
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIdentification of non-conventional small molecule degraders and stabilizers of squalene synthase†
Joseph G. F.
Hoock
 a,
Cecilia
Rossetti
a,
Mesut
Bilgin
b,
Laura
Depta
a,
Kasper
Enemark-Rasmussen
a,
John C.
Christianson
c and
Luca
Laraia
*a
a,
Cecilia
Rossetti
a,
Mesut
Bilgin
b,
Laura
Depta
a,
Kasper
Enemark-Rasmussen
a,
John C.
Christianson
c and
Luca
Laraia
*a
aDepartment of Chemistry, Technical University of Denmark, Kemitorvet 207, Kongens Lyngby 2800, Denmark. E-mail: luclar@kemi.dtu.dk
bLipidomics Core Facility, Danish Cancer Institute, Strandboulevarden 49, Copenhagen 2100, Denmark
cNuffield Department of Rheumatology, Orthopaedics, and Musculoskeletal Sciences, Botnar Research Centre, University of Oxford, Headington, Oxford OX3 7LD, UK
First published on 18th October 2023
Abstract
Squalene synthase (SQS) is an essential enzyme in the mevalonate pathway, which controls cholesterol biosynthesis and homeostasis. Although catalytic inhibitors of SQS have been developed, none have been approved for therapeutic use so far. Herein we sought to develop SQS degraders using targeted protein degradation (TPD) to lower overall cellular cholesterol content. We found that KY02111, a small molecule ligand of SQS, selectively causes SQS to degrade in a proteasome-dependent manner. Unexpectedly, compounds based on the same scaffold linked to E3 ligase recruiting ligands led to SQS stabilization. Proteomic analysis found KY02111 to reduce only the levels of SQS, while lipidomic analysis determined that KY02111-induced degradation lowered cellular cholesteryl ester content. Stabilizers shielded SQS from its natural turnover without recruiting their matching E3 ligase or affecting enzymatic target activity. Our work shows that degradation of SQS is possible despite a challenging biological setting and provides the first chemical tools to degrade and stabilize SQS.
Introduction
Squalene synthase (SQS), also known as farnesyl-diphosphate farnesyltransferase 1 (FDFT1), is an endoplasmic reticulum (ER) resident membrane protein positioned at a unique branch-point between the sterol- and non-sterol arms of the mevalonate pathway. By catalyzing the condensation of two farnesylpyrophosphate (FPP) molecules into squalene, SQS plays a crucial role in the biosynthesis of cholesterol.1 Since SQS commits FPP into the cholesterol branch, it can be regarded as a switch which is utilized by cells to directly dictate the flow of FPP.2 Furthermore, additional non-catalytic functions of SQS in the TGFβ pathway have recently been discovered and roles in early embryonic development have been indicated.3–5 Consequently, SQS has been investigated as a therapeutic target to lower cholesterol levels.6–8 Enzymatic SQS activity has been linked to hypercholesterolemia-associated diseases, as well as lung- and or prostate cancer9 or neurodegenerative disorders.10 A number of known ligands exists, with zaragozic acid A (ZAA, also known as squalestatin 1)11 or TAK-475 (also known as lapaquistat acetate)12 being potent examples of active site binding inhibitors with activity in the nanomolar (nM) range. Despite this, no SQS inhibitor has been successfully brought to the market to date. TAK-475 was the most advanced molecule investigated for the treatment of hypercholesteremia, but development was stopped in phase II and III clinical trials after hepatotoxicity accompanied by elevated bilirubin levels were detected.13With the recent rise of targeted protein degradation (TPD) as a promising therapeutic approach,14 we wanted to revisit SQS as an attractive drug target and develop a probe that could selectively degrade SQS. We hypothesized that a compound able to reduce SQS levels rather than just inhibiting its activity could be an alternative approach to attenuate cholesterol biosynthesis and lower overall cholesterol levels, with possible applications in cancer therapy. Furthermore, SQS degraders could aid the discovery of additional non-catalytic functions of SQS unrelated to the cholesterol biosynthetic pathway.15
Herein, we report the identification of the small molecule SQS degrader KY02111, a recently reported SQS ligand, as well as the serendipitous discovery of SQS stabilizers.3 While compounds designed to function as proteolysis-targeting chimeras (PROTACs) based on ligands with diverse SQS binding sites3,16 led to increased target levels, KY02111 treatment lowered SQS levels in HeLa and U2OS cells in a concentration-, time- and proteasome-dependent manner with excellent selectivity across the proteome. KY02111 did not affect the insertion of SQS in the ER or cause its aggregation, but its degradation could be partially rescued by inhibiting the E3 ligase HRD1. Importantly, partial SQS knockdown using KY02111 led to an overall decrease of cholesterol levels in the form of cholesteryl esters (CE), further supporting its applicability as a tool to study SQS function in a wide range of contexts. Interestingly, envisioned SQS degraders based on the KY02111 scaffold linked to E3 ligase recruiting ligands shielded SQS from its natural degradation by engaging in strong binary interactions with the protein, which might be a relevant strategy for treatment of rare genetic diseases where SQS expression is low.5
Results
We initiated our efforts to identify a small molecule degrader of SQS by synthesizing a small yet diverse degrader library based on two reported SQS-ligands: an active site inhibitor, herein referred to as SQSI, and an unknown-binding site ligand known as KY02111 (Fig. 1A). To cover as large a degrader space as possible, each ligand was diversified by varying linker length and composition as well as by generating different protein of interest (POI) ligand-degrader modality pairings. | ||
| Fig. 1 Full SQS-degrader library. (A) Structure of SQS ligands used to design PROTACs: one active site inhibitor (SQSI, turquoise) and one unknown binding site ligand (KY02111, blue). The final envisioned SQS degraders connect the SQS ligands at positions which are accepted for modification without interfering with target engagement. (B) The final compounds contain one of three different degrader modalities (X): pomalidomide, VH032 and an adamantane tag. The SQS-ligands and the degrader modalities were linked by PEG or alkyl-based linkers of varying lengths. Compound 29 only contains a Boc-protected PEG linker. (C) Full structure of KY02111-based PROTAC 18, negative control 28 and fluorescent probes based on the parent SQS ligands linked to 5-carboxytetramethylrhodamine (TAMRA) via linkers. | ||
SQSI was part of a 2-aminobenzhydrol compound series developed by Daiichi Sankyo, which inhibited SQS activity in rat hepatocytes with an IC50 of 1.3 nM.16 Additional interest in this compound was sparked by the fact that the crystal structure of a close analogue revealed exposure of the piperidine-4-carboxamide to the cytosol (PDB: 3ASX), making it a desirable linker attachment position (Fig. 1A). Together with a readily accessible synthetic route, SQSI was an ideal compound for our efforts. On the other hand, KY02111 was only recently identified as a SQS-binder.3 KY02111 showed no catalytic inhibition in an in vitro activity assay as well as in an in cellulo SREBP reporter gene assay. The authors found that KY02111 likely interferes with a so far unknown protein–protein interaction (PPI) between SQS and TMEM43, which resulted in downregulation of TGFβ-induced signaling. A reduction of SQS levels upon compound treatment was not observed by the authors. Overall, the work suggested that KY02111 likely does not bind SQS at the same site as SQSI. To increase potential ternary complex diversity and thereby the probability of success in identifying an SQS degrader, we based the second half of our library on a KY02111 analogue (SO2093, Fig. 1A).
We implemented additional structural diversity by attaching the two SQS-ligands to three different degrader modalities via a variety of linkers (Fig. 1B). For the formation of PROTACs, we utilized the well described17,18 cereblon (CRBN) and von Hippel Lindau (VHL) ligands pomalidomide19 and VH032,20 respectively. As a less frequently used degrader modality of TPD, we generated hydrophobic tags by attaching an adamantane moiety.21 We linked both ligands via readily available flexible first generation linkers varying in length (8–15 total atoms for pomalidomide and adamantane, 7–16 total atoms for VH032) and composition (PEG and alkyl). Synthesis of the full SQS-degrader library was conducted using procedures reported previously (Fig. 1B and S2 ESI†). While preparing compounds based on SQSI we observed the presence of four different isomers when the samples were dissolved in DMSO. These were fully assigned using variable temperature NMR as two sets of interconvertible atropisomers (Fig. S3 ESI†).
We initially screened the SQS-degrader library for binary target engagement. We first expressed a catalytically active SQS construct (31–370) lacking the C-terminal transmembrane domain in E. coli.22 With the recombinant protein in hand, we developed two individual in vitro screens to evaluate SQS-binding by our compounds – differential scanning fluorimetry (DSF) and fluorescence polarization (FP). DSF measurements (Fig. S4 ESI†) showed dose-dependent stabilization with an increase in melting temperature (ΔTm) of up to 6.3 °C for the active site-based compounds (SQSI, 6, 7, 8). The non-active site compounds inspired by KY02111 also stabilized SQS, yet not to the same extent (ΔTm ≤ 3.6 °C) and with diminished dose-dependency compared to the first half of the compound series.
To confirm the observed target binding, we also developed a fluorescence polarization (FP) assay by synthesizing two 5-TAMRA conjugated probes based on SQSI (26) and KY02111 (27) (Fig. 1C). Protein titration with both fluorescent probes showed strong binding of SQS with similar affinities reflected by Kd values around 100 nM (Fig. 2A). Competition experiments of the SQS-degrader library versus the corresponding FP probe (1–12vs.26, 13–25vs.27) yielded the IC50 and ki values shown (Fig. 2C). All compounds (except 5) were able to outcompete their parent probe. A general trend can be observed where increasing affinity is correlated with attachment of a linker-degrader modality in comparison to the unmodified ligands (e.g.3, 4, 7, 11 compared to SQSI; 14, 18, 19, 22, 23 compared to KY02111). Three out of four PROTACs containing the C9/10 alkyl chain (5, 8, 17) make an exception to this rule due to decreased molecular flexibility and solubility.
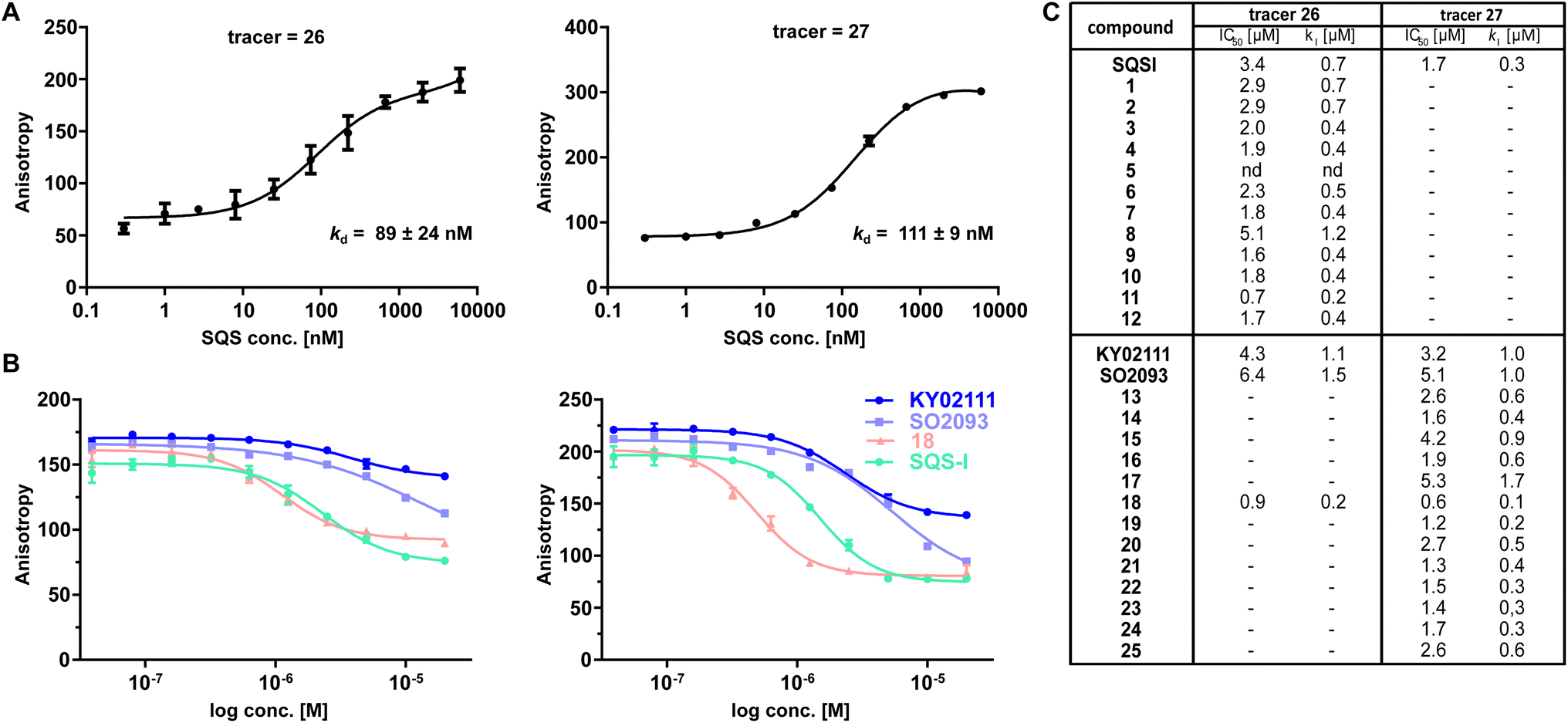 | ||
| Fig. 2 FP assay confirms binding of degrader library to recombinant SQS in vitro. (A) Titration of recombinant SQS vs. active site tracer 26 (c = 20 nM) and unknown site tracer 27 (c = 5 nM) (n = 2; conducted in technical duplicates; mean ± SEM shown). (B) Competition of selected compounds vs.26 and vs.27. 26 can be outcompeted by compounds assumed not to bind the active site (KY02111, SO2093, 18). Vice versa, 27 can be outcompeted by the active-site binding SQSI (n = 4, conducted in technical duplicates, mean ± SEM shown). (C) Competition experiments of the full library vs. either active site FP probe 26 or unknown binding site probe 27 showed that all compounds (except 5) are able to bind SQS in vitro (n = 1–4; mean shown). | ||
Importantly, both the active site (SQSI) and unknown binding site ligands (KY02111, 18), were able to outcompete both active site (26) and unknown site (27) probes (Fig. 2B). For example, SQSI showed slightly stronger displacement of fluorescent probes when competed versus27 (ki = 0.3 μM) than versus26 (ki = 0.7 μM). This was surprising since previously reported data showed that KY02111 did not inhibit the catalytic activity of SQS, leading to the hypothesis that the interaction occurs outside of the active site.3 Since our observations suggested overlapping binding sites of active site based compounds and compounds based on KY02111, we adopted an in vitro activity assay to test possible SQS inhibition by elongated KY02111-based PROTAC 18. SQS requires NADPH as a cofactor for the catalysis of the condensation of two molecules of FPP into squalene. Therefore, the reaction progress can be monitored by measuring a continous decrease in NADPH fluorescence over time (Fig. S5A ESI†).23 Interestingly, both compound 18 (at 3 μM) and KY02111 (at 10 μM), did not significantly inihbit SQS activity, whereas the positive control ZAA (at 1 μM) completety inhibited the reaction (Fig. S5B ESI†). Taking all of our in vitro binding assays into account, we hypothesize that KY02111 binds in the vicinity of the active site, and that its functionalization with a linker and a fluorophore or E3 ligase recruiter, leads to a structural overlap with SQSI-based probes 1–12 and 26. It can however not be excluded that all ligands are also allosteric inhibitors, where binding to a specific site leads to conformational changes in SQS that preclude binding of ligands at other sites.
While in vitro assays showed that linker attachment to both ligands would not interfere with SQS engagement, we probed our library for degradation ability in HeLa cancer cells. Since endogenous SQS is constitutively turned over with a modest half-life (t1/2 ∼ 5 h)24 we incubated the compounds for 18 h to ensure clear correlation between lower SQS abundance and compound treatment. Intensive screening via western blotting identified KY02111 as the most potent SQS-degrader, whereas the designated degrader molecules almost exclusively lead to an increase in SQS levels. PROTACs (1–8, 30 nM to 3 μM) and HyTs (9–12, 2 and 20 μM) derived from SQSI universally lead to an increase in SQS levels at all tested concentrations. Remarkably, KY02111 derived compounds 13 (at 3 μM), 18 (at 3 μM), 21 (at 3 μM) and 25 (2 and 20 μM) all showed a similar trend (Fig. 3A, B, S6A and B ESI†).
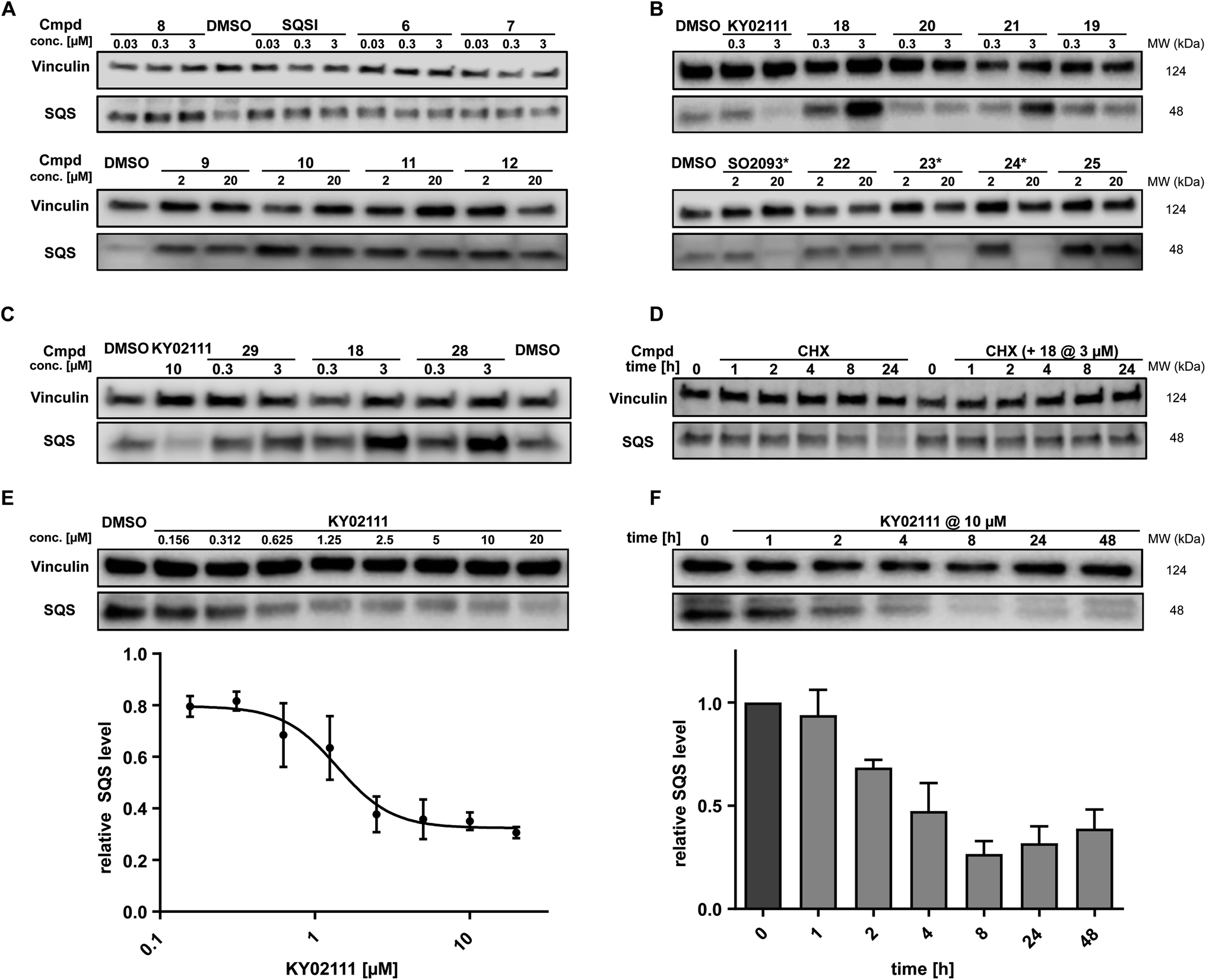 | ||
| Fig. 3 In cellulo SQS-degrader screen reveals KY02111 as only SQS-degrader. (A and B) Western blots showing the changes in SQS protein levels after 18 h treatment of HeLa cells with compounds at indicated concentrations (n = 2 or 3). * compounds precipitated at 20 μM and caused cell death. (C) SQS accumulation by PROTAC 18 is independent of VHL. HeLa cells were treated with either KY02111, compound 29, 18 or 28 for 18 h (n = 3). (D) CHX co-incubation shows that PROTAC 18 stabilizes SQS rather than upregulating it. HeLa cells were treated with 18 (3 μM) and/or CHX (100 μg mL−1) for the indicated time points (n = 3). (E) KY02111 reduces SQS levels in a dose-dependent manner. The concentration to reduce SQS levels by half is reached at DC50 = 1.4 μM and overall SQS levels can be reduced to Dmax = 68%. HeLa cells were treated with the indicated concentrations of KY02111 for 18 h (n = 4; mean ± SEM shown). (F) KY02111 reduces SQS levels in a time-dependent manner. HeLa cells were treated with KY02111 (10 μM) for the indicated time points (n = 3; mean ± SD shown). Please see Fig. S13 ESI† for complete western blots. | ||
In addition to KY02111, we observed a dose-dependent decrease in SQS abundance for three additional hydrophobic molecules: SO0293 and the hydrophobic tag-containing compounds 23 and 24 (Fig. 3B, right). However, during sample preparation, a clear increase in cell death in samples treated with 20 μM of these compounds was seen, which was accompanied by compound precipitation. We confirmed this cytotoxicity for SO2093, and compounds 23 and 24 by assaying cell viability (Fig. S7A and B ESI†). Since KY02111 treatment did not alter cell viability after 48 h, we hypothesized that the decrease in SQS levels by SO2093, compounds 23 and 24 was caused by an unspecific mode of action (MoA) and therefore excluded the compounds from further testing.
In addition to investigating the mechanism behind KY02111-induced SQS degradation, we were also curious about the increase in SQS levels by the bifunctional compounds we originally designed as PROTACs. Since compounds 1–12 are based on an active site inhibitor, we hypothesized that these molecules still act as inhibitors. Instead of forming a productive ternary complex only a binary complex with SQS is formed, thereby blocking the catalytic activity, and leading to compensatory upregulation. We confirmed this by monitoring expression of the sterol-regulated transcription factor SREBP using a reporter assay, which showed increased activity for cells treated with SQSI (Fig. S8 ESI†). Alternatively, another reason for observing increased SQS levels could be a stabilizing ligand-SQS interaction interfering with the natural degradation of SQS (Fig. 3B, right). We hypothesized that this could be the case for the second half of the library based on the degrader KY02111 and utilized the VH032-linked PROTAC 18 to study this phenomenon.
For compound 18, we detected elevated POI levels for incubation at 3 μM compared to 0.3 μM and DMSO. To exclude the possibility of an early Hook-effect25,26 we incubated HeLa cells with even lower concentrations of compound 18, but could not detect any degradation (Fig. S9A ESI†). A recent study by Poirson et al.27 showed that VHL can also act as a protein stabilizer instead of a degrader, possibly due to the formation of a non-productive ternary complex. To test whether VHL recruitment is necessary for SQS stabilization, we synthesized the hydroxyproline epimer of 18, compound 28, which has previously been used as a negative control for VHL-based PROTACs.28 Additionally, we co-incubated compound 18 with a large excess of VH032, the parent VHL-ligand to compete with a potentially stabilizing ternary complex (Fig. S9A ESI†).29 We found that neither incubation with the non-recruiting compound 28, nor the co-incubation with VH032 could prevent SQS stabilization. In fact, treatment with compound 28 resulted in SQS stabilization to a similar degree as for 18 (Fig. 3C), confirming that the VHL protein does not contribute to SQS stabilization. Replacement of the VH032 moiety with adamantane (compound 22) or Boc (compound 29) did not lead to a comparable increase in SQS levels, suggesting that a larger moiety on a similar linker, albeit not any, is required for the observed effect. For the pomalidomide connected derivatives, only compound 13 (Fig. S6 ESI†) was able to increase SQS stability, whereas compound 27, the fluorescent 5-TAMRA probe, did not show any effect on SQS levels (Fig. S6 ESI†).
The nature of the connecting linker also plays an important role in SQS stabilization. Compounds 13, 18 and 28 all contain a 9–10 atom PEG-based linker, whereas shorter (7–8 atoms) alkyl linkers included in compounds 16, 20 and 27 could not induce protein accumulation (Fig. 3B and S6 ESI†). Our SAR analysis indicates that specific binary interactions between SQS and our molecules stabilize, potentially shield, SQS from its natural degradation. This is supported by our finding that co-incubation of compound 18 with the protein biosynthesis inhibitor cycloheximide (CHX)30,31 leads to a stabilization of SQS levels after 8 and 24 h compared to CHX-treated controls alone (Fig. 3D). Of note, 18 showed the highest degree of stabilization (Tm = 3.6 °C, Fig. S4C ESI†) for the compound series based on KY02111 and the overall highest affinity (kI = 0.1 μM) for SQS in our FP assay (Fig. 2B and C) which is in agreement with the above observations. Furthermore, 18 did not inhibit the enzymatic activity of SQS in an in vitro activity assay (Fig. S5 ESI†). Based on this, we believe that bifunctional compounds based on KY02111, connected to an E3 ligase ligand via a 9–10 atom PEG-linker, stabilize SQS through tight binary binding interactions.
To characterize the reduction of SQS levels induced by KY02111 treatment, we performed dose–response and time-dependent experiments in HeLa cells (Fig. 3E and F, respectively). The dose response of KY02111 yielded a DC50 of 1.4 μM and a Dmax of 68%. However, we were not able to induce complete degradation even when increasing the concentration to as high as 20 μM (Fig. 3E). To exclude the possibility of a cell line dependent effect, we treated U2OS cells with KY02111 and observed a similar concentration-dependent reduction in SQS levels (Fig. S9B ESI†). Additionally, we found that the effect on SQS was time-dependent, with maximum levels of degradation reached between 8 and 24 hours, after which protein levels slowly recover (Fig. 3F).
Next, we sought to determine which cellular degradation machineries were responsible for the loss of SQS protein. HeLa cells were pre-incubated for 2 h with either MG132 (proteasomal inhibitor), MLN4924 (neddylation inhibitor) or chloroquine (CQ, lysosomal inhibitor) followed by co-incubation with 10 μM KY02111 for 18 h (Fig. 4B). MG132 was clearly able to block the KY02111-induced SQS degradation, whereas MLN4924 could not rescue protein levels. Interestingly, co-incubation with chloroquine (CQ) led to an increase in the SQS band intensity. However, compared to CQ alone, KY02111 was still able to markedly reduce SQS levels. To the best of our knowledge, lysosomal degradation has not been linked to specific natural degradation of SQS before. However, ER associated autophagy (ER-phagy), which is involved in maintaining the ER, is lysosome dependent.32,33 As SQS is an ER resident enzyme, CQ action could block ER-phagy leading to the accumulation of SQS. Therefore, we believe that KY02111-mediated SQS degradation is unlikely to specifically require the lysosome. Collectively, these observations indicate that KY02111-induced degradation of SQS requires functioning proteasomes, but is not dependent on an E3 ligase which requires activation by neddylation, such as those in the Cullin RING ligase family.34 Having established a correlation between KY02111-treatment, SQS-binding and proteasomal degradation, we wanted to further investigate and understand how KY02111 precisely mediated this process.
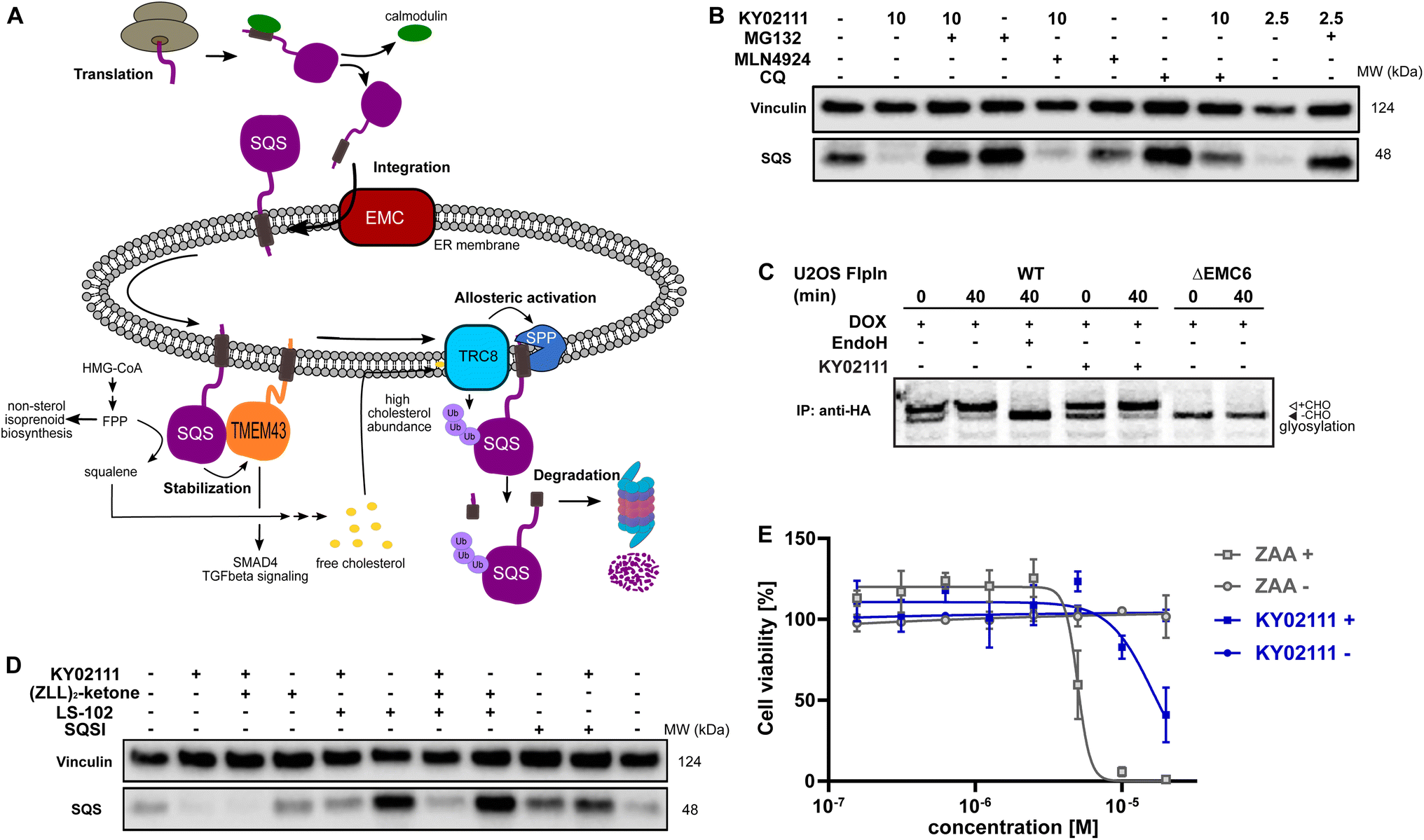 | ||
Fig. 4 Characterization and evaluation of KY02111-mediated SQS degradation. (A) “Lifecycle” of SQS based on literature reports. (B) KY02111-induced SQS reduction is proteasome, and to a lesser degree lysosome, dependent. HeLa cells were pre-incubated with either MG132 (20 μM) or chloroquine (CQ, 20 μM) or MLN4924 (5 μM) for 2 h prior to addition of KY02111 at indicated concentrations (n = 2). (C) 35S-met/cys pulse-chase insertion assay of HA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :SQSopsin inducibly expressed in U2OS Flp-In™ T-Rex™ WT and ΔEMC6 cells. HA:SQSopsin expression was induced by the pretreatment and continuous presence of doxycycline (DOX, 10 ng mL−1, 18 h) throughout the assay. Where indicated, KY02111 (10 μM) was also continuously present during the assay but only after 1 h pretreatment. Radiolabelled HA:SQSopsin from each timepoint (0 and 40 min) was immunoprecipitated and separated by SDS-PAGE. Where indicated, WT eluate was treated with EndoHf to confirm opsin glycosylation. Core glycosylated (+CHO) and unglycosylated (−CHO) HA:SQSopsin are shown. (D) Co-incubation of KY02111 with tool compounds inhibiting natural SQS degradation. HeLa cells were co-incubated with KY02111 (10 μM) and either (ZLL)2-ketone (10 μM), LS-102 (10 μM) or SQSI (1 μM) for 18 h (n = 3). (E) Mild cholesterol auxotrophy after treatment of HeLa cells with KY02111 (20 μM) in the presence of MBCD (3 mM) (n = 3, mean ± SD of one biological replicate shown). Please see Fig. S14 ESI† for complete western blots and membranes. :SQSopsin inducibly expressed in U2OS Flp-In™ T-Rex™ WT and ΔEMC6 cells. HA:SQSopsin expression was induced by the pretreatment and continuous presence of doxycycline (DOX, 10 ng mL−1, 18 h) throughout the assay. Where indicated, KY02111 (10 μM) was also continuously present during the assay but only after 1 h pretreatment. Radiolabelled HA:SQSopsin from each timepoint (0 and 40 min) was immunoprecipitated and separated by SDS-PAGE. Where indicated, WT eluate was treated with EndoHf to confirm opsin glycosylation. Core glycosylated (+CHO) and unglycosylated (−CHO) HA:SQSopsin are shown. (D) Co-incubation of KY02111 with tool compounds inhibiting natural SQS degradation. HeLa cells were co-incubated with KY02111 (10 μM) and either (ZLL)2-ketone (10 μM), LS-102 (10 μM) or SQSI (1 μM) for 18 h (n = 3). (E) Mild cholesterol auxotrophy after treatment of HeLa cells with KY02111 (20 μM) in the presence of MBCD (3 mM) (n = 3, mean ± SD of one biological replicate shown). Please see Fig. S14 ESI† for complete western blots and membranes. | ||
To become functionally active, SQS must be inserted into the ER membrane via its C-terminal transmembrane domain. Insertion of the SQS “tail anchor” occurs post-translationally and is facilitated by the multi-subunit assembly known as ER membrane protein complex (EMC, Fig. 4A).35 Without the EMC, SQS tail anchor insertion into the ER membrane is inefficient and rather than accumulating in the cytoplasm, SQS is degraded by the proteasome in a process differing from the canonical ERAD pathway.24 Given this, we hypothesized that the KY02111-SQS interaction might inhibit its tail anchor insertion into the ER by indirectly preventing it from accessing the EMC, e.g. by causing SQS aggregation (Fig. 4A).
This MoA is consistent with our earlier in vitro binding studies, which showed that KY02111 can interact with a recombinant, soluble form of SQS lacking its tail anchor (aa 31–370). Moreover, polymerization triggered by small molecules has been described previously.36,37 To test this, we performed radiolabeling pulse-chase insertion assays using a doxycycline-inducible (DOX) HA-tagged SQS-construct containing a C-terminal opsin tag after the tail anchor. The opsin tag encodes a sequence that is glycosylated only upon exposure to the ER lumen.24,38 After 40 min, a shift to the glycosylated form of SQS can be observed in wild-type cells which is sensitive to the deglycosidase EndoH (Fig. 4C). Without the EMC (ΔEMC6), SQS remains unglycosylated throughout the chase indicating insertion failure. Pre-treatment (1 h) with KY02111 and its inclusion throughout did not compromise insertion of the SQS tail anchor and resembled untreated cells. This indicates that KY02111 did not cause aggregation and therefore was not acting by attenuating or disrupting normal SQS biogenesis.
Additionally, we investigated whether KY02111 could cause SQS to aggregate and therefore performed in vitro differential light scanning (DLS) experiments as well as size exclusion chromatography in the presence of KY02111 (Fig. S10 ESI†). We determined the diameter (d) of recombinant SQS to be 4–6 nm, which is in agreement with protein sizes generally determined via DLS but could not detect any change in diameter upon incubation with KY02111 (Fig. S10A and B ESI†).
We found that the viability of HeLa cells treated with KY02111 was indistinguishable from DMSO-treated when cells were grown in standard FBS-containing media for up to 48 h (Fig. S7A and B ESI†). However, it has previously been reported that SQS depletion resulting from loss of the EMC led to a cholesterol auxotrophic effect and increased cell death upon cholesterol depletion when using either lipoprotein deficient serum (LPDS) or methyl β-cyclodextrin (MBCD). To test whether a loss of SQS induced by KY02111 (∼65–70%) would compromise cell viability similarly, we grew HeLa cells in FBS-containing media supplemented with MBCD for 72 h and treated with different concentrations of either KY02111 or the potent active site inhibitor ZAA (Fig. 4E). We determined the non-toxic concentration of MBCD to be 3 mM for HeLa cells (Fig. S11A and B ESI†). As reported previously, high concentrations of ZAA (5 and 10 μM) were sufficient to completely abolish cell viability. KY02111 also reduced cell viability at high concentrations (20 μM) although not to the same degree as ZAA. When cells were grown in standard conditions (DMEM 10% FBS), neither ZAA nor KY02111 adversely affected cell viability at 72 h. This indicates that reducing levels of SQS through chemically induced degradation partially phenocopies catalytic inhibition, compromising cholesterol biosynthesis through the mevalonate pathway.
As SQS is a critical component in the mevalonate pathway downstream of FPP, its abundance is tightly controlled. So far, two distinct natural degradation mechanisms of SQS have been described: rapid degradation is facilitated either by signal peptide peptidase (SPP), which cleaves the TMD of SQS at high cholesterol levels sensed by the ER-resident ubiquitin ligase (E3) TRC8, or by HRD1, an important ER-E3 associated with ERAD (Fig. 4A).2 To investigate whether KY02111 treatment might enhance the natural degradation of SQS, we co-incubated HeLa cells with tool compounds targeting these pathways (Fig. 4D). We found that the SPP inhibitor (ZLL)2-ketone2,39 could not prevent a reduction in SQS band intensity in the presence of KY02111, when compared to the individually treated samples. Interestingly, treatment with the HRD1 inhibitor LS-102 (refs. 40 and 41) alone led to a marked accumulation of SQS, which suggests that HRD1 is an important factor facilitating basal turnover. When LS-102 treatment was combined with KY02111, SQS no longer accumulated, however protein levels were still greater than that of KY02111 alone, and similar to the untreated DMSO control. This suggests that KY02111-induced SQS degradation may result from an enhanced basal degradation, though additional contributing mechanisms cannot be excluded at this point.
Since our earlier FP measurements showed competition between active-site inhibitor SQSI and the KY02111-based probe 27, as well as between compound 18 and probe 26 (Fig. 2B), we tested if KY02111-mediated degradation could also be blocked by SQSI. Indeed, SQSI was able to rescue SQS levels in the presence of KY02111, even when incubated at a 10-fold lower concentration (1 μM). Data from Takemoto et al., as well as our own data (Fig. S5 ESI†), suggested that KY02111 does not inhibit the catalytic activity of SQS and therefore, binding should occur outside of the active site.3 We re-confirmed this using a similar SREBP reporter gene assay with KY02111, finding that SREBP target genes are not activated in response to KY02111 treatment (Fig. S8 ESI†).42 SREBP is the transcriptional regulator of SQS and cholesterol biosynthesis. In general, its activity can be correlated to impaired cholesterol synthesis and SQS activity.43,44 Overall, our data suggests that KY02111 binds close to the SQS catalytic site without inhibiting the enzymatic activity. Compounds containing a linker-fluorophore and linker-VH032 part, e.g.26 and 18, could sterically hinder each other, leading to the observed competition in the FP experiments. To fully elucidate the binding interaction between KY02111 and SQS we attempted to generate a crystal structure of the compound–protein complex, but those efforts have been unsuccessful so far.
To further our investigation into the mechanism of degradation induced by KY02111 and determine its proteome-wide selectivity, we performed global proteomic studies using isobaric tandem mass tag labeling (TMT, 16-plex) coupled to mass spectrometry analysis (MS). To this end, we incubated HeLa cells with either KY02111 (10 μM, Fig. 5A), compound 18 (5 μM, Fig. 5B) and SQSI (1 μM, Fig. 5C) for 18 h. The concentrations were chosen with our earlier WB results in mind to ensure robust detection of changes in SQS protein level. We specifically sought to detect changes in protein levels of enzymes within cholesterol/lipid metabolism, ERAD or EMC clients as well as TMEM43, a potential PPI interaction-partner of SQS.
 | ||
| Fig. 5 Global proteomic and lipidomic analysis of KY02111- and 18-treatment in HeLa cells. (A–C) Proteomic profile of HeLa cells treated with KY02111 (10 μM, A), PROTAC 18 (5 μM, B) or SQSI (1 μM, C) for 18 h. The soluble fraction of the lysates was labeled with 16-plex TMT labels and subjected to MS/MS analysis. The analysed data was plotted as −log10p-value vs. log2FC (FC = fold change, n = 3). Please see extended dataset 1 for complete proteomics data and analysis; (D) Changes in lipidomic species of HeLa cells treated with KY02111 (10 μM) or 18 (5 μM) for 18 h. Significantly altered species are highlighted, Holm-Šídák correction was used to determine adjusted p-values (n = 3, p < 0.005). (E) Fold change overview of cholesteryl ester (CE) classes in HeLa cells treated with KY02111 (10 μM) or PROTAC 18 (5 μM) for 18 h. Significantly altered species are highlighted with * (n = 3, p < 0.005), species below limit of detection are highlighted with Δ. Please see extended dataset 2 for complete lipidomics data and analysis. | ||
Importantly, quantitative proteomic analyses were able to reproduce our WB results, identifying SQS (=FDFT1) as significantly reduced (KY02111, log2FC = −1.19, −log10p-value = 4.63) or enriched (18, log2FC = 1.05, −log10p-value = 3.78; SQSI, log2FC = 0.85, −log10p-value = 3.53) in response to their respective treatments (extended dataset 1). Surprisingly, we did not detect a general increase in cholesterol biosynthesis enzyme protein levels as a result of SQS degradation, stabilization or inhibition. It should be noted that we could not detect HMGR in all of our MS data sets. Loss of cholesterol biosynthetic capacity might be compensated for by an increase in cholesterol uptake from the medium (standard growth conditions, 10% FBS) or by utilizing stored cholesteryl esters. KY02111 treatment only reduces SQS protein levels to 30–35% and the remaining fraction may be sufficient to supply cells with cholesterol if needed. We propose that reducing SQS levels by nearly two-thirds could lead to accumulation of its substrate FPP, accompanied by changes to products within the non-sterol isoprenoid pathway like dolichol, ubiquinone and general protein prenylation.45,46 We believe this is indicated by a significant decrease in HMG-CoA synthase (log2FC = −0.51, −log10p-value = 3.54) protein levels, which is consistent with our SREBP reporter gene assay data. Here KY02111 led to a reduction of a luciferase reporter signal connected to a HMGCS promotor region (Fig. S8 ESI†). Moreover this was accompanied by an increase of dehydrodolichyl diphosphate synthase (DHDDS, log2FC = 0.57, −log10p-value = 1.53), which uses FPP as a direct substrate.47,48 This hypothesis is also supported by studies of mice lacking SQS in the liver,4 which show significant increase in FPP levels.15 Since KY02111 was previously reported to inhibit the SQS-TMEM43 PPI, and that knockdown of SQS using siRNAs led to a decrease of TMEM43, we also anticipated a reduction of TMEM43 in cells with chemically reduced SQS levels. To our surprise we did not detect any significant level changes for TMEM43 (log2FC = 0.13, −log10p-value = 0.78) in KY02111-treated samples. This can be attributed to numerous differences in the experimental setup, including the fact that chemical degradation of SQS by KY02111 reaches approximately 70%, whereas siRNA KD produced a greater reduction in SQS levels.
To determine whether KY02111-induced reduction of SQS protein levels may have therapeutic potential by attenuating cholesterol biosynthesis and concomitantly decreasing cholesterol content, we performed a quantitative shotgun lipidomics analysis of HeLa cells, which covered 27 lipid classes and over 446 species (Fig. 5D and S12 ESI† and extended dataset 2). Most notably, we detected a significant reduction in cholesteryl ester levels of CE 16:0 and 18:1 (Fig. 5E) in cells treated with KY02111. CE 16:0 and 18:1 were the two most abundant CE's in HeLa cells, and together, the reduction of these two species alone accounted for an overall change larger than 1 mol% of the whole lipidome. Additional CE species were also detected and affected, albeit at much lower levels and not to a statistically significant level (Fig. S12 ESI†). However, we observed an overall trend in reduction of CE content upon SQS degradation compared to cells treated with DMSO or compound 18 by about 40%. Generally, compound 18 did not have any significant effect on cellular lipid levels (Fig. 5D), which is in line with our in vitro data where we found that 18 did not inhibit enzymatic SQS activity (Fig. S5 ESI†). CE's are the storage form of cholesterol inside cells and are usually produced from acyl-CoA by Sterol-O-acyltransferase (SOAT1).49 As unesterified cholesterol levels are not altered, we believe that the cells compensate for a lower amount of SQS protein and activity not by upregulating cholesterol biosynthesis but by utilizing available cholesteryl esters. This could either occur by hydrolyzing and liberating stored cholesteryl esters or by downregulating ester production. Phosphatidylcholines (PC 32:1) were also altered significantly, which could be increased (≥1 mol%) to compensate for a greater excess of acyl-CoA, since it is not used to produce CE's. We also detected a significant increase in lysophosphatidic acid (LPA 18:0) and lysophosphatidyl glycerol (LPG 16:0). Whether these changes directly relate to KY02111-induced SQS degradation has yet to be determined.
Conclusions
In summary, we unexpectedly identified KY02111 as a degrader of SQS, whereas bifunctional molecules such as 18, which were designed as PROTACs by linking KY02111 to E3 ligase-recruiting ligands, led to an increase in SQS levels by shielding the protein from its natural degradation. KY02111 is a highly selective degrader of SQS, with few additional proteins showing significant changes in expression in response to compound treatment. KY02111-mediated SQS degradation is dose-, time- and proteasome-dependent, however it did not require a cullin-ring ligase, or cause defects in SQS insertion into the ER. SQS degradation could be partially rescued by treatment with an inhibitor of the E3 ligase HRD1, suggesting KY02111 may accelerate the natural turnover of SQS. As such, KY02111 represents a mechanistically new degrader, and the first for SQS. While KY02111 treatment did not reduce free cholesterol, it caused a significant reduction in cholesteryl esters. This makes it a promising tool and potential therapeutic lead in hypercholesterolemia. More generally, this work highlights that SQS degradation is possible despite multiple biological challenges including moderate protein turnover (t1/2 ≈ 5 h) and potential feedback regulation within the cholesterol biosynthesis pathway. This feature has hampered previous attempts to design HMGR PROTACs,50 whereas selectively targeting the sterol sensing domain (SSD) of HMGR with sterol-derivatives yielded HMGR degraders.51 Importantly, SQS lacks a known SSD, impeding the adoption of a similar strategy. In addition to degraders, our newly discovered SQS stabilizer 18 may also have applications in settings where SQS is expressed in low amounts. Recently, Coman et al.5 identified three human patients harboring pathogenic FDFT1 gene variants resulting in dysregulated splicing and transcription with reduced or no expression of SQS. Molecular stabilizers like compound 18 could restore cellular concentrations of SQS without inhibiting enzymatic activity. Overall, the SQS degraders, stabilizers and fluorescent probes reported here, will serve as a toolkit to study the full spectrum of SQS perturbations possible with small molecules.Data availability
Additional experimental details and data are provided in the ESI.† The original MS raw data are deposited to MassIVE with the accession ID MSV000093137. Raw lipidomics data was deposited to the Metabolights server with accession ID MTBLS7990.Author contributions
J. G. F. H. synthesized all compounds, purified recombinant SQS, performed all biophysical and cell based screening, and prepared samples for proteomics and lipidomics. C. R. performed and analysed proteomics experiments and supported cell experiments. M. B. performed lipidomics experiments and analysis. J. C. C. performed EMC experiments and analysis. K. E-R. analyzed NMR data. L. D. supported recombinant protein preparation and structural studies. L. L. designed the project and supervised experiments. J. G. F. H. and L. L. wrote the paper with input from all authors.Conflicts of interest
The authors have no conflicts to declare.Acknowledgements
We thank the Novo Nordisk Foundation (NNF19OC0058183 and NNF21OC0067188) and the Independent Research Fund Denmark (9041-00241B) for funding to L. L. J. C. C. is supported by a Senior Cancer Research Fellowship from Cancer Research UK (C68569/A29217).References
- C. I. Liu, W. Y. Jeng, W. J. Chang, M. F. Shih, T. P. Ko and A. H. J. Wang, Acta Crystallogr., D: Biol. Crystallogr., 2014, 70, 231–241 CrossRef CAS PubMed.
- R. Heidasch, D. Avci, C. Lüchtenborg, D. Kale, H. Beard, T. Mentrup, M. Barniol-Xicota, B. Schröder, S. Verhelst, B. Brügger and M. K. Lemberg, 2022, preprint, DOI:10.1101/2021.07.19.452877.
- Y. Takemoto, S. Kadota, I. Minami, S. Otsuka, S. Okuda, M. Abo, L. L. Punzalan, Y. Shen, Y. Shiba and M. Uesugi, Angew. Chem., Int. Ed., 2021, 60, 21824–21831 CrossRef CAS PubMed.
- S. Nagashima, H. Yagyu, R. Tozawa, F. Tazoe, M. Takahashi, T. Kitamine, D. Yamamuro, K. Sakai, M. Sekiya, H. Okazaki, J. I. Osuga, A. Honda and S. Ishibashi, J. Lipid Res., 2015, 56, 998–1005 CrossRef CAS PubMed.
- D. Coman, L. E. L. M. Vissers, L. G. Riley, M. P. Kwint, R. Hauck, J. Koster, S. Geuer, S. Hopkins, B. Hallinan, L. Sweetman, U. F. H. Engelke, T. A. Burrow, J. Cardinal, J. McGill, A. Inwood, C. Gurnsey, H. R. Waterham, J. Christodoulou, R. A. Wevers and J. Pitt, Am. J. Hum. Genet., 2018, 103, 125–130 CrossRef CAS PubMed.
- M. H. Davidson, Curr. Atheroscler. Rep., 2007, 9, 78–80 CrossRef CAS PubMed.
- M. Ichikawa, M. Ohtsuka, H. Ohki, M. Ota, N. Haginoya, M. Itoh, Y. Shibata, K. Sugita, Y. Ishigai, K. Terayama, A. Kanda and H. Usui, ACS Med. Chem. Lett., 2013, 4, 932–936 CrossRef CAS PubMed.
- T. Ugawa, H. Kakuta, H. Moritani, K. Matsuda, T. Ishihara, M. Yamaguchi, S. Naganuma, Y. Iizumi and H. Shikama, Br. J. Pharmacol., 2000, 131, 63–70 CrossRef CAS PubMed.
- L. Xue, H. Qi, H. Zhang, L. Ding, Q. Huang, D. Zhao, B. J. Wu and X. Li, Front. Oncol., 2020, 10, 1–20 CrossRef PubMed.
- A. P. Kourounakis and E. Bavavea, Arch. Pharm., 2020, 353, 1–8 CrossRef PubMed.
- J. D. Bergstrom, M. M. Kurtz, D. J. Rew, A. M. Amend, J. D. Karkas, R. G. Bostedor, V. S. Bansal, C. Dufresne, F. L. VanMiddlesworth, O. D. Hensens, J. M. Liesch, D. L. Zink, K. E. Wilson, J. Onishi, J. A. Milligan, G. Bills, L. Kaplan, M. N. Omstead and R. G. Jenkins, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 80–84 CrossRef CAS PubMed.
- T. Nishimoto, Y. Amano, R. Tozawa, E. Ishikawa, Y. Imura, H. Yukimasa and Y. Sugiyama, Br. J. Pharmacol., 2003, 139, 911–918 CrossRef CAS.
- J. K. Liao, Circulation, 2011, 123, 1925–1928 CrossRef PubMed.
- A. Mullard, Nat. Rev. Drug Discovery, 2021, 20, 247–250 CrossRef CAS PubMed.
- R. I. Tozawa, S. Ishibashi, J. I. Osuga, H. Yagyu, T. Oka, Z. Chen, K. Ohashi, S. Perrey, F. Shionoiri, N. Yahagi, K. Harada, T. Gotoda, Y. Yazaki and N. Yamada, J. Biol. Chem., 1999, 274, 30843–30848 CrossRef CAS.
- M. Ichikawa, A. Yokomizo, M. Itoh, K. Sugita, H. Usui, H. Shimizu, M. Suzuki, K. Terayama and A. Kanda, Bioorg. Med. Chem., 2011, 19, 1930–1949 CrossRef CAS PubMed.
- D. A. Nalawansha and C. M. Crews, Cell Chem. Biol., 2020, 27, 998–1014 CrossRef CAS PubMed.
- C. Crews, P. Natl and R. Briefly, Nat. Chem. Biol., 2020, 16, 1151 CrossRef.
- C. Steinebach, I. Sosič, S. Lindner, A. Bricelj, F. Kohl, Y. L. D. Ng, M. Monschke, K. G. Wagner, J. Krönke and M. Gütschow, MedChemComm, 2019, 10, 1037–1041 RSC.
- C. Steinebach, S. A. Voell, L. P. Vu, A. Bricelj, I. Sosič, G. Schnakenburg and M. Gütschow, Synth. Ger., 2020, 52, 2521–2527 CrossRef CAS.
- L. M. Luh, U. Scheib, K. Juenemann, L. Wortmann, M. Brands and P. M. Cromm, Angew. Chem., Int. Ed., 2020, 59, 15448–15466 CrossRef CAS.
- J. F. Thompson, D. E. Danley, S. Mazzalupo, P. M. Milos, M. E. Lira and H. J. Harwood, Arch. Biochem. Biophys., 1998, 350, 283–290 CrossRef CAS PubMed.
- D. Stevens, et al., US Pat. US2003O157583A1, 2003.
- N. Volkmar, M.-L. Thezenas, S. M. Louie, S. Juszkiewicz, D. K. Nomura, R. S. Hegde, B. M. Kessler and J. C. Christianson, J. Cell Sci., 2019, 132, jcs223453 Search PubMed.
- M. Békés, D. R. Langley and C. M. Crews, Nat. Rev. Drug Discovery, 2022, 21, 181–200 CrossRef PubMed.
- M. Pettersson and C. M. Crews, Drug Discovery Today Technol., 2019, 31, 15–27 CrossRef PubMed.
- J. Poirson, A. Dhillon, H. Cho, M. H. Y. Lam, N. Alerasool, J. Lacoste, L. Mizan and M. Taipale, bioRxiv, 2022, preprint, DOI:10.1101/2022.08.15.503206.
- A. P. Crew, K. Raina, H. Dong, Y. Qian, J. Wang, D. Vigil, Y. V. Serebrenik, B. D. Hamman, A. Morgan, C. Ferraro, K. Siu, T. K. Neklesa, J. D. Winkler, K. G. Coleman and C. M. Crews, J. Med. Chem., 2018, 61, 583–598 CrossRef CAS PubMed.
- C. Maniaci, S. J. Hughes, A. Testa, W. Chen, D. J. Lamont, S. Rocha, D. R. Alessi, R. Romeo and A. Ciulli, Nat. Commun., 2017, 8, 830 CrossRef PubMed.
- H. L. Ennis, Science, 1964, 146, 1474–1476 CrossRef CAS PubMed.
- M. R. Siegel and H. D. Sisler, Nature, 1963, 200, 675–676 CrossRef CAS.
- F. Reggiori and M. Molinari, Physiol. Rev., 2022, 102, 1393–1448 CrossRef CAS PubMed.
- C. De Leonibus, L. Cinque and C. Settembre, FEBS Lett., 2019, 593, 2319–2329 CrossRef CAS PubMed.
- C. Mayor-Ruiz, M. G. Jaeger, S. Bauer, M. Brand, C. Sin, A. Hanzl, A. C. Mueller, J. Menche and G. E. Winter, Mol. Cell, 2019, 75, 849–858 CrossRef CAS PubMed.
- A. Guna, N. Volkmar, J. C. Christianson and R. S. Hegde, Science, 2018, 359, 470–473 CrossRef CAS PubMed.
- M. Słabicki, H. Yoon, J. Koeppel, L. Nitsch, S. S. Roy Burman, C. Di Genua, K. A. Donovan, A. S. Sperling, M. Hunkeler, J. M. Tsai, R. Sharma, A. Guirguis, C. Zou, P. Chudasama, J. A. Gasser, P. G. Miller, C. Scholl, S. Fröhling, R. P. Nowak, E. S. Fischer and B. L. Ebert, Nature, 2020, 588, 164–168 CrossRef PubMed.
- Y. Ishii, L. Papa, U. Bahadur, Z. Yue, J. Aguirre-ghiso and T. Shioda, Clin. Cancer Res., 2011, 17, 2292–2300 CrossRef CAS.
- S. Brambillasca, M. Yabal, P. Soffientini, S. Stefanovic, M. Makarow, R. S. Hegde and N. Borgese, EMBO J., 2005, 24, 2533–2542 CrossRef CAS.
- M. Irisawa, J. Inoue, N. Ozawa, K. Mori and R. Sato, J. Biol. Chem., 2009, 284, 28995–29004 CrossRef CAS PubMed.
- M. Kikkert, R. Doolman, M. Dai, R. Avner, G. Hassink, S. Van Voorden, S. Thanedar, J. Roitelman, V. Chau and E. Wiertz, J. Biol. Chem., 2004, 279, 3525–3534 CrossRef CAS.
- N. Yagishita, S. Aratani, C. Leach, T. Amano, Y. Yamano, K. Nakatani, K. Nishioka and T. Nakajima, Int. J. Mol. Med., 2012, 30, 1281–1286 CrossRef CAS PubMed.
- T. Schneidewind, A. Brause, B. Schölermann, S. Sievers, A. Pahl, M. G. Sankar, M. Winzker, P. Janning, K. Kumar, S. Ziegler and H. Waldmann, Cell Chem. Biol., 2021, 28, 1780–1794 CrossRef CAS PubMed.
- T. R. Tansey and I. Shechter, Biochim. Biophys. Acta Mol. Cell Biol. Lipids, 2000, 1529, 49–62 CrossRef CAS PubMed.
- Y. Sakakura, H. Shimano, H. Sone, A. Takahashi, K. Inoue, H. Toyoshima, S. Suzuki and N. Yamada, Biochem. Biophys. Res. Commun., 2001, 286, 176–183 CrossRef CAS.
- R. G. Bostedor, J. D. Karkas, B. H. Arison, V. S. Bansal, S. Vaidya, J. I. Germershausen, M. M. Kurtz and J. D. Bergstrom, J. Biol. Chem., 1997, 272, 9197–9203 CrossRef CAS PubMed.
- B. M. Wasko, J. P. Smits, L. W. Shull, D. F. Wiemer and R. J. Hohl, J. Lipid Res., 2011, 52, 1957–1964 CrossRef CAS.
- V. Cantagrel and D. J. Lefeber, J. Inherit. Metab. Dis., 2011, 34, 859–867 CrossRef CAS.
- T. Brandwine, R. Ifrah, T. Bialistoky, R. Zaguri, E. Rhodes-Mordov, L. Mizrahi-Meissonnier, D. Sharon, V. L. Katanaev, O. Gerlitz and B. Minke, Front. Mol. Neurosci., 2021, 14, 693967 CrossRef CAS PubMed.
- T. Y. Chang, C. C. Y. Chang and D. Cheng, Annu. Rev. Biochem., 1997, 66, 613–638 CrossRef CAS.
- M. X. Li, Y. Yang, Q. Zhao, Y. Wu, L. Song, H. Yang, M. He, H. Gao, B. L. Song, J. Luo and Y. Rao, J. Med. Chem., 2020, 63, 4908–4928 CrossRef CAS PubMed.
- S.-Y. Jiang, H. Li, J.-J. Tang, J. Wang, J. Luo, B. Liu, J.-K. Wang, X.-J. Shi, H.-W. Cui, J. Tang, F. Yang, W. Qi, W.-W. Qiu and B.-L. Song, Nat. Commun., 2018, 9, 5138 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc04064j |
| This journal is © The Royal Society of Chemistry 2023 |