
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Borylation directed borylation of N-alkyl anilines using iodine activated pyrazaboles†
C. R. P.
Millet
,
E.
Noone
 ,
A. V.
Schellbach
,
J.
Pahl
,
J.
Łosiewicz
,
G. S.
Nichol
and
M. J.
Ingleson
*
,
A. V.
Schellbach
,
J.
Pahl
,
J.
Łosiewicz
,
G. S.
Nichol
and
M. J.
Ingleson
*
EaStCHEM School of Chemistry, University of Edinburgh, Edinburgh, EH9 3FJ, UK. E-mail: michael.ingleson@edinburgh.ac.uk
First published on 17th October 2023
Abstract
Doubly electrophilic pyrazabole derivatives (pyrazabole = [H2B(μ-C3N2H3)]2) combined with one equiv. of base effect the ortho-borylation of N-alkyl anilines. Initial studies found that the bis(trifluoromethane)sulfonimide ([NTf2]−) pyrazabole derivative, [H(NTf2)B(μ-C3N2H3)]2, is highly effective for ortho-borylation, with this process proceeding through N–H borylation and then ortho C–H borylation. The activation of pyrazabole by I2 was developed as a cheaper and simpler alternative to using HNTf2 as the activator. The addition of I2 forms mono or ditopic pyrazabole electrophiles dependent on stoichiometry. The ditopic electrophile [H(I)B(μ-C3N2H3)]2 was also effective for the ortho-borylation of N-alkyl-anilines, with the primary C–H borylation products readily transformed into pinacol boronate esters (BPin) derivatives. Comparison of borylation reactions using the di-NTf2-and the diiodo-pyrazabole congeners revealed that more forcing conditions are required with the latter. Furthermore, the presence of iodide leads to competitive formation of side products, including [HB(μ-C3N2H3)3BH]+, which are not active for C–H borylation. Using [H(I)B(μ-C3N2H3)]2 and 0.2 equiv. of [Et3NH][NTf2] combines the higher yields of the NTf2 system with the ease of handling and lower cost of the iodide system generating an attractive process applicable to a range of N-alkyl-anilines. This methodology represents a metal free and transiently directed C–H borylation approach to form N-alkyl-2-BPin-aniline derivatives.
Introduction
C–H borylation is a powerful methodology for generating synthetically ubiquitous organoboranes in an efficient manner.1 The use of directing groups (DGs) in C–H borylation reactions enables access to organoboranes with a distinct regiochemistry to that formed from non-directed transformations.2 One specific example of this is in the synthesis of ortho-borylated anilines, which are useful for accessing ortho substituted anilines prevalent in pharmaceuticals, agrochemicals and organic materials.3 Directing groups generally are required for this ortho C–H borylation as in the absence of DGs the electrophilic C–H borylation of anilines leads to para-functionalisation,4 while iridium and cobalt catalysed C–H borylations generally lead to mixtures of meta- and para-borylated products.1b,5 To date, the ortho C–H borylation of anilines has been dominated by approaches requiring the separate installation and removal of a directing group (resulting in “multiple pot” processes).6,7 For example, the electrophilic ortho C–H borylation of aniline derivatives using N-pivaloyl DGs and BBr3 (Fig. 1a, top)8 requires the installation and removal of pivaloyl in separate processes, the latter under forcing conditions.9 The use of transient DGs is preferable as these are installed, direct the C–H borylation and then are removed all in one pot.10 In notable work, the ortho-borylation of anilines using transient DGs has been reported using iridium catalysts and B2Eg2 (Eg = ethylene glycolato).11 This proceeds via in situ formation of an ArylN(H)BEg species (Fig. 1b, inset) that then directs the ortho C–H borylation. The N-BEg unit is then readily cleaved during work-up. While this methodology is highly effective for ArylNH2 species, much lower yields (<30%) are obtained with N-alkyl-anilines.11,12 Given the prevalence of ortho-functionalised N-alkyl-anilines in pharmaceuticals (e.g. Flutemetamol, Entrectinib and Agratroban), the development of a higher yielding, transient DG approach for the ortho-borylation of N-alkyl-anilines is desirable, particularly if the process is precious metal-free.13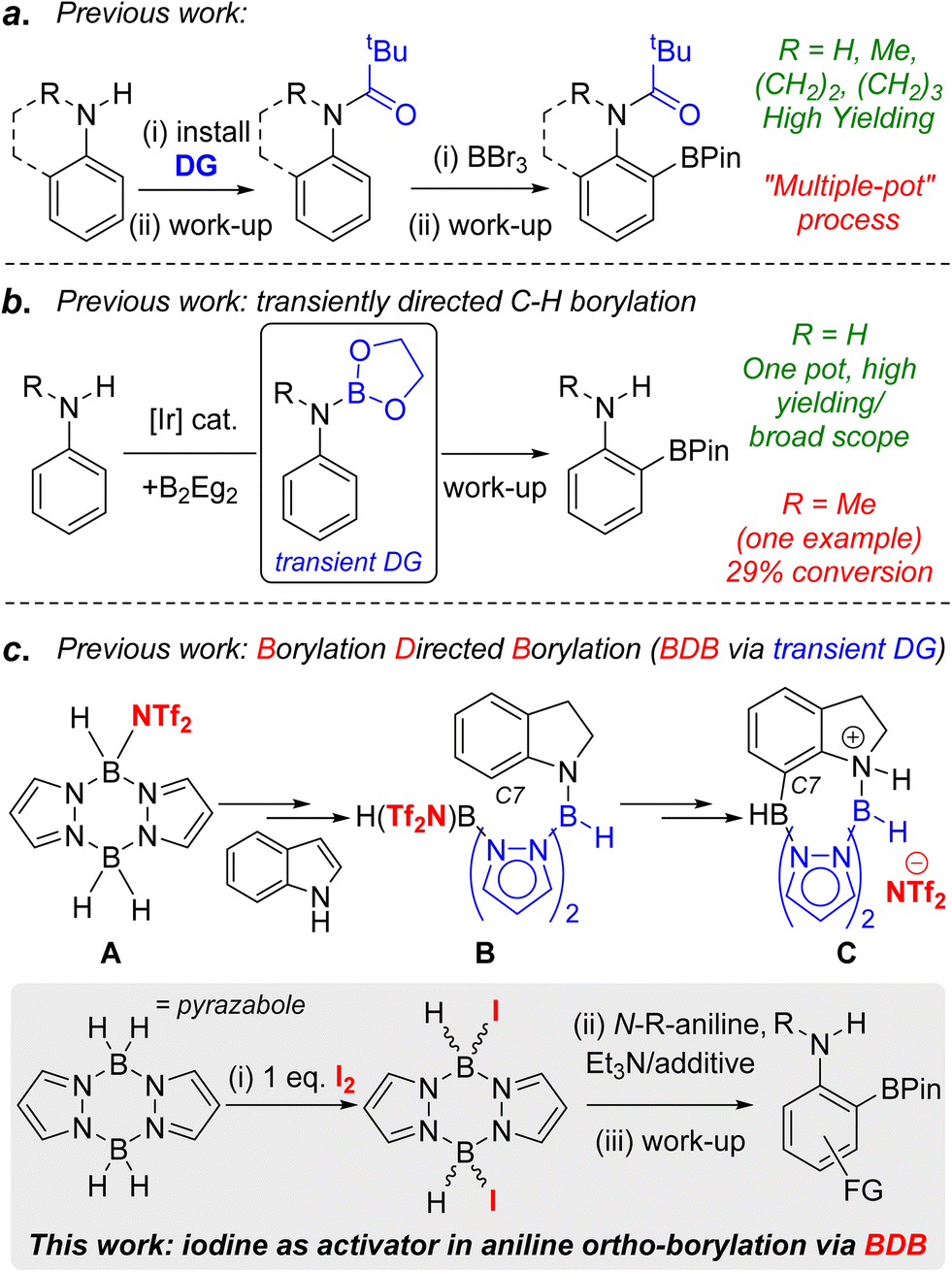 | ||
| Fig. 1 a = pivaloyl directed electrophilic borylation. b = a transient directing group in iridium catalysed ortho-borylation of anilines. c = previous work on indole reduction/C7 borylation via BDB. Inset bottom, this work. | ||
Recently, we reported the borylation directed borylation (BDB) of indoles using pyrazabole electrophile A (Fig. 1c) as a method to install boron units at the C7 position.14,15 In this process reduction of indole to indoline occurs first, with the spectroscopic data indicating that this led to an N-borylated indoline intermediate (e.g.B). The N–B bond and the pyrazabole structure in compound B positions the second boron centre appropriately to borylate the proximal sp2C–H leading to C, a C7 borylated indoline. Protection of the C–B unit and cleavage of the N–B bonds in C during work up formed indolines containing the useful pinacol boronate ester (BPin) group at C7. Therefore, pyrazabole is acting as a transient DG in this BDB process, with transient DGs underexplored in electrophilic C–H borylation.2a,16 Our initial BDB study utilised stoichiometric amounts of bistriflimidic acid (HNTf2 = HN(SO2CF3)2) to form the reactive electrophile A. However, HNTf2 is relatively expensive,17 and it, and NTf2-pyrazabole electrophiles (e.g.A), have to be handled within a glovebox. Therefore, extending the BDB of N-alkyl-aniline derivatives beyond indoline while using an inexpensive and more readily handled activator would be attractive. Herein we report our studies addressing this challenge. This led to the development of iodine as a cheap and easy to handle activator for pyrazaboles that forms ditopic electrophiles that are effective in the transient DG mediated ortho-borylation of N-alkyl-anilines.
Results and discussion
Our first focus was identifying electrophilic pyrazabole – base combinations that achieved the ortho-borylation of our model substrate, N-Me-aniline. Initially, the previously reported 1 (Scheme 1) was added to N-Me-aniline in the presence of 2,6-di-tert-butyl-4-methylpyridine (DBP) as base. At room temperature this led to slow BDB, but on heating to ≥70 °C the BDB product [2]NTf2 was formed as the major product within 18 h. [2]NTf2 was fully characterised, which revealed protonation of the aniline nitrogen occurs during this BDB. A modified (shorter reaction time)14 N–B cleavage/pinacol installation process then led to formation of 3a.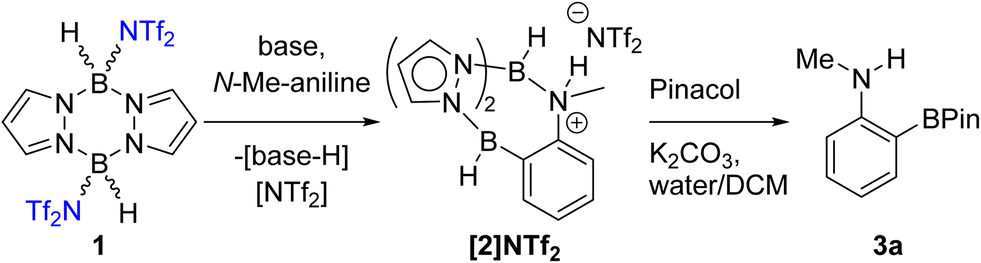 | ||
| Scheme 1 BDB of N-Me-aniline using 1 and an amine base. | ||
DBP is an expensive Brønsted base that was used to simplify initial studies as it does not coordinate to boron electrophiles. In contrast, other Lewis bases (e.g. MeCN) can displace NTf2 anions from 1, and base coordination to boron could retard the BDB reaction.14 Given the aniline substrate also functions as a Brønsted base during BDB (as indicated by the formation of [2]NTf2) only one equivalent of exogenous base is required. Therefore, one equivalent of the inexpensive bases Et3N and Hünigs base were trialled in place of DBP in the BDB of N-Me-aniline using 1. On heating both of these reactions led to the formation of [2]NTf2 and [baseH][NTf2] as a by-product. Pinacol installation/work-up enabled 3a to be isolated in 62 and 65% yield using Et3N and Hünigs base, respectively. Thus cheaper (than DBP) bases can be used in the BDB of N-alkyl-anilines. Our attention turned next to replacing HNTf2 with a simpler to handle and cheaper activator.
Based on the established reactivity of L→BH3 with iodine, which forms reactive boron electrophiles of general formula L→BH2I,18 diiodo-pyrazabole was targeted as an alternative to 1. While dibromo- and dichloro-pyrazaboles are known,19 to our knowledge no B–I containing pyrazaboles have been reported to date. The latter are desirable as iodine is inexpensive, easy to handle and is less coordinating to boron than the lighter halides. Furthermore, L→BH2I species have been demonstrated to react with π nucleophiles to form C–B bonds in a related manner to L→BH2(NTf2) species.20 Therefore, one equivalent of iodine, pyrazabole and Et3N were combined and found to be viable for the BDB of N-Me-aniline (Scheme 2), albeit requiring heating to 100 °C for significant BDB to occur. In contrast, attempts using dibromo-pyrazabole under identical conditions led to no BDB reaction (Scheme 2), indicating that the less coordinating nature of iodide towards boron is vital for this transformation. Despite extensive optimisation studies using iodine activated pyrazabole (see Table S2†) the isolated yield of 3a remained <50% (based on N-Me-aniline) – with Et3N providing the best outcome from the bases explored. Notable points from this optimisation study included: use of >1 equiv. of Et3N retarding the BDB reaction, while using two equiv. of N-Me-aniline and no other base gave only trace amounts of 3a. Given the lower yields of 3a using iodine activated pyrazabole relative to using 1, both systems were analyzed further to determine the origin(s) of this disparity.
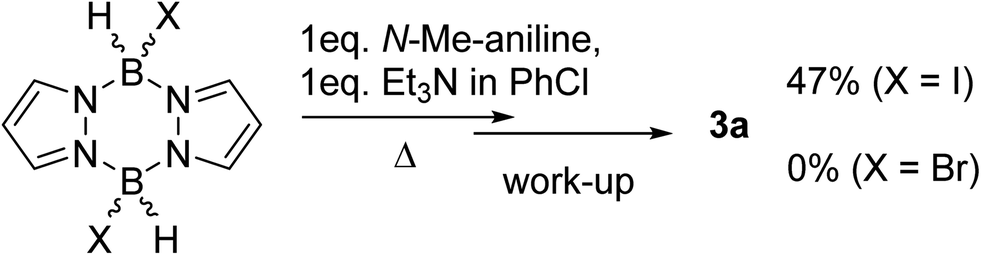 | ||
| Scheme 2 Outcomes using dibromo-versus diiodo-pyrazabole in the BDB of N-Me-aniline. | ||
Mechanistic studies
On analysing the reaction of 1 and one equiv. base (base = DBP or Et3N) with N-Me-aniline by in situ NMR spectroscopy an intermediate was observed. This intermediate, termed 4, could be obtained cleanly by the combination of 1 and the independently synthesised di(N–Me-anilide)-pyrazabole 5 (Scheme 3, see ESI Section 2 and Fig. S14–S20†). Compound 4 displayed two pyrazabole C–H resonances in the 1H NMR spectrum in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio indicating a symmetrically substituted pyrazabole. Further insight into the structure of 4 came from 19F NMR spectroscopy, which revealed NTf2 is not coordinated to boron (δ19F = −78.7, whereas for B-NTf2 systems δ19F ≈ −69),21 and DOSY NMR studies (see ESI, Section 5.3†) which indicated 4 is dimeric. The dimeric structure for 4 presumably is related to the previously reported oxo-bridged dimer D (Scheme 3),22 with an analogous structure fully consistent with the NMR data for 4 (as a single isomer with a cis arrangement of the aniline-N substituents). Compound 4 converted into the BDB product [2]NTf2 slowly at ambient temperature, but more rapidly and in high conversion on heating. While 4 could not be isolated as single crystals suitable for diffraction studies the structure of the dicationic portion of 4 (termed [4]2+) was calculated at the MN15/6-311G(d,p)/PCM (PhCl, PCM = polarizable continuum model) level (inset Scheme 3, note all calculations are performed at this level herein, with the LANL2DZ basis set used for iodide). While the B–N distances in the calculated structure (1.615–1.618 Å) are comparable to related borocations,23 there is evidence for significant distortion in [4]2+ due to steric interactions between the pyrazole rings and the N-Me and N-Ph substituents. For example, the PhC–N–CMe angle is small (102.7° in [4]2+) while the B2N4 core is twisted (in the B2N4 core of D the four nitrogens are co-planar, however in [4]2+ they deviate by upto 0.11 Å above and below the plane made by the four nitrogens). These distortions will destabilise dimeric [4]2+ presumably enabling dissociation into a monomeric form that is required to effect ortho C–H borylation.
:1 ratio indicating a symmetrically substituted pyrazabole. Further insight into the structure of 4 came from 19F NMR spectroscopy, which revealed NTf2 is not coordinated to boron (δ19F = −78.7, whereas for B-NTf2 systems δ19F ≈ −69),21 and DOSY NMR studies (see ESI, Section 5.3†) which indicated 4 is dimeric. The dimeric structure for 4 presumably is related to the previously reported oxo-bridged dimer D (Scheme 3),22 with an analogous structure fully consistent with the NMR data for 4 (as a single isomer with a cis arrangement of the aniline-N substituents). Compound 4 converted into the BDB product [2]NTf2 slowly at ambient temperature, but more rapidly and in high conversion on heating. While 4 could not be isolated as single crystals suitable for diffraction studies the structure of the dicationic portion of 4 (termed [4]2+) was calculated at the MN15/6-311G(d,p)/PCM (PhCl, PCM = polarizable continuum model) level (inset Scheme 3, note all calculations are performed at this level herein, with the LANL2DZ basis set used for iodide). While the B–N distances in the calculated structure (1.615–1.618 Å) are comparable to related borocations,23 there is evidence for significant distortion in [4]2+ due to steric interactions between the pyrazole rings and the N-Me and N-Ph substituents. For example, the PhC–N–CMe angle is small (102.7° in [4]2+) while the B2N4 core is twisted (in the B2N4 core of D the four nitrogens are co-planar, however in [4]2+ they deviate by upto 0.11 Å above and below the plane made by the four nitrogens). These distortions will destabilise dimeric [4]2+ presumably enabling dissociation into a monomeric form that is required to effect ortho C–H borylation.
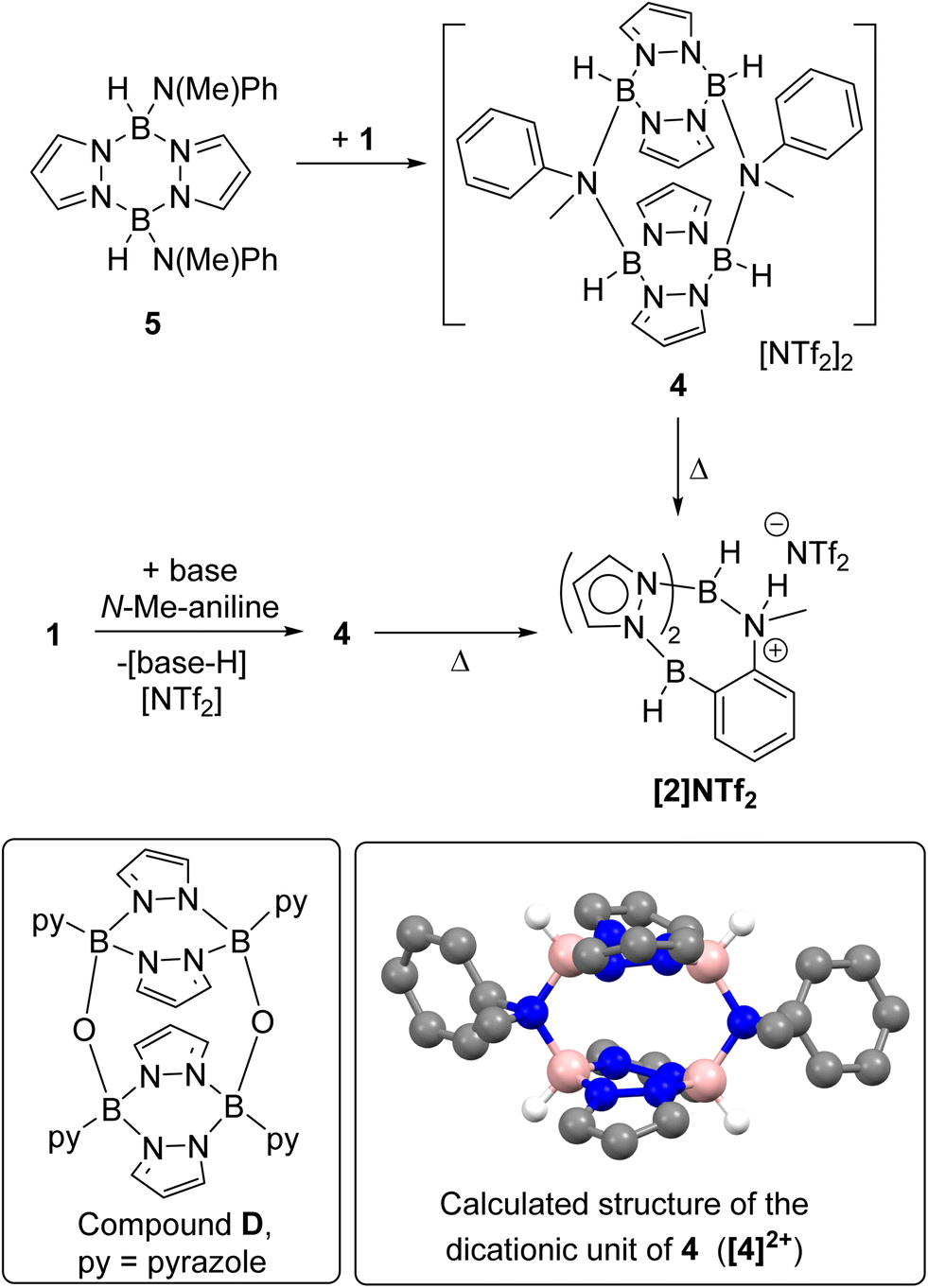 | ||
| Scheme 3 Top, the formation of 4, from combination of 1 and 5; middle, the formation of 4 during BDB and its subsequent conversion into [2]NTf2. Inset bottom left, the previously reported oxo bridged dimeric pyrazabole, D; inset bottom right the calculated structure of [4]2+ showing only the hydrogens bonded to boron for clarity, pink = boron, blue = nitrogen, grey = carbon, white = hydrogen. | ||
Moving to the iodo-pyrazaboles, the reaction of pyrazabole and iodine was investigated first as iodo-pyrazaboles have not been reported previously to our knowledge. The addition of 0.5 equiv. of I2 to pyrazabole led to the rapid formation of the mono-iodo pyrazabole, 6 (Scheme 4) at room temperature (by in situ NMR spectroscopy, Fig. S44†). Addition of a further 0.5 equiv. of iodine led to the full conversion of 6 into the diiodo pyrazabole, 7. Compound 7 is formed as a ca. 1:1 mixture of isomers as indicated by two doublets in the 11B NMR spectrum along with two sets of 2:1 relative integral pyrazole resonances in the 1H NMR spectrum, which is consistent with two symmetrically substituted pyrazaboles. These isomers are assigned as the cis and trans isomers of 7 based on previous reports from the groups of Trofimenko and Nöth on cis and trans isomers being formed for the lighter dihalo pyrazaboles.24,25 Calculations also indicated that the cis and trans isomers of 7 are close in energy (ca. 1 kcal mol−1 calculated free energy difference), consistent with the two species observed in solution being the cis and trans isomers of 7. The addition of one equiv. of I2 in one portion to pyrazabole also led to the formation of 7 and it was isolated in 75% yield. The cis isomer formed single crystals suitable for X-ray diffraction studies. The solid-state structure of the cis isomer of 7 has a B2N4 6-membered core in a flattened boat conformation with the iodide substituents located in the flagpole positions. In 7 the B⋯B distance of 3.031(8) Å is in the expected region and is comparable to a related dihalogenated pyrazabole [H(Br)B(μ-C3N2H2Cl)]2 (3.05 Å).25 The B–I bond distances of 2.290(6) and 2.302(6) Å are at the lower end of B–I bond lengths reported for L→BH2I compounds (L = N-heterocyclic carbenes or PR3).26 Notably, combining equimolar 7 and pyrazabole in chlorobenzene led to formation of the mono-iodo pyrazabole 6 at ambient temperature (by in situ NMR spectroscopy - Fig. S48†), indicating that intermolecular H/I exchange occurs in iodo-pyrazaboles. Finally, it should be noted that 7 has appreciable thermal stability: heating 7 at 100 °C in PhCl for 3 days led to minimal decomposition (<5% by multinuclear NMR spectroscopy), with the only observable new 11B NMR resonance consistent with formation of an L-BI3 compound (based on the δ11B = −34.6, see Fig. S46 and S47†).
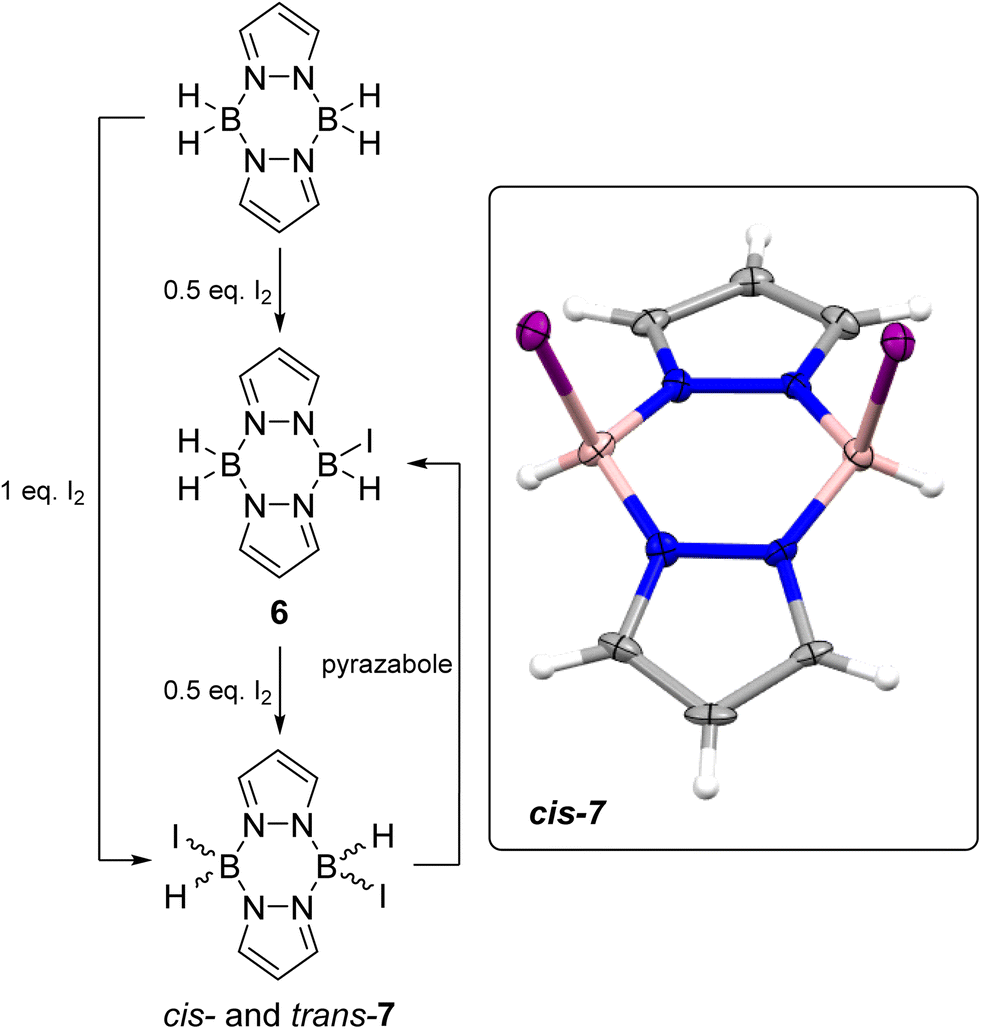 | ||
| Scheme 4 Left, formation of mono- (6) and diiodo-pyrazabole (7). Inset right, the solid state structure of cis-7, ellipsoids at 50% probability. Blue = nitrogen, pink = boron, purple = iodine, grey = carbon, white = hydrogen. | ||
With an understanding of the products formed from combining iodine and pyrazabole in hand the reactivity of 7 towards Et3N was explored, Et3N was selected as it gave the best outcome in our initial optimisation study (see Table S2†). The addition of one equivalent of Et3N to 7 led to formation of the mono-cation 8 (Scheme 5). The identity of 8 was confirmed by single crystal X-ray diffraction analysis (inset, Scheme 5). The solid-state structure of 8 also has a flattened boat conformation for the B2N4 core with the iodide and Et3N moieties being cis in the flagpole positions. The steric demand of Et3N in 8 causes a distortion in the geometry with an increase of the Y–B-Centroid angles (Y = I or NEt3; centroid = calculated centroid of the B2N4 ring) observed on comparing 7 (I–B-centroid = 113.3(3)° and 112.6(3)°) and 8 (I–B-centroid = 118.6(12)°; Et3N–B-centroid = 122.1(14)°). Compound 8 also has a longer B–I bond of 2.36(2) Å vs. the B–I bonds in 7 (2.290(6) and 2.302(6) Å), consistent with greater steric crowding in 8 relative to 7. However, the B–NEt3 bond length in 8 (1.62(2) Å) is in the range of previously reported Et3N-BR3 adducts (1.60–1.69 Å).27
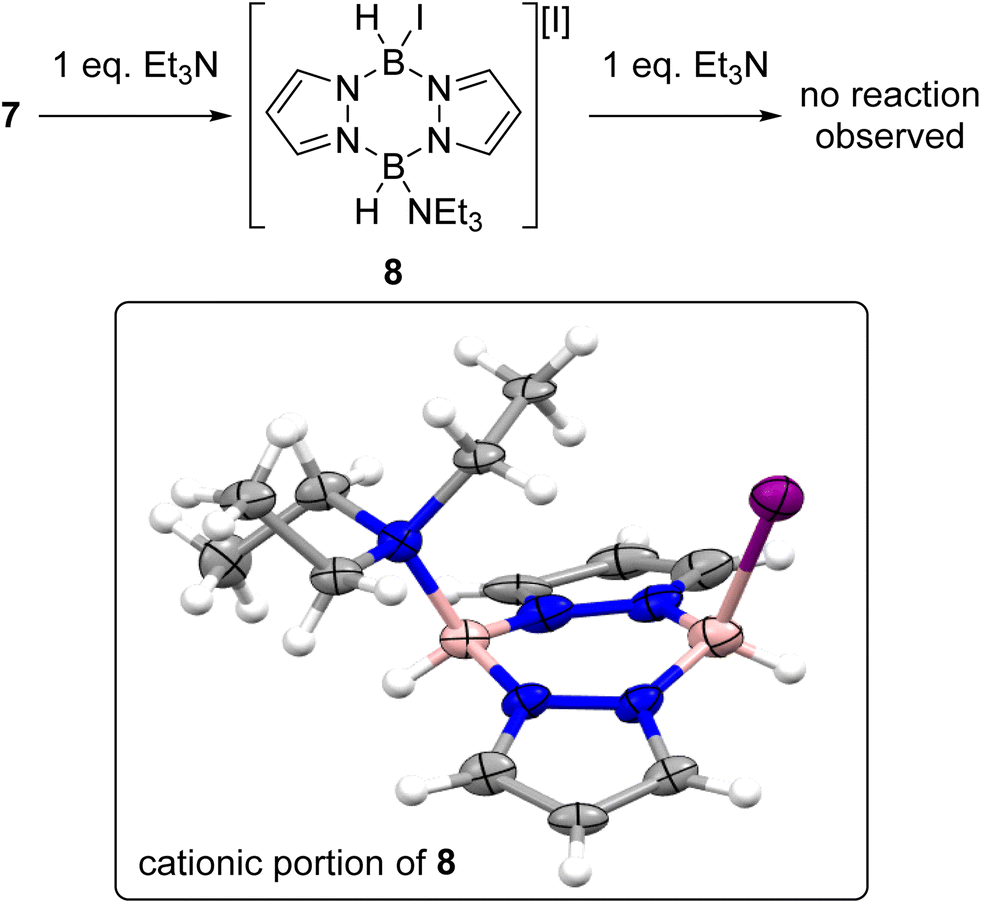 | ||
| Scheme 5 Top, the reaction of 7 towards Et3N. Inset bottom, the solid-state structure of the cationic portion of 8, ellipsoids at 30% probability. Blue = nitrogen, pink = boron, purple = iodine, grey = carbon, white = hydrogen. | ||
In contrast to the di-NTf2 analogue 1 (where both NTf2 anions are displaced by Lewis bases to form dicationic products),14 the addition of further Et3N to 8 did not displace the second iodide (Fig. S49†). This is consistent with the more coordinating nature of iodide relative to [NTf2]−. However, the addition of both N-Me aniline and Et3N (in either order of addition) to 7 led to substitution of both iodides to form the di-anilide product 5 as the major boron containing species. This indicates that Et3N coordination to boron in 8 does not irreversibly block N-Me-aniline from reacting with boron. Next, diiodo-pyrazabole 7 and dianilide-pyrazabole 5 were combined to determine if the iodide analogue of the dimer 4 forms. This led to slow and complex reactivity at room temperature with no iodide analogue of 4 observed. In contrast, the di-NTf2 pyrazabole 1 and compound 5 are completely consumed within minutes of mixing to form 4 cleanly. In the in situ monitored BDB reactions using diiodo-pyrazabole 7, 5 is the only major new pyrazabole product observed, again there is no evidence for the iodide analogue of 4 (by NMR spectroscopy). From the in situ monitoring experiments [2]I forms as one of the major products on heating, but this occurs along with the formation of two other major products. The first of these was assigned as (Me(Ph)N)2BH (δ11B = 29.0 1JB–H = 126 Hz) by comparison to the previous report.28 The second was identified as compound 9 (Scheme 6), which precipitated from the BDB reactions mixtures (along with some [Et3NH][I] precipitating). Compound 9 was independently synthesised and crystallised with X-ray diffraction studies confirming its formulation (inset Scheme 6). These results combined indicate that heating diiodo-pyrazabole 7 in the presence of Et3N/N-Me-aniline leads to competitive (to BDB) break-up of the pyrazabole core and the formation of species that are non-productive for BDB (e.g. compound 9). This contrasts with BDB using the NTf2 derivative 1 (which are much cleaner by in situ NMR spectroscopy with <5% formation of other pyrazole containing products by NMR spectroscopy), indicating that the more coordinating iodide anion plays a crucial role in the cleavage of the pyrazabole core under these conditions. This is presumably the origin of the lower conversions to [2]I (and thus 3a) observed using 7 compared to conversions to [2]NTf2 using the NTf2 analogue 1.
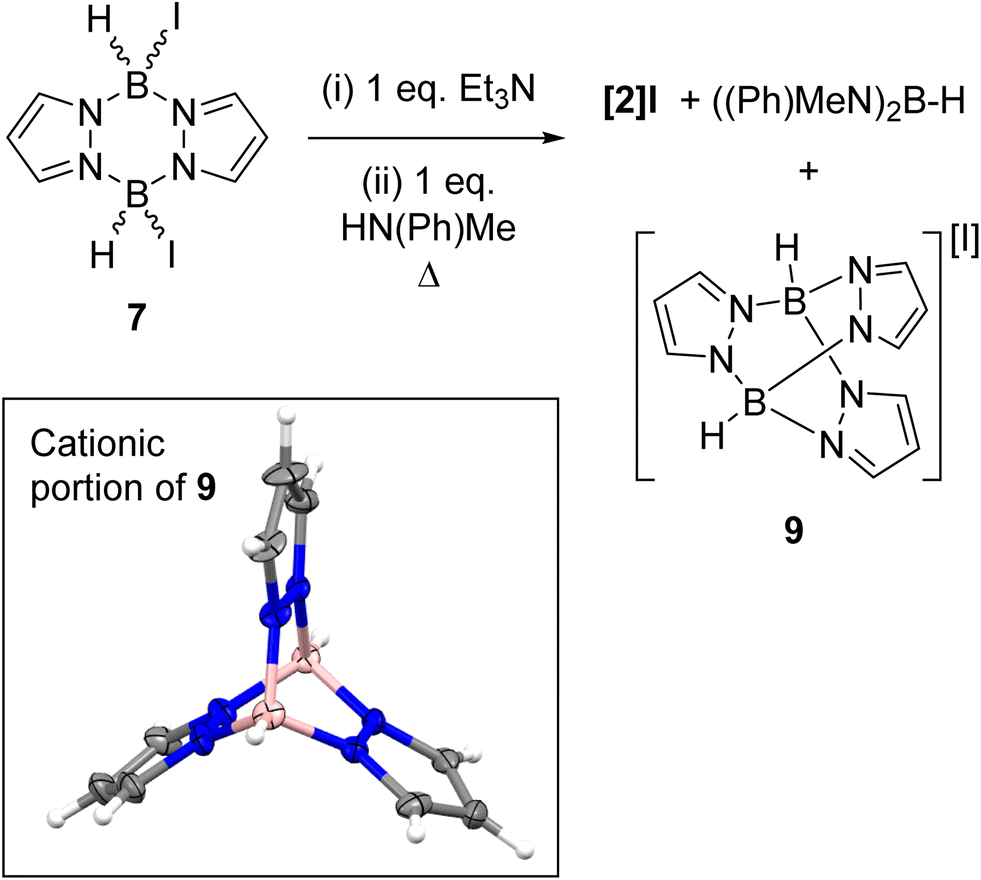 | ||
| Scheme 6 Left, the reactivity of 7 towards Et3N/N-Me-aniline at raised temperatures. Inset, the solid-state structure of compound 9, ellipsoids at 30% probability. Blue = nitrogen, pink = boron, grey = carbon, white = hydrogen. | ||
Given the lower conversion to 3a using 7 relative to that using stoichiometric 1, attempts were made to use sub-stoichiometric HNTf2 (or sub-stoichiometric 1) and stoichiometric pyrazabole in the BDB of N-Me aniline. However, these reactions all led to low yields of 3a, this is consistent with the observation that [Et3NH][NTf2] (the by-product from BDB) and pyrazabole do not react on heating to 100 °C. Therefore alternative approaches were sought to achieve a high yielding, operationally simple and cheaper BDB protocol.
Optimisation of the BDB of N-alkyl-anilines using iodo-pyrazaboles
To combine the best of the NTf2 (higher yields) and iodide (cheaper/easier to handle) systems we considered an in situ anion exchange process that could convert iodo-pyrazaboles into more reactive NTf2-pyrazaboles. The feasibility of iodide/NTf2 exchange initially was explored computationally which indicated that the displacement of iodide from pyrazabole by triflimide is endergonic (by +7.5 kcal mol−1 for the mono-pyrazabole, Scheme 7). This is consistent with the addition of 5 equiv. of [Et3NH][NTf2] to 7 resulting in no observable anion exchange (by NMR spectroscopy). Nevertheless, as the BDB process has a significantly lower overall barrier for the NTf2 system relative to the iodide analogue (1 performs BDB at room temperature, albeit slowly, while 7 requires heating to ≥70 °C for BDB) anion exchange may still lead to an enhanced BDB outcome. Note, a related anion exchange process facilitating an electrophilic C–H borylation with B-trypticenes has been reported recently using stoichiometric Na[B(C6F5)4].29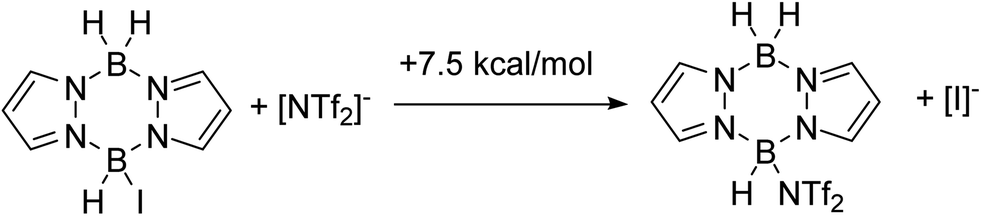 | ||
| Scheme 7 Free energy change for iodide/[NTf2]− exchange. | ||
An initial experiment to assess for any anion exchange derived enhancement in yield used a 0.9:0.1 mix of 7:1 in the BDB of N-Me-aniline with one equiv. of Et3N as base. Notably, this led to comparable yields for the formation of 3a (Scheme 8) to that using 1 equiv. of 1. A significant yield enhancement was also observed using a 0.9:0.1 mix of 7 and 1 in the BDB of tetrahydroquinoline to form 3b post pinacol installation/work-up (Scheme 8). The significant yield enhancement observed using 0.9:0.1 mixtures of 7 and 1 indicates it is not just due to compounds 7 and 1 reacting separately in the BDB process. We tentatively attribute this enhancement to a degree of metathesis of an iodo-pyrazabole with [Et3NH][NTf2] (formed during BDB) leading to a more reactive NTf2-pyrazabole electrophile. Note, during these reactions in chlorobenzene solid precipitates, which on analysis was found to be [Et3NH][I]. Thus the lower solubility of [Et3NH][I] relative to the NTf2 salt under these conditions may be assisting anion exchange. The precipitation of [Et3NH][I] also will reduce the iodide concentration in solution, potentially slowing the formation of decomposition species. This is consistent with the observation that compound 9 is not observed during the reactions using 0.9:0.1 of 7 and 1.
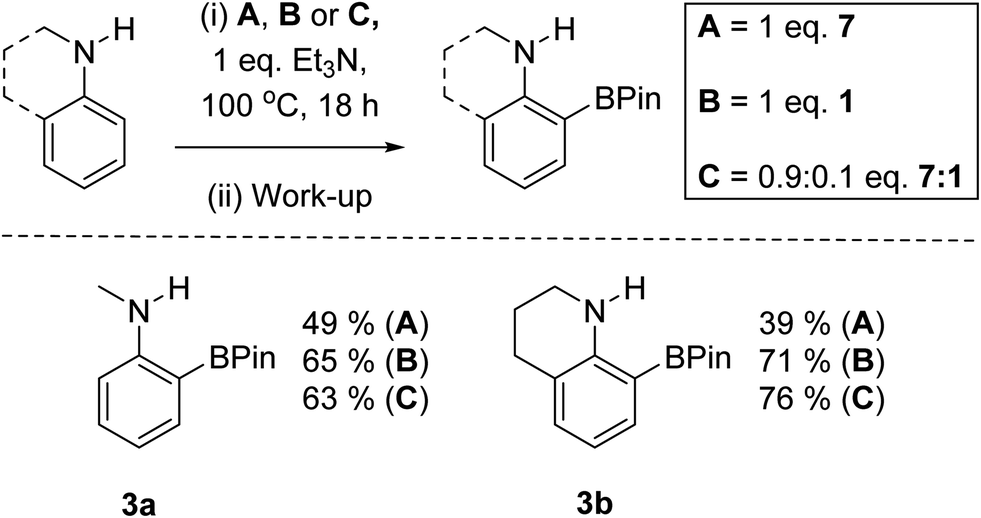 | ||
| Scheme 8 Outcomes from using 1, 7 or 1/7 in the BDB reaction. | ||
Overall, these observations suggested that combining 7 with sub-stoichiometric [cation][NTf2] could result in a similar enhancement in yield. This hypothesis was confirmed by the use of one equiv. of 7 and 0.2 equiv. of [Et3NH][NTf2] in the BDB process leading to a 60% yield of 3a and a 78% yield of 3b (comparable to outcomes from conditions B and C in Scheme 8). This is a notable improvement over the yields reported using iridium catalysed transient DG approaches to form ortho-BPin-N-alkyl-anilines.11,12,30 Note, the use of 0.2 equiv. of LiNTf2 with 7 gave lower yields relative to using [Et3NH][NTf2] under otherwise identical conditions, therefore the latter salt is used hereon. With conditions identified that avoided expensive bases and stoichiometric amounts of anhydrous HNTf2 ([Et3NH][NTf2] can be stored on the bench and is readily accessible from commercial LiNTf2 and [Et3NH][Cl]) a substrate scope exploration was performed (Scheme 9). The scoping study revealed that in addition to 3a and 3b the conditions were amenable to larger alkyl substituents on nitrogen, with the N-iPr derivative, 3c, isolated in 52% yield. Alongside 3b, the seven (3d) and five (3e) membered analogues were also amenable to BDB, indicating the change in positioning of the N-bound pyrazabole unit enforced by the different ring sizes does not significantly influence this BDB reaction. Notably, neither 3d nor any other C9 borylated benzo[b]azepines have been reported previously to our knowledge. This is despite the significant importance of substituted benzo[b]azepines in pharmaceuticals and agrochemicals, including C9-substitued derivatives (e.g. zilpaterol).31 In contrast, the ortho-methyl derivative, 2,N-Me2-aniline, was not amenable to this process. We attribute this to the ortho methyl forcing an orientation that disrupts conjugation between the aniline phenyl ring and the nitrogen lone pair. This was supported by calculations on analogues of 5 containing 2,N-Me2-aniline (twisted away from co-planarity by 44°) and indoline and tetrahydroquinoline (see Table S4†) – with the latter two compounds and 5 having close to co-planar N and phenyl units that maximise conjugation and thus increase the nucleophilicity of the π system (thereby favouring SEAr).
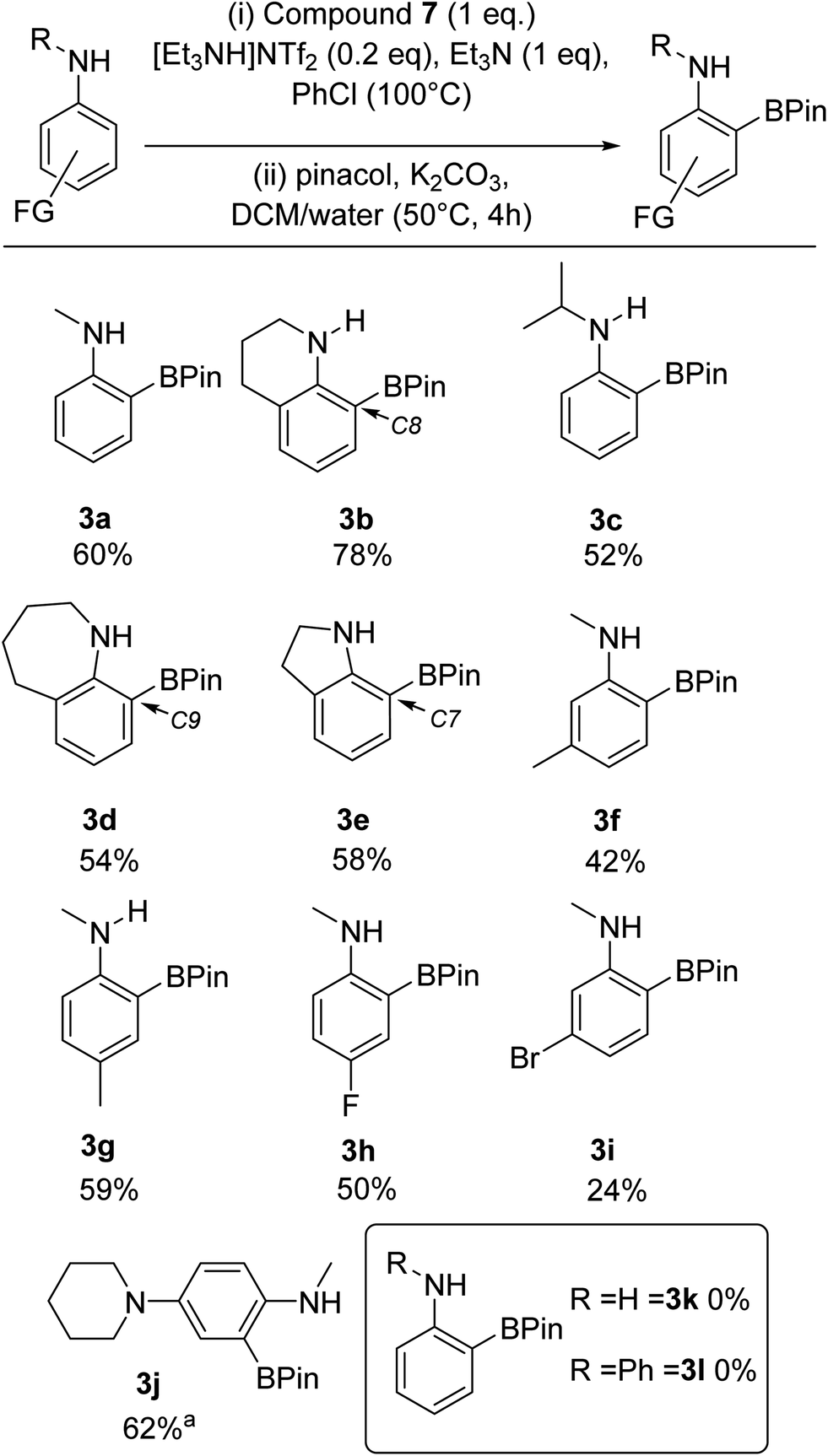 | ||
| Scheme 9 Substrate scope and isolated yields (unless otherwise stated) for the BDB of aniline derivatives using 7/Et3NH[NTf2]. a = conversion versus an internal standard. | ||
Moving to other substituents, as this is an electrophilic borylation using borenium cation equivalents and forcing conditions, functional group tolerance will be limited (as indicated by the p-MeO derivative not being amenable to this process),23 but halides and NR2 groups are tolerated (vide infra). Furthermore, while the ortho methyl aniline derivative was not amenable substituents at the meta (3f and 3i) and para (3g and 3h) positions of N-Me-aniline were tolerated. This BDB process was found to be sensitive to arene electronics, with electron withdrawing groups significantly retarding BDB, requiring longer reaction times for 3h and 3i. Consistent with this observation, an N-Me-aniline substrate substituted with an electron donating group, specifically a para-piperidine unit, performed much better in this BDB process, with 3j isolated in 62% yield. Ortho-substituted anilines containing a para-piperidine unit are important as these motifs are found in approved and developmental bioactives, e.g. Brigatinib and ASP3026.32 Next, we attempted to extend this BDB process to aniline and diphenylamine. However, in both cases no ortho borylated products (3k and 3l) were isolated. While diphenylamine is presumably insufficiently nucleophilic for this BDB reaction (consistent with an SEAr type process), the origin of the incompatibility of aniline with this BDB reaction is currently unclear. Finally, we assessed the amenability of this methodology to scaling and glovebox free conditions: compound 3a was isolated in 62% yield when the BDB process was scaled up ten-fold, while 3a was isolated in 45% yield under glovebox free conditions (making 7in situ from bench stable pyrazabole and iodine, note pyrazabole itself is readily accessed from pyrazole and L→BH3).19
Conclusions
Iodine is an inexpensive activator for pyrazaboles that forms mono- and di-topic pyrazabole electrophiles, with the latter effective in the borylation directed borylation (BDB) of N-alkyl anilines. However, when using diiodo-pyrazabole 7 competitive formation of inactive (for BDB) species occurs that arise from break-up of the B2N4 pyrazabole core. This leads to lower BDB conversions using 7 than when using the di-NTf2 pyrazabole analogue 1 (which reacts with <5% of unwanted side products by NMR spectroscopy). The attractive features of both systems (iodine = cheaper and easy to handle activator, while NTf2-pyrazaboles = higher conversions in BDB) can be combined by using the diiodo-pyrazabole 7 in combination with 0.2. equiv. of [Et3NH][NTf2]. This BDB methodology is operationally simple (no glovebox required) and is applicable to a range of N-alkyl anilines. The primary BDB products can be readily transformed into synthetically ubiquitous pinacol boronates esters, thus this process represents a metal-free transient directed C–H borylation methodology to form desirable N-alkyl-2-BPin-anilines.Data availability
The data supporting this article has been uploaded as part of the ESI,† this includes NMR spectra for all new compounds, in situ NMR spectra for catalytic and mechanistic reactions and Cartesian coordinates for all calculated structures.Author contributions
MI, and CM conceived the research concept and aims and analysed all data. CM performed the majority of the synthetic work and the majority of the analytical components of this project. EN, AS, and JP also performed the synthesis and characterisation of a number of compounds reported in this manuscript. GN and JP collected and solved all the crystal structures. JL performed a number of the calculations. Combined, MI, CM and EN drafted, reviewed and edited the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding from the European Union’s Horizon 2020 research and innovation programme (grant agreement no. 769599). We thank the Mass Spectrometry facility (SIRCAMS) at the University of Edinburgh (UoE). MJI thanks the EPSRC Programme Grant “Boron: Beyond the Reagent” (EP/W007517) for support.References
- (a) Boronic Acids: Preparation and Applications, D. Hall, Wiley-VCH, Weinheim, 2011 Search PubMed; (b) R. Bisht, C. Haldar, M. M. M. Hassan, M. E. Hoque, J. Chaturvedi and B. Chattopadhyay, Chem. Soc. Rev., 2022, 51, 5042 RSC; (c) For electrophilic type C–H borylation see: M.-A. Légaré, M.-A. Courtemanche, E. Rochette and F.-G. Fontaine, Science, 2015, 349, 513 CrossRef PubMed.
- For select reviews on directed C–H borylation see: (a) S. A. Iqbal, J. Pahl, K. Yuan and M. J. Ingleson, Chem. Soc. Rev., 2020, 49, 4564 RSC; (b) A. Ros, R. Fernández and J. M. Lassaletta, Chem. Soc. Rev., 2014, 43, 3229 RSC; (c) M. T. Mihai, G. R. Genov and R. J. Phipps, Chem. Soc. Rev., 2018, 47, 149 RSC.
- T. Kahl, K.-W. Schröder, F. R. Lawrence, W. J. Marshall, H. Höke and F. Jäckh, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, 2011 Search PubMed.
- A. Del Grosso, P. J. Singleton, C. A. Muryn and M. J. Ingleson, Angew. Chem., Int. Ed., 2011, 50, 2102 CrossRef CAS PubMed.
- (a) With iridium see: H. Tajuddin, P. Harrisson, B. Bitterlich, J. C. Collings, N. Sim, A. S. Batsanov, M. S. Cheung, S. Kawamorita, A. C. Maxwell, L. Shukla, J. Morris, Z. Lin, T. B. Marder and P. G. Steel, Chem. Sci., 2012, 3, 3505 RSC; (b) With cobalt see: N. G. Léonard, M. J. Bezdek and P. J. Chirik, Organometallics, 2017, 36, 142 CrossRef; (c) Note, very recently by using bespoke ligands it was possible to significantly improve the selectivity in the iridium catalysed borylation of anilines without using a directing group, see (and references therein): C. Haldar, R. Bisht, J. Chaturvedi, S. Guria, M. M. M. Hassan, B. Ram and B. Chattopadhyay, Org. Lett., 2022, 24, 8147 CrossRef CAS PubMed.
- For an early example of ortho-borylation of aniline derivatives via directed lithiation/trapping with B(OR)3 see: M. A. Siddiqui and V. Snieckus, Tetrahedron Lett., 1988, 29, 5463 CrossRef CAS.
- For select examples using iridium catalysis see: (a) T. A. Boebel and J. F. Hartwig, J. Am. Chem. Soc., 2008, 130, 7534 CrossRef CAS PubMed; (b) P. C. Roosen, V. A. Kallepalli, B. Chattopadhyay, D. A. Singleton, R. E. Maleczka Jr. and M. R. Smith III, J. Am. Chem. Soc., 2012, 134, 11350 CrossRef CAS PubMed; (c) S. M. Preshlock, D. L. Plattner, P. E. Maligres, S. W. Krska, R. E. Maleczka Jr. and M. R. Smith III, Angew. Chem., Int. Ed., 2013, 52, 12915 CrossRef CAS PubMed; (d) M. M. M. Hassan, B. Mondal, S. Singh, C. Haldar, J. Chaturvedi, R. Bisht, R. B. Sunoj and B. Chattopadhyay, J. Org. Chem., 2022, 87, 4360 CrossRef PubMed.
- (a) S. A. Iqbal, J. Cid, R. J. Procter, M. Uzelac, K. Yuan and M. J. Ingleson, Angew. Chem., Int. Ed., 2019, 58, 15381 CrossRef CAS PubMed; (b) J. Lv, X. Chen, X.-S. Xue, B. Zhao, Y. Liang, M. Wang, L. Jin, Y. Yuan, Y. Han, Y. Zhao, Y. Lu, J. Zhao, W.-Y. Sun, K. N. Houk and Z. Shi, Nature, 2019, 575, 336 CrossRef CAS PubMed.
- For example deprotection conditions see: L. Zhang, C. Chen, J. Han, Z.-B. Huang and Y. Zhao, Org. Chem. Front., 2016, 3, 1271 RSC.
- (a) For a recent review on transient DGs in C–H functionalization see: Q. Zhao, T. Poisson, X. Pannecoucke and T. Besset, Synthesis, 2017, 49, 4808 CrossRef CAS; (b) For recent work on transient DGs in electrophilic C–H borylation see: S. Rej and N. Chatani, J. Am. Chem. Soc., 2021, 143, 2920 CrossRef CAS PubMed.
- M. R. Smith III, R. Bisht, C. Haldar, G. Pandey, J. E. Dannatt, B. Ghaffari, R. E. Maleczka Jr. and B. Chattopadhyay, ACS Catal., 2018, 8, 6216 CrossRef PubMed.
- Recent work reported an iridium catalysed C–H borylation route to ortho-BPin-N-Me-aniline in 73% yield, but this value is corrected for recovered starting material, the actual yield of the ortho-borylated product is 27%. See the ESI† in: M. E. Hoque, M. M. M. Hassan and B. Chattopadhyay, J. Am. Chem. Soc., 2021, 143, 5022 CrossRef CAS PubMed.
- While the ortho-borylation of N-alkyl-anilines is an unmet challenge in electrophilic borylation it should be noted that a number of phenols have been successfully ortho-borylated under electrophilic C–H borylation conditions, see: X. Tan, X. Wang, Z. H. Li and H. Wang, J. Am. Chem. Soc., 2022, 144, 23286 CrossRef CAS PubMed.
- (a) J. Pahl, E. Noone, M. Uzelac, K. Yuan and M. J. Ingleson, Angew. Chem., Int. Ed., 2022, 61, e202206230 CrossRef CAS PubMed; (b) C. R. P. Millet, J. Pahl, E. Noone, K. Yuan, G. S. Nichol, M. Uzelac and M. J. Ingleson, Organometallics, 2022, 41, 2638 CrossRef CAS PubMed.
- For seminal earlier work on borylation directed borylation to make 9,10-diboranthracenes see: A. John, M. Bolte, H.-W. Lerner and M. Wagner, Angew. Chem., Int. Ed., 2017, 56, 5588 CrossRef CAS PubMed.
- For other recent reviews covering this topic see: (a) G. Berionni, Angew. Chem., Int. Ed., 2022, 61, e202210284 CrossRef CAS PubMed; (b) S. Rej and N. Chatani, Angew. Chem., Int. Ed., 2022, 61, e202209539 CrossRef CAS PubMed.
- A search carried out on 14th August 2023 found >95% purity HNTf2 was available for £139 for 9 mmol on the website of a leading chemical supplier.
- (a) X. Pan, A. Boussonnière and D. P. Curran, J. Am. Chem. Soc., 2013, 135, 14433 CrossRef CAS PubMed; (b) J. E. Radcliffe, V. Fasano, R. W. Adams, P. You and M. J. Ingleson, Chem. Sci., 2019, 10, 1434 RSC.
- C. I. Nieto, D. Sanz, R. M. Claramunt, I. Alkorta and J. Elguero, Coord. Chem. Rev., 2022, 473, 214812 CrossRef CAS.
- Compare ref. 18a and: A. Prokofjevs, A. Boussonnière, L. Li, H. Bonin, E. Lacôte, D. P. Curran and E. Vedejs, J. Am. Chem. Soc., 2012, 134, 12281 CrossRef CAS PubMed.
- For example see: A. Prokofjevs and E. Vedejs, J. Am. Chem. Soc., 2011, 133, 20056 CrossRef CAS PubMed.
- R. A. Kresiński, J. Chem. Soc., Dalton Trans., 1999, 401 RSC.
- V. Bagutski, A. Del Grosso, J. Ayuso Carrillo, I. A. Cade, M. D. Helm, J. R. Lawson, P. J. Singleton, S. A. Solomon, T. Marcelli and M. J. Ingleson, J. Am. Chem. Soc., 2013, 135, 474 CrossRef CAS PubMed.
- See K. Niedenzu and H. Nöth, Chem. Ber., 1983, 116, 1132 CrossRef CAS , and the ESI† to his manuscript for more discussion on isomerism in pyrazaboles..
- E. Hanecker, T. G. Hodgkins, K. Niedenzu and H. Nöth, Inorg. Chem., 1985, 24, 459 CrossRef CAS.
- (a) A. Solovyev, Q. Chu, S. J. Geib, L. Fensterbank, M. Malacria, E. Lacôte and D. P. Curran, J. Am. Chem. Soc., 2010, 132, 15072 CrossRef CAS PubMed; (b) J. H. Muessig, M. Thaler, R. D. Dewhurst, V. Paprocki, J. Seufert, J. D. Mattock, A. Vargas and H. Braunschweig, Angew. Chem., Int. Ed., 2019, 58, 4405 CrossRef CAS PubMed; (c) G. Kundu, V. S. Ajithkumar, K. V. Raj, K. Vanka, S. Tothadi and S. S. Sen, Chem. Commun., 2022, 58, 3783 RSC.
- (a) H. K. Lingam, C. Wang, J. C. Gallucci, X. Chen and S. G. Shore, Inorg. Chem., 2012, 51, 13430 CrossRef CAS PubMed; (b) R. B. Coapes, F. E. S. Souza, M. A. Fox, A. S. Batsanov, A. E. Goeta, D. S. Yufit, M. A. Leech, J. A. K. Howard, A. J. Scott, W. Clegg and T. B. Marder, J. Chem. Soc., Dalton Trans., 2001, 1201 RSC; (c) K. Takrouri, E. Shalom, I. Goldberg, J. Katzhendler and M. Srebnik, J. Organomet. Chem., 2005, 690, 4150 CrossRef CAS.
- B. Wrackmeyer, E. Molla, P. Thoma, E. V. Klimkina, O. L. Tok, T. Bauer and R. Kempe, Z. Anorg. Allg. Chem., 2011, 637, 401 CrossRef CAS.
- A. Osi, D. Mahaut, N. Tumanov, L. Fusaro, J. Wouters, B. Champagne, A. Chardon and G. Berionni, Angew. Chem., Int. Ed., 2022, 61, e202112342 CrossRef CAS PubMed.
- Note, 3b has been previously synthesised via a directed C–H borylation approach using an iridium catalysis and a traceless DG in 40% isolated yield. This required the DG to be installed in a separate step using a Ru based catalyst. See: D. W. Robbins, T. A. Boebel and J. F. Hartwig, J. Am. Chem. Soc., 2010, 132, 4068 CrossRef CAS PubMed.
- See (and references therein): Q.-Z. Li, Z.-Q. Jia, L. Chen, X. Zhang, H.-J. Leng, R. Zeng, Y.-Q. Liu, W.-L. Zou and J.-L. Li, Org. Lett., 2021, 23, 814 CrossRef CAS PubMed.
- See (and references therein) A. Ono, H. Murakami, T. Seto, T. Shimizu, S. Watanabe, S. Takeshita, K. Takeda, J. Toyoshima, I. Nagase, E. Bahceci, M. Morishita, S. Morita, M. Fukuoka and K. Nakagawa, Drugs in R&D, 2021, 21, 65 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2287754–2287757. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc04269c |
| This journal is © The Royal Society of Chemistry 2023 |