
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA single phosphorylation mechanism in early metabolism – the case of phosphoenolpyruvate†
Joris
Zimmermann
a,
Robert J.
Mayer
 a and
Joseph
Moran
*abc
a and
Joseph
Moran
*abc
aInstitut de Science et d'Ingénierie Supramoléculaires (ISIS), CNRS UMR 7006, Université de Strasbourg, 8 Allée Gaspard Monge, 67000 Strasbourg, France
bInstitut Universitaire de France (IUF), France. E-mail: moran@unistra.fr
cDepartment of Chemistry and Biomolecular Sciences, University of Ottawa, Ottawa, Ontario K1N 6N5, Canada
First published on 4th December 2023
Abstract
Phosphorylation is thought to be one of the fundamental reactions for the emergence of metabolism. Nearly all enzymatic phosphorylation reactions in the anabolic core of microbial metabolism act on carboxylates to give acyl phosphates, with a notable exception – the phosphorylation of pyruvate to phosphoenolpyruvate (PEP), which involves an enolate. We wondered whether an ancestral mechanism for the phosphorylation of pyruvate to PEP could also have involved carboxylate phosphorylation rather than the modern enzymatic form. The phosphorylation of pyruvate with P4O10 as a model phosphorylating agent was found to indeed occur via carboxylate phosphorylation, as verified by mechanistic studies using model substrates, time course experiments, liquid and solid-state NMR spectroscopy, and DFT calculations. The in situ generated acyl phosphate subsequently undergoes an intramolecular phosphoryl transfer to yield PEP. A single phosphorylation mechanism acting on carboxylates appears sufficient to initiate metabolic networks that include PEP, strengthening the case that metabolism emerged from self-organized chemistry.
Introduction
Dynamic self-organized systems exhibiting complex behavior are generally thought to arise when a small number of particular rules or mechanisms simultaneously act on a set of substrates. As the core of microbial metabolism features a small number of repeating reaction mechanisms,1 it has been suggested that it may have initiated through chemical self-organization.2–9 Several experimental reports support the idea that core metabolic pathways, including substantial segments of the central (r)TCA cycle and related pathways, could have initiated in self-organized chemistry, possibly as early as in prebiotic chemistry.2,10–22 However, regardless of whether metabolism emerged enzymatically or nonenzymatically, there is no doubt that phosphorylation is one of the crucial mechanisms within these biological pathways. In addition to introducing phosphoryl groups into the molecular backbone of sugars, phosphorylation can activate functional groups toward reactions that would otherwise be energetically unfeasible.23,24The most common type of phosphorylation in the core pathways of microbial anabolism (the acetyl CoA pathway, the rTCA cycle, and gluconeogenesis) is the phosphorylation of carboxylates to acyl phosphates,25–30 in which nucleotide triphosphates such as adenosine triphosphate (ATP) act as the phosphoryl group donor (Fig. 1). However, the second type of phosphorylation in the anabolic core is quite unique – the conversion of pyruvate or oxaloacetate to form phosphoenolpyruvate (PEP), the metabolite with the most energetic phosphate-bond (ΔG0 of hydrolysis = −61.9 kJ mol−1).31 Unlike the other reactions in Fig. 1, this involves the phosphorylation of an enolate (Fig. 1, red box), rather than a carboxylate. In the enzymatic mechanism, the enolate of pyruvate is generated within the active site of the enzyme (stabilized by Mg2+), which subsequently undergoes phosphoryl transfer from a phosphohistidine intermediate to yield PEP.32–34 In the following steps of anabolic pathways, PEP provides all carbon and phosphoryl groups for the sugar-phosphate intermediates of gluconeogenesis and the pentose phosphate pathway, including ribose-5-phosphate, the building block for ribonucleotide biosynthesis. Thus, enzymatic phosphorylation serves not only a bioenergetic role but also a fundamental structural role in biochemistry through PEP. This dual function suggests that phosphorylation reactions and PEP were both present in the earliest metabolic networks that supported the emergence of life.3 However, if metabolism was the result of chemical self-organization, one would expect that a single phosphorylation mechanism would be sufficient to account for the emergence of the core metabolic network, including the formation of PEP.
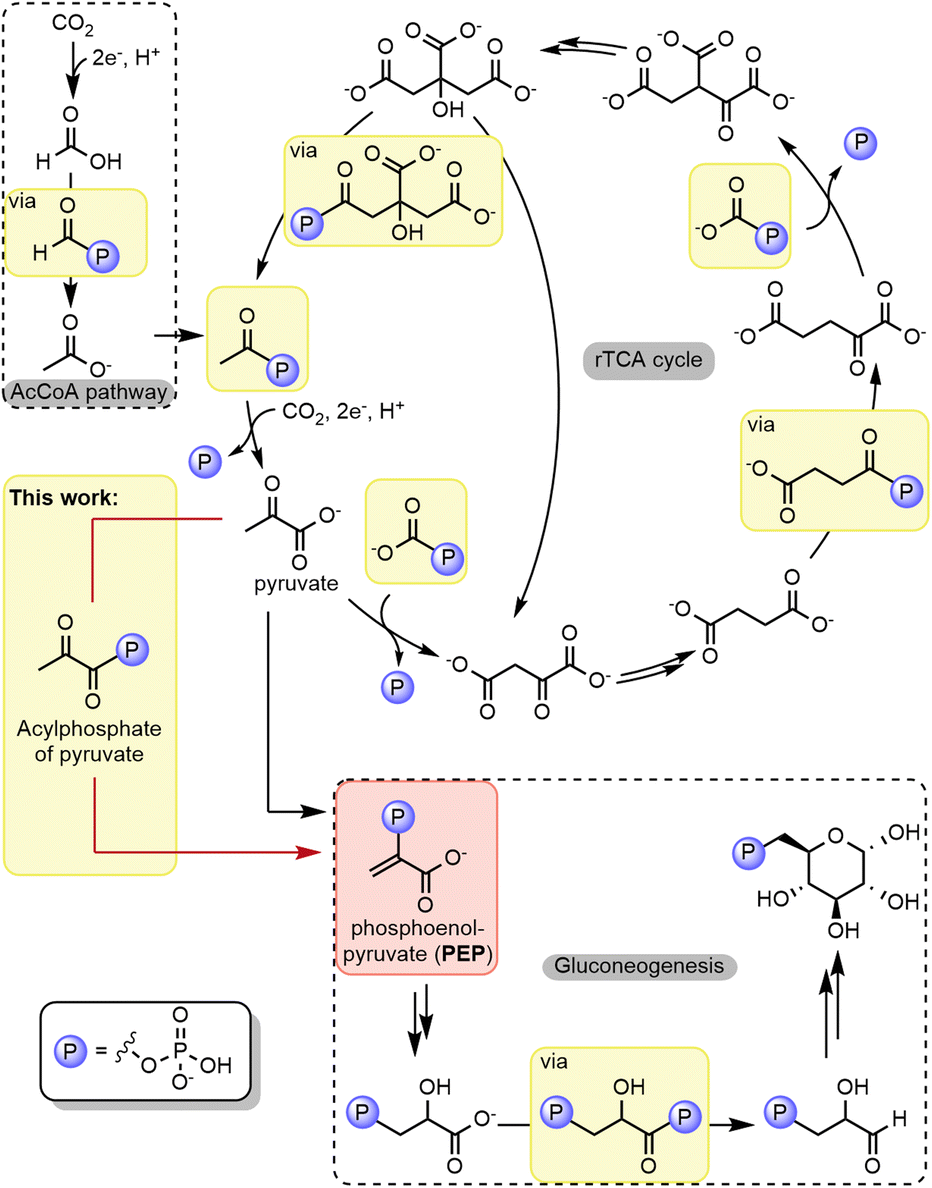 | ||
| Fig. 1 Phosphorylation reactions in core metabolism. Examples of acyl phosphates (highlighted in yellow boxes) involved in different chemical transformations and PEP (highlighted in the red box). | ||
PEP is kinetically stable toward hydrolysis at room temperature but readily hydrolyses at elevated temperatures (pH 1–7) or in the presence of metal ions.35,36 Interestingly, studies on the nonenzymatic hydrolysis of PEP, the reverse reaction of phosphorylation, have concluded that the mechanism proceeds through an acyl phosphate intermediate (Fig. 2).35–37 Initially, the enol phosphate (PEP) was suggested to undergo intramolecular nucleophilic attack by the carboxy group, forming a cyclic pentacoordinate phosphorus intermediate (cPEP·H2O) in equilibrium with the cyclic tetravalent intermediate (cPEP) and the acyl phosphate (AcP-Pyr). Finally, irreversible hydrolysis of the acyclic product leads to the formation of pyruvic acid (Pyr) and inorganic phosphate. It therefore seems plausible that, under the right conditions, phosphorylation of pyruvate to PEP might occur in the reverse way, offering a unified mechanism for all the phosphorylations in core anabolism.
 | ||
| Fig. 2 Nonenzymatic hydrolysis of PEP.37 | ||
We hypothesized that PEP formation via an enolate is the result of biological evolution and that an ancient pathway, whether enzymatic or nonenzymatic, yielded PEP through the direct phosphorylation of the carboxylate of pyruvate followed by intramolecular transfer of the phosphoryl group (Fig. 1, yellow box). Although syntheses of PEP have been reported in various contexts,38–40 the only direct nonenzymatic phosphorylation of pyruvate to PEP was reported by Kiessling using POCl3 in quinoline as a solvent, which, even after further improvements, yielded a maximum of 9% PEP.41,42 However, the mechanism of this reaction is unknown and has never been studied. Considering the energetically highly favorable hydrolysis of PEP, phosphorylation of pyruvate to PEPvia initial carboxylate phosphorylation would be unlikely to happen in bulk water. Indeed, biological phosphorylation and many potentially prebiotic phosphorylation reactions occur in environments where water is largely excluded.43–45 We, therefore, elected to study the mechanism of the Kiessling-type reaction as a model for the potential ancestral enzymatic or nonenzymatic phosphorylation of pyruvate to PEP. Our goal is not to identify prebiotically plausible conditions for such a reaction but simply to determine whether a single phosphorylation mechanism might account for the phosphorylation reactions in core metabolism, strengthening the case for a self-organized metabolic origin.
Results and discussion
Nonenzymatic phosphorylation of pyruvate to PEP, reaction progress, and intermediates
Considering that the free energy of hydrolysis of PEP is 61.9 kJ mol−1,31 the reverse reaction requires a strong phosphate donor to overcome this energy barrier. Although POCl3 was reported to phosphorylate pyruvate,41 we wondered whether simpler oxygen-based phosphorylating agents would be able to achieve the phosphorylation reaction of pyruvate to PEP. After some screening, we found that when Pyr (0.06 mmol) was mixed with one equiv. of P4O10 in paste conditions at 60 °C for 90 minutes, the characteristic 1H NMR signals of PEP were observed after solubilization of the paste in H2O, but only in trace quantities (Table S1, entry 1 and p. S11; also confirmed by mass spectroscopy, see ESI Section VI.A.3 and p. S74†). The pH after solubilization proved to be highly acidic due to the formation of H3PO4 (roughly pH 1). In line with the previous reports of Kiessling and analogous to the formation of the phosphohistidine intermediate found in the biological pathway,32–34 the addition of nitrogenous bases improved the yield of PEP (Table S2 and p. S13†).46 The highest PEP yield (7%) was obtained with the following conditions: P4O10 (1 equiv.), pyridine (2 equiv.), paste conditions, 60 °C, and a reaction time of 1–2 h (Table S3 and p. S23†). P4O10 is known to decompose to various activated phosphate species,47 and to react with pyridine at 70 °C to form bipyridinium phosphates.46,48 Therefore, a series of further phosphorylating agents were evaluated as potential reactive intermediates (Table S4 and p. S24†). Among them, bipyridinium phosphate P2O5L2 (L = pyridine) was found to form PEP in 3% yield after 90 min at 60 °C (Table S4,† entry 9). Polyphosphoric acid gave only traces of PEP (Table S4,† entry 8), and all other polyphosphates were unreactive. The phosphorylation might, therefore, involve a combination of P4O10 and N-phosphorylated adducts as active species.To gain further insight into the mass balance and mechanism, a series of identical reactions were quenched at different time points and examined by 1H and 31P NMR. The three main compounds observed after quenching are shown in Fig. 3A, with a 1H NMR spectrum of the reaction quenched after 3 min depicted in Fig. 3B. The qualitative evolution of the reaction (Table S5 and p. S24†) is shown in Fig. 3C. Immediately after setting up the reaction at 60 °C and quenching, a phosphorylated compound P1-pyr-py is detected prior to significant PEP formation (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of P1-pyr-py/PEP at t = 1 min). Based on the evolution of their relative concentration, P1-pyr-py is likely the key intermediate in the formation of PEP, as it can afford PEP after elimination of HPy+.
:1 ratio of P1-pyr-py/PEP at t = 1 min). Based on the evolution of their relative concentration, P1-pyr-py is likely the key intermediate in the formation of PEP, as it can afford PEP after elimination of HPy+.
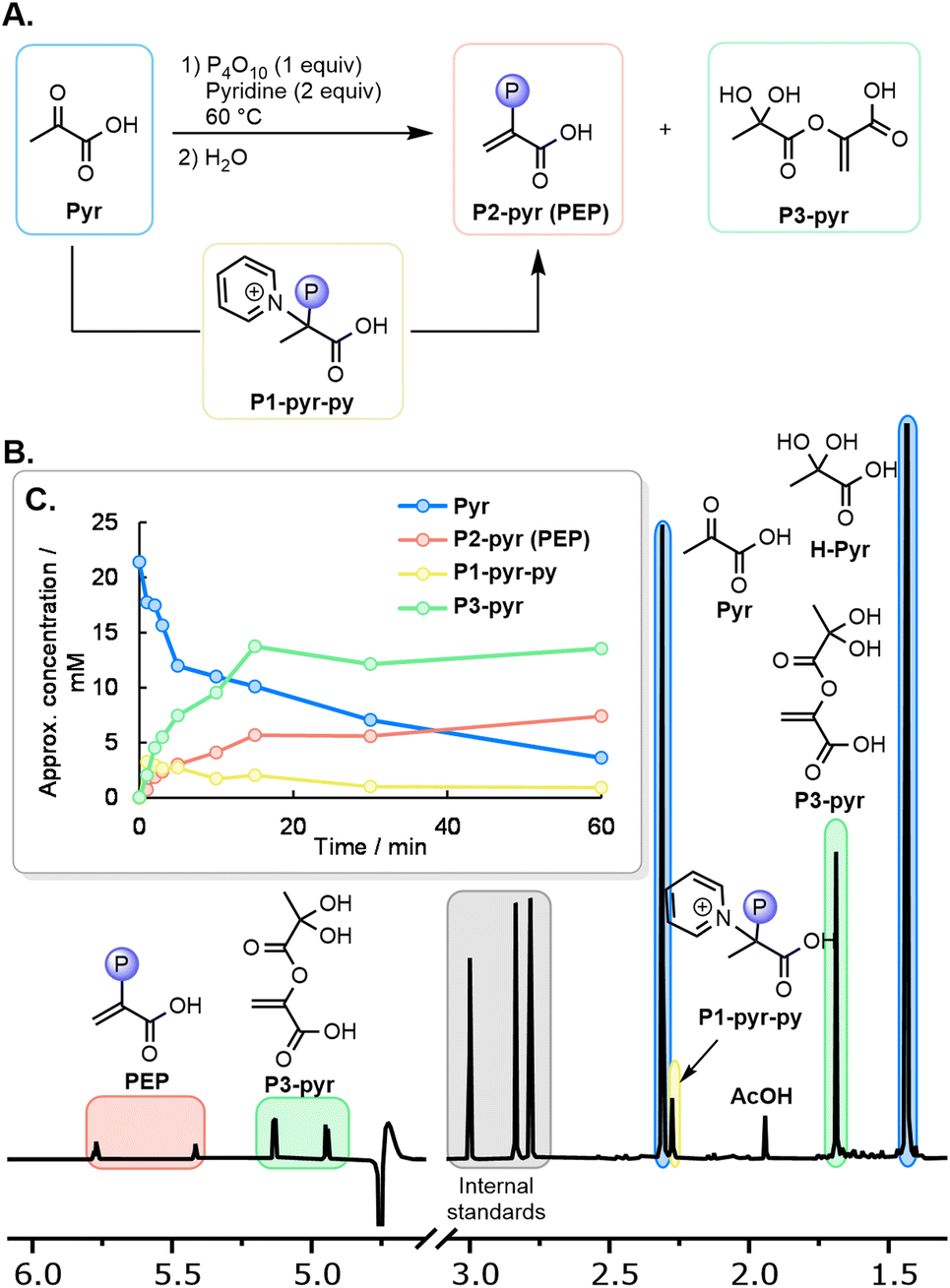 | ||
| Fig. 3 (A) Intermediates and side products in the phosphorylation of pyruvate to PEP with P4O10. (B) Typical 1H NMR spectrum (H2O/D2O 9:1 mixture, 400 MHz) of the quenched reaction mixture after 3 min at 60 °C. (C) Qualitative evolution of the main species in the reaction (for details, see Table S5 and p. S24†). | ||
To determine the structure of P1-pyr-py, its formation was maximized by scaling up and quenching after 2–5 min. A combination of NMR experiments using through-bond (HMBC HSQC, HMQC) and through-space (NOESY, HOESY) correlation techniques, as well as mass spectrometry, allowed P1-pyr-py to be assigned as a phosphorylated covalent adduct of pyruvate and pyridine (ESI, Section VI. A.2 and p. S69†).49 A higher yield of the adduct was observed when 1-methylimidazole (Im) was used as base instead of pyridine (Py), allowing easier characterization (ESI, Section VI. A.1 and p. S61†). Here, P1 refers to the general structure of the phosphorylated covalent adducts formed from a ketoacid and a heterocyclic base (Fig. 3A), whereas P1-pyr-py and P1-pyr-im refer to the specific adducts formed from pyruvate and pyridine (Py) or 1-methylimidazole (Im), respectively. In solution, P1-pyr-py is stable below pH 5, while P1-pyr-im is also stable at basic pH (Table S6 and p. S27†).
Furthermore, the side-product P3-pyr was also identified, which might be formed through the addition of an enol at an activated carboxylic group. To determine the structure of P3-pyr, a similar NMR analysis was performed after suspending the paste in CD3CN, as P3-pyr was observed to be highly soluble under these conditions. The structure of P3-pyr was assigned as a partially enolized dimer of pyruvate (Fig. 3A, ESI, Section VI.A.4 and p. S75†). In water, P3-pyr proved to be stable under acidic conditions but hydrolyzed at elevated temperatures (Table S6 and p. S27†).
Finally, by 1H NMR, pyruvic acid, its hydrate, and acetic acid were also detected (ESI, Section VI.A.5 and p. S79†). Several hydrolysis products of P4O10 were identified by 31P NMR, along with two signals near −5.3 and −4.5 ppm, which were assigned to P1-pyr-py and PEP, respectively (Table S5 and p. S26†).
Paste composition
To differentiate which products are formed directly within the paste versus those that are formed only upon hydrolysis during quenching of the paste with water, solid-state NMR was used to characterize the paste (for details, see ESI, Section VI.B and p. S126†). Magic Angle Spinning NMR (MAS-NMR) allowed the characterization of the paste (phosphorylation conditions with pyridine, 10 min, 60 °C). One-dimensional 1H MAS NMR showed the characteristic 1H NMR signals of PEP and P3-pyr (ESI, Section VI.B.1, p. S127 and S128†), whereas 2D NMR (31P–1H) showed some polyphosphates and pyridinium phosphate (Fig. 4A). Next, the paste (phosphorylation conditions with 1-methylimidazole, 10 min, 60 °C) was analyzed by HR-MAS-NMR spectroscopy. One-dimensional 1H HR-MAS NMR spectra also showed the characteristic 1H NMR signals of PEP and P3-pyr (ESI, Section VI.B.2 and p. S130†). | ||
| Fig. 4 (A) Characterization of the paste by 31P, 1H HETCOR MAS NMR. (B) Characterization of the paste by 1H, 1H, RFDR HR-MAS NMR. | ||
The 2D NMR (1H, 1H RFDR) analysis50 confirmed the presence of PEP and P3-pyr in the paste (Fig. 4B). Additionally, the paste was dissolved in various anhydrous solvents to verify the presence of PEP (Table S13 and p. S131†). In general, the paste was poorly soluble in organic solvents (acetone-d3, CD3CN, CDCl3) and partially soluble in DMSO-d6. Among the different solvents tested, PEP could be detected in CD3CN and DMSO-d6 along with P3-pyr, whereas P1-pyr-py was not detected (Table S13,† entries 2 and 4). Thus, PEP and P3-pyr are formed under the paste conditions and not only after hydrolysis.
Reaction with further substrates
To obtain insights into the mechanistic requirements of the reaction, experiments with additional substrates were performed (Fig. 5, see also Tables S8, S9 and p. S30–S44†). First, reactions of pyruvate derivatives were studied under the established paste conditions after 90 min at 60 °C. The expected phosphoenol products were obtained when one or two –CH3 groups were introduced at the α-position, yielding 9% of P2-1Me and 3% of P2-2Me (21% without base), respectively. Similarly, phenylpyruvic acid was phosphorylated to P2-Ph in 15% yield. Next, α-ketoacids bearing an additional carboxy group were tested. For α-ketoglutarate, P2-glu was only obtained in 0.6% yield; the main phosphorylated product was assigned as the cyclic species P2′-glu (Fig. 6A, 3%). Concerning oxaloacetic acid, the phosphoenol product was obtained in 3% yield (P2-ox, Fig. 5) under base-free conditions in order to avoid decarboxylation of the substrate to pyruvate. Under standard conditions, PEP is also obtained from oxaloacetic acid (Table S9, entry 4 and p. S44†).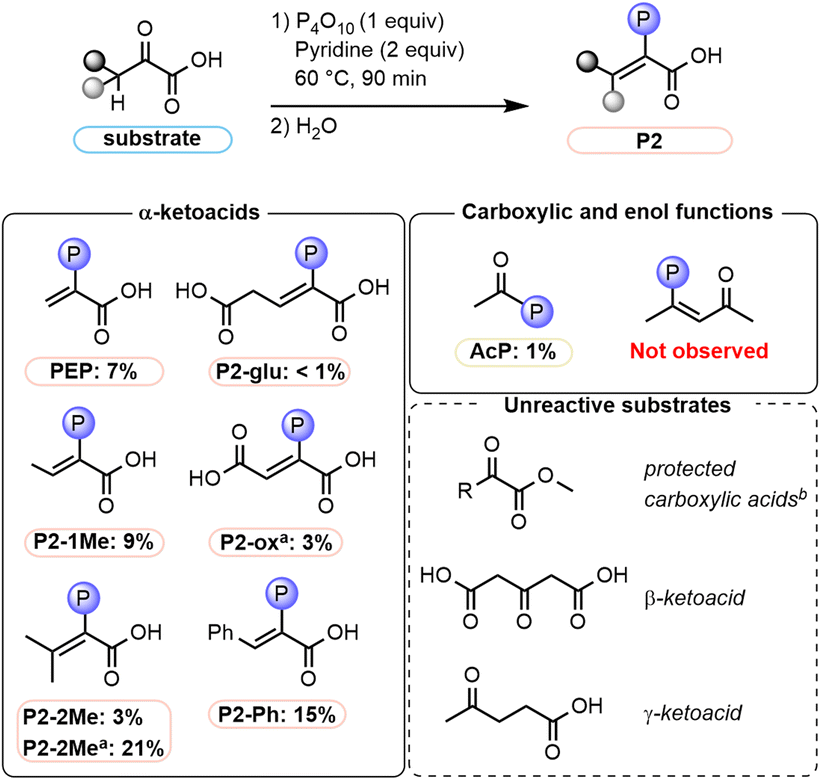 | ||
| Fig. 5 Substrate scope (Table S9†). aThe reaction was run without pyridine. bThe methyl or ethyl esters of pyruvate, phenylpyruvate, oxaloacetate, and α-ketoglutarate were tested (for details, see Table S8, entries 11–14 and p. S31†). | ||
 | ||
| Fig. 6 Structures of intermediates and side products observed in various phosphorylation reactions. (A) Side product arising from α-ketoglutarate. (B) Structures of observed P1-type adducts arising from the reactions of pyruvate (Pyr) with pyridine (Py), Pyr with 1-methylimidazole (Im), and trimethylpyruvic acid (Pyr-3Me) with Im. | ||
To gain insight into whether the carboxylate or enol components of pyruvate are initially phosphorylated, we first submitted two representative model substrates to the reaction conditions. Acetic acid, which features a free carboxylic acid, was phosphorylated to acetyl phosphate AcP (1% yield). In contrast, acetylacetone, which exists to a large extent in its enol form (Table S14, entry 4, p. S135 and S136†), did not produce any enol phosphate under the same reaction conditions. We next investigated the importance of the free carboxylic acid function of α-ketoacids for the phosphorylation reaction. When the methyl or ethyl esters of various ketoacids evaluated in Fig. 5 (pyruvate, phenylpyruvate, oxaloacetate, and α-ketoglutarate) were subjected to the reaction conditions, no phosphorylated products were observed, highlighting the importance of a free carboxylic acid group (Table S8, entries 11–14, p. S31†). As the methyl ester of phenylpyruvate predominantly exists in the enol form (Table S14, entry 5, p. S135 and S137†), this result further suggests that the enol is not phosphorylated directly. Moreover, the influence of the distance between the keto and carboxylic acid moieties was investigated. No products were observed with the β-ketoacid 1,3-acetonedicarboxylic acid or with the γ-ketoacid levulinic acid (Table S8, entries 6–8 and p. S31†), hinting at an intramolecular phosphoryl transfer.
To rationalize the differences in reactivity between α-dimethylpyruvic acid (21% yield of P2-2Me in base-free conditions) and pyruvic acid (traces of PEP in base-free conditions), we compared the reaction mixtures of each substrate after quenching at a short reaction time (3–5 min). Unlike pyruvic acid, even in the presence of pyridine, α-dimethylpyruvic acid did not produce any detectable P1-type adducts (Table S8,† entry 2). Instead, the enol phosphate product was immediately observed. The lack of P1 correlates with the ketoacid's propensity to enolize, with α-dimethylpyruvic acid being more acidic at the α-position.51 Similarly, phenylpyruvic acid produced P2-Ph without any detectable P1-type intermediate (Table S10 and p. S45†). The nucleophilic addition of the base on the ketone is, therefore, less pronounced for substrates that are readily enolizable.
We next explored the influence of the base on the formation and the stability of the P1 adducts. When the reaction was run using Py, P1-pyr-py was formed immediately (Fig. 6B), and its transformation into PEP was almost complete after 1 h (cf.Fig. 3C, Table S5 and p. S24†). In contrast, when Im was used as base, P1-pyr-im was observed in 4% yield (Fig. 6B), but only trace amounts of PEP were observed over the same 1 h period (Table S7, entries 1–6 and p. S29†).
Thus, the phosphorylated adducts generated with different bases (i.e., P1-pyr-py and P1-pyr-im) display different propensities to eliminate, with the formation of PEP from P1-pyr-py being more favorable than from P1-pyr-im. Indeed, increasing the amount of Im used in the reaction (from 2 to 4 equiv.), increased the yield of P1-pyr-im and PEP to 13% and 1.5%, respectively (Table S7, entry 5 and p. S29†), whereas increasing the amount of Py (from 2 to 5 to 10) decreased the yields of both compounds (Table S3, entries 7–8, p. S23†). Even when a stronger base like triethylamine TEA was added to the system 10 min after the formation of P1-pyr-im, the elimination of Im from P1-pyr-im to PEP did not occur (Table S7,† entry 11). Overall, Im appears to be a poorer leaving group than Py for subsequent elimination to the enol phosphate and consequently forms a more stable P1 adduct. Additionally, when the reaction was run with trimethylpyruvic acid Pyr-3Me, a sterically hindered nonenolizable pyruvic acid derivative, the formation of a P1 adduct was only observed when Im was used as base (Fig. 6B, P1-(pyr-3Me)-im; Table S8,† entry 3).
Finally, the reaction of Pyr with P4O10 was studied using triethylamine TEA, a stronger base. In this case, the formation of the phosphorylated intermediate P1 was not observed even at the beginning of the reaction, but PEP was detected directly (Table S7,† entries 15–18). Consequently, the reaction with TEA might not occur through the same mechanism as with pyridine and 1-methylimidazole.
Proposed mechanism
Based on the above observations, a simplified reaction scheme was constructed, which involves two possible pathways for the phosphorylation of α-ketoacids (Fig. 7). Pathway I involves the phosphorylation of the carboxy group and the addition of the base to the ketone to generate the acyl phosphate intermediate Int. A (step I1), which features a free hydroxy group. Subsequent intramolecular phosphoryl transfer to the hydroxy group generates P1 (step I2). P2 is generated by elimination of the base, as seen in the case of pyridine (step I3). The formation of side product P3-pyr can result from the attack of the enol tautomer of pyruvate on the acyl phosphate intermediate (e.g., Int. A). Pathway II involves phosphorylation of the carboxy group to generate the acyl-phosphate intermediate Int. B (step II1), which features a free enol group. | ||
| Fig. 7 Simplified reaction scheme for the phosphorylation of α-ketoacids to products P2. | ||
Subsequently, intramolecular phosphoryl transfer to the enol group generates P2 (step II2). This pathway might be operating in cases where the enol phosphate is formed in the absence of a base. It also seems plausible for α-ketoacids whose enolization is facile, or for reactions run with bases that are unable to add to the keto group (e.g., TEA). The possibility of producing enol phosphates through two different pathways can explain why higher yields are observed for readily enolizable substrates. To verify whether ketoacids with higher enol content are more rapidly phosphorylated, a competition experiment was performed between pyruvic acid and phenylpyruvic acid (Table S8, entry 5 and p. S30†), the latter being more easily enolizable.52,53 Starting from a 1:1 mixture of the two substrates in the presence of pyridine, a product ratio of 20:1 was observed in favor of the enol-phosphate derived from phenylpyruvic acid when the reaction was quenched after 3–5 min. So far, the acyl phosphate intermediate (Int. A or Int. B) has been assumed to be formed in situ, but no direct proof for their formation could be obtained.
Phosphoryl transfer mechanism – alternative synthesis of PEP in CD3CN
To investigate the proposed intramolecular phosphoryl transfer mechanism, we set out to study conditions favoring the formation of the acyl phosphate of pyruvate (AcP-Pyr). To this end, pyruvoyl chloride (Pyr-Cl), featuring an activated carbonyl group, was used instead of Pyr with the corresponding phosphate nucleophile H3PO4. Importantly, in the presence of Py, PEP, P1-pyr-py, and P3-pyr were all observed after 5 min at 60 °C in neat conditions, indicating the similar reactivity of our model system with the standard conditions using Pyr (Table S11 and p. S48†). Next, we investigated the reactivity of AcP-Pyr in homogenous anhydrous conditions. Tetrabutylammonium dihydrogen phosphate (TBAP) was used instead of H3PO4 to avoid water contamination, and TEA was preferred over Py to avoid precipitation (Table S12 and p. S52†).When TEA (0.12 mmol) and TBAP (0.06 mmol) were premixed in CD3CN and Pyr-Cl (0.06 mmol) was added to the mixture, 3% yield of PEP was obtained via its cyclic form cPEP (Fig. 8, Pathway A0, ESI, Section V.F.5 and p. S58†) along with 1% of P3-pyr. To identify the formation of the acyl phosphate intermediate, stepwise experiments were performed. When mixing TBAP and Pyr-Cl in CD3CN, the formation of AcP-Pyr was detected by 1H and 31P NMR along with the formation of pyrophosphate (Fig. 8, Pathway A1; Table S12, entry 5 and p. S52†). In the absence of a base, AcP-Pyr was converted to pyrophosphate and triphosphate in roughly 30 min without formation of PEP (Fig. 8, Pathway A2, Table S12, entry 5 and p. S54–S55†). When trace amounts of H2O were present in the mixture, AcP-Pyr underwent intramolecular phosphoryl transfer to form the corresponding hydroxyphosphate P1-pyr-OH (Fig. 8, Pathway A2′, ESI, Section V.F.4 and p. S57–S58†), accompanied by traces of PEP. When the base TEA (2 equiv.) was added after a reaction time of 5 min, trace amounts (<1%) of PEP along with multiple polyphosphate species were observed (Fig. 8, Pathway A3; Table S12, entry 6 and p. S56†). The low yield in PEP can be explained by the formation of the P1-pyr-OH side product prior to the addition of base. These experiments show that the formation of PEP from AcP-Pyr (through enolization and intramolecular phosphoryl transfer) requires the presence of a base and is in competition with the formation of polyphosphates and the side product P1-pyr-OH (i.e., by a nucleophilic attack of a phosphate or a water molecule on the activated carboxylic acid or on the carbonyl group of AcP-Pyr, respectively).
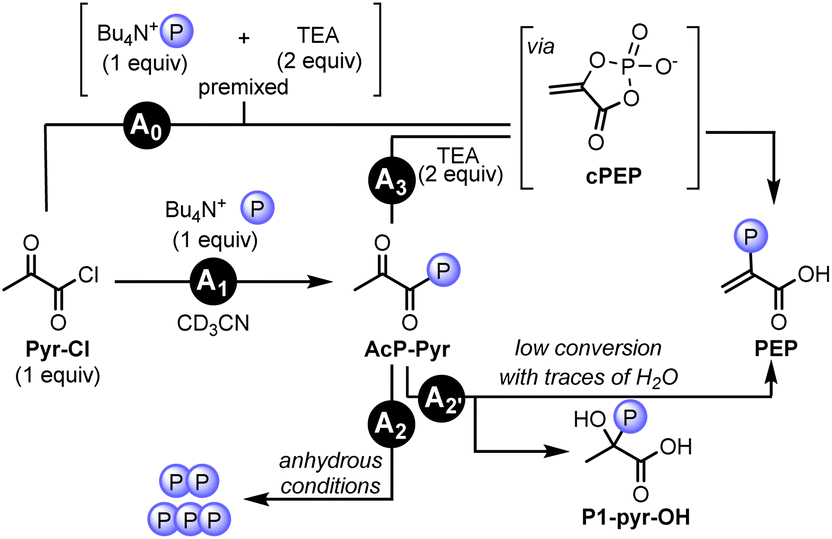 | ||
| Fig. 8 Study of the phosphoryl transfer in aprotic solvent (CD3CN). Reaction A0: reaction of premixed base and phosphate with Pyr-Cl leading to the formation of PEP through its cyclic form cPEP. Reaction A1: the activated carboxylic acid Pyr-Cl is transformed to the corresponding acyl-phosphate (AcP-Pyr), leading to the formation of polyphosphates in anhydrous conditions (A2) or to the formation of PEP by either subsequent addition of TEA (A3) or with trace amount of H2O (along with the formation of the side product P1-pyr-OH (A2′)). | ||
DFT study of the proposed pathways
DFT calculations were performed to evaluate some key aspects of the reaction mechanism. First, we examined the ability of different sites within pyruvate to undergo direct phosphorylation by phosphoryl pyridine (Fig. 9A). N-Phosphoryl pyridine was chosen as the model phosphorylating agent based on our NMR experiments (cf.Fig. 4) and the recent studies of Cummins.46 As shown in Fig. 9A, the computed barrier for the phosphorylation of the carboxylic acid moiety is approx. 60 kJ mol−1 lower than the barrier for the direct phosphorylation of the enol, in line with our experimental observations. Next, we computed the thermochemistry of the overall phosphorylation sequence (Fig. S7 and p. S146†) by analyzing the intermediates identified in aqueous solution (Fig. 9B, P1-pyr-im and P1-pyr-py), along with several possible intermediates (Fig. 9B, Int. A–C and P1-pyr-OH). As expected, without an activated phosphorylating agent, direct phosphorylation of pyruvic acid to AcP-Pyr with phosphoric acid is endergonic by 42.9 kJ mol−1 (ESI, Fig. S7 and p. S146†). From AcP-Pyr, the intramolecular phosphoryl transfer was studied by considering three main pathways by going through nucleophilic addition of the base (Int. A), enolization (Int. B), and addition of H2O (Int. C, Fig. 9B). The latter being unlikely to occur in our paste conditions due to the presence of P4O10, which also acts as a dehydrating agent. Nucleophilic addition of 1-methylimidazole (Im) to the AcP-Pyr ketone was calculated to be favorable by 10.0 kJ mol−1 (Int. A1), whereas the addition of pyridine (Py) or water is slightly disfavored by 15.1 (Int. A2) and 7.5 kJ mol−1 (Int. C), respectively. However, the subsequent intramolecular phosphoryl transfers from the imidazolium hemiaminal (Int. A1), pyridinium hemiaminal (Int. A2), enol (Int. B), or hydrate (Int. C) intermediates derived from AcP-Pyr were all calculated to be exergonic by 16.7, 9.4, 14.6, and 28.6 kJ mol−1, respectively. In line with the experimental observation of intermediates P1, the nucleophile addition is favorable for Im, and subsequent intramolecular phosphoryl transfers from the carboxylate to the enol or alcohol are thermochemically favorable. Interestingly, a pentacoordinate P intermediate (cf.Fig. 2, cPEP·H2O) was not found to be a stable minimum but rather corresponds to the transition state of phosphoryl transfer. However, the computed barrier for the concerted transfer is relatively high, with ≈129–146 kJ mol−1 using explicit molecules of water as a model base for the deprotonation of the enolate and a hydronium ion as a model acid for the protonation of the carboxylate. As the employed computational method is a drastic oversimplification, the absolute numbers of this transformation should only be taken as a very crude estimate. Under the reaction conditions, any other acid or base may promote this transformation, which could result in significantly lower barriers for this reaction. Additionally, the paste conditions will further lower the effective barrier due to the significantly higher concentrations, while the computations were performed for a standard state of 298 K and a concentration of 1 mol L−1 in all reactants. Nevertheless, given that the hydrolysis of PEP to pyruvic acid and phosphoric acid is significantly exergonic (exp.: −61.9 kJ mol−1, comput.: −48.2 kJ mol−1, see Fig. S7 and p. S146†) and irreversible at room temperature, and yet PEP remains relatively stable toward hydrolysis, a high barrier for PEP formation/hydrolysis is to be expected. The thermochemistry for the formation of PEP by the elimination of the tetrahedral intermediates derived from N-methylimidazole (P1-pyr-im) and pyridine (P1-pyr-py) varied significantly (+32.0 vs. −0.4 kJ mol−1), which is in line with the observed resistance of P1-pyr-im to elimination. Overall, DFT calculations confirmed several key aspects of the mechanism: (1) phosphorylation of the carboxylate is favored over that of the enol moiety of pyruvate, (2) AcP-Pyr has a high tendency to undergo nucleophilic addition, (3) the intramolecular phosphoryl transfer is thermochemically favored, and (4) the efficiency of PEP formation by elimination differs for the employed base.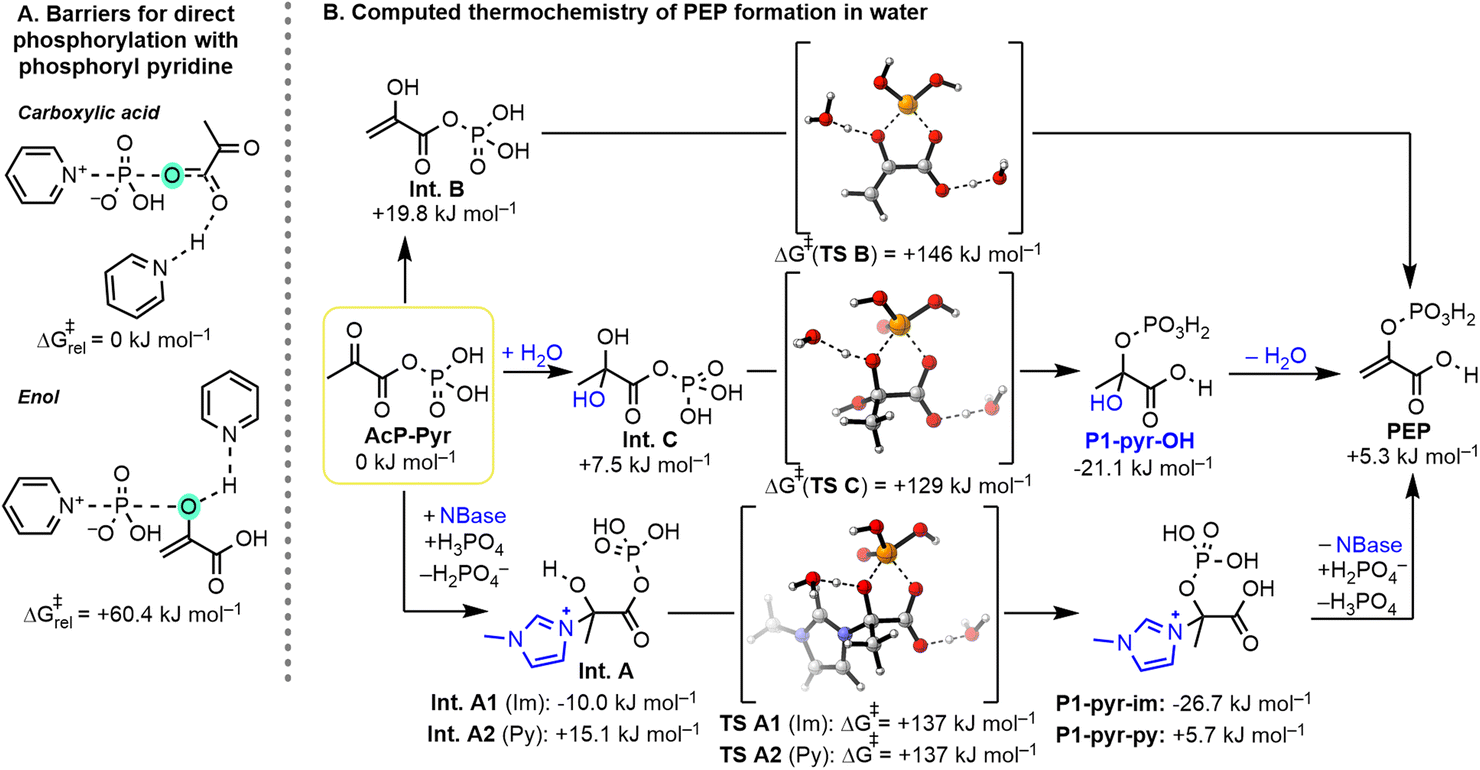 | ||
| Fig. 9 (A) Computational study comparing the phosphorylation of pyruvate at its carboxylate vs. its enol. (B) Computational study showing the thermochemistry of key steps and the barriers for intramolecular phosphoryl transfer. Calculations were performed at the SMD(H2O)M06-2X/def2-TZVPD//SMD(H2O)/M06-2X/def2-SVP level of theory. | ||
Conclusion
The phosphorylation of pyruvate to PEP with P4O10 as a model phosphorylating agent was studied under dry-paste conditions in the presence of nitrogenous bases. After quenching the paste at the very beginning of the reaction, a phosphorylated intermediate P1 was detected. When analyzing the reaction after longer reaction times, PEP was observed along with the side product P3-pyr (Fig. 3). Solid-state NMR spectroscopy gave insights into the composition of the paste, and PEP and P3-pyr were directly observed among polyphosphates and pyridinium phosphates (Fig. 4). The study of several analogous substrates was used to elucidate the mechanism of phosphorylation (Fig. 5). The mechanism strictly requires a free carboxylic acid function, a ketone located alpha to the carboxylic acid, and is more efficient with substrates that are easily enolizable (e.g., phenylpyruvate). Based on these observations, we proposed a mechanism for the phosphorylation of pyruvate to PEP. The carboxy function of pyruvate is initially phosphorylated to an acyl phosphate, which undergoes intramolecular phosphoryl transfer to yield PEP (Fig. 7). Evidence for this mechanism and the formation of an acyl phosphate was obtained by synthesizing PEP starting from pyruvoyl chloride Pyr-Cl in acetonitrile-d3 as a model for aprotic conditions (Fig. 8). Under these conditions, the formation of PEP requires the presence of a base so that intramolecular phosphoryl transfer outcompetes the decomposition of the acyl phosphate intermediate to di- and triphosphates. DFT calculations further supported the mechanistic interpretations. Phosphorylation of the carboxy group was computed to be 60 kJ mol−1 more favorable than phosphorylation of the enol moiety of pyruvate (Fig. 9A). All intramolecular phosphoryl transfers were calculated to be exergonic, although subsequent eliminations of water or base to give the enol phosphate product were found to be mostly endergonic. However, the phosphoryl transfer proceeds through a high-lying transition state, which explains the stability of PEP to hydrolysis (Fig. 9B, Fig. 2). Thus, all phosphorylated core metabolites, including PEP, could potentially be accessible through a single type of phosphorylation mechanism – the phosphorylation of carboxylates. Although we do not claim that the chemistry studied here was carried out under potentially prebiotic conditions, this work nonetheless substantially strengthens arguments for an emergence of metabolism based on chemical self-organization from a small number of simple reaction mechanisms that include phosphorylation, whether it was enzymatic or nonenzymatic.Thinking about the earliest forms of leaving groups and bioenergetics during the evolution of metabolism, De Duve proposed that primitive forms of the metabolic network did not initially rely on the phosphorylation of carboxylates but rather on the direct formation of thioesters from carboxylates, a proposal commonly referred to as the “Thioester World” hypothesis.54–56 Martin and Russell suggested that acetyl phosphate, formed from phosphate and an in situ-generated acetyl thioester during prebiotic CO2 fixation, might have served as a primordial phosphorylating agent for the rest of protometabolism.3 This would make the transition from bioenergetics based on thioesters and acyl phosphates to those based on ATP straightforward since the nonenzymatic phosphorylation of AMP and ADP to ATP by acetyl phosphate is already described.57–60 However, nonenzymatic phosphoryl transfer from acetyl phosphate to other carboxylates is not known. Alternatively, if one assumes that carboxylates could be phosphorylated under prebiotic conditions without invoking thioesters, and one furthermore considers that thiols are essentially catalysts for the anabolic reactions occurring through acyl phosphates (i.e., that these reactions could have originally occurred directly from the acyl phosphate), then invoking thioesters at the earliest stage of protometabolism would not be necessary and the transition to ATP as phosphoryl donor would remain equally straightforward. Several additional lines of reasoning further support acyl phosphates predating thioesters in protometabolism. First, thiols, like phosphates, play an important bioenergetic role throughout metabolism and act as leaving groups, but unlike phosphates, they are not found in the backbone structures of the intermediates of sugar and ribonucleotide biosynthesis. Second, within the anabolic pathways of core metabolism, if one looks beyond the temporary leaving groups used (i.e., acyl phosphates and thioesters), phosphorous is permanently introduced into the structural framework of metabolites much earlier than is sulfur. Phosphate enters directly from pyruvate as PEP during the first step of gluconeogenesis, whereas sulfur enters as sulfide during cysteine biosynthesis, several steps further downstream. Unlike PEP, cysteine is not essential for ribonucleotide biosynthesis. Third, phosphate is geochemically more widely available than simple organic thiols.61–64 Furthermore, whereas acyl phosphates do not contain sulfur, thiol-based leaving groups in metabolism, such as CoA, generally contain phosphoryl groups. Fourth, in water, the formation of acyl phosphates from carboxylates is arguably chemically simpler than the conversion of carboxylates to thioesters – the former requires only a single component (an activated phosphate), whereas the latter requires two (a thiol and an activating agent for the carboxylate). All these points suggest that an early stage of protometabolism was more likely an “Acyl Phosphate World” than a “Thioester World”. Consequently, nonenzymatic carboxylate phosphorylation should be a target of the highest priority for prebiotic chemistry using life as a guide.5
Data availability
Everything is in the ESI.†Author contributions
JZ performed the experiments and wrote the first draft of the paper. RJM performed the DFT calculations. JM conceptualized the project and acquired funding. All authors edited the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Interdisciplinary Thematic Institute ITI-CSC via the IdEx Unistra (ANR-10-IDEX-0002) within the program Investissement d’Avenir. J. Z. thanks the French government for an MRT fellowship. R. J. M. thanks the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) for a fellowship (MA 9687/1-1). This project has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program (grant agreement no. 101001752). J. M. thanks the VW Foundation (no. 96_742) for generous support. Computations were performed at the High-Performance Computing Center of the University of Strasbourg. The authors would like to particulary thank Dr Jesus Raya for his help and expertise in solid-state NMR analyses. Dr Maurice Coppe, and Dr Bruno Vincent are thanked for assistance with NMR analyses. Wahnyalo Kazöne is thanked for MS analyses. Dr Quentin Dherbassy and Emilie Werner are thanked for helpful discussions.Notes and references
- M. C. Weiss, F. L. Sousa, N. Mrnjavac, S. Neukirchen, M. Roettger, S. Nelson-Sathi and W. F. Martin, Nat. Microbiol., 2016, 1, 16116 CrossRef CAS PubMed.
- G. Wächtershäuser, Microbiol. Rev., 1988, 52, 452–484 CrossRef PubMed.
- W. Martin and M. J. Russell, Philos. Trans. R. Soc., B, 2007, 362, 1887–1926 CrossRef CAS.
- S. D. Copley, E. Smith and H. J. Morowitz, Bioorg. Chem., 2007, 35, 430–443 CrossRef CAS PubMed.
- S. A. Harrison and N. Lane, Nat. Commun., 2018, 9, 5176 CrossRef CAS PubMed.
- M. Ralser, Biochem. J., 2018, 475, 2577–2592 CrossRef CAS PubMed.
- J. C. Fontecilla-Camps, Angew. Chem., Int. Ed., 2019, 58, 42–48 CrossRef CAS PubMed.
- K. B. Muchowska, S. J. Varma and J. Moran, Chem. Rev., 2020, 120, 7708–7744 CrossRef CAS PubMed.
- W. F. Martin, Front. Microbiol., 2020, 11, 817 CrossRef PubMed.
- M. A. Keller, A. Zylstra, C. Castro, A. V. Turchyn, J. L. Griffin and M. Ralser, Sci. Adv., 2016, 2, e1501235 CrossRef PubMed.
- C. B. Messner, P. C. Driscoll, G. Piedrafita, M. F. L. D. Volder and M. Ralser, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 7403–7407 CrossRef CAS PubMed.
- P. Laurino and D. S. Tawfik, Angew. Chem., Int. Ed., 2017, 56, 343–345 CrossRef CAS PubMed.
- M. Akouche, M. Jaber, M. Maurel, J. Lambert and T. Georgelin, Angew. Chem., Int. Ed., 2017, 56, 7920–7923 CrossRef CAS PubMed.
- K. B. Muchowska, S. J. Varma, E. Chevallot-Beroux, L. Lethuillier-Karl, G. Li and J. Moran, Nat. Ecol. Evol., 2017, 1, 1716–1721 CrossRef PubMed.
- S. J. Varma, K. B. Muchowska, P. Chatelain and J. Moran, Nat. Ecol. Evol., 2018, 2, 1019–1024 CrossRef PubMed.
- G. Springsteen, J. R. Yerabolu, J. Nelson, C. J. Rhea and R. Krishnamurthy, Nat. Commun., 2018, 9, 91 CrossRef PubMed.
- K. B. Muchowska, S. J. Varma and J. Moran, Nature, 2019, 569, 104–107 CrossRef CAS PubMed.
- L. M. Barge, E. Flores, M. M. Baum, D. G. VanderVelde and M. J. Russell, Proc. Natl. Acad. Sci. U.S.A., 2019, 116, 4828–4833 CrossRef CAS PubMed.
- M. Preiner, K. Igarashi, K. B. Muchowska, M. Yu, S. J. Varma, K. Kleinermanns, M. K. Nobu, Y. Kamagata, H. Tüysüz, J. Moran and W. F. Martin, Nat. Ecol. Evol., 2020, 4, 534–542 CrossRef PubMed.
- R. J. Mayer, H. Kaur, S. A. Rauscher and J. Moran, J. Am. Chem. Soc., 2021, 143, 19099–19111 CrossRef CAS PubMed.
- J. Yi, H. Kaur, W. Kazöne, S. A. Rauscher, L. Gravillier, K. B. Muchowska and J. Moran, Angew. Chem., Int. Ed., 2022, 61, e202117211 CrossRef CAS PubMed.
- S. A. Rauscher and J. Moran, Angew. Chem., Int. Ed., 2022, 134, e202212932 CrossRef.
- F. H. Westheimer, Science, 1987, 235, 1173–1178 CrossRef CAS PubMed.
- S. C. L. Kamerlin, P. K. Sharma, R. B. Prasad and A. Warshel, Q. Rev. Biophys., 2013, 46, 1–132 CrossRef CAS PubMed.
- For formyl phosphate, see: M. R. Mejillano, H. Jahansouz, T. O. Matsunaga, G. L. Kenyon and R. H. Himes, Biochemistry, 1989, 28, 5136–5145 CrossRef CAS PubMed.
- For acetyl phosphate, see: (a) T. D. K. Brown, M. C. Jones-Mortimer and H. L. Kornberg, J. Gen. Microbiol., 1977, 102, 327–336 CrossRef CAS PubMed; (b) B. M. Prüβ and A. J. Wolfe, Mol. Microbiol., 1994, 12, 973–984 CrossRef PubMed.
- For carboxy phosphate, see: (a) P. V. Attwood, Int. J. Biochem. Cell Biol., 1995, 27, 231–249 CrossRef CAS PubMed; (b) P. V. Attwood and J. C. Wallace, Acc. Chem. Res., 2002, 35, 113–120 CrossRef CAS PubMed.
- For succinyl phosphate, see: (a) J. S. Nishimura and A. Meister, Biochem, 1965, 4, 1457–1462 CrossRef CAS; (b) J. S. Nishimura, Biochemistry, 1967, 6, 1094–1099 CrossRef CAS PubMed; (c) F. L. Grinnell and J. S. Nishimura, Biochemistry, 1969, 8, 568–574 CrossRef CAS PubMed.
- For the acyl phosphate of citrate, see: (a) T. Kanao, T. Fukui, H. Atomi and T. Imanaka, Eur. J. Biochem., 2002, 269, 3409–3416 CrossRef CAS PubMed; (b) F. Fan, H. J. Williams, J. G. Boyer, T. L. Graham, H. Zhao, R. Lehr, H. Qi, B. Schwartz, F. M. Raushel and T. D. Meek, Biochemistry, 2012, 51, 5198–5211 CrossRef CAS PubMed.
- For the the acyl phosphate of 1,3-bisphosphoglycerate, see: C. C. F. Blake and D. W. Rice, Philos. Trans. R. Soc. London, 1981, 293, 93–104 CAS.
- A. L. Lehninger, D. L. Nelson and M. M. Cox, Lehninger Principles of Biochemistry, Macmillan, 2005 Search PubMed.
- R. K. Gupta, R. M. Oesterling and A. S. Mildvan, Biochemistry, 1976, 15, 2881–2887 CrossRef CAS PubMed.
- O. Herzberg, C. C. Chen, G. Kapadia, M. McGuire, L. J. Carroll, S. J. Noh and D. Dunaway-Mariano, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 2652–2657 CrossRef CAS PubMed.
- N. E. McCormick and D. L. Jakeman, Biochem. Cell Biol., 2015, 93, 236–240 CrossRef CAS PubMed.
- S. J. Benkovic and K. J. Schray, Biochemistry, 1968, 7, 4097–4102 CrossRef CAS PubMed.
- S. J. Benkovic and K. J. Schray, Biochemistry, 1968, 7, 4090–4096 CrossRef CAS PubMed.
- S. J. Benkovic and K. J. Schray, J. Am. Chem. Soc., 1971, 93, 2522–2529 CrossRef CAS PubMed.
- L. S. White, E. J. Hellman and L. Que, J. Org. Chem., 1982, 47, 3765–3766 CrossRef.
- Mitsubishi Chemical Group Corporation, Jpn Pat., JP2022/138254A, 2022 Search PubMed.
- A. J. Coggins and M. W. Powner, Nat. Chem., 2017, 9, 310–317 CrossRef CAS.
- (a) W. Kiessling, Ber. Dtsch. Chem. Ges. B, 1935, 68, 597–601 CrossRef; (b) W. Kiessling, Ber. Dtsch. Chem. Ges., 1936, 69, 2331–2332 CrossRef; (c) G. Schmidt and S. J. Thannhauser, J. Biol. Chem., 1943, 149, 369–385 CrossRef CAS.
- F. W. Lichtenthaler, Chem. Rev., 1961, 61, 607–649 CrossRef CAS.
- C. Gibard, S. Bhowmik, M. Karki, E.-K. Kim and R. Krishnamurthy, Nat. Chem., 2018, 10, 212–217 CrossRef CAS PubMed.
- E. I. Jiménez, C. Gibard and R. Krishnamurthy, Angew. Chem., Int. Ed., 2021, 60, 10775–10783 CrossRef PubMed.
- O. R. Maguire, I. B. A. Smokers and W. T. S. Huck, Nat. Commun., 2021, 12, 5517 CrossRef CAS PubMed.
- As recently reported, P4O10 reacts with basic N-heterocycles to give activated adducts P2O5L2 and P4O10L4 (L = N-bases): K. Qian, S. M. Shepard, T. Xin, G. Park and C. C. Cummins, J. Am. Chem. Soc., 2023, 145, 6045–6050 CrossRef CAS PubMed.
- Y. Yamagata, H. Watanabe, M. Saitoh and T. Namba, Nature, 1991, 352, 516–519 CrossRef CAS PubMed.
- M. Meisel, H. Bock, B. Solouki and M. Kremer, Angew. Chem., 1989, 101, 1378–1381 CrossRef CAS.
- Baccolini and co-workers previously suggested the structure of PEP to be cyclic based on the observed 31P–13C coupling constants (for details, see: G. Baccolini, C. Boga and G. Micheletti, Phosphorus, Sulfur Silicon Relat. Elem., 2010, 185, 2303–2315 CrossRef CAS .). However, in Fig. S4† (ESI, Section VI.A.1, p. S69) we show that these coupling constants are easily explained by the favored acyclic conformation of PEP. This is further corroborated by the observation of similar coupling constants in the trimethyl ester of PEP, which cannot take on a cyclic form..
- J. Raya, A. Bianco, J. Furrer, J.-P. Briand, M. Piotto and K. Elbayed, J. Magn. Reson., 2002, 157, 43–51 CrossRef CAS PubMed.
- The keto–enol equilibrium constant of pyruvic acid was calculated to be pKE = 5.11 (for details see: A. J. Kresge, Pure Appl. Chem., 1991, 63, 213–221 CrossRef CAS ). However, the enolization equilibria for phenylpyruvic acid and 2-methylpyruvic acid are unknown. An indication of the substituent effect on enolization can be derived from comparing acetaldehyde, 2-methylpropanal, and 2-phenylpropanal which feature an equilibrium constant of 6.23, 3.86, and 3.35 respectively (for details, see: J. R. Keeffe and A. J. Kresge, in Enols, 1990 (Ed.: Z. Rappoport), John Wiley & Sons, Inc., Chichester, UK, 1990, pp. 456)..
- K. Hanai, A. Kuwae, S. Kawai and Y. Ono, J. Phys. Chem., 1989, 93, 6013–6016 CrossRef CAS.
- O. Sciacovelli, A. Dell'atti, A. De Giglio and L. Cassidei, Z. Naturforsch., C: J. Biosci., 1976, 31, 5–11 CrossRef CAS PubMed.
- C. De Duve, Blueprint for a Cell: The Nature and Origin of Life, ed. N. Patterson, 1991, pp. 156–166 Search PubMed.
- J. E. Goldford, H. Hartman, T. F. Smith and D. Segrè, Cell, 2017, 168, 1126–1134 CrossRef CAS PubMed.
- H. Hartman and T. F. Smith, Life, 2019, 9, 69 CrossRef CAS PubMed.
- A. Kitani, S. Tsunetsugu, A. Suzuki, S. Ito and K. Sasaki, Bioelectrochem. Bioenerg., 1995, 36, 47–51 CrossRef CAS.
- A. Kitani, S. Tsunetsugu and A. Suzuki, J. Chem. Soc., Perkin Trans. 2, 1991, 3, 329–331 RSC.
- S. Pinna, C. Kunz, A. Halpern, S. A. Harrison, S. F. Jordan, J. Ward, F. Werner and N. Lane, PLoS Biol., 2022, 20, e3001437 CrossRef CAS PubMed.
- E. Werner, S. Pinna, R. J. Mayer and J. Moran, J. Am. Chem. Soc., 2023, 145(39), 21630–21637 CrossRef CAS PubMed.
- J. D. Toner and D. C. Catling, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 883–888 CrossRef CAS PubMed.
- M. P. Brady, R. Tostevin and N. J. Tosca, Nat. Commun., 2022, 13, 5162 CrossRef CAS PubMed.
- F. Postberg, Y. Sekine, F. Klenner, C. R. Glein, Z. Zou, B. Abel, K. Furuya, J. K. Hillier, N. Khawaja, S. Kempf and L. Noelle, Nature, 2023, 618, 489–493 CrossRef CAS.
- C. R. Walton, S. Ewens, J. D. Coates, R. E. Blake, N. J. Planavsky, C. Reinhard, P. Ju, J. Hao and M. A. Pasek, Nat. Geosci., 2023, 16, 399–409 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc04116f |
| This journal is © The Royal Society of Chemistry 2023 |