
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAccelerating water oxidation – a mixed Co/Fe polyoxometalate with improved turnover characteristics†
Joaquín
Soriano-López‡
 *a,
Friedrich W.
Steuber
a,
Muhamed
Mulahmetović
a,
Maria
Besora
b,
Juan Modesto
Clemente-Juan
c,
Mariah
O'Doherty
a,
Nian-Yong
Zhu
a,
Craig L.
Hill
d,
Eugenio
Coronado
c,
Josep M.
Poblet
b and
Wolfgang
Schmitt
*a
*a,
Friedrich W.
Steuber
a,
Muhamed
Mulahmetović
a,
Maria
Besora
b,
Juan Modesto
Clemente-Juan
c,
Mariah
O'Doherty
a,
Nian-Yong
Zhu
a,
Craig L.
Hill
d,
Eugenio
Coronado
c,
Josep M.
Poblet
b and
Wolfgang
Schmitt
*a
aSchool of Chemistry & SFI AMBER Centre, Trinity College, University of Dublin, Ireland. E-mail: joaquin.soriano@uv.es; schmittw@tcd.ie
bDepartament de Química Física i Inorgànica, Universitat Rovira i Virgili, Marcel·lí Domingo 1, 43007 Tarragona, Spain
cInstitut de Ciència Molecular, Universitat de València, Catedrático José Beltrán 2, 46980 Paterna, Spain
dDepartment of Chemistry, Emory University, Atlanta, Georgia 30322, USA
First published on 31st October 2023
Abstract
Water oxidation is a bottleneck reaction for the establishment of solar-to-fuel energy conversion systems. Earth-abundant metal-based polyoxometalates are promising heterogeneous water oxidation catalysts that can operate in a wide pH range. However, detailed structure–reactivity relationships are not yet comprehensively understood, hampering the design and synthesis of more effective polyoxometalate-based oxidation catalysts. Here we report the synthesis of an ordered, mixed-metal cobalt–iron Weakley archetype [CoII2(H2O)2FeIII2(CoIIW9O34)2]14− (Co2Fe2-WS), which unexpectedly highlights the strong influence of the central, coordinatively saturated metal ions on the catalytic water oxidation characteristics. The resulting species exhibits catalytic turnover frequencies which are up to 4× higher than those of the corresponding archetype tetracobalt-oxo species [CoII2(H2O)2CoII2(PW9O34)2]10− (Co4-WS). It is further striking that the system becomes catalytically inactive when one of the central positions is occupied by a WVI ion as demonstrated by [CoII2(H2O)2CoIIWVI(CoIIW9O34)2]12− (Co3W-WS). Importantly, this study demonstrates that coordinatively saturated metal ions in this central position, which at first glance appear insignificant, do not solely have a structural role but also impart a distinctive structural influence on the reactivity of the polyoxometalate. These results provide unique insights into the structure–reactivity relationships of polyoxometalates with improved catalytic performance characteristics.
Introduction
The production of clean, sustainable and reliable energy to meet the growing global energy demand has arguably become the biggest challenge of the 21st century.1,2 New technologies and energy-storage concepts are required to establish efficient and cost-effective alternatives to fossil fuels.3 An attractive proposition to circumvent anthropogenic CO2 emissions is provided by the hydrogen economy,4 which conceptionally mimics the key attributes of natural photosynthesis and chemically stores solar energy in the form of a two-electron H2 bond.5 In this scheme, water provides a sustainable source of hydrogen, forming a green fuel with the highest gravimetric energy density through the water splitting reaction.6 The resulting O2 and H2 gases can be stored and re-combined to release the stored energy on demand. However, the deployment of this technology is hampered by the lack of a suitable inexpensive, robust, and efficient water oxidation catalysts.7 The water oxidation half-reaction, also classified as the oxygen evolution reaction (OER), is a highly endergonic 4-electron reaction that requires large overpotentials (η) to attain good O2 evolution rates, rendering it as the limiting step for the production of H2.8,9 Therefore, the establishment of stable earth-abundant water oxidation catalysts (WOC) that provide satisfactory oxygen evolution rates at low overpotentials, has become a scientific focus area within the research community.During the last decades, tremendous efforts have been made using homogeneous and heterogeneous Ru- and Ir-based WOCs revealing excellent turnover frequencies at low overpotentials.10,11 However, the high price associated with the rare metal ions and stability issues due to ligand oxidations12,13 prohibit their exploitation in large-scale energy conversion systems. Metal oxides containing abundant elements are promising emerging materials that facilitate high electrocatalytic current densities in alkaline environments.14,15 However among these, only cobalt oxide derived materials sustain satisfactory current densities when the pH value of the reaction media is lowered to neutral conditions.16
During the last decade polyoxometalates (POMs) have attracted significant interest due to their intrinsic redox stability, their synthetic molecular amenability, and processability.17,18 Particularly cobalt-containing POMs (Co-POMs) are nowadays known to represent genuine molecular catalysts19–28 that display high catalytic activity and excellent stability in the solid state as part of modified working electrodes.29–35
Notably, the group of Galán-Mascarós demonstrated that modified carbon paste electrodes containing [Co9(H2O)6(OH)3(HPO4)2(PW9O34)3]16− (Co9) reveal outstanding activity and stability in acidic media exceeding the WOC performance characteristics of IrO2-modified electrodes.36 It is noteworthy that other Co-POMs are active and stable under these acidic conditions.37
In this context, the most studied Co-POM is represented by the Weakley sandwich [Co4(H2O)2(PW9O34)2]10− (Co4-WS), whose OER activity was first reported by Hill and co-workers in 2010.19 Efforts to improve the catalytic properties of Co4-WS and Co9 systems through modification of their components facilitated unique insights into to mechanistic aspect that govern the water oxidation process. For instance, Mo doping at the W addenda atom positions of Co4-WS reduces the onset overpotential by 188 mV.38 Regarding the effect of the heteroatom, the replacement of PV by VV induces a metal-to-metal charge transfer process that decreases the activation energy for the O–O bond formation of the transition state, leading to an overall increase of the oxygen evolution kinetics.39–41 Moreover, increased negative charge densities of the POMs, e.g. achieved through the substitution of PV heteroatoms by GeIV in Co9, lead to energetically higher occupied molecular orbital sets, significantly improving the OER performance.42 Besides, the OER activity of the Weakley sandwich is also strongly influenced by the nature of the four metal ions comprising the central oxo core belt; thus, showing the following trend Ni4 < Mn4 < Fe4 < Co4.43–45 Note that these structures contain PV as heteroatom. These structure–reactivity relationships highlight the impact of molecular modifications, leading to systems with improved performance characteristics.
In spite of these efforts the relations between the POM structure and reactivity are yet to be comprehensively understood. Under this purview, it is somewhat surprising that the role of the at first glance unremarkable inner, coordinatively saturated metal ions that are located in the central oxo-belt of the Weakley sandwich remains unclear and their influence on the catalytic activity is yet to be determined. In this regard, Hill and co-workers reported the structure of a mixed-metal Co2Ni2-WS, analogous to the parent Co4-WS in which the two internal, coordinatively saturated Co ions placed at the central belt have been replaced by two Ni ions.45 Electrochemical measurements, and DFT calculations show a superior OER activity for the mixed-metal Co2Ni2-WS compared to that of Co4-WS (see ESI Section 2† for a more detailed information on the structure–reactivity relationships of Co-based POMs and their analogous structures).
Here we report the synthesis, characterisation, and heterogeneous OER activity of a novel mixed-metal Co–Fe Weakley POM [CoII2(H2O)2FeIII2(B-α-CoIIW9O34)2]14− (Co2Fe2-WS). This novel Co-POM readily assembles and crystalises from its components. Single crystal X-ray diffraction and magnetic measurements are in support of an ordered atom arrangement within the crystal structure. The study of the heterogeneous electrocatalytic OER activity at neutral pH highlights the remarkably improved catalytic performance of this POM that contains central d5 high-spin FeIII ions when compared with that of Co4-WS. Both, a reduction of ca. 80 mV in the overpotential at 1 mA cm−2, and up to 4× faster kinetics demonstrate the success of the alteration of the molecular structure of this POM archetype.
The strong influence of the coordinatively saturated metal ions in central belt on the OER activity is demonstrated. The underlying principle is further exemplified by [CoII2(H2O)2CoIIWVI(CoIIW9O34)2]12− (Co3W-WS) containing a central d0 WVI ion and which drastically decreases the catalytic activity of the POM. The experimental results are supported by DFT calculations, showing that the central, saturated metal ions impart distorted geometries on the external metal centres strongly modulating the OER activity of the POM. These insights can provide the basis for future catalyst design with increased activities.
Results and discussion
Crystal structure of Na[Co2Fe2-WS]
The synthesis of Na[Co2Fe2-WS] was achieved through a condensation reaction involving Co-, Fe- and W- reagents at a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :12.9:9.4 mol ratio at 80 °C in aqueous solution (see ESI Section 4†). The resulting reaction mixture yields olive green crystals. Single-crystal analysis suggests that the crystals are composed of an ordered heterometallic Weakley-type polyoxometalate in which two CoII and two FeIII ions are sandwiched between two B-α-tri-lacunary {CoIIW9} Keggin moieties (Fig. 1 and ESI Section 5†). The latter {CoIIW9} moieties can conceptionally be derived from the classical α-Keggin structure in which one of the four {W3O13} triads which are connected to one another through corner-sharing O-atoms, has been removed to form the tri-lacunary species. The heteroatom positions within the two B-α-tri-lacunary Keggin units are occupied by tetrahedrally coordinated CoII ions. The central rhombic tetranuclear oxo-bridged butterfly moiety is composed of two partially hydrated outer octahedral CoII ions located at the wing-tips, and two inner octahedral, coordinatively saturated FeIII ions. The 14− charge of the polyoxoanion is balanced by Na+ counterions which together with constitutional water molecules surround the cluster entity. The compound can crystallise as rod-shaped (Na[Co2Fe2-WS]_rods) or twinned, plate-shaped crystals (Na[Co2Fe2-WS]_plates). Mixture of both crystals can further be recrystallized (Na[Co2Fe2-WS]_recrystallised) from aqueous solutions. All crystal morphologies were analysed by single-crystal X-ray diffraction (see ESI†).
:12.9:9.4 mol ratio at 80 °C in aqueous solution (see ESI Section 4†). The resulting reaction mixture yields olive green crystals. Single-crystal analysis suggests that the crystals are composed of an ordered heterometallic Weakley-type polyoxometalate in which two CoII and two FeIII ions are sandwiched between two B-α-tri-lacunary {CoIIW9} Keggin moieties (Fig. 1 and ESI Section 5†). The latter {CoIIW9} moieties can conceptionally be derived from the classical α-Keggin structure in which one of the four {W3O13} triads which are connected to one another through corner-sharing O-atoms, has been removed to form the tri-lacunary species. The heteroatom positions within the two B-α-tri-lacunary Keggin units are occupied by tetrahedrally coordinated CoII ions. The central rhombic tetranuclear oxo-bridged butterfly moiety is composed of two partially hydrated outer octahedral CoII ions located at the wing-tips, and two inner octahedral, coordinatively saturated FeIII ions. The 14− charge of the polyoxoanion is balanced by Na+ counterions which together with constitutional water molecules surround the cluster entity. The compound can crystallise as rod-shaped (Na[Co2Fe2-WS]_rods) or twinned, plate-shaped crystals (Na[Co2Fe2-WS]_plates). Mixture of both crystals can further be recrystallized (Na[Co2Fe2-WS]_recrystallised) from aqueous solutions. All crystal morphologies were analysed by single-crystal X-ray diffraction (see ESI†).
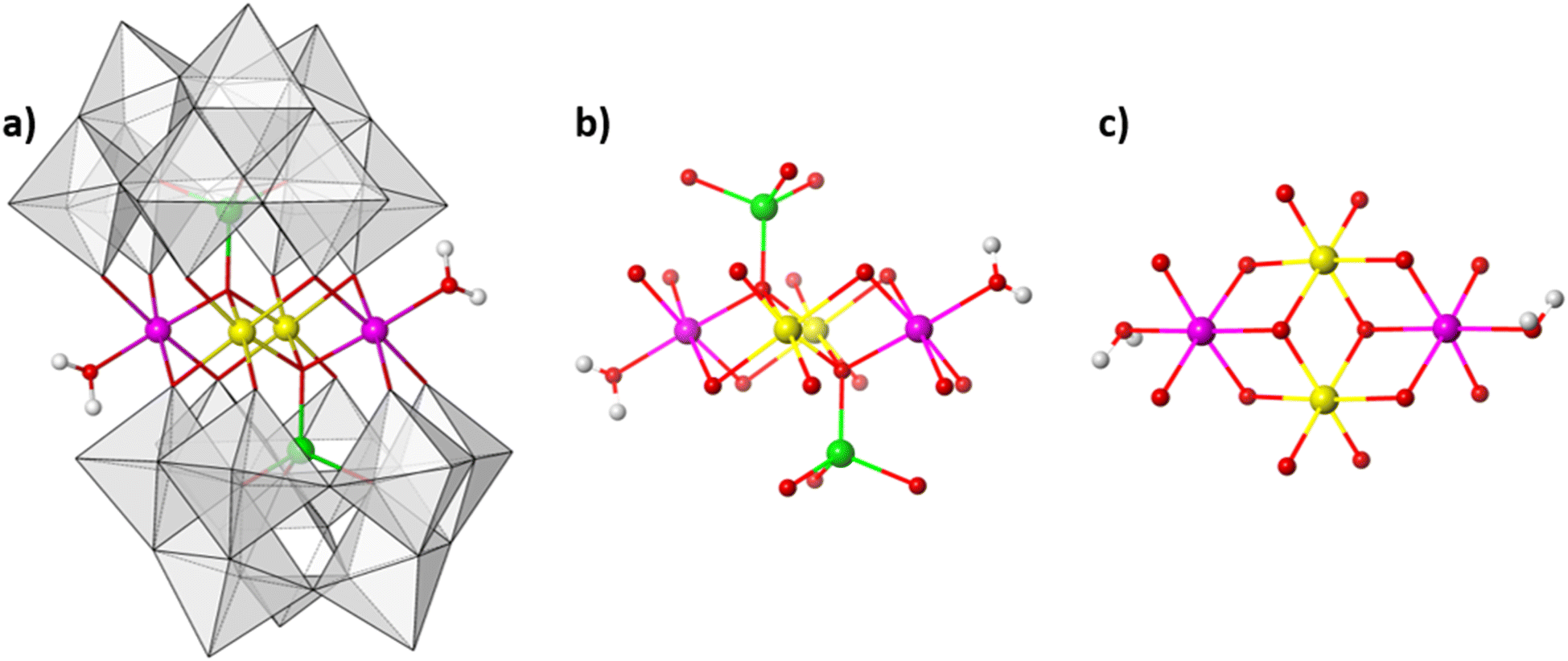 | ||
| Fig. 1 (a) Polyhedral representation of the structure of [Co2Fe2-WS]. (b) Connectivity within the rhombic cobalt–iron-oxo belt and cobalt heteroatoms. (c) Top view of the cobalt–iron-oxo core structure. Colour code: pink: CoII in octahedral geometry; green: CoII in tetrahedral geometry; yellow: FeIII; red: O; white: H; grey polyhedra: {WO6} units. | ||
Bond valence sum analyses and charge balance considerations are indicative of the FeIII and CoII oxidation states. The arrangement within the {(H2O)CoIIFeIII2CoII(H2O)} butterfly moiety is consistent with the observed geometrical parameters whereby CoII–O bond distances range between 2.005(8)–2.160(8) Å and the FeIII–O bond lengths vary between 2.009(8)–2.141(9) Å (for Na[Co2Fe2-WS]_rods). Computationally optimized structures of [Co2Fe2-WS] and the hypothetical structure [Fe2Co2-WS] with the two FeIII ions occupying the external sites in the butterfly arrangement show larger thermodynamic stability of the former (by 10.8 kcal mol−1) as well as better agreement with the crystal structure (RMSD crystal vs. computational is 0.18 for [Co2Fe2-WS] and 0.28 for [Fe2Co2-WS]) further supporting the experimental assignment.
Magnetic properties
The magnetic properties of Na[Co2Fe2-WS] exhibit a continuous decrease of the χT product from room temperature (12.4 emu K mol−1) to a smooth plateau at 22.0 K (see Fig. 2). Below this temperature, a faster decrease is observed until 4.2 emu K mol−1 at 2 K. The decrease in χT can be attributed to a combination of different contributions such as the dominance of antiferromagnetic interactions, the presence of spin frustration, and the first-order spin–orbit coupling of CoII ions in octahedral coordination.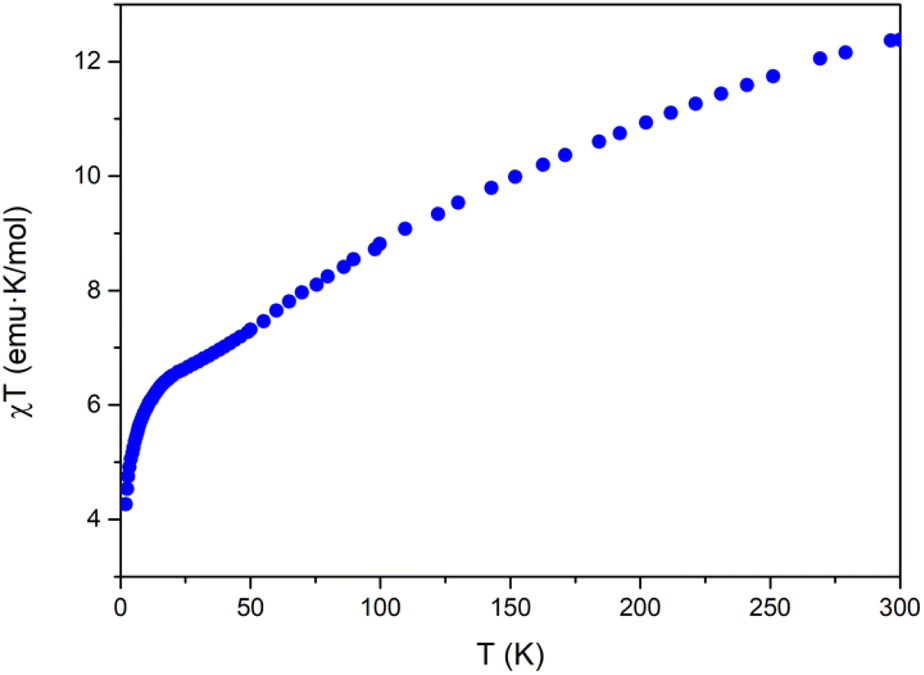 | ||
| Fig. 2 Thermal behavior of χT for Na[Co2Fe2-WS] at 0.1 T field in the range 2–300 K. | ||
A study of these properties has been made considering an effective anisotropic Hamiltonian for both FeIII/CoII distributions in the central rhombic butterfly moiety, i.e. either with the CoII ions occupying the partially hydrated outer octahedral positions and the FeIII ions placed at the two inner octahedral, coordinatively saturated positions, or vice versa, in order to see if the magnetic properties can distinguish between them. Given the large number of independent parameters, some of them have been fixed to well-known values (in sign and magnitude) from previous studies on similar polyanion families. Thus, exchange interactions between two octahedral and between octahedral and tetrahedral CoII ions have been obtained from previous studies extracted from inelastic neutron scattering measurements performed in cobalt based polyoxometalates.46–48 On the other hand, the value for the isotropic interaction between two FeIII ions was obtained from the results reported on a similar polyanion in which cobalt sites were replaced by diamagnetic ions.49 Therefore, only the two exchange parameters between the iron ions and between tetrahedral and octahedral cobalt ions would remain as variables. In addition, all anisotropy axes have been assumed parallel and proportional to the Landé g-factors of both centers (see ESI Section 6†).
A systematic exploration of the parameters and of the anisotropy shows that in the case where the two iron ions are within the short distance of the central rhombus, some solutions compatible with the magnetic behavior are possible. In particular, these situations occur when the two exchange interactions between iron and each type of cobalt are antiferromagnetic and meet the following conditions: (a) the interaction FeIII–CoIIOh is more negative than −10 cm−1, while the FeIII–CoIITd interaction is around −1.2 cm−1, or (b) the interaction FeIII–CoIIOh is around −3.2 cm−1, while the FeIII–CoIITd interaction is more negative than −6 cm−1. Other regions of parameters cannot reproduce the shape of the experimental curve, giving rise to a maximum in the χTvs. T plot, or simply to a continuous drop unable to reproduce the abrupt drop observed below 20 K.
On the other hand, for the other structural isomer with the two cobalt ions placed in the short diagonal of the rhombus, there are no significant regions of parameters that give compatible solutions.
In conclusion, the magnetic properties are more compatible with the presence of the two iron ions placed in the short diagonal of the rhombus.
Heterogeneous OER performance
Solid-state electrochemical studies were performed in a phosphate buffer at pH = 7.2 employing the water-insoluble Ba2+ salt of the POM (Ba[Co2Fe2-WS]), which was obtained through a common metathesis reaction.Initially, the redox behaviour and OER activity was investigated by cyclic voltammetry (CV) and differential pulse voltammetry (DPV) under N2 atmosphere. A Nafion ink containing Ba[Co2Fe2-WS] was deposited on the surface of a glassy carbon working electrode and the setup was completed by a Ag/AgCl reference electrode and a Pt wire counter electrode. All recorded potentials are given versus NHE, unless otherwise stated.
The resulting voltammogram of Ba[Co2Fe2-WS] shows four reversible two-electron redox processes in the −0.80 to +1.20 V potential range (Fig. 3a). In the anodic part, an ill-defined redox pair at E1/2 = +1.010 V can be assigned to the oxidation of the tetrahedral CoII heteroatom to CoIII.50 The reduction of FeIII to FeII can be observed at E1/2 = −0.040 V,51 whereas two different redox pairs associated with the reduction of WVI to WV appear at −0.218 V and −0.584 V.52 The observed FeIII/II and WVI/V redox pairs are fully reversible with peak differences ΔE varying between 13 and 49 mV (Table 1). Interestingly, no O2 to the superoxide O2−˙, reduction peak at ca. −0.33 V (ref. 53) is observed within this potential range. However, when the anodic potential window is increased to +1.60 V, this reduction feature occurs only after the 1st initial cycle when the potential is scanned in the cathodic direction (Fig. 3b).
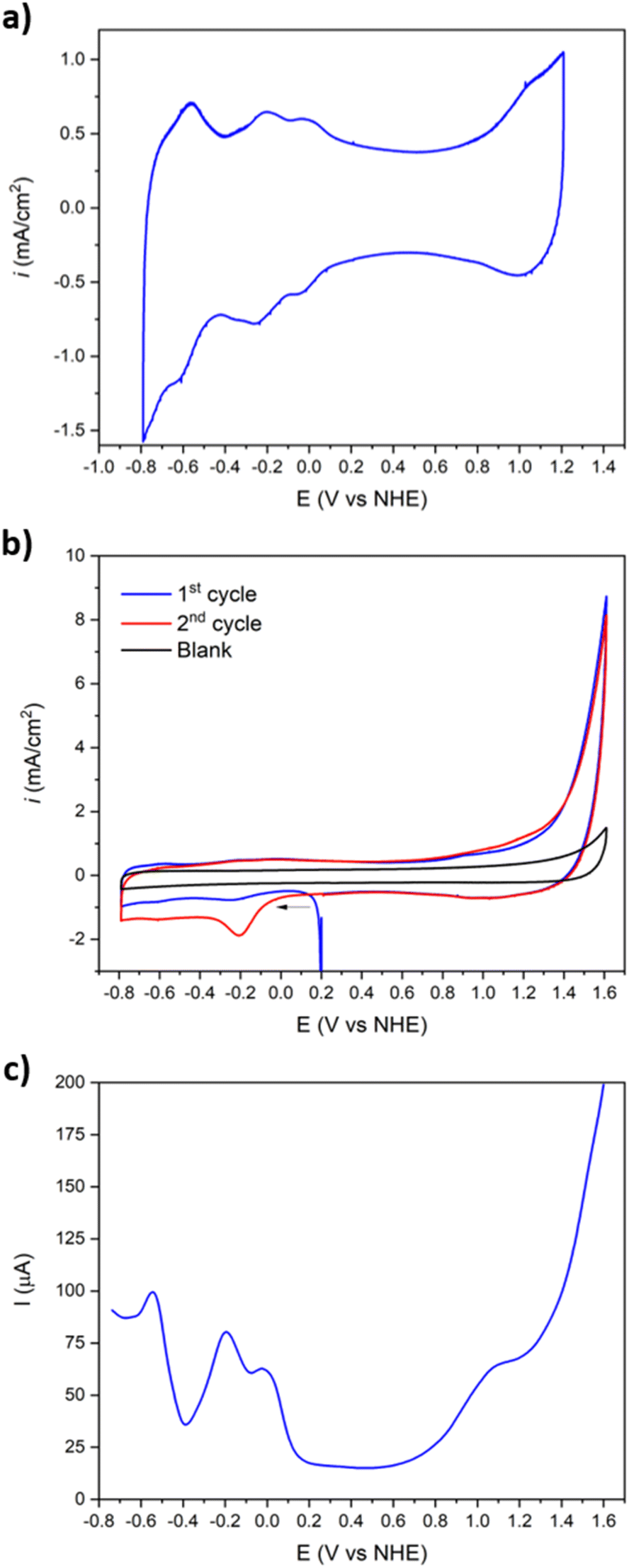 | ||
| Fig. 3 Electrochemical behavior of Ba[Co2Fe2-WS]/Nafion ink (a) CV in the −0.8 to +1.2 V potential range. (b) CV, initial scan direction towards cathodic potentials as indicated by the arrow. The presence of O2 due to OER catalysis is confirmed by the reduction peak appearing at −0.33 V during the second cycle. Note that the blank measurement shown corresponds to the second cycle in which the O2 reduction peak is absent. (c) Differential pulse voltammetry measurement. The experiments were conducted in a single-cell under N2 atmosphere at pH 7.2, 50 mM KPi buffer, using KNO3 (1 M) as electrolyte. The scan rate for the CV was 100 mV s−1. | ||
| Metal centre | CV | DPV | |
|---|---|---|---|
| E 1/2 (V) | ΔE (mV) | E a (V) | |
| a Potentials are given in V vs. NHE. | |||
| CoII/CoIII | +1.010 | — | +1.055 |
| FeII/FeIII | −0.040 | 13 | −0.028 |
| WV/WVI | −0.218 | 35 | −0.196 |
| −0.584 | 49 | −0.544 | |
This behaviour confirms O2 evolution from water oxidation catalysis and is consistent with the observed oxidative waves at positive potentials. That data is consistent with the literature, whereby the oxidation of terminal, water-bonded CoII to CoIII within the central belt of the POM occurs > +1.20 V.40 It is further noteworthy that the O2 reduction wave does not appear in the absence of the Ba[Co2Fe2-WS] POM that acts as catalyst.
Further, the redox behaviour of Ba[Co2Fe2-WS] was investigated by DPV (Fig. 3c), giving rise to peak potentials, Epeak, at +1.055 V for the tetrahedral CoII/CoIII pair, −0.028 V for FeII/FeIII, and −0.196 and −0.544 V for the 1st and 2nd WV/WVI redox pair, respectively. The rapid increase of the current intensity observed after oxidation of the tetrahedral CoII atom is characteristic of a catalytic process.
The electrocatalytic O2 evolution was also confirmed using a rotating ring disk electrode (RRDE), which is composed of a glassy carbon disk and a Pt ring facilitating O2 collection. For this, the Ba[Co2Fe2-WS] Nafion ink composite was drop casted on the glassy carbon disk and the potential was scanned from +0.50 to +1.80 V at a scan rate of 1 mV s−1 at a rotation speed of 700 rpm. A constant potential of −0.33 V was applied at the Pt ring which is sufficient to reduce the O2 that results from the H2O oxidation process to the superoxide, as described by the following redox equations:
| Disk: 2H2O → O2 + 4H+ + 4e− | (1) |
| Ring: O2 + e− → O2−˙ | (2) |
As shown in Fig. 4, O2 evolution due to catalytic water oxidation is demonstrated by an increasing ring current that occurs as soon as the current at the modified glassy carbon electrode starts to deviate from linearity. This behaviour is indeed in sharp contrast to the control conditions employing bare Nafion ink without catalyst and under which no evidence of O2 evolution is observed. Under these conditions, the OER process using Ba[Co2Fe2-WS] as a catalyst commences at an onset overpotential (ηonset) of 580 mV.
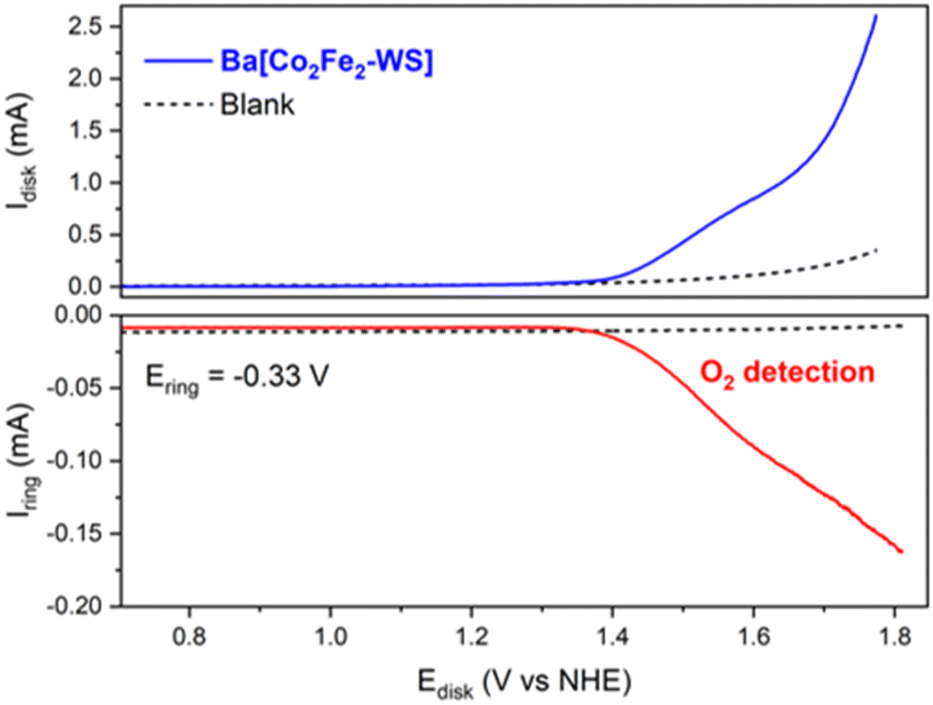 | ||
| Fig. 4 Rotating ring disk electrode (RRDE) experiment allowing the detection of electrocatalytic O2; blue line: Ba[Co2Fe2-WS]/Nafion ink; intersected black line: Nafion ink in the absence of the catalyst. The applied potential at the glassy carbon disk is scanned to positive values. The applied potential at the Pt ring is held constant at −0.33 V to detect the O2 evolved (red line). No O2 could be detected in the absence of catalyst under these conditions. The experiments were conducted in a single-cell under N2 atmosphere using a pH 7.2, 50 mM KPi buffer and KNO3 (1 M) as electrolyte. The scan rate was 1 mV s−1 and the rotation speed was 700 rpm. | ||
The kinetics of the electrocatalytic reaction and stability of Ba[Co2Fe2-WS] under turnover conditions were studied employing modified-carbon paste (CP) electrodes in LSV experiments. The Ba[Co2Fe2-WS]/CP electrodes were used in order to attain higher current densities owing to the increased electric conductivity of CP over Nafion inks. As can be seen in Fig. 5a, the electrodes show best performances at 30–40 wt% catalyst loadings, giving rise to ηonset = 502 mV and an overpotential required to reach 1 mA cm−2 of 625 mV (see Fig. S12†). Higher catalyst content resulted in brittle Ba[Co2Fe2-WS]/CP blends that were difficult to handle. Hence, electrodes with catalyst loadings >40 wt% were not used. LSV data was analysed to deduce the Tafel plots of the catalyst (Fig. 5b). The Tafel slopes for all examined blend compositions are consistent varying closely between 94 mV dec−1 and 99 mV dec−1. It is well known that the Tafel slopes are exclusively dependent on the rate-determining step (RDS) of an electrochemical process. The obtained Tafel values are in-line with other Co-based POMs and are indicative of competing rate-determining chemical and electron-transfer processes.35,36,42
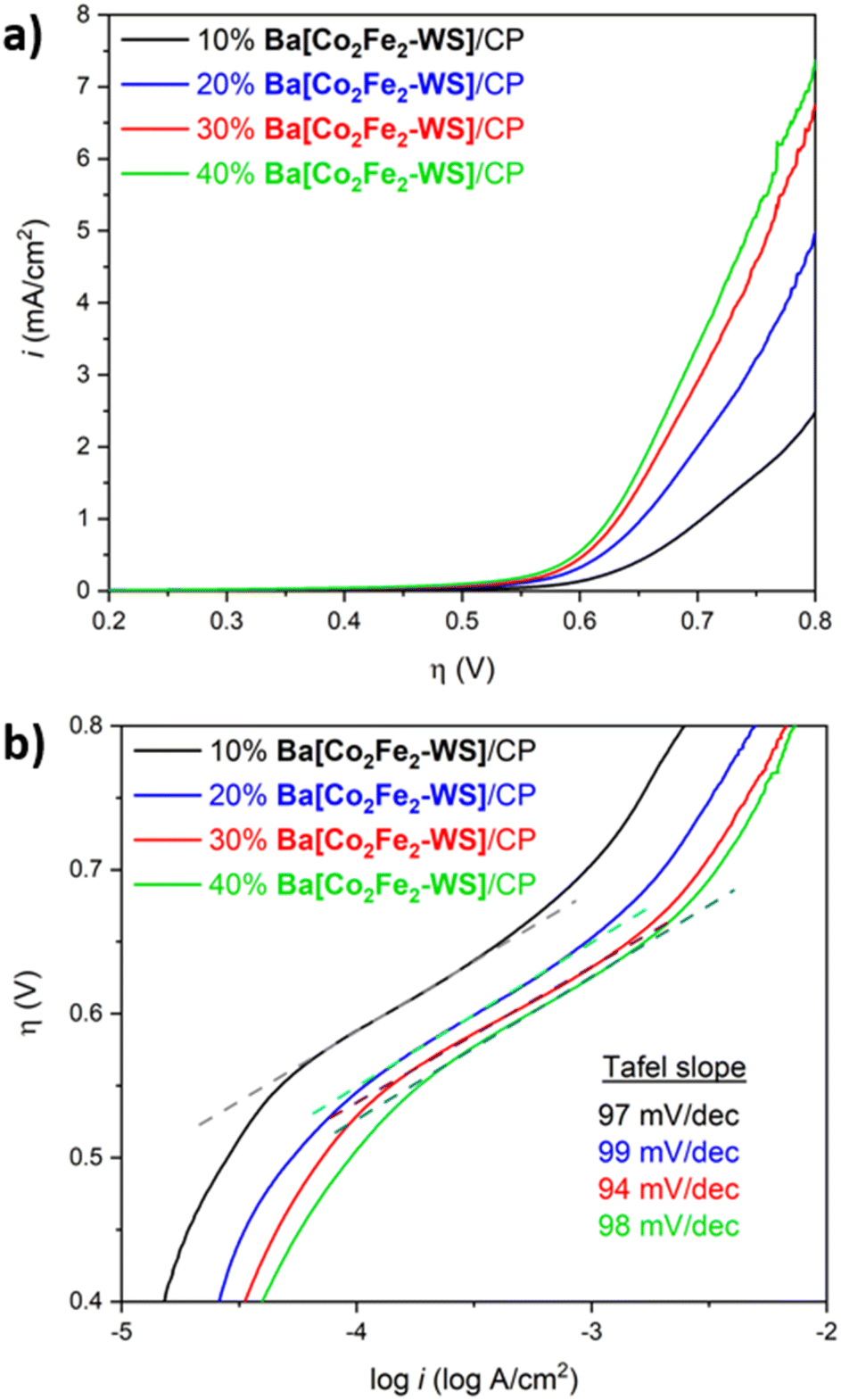 | ||
| Fig. 5 (a) Rotating disk electrode linear sweep voltammetry (RDE-LSV) of Ba[Co2Fe2-WS]/CP electrodes at increasing catalyst loading in the CP blend. (b) Tafel plots obtained from the RDE-LSV data. The dashed lines indicate the Tafel regions. The experiments were conducted in a single-cell using a pH 7.2, 50 mM KPi buffer and KNO3 (1 M) as electrolyte. The scan rate was 1 mV s−1 at a rotation speed of 1600 rpm. | ||
Bulk water electrolysis was performed to test the hydrolytic and oxidative stability under electrocatalytic working conditions (Fig. 6). Chronopotentiometry experiments were conducted at a constant current density of 1 mA cm−2 employing an H-tube with cathode and anode compartments separated by a glass frit. During these experiments the measured overpotential remains stable for >18 hours. A minor increase of the overpotential over time is caused by the accumulation of gas bubbles on the electrode surface, thus decreasing the number of actives sites at the electrode (Fig. S13†).42 Indeed, the removal of the O2 bubbles recovers the initial activity of Ba[Co2Fe2-WS] underlining the excellent long-term stability of the catalyst under heterogeneous conditions. In addition, we quantified the amount of O2 which evolved during chronopotentiometric experiments at a current density of 1 mA cm−2 for 4 hours using a fluorescence probe. We observe a faradaic efficiency of >95% compared with the expected theoretical O2 amount considering a stoichiometric 4e− reaction (see Fig. S14†).
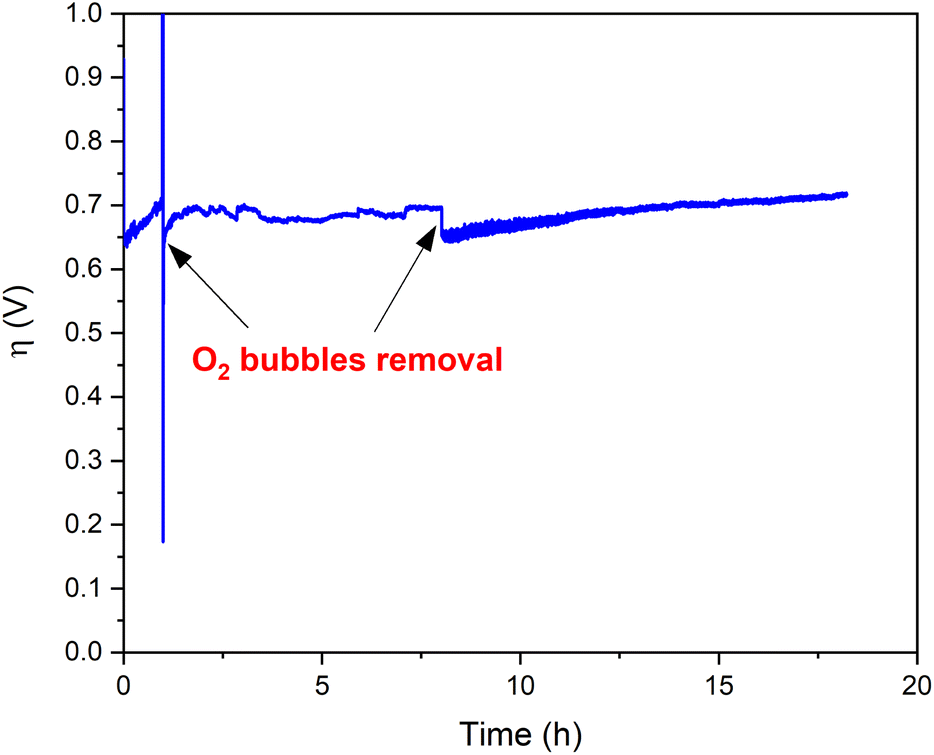 | ||
| Fig. 6 Long-term stability of Ba[Co2Fe2-WS]/CP electrodes. Chronopotentiometry at 1 mA cm−2 employing a 30 wt% Ba[Co2Fe2-WS]/CP electrode. The arrows indicate the removal of the O2 gas bubbles trapped on the electrode surface. The experiments were conducted in an H-cell using a pH 7.2, 50 mM KPi buffer and KNO3 (1 M) as electrolyte. | ||
Post-catalytic characterisation
After the bulk water electrolysis experiments, Ba[Co2Fe2-WS] was recovered from the carbon blend and subjected to a detailed characterisation protocol to confirm the structural stability and to identify of the true catalytic species. Depending on the applied conditions, dissolved Co-containing POMs can undergo hydrolytic decomposition associated with [Co(aq.)]2+ leaching to form electrode-bound CoOx. The latter can act as a competing OER catalyst, thus hampering a detailed characterisation of the Co-POM as a true catalytic species (vide supra). In heterogeneous, solid-state phases, POM-based catalysts have shown high hydrolytic stability, from which several Co-POMs, including Co4-WS, have been undoubtedly identified as the true WOC active species.21–25FT-IR spectra of recovered post-catalytic Ba[Co2Fe2-WS] samples show identical features to those of the freshly prepared samples (Fig. S15†). Raman spectroscopy and X-ray photoelectron spectroscopy (XPS) are excellent surface characterisation techniques that permit the identification of possible additional species formed during electrocatalytic experiments.54 Using these spectroscopic techniques, we could not identify any trace quantities of CoOx in any of the examined samples. The pre- and post-operando Raman spectra are indeed indistinguishable (Fig. S16†). XPS spectra of post-catalytic Ba[Co2Fe2-WS] samples reveal slightly increased binding energies for all examined elements (Fig. S17†) which is characteristic for the oxidation of the catalyst during O2 evolution.37 The binding energy increase mainly stems from the CoII to CoIII oxidation at the tetrahedral sites of the B-α-trilacunary {CoW9} Keggin moieties prior to catalytic O2 evolution. This oxidative process is consistent with the electrochemical analysis. Due to the significant overlap of the Ba 3d and Co 2p XPS absorption bands, Co 2s peaks were analysed in detail (Fig. S17†). The latter experiences an overall binding energy increase of ∼3 eV, whereas the binding energies of the bands of the remaining elements appear increased by ∼1 eV in post-catalytic samples. Further, the edge signals and the number of bands that derive from the Co, Fe and W atoms are identical to those observed in freshly prepared Ba[Co2Fe2-WS] samples. Importantly, all the O1s derived bands appear >530 eV, prohibiting the presence of lattice oxygen atoms that stem form cobalt oxide species.36
These spectroscopic fingerprints are consistent with the reported analyses for Co4-WS,37 confirming that the activity arises from Ba[Co2Fe2-WS] or a closely related molecular structure. This conclusion was further substantiated by elemental analyses which were performed by XPS and energy-dispersive X-ray (EDX) spectroscopy techniques, confirming the expected elemental ratios and stability of Ba[Co2Fe2-WS] after the O2 evolution experiments.
It is noteworthy that the recovered solid samples did not contain detectable residual quantities of Na+ countercations. This is expected, as the oxidation of the tetrahedral CoII heteroatoms to CoIII prior to OER catalysis decreases the overall negative charge of the cluster entity from 14– to 12– (see Fig. 3), whereby trace quantities of the associated smaller Na+ ions are expected to be expelled from the structure. Additionally, elemental analyses (ICP-MS) of the buffer employed during the long-term chronopotentiometry experiment, do no detect Co or Fe species (<1 ppm) that could be a source of competing OER catalysts.
Comparison of the OER activity of Ba[Co2Fe2-WS] with the benchmark Ba[Co4-WS] and Ba[Co3W-WS] systems
In order to evaluate the role of the inner coordinatively, saturated metal ions of the central core of the POM, we compared the activity of Ba[Co2Fe2-WS] with that of the isostructural polyanion [CoII2(H2O)2CoIIWVI(B-α-CoIIW9O34)2]12− (Co3W-WS) (Fig. 7a), in which CoII and WVI ions equally occupy the two central positions of the rhombic, butterfly moiety of the weakley-type POM. Importantly, the results were also compared with the catalytic performance of the parent benchmark system [CoII2(H2O)2CoII2(B-α-PW9O34)2]10− (Co4-WS) (Fig. 7b). These experiments were carried out employing modified carbon paste electrodes containing 30 wt% of the barium salts of the POMs.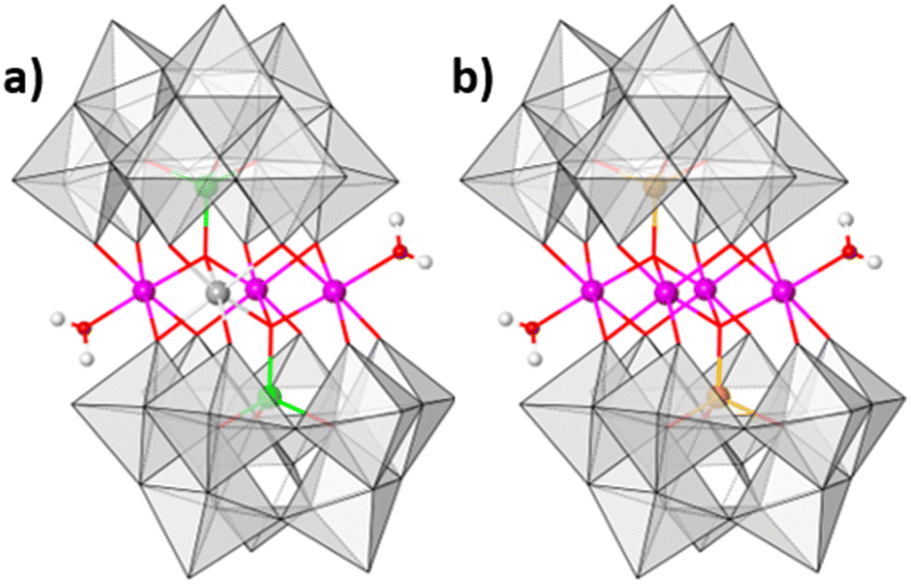 | ||
| Fig. 7 Polyhedral representation of (a) [CoII2(H2O)2CoIIWVI(B-α-CoIIW9O34)2]12− (Co3W-WS) and (b) [CoII2(H2O)2CoII2(B-α-PW9O34)2]10− (Co4-WS). Colour code: pink: CoII in octahedral geometry; green: CoII in tetrahedral geometry; grey: W; orange: P; red: O; white: H; grey polyhedra: {WO6} units. Note that the internal belt positions of Co3W-WS have a 50:50 Co:W equal occupancy. | ||
LSV measurements reveal remarkable differences of the electrocatalytic OER activities despite the similarities of the structures (Fig. 8a). It is striking that the title compound Ba[Co2Fe2-WS] shows a significantly higher catalytic activity than the other two POMs. Ba[Co3W-WS] displays very poor or almost neglectable catalytic activity, whereas Ba[Co4-WS] only surpasses the activity of Ba[Co2Fe2-WS] at very high overpotentials (η > 770 mV).
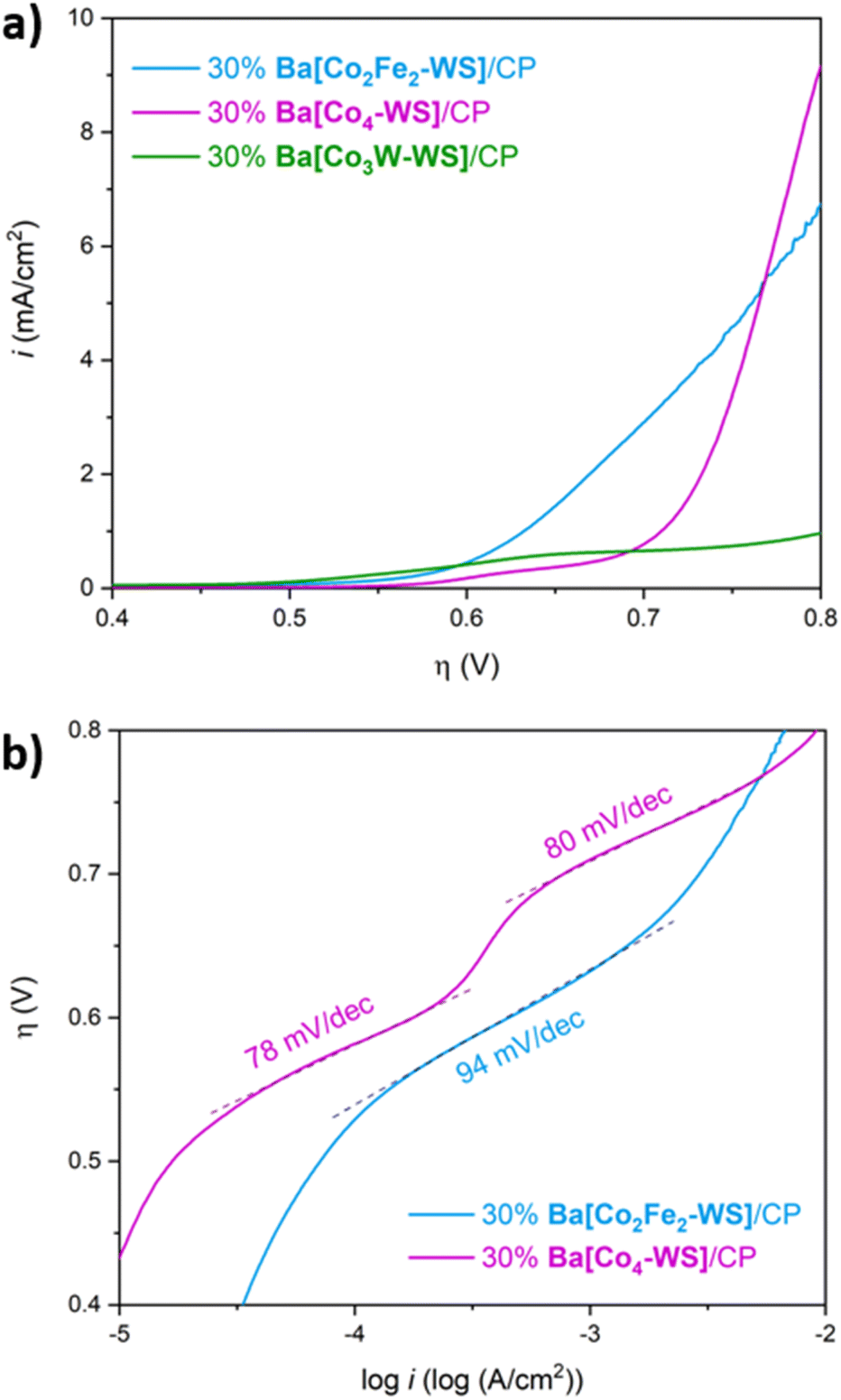 | ||
| Fig. 8 Comparative OER electrocatalytic activity of 30 wt% POM/CP electrodes. Ba[Co2Fe2-WS] (blue), Ba[Co3W-WS] (green), Ba[Co4-WS] (purple). (a) Rotating disk electrode linear sweep voltammetry (RDE-LSV). (b) Tafel plots, as derived from the RDE-LSV data. The experiments were conducted in a single-cell using a pH = 7.2, 50 mM KPi buffer and KNO3 (1 M) as aqueous electrolyte. The scan rate was 1 mV s−1 at a rotation speed of 1600 rpm. | ||
The observed behaviour is maintained when the current densities are normalised by the mole quantities of the POMs (Fig. S18†), which is expected considering the similar molecular weights of the three POMs. Additionally, Ba[Co2Fe2-WS] displays an OER onset overpotential of 528 mV, which is 34 mV lower than that of Ba[Co4-WS] (Fig. S19†). This difference is even more pronounced at a current density of 1 mA cm−2, for which overpotentials of 632 mV and 710 mV are required for Ba[Co2Fe2-WS] and Ba[Co4-WS], respectively.
The active surface areas of the electrodes were considered in order to potentially rationalise the observed activity differences. Although the respective electrocatalytic surface areas (ECSA) cannot directly be assessed due to the unknown parameters of the composite electrodes, the double-layer capacitance (Cdl), which is proportional to the ECSA, can be compared (Fig. S20†). The data clearly demonstrates that Ba[Co2Fe2-WS]/CP possesses the lowest Cdl when compared to Ba[Co4-WS]/CP and Ba[Co3W-WS]/CP. This highlights that the activity differences do not arise form variable electrocatalytic areas but moreover stem from the kinetic profiles that are intrinsic to the individual POMs under OER conditions. Particularly the diminished catalytic activity of Ba[Co3W-WS] directly highlights the influence of the central metal ions on the kinetics of the catalytic OER reaction.
The Tafel plot of Ba[Co2Fe2-WS] further highlights its superior catalytic performance when compared to Ba[Co4-WS] (Fig. 8b). Although both POMs share the same active {CoII–OH2} sites, the different Tafel slopes suggest that their rate-determining step may be governed by their different electronic structures. In the case of Ba[Co4-WS], two well defined Tafel slope regions with 78 mV dec−1 and 80 mV dec−1, are apparent. Differently, Ba[Co2Fe2-WS] exhibits only one single Tafel region with a slope of 94 mV dec−1. Moreover, Ba[Co2Fe2-WS] has an exchange current density (i0) of 1.01 × 10−10 mA cm−2, which is two orders of magnitude higher than i0 for Ba[Co4-WS] (3.64 × 10−12 mA cm−2). This difference can be attributed to a higher electrical conductivity of Ba[Co2Fe2-WS] due to its larger negative charge (14− vs. 10−). The electron charge density has a direct influence on the redox properties of the POMs. Indeed, the q/m ratio, also known as the anion-charge effect (where q is the overall negative charge of the POM and m is the total number of metal centres),55,56 has been used as a chemical descriptor to predict the reactivity of POMs.37,42,57 Due to its higher negative charge, Ba[Co2Fe2-WS] has a q/m ratio of 0.58, whereas Ba[Co4-WS] has a q/m value of 0.45. Hence, the increased anion-charge effect renders a more facile electron transfer within the POM.
The current at the working electrode during RDE-LSV experiments can be described according to the Koutecký–Levich equation,58 from which the kinetics of the catalytic process can be analysed. As water oxidation reactions are known to exhibit sluggish electron transfer processes and as the limiting current is independent of rotational speed of the catalyst-supported electrode within the aqueous system, the current at the electrode can be defined as:
| i = nFAΓkcat(η) | (3) |
Fig. 9 shows the determined turnover frequencies as a function of the applied overpotential for Ba[Co2Fe2-WS] and Ba[Co4-WS] (see ESI Section 3†). These results quantify the improved kinetics of the catalytic OER for Ba[Co2Fe2-WS] compared to Ba[Co4-WS]. At an applied overpotential of 600 mV, Ba[Co2Fe2-WS] displays a TOF of 3.8 × 10−3 s−1, whereas Ba[Co4-WS] shows a TOF of 1.3 × 10−3 s−1. This difference is even more pronounced at an overpotential of 700 mV, giving a TOF = 2.4 × 10−2 s−1 for Ba[Co2Fe2-WS], which is 4× higher than that of Ba[Co4-WS] (TOF = 5.9 × 10−3 s−1).
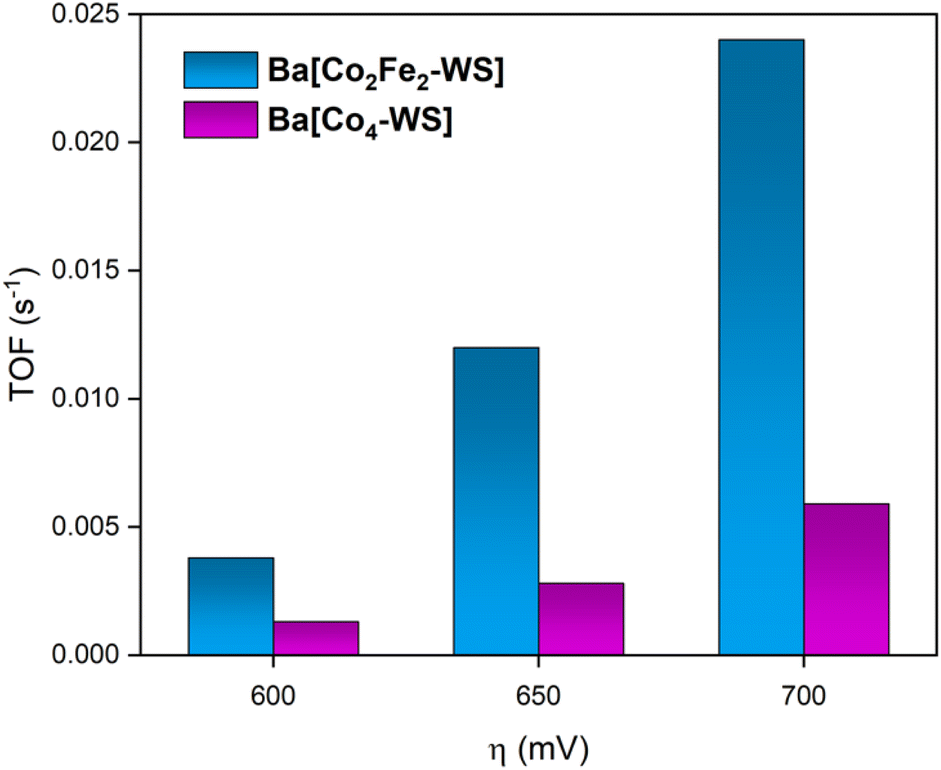 | ||
| Fig. 9 Comparison of the calculated TOFs of Ba[Co2Fe2-WS] and Ba[Co4-WS] at different overpotentials during RDE-LSV measurements. The experiments were conducted using 30 wt% Ba[POM]/CP electrodes at pH = 7.2 using 50 mM KPi buffer and KNO3 (1 M) as aqueous electrolyte. The scan rate was 1 mV s−1 and the rotational speed was 1600 rpm. | ||
Computational study of the Co2Fe2-WS and Co3W-WS systems
We have performed a computational study (B3LYP/6–31g(d,p), LANL2DZ PCM; see ESI Section 8†) to analyse the structures and electrochemical properties of the polyanions [CoII2(H2O)2CoIIWVI(CoIIW9O34)2]12− (A, Co3W-WS) and [CoII2(H2O)2FeIII2(CoIIW9O34)2]14− (Co2Fe2-WS) with the aim of understanding the sharp differences observed on their OER activities. Additionally, these results are compared with our previously reported computational analysis on the OER properties of [CoII4(H2O)2(PW9O34)2]10− (Co4-WS).40We firstly focused on the Co3W-WS species to unravel the origin of the absence of OER activity observed experimentally. Thus, calculations on the Co3W-WS system support that the aqua complex A would rapidly be protonated at two oxygen sites of the POM forming [CoII2(H2O)2CoIIWVI(CoIIW9O34H)2]10− (B) in a water solution at neutral pH. The protonation sites in the POM can be diverse and even though we have not performed an exhaustive analysis of the protonation sites, it was found that the double protonation is computationally spontaneous by more than −0.8 eV (see Fig. 10 and S21†). Afterwards, the two internal tetrahedral CoII heteroatoms in B can be electrochemically oxidized to CoIII at low potentials (≤1 V) via two proton coupled electron transfer (PCET) events, leading to [CoII2(H2O)2CoIIWVI(CoIIIW9O34)2]10−, D. As shown in Fig. 10, the same product D could also be achieved from A species via two electron transfer (ET) processes with even lower potentials (≤0.71 V). However, this seems unlikely to occur in solution due to the high pKa of the POM.
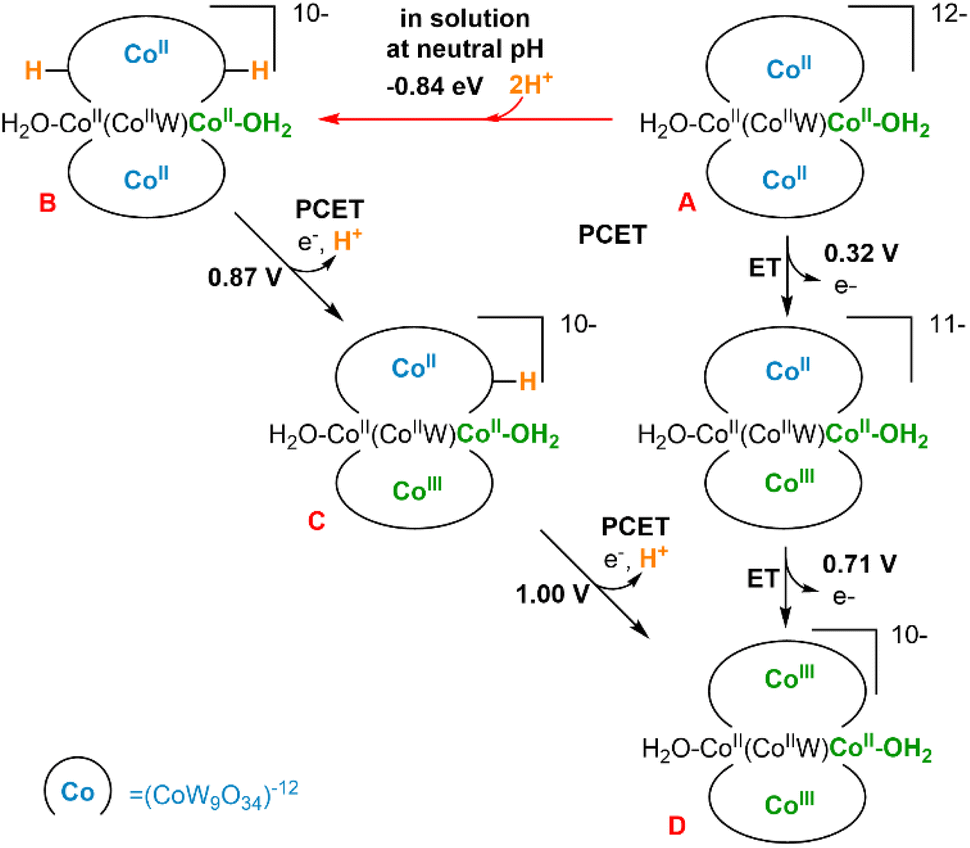 | ||
| Fig. 10 Schematic representation of the double protonation of [CoII2(H2O)2CoIIWVI(CoIIW9O34)2]12− (A, Co3W-WS) and following PCET events, as well as direct ET events. Protonation energies in eV, computed potentials for PCET and ET in V vs. NHE at pH = 7. | ||
Next, we studied the initial steps of the water oxidation reaction mechanism in which, starting from species D (CoII–OH2), two PCET events yield the oxo radical active species F (CoIII–O˙), similar to that observed in the isostructural Co4-WS.40 We have previously demonstrated that one of these two PCET events are the potential-limiting steps of the reaction. Therefore, we can use the computed energy of these two PCET events as a chemical descriptor to obtain a reasonably accurate prediction of the OER activity of Co-POMs. Our calculations show that the first PCET from D (CoII–OH2) takes the electron from the most reactive external cobalt atom leading to species E (CoIII–OH). This step requires an applied potential of 1.82 V (Fig. 11, top). Note that E is computationally found to have an overall multiplicity of 19, with the reactive CoIII atom being a quintet in the ground state. The computed structures with a lower multiplicity are less stable. The second PCET event from E (CoIII–OH) to reach the oxo radical species F (CoIII–O˙) would need an applied potential of 1.37 V. In this case, the ground state of the oxidised external CoIII atom is a triplet and has a marked radical character on the oxygen (1.023e− Mulliken spin density). These results are in good agreement with our experimental results and indicate that the Co3W-WS system would need an overpotential higher than 1 V to catalytically oxidise water.
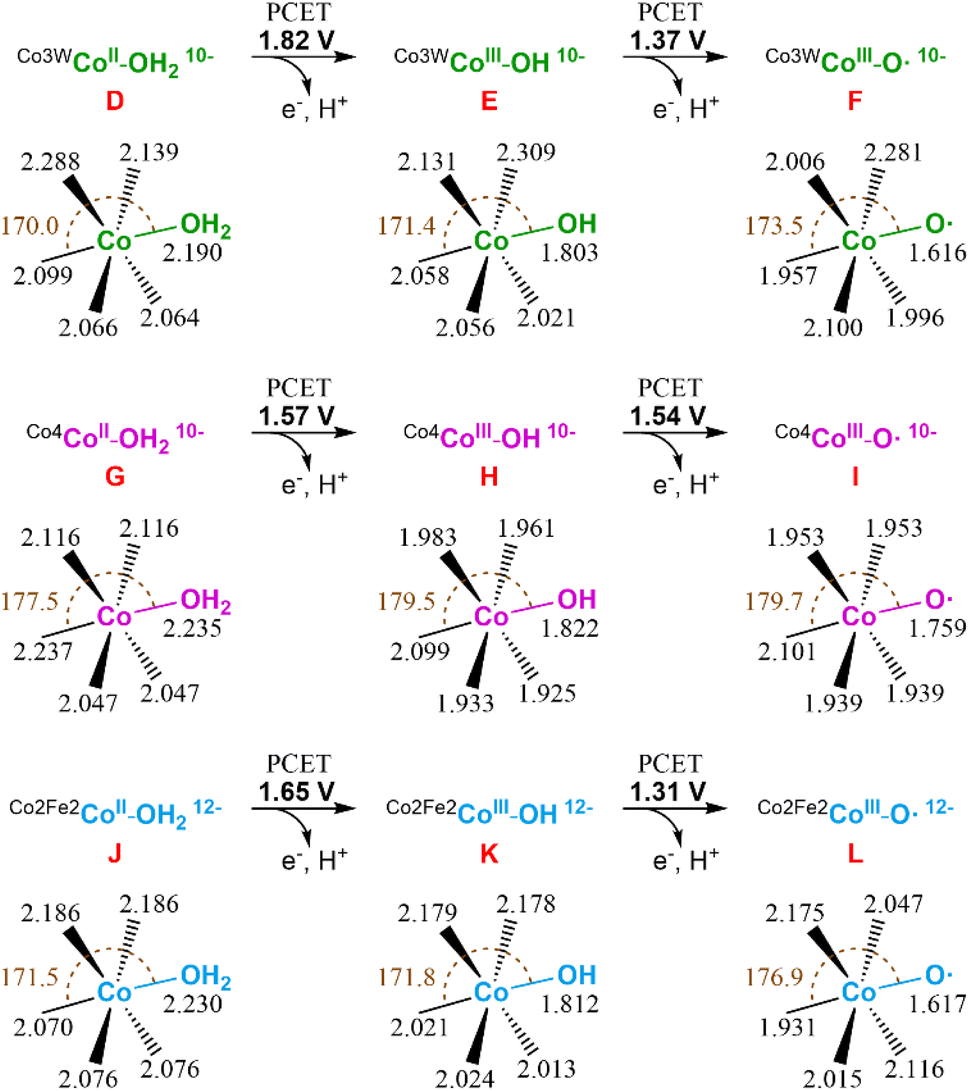 | ||
| Fig. 11 Schematic representation of the first two PCET events of the water oxidation reaction for the Co3W-WS (top), Co4-WS (middle), and Co2Fe2-WS (bottom) systems. Potential energies are given in V vs. NHE at pH = 7. Values for the Co4-WS system are extracted from ref. 44. | ||
It is interesting to see that despite the structural similarity between the Co3W-WS system and the previously studied Co4-WS,40 they display remarkably different reactivity. The structural differences stem from: the replacement of one internal, coordinatively saturated Co atom in the central belt of Co4-WS by a W atom, and two tetrahedral Co heteroatoms instead of the two P heteroatoms. Additionally, the overall charge of the system is the same (10−) once the internal CoII heteroatoms are oxidised to CoIII in Co3W-WS. Whilst the calculations show that the structures of D and G are overall similar, they exhibit some distinct geometrical differences (see Fig. 11). The distances between both externally-located Co atoms located at the wing-tips is 5.587 Å in D and 5.610 Å in G, whereas the distance between the two inner octahedral, coordinatively saturated atoms of the belt is 3.297 Å in D (dW–Co) and 3.347 Å in G (dCo–Co). These values indicate that the replacement of the internal Co atom by a W atom induces a change in the geometry of the central rhombic tetranuclear oxo-bridged butterfly moiety of the POM, thus breaking the symmetry of the system. Taking a closer look at the bond distances of the two water-bounded, external octahedral Co atoms in the Co3W-WS system, we can see that the W atom creates a compressive axial strain that distorts the octahedral symmetry of the external Co atoms (see Fig. S22†). This strain effect strengthens the Co–OH2 bond leading to an increase in the oxidation potential of the metal centre, i.e. the strain effect reduces the OER catalytic activity of the Co3W-WS system.
Following our study, we also computationally explored the initial electrochemical steps of the water oxidation reaction catalysed by the Co2Fe2-WS system. We optimised the structure of the aqua complex [CoII2(H2O)2FeIII2(CoIIW9O34)2]14− (Co2Fe2-WS) as well as the corresponding species with the two CoII heteroatoms oxidised to CoIII, [CoII2(H2O)2FeIII2(CoIIIW9O34)2]12− species J. The difference in energy between both species is just 0.19 V, highlighting an increased ability of Co2Fe2-WS to be oxidised in comparison to Co3W-WS. This process very likely occurs upon protonation of the POM followed by two PCET events, as detailed above for the Co3W-WS system (see Fig. 10). Hence, we consider J as the resting state species in the reaction mechanism, from which the hydroxyl species K (CoIII–OH) and the oxo radical species L (CoIII– O˙) are obtained through the first and second PCET events with associated potentials of 1.65 V and 1.31 V, respectively. As observed for the Co3W-WS system, the CoIII atom in the hydroxyl species K (CoIII–OH) of Co2Fe2-WS also displays a high spin configuration, whereby the quintuplet and the triplet are almost degenerated. The experimentally observed OER overpotential displayed by the Ba[Co2Fe2-WS] salt is slightly smaller than that for Ba[Co4-WS], whereas our calculations show similar values with the Co2Fe2-WS system requiring a slightly larger potential for the first PCET event. This small disagreement may arise from the different overall charge of the systems at the resting state, i.e. 10− vs. 12−, which can influence the outcome of the calculations; thus it will modify the effect of the countercations or the potential protonation of J before its oxidation; which in turn, could result in a small variation of the oxidation mechanism. Overall, two contrary effects can be pointed out as the responsible for the observed similar reactivity: (i) the 12− charge of the Co2Fe2-WS system in front of the 10− charge of the Co4-WS one, which favours the oxidation of the POM and, hence, reduce the OER overpotential;42 and (ii) electron configuration processes going from high to low spin are expected to be advantageous towards OER thanks to the electron reorganization energies. In our calculations we have seen that the presence of the tetrahedral Co heteroatom in Co2Fe2-WS and Co3W-WS generates a structural framework that favours high spin configurations (triplet or quintet) in the external CoIII active centres, such as the hydroxyl (CoIII–OH) and oxo radical (CoIII–O˙) species. On the contrary, this advantage is present within the Co4-WS system in which an electron configuration reorganization from high to low spin occurs in the external, active Co atom after the first PCET event.
Conclusions
In summary, we report a successful strategy to synthesise a mixed-metal cobalt–iron Weakley archetype [CoII2(H2O)2FeIII2(B-α-CoIIW9O34)2]14− (Co2Fe2-WS) POM that forms in good yields in a one-pot, condensation reaction. Single crystal X-ray diffraction and magnetic measurements unambiguously identify the ordered nature of the metal ions within the POM structure. Importantly, the magnetic behaviour could only be reproduced with the proposed structure, whereas the other structural isomer with the two cobalt ions placed in the short diagonal of the rhombus and the iron atoms located in the partially hydrated outer octahedral positions did not find any regions of parameters that yield compatible solutions with the experimental results.The insoluble Ba2+ salt of Co2Fe2-WS was used to study its redox properties and the electrocatalytic water oxidation activity in the solid-state under neutral pH conditions. The novel POM displays high OER activity giving rise to an onset overpotential of 502 mV. The long-term stability under turnover conditions is remarkable. Signs of decomposition could not be detected during the O2 evolution experiments; post-catalytic characterisation identify Ba[Co2Fe2-WS] as the singular active species.
The electrocatalytic performance of Ba[Co2Fe2-WS] was compared to that of two isostructural POMs, namely [CoII4(H2O)2(PW9O34)2]10− (Co4-WS) and [CoII2(H2O)2CoIIWVI(CoIIW9O34)2]12− (Co3W-WS). Interestingly, Ba[Co2Fe2-WS] displays a significantly better OER catalytic activity than Ba[Co4-WS], whereas Ba[Co3W-WS] shows negligible catalytic currents, demonstrating that the coordinatively saturated, central metal ion plays an essential role for the reactivity of the archetype POM-type.
DFT calculations show that the presence of a W atom in the coordinatively saturated central position of the tetraoxo-belt of the Co3W-WS system breaks the symmetry of the POM. This results in a compressive strain effect that distorts the octahedral coordination of the outer CoII ions located at the wing-tips, thus strengthening the Co–OH2 bond and decreasing its OER catalytic activity. Moreover, we have also observed that the presence of the tetrahedral Co heteroatom in Co2Fe2-WS and Co3W-WS generates a structural framework that favours high spin configurations (triplet or quintet) in the external CoII active centres, contrary to the observed in the Co4-WS system-with PV as heteroatom-in which an electron configuration processes going from high to low spin occurs after the first PCET event. We plan to further investigate these systems to study in detail the kinetic steps associated with the oxygen–oxygen bond formation within the catalytic cycle in the close future to completely assess the role of the central, coordinatively saturated metal atoms in the overall reactivity of this type of POMs.
Data availability
CCDC Na[Co2Fe2-WS]_rods: 2068500; Na[Co2Fe2-WS]_plates: 2068501; Na[Co2Fe2-WS]_recrystalized: 2068065; Na[Co3W-WS]: 2068502 contain the supplementary crystallographic data for this paper.Author contributions
J. S.-L. and W. S. conceived the project; J. S.-L. carried out the syntheses, characterisations, analyses, and electrochemical measurements; F. W. S. and N.-Y. Z. performed the X-ray measurements; M. M. carried out syntheses and characterisations; M. O'D. carried out characterisations; M. B. and J. M. P. performed the DFT calculations; J. M. C.-J. and E. C. studied the magnetic behaviour; J. S.-L., and W. S. prepared the manuscript; all authors contributed to discussions throughout the project and the final editing of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
J. S.-L. acknowledges the funding from the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No. 713567, and that from Generalitat Valenciana thought the Plan Gen-T of Excellence (CDEIGENT/2021/037). This research was funded by Science Foundation Ireland (13/IA/1896) and the European Research Council (CoG 2014–647719 and AdG 788222). This work has received support from MCIN/AEI/10.13039/501100011033 (grants CEX2019-000919-M, CNS2022-136079, PID2020-112762GB-I00, PID2020-117152RB-100, and PID2020-117177GB-I00) and by AGAUR (2021-SGR-00110). This study forms also part of the Advanced Materials Program and was supported by MCIN with funding from European Union NextGenerationEU (PRTR-C17.I1) and by Generalitat Valenciana. We thank Dr Brendan Twamley and Dr Tobias Stürzer for their help with the crystallographic analyses.References
- J. R. Galán-Mascarós, Catal. Sci. Technol., 2020, 10, 1967–1974 RSC.
- R. Eisenberg, H. B. Gray and G. W. Crabtree, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 12543–12549 CrossRef CAS PubMed.
- T. R. Cook, D. K. Dogutan, S. Y. Reece, Y. Surendranath, T. S. Teets and D. G. Nocera, Chem. Rev., 2010, 110, 6474–6502 CrossRef CAS PubMed.
- S. A. Sherif, F. Barbir and T. N. Veziroglu, Sol. Energy, 2005, 78, 647–660 CrossRef CAS.
- K. Mazloomi and C. Gomes, Renewable Sustainable Energy Rev., 2012, 16, 3024–3033 CrossRef CAS.
- J. R. McKone, N. S. Lewis and H. B. Gray, Chem. Mater., 2014, 26, 407–414 CrossRef CAS.
- J. Li, C. A. Triana, W. Wan, D. P. Adiyeri Saseendran, Y. Zhao, S. E. Balaghi, S. Heidari and G. R. Patzke, Chem. Soc. Rev., 2021, 50, 2444–2485 RSC.
- J. Soriano-López, W. Schmitt and M. García-Melchor, Curr. Opin. Electrochem., 2018, 7, 22–30 CrossRef.
- C. C. L. McCrory, S. Jung, I. M. Ferrer, S. M. Chatman, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2015, 137, 4347–4357 CrossRef CAS PubMed.
- L. Duan, C. Moyses, M. S. G. Ahlquist and L. Sun, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15584–15588 CrossRef CAS PubMed.
- L. C. Seitz, C. F. Dickens, K. Nishio, Y. Hikita, J. Montoya, A. Doyle, C. Kirk, A. Vojvodic, H. Y. Hwang, J. K. Norskov and T. F. Jaramillo, Science, 2016, 353, 1011–1014 CrossRef CAS PubMed.
- J. D. Blakemore, R. H. Crabtree and G. W. Brudvig, Chem. Rev., 2015, 115, 12974–13005 CrossRef CAS PubMed.
- B. Limburg, E. Bouwman and S. Bonnet, Coord. Chem. Rev., 2012, 256, 1451–1467 CrossRef CAS.
- J. R. Galán-Mascarós, ChemElectroChem, 2015, 2, 37–50 CrossRef.
- I. Roger, M. A. Shipman and M. D. Symes, Nat. Rev. Chem., 2017, 1, 0003 CrossRef CAS.
- M. W. Kanan and D. G. Nocera, Science, 2008, 321, 1072–1075 CrossRef CAS PubMed.
- D. Gao, I. Trentin, L. Schwiedrzik, L. González and C. Streb, Molecules, 2020, 25, 1–20 Search PubMed.
- M. R. Horn, A. Singh, S. Alomari, S. Goberna-Ferrón, R. Benages-Vilau, N. Chodankar, N. Motta, K. Ostrikov, J. MacLeod, P. Sonar, P. Gomez-Romero and D. Dubal, Energy Environ. Sci., 2021, 14, 1652–1700 RSC.
- Q. Yin, J. M. Tan, C. Besson, Y. V. Geletii, D. G. Musaev, A. E. Kuznetsov, Z. Luo, K. I. Hardcastle and C. L. Hill, Science, 2010, 328, 342–345 CrossRef CAS PubMed.
- S. Goberna-Ferrón, L. Vigara, J. Soriano-López and J. R. Galán-Mascarós, Inorg. Chem., 2012, 51, 11707–11715 CrossRef PubMed.
- S. Goberna-Ferrón, J. Soriano-López, J. R. Galán-Mascarós and M. Nyman, Eur. J. Inorg. Chem., 2015, 2015, 2833–2840 CrossRef.
- S. J. Folkman, J. Soriano-Lopez, J. R. Galán-Mascarós and R. G. Finke, J. Am. Chem. Soc., 2018, 140, 12040–12055 CrossRef CAS PubMed.
- J. J. Stracke and R. G. Finke, ACS Catal., 2014, 4, 79–89 CrossRef CAS.
- J. J. Stracke and R. G. Finke, ACS Catal., 2013, 3, 1209–1219 CrossRef CAS.
- J. W. Vickers, H. Lv, J. M. Sumliner, G. Zhu, Z. Luo, D. G. Musaev, Y. V. Geletii and C. L. Hill, J. Am. Chem. Soc., 2013, 135, 14110–14118 CrossRef CAS PubMed.
- M. Natali, S. Berardi, A. Sartorel, M. Bonchio, S. Campagna and F. Scandola, Chem. Commun., 2012, 48, 8808 RSC.
- J. J. Stracke and R. G. Finke, J. Am. Chem. Soc., 2011, 133, 14872–14875 CrossRef CAS PubMed.
- M. Natali, I. Bazzan, S. Goberna-Ferrón, R. Al-Oweini, M. Ibrahim, B. S. Bassil, H. Dau, F. Scandola, J. R. Galán-Mascarós, U. Kortz, A. Sartorel, I. Zaharieva and M. Bonchio, Green Chem., 2017, 19, 2416–2426 RSC.
- J. Wu, L. Liao, W. Yan, Y. Xue, Y. Sun, X. Yan, Y. Chen and Y. Xie, ChemSusChem, 2012, 5, 1207–1212 CrossRef CAS PubMed.
- S.-X. Guo, Y. Liu, C.-Y. Lee, A. M. Bond, J. Zhang, Y. V. Geletii and C. L. Hill, Energy Environ. Sci., 2013, 6, 2654 RSC.
- N. Anwar, A. Sartorel, M. Yaqub, K. Wearen, F. Laffir, G. Armstrong, C. Dickinson, M. Bonchio and T. McCormac, ACS Appl. Mater. Interfaces, 2014, 6, 8022–8031 CrossRef CAS PubMed.
- F. M. Toma, A. Sartorel, M. Carraro, M. Bonchio and M. Prato, Pure Appl. Chem., 2011, 83, 1529–1542 CAS.
- F. M. Toma, A. Sartorel, M. Iurlo, M. Carraro, P. Parisse, C. Maccato, S. Rapino, B. R. Gonzalez, H. Amenitsch, T. Da Ros, L. Casalis, A. Goldoni, M. Marcaccio, G. Scorrano, G. Scoles, F. Paolucci, M. Prato and M. Bonchio, Nat. Chem., 2010, 2, 826–831 CrossRef CAS PubMed.
- M. Blasco-Ahicart, J. Soriano-López and J. R. Galán-Mascarós, ChemElectroChem, 2017, 4, 3296–3301 CrossRef CAS.
- J. Soriano-López, S. Goberna-Ferrón, L. Vigara, J. J. Carbó, J. M. Poblet and J. R. Galán-Mascarós, Inorg. Chem., 2013, 52, 4753–4755 CrossRef PubMed.
- M. Blasco-Ahicart, J. Soriano-López, J. J. Carbó, J. M. Poblet and J. R. Galán-Mascarós, Nat. Chem., 2018, 10, 24–30 CrossRef CAS PubMed.
- J. T. Arens, M. Blasco-Ahicart, K. Azmani, J. Soriano-López, A. García-Eguizábal, J. M. Poblet and J. R. Galán-Mascarós, J. Catal., 2020, 389, 345–351 CrossRef CAS.
- M. Martin-Sabi, J. Soriano-López, R. S. Winter, J.-J. Chen, L. Vilà-Nadal, D.-L. Long, J. R. Galán-Mascarós and L. Cronin, Nat. Catal., 2018, 1, 208–213 CrossRef CAS PubMed.
- H. Lv, J. Song, Y. V. Geletii, J. W. Vickers, J. M. Sumliner, D. G. Musaev, P. Kögerler, P. F. Zhuk, J. Bacsa, G. Zhu and C. L. Hill, J. Am. Chem. Soc., 2014, 136, 9268–9271 CrossRef CAS PubMed.
- J. Soriano-López, D. G. Musaev, C. L. Hill, J. R. Galán-Mascarós, J. J. Carbó and J. M. Poblet, J. Catal., 2017, 350, 56–63 CrossRef.
- B. Liu, E. N. Glass, R.-P. Wang, Y.-T. Cui, Y. Harada, D.-J. Huang, S. Schuppler, C. L. Hill and F. M. F. de Groot, Phys. Chem. Chem. Phys., 2018, 20, 4554–4562 RSC.
- A. Haider, B. S. Bassil, J. Soriano-López, H. M. Qasim, C. Sáenz de Pipaón, M. Ibrahim, D. Dutta, Y.-S. Koo, J. J. Carbó, J. M. Poblet, J. R. Galán-Mascarós and U. Kortz, Inorg. Chem., 2019, 58, 11308–11316 CrossRef CAS PubMed.
- S. Goberna-Ferrón, J. Soriano-López and J. R. Galán-Mascarós, Inorganics, 2015, 3, 332–340 CrossRef.
- K. Azmani, M. Besora, J. Soriano-López, M. Landolsi, A.-L. Teillout, P. de Oliveira, I.-M. Mbomekallé, J. M. Poblet and J. R. Galán-Mascarós, Chem. Sci., 2021, 12, 8755–8766 RSC.
- M. Tao, Q. Yin, A. L. Kaledin, N. Uhlikova, X. Lu, T. Cheng, Y.-S. Chen, T. Lian, Y. V. Geletii, D. G. Musaev, J. Bacsa and C. L. Hill, Inorg. Chem., 2022, 61, 6252–6262 CrossRef CAS PubMed.
- H. Andres, J. M. Clemente-Juan, M. Aebersold, H. U. Güdel, E. Coronado, H. Büttner, G. Kearly, J. Melero and R. Burriel, J. Am. Chem. Soc., 1999, 121, 10028–10034 CrossRef CAS.
- H. Andres, M. Aebersold, H. U. Güdel, J. M. Clemente, E. Coronado, H. Büttner, G. Kearly and M. Zolliker, Chem. Phys. Lett., 1998, 289, 224–230 CrossRef CAS.
- J. M. Clemente-Juan, E. Coronado, A. Gaita-Ariño, C. Giménez-Saiz, H.-U. Güdel, A. Sieber, R. Bircher and H. Mutka, Inorg. Chem., 2005, 44, 3389–3395 CrossRef CAS PubMed.
- C. S. Ayingone Mezui, P. de Oliveira, A.-L. Teillout, J. Marrot, P. Berthet, M. Lebrini and I. M. Mbomekallé, Inorg. Chem., 2017, 56, 1999–2012 CrossRef CAS PubMed.
- E. N. Glass, J. Fielden, A. L. Kaledin, D. G. Musaev, T. Lian and C. L. Hill, Chem. – Eur. J., 2014, 20, 4297–4307 CrossRef CAS PubMed.
- X. Zhang, Q. Chen, D. C. Duncan, R. J. Lachicotte and C. L. Hill, Inorg. Chem., 1997, 36, 4381–4386 CrossRef CAS PubMed.
- P.-E. Car, M. Guttentag, K. K. Baldridge, R. Alberto and G. R. Patzke, Green Chem., 2012, 14, 1680–1688 RSC.
- P. M. Wood, Biochem. J., 1988, 253, 287–289 CrossRef CAS PubMed.
- H. S. Ahn and T. D. Tilley, Adv. Funct. Mater., 2013, 23, 227–233 CrossRef CAS.
- M. T. Pope, Heteropoly and Isopoly Oxometalates, Springer Berlin Heidelberg, Berlin, Heidelberg, 1983, vol. 8 Search PubMed.
- X. López, J. A. Fernández and J. M. Poblet, Dalton Trans., 2006, 1162–1167 RSC.
- A. Solé-Daura, J. M. Poblet and J. J. Carbó, Chem. – Eur. J., 2020, 26, 5799–5809 CrossRef PubMed.
- J. J. Concepcion, R. A. Binstead, L. Alibabaei and T. J. Meyer, Inorg. Chem., 2013, 52, 10744–10746 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2068500–2068502 and 2068065. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc04002j |
| ‡ Current address: Institut de Ciència Molecular, Universitat de València, Catedrático José Beltrán 2, Paterna 46980, Spain. |
| This journal is © The Royal Society of Chemistry 2023 |