. Yan, M. S. Zheng, D. Y. Wu, C. H. Fan, W. Q. Hu, Z. B. Chen, Y. Fang, Q. H. Zhang, Q. F. Dong and B. W. Mao, Nat. Commun., 2018, 9, 1339 CrossRef PubMed.
- X. Cao, X. Ren, L. Zou, M. H. Engelhard, W. Huang, H. Wang, B. E. Matthews, H. Lee, C. Niu, B. W. Arey, Y. Cui, C. Wang, J. Xiao, J. Liu, W. Xu and J.-G. Zhang, Nat. Energy, 2019, 4, 796–805 CrossRef CAS.
- W. Xue, Z. Shi, M. Huang, S. Feng, C. Wang, F. Wang, J. Lopez, B. Qiao, G. Xu, W. Zhang, Y. Dong, R. Gao, Y. Shao-Horn, J. A. Johnson and J. Li, Energy Environ. Sci., 2020, 13, 212–220 RSC.
- S. Liu, Q. Zhang, X. Wang, M. Xu, W. Li and B. L. Lucht, ACS Appl. Mater. Interfaces, 2020, 12, 33719–33728 CrossRef CAS PubMed.
- C. Jin, T. Liu, O. Sheng, M. Li, T. Liu, Y. Yuan, J. Nai, Z. Ju, W. Zhang, Y. Liu, Y. Wang, Z. Lin, J. Lu and X. Tao, Nat. Energy, 2021, 6, 378–387 CrossRef CAS.
- S. Kim, T. K. Lee, S. K. Kwak and N.-S. Choi, ACS Energy Lett., 2021, 7, 67–69 CrossRef.
- G. Zhang, J. Chang, L. Wang, J. Li, C. Wang, R. Wang, G. Shi, K. Yu, W. Huang and H. Zheng, Nat. Commun., 2023, 14, 1081 CrossRef CAS PubMed.
- J. Liu, N. Pei, X. Yang, R. Li, H. Hua, P. Zhang and J. Zhao, Energy Mater., 2023, 3, 300024 Search PubMed.
- R. R. Chianelli, J. Cryst. Growth, 1976, 34, 239–244 CrossRef CAS.
- C. Liao, L. Han, X. Mu, Y. Zhu, N. Wu, J. Lu, Y. Zhao, X. Li, Y. Hu, Y. Kan and L. Song, ACS Appl. Mater. Interfaces, 2021, 13, 46783–46793 CrossRef CAS PubMed.
- E. Peled and S. Menkin, J. Electrochem. Soc., 2017, 164, A1703–A1719 CrossRef CAS.
- Y. Li, W. Huang, Y. Li, A. Pei, D. T. Boyle and Y. Cui, Joule, 2018, 2, 2167–2177 CrossRef CAS.
- Y. Qin, D. Wang, M. Liu, C. Shen, Y. Hu, Y. Liu and B. Guo, ACS Appl. Mater. Interfaces, 2021, 13, 49445–49452 CrossRef CAS PubMed.
- J. G. Zhang, W. Xu, J. Xiao, X. Cao and J. Liu, Chem. Rev., 2020, 120, 13312–13348 CrossRef CAS PubMed.
- Y. Zhou, M. Su, X. Yu, Y. Zhang, J. G. Wang, X. Ren, R. Cao, W. Xu, D. R. Baer, Y. Du, O. Borodin, Y. Wang, X. L. Wang, K. Xu, Z. Xu, C. Wang and Z. Zhu, Nat. Nanotechnol., 2020, 15, 224–230 CrossRef CAS PubMed.
- J. Chen, H. Zhang, M. Fang, C. Ke, S. Liu and J. Wang, ACS Energy Lett., 2023, 8, 1723–1734 CrossRef CAS.
- K. Xu, Chem. Rev., 2004, 104, 4303–4418 CrossRef CAS PubMed.
- J. Wang, Y. Yamada, K. Sodeyama, C. H. Chiang, Y. Tateyama and A. Yamada, Nat. Commun., 2016, 7, 12032 CrossRef CAS PubMed.
- J. Alvarado, M. A. Schroeder, T. P. Pollard, X. Wang, J. Z. Lee, M. Zhang, T. Wynn, M. Ding, O. Borodin, Y. S. Meng and K. Xu, Energy Environ. Sci., 2019, 12, 780–794 RSC.
- I. A. Shkrob, T. W. Marin, Y. Zhu and D. P. Abraham, J. Phys. Chem. C, 2014, 118, 19661–19671 CrossRef CAS.
- J. Qian, W. A. Henderson, W. Xu, P. Bhattacharya, M. Engelhard, O. Borodin and J.-G. Zhang, Nat. Commun., 2015, 6, 6362 CrossRef CAS PubMed.
- X. Ren, L. Zou, X. Cao, M. H. Engelhard, W. Liu, S. D. Burton, H. Lee, C. Niu, B. E. Matthews, Z. Zhu, C. Wang, B. W. Arey, J. Xiao, J. Liu, J.-G. Zhang and W. Xu, Joule, 2019, 3, 1662–1676 CrossRef CAS.
- S. Chen, J. Zheng, L. Yu, X. Ren, M. H. Engelhard, C. Niu, H. Lee, W. Xu, J. Xiao and J. Liu, Joule, 2018, 2, 1548–1558 CrossRef CAS.
- Y. X. Yao, X. Chen, C. Yan, X. Q. Zhang, W. L. Cai, J. Q. Huang and Q. Zhang, Angew. Chem., Int. Ed., 2021, 60, 4090–4097 CrossRef CAS PubMed.
- W. Xue, M. Huang, Y. Li, Y. G. Zhu, R. Gao, X. Xiao, W. Zhang, S. Li, G. Xu, Y. Yu, P. Li, J. Lopez, D. Yu, Y. Dong, W. Fan, Z. Shi, R. Xiong, C.-J. Sun, I. Hwang, W.-K. Lee, Y. Shao-Horn, J. A. Johnson and J. Li, Nat. Energy, 2021, 6, 495–505 CrossRef CAS.
- K. S. Jiang, G. M. Hobold, R. Guo, K.-H. Kim, A. M. Melemed, D. Wang, L. Zuin and B. M. Gallant, ACS Energy Lett., 2022, 7, 3378–3385 CrossRef CAS.
-
M. Ue, Y. Sasaki, Y. Tanaka and M. Morita, Electrolytes for Lithium and Lithium-Ion Batteries, Springer, New York, 2014, ch. 2 Search PubMed.
- X. Chen, X. Li, D. Mei, J. Feng, M. Y. Hu, J. Hu, M. Engelhard, J. Zheng, W. Xu, J. Xiao, J. Liu and J. G. Zhang, ChemSusChem, 2014, 7, 549–554 CrossRef CAS PubMed.
- M. Sina, J. Alvarado, H. Shobukawa, C. Alexander, V. Manichev, L. Feldman, T. Gustafsson, K. J. Stevenson and Y. S. Meng, Adv. Mater. Interfaces, 2016, 3, 1600438 CrossRef.
- Y. Chen, W. Zhao, Q. Zhang, G. Yang, J. Zheng, W. Tang, Q. Xu, C. Lai, J. Yang and C. Peng, Adv. Funct. Mater., 2020, 30, 2000396 CrossRef CAS.
- B. Han, Y. Zhang, C. Liao, S. E. Trask, X. Li, R. Uppuluri, J. T. Vaughey, B. Key and F. Dogan, ACS Appl. Mater. Interfaces, 2021, 13, 28017–28026 CrossRef CAS PubMed.
- B. T. Young, D. R. Heskett, C. C. Nguyen, M. Nie, J. C. Woicik and B. L. Lucht, ACS Appl. Mater. Interfaces, 2015, 7, 20004–20011 CrossRef CAS PubMed.
- K. Yao, J. P. Zheng and R. Liang, J. Power Sources, 2018, 381, 164–170 CrossRef CAS.
- Y.-M. Lin, K. C. Klavetter, P. R. Abel, N. C. Davy, J. L. Snider, A. Heller and C. B. Mullins, Chem. Commun., 2012, 48, 7268–7270 RSC.
- V. Etacheri, O. Haik, Y. Goffer, G. A. Roberts, I. C. Stefan, R. Fasching and D. Aurbach, Langmuir, 2012, 28, 965–976 CrossRef CAS PubMed.
- E. Markevich, G. Salitra and D. Aurbach, ACS Energy Lett., 2017, 2, 1337–1345 CrossRef CAS.
- R. Li, P. Zhang, J. Huang, B. Liu, M. Zhou, B. Wen, Y. Luo and S. Okada, RSC Adv., 2021, 11, 7886–7895 RSC.
- L. Wang, S. Liu, K. Zhao, J. Li, Y. Yang and G. Jia, Ionics, 2018, 24, 3337–3346 CrossRef CAS.
- K. Kim, I. Park, S.-Y. Ha, Y. Kim, M.-H. Woo, M.-H. Jeong, W. C. Shin, M. Ue, S. Y. Hong and N.-S. Choi, Electrochim. Acta, 2017, 225, 358–368 CrossRef CAS.
- D.-T. Nguyen, J. Kang, K.-M. Nam, Y. Paik and S.-W. Song, J. Power Sources, 2016, 303, 150–158 CrossRef CAS.
- N. Xu, Y. Sun, J. Shi, J. Chen, G. Liu, K. Zhou, H. He, J. Zhu, Z. Zhang and Y. Yang, J. Power Sources, 2021, 511, 230437 CrossRef CAS.
- J. Kim, S. Kwak, H. Q. Pham, H. Jo, D.-M. Jeon, A. R. Yang and S.-W. Song, J. Electrochem. Sci. Technol., 2022, 13, 269–278 CrossRef CAS.
- H. Jia, L. Zou, P. Gao, X. Cao, W. Zhao, Y. He, M. H. Engelhard, S. D. Burton, H. Wang, X. Ren, Q. Li, R. Yi, X. Zhang, C. Wang, Z. Xu, X. Li, J. G. Zhang and W. Xu, Adv. Energy Mater., 2019, 9, 1900784 CrossRef.
- G. Yang, S. Frisco, R. Tao, N. Philip, T. H. Bennett, C. Stetson, J.-G. Zhang, S.-D. Han, G. Teeter, S. P. Harvey, Y. Zhang, G. M. Veith and J. Nanda, ACS Energy Lett., 2021, 6, 1684–1693 CrossRef CAS.
- S. Chae, W.-J. Kwak, K. S. Han, S. Li, M. H. Engelhard, J. Hu, C. Wang, X. Li and J.-G. Zhang, ACS Energy Lett., 2021, 6, 387–394 CrossRef CAS.
- D. M. Lutz, A. H. McCarthy, S. T. King, G. Singh, C. A. Stackhouse, L. Wang, C. D. Quilty, E. M. Bernardez, K. R. Tallman, X. Tong, J. Bai, H. Zhong, K. J. Takeuchi, E. S. Takeuchi, A. C. Marschilok and D. C. Bock, J. Electrochem. Soc., 2022, 169, 090501 CrossRef CAS.
- N. von Aspern, G. V. Roschenthaler, M. Winter and I. Cekic-Laskovic, Angew Chem. Int. Ed. Engl., 2019, 58, 15978–16000 CrossRef CAS PubMed.
- E. Markevich, G. Salitra, F. Chesneau, M. Schmidt and D. Aurbach, ACS Energy Lett., 2017, 2, 1321–1326 CrossRef CAS.
- Q. Zhao, S. Stalin and L. A. Archer, Joule, 2021, 5, 1119–1142 CrossRef CAS.
- Y. Yuan, F. Wu, Y. Bai, Y. Li, G. Chen, Z. Wang and C. Wu, Energy Storage Mater., 2019, 16, 411–418 CrossRef.
- J. Lang, Y. Long, J. Qu, X. Luo, H. Wei, K. Huang, H. Zhang, L. Qi, Q. Zhang, Z. Li and H. Wu, Energy Storage Mater., 2019, 16, 85–90 CrossRef.
- M. He, R. Guo, G. M. Hobold, H. Gao and B. M. Gallant, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 73–79 CrossRef CAS PubMed.
- D. Lin, Y. Liu, W. Chen, G. Zhou, K. Liu, B. Dunn and Y. Cui, Nano Lett., 2017, 17, 3731–3737 CrossRef CAS PubMed.
- W. Chang, J. H. Park and D. A. Steingart, Nano Lett., 2018, 18, 7066–7074 CrossRef CAS PubMed.
- J. Xie, L. Liao, Y. Gong, Y. Li, F. Shi, A. Pei, J. Sun, R. Zhang, B. Kong, R. Subbaraman, J. Christensen and Y. Cui, Sci. Adv., 2017, 3, eaao3170 CrossRef PubMed.
- L. Chen, K. S. Chen, X. Chen, G. Ramirez, Z. Huang, N. R. Geise, H. G. Steinruck, B. L. Fisher, R. Shahbazian-Yassar, M. F. Toney, M. C. Hersam and J. W. Elam, ACS Appl. Mater. Interfaces, 2018, 10, 26972–26981 CrossRef CAS PubMed.
- L. Fan, H. L. Zhuang, L. Gao, Y. Lu and L. A. Archer, J. Mater. Chem. A, 2017, 5, 3483–3492 RSC.
- X. Fan, L. Chen, X. Ji, T. Deng, S. Hou, J. Chen, J. Zheng, F. Wang, J. Jiang, K. Xu and C. Wang, Chem, 2018, 4, 174–185 CAS.
- L. Suo, W. Xue, M. Gobet, S. G. Greenbaum, C. Wang, Y. Chen, W. Yang, Y. Li and J. Li, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 1156–1161 CrossRef CAS PubMed.
- G. M. Hobold, J. Lopez, R. Guo, N. Minafra, A. Banerjee, Y. Shirley Meng, Y. Shao-Horn and B. M. Gallant, Nat. Energy, 2021, 6, 951–960 CrossRef CAS.
- S. Ko, T. Obukata, T. Shimada, N. Takenaka, M. Nakayama, A. Yamada and Y. Yamada, Nat. Energy, 2022, 7, 1217–1224 CrossRef CAS.
- Y. Lee, T. K. Lee, S. Kim, J. Lee, Y. Ahn, K. Kim, H. Ma, G. Park, S.-M. Lee, S. K. Kwak and N.-S. Choi, Nano Energy, 2020, 67, 104309 CrossRef CAS.
- Y. Zhao, T. Zhou, T. Ashirov, M. E. Kazzi, C. Cancellieri, L. P. H. Jeurgens, J. W. Choi and A. Coskun, Nat. Commun., 2022, 13, 2575 CrossRef CAS PubMed.
- J. Holoubek, M. Yu, S. Yu, M. Li, Z. Wu, D. Xia, P. Bhaladhare, M. S. Gonzalez, T. A. Pascal, P. Liu and Z. Chen, ACS Energy Lett., 2020, 5, 1438–1447 CrossRef CAS.
- X. Li, J. Liu, J. He, H. Wang, S. Qi, D. Wu, J. Huang, F. Li, W. Hu and J. Ma, Adv. Funct. Mater., 2021, 31, 2104395 CrossRef CAS.
- B. Tong, J. Wang, Z. Liu, L. Ma, P. Wang, W. Feng, Z. Peng and Z. Zhou, J. Power Sources, 2018, 400, 225–231 CrossRef CAS.
- S. Yang, M. Hao, Z. Wang, Z. Xie, Z. Cai, M. Hu, B. Chen, L. Wang and K. Zhou, Chem. Eng. J., 2022, 435, 134897 CrossRef CAS.
- Q. Wu, Y. Qian, X. Tang, J. Teng, H. Ding, H. Zhao and J. Li, ACS Appl. Energy Mater., 2022, 5, 5742–5749 CrossRef CAS.
- Y. Zhang, D. Krishnamurthy and V. Viswanathan, J. Electrochem. Soc., 2020, 167, 070554 CrossRef.
- G. Hernández, R. Mogensen, R. Younesi and J. Mindemark, Batteries Supercaps, 2022, 5, e202100373 CrossRef.
- T. Li, X.-Q. Zhang, P. Shi and Q. Zhang, Joule, 2019, 3, 2647–2661 CrossRef CAS.
- T. Satoh, N. Nambu, M. Takehara, M. Ue and Y. Sasaki, ECS Trans., 2013, 50, 127 CrossRef.
- H. Kwon, J. Baek and H.-T. Kim, Energy Storage Mater., 2022, 55, 708–726 CrossRef.
- R. Schmuch, R. Wagner, G. Hörpel, T. Placke and M. Winter, Nat. Energy, 2018, 3, 267–278 CrossRef CAS.
- Y. Yamada, J. Wang, S. Ko, E. Watanabe and A. Yamada, Nat. Energy, 2019, 4, 269–280 CrossRef CAS.
- Z. Jiang, Z. Zeng, X. Liang, L. Yang, W. Hu, C. Zhang, Z. Han, J. Feng and J. Xie, Adv. Funct. Mater., 2021, 31, 2005991 CrossRef CAS.
- J. T. Frith, M. J. Lacey and U. Ulissi, Nat. Commun., 2023, 14, 420 CrossRef CAS PubMed.
- Y. Huang, R. Li, S. Weng, H. Zhang, C. Zhu, D. Lu, C. Sun, X. Huang, T. Deng, L. Fan, L. Chen, X. Wang and X. Fan, Energy Environ. Sci., 2022, 15, 4349–4361 RSC.
- M. Li, C. Wang, K. Davey, J. Li, G. Li, S. Zhang, J. Mao and Z. Guo, SmartMat, 2023, e1185 CrossRef.
- W.-T. Tsai, J. Hazard. Mater., 2005, 119, 69–78 CrossRef CAS PubMed.
- K. Le Bris, J. DeZeeuw, P. J. Godin and K. Strong, J. Mol. Spectrosc., 2020, 367, 111241 CrossRef CAS.
- B. Flamme, G. R. Garcia, M. Weil, M. Haddad, P. Phansavath, V. Ratovelomanana-Vidal and A. Chagnes, Green Chem., 2017, 19, 1828–1849 RSC.
- L.-N. Wu, J. Peng, F.-M. Han, Y.-K. Sun, T. Sheng, Y.-Y. Li, Y. Zhou, L. Huang, J.-T. Li and S.-G. Sun, J. Mater. Chem. A, 2020, 8, 4300–4307 RSC.
- B. Zhong, J. Wu, L. Ren, T. Zhou, Z. Zhang, W. Liu and H. Zhou, Energy Storage Mater., 2022, 50, 792–801 CrossRef.
- S. S. Zhang, ChemElectroChem, 2020, 7, 3569–3577 CrossRef CAS.
- Y. Liu, D. Lin, Y. Li, G. Chen, A. Pei, O. Nix, Y. Li and Y. Cui, Nat. Commun., 2018, 9, 3656 CrossRef PubMed.
- Q. Zhao, Y. Lu, Z. Zhu, Z. Tao and J. Chen, Nano Lett., 2015, 15, 5982–5987 CrossRef CAS PubMed.
- C. B. Jin, X. Q. Zhang, O. W. Sheng, S. Y. Sun, L. P. Hou, P. Shi, B. Q. Li, J. Q. Huang, X. Y. Tao and Q. Zhang, Angew. Chem., 2021, 133, 23172–23177 CrossRef.
- D. Huang, C. Zeng, M. Liu, X. Chen, Y. Li, S. Hu, Q. Pan, F. Zheng, Q. Li and H. Wang, Chem. Eng. J., 2023, 454, 140395 CrossRef CAS.
- Z. Wen, W. Fang, X. Wu, Z. Qin, H. Kang, L. Chen, N. Zhang, X. Liu and G. Chen, Adv. Funct. Mater., 2022, 32, 2204768 CrossRef CAS.
- T. Hou, K. D. Fong, J. Wang and K. A. Persson, Chem. Sci., 2021, 12, 14740–14751 RSC.
- H. Jia, P. Gao, L. Zou, K. S. Han, M. H. Engelhard, Y. He, X. Zhang, W. Zhao, R. Yi and H. Wang, Chem. Mater., 2020, 32, 8956–8964 CrossRef CAS.
- J. Chen, X. Fan, Q. Li, H. Yang, M. R. Khoshi, Y. Xu, S. Hwang, L. Chen, X. Ji, C. Yang, H. He, C. Wang, E. Garfunkel, D. Su, O. Borodin and C. Wang, Nat. Energy, 2020, 5, 386–397 CrossRef CAS.
- X. Ren, S. Chen, H. Lee, D. Mei, M. H. Engelhard, S. D. Burton, W. Zhao, J. Zheng, Q. Li, M. S. Ding, M. Schroeder, J. Alvarado, K. Xu, Y. S. Meng, J. Liu, J.-G. Zhang and W. Xu, Chem, 2018, 4, 1877–1892 CAS.
- J. Holoubek, H. Liu, Z. Wu, Y. Yin, X. Xing, G. Cai, S. Yu, H. Zhou, T. A. Pascal, Z. Chen and P. Liu, Nat. Energy, 2021, 6, 303–313 CrossRef CAS PubMed.
- Z. Li, H. Rao, R. Atwi, B. M. Sivakumar, B. Gwalani, S. Gray, K. S. Han, T. A. Everett, T. A. Ajantiwalay, V. Murugesan, N. N. Rajput and V. G. Pol, Nat. Commun., 2023, 14, 868 CrossRef CAS PubMed.
- K. Ding, C. Xu, Z. Peng, X. Long, J. Shi, Z. Li, Y. Zhang, J. Lai, L. Chen and Y.-P. Cai, ACS Appl. Mater. Interfaces, 2022, 14, 44470–44478 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Tables S1–S3 and ESI references. See DOI: https://doi.org/10.1039/d3sc03514j |
| ‡ These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Saehun
Kim‡
a,
Saehun
Kim‡